

Abstract
Background
Domestication and selection are processes that alter the pattern of within- and between-population genetic variability. They can be investigated at the genomic level by tracing the so-called selection signatures. Recently, sequence polymorphisms at the genome-wide level have been investigated in a wide range of animals. A common approach to detect selection signatures is to compare breeds that have been selected for different breeding goals (i.e. dairy and beef cattle). However, genetic variations in different breeds with similar production aptitudes and similar phenotypes can be related to differences in their selection history.
Methods
In this study, we investigated selection signatures between two Italian beef cattle breeds, Piemontese and Marchigiana, using genotyping data that was obtained with the Illumina BovineSNP50 BeadChip. The comparison was based on the fixation index (Fst), combined with a locally weighted scatterplot smoothing (LOWESS) regression and a control chart approach. In addition, analyses of Fst were carried out to confirm candidate genes. In particular, data were processed using the varLD method, which compares the regional variation of linkage disequilibrium between populations.
Results
Genome scans confirmed the presence of selective sweeps in the genomic regions that harbour candidate genes that are known to affect productive traits in cattle such as DGAT1, ABCG2, CAPN3, MSTN and FTO. In addition, several new putative candidate genes (for example ALAS1, ABCB8, ACADS and SOD1) were detected.
Conclusions
This study provided evidence on the different selection histories of two cattle breeds and the usefulness of genomic scans to detect selective sweeps even in cattle breeds that are bred for similar production aptitudes.
Electronic supplementary material
The online version of this article (doi:10.1186/s12711-015-0128-2) contains supplementary material, which is available to authorized users.
Background
After domestication, natural and artificial selection have led to different animal strains. During the industrial revolution (200 to 250 years ago), these animal strains were artificially clustered into breeds based on their phenotypic characteristics and the environmental conditions in which they are raised. Animals have been selected for traits that are important for various human communities [1, 2]. Domestication and subsequent natural and artificial selection have changed the frequency of mutations that affect phenotypic traits [3]. Understanding how and where selection has shaped the patterns of genetic variation remains one of the most challenging topics in genetics [3]. Thanks to their great phenotypic variability, domestic animals offer the opportunity to explore genotype-phenotype relationships and represent excellent models for studies on evolutionary biology [4–6]. According to the theory of genetic hitch-hiking [7], when the favourable allele of a gene spreads in a population, the sequences that are upstream and downstream of this gene also undergo an increase in frequency until fixation [8]. The integration of disciplines such as quantitative genetics and population genetics has made the study of species’ variability easier and more reliable [5]. For example, association and hitch-hiking mapping allow the identification of quantitative trait nucleotides (QTN) that are responsible for differences in economically important traits [9].
The recent advances in genetics and statistical methodologies contribute to the characterization of biological diversity, animal domestication, and breed development [4]. In particular, the availability of high-density single nucleotide polymorphism (SNP) panels provides an exciting opportunity to identify genomic regions under selection [10]. The abundance of SNPs throughout the genome makes these genetic makers particularly suitable for the detection of genomic regions where a reduction in heterozygosity (selective sweep) occurred [11]. In mammals, several methods based on the measurement of differences in allele frequency, and on linkage disequilibrium (LD) patterns and haplotype structures have been used to examine selective sweeps or patterns of diversity [12–16]. In population genetics, the most commonly used statistics to detect signatures of selection are the calculation of the fixation index (Fst) [17], composite log likelihood (CLL) [18] and extended haplotype homozygosity (EHH) [19]. Recently, in humans, intra- and inter-population genetic diversity was investigated by comparing continuous stretches of diploid DNA sequences that are identical on each strand [20] and that are called runs of homozygosity (ROH) [21].These methods can help to identify genomic regions that have undergone selection (natural or artificial) and to detect associations between traits of economic interest and genes present in these regions. In cattle, most studies have compared breeds with different production aptitudes, for example dairy and beef breeds [18, 22, 23], in order to detect signatures of selective breeding [22, 24, 25]. As expected, these comparisons highlighted genes that have a huge effect on phenotypes (i.e. DGAT1 or ABCG2 for dairy and MSTN for beef cattle, respectively). However, there is a wide range of bovine breeds with different selection histories some of which have the same production aptitudes. For example, some dairy breeds have been selected mainly to improve milk yield, whereas other breeds have been privileged for milk composition or functional traits. Therefore, studies that compare breeds with similar production aptitudes [13, 26–29] can be considered highly informative to investigate their genetic variability for breeding purposes. Here, we studied the genetic differences between two Italian beef cattle breeds, Piemontese and Marchigiana. These two populations exhibit similar morphological and productive traits, but have different origins and selection histories. We analyzed the genetic variation between these two breeds that were genotyped with the Illumina BovineSNP50 BeadChip assay (http://www.illumina.com) by comparing the SNP allele frequencies. We used smoothed fixation indices (Fst) [30] and their interpretation followed the approach of Pintus et al. [23]. In order to confirm the results obtained with the LOWESS/control chart procedure, we performed an analysis with the varLD software that measures the genetic variability between populations by comparing the differences in regional LD patterns [31].
Methods
Samples, genotyping and data editing
A total of 364 Piemontese and 410 Marchigiana bulls were sampled for this study. Animals were genotyped using the Illumina Infinium Bovine BeadChip that includes 54 001 SNPs (http://www.illumina.com). SNPs that were not located on the 29 autosomes of the Bos taurus UMD 3.1/bosTau6 build of the bovine genome assembly were excluded. Quality controls removed SNPs that were monomorphic in both breeds and that had more than 2.5% missing data or a minor allele frequency less than 1%. Missing data were replaced with the most frequent allele at that specific locus for each breed. After quality control, 43 009 SNPs were retained for the analysis.
Detection of relevant signals using the LOWESS/control chart procedure
First, allele frequencies and observed and expected heterozygosities were calculated separately for each breed. Then, total allele frequencies at each locus, fp and fq, were calculated by considering all animals as a single population as follows:
where fPm and fMa and nPm and nMa are allele frequencies and number of individuals in the Piemontese and Marchigiana breeds, respectively.
Expected heterozygosity in the populations (Hs) and overall heterozygosity (Ht) were calculated. Finally, Fst was calculated according to Weir and Cockerham [30] as:
Fst computation generates Fst data patterns along the chromosome that are usually highly variable and thus difficult to interpret.
In this study, in order to smooth Fst data patterns and to simplify graphical presentations, values were fitted with a locally weighted scatterplot smoothing (LOWESS) regression [32], separately for each autosome, using the PROC LOESS procedure in SAS 9.2 (SAS/STAT® Software version 9.2, SAS Institute, Inc., Cary, NC, USA) as suggested by Pintus et al. [23]. The number of local regressions varied between chromosomes due to differences in chromosome length. The data interval included in the analysis was defined by the smoothing parameter S used for the LOWESS regression [33]. We applied the same smoothing parameter as applied in [23] that corresponds to an interval of 20 SNPs for each separate regression. Additional file 1: Table S1 contains the different smoothing parameters that were used in the analysis for each chromosome.
Since Fst values that deviate from the average pattern can be considered as signatures of selection, LOWESS-smoothed data were analysed using a control chart approach. Control charts are graphically represented as a flow of data between two control limits. In this study, LOWESS-smoothed Fst values were plotted against their position along the chromosome and the limits of the control chart were set to three standard deviations from the mean Fst value. LOWESS-smoothed Fst values that exceeded these limits were flagged as outlier signals.
Detection of relevant signals using the varLD software
In order to confirm the results obtained with the LOWESS/control chart procedure, the varLD method was applied to our dataset according to Teo et al. [34]. We chose this method because it is based on assumptions that differ from those of the LOWESS/control chart approach and it has already been used to detect selection signatures in cattle [35]. In particular, it is based on LD and compares patterns of LD across populations. Data editing parameters (minor allele frequency (MAF) and % of missing data) and haplotype length (number of SNPs) for the VarLD method were set equal to those of the LOWESS procedure.
Annotation and functional analysis
Annotated genes in the genomic regions that corresponded to peaks that were above the upper limit of the control chart were identified from the UCSC Genome Browser Gateway (http://genome.ucsc.edu./) and National Centre for Biotechnology Information (NCBI) (www.ncbi.nlm.nih.gov) databases. Intervals of 0.25 Mb upstream and downstream of each significant SNP were considered. Gene-specific functional analyses were performed by GeneCards (www.genecards.org) and NCBI databases consultation. In addition, functional interactions between proteins that were encoded by some of the candidate genes were investigated using the database STRING 9.0 of functional protein association networks (http://string-db.org/) [36]. Finally, to investigate the biological function of each annotated gene (and related proteins) contained in the significant genomic regions, an accurate literature search was conducted. Gene names and symbols were derived from the HUGO Gene nomenclature database (www.genenames.org).
