

Abstract
The tumor response to most therapeutic agents in cancer is highly unpredictable. Cancer models which can adequately represent tumor heterogeneity and predict in vivo drug sensitivity are intense areas of investigation. Cancer cell lines and patient-derived xenograft models are the most frequently used models in cancer research and anticancer drug screening. Recently, cancer “organoid” culture conditions have been developed to establish in vitro growth of patient-derived samples at higher efficiency and they are very promising for large scale drug screening and fundamental cancer biology research. Here, we leverage our experience in prostate cancer to discuss the advantages and limitations of these cancer models and summarize the development of cancer organoid culture—a development which may provide a new path towards personalized medicine in the future.
Introduction
The current drug development paradigm where all patients afflicted with a particular cancer type are enrolled without biomarker selection has an unacceptable failure rate. In many “failed” trials that did not show a statistically significant benefit to the overall trial population, a small subset of patients derived significant clinical benefit. This is best illustrated by the FDA withdrawal of approval for gefitinib—the first clinically tested EGFR inhibitor—after its failure to improve overall survival in unselected patients with advanced lung cancer [1]. After identification of EGFR mutations as a predicative biomarker for tumor response, multiple positive trails in this subset of patients have led to the approval and use of EGFR inhibitors [2-5]. Following this important concept, subsequent trials of molecularly-defined patient subsets (e.g., crizotinib in ALK and ROS1 rearranged lung cancer) were highly encouraging [6,7].
With the rapid development of multiple therapies with specific molecular targets, the identification of molecular biomarkers of drug sensitivity is a critical step. In order to discover therapeutic biomarkers, the tumor models must recapitulate the original tumor, predict the in vivo treatment response in the patient, and suit to high-throughput screening. In this review, we discuss recent advances in in vitro culture technology and their applicability to precision medicine.
Cancer cell lines
Ever since the HeLa cell line was successfully developed [8], cancer cell lines have been invaluable for the mechanistic study of tumorigenesis as well as the identification of markers of therapeutic response. There are several advantages of using cancer cell lines. First, they grow indefinitely; second, the maintenance of cell lines is straightforward; third, screening of a large repertoire of cell lines can identify biomarkers of drug sensitivity. Indeed, studies initiated using cell lines have led to the discovery of predictive biomarkers to targeted agents, including EGFR inhibitors, BRAF and MEK inhibitors, and PARP inhibitors [9-13]. Currently, there are ∼1,500 cancer cell lines available worldwide. Large-scale efforts led by the Broad Institute and the Sanger Institute aim to combine genetic characterization of these lines and high throughput drug testing to identify potential molecular biomarkers of therapeutic response [9,14].
However, the currently available cancer cell lines have a number of limitations. Foremost, most cancer types generate cell lines with a very low efficiency and the established lines represent a selection of particular subsets of tumor that can grow in vitro. This selection process results in cancer cell lines that do not represent the diversity of human cancer. Prostate cancer represents the most extreme example—despite being the most common cancer in men, only seven lines have been established. Second, extensive passaging commonly results in lost heterogeneity during adaption to the culture conditions in vitro by epigenetic or genetic mechanisms [15] (Table 1). Cancer cells lose their differentiation characteristics with increased proliferation capacity, and gene expression profiles change within several passages. For example, the gene expression profiles of MIN-6 cell have global changes between the low passage and high passage cells [16]. Third, most lines were derived from a time when germline DNA and clinical annotation was unavailable, making identification of somatic mutations and correlation with patient disease course and therapeutic responses difficult.
Table 1. Characteristics of prostate cancer cell lines, PDX models and 3D organoids.
| Drug screens | ||||
| Genetic manipulation | ||||
| 2D culture cell lines | Heterogeneity | |||
| Initiation efficiency | Amenable | High throughput | ||
| Low | ||||
| PDX models | Loss | |||
| Low | ||||
| 3D organoids | Maintained | Low throughput | ||
| High | Not amenable | |||
| Maintained | ||||
| Amenable | ||||
| High throughput |
Patient-derived xenograft
Patient-derived xenograft (PDX) models are derived from tumor chunks directly implanted into immunocompromised mice without dissociation. Recently, the development and characterization of PDX models has become an increasing interest for cancer research. The main advantage of PDX models is that they retain the donor tumor heterogeneity and remain stable across passages in vivo [17] (Table 1). These models have been proven to be predictive of clinical outcomes and are being used for preclinical drug testing and personalized medicine strategies [18,19]. Although the development of PDX cancer models brings some improvement compared to the cancer cell line models, the PDX models still have important limitations that hinder their use in targeted cancer therapy. First, the engraftment failure is still very high for some cancer types, such as prostate cancer [20] and estrogen receptor positive breast cancer [21]. Second, in most of the PDX models, engraftment relies on large quantities of surgical tumor tissues, and needle biopsies are often insufficient. Third, the engraftment and drug validation time in mice usually requires over 6 months. This time delay limits the applicability of PDX models for real-time patient treatment. Fourth, while PDX models may be suitable for a limited number of drug combinations, they are not amenable to high throughput screening. Finally, PDX models are not amenable to genetic manipulation, such as introduction of transgenes or knockdown or knockout studies.
