


Abstract
With the aim of broadening the versatility of lentiviral vectors as a tool in nucleic acid research, we expanded the genetic code in the propagation of lentiviral vectors for site-specific incorporation of chemical moieties with unique properties. Through systematic exploration of the structure–function relationship of lentiviral VSVg envelope by site-specific mutagenesis and incorporation of residues displaying azide- and diazirine-moieties, the modifiable sites on the vector surface were identified, with most at the PH domain that neither affects the expression of envelope protein nor propagation or infectivity of the progeny virus. Furthermore, via the incorporation of such chemical moieties, a variety of fluorescence probes, ligands, PEG and other functional molecules are conjugated, orthogonally and stoichiometrically, to the lentiviral vector. Using this methodology, a facile platform is established that is useful for tracking virus movement, targeting gene delivery and detecting virus–host interactions. This study may provide a new direction for rational design of lentiviral vectors, with significant impact on both basic research and therapeutic applications.
INTRODUCTION
Lentiviral vectors derived from the human immunodeficiency virus (HIV-1) have become major tools for gene delivery in mammalian cells. They consist of the structural and enzymatic components of HIV-1 enveloped by vesicular stomatitis virus glycoprotein (VSVg) (1–5). Such lentiviral vectors have been received particular attention among many different viruses due to its ability to infect both dividing and nondividing cells and integrate the delivered gene into the host genome (6). Currently the lentiviral vectors have been frequently used by biologists to deliver genetic material into a variety of mammalian cells (7). This process can also be performed inside living organisms (8) and has been explored in clinical trials for gene therapy (9–11). In particular, two successful lentiviral vector-based clinical trials in 2013 for treatment of Wiskott-Aldrich syndrome (WAS) and Metachromatic leukodystrophy (MLD) have further strengthened the potential use and impact of the lentiviral vector, especially for boosting lentiviral gene therapy (10–12).
The component that determines the tissue/cell tropism of the lentiviral vector and the gene delivery efficiency is the envelope glycoprotein of VSV (VSVg) (Figure 1A), which mediates host receptor binding, internalization and trafficking within the targeted cells (13,14). A deeper understanding of the properties of the VSVg envelope via structure–function exploration may expand the potential of diverse applications of lentiviral vectors in biomedicine, bio-technology and even nanotechnology (15). Incorporation of chemical moieties such as an azide group on the proper position of the vector surface (Figure 1B) would facilitate click chemistry-mediated introduction of unique properties that enable single-virus tracking, targeted nucleic acid delivery, vector developing for gene therapy and block building for the construction of nano-structured materials. For many of these contemplated applications, the site-specific display of chemical moieties may be crucial, since random or improper modifications of viruses may abrogate the propagation and even the infectivity of progeny viruses (16,17).
Figure 1.
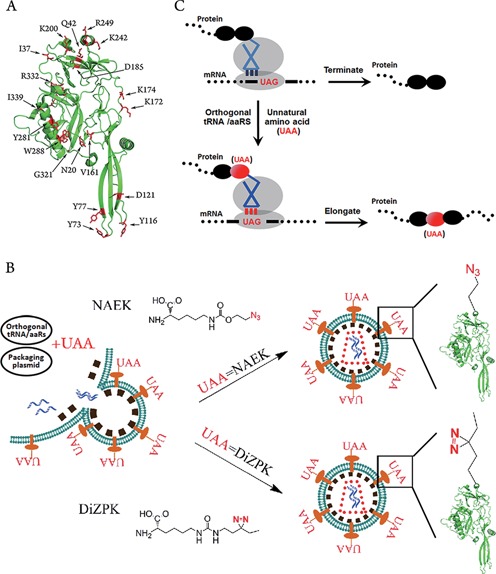
Genetic and site-specific display of unnatural amino acids (UAAs) on the envelope of lentiviral vectors. (A) The structure of lentiviral VSVg protein. Total 33 residues, marked in red, within VSVg were mutated in this study. (B) Schematic representative of lentiviral vector engineering via genetic code expansion for site-specific incorporation of UAAs (NAEK or DiZPK) on the vector surface. (C) Schematic representation of genetic code expansion for site-specific incorporation of UAAs. The UAAs were incorporated into VSVg protein via UAG-code and an orthogonal aaRS/tRNA.
Here, we report the development of a viable and facile platform for site-specific incorporation of unnatural amino acids into lentiviral vectors via genetic code expansion. Genetic code expansion is an artificial process that usually uses a stop codon to encode an unnatural amino acid (UAA) into a protein of interest (18–22) (Figure 1C). The high fidelity of genetic code expansion plus the facile technique of mutagenesis allows precise control of the placement of the chemical moiety-containing UAA and the following orthogonal modification into the lentiviral vector, providing a foundation for the systematic structure–function exploration of such a vehicle for delivery of a variety of functional nucleic acid molecules. Via the display of azide- and diazirine-moieties, modifiable sites on the surface of the lentiviral vector were identified that have a minimal effect on propagation and infectivity, even upon tailing with proper ligands. Using this approach, site-specific and stoichiometric conjugation of a variety of biophysical probes, tags and novel functional molecules on the envelope of lentiviral vectors is realized, which has a significant impact on single-virus tracking, targeted gene delivery and other biological applications.
MATERIALS AND METHODS
Material
Plasmids used to propagate the lentiviral vector included pCMV-VSVG (Addgene), pNL 4–3 (NIH AIDS Reagent Program) and pAdvantage (Promega). The methanosarcina barkeri MS pyrrolysyl tRNA synthetase/tRNACUA pair (MbPylRS/tRNACUA) and pSupAR-Mb-DiZPK-RS for site-specific incorporation of UAAs were developed in-house as previously reported (23,24), The mutant plasmids (pCMV-VSVG-TAG) containing an amber codon within the open reading frame were obtained from the wild-type pCMV-VSVG via site-directed mutagenesis (Agilent Technologies) and confirmed by gene sequencing (BGI Beijing). All plasmids used for transfection were amplified using a Maxiprep kit (Promega), according to the manufacturer's instructions. Antibodies used included a mouse monoclonal anti-VSVg (Sigma), rabbit monoclonal anti-GAPDH (Cell Signaling Technology), goat anti-mouse IgG-HRP or goat anti-rabbit IgG-HRP (ZSGB-BIO), monoclonal anti-integrin αvβ3 (Millipore), normal rabbit IgG (Santacruz), normal mouse IgG (Santacruz), Alexa 488-labeled secondary antibody (Cell Signaling Technology) and Alexa 594 Donkey Anti-Mouse IgG (Invitrogen).
Nϵ-2-azidoethyloxycarbonyl-L-lysine (NAEK) and ([3-(3-methyl-3H-diazirine-3-yl)-propamino] carbonyl)-Nϵ–lysine (DiZPK) were synthesized as previously reported (23,24) and characterized by mass spectroscopy (Supplementary Figure S1). Dibenzocyclooxtyne -arginine-glycine-aspartic acid (DIBO-RGD) and Dibenzocyclooxtyne-arginine-alanine -aspartic acid (DIBO-RAD) were purchased from GL Biochem (Shanghai) Ltd and dissolved in phosphate buffered saline (PBS) to a desired concentration. DIBO-Alexa 488, 1, 1′-dioctadecyl-3,3,3′,3′ -tetramethylindocarbocyanine perchlorate (DiI) and 4′,6-diamidino-2-phenylindole (DAPI) was purchased from Life Technologies; Dibenzocyclooxtyne- Polyethylene glycol (DIBO-PEG) was synthesized in-house.
Cell culture
Cell lines Human Embryonic Kidney 293T (HEK-293T), HeLa, MDA-MB-435S, U-87 MG (U87) and Michigan Cancer Foundation-7 (MCF-7) were obtained from the National Platform of Experimental Cell Resources for SCI-Tech (Beijing, China). HEK-293T, HeLa and MCF-7 cells were maintained in Dulbecco's Modified Eagle Medium (DMEM, Macgene), Roswell Park Memorial Institute medium 1640 (RPMI1640, Macgene) medium and Modified Eagle Medium (MEM; Macgene), respectively, were supplemented with 10% (v/v) fetal bovine serum (FBS; Gibco), 100-IU/ml penicillin and 100-μg/mL streptomycin (Macgene). These cells were cultured at 37°C in a humidified incubator under 5% CO2 and passaged every 2 days at ∼80–90% confluency. U87 cells were maintained in MEM supplemented with 10% (v/v) FBS, 100-IU/ml penicillin and 100-μg/ml streptomycin. MDA-MB-435S cells were maintained in Leibovitz L-15 medium (Macgene) supplemented with 10% (v/v) FBS, 0.01-mg/mL bovine insulin (Macgene), 0.01-mg/ml glutathione (Gibco), 100-IU/ml penicillin and 100-μg/ml streptomycin. These cell lines were also cultured at 37°C in a humidified incubator under 5% CO2 and passaged every 3 days at ∼80–90% confluency.
