

Abstract
As modern and ancient DNA sequence data from diverse human populations accumulate1–4, evidence is increasing in support of the existence of beneficial variants acquired from archaic humans that may have accelerated adaptation and improved survival in new environments — a process, known as adaptive introgression (AI). Within the past couple of years, a series of studies5–8 have identified genomic regions showing strong evidence for archaic adaptive introgression. In this Review, we provide an overview of the statistical methods developed to identify archaic introgressed fragments in the genome sequences of modern humans, and to determine whether positive selection has acted on these fragments. We discuss recently reported examples of adaptive introgression and consider the level of supporting evidence for each, grouped by selection pressure. We discuss challenges and recommendations for inferring selection on introgressed regions.
Introduction
The relationship between modern humans and other, now extinct, archaic hominin groups has been a subject of controversy since the 1970s. Two competing hypotheses were originally proposed: the Multiregional model9 posited that modern humans evolved in parallel throughout Africa and Eurasia from different archaic groups, while exchanging migrants, whereas the Out of Africa model proposed that all present day humans had a recent origin in the African continent, from which they expanded across the world10. Over the past 30 years, however, these two hypotheses were increasingly seen as an over-simplification. Other intermediate models emerged, involving a recent origin in Africa with limited amounts of admixture from Eurasian archaic groups11, or considerable assimilation of Neanderthals into the modern human gene pool during modern human expansion into Europe12.
Until recently, analyses of whole-genome sequences from modern human populations seemed to support the Out of Africa model, although certain studies observed genomic patterns that might suggest local gene flow between modern and archaic populations13, 14. Archaeological evidence also suggests Neanderthal presence up to 40,000 years ago in Europe and Western Asia15, which means that they could have co-existed with modern human populations for a period of at least 2,600 years. In the past five years, however, whole-genome sequences from two archaic human groups, Neanderthals1, 4, 16 and Denisovans2, 3, have provided direct insights regarding the extent of gene flow between archaic humans and modern humans.
Although it is now widely accepted that admixture occurred between different human groups, little is known about the adaptive contribution of the introgressed segments. Cases of introgression enabling adaptation in animals and plants have been extensively documented (for reviews, see Arnold and Martin 200917, Rieseberg 200918 and Hedrick 201319), although surprisingly little attention has been devoted to adaptive introgression in humans20, 21.
We review here recent human genetic studies that have identified several examples of archaic adaptive introgression of modern humans with Neanderthals or Denisovans. First, we provide a brief introduction to the statistical methods that have been employed to detect adaptive introgression based on the wholegenome sequences of modern and archaic humans. We then review the evidence in support of particular proposed instances of archaic adaptive introgression. Finally, we discuss several unanswered questions in the field and propose possible avenues of future research, such as the development of methods to jointly model selection and introgression.
Archaic gene flow
While the majority of non-African human ancestry is shared with Africans, non- Africans also possess a small amount of DNA (1.5–2.1%) from Neanderthals1. The level of Neanderthal admixture varies in non-Africans; for example it was recently found that the whole-genome sequences of Asian individuals show a larger proportion of Neanderthal ancestry segments than sequences from Europeans3, 22, 23. In addition, a small portion of Melanesian, Papuan and Australian ancestry (3–6%) derives from Denisovans2, 3, 24 and lower amounts of Denisovan ancestry (0.2%) are also found in East Asia4, 25. In addition, recent analyses of whole-genome sequences of individuals from different populations in Africa suggest that some African populations may have also exchanged genetic material with as yet undetermined archaic human groups13, 26, 27. For a review of recent demographic inferences from analysis of modern and archaic human genome sequences, see reference 28.