Results
Detection of selection signatures using control chart and varLD approaches
The average genome-wide observed heterozygosity was lower for the Marchigiana breed (0.327) than for the Piemontese breed (0.346). The largest and smallest differences between the two breeds were observed on BTA25 (0.033) (BTA for Bos taurus chromosome) and BTA2 (0.009), respectively (Fig. 1). The overall genetic differentiation between the two breeds was weak (mean Fst = 0.0285 ± 0.004 SD). This value suggests that 2.85% of the genetic variation observed in the sample is explained by population differences, whereas the remaining (97.15%) is due to individual differences within the population.
Fig. 1.
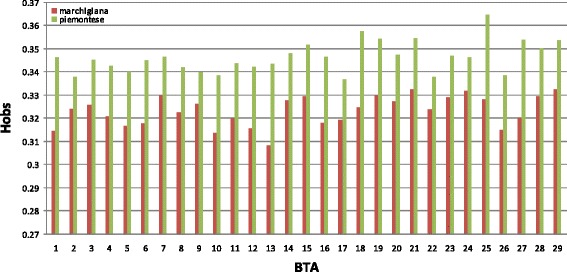
Comparison of average heterozygosity (Hobs) per chromosome (BTA) between the two breeds. Green = Piemontese and red = Marchigiana
The LOWESS/control chart analysis detected 138 outliers in the whole genome. Additional file 2: Figure S1 shows that the largest number of significant peaks (n = 10) was found on BTA6 whereas on BTA25, 28 and 29, only one peak was detected. No significant peak was observed on BTA27. Moreover, several intriguing peaks on BTA2, 5, 8, 9, 12, 14, 23, 26 and 29 were detected [See Additional file 2: Figure S1] but because they did not exceed the upper limit of the control chart they were considered as borderline. Figure 2 shows an example of a borderline peak on BTA26 at about 7 Mb. The varLD method (Fig. 3) detected 67 significant outlier SNPs at the genome-wide level whereas less than half of these were found by the LOWESS/control chart approach. The maximum number of significant SNPs observed with varLD was on BTA1 (n = 4) followed by BTA2 to 11 with three, BTA12 to 24 and BTA26 and 29 with two and BTA25, 27 and 28 with one significant SNP. A total of 933 and 189 annotated genes, derived from the bovine genome assembly (Bos taurus UMD 3.1/bosTau6) UCSC Genome Browser Gateway and NCBI databases, were detected in the genomic regions that surrounded peaks that exceeded the upper limit of the control chart [See Additional file 3: Table S2] and varLD method, respectively [See Additional file 4: Table S3].
Fig. 2.
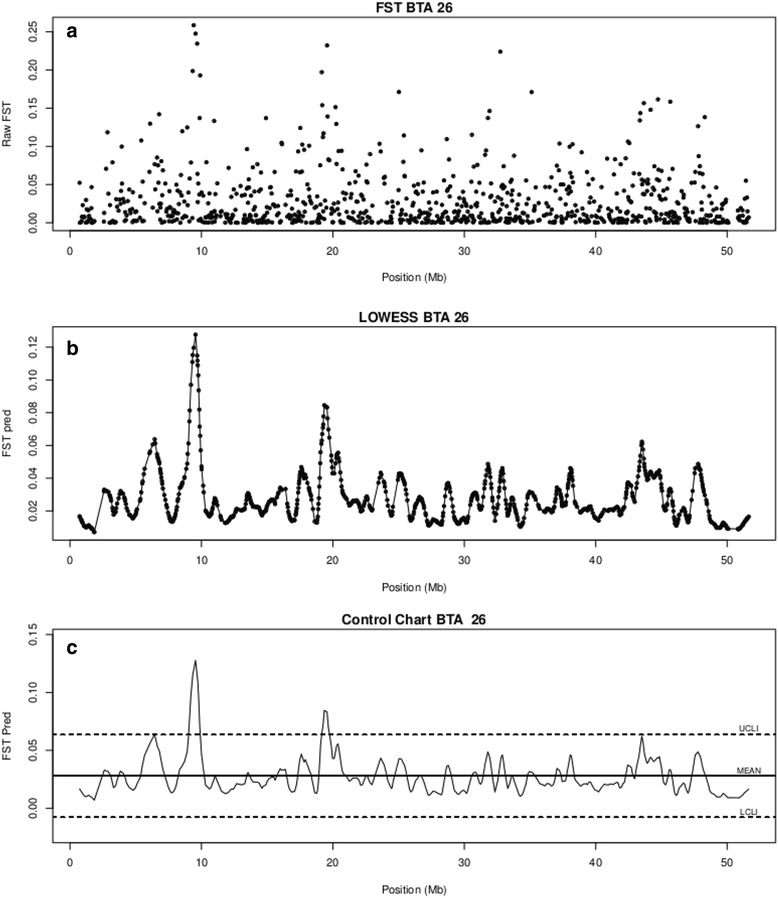
a Pattern of raw Fst data calculated for SNPs located on BTA 26. b Predicted Fst values for SNPs located on BTA 26 using the LOWESS regression with the smoothing parameter set at 0.022. c Control chart of predicted Fst values for BTA 26. Solid line = mean, dotted lines are upper (UCLI) and lower (LCLI) control limits. These control limits are three standard deviations apart from the mean value. The borderline peak is at about 7 Mb
Fig. 3.
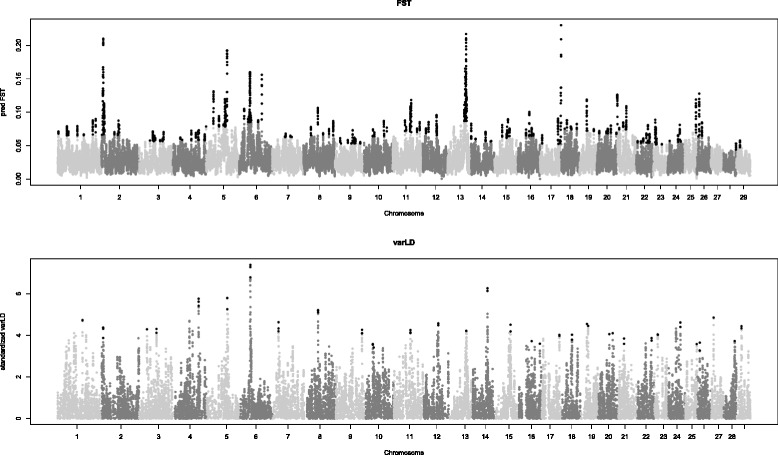
Comparison between genome-wide Fst and varLD analysis for the two breeds. Manhattan plots demonstrate the presence of significant signals in the same regions on several BTA chromosomes between genome-wide Fst and varLD analyses. Black dots represent significant signals with whole-genome significance thresholds set at three standard deviations apart from the mean value
Identification of genes that are known to affect bovine production traits using control chart and varLD approaches
The reliability of the LOWESS/control chart analysis was confirmed by the detection of outlier signals that were located in the genomic regions that contain genes known to affect production traits in cattle. Among these genes, MSTN on BTA2, ABCG2 on BTA6, DGAT1 on BTA14 and FTO on BTA18 should be noted (Table 1). Analyses of the genome-wide Manhattan plots of Fst and LD showed that, overall, the results obtained with the varLD and the LOWESS/control chart approaches were comparable (Fig. 3). Overlapping outlier signals were detected on eight autosomes by both methods. For example, both procedures detected significant outlier signals on BTA2 and 6 [See Additional file 5: Figure S2] in regions that contain annotated genes known to affect bovine production traits (Table 1). Furthermore, significant signals that were detected by both methods were identified at the same positions on BTA4, 13, 17, 19, 25 and 26. Obviously, both methods identified the same annotated genes in these regions (Table 2). Moreover, eight chromosomes (BTA5, 9, 11, 12, 15, 18, 21 and 23) showed peaks at positions that did not correspond exactly in terms of base pairs, but were located within the same autosomal region as several annotated genes known to affect bovine production traits [See Additional file 5: Figure S2]. For 13 chromosomes, no common outlier signals were detected with both methods. Finally, regarding BTA27, no significant signal was detected using the smoothed Fst, whereas one significant SNP at 8.4 Mb was observed with the regional LD variation method but there was no annotated gene in the 0.5 Mb region that surrounded that SNP (see Methods section). Moreover, BTA1, 8, 14 and 28 exhibited significant signals [See Additional file 5: Figure S2] in regions for which no annotated gene was found.