Expression of VSVg proteins bearing NAEK or DiZPK in HEK 293T cells
Each well of a 6-well plate was seeded with 3 × 105 HEK-293T cells. Twenty-four-hours later, 1.2-μg of pCMV-VSVG-TAG or wild-type pCMV-VSVG plasmid was co-transfected into HEK 293T cells with 0.6-μg MbPylRS/tRNACUA plasmid for NAEK or pSupAR-Mb-DiZPK-RS plasmid for DiZPK in a single well using Mega Tran 1.0 (Origene) as the transfection reagent, according to the manusfacturer's instructions. The medium was replaced by fresh medium supplemented with 10% (v/v) FBS; GIBCO), 100-IU/ml penicillin, 100-mg/ml streptomycin (Macgene) and 1-mM NAEK or DiZPK 6 h post-transfection. The total protein was harvested 48 h post-transfection using Radio-Immunoprecipitation Assay (RIPA) buffer [50-mM Tris-HCl pH:8.8, 150-mM NaCl, 0.1% sodium dodecyl sulfate (SDS), 0.5% deoxycholate, and 0.5% NP-40], and protein concentrations were quantified using the BCA Protein Assay kit (Thermo), according to the manufacturer's instructions. The samples were separated on 9% SDS-polyacrylamide gel and transferred to PVDF membrane (Millipore) for immunoblotting detection. VSVg was detected using a mouse monoclonal anti-VSVg (Sigma, V5507); while GAPDH was used as a loading control and was visualized using a rabbit monoclonal anti-GAPDH (Cell Signaling Technology). The secondary antibodies were goat anti-mouse IgG-HRP and goat anti-rabbit IgG-HRP (ZSGB-BIO). The chemiluminescent signal was captured with a digital imaging system (ChemiDoc™ XRS+ System, BIO-RAD).
Conjugation of Alexa 488 with NAEK-containing VSVg was carried out through copper-free click chemistry. The wild-type VSVg proteins and NAEK bearing VSVg at Y77 or I339 were purified by immunoprecipitation (IP) using mouse monoclonal anti-VSVg (Sigma) immobilized on Protein A/G PLUS-Agarose (SantaCruzBio). The IP products were then reacted with DIBO-Alexa 488 at 4°C for 2 h; the reaction products were characterized by 9% SDS-PAGE. After the electrophoresis, the fluorescent signal of the band was detected at the desired location through fluorescent scanning using a digital imaging system (ChemiDoc™ XRS+ System, BIO-RAD). The proteins in the gel were transferred to PVDF membrane (Millipore) for immunoblotting detection. VSVg was detected using a mouse monoclonal anti-VSVg (Sigma Aldrich) (1:5000) and goat anti-mouse IgG-HRP (ZSGB-BIO). The chemiluminescent signal was captured by a digital imaging system (ChemiDoc™ XRS+ System, BIO-RAD).
Mass spectrometry measurement of VSVg-bearing NAEK
VSVg proteins with or without NAEK incorporated at different positions were purified by IP, which was performed using mouse monoclonal anti-VSVg (Sigma Aldrich) and Protein A/G PLUS-Agarose (SantaCruzBio), following the manufacturer's instructions. The IP products were separated by 9% SDS-PAGE and visualized with Coomassie blue staining. The corresponding protein bands were sliced and digested in gel with sequencing-grade trypsin overnight at 37°C. The resulting peptides were extracted twice with 5% TFA/60% acetonitrile and once with acetonitrile, and then concentrated. Peptide mixtures were suspended in 1% FA and separated by online reversed-phase (RP) nanoscale capillary liquid chromatography (Easy-nLC II, Thermo Fisher Scientific). The peptide solution was autosampled directly and bound onto a trapping column packed with 5-μm C18 reversed-phase material. The peptide mixtures were separated on an analytical column (75 μm–10 cm) packed with 3-μm C18 reversed-phase material with a 40-min effective linear gradient from 5% to 40% of 100% (v/v) CAN, 0.1% (v/v) formic acid. The data-dependent mass spectra were acquired with an LTQ-Orbitrap mass spectrometer (Thermo Fisher) equipped with a nanoelectrospray ion source. An electrospray voltage of 2.2 kV was used. Full-scan MS spectra (m/z 350−1800) were acquired in the Orbitrap analyzer, with a resolution of 60 000 (FWHM). The top 15 most abundant precursor ions from each MS scan with charge ≥2 were selected for MS/MS scans in the linear ion trap analyzer. Database searches were performed by an in-house Mascot server (Matrix Science Ltd) on a Swiss Prot database. Tolerances were adjusted to 10 ppm (precursor) and 0.8 Da (fragment).
Propagation of wild-type, and NAEK- and DiZPK-containing lentiviral vectors
The detailed procedure for propagation of wild-type lentiviral vectors has been described previously (25). Briefly, 3 × 105 HEK 293T cells per well were seeded into 6-well plates 24 h before transfection. HEK 293T cells were co-transfected with 1.0 μg plasmid pNL4–3, 0.7 μg pCMV-VSVG and 0.3 μg pAdvantage. After changing the medium 6 h post-transfection, the cells were continuously cultured for 48 h and the virus-containing supernatant was collected, concentrated by ultracentrifugation (Avanti J-30I, 82700 g, 4°C for 2 h) and re-suspended in 50-mM Tris•HCl (pH 7.40). For production of NAEK- or DiZPK-containing lentiviral vectors, an almost identical procedure was carried out, but pCMV-VSVG plasmid was changed to mutant pCMV-VSVG-TAG and aaRS/tRNA plasmid (MbPylRS/tRNACUA or pSupAR-Mb-DiZPK-RS) (0.35 μg) and the medium was supplemented with 1-mM NAEK or DiZPK.
Conjugation of Alexa 488, RGD/RAD ligands and PEG molecules with NAEK-containing lentiviral vectors
The general procedure for conjugation of Alexa 488, arginine-glycine-aspartic acid (RGD) and arginine-alanine-aspartic acid (RAD) is as follows: 100-μL of concentrated NAEK-containing lentiviral vector was separately conjugated using the same volume of Alexa Fluor® 488 DIBO alkyne (150 μM) (Life Technologies), RGD-DIBO alkyne and RAD-DIBO alkyne (200 μM) dissolved in PBS buffer at 4°C for 1 h. Afterward, the unreacted ligands were excluded by buffer exchange using 100-kDa filters (Amicon Ultra-0.5-ml Centrifugal Filters). The conjugated products were assayed with luciferase assay and western blotting. A parallel experiment was performed using the same procedure, except that the wild-type vector was substituted for the NAEK-containing lentiviral vector. Vectors thus obtained from such treatment acted as controls for each conjugated vector in an infectivity test and for normalization.
PEG conjugation of NAEK-containing lentiviral vectors was carried out as follows: the representative mutant lentiviral vector A246NAEK was incubated with 5K PEG-DIBO (200 μM) at RT for 2 h. Afterward, the conjugated products were purified by ultrafiltration using proper filter and PBS as a buffer to remove unreacted PEG-DIBO. Using the same approach, different sizes of PEG-DIBO were conjugated to the NAEK-containing lentiviral vector. The PEG-conjugated lentiviral vectors were then mixed with loading buffer (Applygen), boiled for 5 min and resolved on 9% SDS-polyacrylamide gels for immunoblotting analysis.
Vector infectivity test
Each well of a 96-well plate was seeded with 5 × 103 HeLa cells, which were grown in 5% CO2 at 37°C overnight. Three-hundred-microliters of vector-containing supernatant was added in triplicate wells of a 96-well plate. After 24 h, the culture medium was replaced by DMEM supplemented with 10% FBS and incubated at 37°C under 5% CO2 for 48 h. After removal of the medium, 40-μL of Bright-Glo reagent (Promega) was added to each well and then the relative fluorescence was detected by an illuminometer (Berthold, Centro XS3 LB 961).
Immunofluorescence assay of lentiviral vectors
HeLa cells were plated in 35-mm glass-bottom culture dishes one day prior to the performance of the immunofluorescence assay. Upon reaching 50% confluence, cells were incubated with unconjugated or conjugated lentiviral vectors at 4°C for 30 min to synchronize binding to lentiviral vectors. After removal of excess lentiviral vectors, the infected cells were fixed with 4% paraformaldehyde (PFA) for 15 min at RT, permeabilized for 10 min by PBST (0.5% (v/v) Triton X-100 in PBS). The treated cells were incubated with blocking buffer (10% (v/v) normal goat serum, 1% (m/v) BSA in PBT) for 1 h at RT, and then sequentially incubated with 2 μg/ml anti-VSVG antibody overnight at 4°C, 0.4 μg/ml Alexa Fluor® 594 Donkey Anti-Mouse IgG (Invitrogen) for 1 h in the dark and 2 μg/ml (DAPI) (Life Technologies) for 10 min in the dark at RT. The samples were rinsed three times with 1 × PBS between the successive steps. The fluorescent images of cells were recorded by a confocal microscope (Leica, TCS SP8, Germany).