The presence of Neanderthal and Denisovan DNA in the whole-genome sequences of present-day non-African individuals is generally accepted to be a consequence of admixture, likely due to limited interbreeding between modern and archaic humans29. An alternative explanation is ancestral population structure. Under this model, population subdivision in the ancestral population of archaic and modern humans may have resulted in some modern human groups – like the ancestors of Eurasians – being more closely related to Neanderthals than other groups that stayed in Africa30 (Figure 1). However, the date of the last Neanderthal gene exchange into present-day Eurasians supports the admixture scenario. This event can be dated with some accuracy by examining the distribution of the length of tracts of introgressed DNA. Recombination breaks down haplotypes into shorter and shorter fragments. DNA from recent introgression events should, therefore, fall into longer contiguous tracts than DNA resulting from old introgression events31–33 or from ancient population structure (Figure 1). Measurements of introgression tract lengths indicate that the last gene flow event occurred 50–60 thousand years ago (kya)34–36. This timeframe is too recent to support a scenario of ancestral structure, as the divergence time between humans and Neanderthals is thought to have occurred 550–765 kya (assuming a human mutation rate of 0.5*10−9 per bp per year) or 275–383 kya (assuming a mutation rate of 10−9 per bp per year)4. Furthermore, it also largely postdates the African-Eurasian population split, estimated to have occurred 100– 160 kya (under the slow mutation rate) or 50–80 kya (under the fast rate)37–39. Additional analyses based on the distribution of SNP allele frequencies in Neanderthals and present-day humans and of allelic configurations in short genomic blocks also support the hypothesis of post-split admixture40, 41. Finally, all of these studies provided evidence for archaic human gene flow into modern humans1, 3, 4, but not in the reverse direction. The reason for this may be that all archaic humans for whom genome sequences are available so far likely pre-date the time of contact with modern humans.
Figure 1. Expected length of archaic tracts under admixture and ancestral structure scenarios.

Schematic comparison of expected tract length of archaic-like tracts for a scenario of admixture (left panel) and a scenario of ancestral population structure (right panel). Because there is less time for recombination to break down the migrant tracts (red) in the admixed population, the expected tract length in this case will be longer than in the case of ancestral structure.
Methods to infer introgressed segments
The challenge for methods seeking to identify introgression is to distinguish true introgression from shared ancestral genetic variation (Figure 1). Any two populations will always share some segments of DNA that originated in their common ancestor because both populations descend from the same population and might therefore have inherited some of the same segments of DNA from the ancestral population. For this reason, two DNA segments sampled from different populations may share a most recent common ancestor (MRCA) more recently than two DNA sequences sampled from the same population. The same argument is true for species, and we will in general here not distinguish between species and populations, in part to avoid entering into discussions of species concepts and definitions in hominins.
Patterson’s D and genome-wide data
There are several statistical methods available for identifying introgression from genome-wide data1, 42, 43, 44. The most well known is Patterson’s D1, 42, 43, a statistic that measures the excess sharing of derived alleles between each of two populations in a pair (ingroup populations) and an outgroup population (Supplementary information S1 (box)). Patterson’s D takes advantage of a phylogenetic argument: if neither of the ingroup populations have had any gene flow from the outgroup population, then each of the two ingroup populations should share approximately the same number of derived alleles with the outgroup. The significance of deviations from the symmetric pattern expected in the absence of introgression is evaluated using a blockbootstrap or jack-knife method applied to genome-wide data. This statistic was used as one of the primary lines of evidence for identifying human-Neanderthal introgression by showing that populations outside Africa share more derived alleles with Neanderthals than African populations do1.
Identifying specific genes or segments of the genome that are introgressed is even more challenging, because the simple re-sampling methods for evaluating the significance of Patterson’s D statistic are inapplicable to shorter genomic regions44. Inferences regarding specific regions must instead rely on demographic models that include assumptions about parameters such as divergence times, effective population sizes, and recombination rates.
