

Abstract
Experimental techniques which interface single biomolecules directly with microelectronic systems are increasingly being used in a wide range of powerful applications, from fundamental studies of biomolecules to ultra-sensitive assays. Here we review several technologies which can perform electronic measurements of single molecules in solution: ion channels, nanopore sensors, carbon nanotube field-effect transistors, electron tunneling gaps, and redox cycling. We discuss the shared features among these techniques that enable them to resolve individual molecules, and discuss their limitations. Recordings from each of these methods all rely on similar electronic instrumentation, and we discuss the relevant circuit implementations and potential for scaling these single-molecule bioelectronic interfaces to high-throughput arrayed sensing platforms.
Keywords: single molecule, nanopore, carbon nanotube, redox cycling, low-noise amplifier
I. Introduction
Living organisms are extraordinarily complex systems, possessing emergent features which arise from a deep hierarchy of chemical, mechanical, and electrical interactions among the tissues, cells, and molecules from which they are composed. Understanding each structural level poses unique practical challenges. At the smallest end of the scale, it can be challenging to acquire any useful information at all about an individual molecule, owing to its small physical size. Nanoscale dimensions make single molecules very susceptible to interactions with their environment and often lead to weak, stochastic signals that defy classical measurement techniques.
Despite these challenges, interfacing with single molecules offers unique opportunities to understand the basis of larger living systems, as well as to take advantage of the inherent spatial localization and heterogeneity that single-molecule data can offer. Equally valuable, this can lead to molecular assays using extremely small analyte volumes at low concentration. Applications to DNA sequencing have tended to make headlines1,2, but at least as exciting are possibilities for single-molecule assays for non-genomic analytes for which no straightforward amplification protocol exists.
Single-molecule measurements reduce large continuous phenomena to discrete quantized systems. Molecules exist in integer numbers, and can be heterogeneous; molecules may exhibit discrete identifiable chemical or physical states, and they may shift between these states at discrete moments in time. Single molecules typically operate on faster timescales than ensemble systems, and the continuous output of ensemble measurements often represent the average of discrete molecular states, rather than a fundamentally continuous quantity.
Working with discrete variables can relax some of the experimental challenges brought on by weak signals; as long as discrete states can be distinguished from one another, there is complete information. However, the timing of signals remains a continuous variable, and in all recordings there will be a fundamental limit to the precision with which the timing of state transitions can be known.
Although many of the most popular single-molecule techniques are based on light microscopy, non-optical pathways can offer considerable advantages. Electrochemical instrumentation can be cheap, fast, and small compared to most optical imaging systems; modern transistors have dimensions smaller than many biomacromolecules. Signal levels are often much larger for electrochemical measurements as compared to optical techniques, because chemical and electronic transport can be much faster than photon generation in biochemical dyes. However, it can be more difficult to isolate single-molecule electrochemical signals from background activity.
Successful single-molecule transducers generally have the two complementary characteristics of high gain and nanoscale spatial localization. First, the transducers deliver some implicit amplification in the biophysical operation of a sensor, whereby single molecules influence nearby fluorophores, mobile ions, or electron channels. Second, this gain is spatially localized, reducing interference and background noise levels. Information regarding the state of a single molecule is converted into a stream of photons, ions, or electrons, and this transducer output is collected and amplified electronically, as illustrated in Figure 1. The weak signal levels mean that statistical errors and noise are primary concerns, and electronic measurement systems for single-molecule sensors tend to converge on a few circuit topologies which minimize measurement noise.
Figure 1.

An overview of available classes of single-molecule measurements. The signal path of all of these systems begins with conversion of a molecular state into a stream of ions, photons, electrons, or force, and the strength of this initial transduction often determines the overall signal quality. Eventually, the transduced signal is converted into an electronic current, which is them amplified and connected with other electronic systems. This review focuses on the highlighted platforms, which are physically small and entirely electrochemical.
In this Review, we discuss several varieties of single-molecule bioelectronic sensors, focusing on aqueous non-optical platforms. We describe both the mechanisms of their interfaces with traditional microelectronics and the origin of the sensors’ signal amplitudes, temporal properties, noise performance, and potential for future large-scale integration. We exclude mass spectrometry3 and atomic force microscopy4, reviews of which can be found elsewhere. In Section II, we discuss ion channels and nanopore sensors. In Section III, we review single-molecule field-effect transistors, primarily focusing on those based on single carbon nanotubes. Section IV presents tunneling gaps for electron transport through single molecules. In Section V, single-molecule electrochemical methods based on oxidation-reduction reactions are reviewed. Section VI discusses the low-noise electronic current measurement systems traditionally employed for single-molecule bioelectronics. Section VII concludes and comments on possibilities for future enhancements and applications.
II. Ion channels and nanopores
i. Single ion channel recordings
Ion channel proteins are found in the cellular membranes of all organisms, and they are involved in a wide range of critical cellular processes5. In a historical context, single-ion-channel recordings6 may be considered the first successful single-molecule bioelectronic recordings of any type, and they are central to modern physiology.
As the field of electrophysiology progressed over the course of the 20th century, it was found that nerve cells can transmit signals via action potentials, electrical impulses which are propagated by a coordinated flux of dissolved ions in and out of a cell. Ions are not transported across cellular membranes uniformly; rather, the flux is concentrated through discrete protein ion channels which open and close (“gate”) as a function of time. A wide range of ion channels have evolved which selectively transmit certain types of ions, and whose gating is sensitive to different environmental conditions such as electric fields,7 ligand binding,8 pH,9 temperature,10 or mechanical forces.11
The fact that the activity of single ion channels can be observed at all should not be taken for granted; even the existence of discrete ion channels was not definitively proven until the direct observation of single-channel gating currents12 in the 1970s, and there are many proteins which still have not yet been observed as single channels. Even when a single channel can be isolated, recordings remain constrained by the noise floor of available experimental methods. In physiological conditions, single channels often have conductance on the order of femtosiemens (fS) or picosiemens (pS), and operate at membrane potentials of tens of millivolts (mV), producing single-channel currents on the order of picoamperes (pA) or lower. While single-channel recordings have been made of many higher-conductance ion channels, proteins which produce ion currents less than 0.1 pA are generally impractical to record at single-channel resolution.
For detailed treatments of ion channels the reader is referred to numerous texts5,6 and reviews7–11 on these diverse molecules; here we simply wish to emphasize measurement considerations that make single channels electrically detectable. Several laboratory arrangements can facilitate recording ionic currents through single membrane proteins, including classical small-diameter glass pipettes13, microfabricated glass14 and polymer15 apertures, and scanning electrochemical microscopes (SECM) 16. From an electronic instrumentation perspective, even the largest single-channel currents are considered very small. Nonetheless, the picoampere currents through these channels represent a flux of millions ions per second, which is a huge amplification of the conformational change of a single transmembrane protein. (This rate is thousands of times larger, for instance, than the number of photons emitted by the brightest organic fluorophores.) The biophysical properties that make an ion channel such an efficient modulator of localized ion flux are the same properties that allow its activity to be observed at single-molecule resolution.
ii. Nanopore Sensors
Nanopore sensors utilize similar physical principles as ion channel recordings, except that the ion channel itself is not under interrogation. Instead, ion flux is used as a proxy to detect other analyte molecules which may occupy the channel of the nanopore. Ideally, the shape of the ion channel (Fig. 2a–b) does not fluctuate; rather, changes in its conductance can be attributed to the arrival of analyte molecules from the bulk solution (Fig. 2c). This principle bears strong resemblance to Coulter counters17 which are routinely employed for automated cell counting and sorting. However, working with nanoscale molecules rather than microscale cells brings a new set of challenges.18
Figure 2.

a. Protein structure of alpha hemolysin (α-HL), a bacterial toxin which self-assembles through cell membranes. b. Top view of the α-HL structure, showing its open center which allows ions to pass through cell membranes and can be used as a nanopore sensor. c. Electric fields can induce single-stranded DNA molecules to pass through the α-HL pore, transiently reducing the ionic conductance. d. Enzymes such as DNA polymerase can be paired with nanopores to control the translocation velocity. e. Polymerase rachets a DNA molecule through a nanopore one base at a time, leaving behind a stepwise residual current. f. The residual current is sensitive to the DNA base composition, such as the methylated-C substitution shown here. g. Other biomolecular structures can also be used to construct nanopores, such as DNA origami.
Images a+b are from 127, with permission.
Image c is from 20, with permission.
Images d–f are from 128, with permission.
Image g is from 129, with permission.