Dynamic tracking of lentiviral vectors
HeLa cells plated in 35-mm glass-bottom culture dishes were stained with 5-μg/ml Hoechst 33342 (λex 350 nm, λem 461 nm) (Life Technologies) at 37°C for 30 min in the dark to label nucleic acids, and were then stained with 2-μg/ml DiI (λex 549 nm, λem 565 nm) (Life Technologies) at 4°C for 10 min in the dark to label the plasma membrane. Then cells were incubated with Alex488-labeled or -unlabeled lentiviral vector for 30 min at 4°C to allow virus binding. The excess lentiviral vector were discarded by washing in PBS and then the treated cells were moved to an incubator at 37°C. The dynamic behavior of the viruses was tracked fluorescently with a spinning-disk confocal microscope equipped with a CO2 online culture system (Perkin Elmer). The Alexa 488 was detected using continuous excitation with a 488-nm laser and DiI was detected using continuous excitation with a 555-nm laser.
Flow-cytometry analysis
The expression of integrin αvβ3 on the surface of MDA-MB-435S, U87 and MCF-7 cells was determined by flow cytometry. Briefly, 5 × 105 cells were trypsinized and fixed with 4% paraformaldehyde (PFA) at RT for 15 min, washed with PBST (0.1% Triton in PBS) and centrifuged for 5 min at 300 g three times. The treated cells were blocked with 10% BSA in PBST for 1 h, washed and incubated with monoclonal anti-integrin αvβ3 (Millipore) (1:1000) at 4°C overnight. Afterward, the cells were washed with PBST three times, incubated with Alexa 488-labeled secondary antibody (1:1000) (Cell Signaling Technology) at RT for 1 h, washed with PBST three times and analyzed by Guava EasyCyte Plus (Guava Technologies). Measurements in which the primary antibody was replaced by an irrelevant antibody (normal rabbit IgG, 1:1000) (Santacruz) were used as controls. For each experiment, 5000 cells were analyzed.
Targeting delivery of modified lentiviral vectors
Concentrated wild-type and mutant lentiviral vectors bearing NAEK at 192, 243, 246, 247 and 252, respectively, were used for targeted delivery of modified lentiviral vectors. The lentiviral vectors were labeled as described above. MDA-MB-435S, U87 and MCF-7 were seeded in 96 plants one day before infection, and the infectivity was validated as previously described.
Photocrosslinking of DiZPK-bearing lentiviral vectors and host cells
HEK 293T cells were plated in 100-mm culture dishes one day prior to photocrosslinking. When HEK 293T cells had reached 80% confluence, they were incubated with DiZPK-bearing or wild-type lentiviral vectors in 1-mL PBS for 30 min at 4°C to synchronize lentiviral vectors-binding to the cell membrane. Photocrosslinking was performed by irradiation of the cells with UV light (365 nm) for 10 min using a Hoefer UVC 500 Crosslinker installed with 365-nm-wavelength UV lamps (Amersham Biosciences) at a distance of 3 cm on ice. After removal of the PBS, the protein was extracted using RIPA buffer and centrifugation at 12 500 g for 15 min at 4°C, then the protein extract was enriched by IP and analyzed by Tricine-SDS-PAGE.
RESULTS
Evaluating the compatibility of genetic code expansion for site-specific incorporation of an azide moiety into a viral envelope protein
We first investigated the compatibility of the amber suppressor with the orthogonal MbPylRS/MbtRNACUA pair, a shuttle system for encoding UAAs in bacterial and mammalian cells (23), in the expression of the lentiviral envelope protein VSVg. A VSVg-coding plasmid, pCMV-VSVG, was mutated with the codon of residues Y77 or I339 (13) (Figure 1A), changed to the amber codon (TAG). Then the mutant plasmids, pCMV-VSVG-TAG77 and pCMV-VSVG-TAG339, were respectively transfected into HEK 293T cells to test their ability to display Nϵ-2-azidoethyloxycarbonyl-l-lysine (NAEK; Figure 1B), an azide-bearing amino acid (23), at the defined position of VSVg. Western blotting detected a complete VSVg protein when the plasmid encoding the MbPylRS/MbtRNACUA pair (23) was co-transfected into HEK 293T cells in the presence of 1-mM UAA (Figure 2A). No VSVg expression was detected in the absence of UAA, which suggests that UAA is essential for the expression of mutant viral VSVg protein during genetic code expansion. The presence of NAEK in the VSVg protein was confirmed by a click reaction between the mutant VSVg protein and DIBO-Alexa 488, a fluorescence probe, by which azide-bearing VSVg became visible through fluorescence scanning (Figure 2B). Further confirmation of the NAEK display at the defined site was carried out by LC-MS/MS peptide sequencing as shown in Figure 2C.
Figure 2.
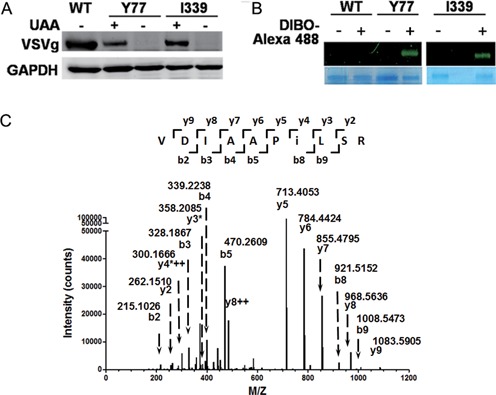
Site-specific incorporation of NAEK VSVg envelope protein. Y77 and I339 were chosen as representatives to demonstrate the compatibility of genetic code expansion with the expression of viral protein. (A) The unnatural amino acid (UAA)-dependent expression of VSVg proteins carrying NAEK at sites Y77 and I339, respectively. The absence (–) and presence (+) of UAA had different effects on VSVg expression. The expression level of VSVg was detected by western blotting analysis and GAPDH acted as an internal loading control. (B) Verification of NAEK incorporation into VSVg protein via Alexa 488 probe. Incorporation of NAEK into VSVg protein makes it possible to conjugate with DIBO-Alexa 488 via Click reaction and is thus visible. No fluorescent signal was detected for wild-type VSVg (WT) due to lack of NAEK residue. The VSVg protein, purified by IP, was incubated with (+) or without (–) DIBO-Alexa 488 at 4°C for 1 h and was then characterized by 9% SDS-PAGE. The upper bands show the corresponding fluorescent scanning results; the lower bands show the loading of VSVg protein as detected by Coomassie blue staining. (C) Verification of NAEK incorporation at the defined site (I339 as a representative) by LC-MS/MS peptide sequencing. The MS/MS fragmentation spectrum of a tryptic peptide derived from the purified protein confirm the incorporation of NAEK at I339. The mass difference of 2y4*++-y3*[2×300.1666–2-(358.2085–1) = 241.1247] corresponds well to the mass of NAEK residue (259.13–18.01 = 241.12).
The facile incorporation of the azide-bearing amino acid at sites Y77 and I339 suggests the broad utility of this approach for the systematic study of VSVg protein. Thirty-three representative residual sites, most of which are located on the vector surface (13) (Figure 1A), were chosen with the aim of individual incorporation of azide-bearing UAA. The choice of these sites was dictated by their locations being either potentially involved or not involved in host recognition, binding, or virus–host membrane fusion. Similar to the display of UAA at sites Y77 and I339, all codons for the chosen amino acids were separately changed to TAG using their corresponding primers (Supplementary Table S1). Next, the mutant plasmids were tested in parallel for their ability to incorporate NAEK at the defined positions of VSVg. Western blotting indicated that a full-length VSVg protein was detected for most mutant sites except Y73, K200, Y281, W288, D344 and G345 (Supplementary Figure S2), which are located at the trimerization domain or the rigid block domain. Quantification of UAA at all successful sites indicated that the most efficient sites were D35, Y77, E201, K225, R249, I339 and R342, which displayed approximately 50–70% of the expression level of the wild-type VSVg. These were followed by sites I37, Q42, D121, V161, K172, K174, D185, D192, A240, K242, D243, A246, A247 and M346, which were about 20–50% as efficient as the wild-type VSVg (Figure 3). The remaining sites displayed poor UAA incorporation compared with the wild-type VSVg: approximately 5–20%.
Figure 3.