Phylogenetic information and sequence divergence
A number of statistics commonly used in genetic analyses may capture information regarding introgression. In combination with parametric simulations for evaluating significance, these statistics may be used to distinguish incomplete lineage sorting (ILS) from introgression. While Patterson’s D statistic (see above) captures phylogenetic information, an alternative is to use statistics based on sequence divergence. An introgressed haplotype should have low sequence divergence to the putative archaic source population, but high sequence divergence to other present-day human individuals. One approach to identify an introgressed segment6 is to calculate the most likely time of the most recent common ancestor (TMRCA) of the test haplotype and the archaic haplotype, as well as the TMRCA of the test haplotype and a second modern human haplotype. 6 A test human haplotype that has a very recent TMRCA with an archaic population, but a very ancient TMRCA with other human haplotypes, is likely to have been introgressed from the archaic population. We note that to formalize this into a test of introgression, it is necessary to make specific assumptions regarding divergence times and population sizes. Simulations can then be used to determine statistical significance.
Tract length and linkage disequilibrium (LD), S*
As previously mentioned, the expected length of an introgressed tract depends on the time since introgression. In fact, under simple assumptions, the lengths of introgressed tracts should approximately follow an exponential distribution with mean length ([1−m]r[t−1])−1, where t is the number of generations since a proportion m of the population was replaced by migrants from another population, and r is the recombination rate (in units of Morgans) per bp. See also reference 31 for conditions under which this approximation breaks down. A defining feature of introgression is that it should on average generate longer tracts than ILS. Furthermore, as the length of the tracts only depends (to a first approximation) on r, m, and t, and not on effective population sizes, using the length of the introgressed haplotype provides a more robust method for distinguishing between introgression and shared ancestral variation (Supplementary information S2 (box)). The only caveat is that introgressed haplotypes are not directly observable but must be inferred from the data. Alternatively, statistics that summarize information related to haplotype length without directly inferring haplotypes can be used. In particular, the existence of a long introgressed haplotype should increase long-range linkage disequilibrium (LD). Examining patterns of LD therefore provides an alternative method for identifying introgression. The S* statistic (Box 1, Supplementary information S3 (box)) provides a commonly used method for extracting this information, although it also incorporates information regarding divergence13, 14, 23, 45. S* was originally derived to identify genome-wide evidence of introgression similarly to D, but without knowledge of the donor population. In subsequent studies, it was also used locally to identify highly divergent haplotypes harboring variants in strong linkage disequilibrium, to search for regions introgressed from Neanderthals into non-Africans23.
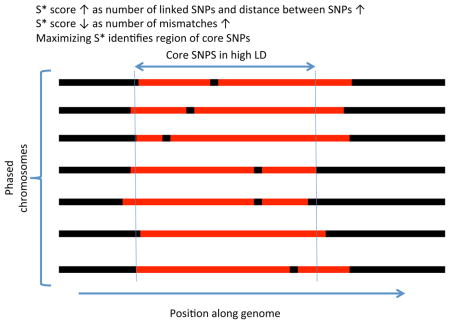
BOX 1. Description of the S* statistic.
S* is a both a measure of linked disequilibrium in a set of phased chromosomes and a method to discover linked SNPs. We show a region within a sample of phased chromosomes and denote a shared haplotype in red. S* is calculated by optimizing the sum of scores S(i,j) where i and j are two consecutive SNP positions being considered for inclusion in the final haplotype (see Supplementary information S3 (box) for an example). S(i,j) is an heuristic score but it rewards fully linked pairs (no mismatches) and it is higher the further the distance between linked SNPs, thereby rewarding longer haplotypes. Mismatches within the haplotype are noted as black segments internal to the red region, and regions where there is no longer a shared haplotype are noted as black flanking regions. In the process of calculating S* via a dynamic programming method, not only is the optimal score calculated but also the set of SNPs that yield the optimum score, denoted by the two vertical lines defining the boundary of the region determined by S*. Although there is some sharing of haplotypes outside this region, the number of mismatches would make S* suboptimal, and they are therefore excluded.