The basis of many of the limitations of nanopore sensors derives from the finite ionic currents that they produce; as with single-ion-channel recordings, nanoscale dimensions lead to weak signals which are challenging to measure. The simplest electrical model for a nanopore considers a cylindrical aperture through an insulating membrane, in which case the measured steady-state current (IBIAS) can be described by:
| (1) |
where VBIAS is the applied voltage, d is the diameter of the aperture, q is the elementary charge, and ni and μi are the number density and electrophoretic mobility, respectively, of dissolved cations and anions present in the electrolyte. Fits to measured data often find pores have an effective thickness h somewhat smaller than the actual membrane, since pores tend to have an ‘hourglass’ shape rather than a perfect cylinder. The 1/d term arises from the hemispherical electrolyte “access resistance” (RA) at the two sides of the pore19 (Fig. 3d). Although some implementations of nanopore sensors are actually repurposed ion channel proteins, nanopore sensors are not part of a living cell. As a consequence, they can be measured outside physiological conditions and, for practical reasons, are often tuned to achieve larger signal levels (high salt concentration and higher voltages). In addition, since nanopores are intended to be non-gating and described primarily by their geometry, a range of pore constructions can be considered, including reconstituted ion channel proteins20–22, fabricated apertures in solid-state materials23–25 (Fig. 3), and other nanoscale constructs such as DNA origami26 (Fig. 2g). With these enhancements, the signals captured from nanopores can be considerably larger than for physiological ion channels. Bias currents greater than 100 pA are common in protein nanopores27, while solid-state nanopore currents typically exceed 1 nA28.
Figure 3.

a. An illustration of a double-stranded DNA molecule passing through a solid-state nanopore. Inset, a TEM image of a silicon nitride nanopore. b. Current signals from translocations through SiNx nanopores can be as large as several nanoamperes. c. Nanopore current blockades are sensitive to the orientation and position of a translocating molecule. d. Simplified electrical equivalent circuit of a solid-state nanopore, highlighting the access resistance RA and the membrane capacitance CM.
Images a+b are from 28, with permission.
Images c+d are from 45, with permission.
There are many potential applications of nanopore sensors, including single-molecule detection of toxins29, proteins30, and neurotransmitters31. Despite this richness of potential applications, in much of the literature, the word ‘nanopore’ has become synonymous with DNA sequencing. It has been the hope that nanopores can serve as the foundation of a future generation of single-molecule sequencing platforms, in which single intact nucleic acids are threaded through nanopores while their base sequence is determined in real-time32. There is real basis for this vision; the molecular-scale geometry of a nanopore can confine polymers to pass through the channel single-file, unfolded and in sequence. Recent commercial announcements suggest that the vision of nanopore sequencing has nearly been achieved 2. However, while signal-to-noise ratios for counting single molecules can be quite robust, resolving features within individual molecules is often somewhat less reliable.33
Researchers continue to work on strategies to improve nanopores’ sensing performance, and these efforts can be categorized into several complementary goals: (1) improving chemical selectivity, (2) slowing transport or extending the time a molecule occupies a pore, (3) increasing signal levels, and (4) reducing noise levels.
Improving nanopores’ chemical selectivity includes a wide scope of activities which emphasize chemical interactions between the pore and analyte molecules. Chemical probes can be attached to the openings34 or inner walls of nanopores35 which selectively bind target molecules, extending their translocation times relative to other molecules. There can also be significant chemical interactions between a molecule and the surfaces of the nanopore, which depend on the geometry and material of the pore.
The time which an analyte molecule spends in the channel determines the required temporal resolution of a nanopore recording, but improving time resolution involves a tradeoff with measurement noise. Electrophoretically-driven DNA can travel through nanopores in less than 1 microsecond per base, while noise-limited temporal resolution has often been on the order of tens of microseconds. All else being equal, slowing down translocation speeds would permit recordings to be made slower and with less error. Molecular motion can be slowed down by a number of mechanisms. One can decrease the temperature 36, which both increases the viscosity of the electrolyte and slows chemical kinetics. Electrolyte viscosity can also be increased with glycerol or other hygroscopic chemicals. Translocation times have been found to be extended for very small-diameter nanopores37, as polymers’ conformations are limited by steric confinement and stronger van der Waals interactions with the surface of the pore. Molecular motion can also be modulated by improving the electrostatic conditions, through the modification of static charges on the walls of the nanopore, or introduction of tightly-bound counterions38 to modify the effective charge of the translocating molecule. Electronic control schemes involving biasing of integrated electrodes have also been proposed39, but fabricating these structures has proven difficult. Perhaps most promisingly, polymer translocation times have been extended dramatically through the use of enzymes such as polymerase, which can “ratchet” a long DNA molecule through the pore one base at a time40 (Fig. 2d–f). This approach offers the intriguing combination of the sensitivity of nanopore sensors with the chemical specificity and slower kinetics of natural enzymes.
Increasing the signal levels from nanopores is largely a matter of modifying their geometries. The signal amplitude (ΔI) is distinct from the bias current (IBIAS), although often the signal amplitude is a fraction of the bias current, and the two cannot be adjusted independently. Increasing the bias voltage and electrolyte concentration are two obvious means of increasing the signal amplitude, but higher voltages can counterproductively increase the translocation velocities, and high salt conditions can be undesirable if they unstabilize necessary polymer or enzyme structures. Increasing the diameter d of a nanopore can increase IBIAS, but is less likely to improve ΔI. The one geometrical change that tends to have unequivocally positive effects is the reduction of the effective thickness h; this tends to improve both IBIAS and ΔI, while also improving capture efficiency and spatial resolution28 (Fig. 3b).
Efforts to decrease noise in nanopore recordings have several components, whose relative importance can change depending on the application. Interference from external energy sources is always a concern, but can generally be addressed with proper electronic isolation and electromagnetic shielding. Low-frequency 1/f noise can be quite problematic in solid-state nanopores, though it can be mitigated to some degree with surface coatings, harsh acid exposure35, or high-voltage electrochemical cleaning41. However, fluctuating surface charges on the walls of the pore42,43 may represent the limiting noise floor after other sources are reduced. Higher-frequency broadband noise is related to capacitive parasitics present in nanopore measurements, namely the capacitance of the thin dielectric membrane which supports the nanopore. Decreasing sensor parasitic capacitances can have a drastic impact on the noise amplitude for high-bandwidth nanopore recordings 44. As the noise properties of nanopores themselves have improved, the noise contributions of the recording electronics have started to become a limiting factor, leading to increased interest in custom low-noise electronics for nanopore recordings45,46,47.
Both ion channels and nanopore sensors are able to produce impressively large signals from single-molecule activity thanks to their nanoscale dimensions and the fact that the positions of individual molecules affect the density of nearby mobile dissolved ions. Still, it is worth observing that the electrophoretic mobility of aqueous ions (for example, at room temperature potassium ions have μK+ ≈ 7 × 10−4 cm2 V−1 s−1) can be roughly six orders of magnitude slower than the mobility of electrons in solid-state materials (μe− ≈ 1400 cm2 V−1 s−1 for silicon). If it could be arranged for single biomolecules to have electrostatic influence over the carrier density in an electronic conductor, the resulting electronic current signals could be much larger than for an ionic channel.
III. Nanowire and Nanotube Field-Effect Transistors
There have been a number of demonstrations of electronic transistors whose gates are exposed to ionic solutions, allowing molecules from solution to have electrostatic influence over the transistor channel48,49. However, most available devices do not offer single-molecule sensitivity because their measured signals are the sum of influences from many uncorrelated molecules in solution. However, as nanoscale transistors approach single-molecule dimensions, opportunities emerge for direct field-effect detection of the electrical charge of individual analyte molecules.
Semiconducting nanowires have been employed as field-effect biosensors50, where an electronic channel is sensitive to nearby charges in solution. Such nanowire field-effect transistors (NW-FETs) can be implemented using inorganic semiconductors either through bottom-up growth methods 51 or top-down lithography. 52 If these devices are appropriately functionalized they can be used for specific chemical detection 50. In this arrangement, however, the entire exposed surface of the nanowire is equally sensitive, and background surface charge fluctuations can conceal signatures of analyte molecule binding. As a result, more elaborate frequency domain techniques have been employed to try to discern single (or few) molecule behavior in a complex 1/f noise background53. Alternately, nanowires can be constructed within or adjacent to a nanochannel to provide spatial localization, responding much like solid-state nanopores to local charges of single confined biomolecules54.
Another nanowire device which can reach single-molecule sensitivities is the carbon-nanotube field-effect transistor (CNT-FET) 55. Carbon nanotubes can act as near-ideal one-dimensional electronic conductors, and can be either metallic or semiconducting depending on the particulars of the nanotube’s carbon lattice 56. Similarly to inorganic nanowire FETs, the geometry of a CNT-FET is particularly appealing for chemical sensors because the conducting channel can be directly exposed to aqueous electrolyte without any insulating layer. In addition, carbon nanotubes can be readily functionalized using organic chemistries 57. Like a nanowire, a solution-gated CNT-FET can be sensitive to chemical conditions along its entire length, and detecting single molecules is aided by confining the transistor’s sensitivity to a single point along the exposed channel. Fortunately, unlike the nanowire, this happens naturally when a nanotube is point-functionalized to tether a molecule under study because the tether directly affects the conducting channel of the device. In this configuration, with a defect tethered molecule, we refer to the device as a single-molecule field-effect transistor (smFET).