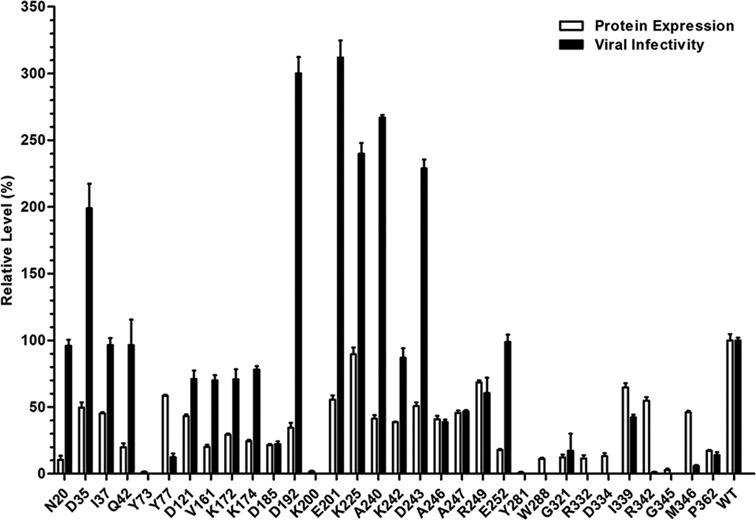
Comparisons of the effect of UAA incorporation at different sites on expression of VSVg protein and propagation/infectivity of progeny vectors. The expression levels of VSVg, which reflect the efficiency of UAA incorporation, were analyzed by western blotting (Supplementary Figure S2), quantitated by scanning the VSVg band and then normalized to that of wild-type VSVg. The infectivity of lentiviral vectors containing NAEK was detected by luciferase assay and normalized to that of the wild-type lentiviral vector. All quantitative data shown are average values with standard deviations from triplicate experiments.
Establishing a facile platform for site-specific engineering of a lentiviral vector
After the successful incorporation of NAEK into VSVg protein, we next determined whether it could be displayed on the lentiviral vector. To produce NAEK-bearing lentiviral vectors (Figure 1B), pCMV-VSVG-TAG77 or pCMV-VSVG-TAG339 was transfected into HEK 293T, together with other three plasmids, MbPylRS/MbtRNACUA pair, pNL4–3 (the plasmid for expression of gag, pol, rev enzymes and luciferase-coding lentiviral genome) and pAdvantage (the plasmid for enhancing packaging efficiency). The production and infectivity of the progeny vectors was verified and quantified by a luciferase assay, that is, transduction of putatively produced viral vectors into HeLa cells. From the transduced HeLa cells, we detected luciferase signals (Figure 4A), suggesting the success of propagation of lentiviral vectors. The packaging condition, especially the ratio among the four plasmids, was then screened and optimized for propagation of lentiviral vectors. We found that the maximum reading was detected when the mass ratio of pCMV-VSVG-TAG:pPylRS/tRNACUA:pAdvantage:pNL4–3 was 0.7:0.35:0.3:1. We also found that 0.25 mM UAA in the culture medium was sufficient for the propagation of progeny viruses. A further increase in UAA concentration produced no increase in luciferase readings, suggesting that UAA is saturated for viral vector production. The absence of UAA in the packaging system produced only a background level of luciferase signal (Figure 4A), indicating that no viral vector was produced in the absence of UAA. This demonstrates the critical role of NAEK in the propagation of lentiviral vectors.
Figure 4.
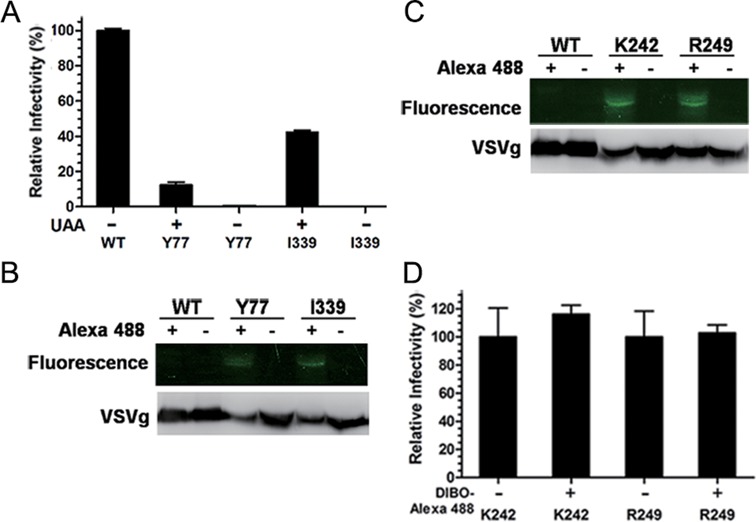
Site-specific display of NAEK on the surface of lentiviral vectors. (A) Test of the infectivity of the lentiviral vector containing NAEK at site Y77 or I339, respectively, by luciferase assay using HeLa cells as the host. The infectivity of lentiviral vectors was quantified by luminescence measurement and normalized to that of the wild-type lentiviral vectors (WT). (B) Verification of NAEK expression on lentiviral vectors by their capability to conjugate with the DIBO-Alexa 488 probe via click reaction, which made NAEK-bearing protein visible by fluorescence upon conjugation with Alexa 488. The wild-type (WT), acting as a negative control, and mutant lentiviral vectors were separately incubated with (+) or without (–) DIBO-Alexa 488 and then analyzed by fluorescence scanning (upper bands) and western blotting (lower bands) via 9% SDS-PAGE. (C) Generation of highly infectious NAEK-containing lentiviral vectors with (+) or without (–) Alexa 488 at site K242 and R249, respectively, which were analyzed by fluorescent scanning (upper bands) and western blotting(lower bands). (D) Characterizations of the infectivity of lentiviral vectors with (+) or without (–) Alexa 488 at site K242 and R249, respectively by luciferase assay. All quantitative data shown are average values with standard deviations from triplicate experiments. These lentiviral vectors produced from the same transfection were split, treated and compared in parallel.
Display of NAEK onto progeny lentiviral vectors was confirmed by a bio-orthogonal reaction between the progeny lentiviral vectors and the probe DIBO-Alexa 488. The term bio-orthogonal reaction refers a chemical reaction that occurs inside of living systems without interfering with the native biochemical processes. A green fluorescent band was detected upon incubation of the progeny lentiviral vectors with the probe (at 25°C for 1 h) and analysis by SDS-PAGE electrophoresis (Figure 4B). As a control, there was no fluorescence detected for both wild-type lentiviral vectors and mutant lentiviral vectors. LC-MS/MS peptide sequencing of the fluorescently labeled gel bands indicated that Alexa 488 was linked to lentiviral vectors via the displayed NAEK (Supplementary Figure S3). We thus concluded that genetic code expansion enabled the display of NAEK at the defined sites of the lentiviral vector. However, the remaining luminescence readings of only approximately 10% and 40% of the wild-type viral vector readings (Figure 4A) indicated that the yield/infectivity of the chimera vectors was significantly lowered. We concluded that both Y77 and I339 within the lentiviral vectors might not be the proper sites for the display of UAA; incorporation of UAA at these sites significantly affects virion assembly and/or infectivity.
Exploring the structure–function relationship of lentiviral vectors
Having systematically displayed UAAs on the viral envelope protein, we next determined whether such NAEK-carrying VSVg proteins still have the ability to partake in virion assembly. If so, which sites displaying UAA have the minimal deleterious effects on virion assembly and infectivity? Similar to displaying UAA at Y77 and I339, all 33 mutant pCMV-VSVG-TAG plasmids were co-transfected separately with the set of package plasmids into HEK 293T cells, and the putative harvest lentiviral vectors were tested by luciferase assay. As shown in Figure 3, we had detected the luminescence of cells infected by lentiviral vectors, and normalized the luminescence of mutant lentiviral vectors to that of wild type lentiviral vector. High luminescence was detected when UAA was displayed at N20, D35, I37, Q42, D192, E201, K225, A240, K242, D243 and E252, comparable with (>95%) and higher than that of the wild-type lentiviral vector. This suggests that these surface sites have a high tolerance to UAA display; replacement of the natural amino acid with NAEK at these sites has no effect on virion propagation and infectivity. To display UAA at sites D121, V161, K172, K174, A246, A247, R249 and I339, an approximately 40–85% of the luminescence was maintained for the mutant lentiviral vectors (Figure 3). This suggests that these sites are also tolerant to UAA engineering, although the efficiency of virion assembly and/or infectivity somehow decreased. For the remaining sites, a < 25% luminescence was detected, suggesting that UAA at those sites had a significantly deleterious effect on progeny virus production and/or infection.