Conditional reprogrammed cells
Recently, several groups have investigated novel culture methods to address the current limitations. These work started with the goal of culturing normal benign cells. Historically, when normal cells are cultured in traditional serum-containing media, they divide for a limited number of passages prior to reaching “senescence”, a phenomenon known as the “Hayflick limit” [22].
Recently, Alison McBride and Richard Schlegel's laboratories have developed serum-free conditions that employ the combination of the Rho kinase (ROCK) inhibitor, Y-27632, and irradiated fibroblast feeder cells. Using these conditions, a number of different normal epithelial cell types, including keratinocytes, prostate, breast, and lung cells can propagate indefinitely without acquisition of genetic defects. These cells harbor a stem-like phenotype in vitro but maintain the capability to differentiate and have been termed “conditional reprogrammed cells” or CRCs. CRCs activate endogenous telomerase [23,24]. CRCs have a number of potentially important applications. In genetic disease, CRCs can theoretically be isolated from an affected patient, the defective gene can be corrected in vitro, and the corrected cells can be reintroduced into the patient. In cancer, cells can be isolated and tested for chemosensitivity. In a proof of principle experiment, Schlegel and colleagues isolated tumor CRCs from a patient with respiratory papillomatosis, identified that the cells were sensitive to the histone deacetylase inhibitor, vorinostat, and treated the patient resulting in a 15 month disease stabilization [25]. Using CRC technology, Engelman and colleagues established cell culture models from non-small cell lung cancer patients who have progressed on EGFR and ALK kinase inhibitors to discover patient-specific resistance mutations [26*].
Benign organoid cultures
Over the past 5 years, Hans Clevers and colleagues developed 3D culture conditions where single epithelial stem cells grow to form the physiological architecture of the organ. After the observation that intestinal epithelial stem cells require active WNT pathway signaling for survival and that they express the LGR5 receptor which further amplify WNT signaling. The group first grew intestinal organoids using serum-free media supplemented with WNT3a, and R-spondin, the ligand of LGR5. Amazingly, single LGR5 positive stem cells were found to form organoids with the correct crypt-villus structure with all of the component cell types, including stem cells, goblet cells, transiently amplifying cells, and villus cells. Like normal intestinal tissue, cell division is restricted to the crypt and the differentiating cells move up the villi, eventually sloughing off [27**]. Because these 3D epithelial structures recapitulate the histology and differentiation of the intestinal epithelium in vivo, they were termed “organoids.” Lancaster and Knoblich defined an organoid as containing several cell types that develop from stem cells or organ progenitors and self-organize through cell sorting and spatially-restricted lineage commitment, similar to the process which occurs in vivo [28].
Like CRC, the intestinal organoids grow indefinitely without senescence and can be genetically manipulated. The power of this technology in genetic diseases was illustrated when Hans Clevers' group showed that intestinal organoids derived from patients with cystic fibrosis can be isolated, and the mutant CFTR gene can be repaired using CRISPR/Cas9 system, and normal secretory function can be restored [29].
Remarkably, basal organoid conditions with tissue-specific modifications can be used to generate organoids from almost any epithelial organ, including the colon [30*], stomach [31], liver [32], kidney [33-35], thyroid [36], inner ear [37], retina [38], pituitary gland [39], brain [40] prostate [41**] and pancreas [42**].
Organoid culture can also further identify stem cell compartments and lineage plasticity. For example, the prostate epithelium is comprised of two layers, a basal layer and a luminal layer (Figure 1A). The lineage hierarchy of the prostate epithelium has been controversial: some data suggests stem cells reside only in the basal layer and differentiate into luminal cells (analogous to the basal cells of the epidermis) [43] while other data suggests that luminal and basal layers are self-sustaining lineages and have independent stem cells [44,45]. Using FACS sorting to isolate single basal and luminal cells, Karthaus and colleagues succeeded in establishing prostate organoids from both prostate basal and luminal progenitor cells [41**]. This was the first reported in vitro propagation of luminal cells and confirmed the coexistence of basal and luminal stem cells.