Fabrication of CNT-FETs begins with growth of carbon nanotubes using chemical vapor deposition. Long parallel nanotubes can be routinely grown, but it is difficult to predict their diameter and chirality. Atomic force microscopy can be used to identify nanotubes of a desired diameter (typically < 2 nm for this application), followed by photolithography to pattern metal contacts and etch away extraneous nanotubes. Several dozen transistors can be created along the length of a single carbon nanotube, increasing fabrication throughput 58; however, the stochastic nature of the nanotubes’ chirality and exact locations means that reliably fabricating these devices can be challenging.
The basic exposed gate device structure is shown in Fig. 4a. The electronic carrier density in the nanotube can be modulated by applying a voltage to the supporting silicon wafer, commonly referred to as the “back gate”. The carbon nanotube can also be gated by controlling the electrochemical potential of the liquid electrolyte using an Ag/AgCl or platinum pseudo-reference electrode59. When a solution potential is applied, mobile ions in the electrolyte accumulate at the nanotube surface and induce carriers in the electronic channel. The distance over which the ions accumulate is described by the Debye length, which is < 1 nm for typical physiological buffer concentrations. This can be modeled as an electrochemical capacitance, and because the liquid gate capacitance is generally an order of magnitude larger than the back-gate capacitance, the nanotube is generally much more sensitive to the liquid gate.
Figure 4.
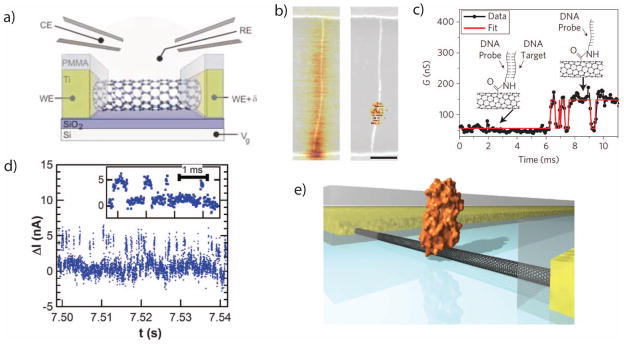
a. An illustration of a liquid-gated field-effect transistor made from a single carbon nanotube. b. Scanning gate microscopy images of a CNT-FET before and after point functionalization. The CNT is chemically modified so that its conductance is predominantly controlled by the electrostatics at a single defect site. c. A time trace from a carbon nanotube modified with a DNA probe molecule. The CNT exhibits two discrete conductance levels, corresponding to the hybridization and melting of a complementary DNA molecule. d. An SMFET trace with an attached enzyme, showing discrete conductance levels that correspond to the states within the activity of the enzyme. e. A rendering of an enzyme tethered to an SMFET.
Image a is from 60, with permission.
Images b+c are from 62, with permission.
Images d+e are from 61, with permission.
Two techniques have been demonstrated which create a localized region of charge sensitivity along the nanotube. Each of these techniques locally disturb a nanotube’s electronic structure through chemical modifications. The first approach electrochemically removes atoms from the carbon lattice60, disturbing its symmetry and creating a point defect with reduced conductance. In the second approach a pyrene molecule tightly adsorbs to the surface of the nanotube through pi-pi stacking61. In each case, the chemical structure of the modification is then used to attach probe molecules which provide chemical specificity for analyte molecules of interest.
The point-defect approach is accomplished using conductance-controlled electrochemical etching of the tube in sulfuric acid. When the liquid gate voltage applied to a platinum solution electrode is reduced below a threshold voltage (approximately −1 V), the nanotube conductance initially decreases very slowly, followed by an abrupt jump. The applied voltage is immediately removed when the abrupt jump is detected, stopping the oxidation. The oxidized device is exposed to potassium permanganate to create a carboxylic acid functional group on the defect site, which can be used to attach a known probe molecule.
The non-covalent functionalization is performed with exposure of the CNT-FETs to a solution of pyrene linker molecules, which stochastically adsorb to the nanotubes. Pyrene forms a tight non-covalent bond with the surface of the carbon nanotube, creating a localized electronic scattering site. The linker molecules also contain attachment chemistries for probe molecules or enzymes to be recorded.
Neither functionalization approach controls the probe location a priori, although it can be characterized afterwards by scanning gate microscopy (SGM), which uses an electrically-biased tip of an atomic force microscope as a liquid gate while recording the conductance of the nanotube. Fig. 4b shows SGM images before and after functionalization; in a a pristine carbon nanotube, transport is limited by Schottky barriers at the contacts, while after functionalization the gating sensitivity localized to a small region near the center of the device.
smFETs have been used to study single-molecule DNA hybridization62 (Fig. 4c) as well as lysozyme protein dynamics61 (Fig. 4d–e). For DNA studies, a single-stranded probe was attached to a point defect using a covalent amine to carboxylic acid coupling reaction, and the binding of complementary DNA target molecules resulted in two-level fluctuations in the tube conductance63. For enzyme recordings, lysozyme variants were genetically engineered with a cysteine residue which was then attached to a pyrene-maleimide linker molecule. Charge movements during the enzyme activity produced discrete conductance fluctuations in the smFET which were used to track the enzyme’s activity.
Since these signals are produced by the position or absence of individual analyte molecules, they tend to fluctuate between discrete states. The statistics of these transitions can resemble those of other systems, such as data from single-molecule FRET studies64 or random telegraph noise in nanoscale semiconductor devices.65 These traces are naturally stochastic, but time traces can be analyzed using established techniques such as hidden Markov models.66
The signals produced by smFETs are remarkably large for single-molecule biosensors, thanks to the high mobility of the electrons travelling through the nanotube. The conductance of a CNT-FET can be hundreds of nS, producing signals as large as tens of nanoamperes (nA).
Unfortunately, noise levels in smFETs tend also to be fairly high, limiting the usable dynamic range of the sensors. Most of the reported noise is flicker or 1/f noise, which could either be a result of charges fluctuating in the electrolyte or in the insulator beneath the nanotube 67. Studies have shown that 1/f noise in carbon nanotube transistors is a strong function of device length 68, suggesting simple scaling of the transistor geometry may reduce noise levels. The noise spectrum would be expected to reach a shot noise floor and a capacitive noise regime 69, but this may be negligible until flicker noise levels are reduced.
IV. Electron Tunneling Gaps
In the single-molecule transducers discussed so far, the measured signal comes from a molecule’s influence on adjacent ions (in the case of nanopores and ion channels) or an adjacent electron channel (in the case of NW-FETs). In the case of a CNT-FET the electron channel is itself a single carbon nanotube molecule; one might intuit that another class of transducers might be possible in which electrons travel through the analyte molecule itself, rather than through adjacent conducting channels.
Electron transport through single organic molecules has been investigated for several decades, often with ambition to use single molecules as building blocks in electronic circuits70. Although large-scale assembly of single-molecule circuits is not yet possible, scientific progress has been made towards understanding electron transport through molecular junctions71. The most common experiments utilize scanning probe microscopy (SPM), in which a sharp conductive tip is brought atomically close to a surface while the current through the tip is monitored. This technique can be used to measure electronic currents through single molecules, although the observed current is inevitably a function of the overall system, including the physics of the contacts between the metal tip and the molecule of interest72,73.
At the length scale of single molecules, electron transport is a quantum mechanical phenomenon, and electrons can interact with the energetic structure of the molecule. Metallic wires smaller than the Fermi wavelength of an electron have quantized conductance G0 = 2e2/h (approximately 77 microsiemens), and small organic molecules often exhibit conductances on the order of 10−6 G0 to 10−2 G0. The upper ends of this range compare favorably to nanopores and NW-FETs, and could yield impressively strong signals if these structures can be reliably implemented.
Single-molecule electronic transport is often investigated using repeated formation and mechanical breaking of a nanoscale metallic wire74. Figure 5a shows this arrangement schematically; a nanoscale probe on a cantilever approaches a conducting surface, while electrically monitoring the current through the probe and optically measuring the deflection of the cantilever. When the tip is brought into contact with the surface and withdrawn (Fig. 5b), it transiently forms a metal nanowire, with quantized conductance levels. Soon after the junction breaks, analyte molecules can span the gap, which is detected as nonzero conductances lower than G0 (Fig. 5c). Since this wire-pull can be repeated many times, histograms of these molecular currents can be made, and these histograms can correlate to the chemical structures and geometries of the molecules in solution75. In addition to using scanning probe platforms, tunneling gaps can be fabricated using mechanically-controlled break junctions (MCBJ) 76,77, as well as with electromigration 78,79, and these junctions can similarly measure single-molecule conductances.
Figure 5.

a. A schematic of a typical scanning probe microscopy (SPM) experiment for single-molecule conductance measurements. A conductive cantilever is brought in contact with a conductive surface, under an electrical bias voltage (Vb), while the current (I) through the probe is monitored. The deflection of the cantilever can be monitored using a reflected laser to infer the applied mechanical force. b. The probe is brought repeatedly into contact with the surface and withdrawn. As the probe withdraws, it forms a transient single-atom wire junction. c. Monitoring the current through the probe during its withdrawal displays quantized conductance, and after the metal junction breaks, analyte molecules can enter the junction. Histograms from many wire-pull experiments can highlight the conductances of single analyte molecules. d. If the SPM tip is held at a constant distance from the surface, analyte molecules can diffuse into the junction, creating transient spike trains. e. A rendering of a proposed DNA sequencing platform utilizing nanoelectrodes functionalized with recognition molecules that record tunneling currents through a DNA molecule as it passes through a nanopore.