Table 1.
Genes known to affect bovine traits identified by the control chart approach
| BTA | Position (bp) | Gene symbol and name |
|---|---|---|
| 2 | 6213566-6220196 | MSTN myostatin |
| 6 | 37959536-38030586 | ABCG2 ATP-binding cassette, subfamily G (white), member 2 |
| 10 | 37829007-37885645 | CAPN3 calpain 3 |
| 13 | 66863225-66872531 | GHRH growth hormone releasing hormone |
| 14 | 1795425-1804838 | DGAT1 diacylglycerol O-acyltransferase 1 |
| 18 | 22118201-22541539 | FTO fat mass and obesity associated |
Table 2.
List of common candidate genes that were detected by both the control chart and varLD methods
| BTA | Position (Mb) | Gene symbol |
|---|---|---|
| 2 | 5.6 to 5.8 | NABP1, INPP1 |
| 4 | 92.2 to 92.3 | GRM8, ZNF800, MIR592 |
| 6 | 37.9 to 38.1 | HERC6, PYURF, NAP1L5, HERC5, FAM13A, PPM1K, ABCG2, PKD2, IBPS, MEPE |
| 13 | 63.2 to 63.6 | SNTA1, E2F1, ZNF341, CHMP4B, PXMP4, NECAB3 |
| 17 | 64.8 to 65.0 | SIRT4, RPLP0, RAB35, PLA2G1B, ALDH2, COX6A1, GATC, ACADS, POP5, RNF10, MAPKAPK5, TRIAP1, SRSF9,DYNLL1, MLEC,UNC119B, CABP1 |
| 18 | 35.7 to 35.8 | CDH3 |
| 19 | 28.4 | PER1, HSE, ALOX15B, GUCY2D, PFAS, RANGRF, RPL26, KCNAB3, TMEM, CYB5D1, TRAPCC1, VAMP2, AURKB, ARHGEF15, ODF4 |
| 25 | 41.0 to 41.1 | CARD11, GNA12 |
| 26 | 9.5 to 9.8 | PAPSS2, ATAD1, RNSL |
Detection of putative candidate genes using control chart and varLD methods
Using the LOWESS/control chart approach at the whole-genome level led to the identification of several candidate genes that are involved in numerous biological processes, i.e. lipid and carbohydrate metabolisms (ACAD11, ACADVL, ADIG, DGAT2, ALDOB, HADH, GAA and GPT), reproduction (DNAH2, AMHR2, ABCD3, SPEM1, ZAR1 and SHBG), cartilage/bone morphogenesis (LECT2, FLNB, CRTAC1, GDF5, RARG, UQCC, TGFB and GLOI) and biology of the muscle (PVALB, MYL9, FBXO32 and CHN2) (Table 3). Several apoptosis regulatory genes (APAF1, CARD11, CARD14 and WDR92) (Table 3) and several genes that are involved in immune functions and encode proteins that are active in the immune and acute inflammatory responses (DEFB, CXCL12, PROC and PROCR) (Table 3) were detected. Three signatures of positive selection were identified in regions that contained genes that play a role in heme biosynthesis and transport i.e. NRF1 on BTA4 at 94 Mb, ABCB8, ABCF2 and SMARCD3 on BTA4 at 114 Mb, and ALAS1 on BTA22 at 49 Mb. [See Additional file 3: Table S3]. Two genes that are involved in the response to oxidative stress (SOD1 on BTA1 and VNN2 on BTA9) were detected. Moreover, four different gene families i.e. ALOX and MYH on BTA19, HB on BTA15, and KRT on BTA5 were also highlighted. Finally, many genes associated with neurological development and behavioural disorders were pinpointed (CACNG2, CALN1, ACCN3, EFNB3, DLGAP1 and ATP1B2). A complete list of the genes identified by the control chart approach for all 29 bovine autosomes is in Table 3. Using the varLD approach, many candidate genes were detected, among which APOL3 on BTA5 (between 74 974 805 and 74 986 756 bp) and LCAT on BTA18 were the most interesting.
Table 3.
List of candidate genes identified based on control chart outliers values
| BTA | Gene symbol | Gene name | Biological function |
|---|---|---|---|
| 1 | ACAD11 | acyl-CoA dehydrogenase family, member 11 | lipid metabolism |
| CHST2 | carbohydrate (N-acetylglucosamine-6-O) sulfotransferase 2 | cartilage morphogenesis | |
| SOD1 | superoxide dismutase 1, soluble | oxidative stress | |
| 2 | PROC | protein C (inactivator of coagulation factors Va and VIIIa) | immune response |
| 3 | ABCD3 | ATP-binding cassette, sub-family D (ALD), member 3 | lipid metabolism |
| 4 | ABCB8 | ATP-binding cassette, sub-family B (MDR/TAP), member 8 | transport of heme |
| ABCF2 | ATP-binding cassette, sub-family F (GCN20), member 2 | transport of heme | |
| CHN2 | chimerin 2 | smooth muscle cell proliferation | |
| CHPF2 | chondroitin polymerizing factor 2 | cartilage biosynthesis | |
| TWIST1 | twist family bHLH transcription factor 1 | bone and muscle development | |
| NRF1 | nuclear respiratory factor 1 | heme biosynthesis/transport | |
| ACCN3 | acid-sensing (proton-gated) ion channel 3 | Sensory neuron physiology pain induced by acidosis | |
| SMARCD3 | SWI/SNF related, matrix associated, actin dependent regulator of chromatin, subfamily d, member 3 | heme biosynthesis/transport | |
| 5 | CACNG2 | calcium channel, voltage-dependent, gamma subunit 2 | behavioural disorders |
| CALCOCO1 | calcium binding and coiled-coil domain 1 | cell growth | |
| CHST11 | carbohydrate (chondroitin 4) sulfotransferase 11 | cartilage biosynthesis | |
| RARG | retinoic acid receptor, gamma | skeletal development | |
| PVALB | parvalbumin | muscle relaxation | |
| APAF1 | apoptotic peptidase activating factor 1 | apoptosis | |
| SOAT2 | sterol O-acyltransferase 2 | cholesterol metabolism | |
| KRTs | keratins | epithelia development | |
| AMHR2 | Anti-Mullerian hormone receptor, type II | male sex differentiation | |
| 6 | GBA3 | glucosidase, beta, acid 3 (gene/pseudogene) | flavonoids metabolism |
| HADH | hydroxyacyl-CoA dehydrogenase | lipid metabolism | |
| ZAR1 | zygote arrest 1 | reproduction | |
| 7 | LECT2 | leukocyte cell-derived chemotaxin 2 | cartilage/bone differentiation |
| TGFBI | transforming growth factor, beta-induced, 68kDa | cartilage/bone development | |
| 8 | ALDOB | aldolase B, fructose-bisphosphate | carbohydrates metabolism |
| LOXL2 | lysyl oxidase-like 2 | biogenesis of connective | |
| 9 | CTGF | connective tissue growth factor | connective morphogenesis |
| VNN2 | vanin 2 | stress oxidative response | |
| 11 | PROKR1 | prokineticin receptor 1 | Ca+2 mobilization |
| WDR92 | WD repeat domain 92 | apoptosis modulator | |
| 13 | UQCC | ubiquinol-cytochrome c reductase complex assembly factor 1 | skeletal development |
| MYL9 | myosin, light chain 9, regulatory | muscle metabolism | |
| DEFBs | defensin beta | immune response | |
| GDF5 | growth differentiation factor 5 | skeletal development | |
| ACSS2 | acyl-CoA synthetase short-chain family member 2 | lipid metabolism | |
| ADIG | adipogenin | adipocyte development | |
| PROCR | protein C receptor, endothelial | immune response | |
| BPIFB2 BPIF 3 BPIF6 | BPI fold containing family B, members 2, 3 and 6 | innate immune response | |
| 14 | FBXO32 | F-box protein 32 | muscular diseases |
| GPT | glutamic-pyruvate transaminase (alanine aminotransferase) | liver gluconeogenesis | |
| 15 | DGAT2 | diacylglycerol O-acyltransferase 2 | lipid metabolism |
| TUB | tubby bipartite transcription factor | obesity | |
| HBs | haemoglobins | oxygen binding/transport | |
| 16 | ACBD3 | acyl-CoA binding domain containing 3 | reproduction |
| DHRS3 | dehydrogenase/reductase (SDR family) member 3 | steroids metabolism | |
| PLOD1 | procollagen-lysine, 2-oxoglutarate 5-dioxygenase 1 | connective synthesis | |
| 17 | ACADS | acyl-CoA dehydrogenase, C-2 to C-3 short chain | lipid metabolism |
| 18 | ACSF3 | acyl-CoA synthase family member 3 | lipid metabolism |
| MVD | mevalonate (diphospho) decarboxylase | cholesterol biosynthesis | |
| 19 | SPEM1 | spermatid maturation 1 | reproduction |
| SHBG | sex hormone-binding globulin | steroids metabolism | |
| DNAH2 | dynein, axonemal, heavy chain2 | sperm motility | |
| GAA | glucosidase, alpha; acid | carbohydrates metabolism | |
| ATP1B2 | ATPase, Na+/K+ transporting, beta 2 polypeptide | osmoregulation | |
| ACADVL | acyl-CoA dehydrogenase, very long chain | lipid metabolism | |