Figure 1.
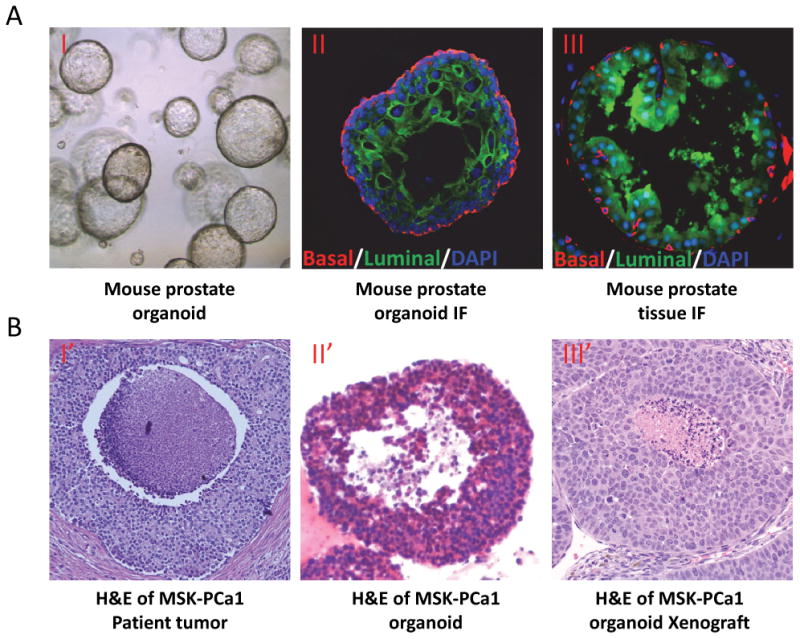
Examples of mouse prostate organoid and human prostate cancer organoid. A. Mouse prostate organoid (phase contrast)(I), mouse prostate organoid (II) and mouse prostate gland (III) immunofluorescent staining with cytokeratin 5 positive basal cells (Red) and cytokeratin 8 positive luminal cells (Green). B. H&E of MSK-PCa1 in situ tumor (I′), MSK-PCa1 organoid (IT) and xenograft of MSK-PCa1 organoid (III′).
Cancer organoid cultures
The new CRC and organoid conditions that allow indefinite propagation of multiple benign epithelial lineages has led to great excitement in that they may allow for generation of novel in vitro patient-derived cancer models. Currently, multiple efforts are underway in a number of different cancer types, including colorectal, pancreas, and lung cancer. Recently, Tuveson and colleagues used organoid conditions to establish benign and cancer organoids from patients and mouse models [42**]. These organoids can be cultured indefinitely and grafted into immunocompromised mice. Here, we will focus on our published work on prostate cancer organoid cultures. In order to maximize the utility of these models, it is critical that they are clinically and molecularly annotated, including information on patient prior and future treatment response and model mutational and transcriptional profiles.
Prostate cancer organoids
Prostate cancer is the second leading cause of cancer death in western men. Prostate cancer has proven very difficult to culture in vitro, with only seven publically available cell lines. Furthermore, many recently identified recurrent genetic lesions in prostate cancer, such as SPOP mutation, FOXA1 mutation TMPRSS2-ERG interstitial deletion, and CHD1 deletion are not represented in the available prostate cancer cell lines. These genetic lesions are also highly specific to prostate cancer and are not found in other malignancies. This limits both mechanistic studies of these genetic lesions and determination of their role in therapeutic response.
Recently, using the organoid culture system optimized for benign prostate epithelial cells [41], we succeeded in establishing seven organoid cultures derived from biopsies of metastatic prostate cancer and from circulating tumor cells (Figure 2). The cultures were annotated with detailed clinical history, including initial tumor histology, grade, stage, and treatment course.
Figure 2.

Schematic diagram of cancer organoid cultures and drug sensitivity test. Isolate prostate cancer cells from freshly collected tissue biopsies or circulating tumor cells; seed the tumor cells into prostate organoid culture system; analyze the organoid cell at the histological, genomic and transcriptomic level; compare with the original tumor; predict potential therapeutic drugs using the information from genomic and transcriptional analysis; test the drug sensitivity using the organoid models.
These seven organoid lines were molecularly characterized in detail, including whole exome sequencing of germine and organoid DNA to identify somatic mutations, array comparative genomic hybridization to determine copy number alterations, and paired-end RNA-sequencing to determine the transcriptional landscape and identify fusion transcripts. Remarkably, in 3D culture, the organoid lines adapted a histology which was highly reminiscent of the tumor histology (Figure 1B). Overall, the organoid lines harbor a low number of somatic mutations, but a large number of copy number alterations, both characteristic of prostate cancer. The seven lines harbor many recurrent genomic alterations typical of metastatic prostate cancer, including PTEN loss, TMPRSS2-ERG interstitial deletion, SPOP mutation, FOXA1 mutation, and CHD1 loss [46**]. Transcriptome analysis indicates that the seven lines are highly diverse, recapitulating the phenotypic diversity of castrate-resistant prostate cancer [47].