Image a+b are from 130, with permission
Image c is from 131, with permission
Image d is from 126, with permission
Image e is from 32, with permission
Experimentally observed single-molecule conductances are highly variable, due to difficulties in reliable nanoelectrode formation, variable metal-to-molecule electrical contacts, and thermal fluctuations of the molecules themselves 77. The mechanisms of the break junction procedure mean that measured currents can change by eight orders of magnitudes within seconds, making accurate instrumentation a challenge 80. Molecular contacts vary with the atomic arrangement of the surface and the orientation of the molecule; although some molecules’ conductance histograms can have well-defined peaks, they tend to have fairly wide variances. Covalent bonds to the metal surface can improve these contacts 81, as can using surface-bound probe molecules which in turn bind the analyte 82. In experiments which maintain tunneling probes at a constant distance for extended periods of time, stochastic current spikes are observed, which are attributable to molecules diffusing in and out of the sensing region, and reorienting within the gap (Fig. 5d). Interestingly, statistical features of these stochastic spikes can be used to distinguish between molecules 83,84.
Molecular conductance has been proposed as a mechanism for DNA sequencing 32,85,86, using transverse tunneling currents through nucleotides in a larger intact DNA molecule (Fig. 5e). While this remains an open area of research, tunneling currents have been demonstrated to differentiate nucleotide monomers in solution 76,82, and nucleotides within oligomers 87,88. Electron transport can also detect amino acids83 and proteins spanning nanogaps 79.
V. Redox Cycling and Electrocatalysis
A final electronic single-molecule detection strategy becomes available when the analyte molecule itself is electrochemically active. Under the right conditions, electrochemical reduction or oxidation (redox) reactions of single analyte molecules can be converted into a measurable current at electrodes which connect to external electronic circuitry.
A typical reversible redox reaction involves the addition of one or a few electrons to a molecule in solution (reduction) or the reverse process of withdrawing electrons (oxidation), as summarized by the reaction
| (2) |
Here O and R represent different charge states of the analyte and are referred to as the oxidized and reduced forms of the molecule, respectively, while n is the number of electrons transferred (typically, n = 1 or 2). Eq (A) highlights the challenge inherent to single-molecule measurements based on electrochemistry: in order to electronically resolve individual electron-transfer processes, one would need to directly detect n electrons, an impossible feat in near-physiological conditions at room temperature. Thus, in order electrochemically detect a single redox molecule, one analyte molecule must participate in many redox reactions. To date, two main schemes have emerged to achieve this charge amplification, each suitable for a specific class of analytes.
The first approach, redox cycling, can be applied to small analyte molecules that undergo reversible electrochemical reactions, as typified by Eq. (A); this includes several important hormones and neurotransmitters. The core concept of redox cycling is to repeatedly reduce and oxidize each target molecule such that it oscillates between the O and R states. This repeated oxidation and reduction can be considered to be amplification, and the total amount of charge transferred is ne times the number of oxidation-reduction cycles, where −e is the electron charge. The number of oxidation and reduction reactions can in principle be arbitrarily large, but the usefulness of redox cycling will depend on achieving rapid cycling between the two states.
Efficient redox cycling can be achieved using two closely-spaced electrodes biased at different potentials, such that the reduction reaction is favored at one electrode and the oxidation is favored at the second electrode. Each target molecule then acts as an electron shuttle, removing charge from one electrode and delivering it to the second electrode during each oxidation-reduction cycle. The resulting current per molecule is
| (3) |
where τ is the average time for performing one cycle 89. Clearly, in order to achieve large signal levels the cycling time must be made as small as possible. Since under physiological conditions electric fields are predominantly screened by mobile salt ions, the only means of transport between electrodes for individual molecules is Brownian motion (diffusion). This dictates the geometry required for effective redox cycling: because the expected diffusion time between two (parallel, planar) electrodes is given by τ/2= z2/2D, where D is the diffusion coefficient of the analyte and z is the distance between the electrodes, the spacing between the two electrodes should be made as small as possible. For a typical small molecule with n = 1 and D = 0.5×10−9 m2/s, coupled with an electrode spacing of 10 nm, Eq (3) thus predicts an average cycle time of ~0.2 μs and a corresponding diffusion-limited current of ~1 pA. This signal level is readily measurable with a bandwidth of order 1 kHz. For an electrode spacing of 1 μm, on the other hand, the current drops to ~0.1 fA, well below the noise floor of most instrumentation. This simple analysis thus highlights the key requirement for single-molecule detection by redox cycling: electrode spacing must be maintained on nanometer scales.
Early realizations of single-molecule detection by redox cycling focused on scanning probe electrochemical methods, in which a flat electrode tip was brought to within 10 nm of a metallic surface, with the tip and surface serving as the two electrodes 89–91. Step-like features in the current were observed which were attributed to individual redox molecules entering the detection region and undergoing redox cycling before exiting again. These first experiments were more recently extended to recessed electrodes immersed in liquid mercury, which allows trapping a very small volume of solution between a solid electrode and the mercury bath 92. In this case, current levels were observed which were consistent with single or small numbers of molecules being present in the trapped volume.
Over recent years, a parallel effort has also been underway to develop microfabricated solid-state devices that create the conditions required for efficient redox cycling 93. These so-called nanogap transducers consist of a channel or chamber whose floor and roof host independently biased electrodes separated by <100 nm (Fig. 6a). Thanks to their reliance on modern lithographic techniques, these devices are suitable for parallelization and integration into more complex systems. Using nanogap devices, discrete signals whose dynamics were consistent with Brownian random walks were reported, providing quantitative evidence for single-molecule sensitivity 93–95 (Fig. 6b). The current per molecule was however found to be typically a factor 4–10 lower than predicted from pure diffusion. This discrepancy was attributed to transient adsorption of the analyte molecules to the electrodes, increasing the shuttling time beyond the time expected for free diffusive motion. While adsorption can be mitigated by adjusting the surface properties of the electrode, it remains unclear whether it can be fully eliminated; at the present time, currents on the order of ~10 fA per molecule remain typical, a significant challenge for subsequent electronic circuitry.
Figure 6.

a. An illustration of a redox cycling measurement, in which a single analyte molecule diffuses between two closely spaced electrodes and is alternatively oxidized and reduced. b. Representative single-molecule data from a redox-cycling experiment. As molecules diffuse in and out of the region between the two electrodes, the measured current changes. c. An illustration of electrocatalysis, in which surface-bound catalysts enable electrochemical reactions at rates far exceeding those with a bare electrode surface. d. Current traces from the arrival of single nanoparticle catalysts at an electrode surface.
Images a is from 94, with permission.
Image b is adapted from 95, with permission.
Images c+d are from 98, with permission.
Given these constraints, what sets the noise level and SNR for redox-cycling detection? An unavoidable limitation is of course the shot noise generated by the redox cycling process itself. It is important to note, however, that the sequential nature of redox cycling insures that it is not a Poisson process, and therefore that the normal prescription for shot noise, S = 2neI, where I is the total current, only represents an upper bound for the shot noise. Other important noise sources may involve the variable electrochemical potentials of single molecules. To our knowledge, and despite ongoing efforts, no detailed theoretical prediction yet exists for the noise spectral density resulting from redox cycling.
A second, potentially more damaging source of noise results from the reliance on Brownian motion for charge transport. Freely diffusing molecules can repeatedly enter and leave a nanogap detection volume, leading to corresponding telegraph-like noise in the measured redox cycling current 93,96. Dominating this process are very short events that cannot be resolved within the bandwidth available for fA-level measurements, and which therefore manifest themselves as background noise. It has been argued 97 that this so-called diffusion noise can be suppressed using advective flow: molecules transiting through a nanogap detection volume of sufficient length necessarily undergo redox cycling for enough time to allow detection. Experimental verification of this analysis has not, however, yet been realized.
Another means of achieving electronic single-molecule detection may come through electrocatalysis. Rather than detecting redox molecules themselves, electrocatalysis offers an opportunity to detect molecules which catalyze redox reactions involving other nearby species. Many redox reactions are very slow at bare electrodes and require the presence of a suitable catalyst to achieve significant turnover. A conducting electrode immersed in a solution that contains an electrochemically-active species may, therefore, not record a current until a catalyst is also present. This therefore provides a route for detecting the presence of individual catalysts at the surface of the electrode via the catalytic electrochemical current, as illustrated in Fig. 6c. Although electrocatalysis also quantifies redox currents at an electrode, it is different from redox cycling; rather than relying on the transport of redox molecules between two electrodes, the catalyst instead facilitates electrochemical reactions of other molecules in solution. One catalyst can interact with a great many redox molecules, without relying on transport between two electrodes.