Developing azide-containing lentiviral vectors as a tool for single-virus tracking
Having established the approach to incorporate NAEK site-specifically into lentiviral vectors, we then demonstrated its usefulness in virology. The chemically engineered lentiviral vectors with NAEK at sites K242 or R249, which have a minimal effect on both VSVg expression and virion infectivity (Figure 3), were separately incubated with DIBO-Alexa 488 to generate visible vectors. As a result of this treatment, visible particles were obtained (Figure 4C), indicating that the fluorophore probe was successfully coupled to the lentiviral vectors via a strain-promoted alkyne-azide cycloaddition. Furthermore, we found that the chimera lentiviral vector showed almost the same luminescence reading as its parental mutant lentiviral vector (Figure 4D), demonstrating that introduction of Alexa 488 on lentiviral vector by copper-free bio-orthogonal reaction had very little, if any, effect on the infectivity. This is significantly different from the copper-catalyzed azide-alkyne cycloaddition, which usually caused irreversible oxidative damage due to the copper (I) catalyst (26).
We then used this visible lentiviral vector for single-virus tracking, to demonstrate its significance. The visible chimera lentiviral vector, its parental mutant lentiviral vector and the wild-type lentiviral vector were added to HeLa cells in parallel. After incubation at 4°C for 30 min to synchronize binding, these three samples were fixed and analyzed by immunofluorescence using confocal microscopy. The chimera vector, detected by both VSVg antibody-staining (red color) and Alexa 488 (green color), was clearly observed to be localized on the cell surface (yellow color) (Figure 5A). As a control, no Alexa 488 signaling, but only VSVg antibody-staining was detected for both the wild-type and the parental mutant lentiviral vector. The real-time movements of fluorescently labeled single lentiviral vector were then tracked on a spinning-disk confocal microscope platform by shifting infected HeLa cells to 37°C in a CO2 online culture system. Tracking a single virus is of importance in the understanding of the mechanism of viral entry and transport (27), and is also important for demonstrating the viability and utility of this method. Our experiment was focused on monitoring the movement of the enveloped single virus from cell membrane to cytoplasm, via either fusion or endocytosis, a key step in successful infection.
Figure 5.
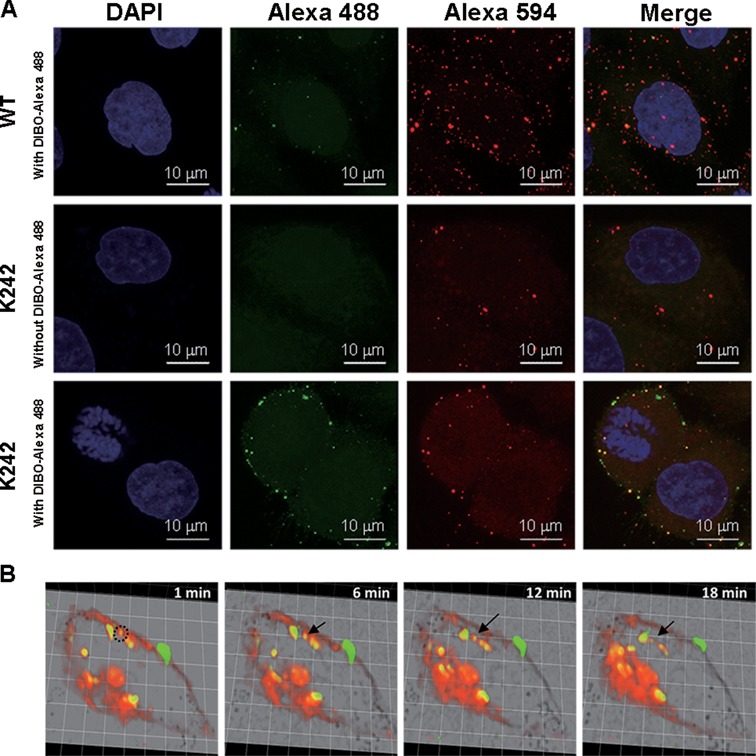
Development of a NAEK-containing lentiviral vector as a tool for single-virus tracking. (A) Characterization of the visibility of NAEK-containing lentiviral vectors at K242 upon treatment with DIBO-Alexa 488. The wild-type vectors (WT) treated by DIBO-Alexa 488 and NAEK-containing lentiviral vector without treating by DIBO-Alexa 488 act as the negative control. (B) Real-time monitoring of the dynamic process of entry and transport of Alexa 488-conjugated lentiviral vector. HeLa cells were stained with Hoechst 33342 and DiI, respectively, to label host nucleic acids and the plasma membrane. After incubation of HeLa cells with lentiviral vector at 4°C for 30 min to synchronize binding, the infected HeLa cells were placed at 37°C in a CO2 online culture system and tracked using a spinning-disk confocal microscope platform. The Alexa 488 was detected at 488 nm, and DiI was detected at 555 nm. A series of images were selected from 1 to 18 min that reflect the dynamic process of endocytosis of lentiviral vectors.
Figure 5B shows the series of images that were selected from the real-time movement of lentiviral vectors (Supplementary Movie S1). Visualization revealed that the green vector remained bound in the 1–6 min of co-culture with HeLa cells at 37°C, and had started separating from the cell membrane (shown in red; DiI staining; Figure 5B). The vector subsequently traveled into the cytoplasm, clearly via endocytosis, as seen by the green vector enclosed by the red cell membrane, and separated completely from the cell membrane in 12 min. Furthermore, the viral motility in different pathways within the infected cell was quantitatively analyzed (Supplementary Figure S4); the success of viral particle tracking was attributed to the long life (>30 min) of the Alexa 488 signal. Our observation was generally consistent with previous experiments tracking lentiviral vectors (28). However, in our study the endocytosis of lentiviral vector required only 12 min (Figure 5B), significantly less than that required by Quantum Dots-labeled lentiviral vectors, which takes more than 30 min (28). This may reflect a size difference of the probe, which is only 2% of the size of a single lentiviral vector for Alexa 488, and thus it has a different effect on viral velocity.
Developing an azide-containing lentiviral vector as a vehicle for targeted gene delivery
To broaden the versatility of lentiviral vectors in therapeutics, the lentiviral vector was tethered to RGD, a tripeptide composed of l-arginine (Arg), glycine (Gly) and l-aspartic acid (Asp), via the azide moiety to generate a targeted delivery vehicle. RGD is a motif recognized by a subset of integrins, which are cell surface adhesion molecules that mediate both cell–substratum and cell–cell interactions (29). Thus, the RGD motif might have potential use for coating lentiviral vectors to improve their delivery efficiency and selectivity in targeting integrin αvβ3-expressing tumor cells. The lentiviral vectors containing azide moiety at certain sites that have less effect on virion assembly and infectivity (Figure 3) were separately conjugated with ligand DIBO-RGD or DIBO-RAD under the same conditions as that for DIBO-Alexa 488. RAD is a mutated motif of RGD in which glycine is changed to alanine, and thus acts as a negative control (30). In addition, a cyclic RGD (Supplementary Figure S5), instead of its linear form, was utilized because of its greater stability and selectivity as previously reported (31).
These obtained vectors were then transfected into MDA-MB-435S cells (32), which have roughly 3-fold higher levels of integrin αvβ3-expression, analyzed by flow cytometry, than in MCF-7 cells (33), which acted as controls (Figure 6A). We found that conjugation of RGD at different sites of lentiviral vectors led to different threshold effect on infectivity toward MDA-MB-435S. A significant enhancement was observed for site 246, an almost 10-fold increase compared with its RAD-carrying counterpart and azide-containing lentiviral vector (control). Site 192 showed an approximately 7-fold enhancement, and sites 243 and 247 exerted only a mild (3–5-fold) enhancement (Figure 6B). No improvement was observed upon conjugation of RGD at site 252. In contrast, tethering RAD rather than RGD to the lentiviral vector resulted in little improvement in infectivity for all these sites, disclosing the critical role of RGD in the enhanced infectivity. However, for MCF-7 cells, the efficiency of lentiviral infection was not affected by either RGD or RAD conjugation (Figure 6C), presumably due to the low expression of integrin αvβ3 on the cell surface. Therefore, not random, but only site-specific conjugation of RGD at the proper positions of the lentiviral vector is an effective way to improve the infectivity toward integrin-highly expressed cells.
Figure 6.