Probabilistic Models: CRF and HMM
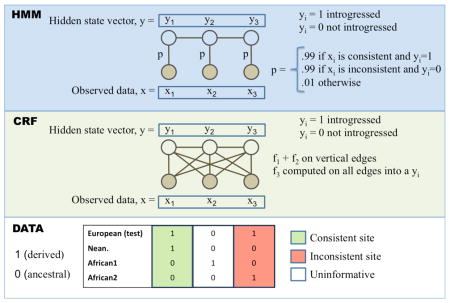
An alternative to using simulations to determine the significance of a summary statistic is to incorporate the parametric assumptions directly into a probabilistic framework. For example, both Prüfer et al.4 and Seguin-Orlando et al.35 developed a Hidden Markov Model (HMM) to detect archaic introgressed fragments. In both cases, the authors used information from non-admixed Africans as well as an archaic (Denisovan or Neanderthal) genome sequence and a test phased genome sequence with resolved haplotypes from a non-African population that may contain introgressed fragments. Under the HMM framework, the ancestry of each SNP in the genome is a hidden random variable with two states, archaic (that is, introgressed) or modern human, which are estimated from the genomic data. For each site in the test genome, the most likely state (archaic ancestry or modern human ancestry) giving rise to the observed data is then inferred probabilistically, with the posterior decoding of the HMM46. This approach allowed the authors of both studies to infer the probability of introgression at different regions of the genomes of non-African individuals. The primary difference between the two methods is that Prüfer et al.4 used a priori chosen parameters, while Seguin-Orlando et al.35 estimated parameters from a reference data set.
A similar method – called a conditional random field (CRF) – models the ancestry of a set of contiguous SNPs along windows of the genome.47 Under this framework, the SNP ancestry is also a random variable with two states, archaic or modern. However, unlike the HMM methods, the CRF model contains one or more emission functions which incorporate information about linkage disequilibrium, haplotype structure and allele configurations from nearby SNPs simultaneously from multiple individuals (Box 2). The parameters of the method are calibrated using parametric simulations with specific demographic assumptions such as divergence times and effective population sizes.
BOX 2. Hidden Markov Model (HMM) and Conditional Random Field (CRF) frameworks.
These are two related probabilistic models for estimating the ancestry, yi, of SNPs across a genome sequence. Each yi takes on two possible states, 1 for introgressed (i.e. archaic ancestry) and 0 for not introgressed (i.e. modern human ancestry). The observed data, xi, is a matrix of haplotypes, that in the example consist of whole-genome sequences of individuals from a European test population, an African population (2 haplotypes) and Neanderthals, examined at 3 SNP positions. x1 is a site that is consistent with introgression as the derived allele is seen in the test and Neanderthal sequence but not from Africans. Likewise, x3 is an inconsistent site as the derived allele seems to be of modern human origin, while the site x2 is uninformative. The ancestry states and observed data are connected through the emission probabilities (p) or emission functions (fi) for the HMM or CRF, respectively, denoted by the edges connecting the xi and the yi. The CRF can have more general relationships as denoted by the diagonal edges; in ref. 46, the authors use the sum of three emission functions f1, f2, f3 - f1 scores “consistent” sites 1, f2 scores “inconsistent” sites 1, and f3 evaluates to 1 if the entire test haplotype is relatively closer to the Neanderthal sequences than to the African haplotype. In contrast, the HMM in ref. 4 have fixed emission probabilities, p, for observed states that are either consistent or inconsistent. Edges between the yi and yi+1 states represent transition probabilities (HMM) or more general transition functions (CRF). The transition probabilities and functions model linkage between ancestral states along the genome, and the transition parameters depend on the recombination distance between sites, the admixture proportion and admixture time. Crucially, both of these frameworks have efficient algorithms for inferring the most likely sequence of ancestral states.