This concept has been explored in recent years in the form of real-time study of the binding of (catalytic) nanoparticles to an electrode surface: each nanoparticle binding to the surface increases the catalytic ability of that surface, leading to a step in the electrochemical current detected at the electrode 98–101. To date, the main focus has been on proof-of-concept demonstrations based on metal or metal-oxide nanoparticles, which can catalyze currents of order 10–100 pA upon attachment to a (non-catalytic) electrode (Fig. 6d).
In theory, the same concept could be extended to observe individual catalytic molecules, such as redox enzymes, which are proteins that catalyze electrochemical reactions. The enzyme glucose oxidase is a particularly well-known example of this class of biocatalysts due to its broad deployment in glucose sensing applications. Through appropriate chemical modification, redox enzymes can be “wired” to an electrode surface, the electrode then providing (or withdrawing) electrons resulting from a catalytic reaction taking place in solution and providing a direct amperometric measurement of the enzymatic activity. This approach has been successfully applied in bulk toward fundamental understanding of the mechanistic properties of redox enzymes, a technique known as protein film voltammetry 102,103. Attempts to scale this approach down to single molecules, however, have proven challenging. The difficulty arises from the fact that the catalytic rate of redox enzymes is rather slow, reaching only 101 – 104 catalytic cycles per second even under optimal conditions, which offers little or no advantage over fluorescent methods104. This corresponds to maximum currents of only ~1 fA per molecule for even the fastest redox enzymes, making electronic measurement impractical. So far, single-enzyme detection using this approach has not been demonstrated, and the lowest detection level has been in the range of 10–50 enzymes (hydrogenase molecules reducing protons to molecular hydrogen and collectively generating ~20 fA of electrocatalytic current 105).
VI. Low-noise current amplifiers
Almost without exception, the signals produced by single-molecule electronic sensors are amplified with voltage-clamp current preamplifiers, which apply a constant voltage across the sensor while measuring the resulting current (Fig. 7a). This holds true for ion channels, nanopores, smFETs, electron tunneling junctions, and redox cycling, as discussed in this review, as well as many photodiode-based optical systems. All of these transducers share the properties of having very low electrical conductance, and producing weak current signals106.
Figure 7.

a. Electronic nanosensors are often arranged in a configuration which measures their current (ISENSOR) while applying a constant voltage (VBIAS) across the device. Variance in the measurement can be represented by an input-referred equivalent noise source, whose current (INOISE) is added to the desired value. b. A simplified schematic of a low-noise current preamplifier connected to a device modeled as a resistor, RS, with parasitic capacitances (CSP and CSS). The operational amplifier has its own input capacitance (CI) and feedback capacitance (CF). Noise sources are also noted in the figure including the input-referred voltage noise of the amplifier (vn-A), the thermal noise of the feedback resistor (in-Rf), and the noise of the sensor itself (in-S). c. Representative current noise power spectral density at the input of the current preamplifier. Below the frequency f1, the noise is dominated by the flicker noise of the transducer. Between f1 and f2, the thermal noise of the transducer and the feedback resistor of the preamplifier set the noise floor. Flicker noise in the amplifier or imperfect dielectrics in the transducer provide a ∝f slope between f2 and f3. Above f3, the input-referred noise acquires a ∝f2 slope due to the noise of the amplifier acting through ΣCIN. This noise can be greatly reduced by scaling down the physical dimensions of the sensors, fluidics, interconnects, and electronic circuits. d. A micrograph of a low-noise current preamplifier implemented in a modern CMOS process. e. An illustration of a lipid bilayer reconstituted on the surface of an integrated circuit amplifier chip, reducing interconnects and relevant capacitances.
Images c+d are from 45, with permission.
Image e is from 112, with permission.
These current preamplifiers are based on an operational amplifier with a high-impedance feedback network (Fig. 7b). The amplifier maintains a constant voltage at the negative op amp terminal, while the output voltage is linearly proportional to the signal current I. This makes the system more tolerant of stray input capacitances (CIN); for a capacitor, , and thus the current through CIN is nominally zero, although CIN will interact with the amplifier to contribute higher-frequency noise density (Fig. 7c).
The high-impedance feedback network may consist of a resistor, capacitor, or both in parallel. Since all resistors contribute thermal current noise (SI = 4kT/R A2/Hz), the largest tolerable RF is preferable. Feedback resistances ranging from several MΩ up to tens of GΩ are common, although these high-value resistors tend to be physically large and limit the largest measurable signal to I < VDD/RF, where VDD is the supply voltage. Purely capacitive feedback yields the lowest noise, but the accumulated charge on CF must be periodically discharged through a reset switch.
In a well-optimized system, at kHz bandwidths the input-referred noise spectral density tends to be on the order of 1–100 fA/√Hz, implying fA standard deviations for slow (<100Hz) signals, pA standard deviations for moderately fast (<100kHz) signals, and pA-to-nA deviations for MHz bandwidths. 46,107
Any vision of a small parallelized single-molecule bioelectronic sensor array will need an appropriately-sized multichannel electronic interface108. Discrete-component current amplifiers are generally on the scale of several cubic centimeters or more per channel, and systems with as many as 384 parallel low-noise current measurements are commercially available as large rack-mount instruments109. Modern CMOS technology offers a relatively straightforward path to shrinking the full electronic signal chain to sub-millimeter dimensions110 (Fig. 7d) This can lead to shorter interconnects, less capacitance (leading to lower noise), and large arrays of closely-integrated sensors111. To take full advantage of these reduced interconnects can demand re-configuring the fluidics and sensors around the integrated circuits themselves; for example, introducing the amplifier into a nanopore fluid cell or assembling sensors directly onto the surface of the semiconductor chip. One such arrangement is shown in Figure 7e, with lipid bilayers reconstituted directly onto microfabricated supports on the surface of a CMOS amplifier chip112.
There have been several demonstrated integrated CMOS current preamplifiers for single-molecule bioelectronic sensors 45,47,113–115, in addition to the long and established history of integrated low-noise preamplifiers for photodiodes 116 and high-energy particle detectors 117. (Additionally, there are ongoing commercial efforts which incorporate ASICs with nanopore measurements2,33, although at this time few specific technical details are publically available.) Integrated amplifier designs face similar tradeoffs as discrete designs, with different constraints. Integrated semiconductor processes often do not offer JFET transistors, leaving MOS transistors as a next-best substitute. Integrated semiconductors also do not have high-value resistors available for the feedback network. There are several imperfect resistive-feedback substitutes, such as weakly-inverted transistors or active low-tranconductance stages115, but these devices introduce shot noise which is not present in discrete resistors. Capacitive feedback is still the lowest-noise topology, if an appropriate reset scheme can be implemented. The simplest arrangement is a single reset switch 107, which is also the arrangement of discrete capacitive-feedback amplifiers such as the popular Axopatch 200B (Molecular Devices). Discharging the feedback network through a switched-capacitor network is also a possibility, as is arranging a more complicated system which alternates the polarity of the first stage in order to charge and discharge a feedback capacitor118.
Integrated electronics provide a path for higher throughput and smaller dimensions, but ultimately they can do no better than the signal-to-noise ratio of the single-molecule transducer. Reductions in parasitic capacitance and careful customization of preamplifier performance may see an additional order of magnitude reduction in noise floors from today’s state-of-the-art; beyond this, we will need to look for improvements in the specificity, sensitivity, and gain of the sensors themselves.
VII. Discussion
The usefulness of single-molecule data and the low cost of non-optical platforms will continue to drive investments and advances in single-molecule bioelectronic platforms. One path for improvement is simply scaling up single-molecule electronic platforms for higher throughput and higher levels of automation. Single-molecule data tend to be noisy and stochastic and often require more manual analysis than is tolerable outside a research environment. Parallelized systems, along with appropriate software and algorithms, will help to reduce that tradeoff, allowing more data to be collected and making lower-yield experiments more feasible.
Although in the abstract, single-molecule measurements represent the ultimate in sensitivity, in many applications they are impractical or unnecessary. Single-molecule sensing will see its growth as a key tool for applications that work with dilute analytes; applications with subtly heterogeneous samples; and studies of the biophysics of single-molecule interactions. One of the main limitations of single-biomolecule measurements is that they often support a relatively small range of analyte concentrations. At low concentrations the analyte molecules are observed infrequently, and at high concentrations molecules appear too often to be differentiated from one another.
DNA sequencing remains the highest-profile commercial application in this space, and the scientific community has come to appreciate the value of long-read single-molecule sequence data119. Nanopores appear poised to be the first electronic single-molecule platforms to be widely used outside of a laboratory; there are a number of companies working to commercialize nanopore DNA sequencing, to varying degrees of success2,120,121. Ultimately, though, nanopore DNA sequencing may prove to be just the leading edge of a broader class of single-molecule electronic biosensors.
CMOS-integrated smFETs naturally lend themselves to large dense arrays, with close integration of the measurement electronics. In some sense, the smFET is a nanoscale equivalent to microscale ion-sensitive field-effect transistors (ISFETs), which have seen recent success in the DNA sequencing market.122 smFETs are smaller and faster than ISFETs, but their construction in large arrays remains unproven. The primary limitation to achieving arrays of smFETs comes from the initial growth and positioning of the carbon nanotubes, and from high flicker noise. Fabricating these devices remains a challenging and manual process.