In tumors with sufficient tissue, we performed whole exome sequencing and RNA-sequencing of tumor tissue from which the organoid lines were derived. This analysis showed that the lines harbored identical somatic mutations as the tumor after 3 months of in vitro passage and shared a similar transcriptome, confirming that the tumor organoids molecularly represented the tumor tissue in vivo.
One of the seven organoid lines was derived from circulating tumors cells (CTC). CTCs are rare cancer cells in the peripheral blood of patients with solid tumors. CTCs promise a non-invasive real-time biomarkerfor diagnosis, prognosis and therapeutic response monitoring [48]. Using the “CTC-iChip” technology which efficiently removes the normal blood cells [49], Yu and colleagues successfully established six CTCs cell lines out of thirty six patient blood samples [50*]. Using the simple Ficoll-Paque with CD45 depletion cocktail to isolate CTCs, we successfully established one CTC organoid culture from a patient with metastatic prostate cancer [46]. While challenges remain, cancer organoid culture derived from CTCs may help predict the effective therapies for patients with a simple blood draw instead of invasive biopsies or imaging over the course of their disease progression.
Using the seven organoid lines, we showed in proof of concept studies that they exhibit highly differential sensitivities to drugs that target the androgen receptor and the phosphoinositide-3-kinase pathways.
Future directions and unresolved questions
The clinical practice of oncology is changing rapidly towards precision medicine. The biopsy and molecular analysis of metastatic specimens has become a more common practice. At the same time, improvement of culture technology including organoid and CRC affords the opportunity to generate next generation well-annotated models at an unprecedented rate. This leads to exciting questions for the field to address.
First, what is the best culture technology? Organoids maintain a 3D structure more representative of human tumor, but whether organoid culturing leads to better assessment of therapeutic response is unknown. In addition, the culture conditions may significantly affect therapeutic response. Since conditions are rich in growth factors that allow growth of even normal cells, cancer cells may lose dependence on driver oncogenes. For example, the inclusion of EGF may affect response to EGFR inhibitors. Next, how representative is in vitro treatment response to actual clinical therapeutic response? This needs to be validated in a number of lines from patients with known treatment response. Third, will in vitro high-throughput screening discover molecular markers of response? Given the diversity of each tumor type, a large number of lines with each genetic lesion are required to address the role of uncommon genetic lesions. This highlights the need for a large-scale effort to develop reagents and share reagents and lines. Finally, the optimal implementation of these culturing techniques is envisioned to involve the rapid generation of in vitro models from a patient, the testing of a number of drugs on these models, and use of these results to guide treatment for the individual patient.
Acknowledgments
We thank Dr. Samuel Kaffenberger for providing editorial input. Funding was providing by the National Institutes of Health (5K08CA140946), US Department of Defense (W81XWH-10-1-0197), the Geoffrey Beene Cancer Center the STARR Cancer Consortium for funding (I8-A722), and the Stand Up To Cancer - Prostate Cancer Foundation Prostate Dream Team Translational Research Grant (SU2CAACR-DT0712), Prostate Cancer Foundation Movember Challenge Grant, NIH Cancer Center Core Grant (P30 CA008748).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Thatcher N, Chang A, Parikh P, Rodrigues Pereira J, Ciuleanu T, von Pawel J, Thongprasert S, Tan EH, Pemberton K, Archer V, Carroll K. Gefitinib plus best supportive care in previously treated patients with refractory advanced non-small-cell lung cancer: Results from a randomised, placebo-controlled, multicentre study (iressa survival evaluation in lung cancer) Lancet. 2005;366(9496):1527–1537. doi: 10.1016/S0140-6736(05)67625-8. [DOI] [PubMed] [Google Scholar]