Inferring selection on introgressed DNA
If a haplotype was introduced by introgression from archaic humans, the reason for its continued survival in present-day humans may be that it was affected by positive selection or balancing selection, or that it was not removed from the population by negative selection or genetic drift. Distinguishing between these hypotheses requires additional statistical analyses, because when a genomic region contains introgressed DNA, the pattern of polymorphism may be different than expected under a neutral model with no introgression. As mentioned previously, in regions with introgressed DNA, the haplotype structure will change, there will be an increase in LD48, and the distribution of allele frequencies will change from that expected without introgression40. These patterns are exactly the signals that many standard methods for detecting selection employ. These include the Integrated Haplotype Score (iHS)49, Extended Haplotype Homozygosity (EHH)50, Cross-Population Extended Haplotype Homozygosity (XP-EHH)51, Tajima’s D52, Fay and Wu’s H53, Composite of Multiple Signals (CMS)54, Fst 55, 56 or variations on Fst such as the Locus Specific Branch Length (LSBL)57 or the Population Branch Statistic (PBS)58. Naïve use of these methods can therefore easily lead to false inferences of selection as the pattern generated by introgression alone may be incorrectly interpreted as evidence of selection.
High archaic haplotype frequency
Positive selection produces an increase in selected allele frequencies over time. A direct way of inferring selection is therefore the detection of significantly high frequencies of an introgressed fragment in a specific population, relative to other populations. This approach is essentially the information that FST-type methods rely on. These approaches may be somewhat less likely to be misled by introgression than many other methods for detecting selection. Introgression introduces new genetic variation, but as long as the level of introgression is low, a previously rare allele introduced by introgression will not usually segregate at high frequencies—genetic drift or selection must act on the allele to increase its frequency. Therefore, allelefrequency differentiation methods for distinguishing between selection and pure genetic drift are largely applicable. However, care must be taken to ensure that the strong LD characteristic of introgressed regions48, 59 does not create false positives. For example, methods that summarize allele frequency differences from many linked SNPs within a region using sliding-window approaches could be biased, or have increased variance, because the allele frequencies of the introgressed alleles are more correlated than expected in the absence of introgression.
However, a telltale signature of adaptive introgression is the presence of mutations in strong LD that exist at high frequency in a particular population and that are only present in the archaic source population, while absent or at very low frequencies in other present-day human populations (Figure 2). For example, a set of 5 such mutations cluster tightly together in the EPAS1 (endothelial PAS domain protein 1) region in Tibetans, suggesting archaic adaptive introgression has occurred.8 Other candidates for adaptive introgression also display similar patterns of archaic allele sharing, including those found in refs. 23, 47.
Figure 2. Example coalescent genealogy of uniquely shared mutations.

Several DNA fragments from two modern populations (pink and light-blue chromosomes) are sequenced. A diploid sequence is also obtained from an extinct archaic population (yellow chromosomes) that split from the population tree more anciently than the two modern populations split from each other. Uniquely shared mutations (red stars) occur in the archaic population, but are passed on to the ancestors of the light-blue modern population via admixture (dashed line). These are then are swept to high frequency by selection, producing a shallow local coalescent genealogy. This process results in sites with high-frequency derived alleles in the light-blue samples that are present in the archaic sample but not the pink samples from the other modern population. Mutations in the genealogy that are not uniquely shared are shown as green stars.
Balancing selection
The identification of introgressed regions that persist in humans owing to balancing selection can be a complicated task. Local admixture and balancing selection both generate increased within-population variability and deep coalescent genealogies, which makes it difficult to decipher whether one or both of these events affected a particular region of the genome. In one study,5 the authors looked for adaptive introgression via balancing selection in two ways. First, they searched for long HLA haplotypes that are deeply divergent (showing a large number of differences with other haplotypes in the same locus), that have high sequence homogeneity in individuals that possess them, and that show extreme LD only in non-African populations. The authors argued that these are unusual features that are unlikely to be predicted exclusively by a model of balancing selection: even though this type of selection generally acts to preserve sequence diversity, it should not produce long homogenous haplotypes that are deeply divergent from other haplotypes, especially in the HLA region, which is known to undergo rapid diversification by recombination. One variant fulfilling the authors’ criteria (HLA-B*73:01) is also in strong linkage disequilibrium with variants that are present in the Denisovan genome, suggesting it may have introgressed from an archaic source. Simulations rejected a scenario of ancestral polymorphism, but assumed a model without ancestral structure and without gene flow, which may not be realistic given the complexities of human demographic history. Their second approach was to computationally infer modern human HLA haplotypes that may have been present in the Neanderthal and Denisovan genomes, and then analyze the global distribution of these haplotypes in present-day humans. The authors could not determine directly if the haplotypes were actually present in the archaic genomes, as it is currently impossible to phase these sequences. Many of the haplotypes they inferred to be present in Neanderthals and Denisovans were found at high frequency in different populations of Eurasia, but at low frequency or absent in Africa, again suggestive of adaptive introgression.