Redox-cycling has at least one practical advantage, in that it is a direct extension of established electrochemical methods. The lower gain of redox cycling and electrocatalysis may provide an advantage in dynamic range, allowing a single platform to scale from single-molecule to ensemble measurements. Unfortunately, achieving high cycling efficiency requires single-nanometer fluidic channels which are challenging to fabricate.
For tunneling gaps, at the moment their extreme sensitivity is as much of a detriment as an asset. Like many nanoscale devices, they suffer from fabrication and reliability challenges, but single-molecule tunneling measurements may yield insights into molecular electronics more broadly73. Tunneling sensors may also be combined with nanopores78,123 or nanochannels124 which confine analyte molecules’ locations and orientations. As tunneling currents depend strongly on the metal-to-molecule interface, future advances in tunneling gaps may incorporate as much chemistry as nanofabrication125,126.
In concept, electronic single-molecule platforms are not limited spatially by light diffraction, and are not limited temporally by photon emission. However, in practice, their implementation can be held back by optical lithography and the dimensions of metal interconnects and fluidic channels. Above all, yield and reliability remain the primary obstacles. Their future is bright, but nanoscale electronic single-molecule biosensors can be just as fickle as the single molecules that they measure.
Acknowledgments
This work was supported in part by the National Institutes of Health under grants U19-AI109761, R01-HG006882, R01-HG006879, and R01-GM107417.
References
- 1.Eid J, Fehr A, Gray J, Luong K, Lyle J, Otto G, Peluso P, Rank D, Baybayan P, Bettman B, et al. Science. 2009;323:133. doi: 10.1126/science.1162986. [DOI] [PubMed] [Google Scholar]
- 2.Eisenstein M. Nature biotechnology. 2012;30:295. doi: 10.1038/nbt0412-295. [DOI] [PubMed] [Google Scholar]
- 3.Fenn J, Mann M, Meng C. Science. 1989 doi: 10.1126/science.2675315. [DOI] [PubMed] [Google Scholar]
- 4.Hansma H, Hoh J. Annual review of biophysics and biomolecular structure. 1994 doi: 10.1146/annurev.bb.23.060194.000555. [DOI] [PubMed] [Google Scholar]
- 5.Hille B. 2001 [Google Scholar]
- 6.Sakmann B, Neher E. 2009 [Google Scholar]
- 7.Armstrong CM, Hille B. Neuron. 1998;20:371. doi: 10.1016/s0896-6273(00)80981-2. [DOI] [PubMed] [Google Scholar]
- 8.Changeux JP, Devillers-Thiéry a, Chemouilli P. Science (New York, NY) 1984;225:1335. doi: 10.1126/science.6382611. [DOI] [PubMed] [Google Scholar]
- 9.Chen CC, England S, Akopian aN, Wood JN. Proceedings of the National Academy of Sciences of the United States of America. 1998;95:10240. doi: 10.1073/pnas.95.17.10240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Clapham DE. 2003;426:517. doi: 10.1038/nature02196. [DOI] [PubMed] [Google Scholar]
- 11.Martinac B. Journal of Cell Science. 2004;117:2449. doi: 10.1242/jcs.01232. [DOI] [PubMed] [Google Scholar]
- 12.Neher E, Sakmann B. Nature. 1976 doi: 10.1038/260799a0. [DOI] [PubMed] [Google Scholar]
- 13.Hamill OP, Marty A, Neher E, Sakmann B, Sigworth FJ. 1981:85. doi: 10.1007/BF00656997. [DOI] [PubMed] [Google Scholar]
- 14.Fertig N, Meyer C, Blick R, Trautmann C, Behrends J. Physical Review E. 2001;64:1. doi: 10.1103/PhysRevE.64.040901. [DOI] [PubMed] [Google Scholar]
- 15.Mayer M, Kriebel JK, Tosteson MT, Whitesides GM. Biophysical journal. 2003;85:2684. doi: 10.1016/s0006-3495(03)74691-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Novak P, Gorelik J, Vivekananda U, Shevchuk AI, Ermolyuk YS, Bailey RJ, Bushby AJ, Moss GWJ, Rusakov Da, Klenerman D, Kullmann DM, Volynski KE, Korchev YE. Neuron. 2013;79:1067. doi: 10.1016/j.neuron.2013.07.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Graham M. Journal of the Association for Laboratory Automation. 2003;8:72. [Google Scholar]
- 18.Wanunu M. Physics of life reviews. 2012;9:125. doi: 10.1016/j.plrev.2012.05.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hall JE. The Journal of general physiology. 1975;66:531. doi: 10.1085/jgp.66.4.531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kasianowicz JJ, Brandin E, Branton D, Deamer DW. Proceedings of the National Academy of Sciences of the United States of America. 1996;93:13770. doi: 10.1073/pnas.93.24.13770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Derrington IM, Butler TZ, Collins MD, Manrao E, Pavlenok M, Niederweis M, Gundlach JH. Proceedings of the National Academy of Sciences. 2010:1. doi: 10.1073/pnas.1001831107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wendell D, Jing P, Geng J, Subramaniam V, Lee TJ, Montemagno C, Guo P. Nature Nanotechnology. 2009;4:765. doi: 10.1038/nnano.2009.259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Healy K, Schiedt B, Morrison AP. Nanomedicine (London, England) 2007;2:875. doi: 10.2217/17435889.2.6.875. [DOI] [PubMed] [Google Scholar]
- 24.Dekker C. Nature Nanotechnology. 2007;2:209. doi: 10.1038/nnano.2007.27. [DOI] [PubMed] [Google Scholar]
- 25.Howorka S, Siwy Z. Chemical Society reviews. 2009;38:2360. doi: 10.1039/b813796j. [DOI] [PubMed] [Google Scholar]
- 26.Bell N, Engst C, Ablay M, Divitini G, Ducati C, Liedl T, Keyser UF. Nano Letters. 2011 doi: 10.1021/nl204098n. [DOI] [PubMed] [Google Scholar]
- 27.Meller a, Nivon L, Brandin E, Golovchenko J, Branton D. Proceedings of the National Academy of Sciences of the United States of America. 2000;97:1079. doi: 10.1073/pnas.97.3.1079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Wanunu M, Dadosh T, Ray V, Jin J, McReynolds L, Drndić M. Nature Nanotechnology. 2010;5:807. doi: 10.1038/nnano.2010.202. [DOI] [PubMed] [Google Scholar]
- 29.Halverson KM, Panchal RG, Nguyen TL, Gussio R, Little SF, Misakian M, Bavari S, Kasianowicz JJ. The Journal of biological chemistry. 2005;280:34056. doi: 10.1074/jbc.M507928200. [DOI] [PubMed] [Google Scholar]
- 30.Rodriguez-Larrea D, Bayley H. Nature Nanotechnology. 2013:1. doi: 10.1038/nnano.2013.22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Boersma AJ, Brain KL, Bayley H. ACS nano. 2012;6:5304. doi: 10.1021/nn301125y. [DOI] [PubMed] [Google Scholar]
- 32.Branton D, Deamer DW, Marziali A, Bayley H, Benner Sa, Butler T, Di Ventra M, Garaj S, Hibbs A, Huang X, Jovanovich SB, Krstic PS, Lindsay S, Ling XS, Mastrangelo CH, Meller A, Oliver JS, Pershin YV, Ramsey JM, Riehn R, Soni GV, Tabard-Cossa V, Wanunu M, Wiggin M, Schloss Ja. Nature Biotechnology. 2008;26:1146. doi: 10.1038/nbt.1495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Thompson JF, Oliver JS. Electrophoresis. 2012;33:3429. doi: 10.1002/elps.201200136. [DOI] [PubMed] [Google Scholar]
- 34.Howorka S, Cheley S, Bayley H. Nature Biotechnology. 2001;19:636. doi: 10.1038/90236. [DOI] [PubMed] [Google Scholar]
- 35.Wanunu M, Meller A. Nano Letters. 2007;7:1580. doi: 10.1021/nl070462b. [DOI] [PubMed] [Google Scholar]