- 2.Rosell R, Carcereny E, Gervais R, Vergnenegre A, Massuti B, Felip E, Palmero R, Garcia-Gomez R, Pallares C, Sanchez JM, Porta R, et al. Erlotinib versus standard chemotherapy as first-line treatment for european patients with advanced egfr mutation-positive non-small-cell lung cancer (eurtac): A multicentre, open-label, randomised phase 3 trial. The Lancet Oncology. 2012;13(3):239–246. doi: 10.1016/S1470-2045(11)70393-X. [DOI] [PubMed] [Google Scholar]
- 3.Mok TS, Wu YL, Thongprasert S, Yang CH, Chu DT, Saijo N, Sunpaweravong P, Han B, Margono B, Ichinose Y, Nishiwaki Y, et al. Gefitinib or carboplatin-paclitaxel in pulmonary adenocarcinoma. The New England journal of medicine. 2009;361(10):947–957. doi: 10.1056/NEJMoa0810699. [DOI] [PubMed] [Google Scholar]
- 4.Sequist LV, Yang JC, Yamamoto N, O'Byrne K, Hirsh V, Mok T, Geater SL, Orlov S, Tsai CM, Boyer M, Su WC, et al. Phase iii study of afatinib or cisplatin plus pemetrexed in patients with metastatic lung adenocarcinoma with egfr mutations. J Clin Oncol. 2013;31(27):3327–3334. doi: 10.1200/JCO.2012.44.2806. [DOI] [PubMed] [Google Scholar]
- 5.Wu YL, Zhou C, Hu CP, Feng J, Lu S, Huang Y, Li W, Hou M, Shi JH, Lee KY, Xu CR, et al. Afatinib versus cisplatin plus gemcitabine for first-line treatment of asian patients with advanced nonsmall-cell lung cancer harbouring egfr mutations (lux-lung 6): An open-label, randomised phase 3 trial. The Lancet Oncology. 2014;15(2):213–222. doi: 10.1016/S1470-2045(13)70604-1. [DOI] [PubMed] [Google Scholar]
- 6.Shaw AT, Kim DW, Nakagawa K, Seto T, Crino L, Ahn MJ, De Pas T, Besse B, Solomon BJ, Blackhall F, Wu YL, et al. Crizotinib versus chemotherapy in advanced alk-positive lung cancer. The New England journal of medicine. 2013;368(25):2385–2394. doi: 10.1056/NEJMoa1214886. [DOI] [PubMed] [Google Scholar]
- 7.Shaw AT, Ou SH, Bang YJ, Camidge DR, Solomon BJ, Salgia R, Riely GJ, Varella-Garcia M, Shapiro GI, Costa DB, Doebele RC, et al. Crizotinib in ros1-rearranged non-small-cell lung cancer. The New England journal of medicine. 2014 doi: 10.1056/NEJMoa1406766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Skloot R. The immortal life of henrietta lacks. Crown Publishers; New York: 2010. [Google Scholar]
- 9.Barretina J, Caponigro G, Stransky N, Venkatesan K, Margolin AA, Kim S, Wilson C, Lehar J, Kryukov GV, Sonkin D, Reddy A, et al. The cancer cell line encyclopedia enables predictive modelling of anticancer drug sensitivity. Nature. 2012;483(7391):603–607. doi: 10.1038/nature11003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lynch TJ, Bell DW, Sordella R, Gurubhagavatula S, Okimoto RA, Brannigan BW, Harris PL, Haserlat SM, Supko JG, Haluska FG, Louis DN, et al. Activating mutations in the epidermal growth factor receptor underlying responsiveness of non-small-cell lung cancer to gefitinib. The New England journal of medicine. 2004;350(21):2129–2139. doi: 10.1056/NEJMoa040938. [DOI] [PubMed] [Google Scholar]
- 11.Pao W, Miller V, Zakowski M, Doherty J, Politi K, Sarkaria I, Singh B, Heelan R, Rusch V, Fulton L, Mardis E, et al. Egf receptor gene mutations are common in lung cancers from “never smokers” and are associated with sensitivity of tumors to gefitinib and erlotinib. Proc Natl Acad Sci USA. 2004;101(36):13306–13311. doi: 10.1073/pnas.0405220101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Solit DB, Garraway LA, Pratilas CA, Sawai A, Getz G, Basso A, Ye Q, Lobo JM, She Y, Osman I, Golub TR, et al. Braf mutation predicts sensitivity to mek inhibition. Nature. 2006;439(7074):358–362. doi: 10.1038/nature04304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.McDermott U, Sharma SV, Dowell L, Greninger P, Montagut C, Lamb J, Archibald H, Raudales R, Tarn A, Lee D, Rothenberg SM, et al. Identification of genotype-correlated sensitivity to selective kinase inhibitors by using high-throughput tumor cell line profiling. Proc Natl Acad Sci USA. 2007;104(50):19936–19941. doi: 10.1073/pnas.0707498104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Garnett MJ, Edelman EJ, Heidorn SJ, Greenman CD, Dastur A, Lau KW, Greninger P, Thompson IR, Luo X, Soares J, Liu Q, et al. Systematic identification of genomic markers of drug sensitivity in cancer cells. Nature. 