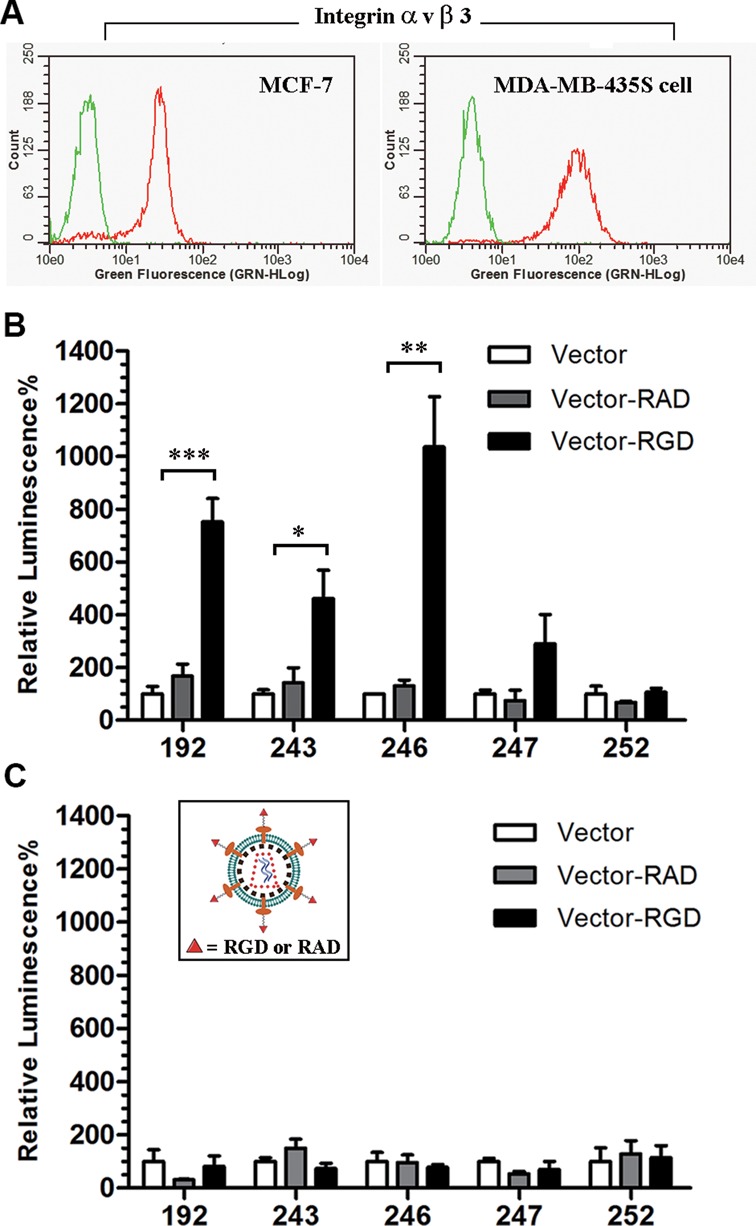
Development of a NAEK-containing lentiviral vector as a vehicle for the selective delivery of nucleic acid macromolecules into cells with high expression of integrins. (A) Expression of integrin αvβ3 in MDA-MB-435S vs. MCF-7 cells as detected by flow cytometry. The trypsinized cells, after sequential blocking by 10% BSA and washing, were incubated with monoclonal integrin αvβ3 antibody (red line) and an isotype control IgG antibody (green line) overnight in parallel and then incubated with Alexa 488 labeled secondary antibody for 1 h. After washing three times with PBST, the treated cells were analyzed by Guava EasyCyte Plus. The level of integrin αvβ3 expression on the cell surface of MDA-MB-435S cells is almost 3-fold higher than that on MCF-7 cells according to flow cytometry. (B) Characterization of the enhanced infectivity of NAEK-containing lentiviral vectors (Vector) and vectors upon conjugation with RGD (Vector-RGD) or RAD (Vector-RAD) at different sites towards MDA-MB-435S cells. The significance was confirmed by one-way ANOVA test (*P < 0.05, **P < 0.01, ***P < 0.001). (C) Characterization of the infectivity of the NAEK-containing vector and the RGD- or RAD-conjugated lentiviral vectors in MCF-7 cells. All quantitative data shown are average values with standard deviations from triplicate experiments.
A similar trend of enhanced infectivity was observed in U87 cells, a human primary glioblastoma cell line that also exhibit high levels of integrin αvβ3-expression (34) (Supplementary Figure S6A). Furthermore, when the reporter luciferase gene in the lentiviral genome was replaced by a visible green fluorescent protein (GFP) gene, we also found that the RGD-conjugated lentiviral vector was much more efficient than the non-conjugated and the corresponding RAD-conjugated vector in the delivery of these genes into U87 cells that have high integrin expression (Supplementary Figure S6B). For MCF-7 cells with low integrin expression, no significant differentiation was found for these three vectors. Our results suggest that site-selective conjugation of RGD as a method of enhancing lentiviral vector tropism is an attractive way to modify lentiviral vector tropism and improve gene delivery; this has a significant role to play in expansion of the versatility of lentiviral vector as a tool for targeting delivery of therapeutic genes.
Developing an azide-containing lentiviral vector as an anchor for site-specific PEGylation
One potential issue that exists in lentiviral vector-based gene therapy is immunogenicity, which is caused by certain peptide residues present on the vector surface leading to adaptive immune responses. Masking antigenicity domains with polyethyleneglycol (PEG) chains is an effective way to decrease immunogenicity of therapeutic proteins (35). For proof of this concept, the azide-containing lentiviral vector at site 246, which has been successfully tethered with RGD ligand, was chosen for conjugating with DIBO-PEG via the same azide-alkyne cycloaddition. We found that incubation of the azide-containing lentiviral vectors with 5-kDa DIBO-PEG generated an envelope protein with an increased molecular weight, as characterized by western blotting (Supplementary Figure S7A). In a control experiment, wherein wild-type lentiviral vector was incubated with DIBO-PEG, no new VSVg protein was detected. This suggests that the lentiviral vector was indeed PEGylated via the incorporated azide moiety. We also found that this coupling reaction was quick, being usually accomplished within 2 h (Supplementary Figure S7B). Using the same methodology, linear 10- and 20-kDa PEG moieties were also separately coupled to the azide-containing lentiviral vector (Supplementary Figure S7C). Clearly, our approach is an effective route for site-specific PEGylation of lentiviral vector. The PEGylation at these specific sites might be useful at alleviating lentiviral vector-mediated immunogenicity in gene therapy.
Exploring the compatibility of site-specific incorporation of a diazirine-bearing amino acid in lentiviral vector
Having established the approach to genetically incorporate an azide moiety into a lentiviral vector, we next determined whether other chemical moieties synthesized in laboratories with unique properties could also be site-specifically incorporated using this approach. A diazirine-bearing photo-crosslinking amino acid (3-(3-methyl-3H-diazirine-3-yl)-propamino)carbonyl)-Nϵ-L-lysine (DiZPK, Figure 1C) (24) was chosen as a representative. Display of such a diazirine-bearing photo-crosslinking moiety on the lentiviral vector surface may help to probe virus–host interactions (Supplementary Figure S8A) and even capture the host factors involved in the viral life cycle. Incorporation of DiZPK into a lentiviral vector was carried out by an identical procedure, but replacing UAA NAEK with DiZPK in the culture media. Successful incorporation of DiZPK at Y77 or Y116 was demonstrated by western blot analysis (Supplementary Figure S8B). The high efficiency (Y116) of incorporation of DiZPK into lentiviral vectors was evaluated according to the luciferase assay readings (Supplementary Figure S8C). This indicated that photo-sensitive diazirine moieties were also able to be incorporated into lentiviral vectors site-specifically and highly efficiently. Two cellular proteins have been captured upon photo-crossing activation of lentiviral vectors carrying DizPK at Y77 and Y116 during transfection (Supplementary Figure S8D), and their potential sequences and function are under exploration. The successful incorporation of NAEK and DiZPK at any defined position in a lentiviral vector suggests that this approach may be viable and useful for facile incorporation of other UAAs, which might carry a variety of novel chemical, physical, and biological properties, and thus further broaden the versatility of lentiviral vectors.
DISCUSSION
Optimization of the genome backbone, one major component of lentiviral vectors, has given rise to a powerful vehicle for carrying and delivering a variety of genetic materials (36–39). Exploration of envelope properties, another major component of lentiviral vectors, might further broaden its versatility in biomedical research. As demonstrated in this study, the introduction of a chemical moiety on some sites of the fragile envelope disrupts the propagation of progeny viruses and their infectivity (Figure 3). Therefore, it is necessary to develop a nondestructive approach for site-specific engineering of the lentiviral vector. The emerging technique of genetic code expansion, that is, allocating a stop codon to encode an amino acid that is not among the 20 natural amino acids, may exhibit many relevant, promising features (18,20,23,24).
We developed a facile platform via this technique in the propagation of lentiviral vectors for site-specific incorporation of a series of chemical moieties synthesized in the laboratory. The genetic code expansion plus the mutagenesis technique allowed precise control of the placement of the azide, diazirine, and other chemical moieties, and the subsequent conjugation of a variety of functional molecules, orthogonally and stoichiometrically, on the fragile envelope of the lentiviral vector (Figures 2–6). This is superior to posttranslation-like modifications that generally target the active groups of all constitutive amino acids with a complex mixture of generated conjugates (40,41).