Genotype-phenotype associations
If an introgressed variant is associated with a phenotype known to confer an advantage to a particular population, the variant may have undergone selection in that population. For example, the introgressed EPAS1 gene in Tibetans contains SNP variants associated with statistically significantly reduced hemoglobin levels, which possibly served as an adaptation to high-altitude hypoxia60. Similarly, a relationship between geographical location and genotype frequencies can be suggestive of environmental selection, as with haplotype frequency in the UV-B response gene, HYAL2, and latitude7. However, a genotype-phenotype association by itself does not constitute enough support for adaptation61, and additional tests must be performed to assess if the genotype was actually driven to high frequencies due to selection.
Candidates for adaptive introgression
Here, we review the evidence for adaptive introgression to date, in the context of the methods described in the previous two sections. These recent studies either test single loci, or characterize adaptive introgression genome-wide, based on analyses of the available archaic human genomes (Table 1). We omit studies that had proposed adaptive introgression for some human haplotypes before the whole-genome sequences of Neanderthals and Denisovans62, 63 became available, as many of the early candidates were subsequently found to be absent in the genome sequences1, 64 (see John Hawk’s blog post, Online Resources).
Table 1. Candidates for adaptive introgression.
A summary of recent studies reporting adaptive introgression with a comparison of the evidence provided in support of positive selection acting on this region. The table lists the location of the introgressed haplotype (noting the actual haplotype where known), the most likely archaic source population for each introgressed fragment, and the modern human populations that possess this introgressed fragment. The next columns list the form of evidence provided to demonstrate positive selection at this locus, and the most likely population in which selection occurred. A question mark denotes that the source population, the receiving population or the population where selection occurred remains unresolved and/or has conflicting reports from different studies.
| Location of introgressed haplotype | Closest putative archaic source population | Populations showing evidence of introgression | Selection tests | Most likely population where selection occurred | Reference(s) |
|---|---|---|---|---|---|
| HLA-A, HLA-B, HLA-C | Neanderthal, Denisova | Europeans, East Asians, Melanesians | Extreme allelic and haplotypic diversity in the HLA region indicative of balancing selection | Europeans, East Asians, Melanesians | 5 |
| HLA-DPB1 | Neanderthal (?), but this questioned in ref. 73) | Europeans (?) | No formal test of neutrality performed72, but a phylogenetic analysis suggests the haplotype is not introgressed73 | - | 72, 73 |
| STAT2 (haplotype N) | Neanderthal | Non-Africans | One-tailed test for elevated frequency of diagnostic SNP in introgressed haplotype, based on empirical distribution of SNP frequencies | Papuans | 6 |
| STAT2 (haplotype D) | Denisova (?) | Papuans | No formal test of neutrality performed, but haplotype is only present in Papuans at a low frequency | - | 6 |
| OAS1 | Denisova, but extremely ancient coalescence with human reference suggests direct source was a different archaic group | Melanesians | - | 75 | |
| OAS gene cluster | Neanderthal | Non-Africans | 47, 74 | ||
| 3p21.31/HYAL2 | Neanderthal | East Asians | iHS49, EHH50, CMS54 | East Asians | 7 |
| MC1R | Neanderthal | Non-Africans | Tajima’s D52, Fu and Li’s test82, iHS49 | Taiwanese (?) | 78 |
| SLC16A11 and SLC16A13 | Neanderthal | Native Americans | Genotype-phenotype association | Native Americans (?) | 90 |
| DMD | Neanderthal | Non-Africans | No tests performed | - | 91, 92 |
| EPAS1 | Denisova | East Asians or Tibetans only | High population differentiation58 | Tibetans | 8 |
| Various regions identified via S*, containing genes involved in the integumentary system | Neanderthal | Europeans, East Asians |
|