2012;483(7391):570–575. doi: 10.1038/nature11005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Olivotto M, Dello Sbarba P. Environmental restrictions within tumor ecosystems select for a convergent, hypoxia-resistant phenotype of cancer stem cells. Cell Cycle. 2008;7(2):176–187. doi: 10.4161/cc.7.2.5315. [DOI] [PubMed] [Google Scholar]
- 16.O'Driscoll L, Gammell P, McKiernan E, Ryan E, Jeppesen PB, Rani S, Clynes M. Phenotypic and global gene expression profile changes between low passage and high passage min-6 cells. J Endocrinol. 2006;191(3):665–676. doi: 10.1677/joe.1.06894. [DOI] [PubMed] [Google Scholar]
- 17.Hidalgo M, Amant F, Biankin AV, Budinska E, Byrne AT, Caldas C, Clarke RB, de Jong S, Jonkers J, Maelandsmo GM, Roman-Roman S, et al. Patient-derived xenograft models: An emerging platform for translational cancer research. Cancer discovery. 2014;4(9):998–1013. doi: 10.1158/2159-8290.CD-14-0001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Tentler JJ, Tan AC, Weekes CD, Jimeno A, Leong S, Pitts TM, Arcaroli JJ, Messersmith WA, Eckhardt SG. Patient-derived tumour xenografts as models for oncology drug development. Nat Rev Clin Oncol. 2012;9(6):338–350. doi: 10.1038/nrclinonc.2012.61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hidalgo M, Bruckheimer E, Rajeshkumar NV, Garrido-Laguna I, De Oliveira E, Rubio-Viqueira B, Strawn S, Wick MJ, Martell J, Sidransky D. A pilot clinical study of treatment guided by personalized tumorgrafts in patients with advanced cancer. Mol Cancer Ther. 2011;10(8):1311–1316. doi: 10.1158/1535-7163.MCT-11-0233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lawrence MG, Taylor RA, Toivanen R, Pedersen J, Norden S, Pook DW, Frydenberg M, Australian Prostate Cancer B, Papargiris MM, Niranjan B, Richards MG, et al. A preclinical xenograft model of prostate cancer using human tumors. Nat Protoc. 2013;8(5):836–848. doi: 10.1038/nprot.2013.043. [DOI] [PubMed] [Google Scholar]
- 21.DeRose YS, Wang G, Lin YC, Bernard PS, Buys SS, Ebbert MT, Factor R, Matsen C, Milash BA, Nelson E, Neumayer L, et al. Tumor grafts derived from women with breast cancer authentically reflect tumor pathology, growth, metastasis and disease outcomes. Nat Med. 2011;17(11):1514–1520. doi: 10.1038/nm.2454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hayflick L, Moorhead PS. The serial cultivation of human diploid cell strains. Exp Cell Res. 1961;25:585–621. doi: 10.1016/0014-4827(61)90192-6. [DOI] [PubMed] [Google Scholar]
- 23.Liu X, Ory V, Chapman S, Yuan H, Albanese C, Kallakury B, Timofeeva OA, Nealon C, Dakic A, Simic V, Haddad BR, et al. Rock inhibitor and feeder cells induce the conditional reprogramming of epithelial cells. Am J Pathol. 2012;180(2):599–607. doi: 10.1016/j.ajpath.2011.10.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Chapman S, Liu X, Meyers C, Schlegel R, McBride AA. Human keratinocytes are efficiently immortalized by a rho kinase inhibitor. J Clin Invest. 2010;120(7):2619–2626. doi: 10.1172/JCI42297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Yuan H, Myers S, Wang J, Zhou D, Woo JA, Kallakury B, Ju A, Bazylewicz M, Carter YM, Albanese C, Grant N, et al. Use of reprogrammed cells to identify therapy for respiratory papillomatosis. The New England journal of medicine. 2012;367(13):1220–1227. doi: 10.1056/NEJMoa1203055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26*.Crystal AS, Shaw AT, Sequist LV, Friboulet L, Niederst MJ, Lockerman EL, Frias RL, Gainor JF, Amzallag A, Greninger P, Lee D, et al. Patient-derived models of acquired resistance can identify effective drug combinations for cancer. Science. 2014;346(6216):1480–1486. doi: 10.1126/science.1254721. Yuan and colleagues first use tumor CRCs which result in a 15 month disease stabilization. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27**.Sato T, Vries RG, Snippert HJ, van de Wetering M, Barker N, Stange DE, van Es JH, Abo A, Kujala P, Peters PJ, Clevers H. Single Igr5 stem cells build crypt-villus structures in vitro without a mesenchymal niche. Nature. 2009;459(7244):262–265. doi: 10.1038/nature07935. In this paper, Sato and colleagues established the basal murine intestinal organoids culture condition which became a powerful technology for cancer research. [DOI] [PubMed] [Google Scholar]