- 36.Wanunu M, Sutin J, McNally B, Chow A, Meller A. Biophysical Journal. 2008;95:4716. doi: 10.1529/biophysj.108.140475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Venta K, Shemer G, Puster M, Rodriguez-Manzo JA, Balan A, Rosenstein JK, Shepard KL, Drndic M. ACS nano. 2013:4629. doi: 10.1021/nn4014388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kowalczyk SW, Wells DB, Aksimentiev A, Dekker C. Nano letters. 2012;12:1038. doi: 10.1021/nl204273h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Polonsky S, Rossnagel S, Stolovitzky G. Applied Physics Letters. 2007;91:153103. [Google Scholar]
- 40.Lieberman KR, Cherf GM, Doody MJ, Olasagasti F, Kolodji Y, Akeson M. 2010:995. doi: 10.1021/ja1087612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Beamish E, Kwok H, Tabard-Cossa V, Godin M. Nanotechnology. 2012;23:405301. doi: 10.1088/0957-4484/23/40/405301. [DOI] [PubMed] [Google Scholar]
- 42.Hoogerheide D, Garaj S, Golovchenko J. Physical Review Letters. 2009:102. doi: 10.1103/PhysRevLett.102.256804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Bezrukov SM, Kasianowicz JJ. Physical review letters. 1993;70:2352. doi: 10.1103/PhysRevLett.70.2352. [DOI] [PubMed] [Google Scholar]
- 44.Tabard-Cossa V, Trivedi D, Wiggin M, Jetha NN, Marziali A. Nanotechnology. 2007;18:305505. [Google Scholar]
- 45.Rosenstein JK, Wanunu M, Merchant CA, Drndic M, Shepard KL. Nature Methods. 2012:9. doi: 10.1038/nmeth.1932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Kim D, Goldstein B, Member GS, Tang W, Member S, Sigworth FJ, Culurciello E, Member S. 2012:1. [Google Scholar]
- 47.Chen C, Yemenicioglu S, Uddin A, Corgliano E, Theogarajan L. IEEE Engineering in Medicine and Biology Conference. 2013;2013:164. doi: 10.1109/EMBC.2013.6609463. [DOI] [PubMed] [Google Scholar]
- 48.Besteman K, Lee JO, Wiertz FGM, Heering HA, Dekker C. Nano Letters. 2003;3:727. [Google Scholar]
- 49.Lee CS, Kim SK, Kim M. Sensors (Basel, Switzerland) 2009;9:7111. doi: 10.3390/s90907111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Cui Y, Wei Q, Park H, Lieber CM. Science (New York, NY) 2001;293:1289. doi: 10.1126/science.1062711. [DOI] [PubMed] [Google Scholar]
- 51.Cui Y, Lauhon LJ, Gudiksen MS, Wang J, Lieber CM. Applied Physics Letters. 2001;78:2214. [Google Scholar]
- 52.Stern E, Klemic JF, Routenberg Da, Wyrembak PN, Turner-Evans DB, Hamilton AD, LaVan Da, Fahmy TM, Reed Ma. Nature. 2007;445:519. doi: 10.1038/nature05498. [DOI] [PubMed] [Google Scholar]
- 53.Zheng G, Gao XPA, Lieber CM. Nano Letters. 2010;10:3179. doi: 10.1021/nl1020975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Xie P, Xiong Q, Fang Y, Qing Q, Lieber CM. Nature Nanotechnology. 2011:1. doi: 10.1038/nnano.2011.217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.McEuen PL, Fuhrer MS. IEEE Transactions On Nanotechnology. 2002;1:78. [Google Scholar]
- 56.Dénervaud N, Becker J, Delgado-gonzalo R, Damay P, Rajkumar AS, Unser M. 2013:110. doi: 10.1073/pnas.1308265110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Cai H, Cao X, Jiang Y, He P, Fang Y. Analytical and bioanalytical chemistry. 2003;375:287. doi: 10.1007/s00216-002-1652-9. [DOI] [PubMed] [Google Scholar]
- 58.Zhang X, Chenet D, Kim B, Yu J, Tang J, Nuckolls C, Hone J. Journal of Vacuum Science & Technology B: Microelectronics and Nanometer Structures. 2013;31:06FI01. [Google Scholar]
- 59.Rosenblatt S, Yaish Y, Park J, Gore J, Sazonova V, McEuen PL. Nano Letters. 2002;2:869. [Google Scholar]
- 60.Goldsmith BR, Coroneus JG, Khalap VR, Kane AA, Weiss GA, Collins PG. Science. 2007;315:77. doi: 10.1126/science.1135303. [DOI] [PubMed] [Google Scholar]
- 61.Choi Y, Moody IS, Sims PC, Hunt SR, Corso BL, Perez I, Weiss GA, Collins PG. Science. 2012;335:319. doi: 10.1126/science.1214824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Sorgenfrei S, Chiu CY, Gonzalez RL, Yu YJ, Kim P, Nuckolls C, Shepard KL. Nature Nanotechnology. 2011;6:125. doi: 10.1038/nnano.2010.275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Sorgenfrei S, Chiu C-y, Gonzalez RL, Yu Y-J, Kim P, Nuckolls C, Shepard KL. Nat Nano. 2011;6:126. doi: 10.1038/nnano.2010.275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Fei J, Bronson JE, Hofman JM, Srinivas RL, Wiggins CH, Gonzalez RL. Proceedings of the National Academy of Sciences. 2009;106:15702. doi: 10.1073/pnas.0908077106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Realov S, Shepard KL. Electron Devices Meeting (IEDM), 2010 IEEE International. 2010:28.2.1. [Google Scholar]
- 66.Bronson JE, Fei J, Hofman JM, Gonzalez RL, Jr, Wiggins CH. Biophysical Journal. 2009;97:3196. doi: 10.1016/j.bpj.2009.09.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Männik J, Heller I, Janssens AM, Lemay SG, Dekker C. Nano letters. 2008;8:685. doi: 10.1021/nl073271h. [DOI] [PubMed] [Google Scholar]
- 68.Ishigami M, Chen JH, Williams ED, Tobias D, Chen YF, Fuhrer MS. Applied Physics Letters. 2006;88:203116. [Google Scholar]
- 69.Carminati M, Vergani M, Ferrari G, Caranzi L, Caironi M, Sampietro M. Sensors and Actuators B: Chemical. 2012;174:168. [Google Scholar]
- 70.Aviram A, Ratner M. Chemical Physics Letters. 1974:29. [Google Scholar]
- 71.Tao NJ. Nature nanotechnology. 2006;1:173. doi: 10.1038/nnano.2006.130. [DOI] [PubMed] [Google Scholar]
- 72.Aradhya SV, Venkataraman L. Nature nanotechnology. 2013;8:399. doi: 10.1038/nnano.2013.91. [DOI] [PubMed] [Google Scholar]
- 73.McCreery RL, Yan H, Bergren AJ. Physical chemistry chemical physics: PCCP. 2013;15:1065. doi: 10.1039/c2cp43516k. [DOI] [PubMed] [Google Scholar]
- 74.Venkataraman L, Klare JE, Tam IW, Nuckolls C, Hybertsen MS, Steigerwald ML. Nano. 2006:2. doi: 10.1021/nl052373+. [DOI] [PubMed] [Google Scholar]
- 75.Venkataraman L, Klare JE, Nuckolls C, Hybertsen MS, Steigerwald ML. Nature. 2006;442:1. doi: 10.1038/nature05037. [DOI] [PubMed] [Google Scholar]
- 76.Tsutsui M, Taniguchi M, Yokota K, Kawai T. Nature Nanotechnology. 2010:1. doi: 10.1038/nnano.2010.42. [DOI] [PubMed] [Google Scholar]
- 77.Stava E, Yu M, Shin HC, Shin H, Kreft DJ, Blick RH. Lab on a chip. 2013;13:156. doi: 10.1039/c2lc40925a. [DOI] [PubMed] [Google Scholar]
- 78.Tsutsui M, Rahong S, Iizumi Y, Okazaki T, Taniguchi M, Kawai T. Scientific Reports. 2011:1. doi: 10.1038/srep00046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Ilyas A, Asghar W, Billo Ja, Syed EaQ, Iqbal SM. 2011 IEEE/NIH Life Science Systems and Applications Workshop (LiSSA) 2011:79. [Google Scholar]
- 80.Mészáros G, Li C, Pobelov I, Wandlowski T. Nanotechnology. 2007;18:424004. doi: 10.1088/0957-4484/18/42/424004. [DOI] [PubMed] [Google Scholar]
- 81.Cheng Z, Skouta R, Vazquez H, Widawsky JR, Schneebeli S, Chen W, Hybertsen MS. Nature Nanotechnology. 2011;6:353. doi: 10.1038/nnano.2011.66. [DOI] [PubMed] [Google Scholar]
- 82.Chang S, Huang S, He J, Liang F, Zhang P, Li S, Chen X, Sankey O, Lindsay S. Nano letters. 2010;10:1070. doi: 10.1021/nl1001185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Zhao Y, Ashcroft B, Zhang P, Liu H, Sen S, Song W, Im J, Gyarfas B, Manna S, Biswas S, Borges C, Lindsay S. Nature Nanotechnology. 2014 doi: 10.1038/nnano.2014.54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Tsutsui M, Taniguchi M, Kawai T. Nature communications. 2010;1:138. doi: 10.1038/ncomms1141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Zwolak M, Di Ventra M. Nano letters. 2005;5:421. doi: 10.1021/nl048289w. [DOI] [PubMed] [Google Scholar]