| ALOXs | arachidonate-lipoxygenases | lipid metabolism | |
| MYHs | myosin, heavy chain skeletal muscle | muscle biology | |
| CARD14 | caspase recruitment domain family, member 14 | apoptosis | |
| EFNB3 | ephrin B3 | brain development | |
| ATP1B2 | ATPase, Na/K transporting beta 2 polypeptide | neurite outgrowth | |
| 22 | PDHB | pyruvate dehydrogenase (lipoamide) beta | tricarboxylic acid cycle |
| FLNB | filamin B, beta | cartilage/bone morphogenesis | |
| TLR9 | toll-like receptor 9 | immune response | |
| OXTR | oxytocin receptor | parturition | |
| CAV3 | caveolin 3 | muscle biology | |
| ALAS1 | aminolevulinate, delta-, synthase 1 | biosynthesis of heme | |
| ACOX2 | acyl-CoA oxidase 2, branched chain | lipid metabolism | |
| 23 | MLN | motilin | control of peristalsis |
| GLO1 | glyoxalase | osteoclastogenesis | |
| 24 | DLGAP1 | disc, large (Drosophila) homolog-associated protein 1 | postsynaptic scaffold in neuronal cell |
| 25 | CARD11 | caspase recruitment domain family, member 11 | apoptosis |
| CALN1 | calneuron 1 | memory and learning | |
| 26 | LOXL4 | lysyl oxidase-like 4 | biogenesis of connective tissue |
| CRTAC1 | cartilage acidic protein 1 | cartilage production | |
| 28 | CXCL12 | chemokine (C-X-C motif) ligand 12 | immune response |
Moreover, several members of the LT/LBP gene family were identified by both methods on BTA13 with seven isoforms detected by varLD (BPIFA2A, BPIFA2C, BPIFA2B, BPIFA3, BPIFA1, BPIFB1 and BPIFB5) and three by control chart (BPIFB2, BPIFB6 and BPIFB3).
Discussion
The two bovine breeds, Piemontese and Marchigiana, that were included in this study share similar phenotypic and production characteristics, but have different origins and breeding histories [1, 37]. The Piemontese breed is raised in Northern Italy and derives mainly from Bos brachyceros [38]. In the past, it was considered as a triple aptitude breed (draught, milk and meat) and, over time, its ability to produce meat has improved while maintaining a high level of milk production. The Marchigiana breed is raised in Central Italy and its origin can be traced backed to Bos primigenius [37]. Initially, it was considered as a dual-purpose breed (draught and meat) but because of its high capacity for muscle growth, it has become specialized as a beef breed. It was recognized as a separate breed at the beginning of the last century when crosses with Chianina bulls were stopped.
In our study, the Marchigiana breed showed a slightly lower level of chromosome heterozygosity than the Piemontese breed (Fig. 1), which confirms the findings reported by Bozzi et al. [39] who analyzed the genetic diversity of several beef cattle breeds and found that the Marchigiana breed had the lowest level of genetic diversity. This is probably due to the breeding policy conducted by farmers, but also to the small size of the breed. The genetic distances between the Piemontese and Marchigiana breeds that we report here are compatible with data from the literature. In fact, several studies showed that cattle breeds with similar production aptitudes display some genetic variability [40–45]. In our case, although the Piemontese and Marchigiana breeds share similar phenotypic and production characteristics, the genetic architectures of the traits differ. Indeed, we identified genes that are usually found when breeds with divergent artificial selection are compared (DGAT1 on BTA14 and ABCG2 on BTA6) [22, 46], but also polymorphisms in genes that control phenotypes that are specific to each breed (MSTN and FTO) (Table 1).
Another interesting finding of our study on these two breeds is the identification of selection signatures in genomic regions that are known to harbour candidate genes for dairy traits, such as DGAT1 and ABCG2. However, some authors suggested that these genes may also have a role in beef cattle breeds [47–49]. Different causative polymorphisms may explain the detection of DGAT1 and ABCG2. Regarding DGAT1, allele K is fixed in the Piemontese breed whereas allele A is most probably present in the Marchigiana breed [50] since in the past it was crossed with the Chianina breed that possesses this allele. Finally, Ron et al. [51] showed that the polymorphism of the ABCG2 gene differs among beef cattle breeds.
The detection of MSTN was rather unexpected in this comparison between the Piemontese and Marchigiana breeds. Two possible explanations are: (1) muscular hypertrophy in each of these breeds is caused by different mutations in the MSTN gene, i.e. a G > T transversion that introduces an early stop codon in the third exon of the gene [52] is present in the Marchigiana breed whereas a G > A transition in the same exon [53] is found in the Piemontese breed; or (2) the mutation responsible for muscle hypertrophy is fixed in the Piemontese breed [54] while its frequency is low in the Marchigiana breed [52] because of different selection strategies applied in each breed. The two candidate genes, UQCC and GDF5 detected on BTA13 contain polymorphisms that are associated with stature in humans [55] and with body size determination in European Bos taurus cattle [56] (Table 4). In general, Piemontese bulls are smaller (height = 135 cm, and weight = 850 kg) (www.anaborapi.it) than many other beef breeds [57], including the Marchigiana breed (height = 140 cm, and weight = 1.200 kg) (www.anabic.it). Moreover, the GHRH gene, which is responsible for the release of growth hormone, is located in the same region of BTA13 (Table 1), which is consistent with the identification of a QTL (quantitative trait locus) in this chromosomal region in a genome-wide association study on several beef cattle breeds [58].
Table 4.
Genes detected in this work and previously reported by other authors for beef cattle
A primary goal for livestock industry is to produce high-quality meat for human consumption. Thus, it is necessary to know and understand which factors influence the transformation of muscle to meat, which depends mainly on the intrinsic properties of the myofibers (type, composition and size), enzymatic proteolytic activities (cathepsin and calpain-calpastatin system) [59, 60] but also on structural features such as connective tissue and intramuscular fat deposition [61]. It is generally recognized that the process of meat tenderization is the result of biochemical reactions that involve both proteolytic and apoptotic pathways [60, 61]. In our study, we identified several genes that are involved in the positive regulation of cellular apoptosis in skeletal muscle such as APAF1, CARD11, CARD14 and WDR92. Numerous genes that have a role in lipid metabolism (ACAD11, ABCD3, HADH, ACSS2, DGAT2 and ACADS), cholesterol and steroids synthesis (SOAT2, DHRS3, SHBG and MVD), and in the biology of adipose tissue (ADIG and TUB) were detected. One possible explanation may be related to the distinctive metabolism of adipose tissue in the Piemontese breed compared to other beef breeds. A comparative analysis of young bulls from various European breeds (including Marchigiana) showed that the Piemontese breed had the lowest scores for carcass traits such as fatness and fat percentage [62]. Another comparison between Piemontese, Red Angus and Gelbvieh breeds revealed that the Piemontese breed also had the lowest score for fat thickness [63]. Moreover, the cholesterol level of Piemontese meat is lower than that of other cattle breeds (Piemontese Breed Consortium www.coalvi.it) or other livestock species (Piemontese Association of the United States).