- 28.Lancaster MA, Knoblich JA. Organogenesis in a dish: Modeling development and disease using organoid technologies. Science. 2014;345(6194):1247125. doi: 10.1126/science.1247125. [DOI] [PubMed] [Google Scholar]
- 29.Schwank G, Koo BK, Sasselli V, Dekkers JF, Heo I, Demircan T, Sasaki N, Boymans S, Cuppen E, van der Ent CK, Nieuwenhuis EE, et al. Functional repair of cftr by crispr/cas9 in intestinal stem cell organoids of cystic fibrosis patients. Cell Stem Cell. 2013;13(6):653–658. doi: 10.1016/j.stem.2013.11.002. [DOI] [PubMed] [Google Scholar]
- 30*.Sato T, Stange DE, Ferrante M, Vries RG, Van Es JH, Van den Brink S, Van Houdt WJ, Pronk A, Van Gorp J, Siersema PD, Clevers H. Long-term expansion of epithelial organoids from human colon, adenoma, adenocarcinoma, and barren's epithelium. Gastroenterology. 2011;141(5):1762–1772. doi: 10.1053/j.gastro.2011.07.050. This paper describes the patient-derived organoid cultures from normal and cancerous colon. [DOI] [PubMed] [Google Scholar]
- 31.Barker N, Huch M, Kujala P, van de Wetering M, Snippert HJ, van Es JH, Sato T, Stange DE, Begthel H, van den Born M, Danenberg E, et al. Lgr5(+ve) stem cells drive self-renewal in the stomach and build long-lived gastric units in vitro. Cell Stem Cell. 2010;6(1):25–36. doi: 10.1016/j.stem.2009.11.013. [DOI] [PubMed] [Google Scholar]
- 32.Huch M, Dorrell C, Boj SF, van Es JH, Li VS, van de Wetering M, Sato T, Hamer K, Sasaki N, Finegold MJ, Haft A, et al. In vitro expansion of single Igr5+ liver stem cells induced by wnt-driven regeneration. Nature. 2013;494(7436):247–250. doi: 10.1038/nature11826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Taguchi A, Kaku Y, Ohmori T, Sharmin S, Ogawa M, Sasaki H, Nishinakamura R. Redefining the in vivo origin of metanephric nephron progenitors enables generation of complex kidney structures from pluripotent stem cells. Cell Stem Cell. 2014;14(1):53–67. doi: 10.1016/j.stem.2013.11.010. [DOI] [PubMed] [Google Scholar]
- 34.Xia Y, Nivet E, Sancho-Martinez I, Gallegos T, Suzuki K, Okamura D, Wu MZ, Dubova I, Esteban CR, Montserrat N, Campistol JM, et al. Directed differentiation of human pluripotent cells to ureteric bud kidney progenitor-like cells. Nat Cell Biol. 2013;15(12):1507–1515. doi: 10.1038/ncb2872. [DOI] [PubMed] [Google Scholar]
- 35.Takasato M, Er PX, Becroft M, Vanslambrouck JM, Stanley EG, Elefanty AG, Little MH. Directing human embryonic stem cell differentiation towards a renal lineage generates a self-organizing kidney. Nat Cell Biol. 2014;16(1):118–126. doi: 10.1038/ncb2894. [DOI] [PubMed] [Google Scholar]
- 36.Antonica F, Kasprzyk DF, Opitz R, Iacovino M, Liao XH, Dumitrescu AM, Refetoff S, Peremans K, Manto M, Kyba M, Costagliola S. Generation of functional thyroid from embryonic stem cells. Nature. 2012;491(7422):66–71. doi: 10.1038/nature11525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Koehler KR, Mikosz AM, Molosh AI, Patel D, Hashino E. Generation of inner ear sensory epithelia from pluripotent stem cells in 3d culture. Nature. 2013;500(7461):217–221. doi: 10.1038/nature12298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Eiraku M, Takata N, Ishibashi H, Kawada M, Sakakura E, Okuda S, Sekiguchi K, Adachi T, Sasai Y. Self-organizing optic-cup morphogenesis in three-dimensional culture. Nature. 2011;472(7341):51–56. doi: 10.1038/nature09941. [DOI] [PubMed] [Google Scholar]
- 39.Suga H, Kadoshima T, Minaguchi M, Ohgushi M, Soen M, Nakano T, Takata N, Wataya T, Muguruma K, Miyoshi H, Yonemura S, et al. Self-formation of functional adenohypophysis in three-dimensional culture. Nature. 2011;480(7375):57–62. doi: 10.1038/nature10637. [DOI] [PubMed] [Google Scholar]