- 86.Albrecht T. Nature Communications. 2012;3:810. doi: 10.1038/ncomms1791. [DOI] [PubMed] [Google Scholar]
- 87.Huang S, He J, Chang S, Zhang P, Liang F, Li S, Tuchband M, Fuhrmann A, Ros R, Lindsay S. Nature nanotechnology. 2010:1. doi: 10.1038/nnano.2010.213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Tsutsui M, Matsubara K, Ohshiro T, Furuhashi M, Taniguchi M, Kawai T. Journal of the American Chemical Society. 2011;133:9124. doi: 10.1021/ja203839e. [DOI] [PubMed] [Google Scholar]
- 89.Fan FRF, Bard AJ. Science. 1995;267:871. doi: 10.1126/science.267.5199.871. [DOI] [PubMed] [Google Scholar]
- 90.Fan FRF, Kwak J, Bard AJ. Journal of the American Chemical Society. 1996;118:9669. [Google Scholar]
- 91.Bard AJ, Fan FRF. Accounts of Chemical Research. 1996;29:572. [Google Scholar]
- 92.Sun P, Mirkin MV. Journal of the American Chemical Society. 2008;130:8241. doi: 10.1021/ja711088j. [DOI] [PubMed] [Google Scholar]
- 93.Zevenbergen MAG, Krapf D, Zuiddam MR, Lemay SG. Nano Letters. 2007;7:384. doi: 10.1021/nl062571g. [DOI] [PubMed] [Google Scholar]
- 94.Zevenbergen MAG, Singh PS, Goluch ED, Wolfrum B, Lemay SG. Nano Letters. 2011:2881. doi: 10.1021/nl2013423. [DOI] [PubMed] [Google Scholar]
- 95.Kang S, Nieuwenhuis AF, Mathwig K, Mampallil D, Lemay SG. ACS nano. 2013;7:10931. doi: 10.1021/nn404440v. [DOI] [PubMed] [Google Scholar]
- 96.Zevenbergen MAG, Singh PS, Goluch ED, Wolfrum BL, Lemay SG. Analytical Chemistry. 2009;81:8203. doi: 10.1021/ac9014885. [DOI] [PubMed] [Google Scholar]
- 97.Singh PS, Katelhon E, Mathwig K, Wolfrum B, Lemay SG. ACS nano. 2012;6:9662. doi: 10.1021/nn3031029. [DOI] [PubMed] [Google Scholar]
- 98.Kleijn SEF, Lai SCS, Miller TS, Yanson AI, Koper MTM, Unwin PR. Journal of the American Chemical Society. 2012;134:18558. doi: 10.1021/ja309220m. [DOI] [PubMed] [Google Scholar]
- 99.Xiao X, Bard AJ. Journal of the American Chemical Society. 2007;129:9610. doi: 10.1021/ja072344w. [DOI] [PubMed] [Google Scholar]
- 100.Xiao X, Fan FRF, Zhou J, Bard AJ. Journal of the American Chemical Society. 2008;130:16669. doi: 10.1021/ja8051393. [DOI] [PubMed] [Google Scholar]
- 101.Kwon SJ, Zhou H, Fan FRF, Vorobyev V, Zhang B, Bard AJ. Physical Chemistry Chemical Physics. 2011;13:5394. doi: 10.1039/c0cp02543g. [DOI] [PubMed] [Google Scholar]
- 102.Bernhardt PV. Aust J Chem. 2006;59:233. [Google Scholar]
- 103.Armstrong FA, Heering HA, Hirst J. Chemical Society reviews. 1997;26:169. [Google Scholar]
- 104.Bard AJ. ACS nano. 2008;2:2437. doi: 10.1021/nn800801z. [DOI] [PubMed] [Google Scholar]
- 105.Hoeben FJM, Meijer FS, Dekker C, Albracht SPJ, Heering HA, Lemay SG. ACS nano. 2008;2:2497. doi: 10.1021/nn800518d. [DOI] [PubMed] [Google Scholar]
- 106.Rosenstein J, Sorgenfrei S, Shepard KL. Custom Integrated Circuits Conference (CICC) 2011 [Google Scholar]
- 107.Crescentini M, Bennati M, Carminati M, Tartagni M. IEEE Transactions on Biomedical Circuits and Systems. 2013:1. doi: 10.1109/TBCAS.2013.2262998. [DOI] [PubMed] [Google Scholar]
- 108.Nazari M, Mazhab-Jafari H. IEEE Transactions on Biomedical Circuits and Systems. 2012 doi: 10.1109/TBCAS.2012.2203597. [DOI] [PubMed] [Google Scholar]
- 109.Farre C, Fertig N. Expert opinion on drug discovery. 2012;7:515. doi: 10.1517/17460441.2012.682056. [DOI] [PubMed] [Google Scholar]
- 110.Sigworth FJ, Klemic KG. IEEE transactions on nanobioscience. 2005;4:121. doi: 10.1109/tnb.2004.842471. [DOI] [PubMed] [Google Scholar]
- 111.Lindsay S. 2012:164201. doi: 10.1088/0953-8984/24/16/164201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Rosenstein JK, Ramakrishnan S, Roseman J, Shepard KL. Nano letters. 2013:1. doi: 10.1021/nl400822r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Kim J, Pedrotti K, Dunbar WB. Sensors and Actuators B: Chemical. 2012 doi: 10.1016/j.snb.2012.11.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Goldstein B, Member GS, Kim D, Xu J, Vanderlick TK, Culurciello E, Member S. 2012;6:111. doi: 10.1109/TBCAS.2011.2182512. [DOI] [PubMed] [Google Scholar]
- 115.Ferrari G, Gozzini F, Molari A, Sampietro M. IEEE Journal of Solid State Circuits. 2009;44:1609. [Google Scholar]
- 116.Vanisri T, Toumazou C. IEEE Journal of Solid-State Circuits. 1995;30:677. [Google Scholar]
- 117.Radeka V. Annual Review of Nuclear and Particle Science. 1988 [Google Scholar]
- 118.Denison TA. Massachusetts Institute of Technology. 2000. [Google Scholar]
- 119.English AC, Richards S, Han Y, Wang M, Vee V, Qu J, Qin X, Muzny DM, Reid JG, Worley KC, Gibbs Ra. PloS one. 2012;7:e47768. doi: 10.1371/journal.pone.0047768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Maitra RD, Kim J, Dunbar WB. Electrophoresis. 2012 doi: 10.1002/elps.201200272. n/a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Pennisi E. Science. 2014;343:829. doi: 10.1126/science.343.6173.829. [DOI] [PubMed] [Google Scholar]
- 122.Rothberg JM, Hinz W, Rearick TM, Schultz J, Mileski W, Davey M, Leamon JH, Johnson K, Milgrew MJ, Edwards M, Hoon J, Simons JF, Marran D, Myers JW, Davidson JF, Branting A, Nobile JR, Puc BP, Light D, Clark TA, Huber M, Branciforte JT, Stoner IB, Cawley SE, Lyons M, Fu Y, Homer N, Sedova M, Miao X, Reed B, Sabina J, Feierstein E, Schorn M, Alanjary M, Dimalanta E, Dressman D, Kasinskas R, Sokolsky T, Fidanza JA, Namsaraev E, McKernan KJ, Williams A, Roth GT, Bustillo J. Nature. 2011;475:348. doi: 10.1038/nature10242. [DOI] [PubMed] [Google Scholar]
- 123.Ivanov AP, Instuli E, McGilvery CM, Baldwin G, McComb DW, Albrecht T, Edel JB. Nano letters. 2011;11:279. doi: 10.1021/nl103873a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Liang X, Chou S. Nano letters. 2008;8:1472. doi: 10.1021/nl080473k. [DOI] [PubMed] [Google Scholar]
- 125.Park YS, Whalley AC, Kamenetska M, Steigerwald ML, Hybertsen MS, Nuckolls C, Venkataraman L. 2007:15768. doi: 10.1021/ja0773857. [DOI] [PubMed] [Google Scholar]
- 126.Lindsay S, He J, Sankey O, Hapala P, Jelinek P, Zhang P, Chang S, Huang S. Nanotechnology. 2010;21:262001. doi: 10.1088/0957-4484/21/26/262001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Song L, Hobaugh MR, Shustak C, Cheley S, Bayley H, Gouauxt JE. 1996:10009. doi: 10.1126/science.274.5294.1859. [DOI] [PubMed] [Google Scholar]
- 128.Laszlo AH, Derrington IM, Brinkerhoff H, Langford KW, Nova IC, Samson JM, Bartlett JJ, Pavlenok M, Gundlach JH. Proceedings of the National Academy of Sciences of the United States of America. 2013:1. doi: 10.1073/pnas.1310240110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Langecker M, Arnaut V, Martin TG, List J, Renner S, Mayer M, Dietz H, Simmel FC. Science. 2012;338:932. doi: 10.1126/science.1225624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Nef C, Frederix PLTM, Brunner J, Schönenberger C, Calame M. Nanotechnology. 2012;23:365201. doi: 10.1088/0957-4484/23/36/365201. [DOI] [PubMed] [Google Scholar]
- 131.Xu B, Tao NJ. Science (New York, NY) 2003;301:1221. doi: 10.1126/science.1087481. [DOI] [PubMed] [Google Scholar]