The Marchigiana breed is considered as a hardy breed with excellent adaptability to pasture in harsh environments (www.agraria.org). Genes that are involved in the triggering and regulation of innate immune responses such as chemokines (CXCL12) [64], defensins (DEFB119, 122, 122A) and toll-like receptors (TLR9) were detected in our study, which is consistent with the high level of resistance to diseases and endoparasites of this breed. Among these genes, TLR9 was previously reported by Ramey et al. [6] (Table 3) and a comparative analysis of 16 European breeds showed that three TLR genes contained fixed polymorphisms in the Marchigiana breed whereas only rare alleles were found at the same SNPs in the Piemontese breed [65]. Meat quality can be modulated by genes that are involved in acute inflammatory response processes [66]. Cytokines are a large family of soluble molecules (that also include chemokines) that regulate the inflammatory response. Recently, it was shown that genes that control immune and acute stress responses also play a role in the determination of beef tenderness [61, 67, 68]. A comparison between different types of beef cattle revealed that genes that encode immune response proteins are deregulated in situations of stress such as diseases, which suggests that they play an important role in muscle metabolism and influence final beef tenderness [67, 69]. Meat tenderness is measured by the force required to cut through a piece of meat i.e. the greater is the force required, the tougher is the meat. This is known as the Warner-Bratzler shear force test, which is the most popular method to measure the tenderness of the meat [70, 71]. A comparison of the tenderness of the longissimus dorsii muscle from Chianina, Piemontese, Marchigiana, Limousine and Charolais bulls showed that the Warner-Bratzler shear force values were lowest for the Piemontese samples [72]. Most interesting was the identification of genes that play a role in the biology of erythrocytes. Hemoglobin and myoglobin are the two main heme proteins that are responsible for oxygen binding and transport [69]. In this study, six genes (NRF1, SMARCD3, ALAS1, HB, ABCF2 and ABCB8) involved in the metabolism of heme were identified (Table 3). ALAS1 is a housekeeping gene that encodes a mitochondrial enzyme that catalyzes heme biosynthesis. Fig. 4 shows the relationships between bovine ALAS1 and other proteins that were determined by data integration in the STRING 9.0 database. The highest confidence values (0.91 and 0.89) were found between ALAS1 and NRF1 and between ALAS1 and SMARCD3, respectively, which indicate a direct interaction. This is in agreement with Chambaz and co-workers [73] who reported greater heme/iron content in Piemontese meat than in the meat of other European beef cattle breeds. In addition to the genes that control heme content, or modulate the response to various stressors or determine the structure and composition of myofibers, particular attention should be addressed to those that influence oxidative stress. Oxidative stress is considered as a metabolic disturbance that affects the health status but also the quality of the final animal products [74]. Overall, information on the link between oxidative stress and meat quality is scarce and conflicting and, for cattle, there is no direct evidence that the genetic background has an influence on oxidative stress and meat characteristics [75]. Oxidative damage to tissues is prevented by various factors such as non enzymatic antioxidant molecules (vitamins, polyphenols and thiols) and enzymes that are incorporated within the cell membranes [76] with superoxide dismutase, catalase and glutathione peroxidase being the most important antioxidative enzymes [74]. In our study, selective sweeps were highlighted in the chromosomal region that contains the SOD1 gene.
Fig. 4.

Protein network of bovine ALAS1 according to STRING 9.0 action view. Nodes are proteins; lines indicate interactions between proteins with: pink lines for post-translational, yellow lines for expression, black lines for reaction, blue lines for binding and light blue lines for phenotype. Protein interactions include direct (physical) and indirect (functional) associations derived from different sources (genomic context, high through-put experiments, conserved coexpression, previous knowledge). 0.91 and 0.89 are the confidence values for the products of NRF1 and SMARCD3, respectively
The presence of smoothed Fst peaks that reached but did not exceed the upper limit of the control chart (borderline peaks) on several autosomes [See Additional file 2: Figure S1] suggests recent selection. In general, selection acts in genomic regions that have a functional importance, thus, identification of genes that have undergone recent selection or that are currently under selection is relevant [77]. An incomplete selective sweep appears in a population when the frequency of a favourable allele of a gene is increasing but has not reached fixation yet. In addition, Schwarzenbacher et al. [5] and Ramey et al. [6] identified selective sweeps, which, although they had not reached fixation, represented potentially interesting regions for cattle. Our comparison between the LOWESS/control chart and the varLD approaches to detect selection signatures provided concordant results for 16 of the 29 bovine autosomes. Such incomplete agreement is rather frequent in studies on the detection of selection signatures [78]. The LOWESS/control chart method detected more significant markers than the varLD method. In addition, when both methods detected the same chromosomal regions and candidate genes, as for MSTN on BTA2 and ABCG2 on BTA6, some of the significant markers did not coincide. The reasons for these discrepancies are probably due to differences between the two methods, in particular in the metrics used. On the one hand, the LOWESS/control chart approach is based on the analysis of differences in allele frequencies at a single locus, although the LOWESS smoothing corrects for local variation. On the other hand, the varLD method relies on the difference in correlation structures among markers located in the same window between two populations and the SNP that is flagged as the most significant is the SNP that is located nearest to the centre of the window [31]. Therefore, for the same genomic region, the LOWESS/control chart approach can detect a larger number of significant markers than the varLD method. For example, for the region that contains MSTN on BTA2, the LOWESS/control chart approach detected 10 markers (between 4.9 and 6.6 Mb), whereas only three significant SNPs (between 5.6 and 5.8 Mb) were identified by the varLD method. Another discrepancy between the two methods was the different positions of some detected peaks, which may be due to how signals are averaged or smoothed and/or to the rationale used to assess significant markers.
Conclusions
Phenotypic variability is the basis of most comparative studies conducted in all living beings. In this study, we showed that, even when phenotypic diversity is not sufficiently large to be detected, investigating the polymorphisms that are present in the regions of the genome that are involved in breeding traits can be very useful in terms of genetic improvement. Our results highlight interesting genomic differences between two cattle breeds that share the same production aptitudes. These variations were located in regions that contain both genes known to affect production traits (in both beef and dairy cattle) and new candidate genes. In many cases, Fst revealed clear differences but borderline values also flagged regions where selection is currently acting. We detected genes that are involved in different metabolic pathways. This finding confirms the great complexity of the mechanisms that underlie quantitative traits, which can show genetic variation even among breeds that are phenotypically similar. With the aim of increasing both the quality and quantity of meat in bovine breeds, it would be interesting to analyze in detail the genes that we identified (such as SOD1, LOXL2, CAV3, ACADS, CXCL12, MYL9, MVD, TLR9 and ALAS1) in order to include them in selection programs. Therefore, to increase breed performance and reveal how selection shapes the genome, it is essential to dissect the genetic architecture of each population.
Additional files
List of chromosomal specific smoothing parameters S. Table S1 provides the LOWESS smoothing parameters that were calculated for each chromosome.
Patterns of raw Fst data, predicted Fst values and control chart of predicted Fst values on BTA1 to 29. Description: The plots represent the pattern of raw Fst data calculated for SNPs located on each chromosome (BTA), the predicted Fst values for SNPs located on each BTA using the LOWESS regression with chromosomal specific smoothing parameter and the control chart of predicted Fst values for each BTA. Upper control limit (UCLI) and lower control limit (LCLI) are three standard deviations apart from the mean value.
List of 933 genes annotated in cattle detected using the control chart method. Description: This list includes all the bovine annotated genes derived from Bos taurus UMD 3.1/bosTau6 assembly that are present in the 0.5 Mb interval (0.25 Mb upstream and downstream) considered for each significant SNP using the control chart method.
List of 189 genes annotated in cattle detected using varLD. Description: This list includes all the bovine annotated genes derived from Bos taurus UMD 3.1/bosTau6 assembly that are present in the 0.5 Mb interval (0.25 Mb upstream and downstream) considered for each significant SNP using the varLD approach.
Plots obtained with the Fst method vs the varLD method. Description: Chromosome-wide plots obtained in the Fst and varLD analyses for BTA1 to 29. Solid lines represent the threshold set at three standard deviations apart from the mean value.
Acknowledgements
This work was supported by the Italian Ministry of Agriculture (grant INNOVAGEN). The authors wish to acknowledge the Italian Piemontese (ANABORAPI) and Central Italy Beef Cattle (ANABIC) associations.
Footnotes
Competing interests
The authors declare that they have no competing interests.
Authors’ contributions
SS, GM and NPPM designed the experiments. SS, GM, GG, CD and MC analysed data. AV provided genotypes and helped to draft the manuscript. SS and NPPM drafted the manuscript. All authors read and approved the final manuscript.
Contributor Information
Silvia Sorbolini, Email: ssorbolini@uniss.it.
Gabriele Marras, Email: gbmarras@uniss.it.
Giustino Gaspa, Email: gigaspa@uniss.it.