One advantage of substituting a natural amino acid with a similarly sized UAA is that structural disruption of the lentiviral vector is minimal, and therefore the propagation and innate infectivity is unlikely to change. In this study we found that, for sites D35 (located at the trimerization domain), I37, E201, K225, A240, K242, D243 and R249 (all located at the PH domain) (13), UAA incorporation had almost no negative effect on both envelope protein expression and virion propagation. We also found that, for sites N20 (located at the trimerization domain), Q42 and E252 (located at the PH domain), and V161, K172 and K174 (all located at the fusion domain) (13), despite some deleterious effects upon UAA incorporation on the expression of VSVg protein, significant quantities of lentiviral vectors were produced (Figure 3). This suggests that replacement of natural amino acids with UAA at certain sites of the trimerization and fusion domains, and most sites of the PH domain, has less effect on virion assembly and/or infectivity. Untouchable sites were also identified, including R342 and M346 (located at the lateral domain), and Y77 (located at the fusion domain), wherein UAA incorporation led to rather fewer or even no lentiviral vector production (Figure 3). This suggests that these amino acids are conserved for propagation or infection of progeny vectors. For sites Y73, D185, K200, Y281, G321, G345 and P362, due to poor expression of viral envelope proteins upon UAA incorporation (Figure 3), their involvement in the propagation of lentiviral vectors remains undetermined. Overall, the high fidelity of genetic code expansion makes it possible to explore the structure–function relationship of lentiviral vectors, by which around 14 modifiable sites on the envelope have been identified that affect neither its propagation nor its infectivity.
The high fidelity of genetic code expansion also makes it possible for orthogonal and stoichiometric introduction of Alexa 488, RGD and PEG ligands at the proper position of lentiviral vectors, which is otherwise difficult. This has a significant impact on broadening the versatility of lentiviral vectors in tracking single-virus movement, targeted gene delivery, detecting virus–host interaction, and other biological applications. In particular, the much shorter time observed in endocytosis (Figure 5) verifies the major advantage of this platform—neither the infectious capability nor the velocity of the lentiviral vector is sacrificed upon site-selective and -specific introduction of a small-sized probe. Consistently, approximately 10-fold infectivity enhancement upon conjugation of RGD at site 246 of the lentiviral vector toward integrin αvβ3-positive cells (Figure 6) also demonstrated the viability and utility of this platform for site-selective and -specific engineering of lentiviral vectors. Clearly, using this platform, other chemical moieties, functional ligands, DNA/RNA aptamers, and even antibodies might also be site-specifically engineered or conjugated onto lentiviral vectors, to enhance viral infectivity and specificity or alter the tissue/cell tropism. Therefore, our report may provide a new and facile platform for the development of lentiviral vectors as a versatile tool, such as remodeling the surface structure of viruses, probing virus–host interactions, improving immunogenicity of infectious viruses, increasing the host tolerance toward therapeutic vectors.
In conclusion, with the aim of broadening the versatility of lentiviral vectors as a powerful tool, we expanded the genetic code in propagation of lentiviral vectors for site-specific incorporation of chemical moieties synthesized in laboratories. Through systematic structure–function explorations, displaying azide, diazirine and other moieties, the modifiable sites on lentiviral vector surfaces were identified that neither affected its propagation nor its infectivity. The ability to direct chemical moieties synthesized in the laboratory to any site of a lentiviral vector in vivo resulted in excellent imaging upon fluorescence labeling, significant improvement of target delivery upon RGD proper conjugation, and real-time detection of the virus-host interactions. This study may provide a foundation for the rational design of lentiviral vectors, with a significant impact on both basic research and therapeutic applications. Although the study here was focused on the VSVg-enveloped lentiviral vector in order to demonstrate the viability and utility of this method, the method could be easily applied to other types of enveloped viruses.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
Acknowledgments
The authors thank Bo Xu, Jun Wu, Dan Liu, Lan Yuan, Qihua He, and the Peking University medical and health analysis center Facility for expert assistance.
Footnotes
Present address: Demin Zhou, School of Pharmaceutical Sciences, Peking University, Beijing 100191, China.
FUNDING
National Basic Research Program of China (973 Program; Grant No. 2010CB912300); National Natural Science Foundation of China (Grants Nos. 81101239, 20932001, 91029711, 20852001). Funding for open access charge: National Basic Research Program of China (973 Program; Grant No. 2010CB912300).
Conflict of interest statement. None declared.
REFERENCES
- 1.Papayannakos C., Daniel R. Understanding lentiviral vector chromatin targeting: working to reduce insertional mutagenic potential for gene therapy. Gene Ther. 2013;20:581–588. doi: 10.1038/gt.2012.88. [DOI] [PubMed] [Google Scholar]
- 2.Arya S.K., Zamani M., Kundra P. Human immunodeficiency virus type 2 lentivirus vectors for gene transfer: expression and potential for helper virus-free packaging. Hum. Gene Ther. 1998;9:1371–1380. doi: 10.1089/hum.1998.9.9-1371. [DOI] [PubMed] [Google Scholar]
- 3.Poeschla E.M., Wong-Staal F., Looney D.J. Efficient transduction of nondividing human cells by feline immunodeficiency virus lentiviral vectors. Nat. Med. 1998;4:354–357. doi: 10.1038/nm0398-354. [DOI] [PubMed] [Google Scholar]
- 4.Olsen J.C. Gene transfer vectors derived from equine infectious anemia virus. Gene Ther. 1998;5:1481–1487. doi: 10.1038/sj.gt.3300768. [DOI] [PubMed] [Google Scholar]
- 5.Schnell T., Foley P., Wirth M., Munch J., Uberla K. Development of a self-inactivating, minimal lentivirus vector based on simian immunodeficiency virus. Hum. Gene Ther. 2000;11:439–447. doi: 10.1089/10430340050015905. [DOI] [PubMed] [Google Scholar]
- 6.Naldini L., Blomer U., Gallay P., Ory D., Mulligan R., Gage F.H., Verma I.M., Trono D. In vivo gene delivery and stable transduction of nondividing cells by a lentiviral vector. Science. 1996;272:263–267. doi: 10.1126/science.272.5259.263. [DOI] [PubMed] [Google Scholar]
- 7.Moffat J., Grueneberg D.A., Yang X., Kim S.Y., Kloepfer A.M., Hinkle G., Piqani B., Eisenhaure T.M., Luo B., Grenier J.K., et al. A lentiviral RNAi library for human and mouse genes applied to an arrayed viral high-content screen. Cell. 2006;124:1283–1298. doi: 10.1016/j.cell.2006.01.040. [DOI] [PubMed] [Google Scholar]
- 8.Jakobsen M., Stenderup K., Rosada C., Moldt B., Kamp S., Dam T.N., Jensen T.G., Mikkelsen J.G. Amelioration of psoriasis by anti-TNF-alpha RNAi in the xenograft transplantation model. Mol. Ther. 2009;17:1743–1753. doi: 10.1038/mt.2009.141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Cartier N., Hacein-Bey-Abina S., Bartholomae C.C., Bougneres P., Schmidt M., Kalle C.V., Fischer A., Cavazzana-Calvo M., Aubourg P. Lentiviral hematopoietic cell gene therapy for X-linked adrenoleukodystrophy. Methods Enzymol. 2012;507:187–198. doi: 10.1016/B978-0-12-386509-0.00010-7. [DOI] [PubMed] [Google Scholar]