- 40.Lancaster MA, Renner M, Martin CA, Wenzel D, Bicknell LS, Hurles ME, Homfray T, Penninger JM, Jackson AP, Knoblich JA. Cerebral organoids model human brain development and microcephaly. Nature. 2013;501(7467):373–379. doi: 10.1038/nature12517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41**.Karthaus WR, Iaquinta PJ, Drost J, Gracanin A, van Boxtel R, Wongvipat J, Dowling CM, Gao D, Begthel H, Sachs N, Vries RG, et al. Identification of multipotent luminal progenitor cells in human prostate organoid cultures. Cell. 2014;159(1):163–175. doi: 10.1016/j.cell.2014.08.017. This is the first reported in vitro propagation of luminal cells and confirmed the coexistence of basal and luminal stem cells. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42**.Boj SF, Hwang CI, Baker LA, Chio II, Engle DD, Corbo V, Jager M, Ponz-Sarvise M, Tiriac H, Spector MS, Gracanin A, et al. Organoid models of human and mouse ductal pancreatic cancer. Cell. 2015;160(1-2):324–338. doi: 10.1016/j.cell.2014.12.021. This paper reported the propagtion of pancreatic orgnaoids from mouse and from human, benign and malignant. The tumor organoids can be serially passaged, retained gentetic lesions, and can be grafted into immunocompromised mice. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Goldstein AS, Huang J, Guo C, Garraway IP, Witte ON. Identification of a cell of origin for human prostate cancer. Science. 2010;329(5991):568–571. doi: 10.1126/science.1189992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Wang X, Kruithof-de Julio M, Economides KD, Walker D, Yu H, Halili MV, Hu YP, Price SM, Abate-Shen C, Shen MM. A luminal epithelial stem cell that is a cell of origin for prostate cancer. Nature. 2009;461(7263):495–500. doi: 10.1038/nature08361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Choi N, Zhang B, Zhang L, Ittmann M, Xin L. Adult murine prostate basal and luminal cells are self-sustained lineages that can both serve as targets for prostate cancer initiation. Cancer cell. 2012;21(2):253–265. doi: 10.1016/j.ccr.2012.01.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46**.Gao D, Vela I, Sboner A, Iaquinta PJ, Karthaus WR, Gopalan A, Dowling C, Wanjala JN, Undvall EA, Arora VK, Wongvipat J, et al. Organoid cultures derived from patients with advanced prostate cancer. Cell. 2014;159(1):176–187. doi: 10.1016/j.cell.2014.08.016. This study describes the organoid cultures derived from patients with advanced prostate cancer, including the first CTC organoid from prostate cancer patient. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Grasso CS, Wu YM, Robinson DR, Cao X, Dhanasekaran SM, Khan AP, Quist MJ, Jing X, Lonigro RJ, Brenner JC, Asangani IA, et al. The mutational landscape of lethal castration-resistant prostate cancer. Nature. 2012;487(7406):239–243. doi: 10.1038/nature11125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.De Mattos-Arruda L, Cortes J, Santarpia L, Vivancos A, Tabernero J, Reis-Filho JS, Seoane J. Circulating tumour cells and cell-free DNA as tools for managing breast cancer. Nat Rev Clin Oncol. 2013;10(7):377–389. doi: 10.1038/nrclinonc.2013.80. [DOI] [PubMed] [Google Scholar]
- 49.Ozkumur E, Shah AM, Ciciliano JC, Emmink BL, Miyamoto DT, Brachtel E, Yu M, Chen PI, Morgan B, Trautwein J, Kimura A, et al. Inertial focusing for tumor antigen-dependent and -independent sorting of rare circulating tumor cells. Sci Transl Med. 2013;5(179):179ra147. doi: 10.1126/scitranslmed.3005616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50*.Yu M, Bardia A, Aceto N, Bersani F, Madden MW, Donaldson MC, Desai R, Zhu H, Comaills V, Zheng Z, Wittner BS, et al. Cancer therapy. Ex vivo culture of circulating breast tumor cells for individualized testing of drug susceptibility. Science. 2014;345(6193):216–220. doi: 10.1126/science.1253533. Yu and colleagues first successfully established six CTCs cell lines out of thirty six breast cancer patients' blood samples. [DOI] [PMC free article] [PubMed] [Google Scholar]