Corrado Dimauro, Email: dimauro@uniss.it.
Massimo Cellesi, Email: mcellesi@uniss.it.
Alessio Valentini, Email: alessio@unitus.it.
Nicolò PP Macciotta, Email: macciott@uniss.it.
References
- 1.Blott SC, Williams JL, Haley CS. Genetic relationships among European cattle breeds. Anim Genet. 1998;29:273–82. doi: 10.1046/j.1365-2052.1998.00327.x. [DOI] [PubMed] [Google Scholar]
- 2.Felius M. Cattle breeds: an encyclopedia. 1. Misset: Doetinchem; 1995. [Google Scholar]
- 3.Andersson L. How selective sweeps in domestic animals provide new insight into biological mechanisms. J Intern Med. 2012;271:1–14. doi: 10.1111/j.1365-2796.2011.02450.x. [DOI] [PubMed] [Google Scholar]
- 4.Wiener P, Wilkinson S. Deciphering the genetic basis of animal domestication. Proc R Soc Lond B Biol Sci. 2011;278:3161–70. doi: 10.1098/rspb.2011.1376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Schwarzenbacher H, Dolezal M, Flisikowski K, Seefried F, Wurmser C, Schlötterer C, et al. Combining evidence of selection with association analysis increases power to detect regions influencing complex traits in dairy cattle. BMC Genomics. 2012;13:48. doi: 10.1186/1471-2164-13-48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ramey HR, Decker JE, McKay SD, Rolf MM, Schnabel RD, Taylor JF. Detection of selective sweeps in cattle using genome-wide SNP data. BMC Genomics. 2013;14:382. doi: 10.1186/1471-2164-14-382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Maynard-Smith J, Haigh J. The hitch-hiking effect of a favourable gene. Genet Res. 1974;23:23–35. doi: 10.1017/S0016672300014634. [DOI] [PubMed] [Google Scholar]
- 8.Przeworski M, Coop G, Wall JD. The signature of positive selection on standing genetic variation. Evolution. 2005;59:2312–23. doi: 10.1554/05-273.1. [DOI] [PubMed] [Google Scholar]
- 9.Ron M, Weller JI. From QTL to QTN identification in livestock-winning points rather than knock-out: a review. Anim Genet. 2007;38:429–39. doi: 10.1111/j.1365-2052.2007.01640.x. [DOI] [PubMed] [Google Scholar]
- 10.Akey JM, Zhang G, Zhand K, Jin L, Shriver MD. Interrogating a high-density SNP map for signatures of natural selection. Genome Res. 2002;12:1805–14. doi: 10.1101/gr.631202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Andersson L, Georges M. Domestic-animal genomics: deciphering the genetics of complex traits. Nat Rev Genet. 2004;5:202–12. doi: 10.1038/nrg1294. [DOI] [PubMed] [Google Scholar]
- 12.Nielsen R, Bustamante CD, Clark AG, Glanowski S, Sackton TB, Hubisz MJ, et al. A scan for positively selected genes in the genomes of humans and chimpanzees. PLoS Biol. 2005;3:e170. doi: 10.1371/journal.pbio.0030170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Flori L, Fritz S, Jaffrézic F, Boussaha M, Gut I, Heath S, et al. The genome response to artificial selection: a case study in dairy cattle. PLoS One. 2009;4:e6595. doi: 10.1371/journal.pone.0006595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Boyko AR, Quignon P, Li L, Schoenebeck JJ, Degenhardt JD, Lohmueller KE, et al. A simple genetic architecture underlies morphological variation in dogs. PLoS Biol. 2010;8:e1000451. doi: 10.1371/journal.pbio.1000451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kijas JW, Townley D, Dalrymple BP, Heaton MP, Maddox JF, McGrath A, et al. A genome wide survey of SNP variation reveals the genetic structure of sheep breeds. PLoS One. 2009;4:e4668. doi: 10.1371/journal.pone.0004668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wilkinson S, Lu ZH, Megens HJ, Archibald AL, Haley C, Jackson IJ, et al. Signatures of diversifying selection in European pig breeds. PLoS Genet. 2013;9:e1003453. doi: 10.1371/journal.pgen.1003453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wright S. Coefficient of inbreeding and relationship. Am Nat. 1922;56:330–8. doi: 10.1086/279872. [DOI] [Google Scholar]
- 18.Stella A, Ajmone-Marsan P, Lazzari B, Boettcher P. Identification of selection signatures in cattle breeds selected for dairy production. Genetics. 2010;185:1451–61. doi: 10.1534/genetics.110.116111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Sabeti PC, Reich DE, Higgins JM, Levine HZP, Richter DJ, Schaffner SF, et al. Detecting recent positive selection in the human genome from haplotype structure. Nature. 2002;419:832–7. doi: 10.1038/nature01140. [DOI] [PubMed] [Google Scholar]
- 20.Gibson J, Morton NE, Collins A. Extended tracts of homozygosity in outbred human populations. Hum Mol Genet. 2006;15:789–95. doi: 10.1093/hmg/ddi493. [DOI] [PubMed] [Google Scholar]
- 21.Ku CS, Naidoo N, Teo SM, Pawtain Y. Regions of homozygosity and their impact on complex diseases and traits. Hum Genet. 2011;129:1–15. doi: 10.1007/s00439-010-0920-6. [DOI] [PubMed] [Google Scholar]
- 22.Hayes BJ, Chamberlain AJ, MacEachern S, Savin K, McPartlan H, MacLeod I. A genome map of divergent artificial selection between Bos taurus dairy cattle and Bos taurus beef cattle. Anim Genet. 2009;40:176–84. doi: 10.1111/j.1365-2052.2008.01815.x. [DOI] [PubMed] [Google Scholar]
- 23.Pintus E, Sorbolini S, Albera A, Gaspa G, Dimauro C, Steri R, et al. Use of locally weighted scatterplot smoothing (LOWESS) regression to study selection signatures in Piedmontese and Italian Brown cattle breeds. Anim Genet. 2014;45:1–11. doi: 10.1111/age.12076. [DOI] [PubMed] [Google Scholar]
- 24.Qanbari S, Pimentel ECG, Tetens J, Thaller G, Lichtner P, Sharifi AR, et al. A genome-wide scan for signatures of recent selection in Holstein cattle. Anim Genet. 2010;41:377–89. doi: 10.1111/j.1365-2052.2009.02016.x. [DOI] [PubMed] [Google Scholar]
- 25.Mancini G, Gargani M, Chillemi G, Nicolazzi EL, Ajmone Marsan P, Valentini A, et al. Signatures of selection in five Italian cattle breeds detected by a 54K SNP panel. Mol Biol Rep. 2014;41:957–65. doi: 10.1007/s11033-013-2940-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Munilla S, Gonzalez-Rodriguez A, Mouresan EF, Canas-Alvarez JJ, Altariba J, Diaz CJ, et al. Selection signatures in autochthonous Spanish cattle breeds using site frequency spectrum statistics. In Proceedings of the 10th World Congress of Genetic Applied to Livestock Production: 17–22 August 2014; Vancouver. 2014:256. https://asas.org/docs/default-source/wcgalp-proceedings-oral/256_paper_9555_manuscript_732_0.pdf?sfvrsn=2.