- 10.Aiuti A., Biasco L., Scaramuzza S., Ferrua F., Cicalese M.P., Baricordi C., Dionisio F., Calabria A., Giannelli S., Castiello M.C., et al. Lentiviral hematopoietic stem cell gene therapy in patients with Wiskott-Aldrich syndrome. Science. 2013;341:1233151. doi: 10.1126/science.1233151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Biffi A., Montini E., Lorioli L., Cesani M., Fumagalli F., Plati T., Baldoli C., Martino S., Calabria A., Canale S., et al. Lentiviral hematopoietic stem cell gene therapy benefits metachromatic leukodystrophy. Science. 2013;341:1233158. doi: 10.1126/science.1233158. [DOI] [PubMed] [Google Scholar]
- 12.Flight M.H. Trial watch: Clinical trial boost for lentiviral gene therapy. Nat. Rev. Drug Discov. 2013;12:654. doi: 10.1038/nrd4111. [DOI] [PubMed] [Google Scholar]
- 13.Roche S., Rey F.A., Gaudin Y., Bressanelli S. Structure of the prefusion form of the vesicular stomatitis virus glycoprotein G. Science. 2007;315:843–848. doi: 10.1126/science.1135710. [DOI] [PubMed] [Google Scholar]
- 14.Roche S., Albertini A.A., Lepault J., Bressanelli S., Gaudin Y. Structures of vesicular stomatitis virus glycoprotein: membrane fusion revisited. Cell. Mol. Life Sci. 2008;65:1716–1728. doi: 10.1007/s00018-008-7534-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Mateu M.G. Virus engineering: functionalization and stabilization. Protein Eng. Des. Sel. 2011;24:53–63. doi: 10.1093/protein/gzq069. [DOI] [PubMed] [Google Scholar]
- 16.Everts M., Saini V., Leddon J.L., Kok R.J., Stoff-Khalili M., Preuss M.A., Millican C.L., Perkins G., Brown J.M., Bagaria H., et al. Covalently linked Au nanoparticles to a viral vector: potential for combined photothermal and gene cancer therapy. Nano Lett. 2006;6:587–591. doi: 10.1021/nl0500555. [DOI] [PubMed] [Google Scholar]
- 17.Jiang W., Kim B.Y., Rutka J.T., Chan W.C. Nanoparticle-mediated cellular response is size-dependent. Nat. Nanotechnol. 2008;3:145–150. doi: 10.1038/nnano.2008.30. [DOI] [PubMed] [Google Scholar]
- 18.Wang L., Brock A., Herberich B., Schultz P.G. Expanding the genetic code of Escherichia coli. Science. 2001;292:498–500. doi: 10.1126/science.1060077. [DOI] [PubMed] [Google Scholar]
- 19.Wang Q., Parrish A.R., Wang L. Expanding the genetic code for biological studies. Chem. Biol. 2009;16:323–336. doi: 10.1016/j.chembiol.2009.03.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Chin J.W., Cropp T.A., Anderson J.C., Mukherji M., Zhang Z., Schultz P.G. An expanded eukaryotic genetic code. Science. 2003;301:964–967. doi: 10.1126/science.1084772. [DOI] [PubMed] [Google Scholar]
- 21.Liu C.C., Schultz P.G. Adding new chemistries to the genetic code. Annu. Rev. Biochem. 2010;79:413–444. doi: 10.1146/annurev.biochem.052308.105824. [DOI] [PubMed] [Google Scholar]
- 22.Niu W., Schultz P.G., Guo J. An expanded genetic code in mammalian cells with a functional quadruplet codon. ACS Chem. Biol. 2013;8:1640–1645. doi: 10.1021/cb4001662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Nguyen D.P., Lusic H., Neumann H., Kapadnis P.B., Deiters A., Chin J.W. Genetic encoding and labeling of aliphatic azides and alkynes in recombinant proteins via a pyrrolysyl-tRNA Synthetase/tRNA(CUA) pair and click chemistry. J. Am. Chem. Soc. 2009;131:8720–8721. doi: 10.1021/ja900553w. [DOI] [PubMed] [Google Scholar]
- 24.Lin S., Zhang Z., Xu H., Li L., Chen S., Li J., Hao Z., Chen P.R. Site-specific incorporation of photo-cross-linker and bioorthogonal amino acids into enteric bacterial pathogens. J. Am. Chem. Soc. 2011;133:20581–20587. doi: 10.1021/ja209008w. [DOI] [PubMed] [Google Scholar]
- 25.Yang J.P., Zhou D., Wong-Staal F. Screening of small-molecule compounds as inhibitors of HCV entry. Methods Mol. Biol. 2009;510:295–304. doi: 10.1007/978-1-59745-394-3_22. [DOI] [PubMed] [Google Scholar]
- 26.Lin S., Yan H., Li L., Yang M., Peng B., Chen S., Li W., Chen P.R. Site-specific engineering of chemical functionalities on the surface of live hepatitis D virus. Angew. Chem. Int. Ed. Engl. 2013;52:13970–13974. doi: 10.1002/anie.201305787. [DOI] [PubMed] [Google Scholar]
- 27.Brandenburg B., Zhuang X. Virus trafficking - learning from single-virus tracking. Nat. Rev. Microbiol. 2007;5:197–208. doi: 10.1038/nrmicro1615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Joo K.I., Lei Y., Lee C.L., Lo J., Xie J., Hamm-Alvarez S.F., Wang P. Site-specific labeling of enveloped viruses with quantum dots for single virus tracking. ACS Nano. 2008;2:1553–1562. doi: 10.1021/nn8002136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ruoslahti E., Pierschbacher M.D. New perspectives in cell adhesion: RGD and integrins. Science. 1987;238:491–497. doi: 10.1126/science.2821619. [DOI] [PubMed] [Google Scholar]
- 30.Cherny R.C., Honan M.A., Thiagarajan P. Site-directed mutagenesis of the arginine-glycine-aspartic acid in vitronectin abolishes cell adhesion. J. Biol. Chem. 1993;268:9725–9729. [PubMed] [Google Scholar]
- 31.Muller G., Gurrath M., Kessler H. Pharmacophore refinement of gpIIb/IIIa antagonists based on comparative studies of antiadhesive cyclic and acyclic RGD peptides. J. Comput. Aided Mol. Des. 1994;8:709–730. doi: 10.1007/BF00124017. [DOI] [PubMed] [Google Scholar]
- 32.Crisp J.L., Savariar E.N., Glasgow H.L., Ellies L.G., Whitney M.A., Tsien R.Y. Dual targeting of integrin alphavbeta3 and matrix metalloproteinase-2 for optical imaging of tumors and chemotherapeutic delivery. Mol. Cancer Ther. 2014;13:1514–1525. doi: 10.1158/1535-7163.MCT-13-1067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Menendez J.A., Vellon L., Mehmi I., Teng P.K., Griggs D.W., Lupu R. A novel CYR61-triggered ‘CYR61-alphavbeta3 integrin loop’ regulates breast cancer cell survival and chemosensitivity through activation of ERK1/ERK2 MAPK signaling pathway. Oncogene. 2005;24:761–779. doi: 10.1038/sj.onc.1208238. [DOI] [PubMed] [Google Scholar]
- 34.Xiong Z., Cheng Z., Zhang X., Patel M., Wu J.C., Gambhir S.S., Chen X. Imaging chemically modified adenovirus for targeting tumors expressing integrin alphavbeta3 in living mice with mutant herpes simplex virus type 1 thymidine kinase PET reporter gene. J. Nucl. Med. 2006;47:130–139. [PMC free article] [PubMed] [Google Scholar]
- 35.Hinds K.D., Kim S.W. Effects of PEG conjugation on insulin properties. Adv. Drug Deliv. Rev. 2002;54:505–530. doi: 10.1016/s0169-409x(02)00025-x. [DOI] [PubMed] [Google Scholar]
- 36.Zennou V., Serguera C., Sarkis C., Colin P., Perret E., Mallet J., Charneau P. The HIV-1 DNA flap stimulates HIV vector-mediated cell transduction in the brain. Nat. Biotechnol. 2001;19:446–450. doi: 10.1038/88115. [DOI] [PubMed] [Google Scholar]
- 37.Firat H., Zennou V., Garcia-Pons F., Ginhoux F., Cochet M., Danos O., Lemonnier F.A., Langlade-Demoyen P., Charneau P. Use of a lentiviral flap vector for induction of CTL immunity against melanoma. Perspectives for immunotherapy. J. Gene Med. 2002;4:38–45. doi: 10.1002/jgm.243. [DOI] [PubMed] [Google Scholar]
- 38.ter Brake O., Westerink J.T., Berkhout B. Lentiviral vector engineering for anti-HIV RNAi gene therapy. Methods Mol. Biol. 2010;614:201–213. doi: 10.1007/978-1-60761-533-0_14. [DOI] [PubMed] [Google Scholar]
- 39.Kymalainen H., Appelt J.U., Giordano F.A., Davies A.F., Ogilvie C.M., Ahmed S.G., Laufs S., Schmidt M., Bode J., Yanez-Munoz R.J., et al. Long-term episomal transgene expression from mitotically stable integration-deficient lentiviral vectors. Hum. Gene Ther. 2014;25:428–442. doi: 10.1089/hum.2013.172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Wang Y.S., Youngster S., Grace M., Bausch J., Bordens R., Wyss D.F. Structural and biological characterization of pegylated recombinant interferon alpha-2b and its therapeutic implications. Adv. Drug Deliv. Rev. 2002;54:547–570. doi: 10.1016/s0169-409x(02)00027-3. [DOI] [PubMed] [Google Scholar]
- 41.Dhalluin C., Ross A., Leuthold L.A., Foser S., Gsell B., Muller F., Senn H. Structural and biophysical characterization of the 40 kDa PEG-interferon-alpha2a and its individual positional isomers. Bioconjug. Chem. 2005;16:504–517. doi: 10.1021/bc049781+. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.