- 27.Yang S, Li X, Fan B, Tang Z. A genome scan for signatures of selection in Chinese indigenous and commercial pig breeds. BMC Genet. 2014;15:7. doi: 10.1186/1471-2156-15-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ryu J, Lee C. Identification of contemporary selection signatures using composite likelihood and their associations with marbling score in Korean cattle. Anim Genet. 2014;45:765–70. doi: 10.1111/age.12209. [DOI] [PubMed] [Google Scholar]
- 29.Porto-Neto LR, Lee SH, Sonstegard T, van Tassel C, Lee HK, Gibson JP, et al. Genome-wide detection of signatures of selection in Korean Hanwoo cattle. Anim Genet. 2014;45:180–90. doi: 10.1111/age.12119. [DOI] [PubMed] [Google Scholar]
- 30.Weir BS, Cockerham CC. Estimating F-statistics for the analysis of population structure. Evolution. 1984;38:1358–70. doi: 10.2307/2408641. [DOI] [PubMed] [Google Scholar]
- 31.Ong RT, Teo YY. varLD a program for quantifying variation in linkage disequilibrium patterns between populations. Bioinformatics. 2010;26:1269–70. doi: 10.1093/bioinformatics/btq125. [DOI] [PubMed] [Google Scholar]
- 32.Cleveland WS. Robust locally weighted fitting and smoothing scatterplots. J Am Stat Ass. 1979;74:829–36. doi: 10.1080/01621459.1979.10481038. [DOI] [Google Scholar]
- 33.Choen RA. An introduction to PROC LOESS for local regression. Cary: SAS Institute INC; 1999. [Google Scholar]
- 34.Teo YY, Fry AE, Bhattacharya K, Small KS, Kwiatkowski DP, Clark TG. Genome-wide comparisons of variation in linkage disequilibrium. Genome Res. 2009;19:1849–60. doi: 10.1101/gr.092189.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Perez O’Brien AM, Utsunomiya YT, Meszaros G, Bickhart DM, Liu GE, Van Tassel CP, et al. Assessing signatures of selection through variation in linkage disequilibrium between taurine and indicine cattle. Genet Sel Evol. 2014;46:19. doi: 10.1186/1297-9686-46-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Szklarczyk D, Franceschini A, Kuhn M, Siminovic M, Roth A, Minguez P, et al. The STRING database in 2011: functional interaction networks of proteins, globally integrated and scored. Nucleic Acids Res. 2011;39(Database issue):D561–8. doi: 10.1093/nar/gkq973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ciampolini R, Moazami-Goudarzi K, Vaiman D, Dillmann C, Mazzanti E, Foulley JL, et al. Individual multilocus genotypes using microsatellite polymorphisms to permit the analysis of the genetic variability within and between Italian beef cattle breeds. J Anim Sci. 1995;73:3259–68. doi: 10.2527/1995.73113259x. [DOI] [PubMed] [Google Scholar]
- 38.Moioli B, Napolitano F, Catillo G. Genetic diversity between Piedmontese, Maremmana, and Podolica cattle breeds. J Hered. 2004;95:250–6. doi: 10.1093/jhered/esh032. [DOI] [PubMed] [Google Scholar]
- 39.Bozzi R, Franci O, Forabosco F, Pugliese C, Crovetti A, Filippini F. Genetic variability in three Italian beef cattle breeds derived from pedigree information. Ital J Anim Sci. 2006;5:129–37. [Google Scholar]
- 40.Kantanen J, Olsaker I, Holm LE, Lien S, Vilkki J, Brusgaard K, et al. Genetic diversity and population structure of 20 North European cattle breeds. J Hered. 2000;91:446–57. doi: 10.1093/jhered/91.6.446. [DOI] [PubMed] [Google Scholar]
- 41.Cañón J, Alexandrino P, Bessa I, Carleos C, Carretero Y, Dunner S, et al. Genetic diversity measures of local European beef cattle breeds for conservation purposes. Genet Sel Evol. 2001;33:311–32. doi: 10.1186/1297-9686-33-3-311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Maudet C, Luikart G, Taberlet P. Genetic diversity and assignment tests among seven French cattle breeds based on microsatellite DNA analysis. J Anim Sci. 2002;80:942–50. doi: 10.2527/2002.804942x. [DOI] [PubMed] [Google Scholar]
- 43.Ciampolini R, Cetica V, Ciani E, Mazzanti E, Fosella X, Marroni F, et al. Statistical analysis of individual assignment tests among four cattle breeds using fifteen STR loci. J Anim Sci. 2006;84:11–9. doi: 10.2527/2006.84111x. [DOI] [PubMed] [Google Scholar]
- 44.Li MH, Kantanen J. Genetic structure of Eurasian cattle (Bos taurus) based on microsatellites: clarification for their breed classification. Anim Genet. 2010;41:150–8. doi: 10.1111/j.1365-2052.2009.01980.x. [DOI] [PubMed] [Google Scholar]
- 45.Amigues Y, Boitard S, Bertrand C, SanCristobal M, Rocha D. Genetic characterization of the Blonde d’Aquitaine cattle breed using microsatellite markers and relationship with three other French cattle populations. J Anim Breed Genet. 2011;128:201–8. doi: 10.1111/j.1439-0388.2010.00890.x. [DOI] [PubMed] [Google Scholar]
- 46.Qanbari S, Gianola D, Hayes B, Schenkel F, Miller S, Moore S, et al. Application of site haplotype-frequency based approaches for detecting selection signatures in cattle. BMC Genomics. 2011;12:318. doi: 10.1186/1471-2164-12-318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Gorlov IF, Fedunin AA, Randelin DA, Sulimova GE. Polymorphisms of bGH, RORC, and DGAT1 genes in Russian beef cattle breeds. Russian J Genet. 2014;50:1302–7. doi: 10.1134/S1022795414120035. [DOI] [PubMed] [Google Scholar]
- 48.Abo-Ismail MK, Vander Voort G, Squires JJ, Swanson KC, Mandell IB, Liao X, et al. Single nucleotide polymorphisms for breed efficiency and performance in crossbred beef cattle. BMC Genet. 2014;15:14. doi: 10.1186/1471-2156-15-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Yuan Z, Li J, Li J, Gao X, Gao H, Xu S. Effects of DGAT1 gene on meat and carcass fatness quality in Chinese commercial cattle. Mol Biol Rep. 2013;40:1947–54. doi: 10.1007/s11033-012-2251-2. [DOI] [PubMed] [Google Scholar]
- 50.Kaupe B, Winter A, Fries R, Erhardt G. DGAT1 polymorphism in Bos indicus and Bos taurus cattle breeds. J Dairy Res. 2004;71:182–7. doi: 10.1017/S0022029904000032. [DOI] [PubMed] [Google Scholar]
- 51.Ron M, Cohen-Zinder M, Peter C, Weller JI, Erhardt G. A polymorphism in ABCG2 in Bos indicus and Bos taurus cattle breeds. J Dairy Sci. 2006;89:4921–3. doi: 10.3168/jds.S0022-0302(06)72542-5. [DOI] [PubMed] [Google Scholar]
- 52.Marchitelli C, Savarese MC, Crisà A, Nardone A, Ajmone-Marsan P, Valentini A. Double muscling in Marchigiana beef breed is caused by a stop codon in the third exon of myostatin gene. Mamm Genome. 2003;14:392–5. doi: 10.1007/s00335-002-2176-5. [DOI] [PubMed] [Google Scholar]
- 53.Kambadur R, Sharma M, Smith TPL, Bass JJ. Mutations in myostatin (GDF8) in double-muscled Belgian Blue and Piedmontese cattle. Genome Res. 1997;7:910–5. doi: 10.1101/gr.7.9.910. [DOI] [PubMed] [Google Scholar]
- 54.Casas E, Keele JW, Fahrenkrug SC, Smith TPL, Cundiff LV, Stone RT. Quantitative analysis of birth, weaning, and yearling weights and calving difficulty in Piedmontese crossbreds segregating an inactive myostatin allele. J Anim Sci. 1999;77:1686–92. doi: 10.2527/1999.7771686x. [DOI] [PubMed] [Google Scholar]
- 55.Sanna S, Jackson AV, Nagaraja R, Willer CJ, Chen WM, Bonnycastle LL, et al. Common variants in the GDF5-UQCC region are associated with variation in human height. Nat Genet. 2008;40:198–203. doi: 10.1038/ng.74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Randhawa IAS, Khatkar MS, Thomson PC, Raadsma HW. Composite signatures of directional selection identified multiple genes for stature on bovine chromosome 13 and 14. Proc Assoc Advmt Anim Breed Genet. 2013;20:445–58. [Google Scholar]
- 57.Arango JA, Cundiff LV, Van Vleck LD. Comparisons of Angus, Charolais, Galloway, Hereford, Longhorn, Nellore, Piedmontese, Salers, and Shorthorn breeds for weight, weight adjusted for condition score, height, and condition score of cows. J Anim Sci. 2004;82:74–84. doi: 10.2527/2004.82174x. [DOI] [PubMed] [Google Scholar]
- 58.McClure MC, Ramey HR, Rolf MM, McKay SD, Decker JE, Chapple RH, et al. Genome-wide association analysis for quantitative trait loci influencing Warner-Bratzler shear force in five taurine cattle breeds. Anim Genet. 2012;43:662–73. doi: 10.1111/j.1365-2052.2012.02323.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Koomharaie M. The role of (Ca2+)-dependent proteases (calpains) in post-mortem proteolysis and meat tenderness. Biochimie. 1992;74:239–45. doi: 10.1016/0300-9084(92)90122-U. [DOI] [PubMed] [Google Scholar]
- 60.Ouali A, Herrera-Mendez CH, Coulis C, Becila S, Boudjellal A, Aubry L, et al. Revisiting the conversion of muscle into meat and the underlying mechanisms. Meat Sci. 2006;74:44–58. doi: 10.1016/j.meatsci.2006.05.010. [DOI] [PubMed] [Google Scholar]
- 61.Zoico E, Roubenoff R. The role of cytokines in regulating protein metabolism and muscle function. Nutr Rev. 2002;60:39–51. doi: 10.1301/00296640260085949. [DOI] [PubMed] [Google Scholar]
- 62.Albertì P, Panea B, Sanudo C, Olleta JL, Ripoll G, Ertbjerg P, et al. Live weight, body size and carcass characteristics of young bulls of fifteen European breeds. Livest Sci. 2008;114:19–30. doi: 10.1016/j.livsci.2007.04.010. [DOI] [Google Scholar]
- 63.Tatum JD, Gronewald KW, Seideman SC, Lamm WD. Composition and quality of beef from steers sired by Piedmontese, Gelbvieh and Red Angus bulls. J Anim Sci. 1990;68:1049–60. [Google Scholar]
- 64.Rot A, von Adrian VH. Chemokines in innate and adaptive host defense: basic chemokinese grammar for immune cells. Annu Rev Immunol. 2004;22:891–928. doi: 10.1146/annurev.immunol.22.012703.104543. [DOI] [PubMed] [Google Scholar]
- 65.Mariotti M, Williams JL, Dunner S, Valentini A, Pariset L. Polymorphisms within the toll-like receptor (TLR)-2,-4,- and 6 genes in cattle. Diversity. 2009;1:7–18. doi: 10.3390/d1010007. [DOI] [Google Scholar]
- 66.Ferguson DM, Warner RD. Have we underestimated the impact of pre-slaughter stress on meat quality in ruminants? Meat Sci. 2008;80:12–9. doi: 10.1016/j.meatsci.2008.05.004. [DOI] [PubMed] [Google Scholar]
- 67.Zhao C, Tian F, Yu Y, Luo J, Mitra A, Zhan F, et al. Functional genomic analysis of variation on beef tenderness induced by acute stress in Angus cattle. Comp Funct Genomics. 2012;2012:756284. doi: 10.1155/2012/756284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Utsunomiya YT, Pérez O’Brien AM, Sonstegard TS, Van Tassel CP, Santana Do Carmo A, Mészàros G, et al. Detecting loci under recent positive selection in dairy and beef cattle by combining different genome-wide scan methods. PLoS ONE. 2013;8:e64280. doi: 10.1371/journal.pone.0064280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Kranen RW, van Kuppevelt TH, Goedhart HA, Veerkamp CH, Lambooy E, Veerkamp JH. Hemoglobin and myoglobin content in muscles of broiler chickens. Poult Sci. 1999;8:467–76. doi: 10.1093/ps/78.3.467. [DOI] [PubMed] [Google Scholar]
- 70.Bratzler LJ. Determining the tenderness of meat by use of the Warner-Bratzler method. Proc Recip Meat Conf. 1949;2:117–21. [Google Scholar]
- 71.Wheeler TL, Koohmaraie M, Cundiff LV, Dikeman ME. Effects of cooking and sharing methodology on variation in Warner-Bratzler shear force values in beef. J Anim Sci. 1994;72:2325–30. doi: 10.2527/1994.7292325x. [DOI] [PubMed] [Google Scholar]
- 72.Borghese A, Romita A, Gigli S. Friesian crossbred bulls productivity in comparison with Friesian purebreds. Carcass and meat evaluation. Ann Ist Super Zootec. 1978;11:71–91. [Google Scholar]
- 73.Chambaz A, Kreuzer M, Scheeder MRL, Dufey PA. Characteristics of steers of six beef breeds fattened from eight months of age and slaughtered at a target level of intramuscular fat: II. Meat quality Arch Tierz. 2001;44:473–88. [Google Scholar]
- 74.Castillo C, Pereira V, Abuelo A, Hernàndez J. Effect of supplementation with antioxidants on the quality of bovine milk and meat production. Sci World J. 2013;2013:616098. doi: 10.1155/2013/616098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Bekhit AEDA, Hopkins DL, Fahri FT, Ponnampalam EN. Oxidative processes in muscle systems and fresh meat: Sources, markers, and remedies. Comp Rev Food Sci F. 2013;12:565–97. doi: 10.1111/1541-4337.12027. [DOI] [PubMed] [Google Scholar]
- 76.Descalzo AM, Sancho AM. A review of natural antioxidants and their effects on oxidative status, odour and quality of fresh beef produced in Argentina. Meat Sci. 2008;79:423–36. doi: 10.1016/j.meatsci.2007.12.006. [DOI] [PubMed] [Google Scholar]
- 77.Nielsen R, Hellmann I, Hubisz M, Bustamante C, Clark AG. Recent and ongoing selection in the human genome. Nat Rev Genet. 2007;8:857–68. doi: 10.1038/nrg2187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Akey JM. Constructing genomic maps of positive selection in humans: where do we go from here? Genome Res. 2009;19:711–22. doi: 10.1101/gr.086652.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Bongiorni S, Mancini G, Chillemi G, Pariset L, Valentini A. Identification of a short region on chromosome 6 affecting direct calving ease in Piedmontese cattle breed. PLoS One. 2012;7:e50137. doi: 10.1371/journal.pone.0050137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Barendse W, Harrison BE, Bunch RJ, Thomas MB, Turner LB. Genome wide signatures of positive selection: the comparison of independent samples and identification of regions associated to traits. BMC Genomics. 2009;10:178. doi: 10.1186/1471-2164-10-178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Zhao C, Tian F, Yu Y, Luo J, Hu K, Bequette BJ, et al. Muscle transcriptomic analyses in Angus cattle with divergent tenderness. Mol Biol Rep. 2012;39:4185–93. doi: 10.1007/s11033-011-1203-6. [DOI] [PubMed] [Google Scholar]
- 82.Wang YH, Bower NI, Reverter A, Tan SH, De Jager N, Wang R, et al. Gene expression patterns during intramuscular fat development in cattle. J Anim Sci. 2009;87:119–30. doi: 10.2527/jas.2008-1082. [DOI] [PubMed] [Google Scholar]
- 83.Connor EE, Kahl S, Elsasser TH, Parker JS, Li RW, Van Tassel CP, et al. Enhanced mitochondrial complex gene function and reduced liver size may mediate improved feed efficiency of beef cattle during compensatory growth. Funct Integr Genomics. 2010;10:39–51. doi: 10.1007/s10142-009-0138-7. [DOI] [PubMed] [Google Scholar]
- 84.Boitard S, Rocha D. Detection of signatures of selective sweeps in the Blonde d’Aquitaine cattle breed. Anim Genet. 2013;44:579–83. doi: 10.1111/age.12042. [DOI] [PubMed] [Google Scholar]
- 85.Barrichello Mokry F, Higa RH, De A, Mudadu M, Oliveira De Lima A, Laguna Conceinção Meirelles S, et al. Genome-wide association study for backfat thickness in Canchim beef cattle using Random Forest approach. BMC Genet. 2013;14:47. doi: 10.1186/1471-2156-14-47. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
List of chromosomal specific smoothing parameters S. Table S1 provides the LOWESS smoothing parameters that were calculated for each chromosome.
Patterns of raw Fst data, predicted Fst values and control chart of predicted Fst values on BTA1 to 29. Description: The plots represent the pattern of raw Fst data calculated for SNPs located on each chromosome (BTA), the predicted Fst values for SNPs located on each BTA using the LOWESS regression with chromosomal specific smoothing parameter and the control chart of predicted Fst values for each BTA. Upper control limit (UCLI) and lower control limit (LCLI) are three standard deviations apart from the mean value.
List of 933 genes annotated in cattle detected using the control chart method. Description: This list includes all the bovine annotated genes derived from Bos taurus UMD 3.1/bosTau6 assembly that are present in the 0.5 Mb interval (0.25 Mb upstream and downstream) considered for each significant SNP using the control chart method.
List of 189 genes annotated in cattle detected using varLD. Description: This list includes all the bovine annotated genes derived from Bos taurus UMD 3.1/bosTau6 assembly that are present in the 0.5 Mb interval (0.25 Mb upstream and downstream) considered for each significant SNP using the varLD approach.
Plots obtained with the Fst method vs the varLD method. Description: Chromosome-wide plots obtained in the Fst and varLD analyses for BTA1 to 29. Solid lines represent the threshold set at three standard deviations apart from the mean value.