Europeans, East Asians | 23 |
| Various regions identified via CRF, containing genes involved in keratin filament, sugar metabolism, muscle contraction and oocyte meiosis | Neanderthal | Europeans, East Asians | Windows where the population frequency of the Neanderthal genetic material is too high to be explained by neutral drift | Europeans, East Asians | 47 |
| Various regions identified via HMM | Neanderthal, Denisova | Europeans, East Asians, Melanesians | No claims about selection and no formal tests of neutrality performed | - | 4, 35 |
The divergence times estimated from genome sequence data in several of the studies below required use of a parameter for the human mutation rate. The precise estimate of the human mutation rate has been controversial and remains under study, and a range of accepted values are routinely used in current studies65. Therefore, whenever possible, we state the human mutation rate assumed in each paper when reporting divergence times and note that divergence times obtained from the more commonly used faster rate (10−9 per bp per year) can be converted to those that would be obtained from the slower rate (0.5*10−9 per bp per year) by simply doubling the original time.
Genome-wide admixture maps
Two studies have produced Neanderthal genome-wide admixture maps of European and Asian genomes, based on the whole-genome sequence of a female Neanderthal from Siberia4 and present-day human data from the 1000 Genomes Project 66. In one study, the authors employed a CRF model47 (see Box 2) while in the in the other study, the authors relied on the S* statistic to identify introgressed segments23 (Box 1, Supplementary information S3 (box)). While these two studies find notable signatures of negative selection pushing introgressed variants to low frequencies and a depletion of introgressed tracts in functional genomic regions, possibly due to hybrid sterility, the authors of both studies found considerable evidence of adaptive introgression. Several examples are outlined below. Additionally, one of the studies performed a functional enrichment analysis and found that genes involved in keratin filament, sugar metabolism, muscle contraction and oocyte meiosis have been involved in archaic human adaptive introgression47.
A different genome-wide study (reviewed in the Metabolism section) employed the D statistic locally to detect Neanderthal-like regions at specific genes.67 The authors found that lipid catabolism genes tended to have significantly higher D values in Europeans (supporting a tree clustering Neanderthals with Europeans, to the exclusion of Africans) than the rest of the genome. Additionally, signals of recent positive selection were reported in the same regions with high D in modern Europeans.67
Finally, HMMs identified within present-day human genome data were used to identify putatively introgressed haplotypes in two other studies4,35. These methods identified present-day human haplotypes that are phylogenetically consistent with forming a clade with an archaic individual (Neanderthal or Denisova). The genes contained in the identified regions were not characterized, and tests of selection were not performed in these cases. However, the divergence between the introgressing Neanderthal in Eurasians and the sequenced Neanderthal genome (77–114 kya assuming a mutation rate of 0.5*10−9 per bp year or 38–57 kya assuming a rate of 10−9 per bp per year) was much younger than the divergence between the introgressing Denisovan in the Sahul people (Oceanians, including native Papuans and Australians) and the sequenced Denisovan genome (276–403 kya assuming the slower mutation rate or 138–202 kya assuming the faster rate).4 This may indicate that the Denisovan population was more diverse and/or more structured than the Neanderthal population.
Examining the candidate genes identified from both the genome-wide and single loci studies, we can speculate on the plausible selection pressures and mechanisms underlying examples of adaptive introgression. We organize our discussion of candidate genes below by broad functional categories.
Defense against pathogens
STAT2 is an innate immune gene that is involved in interferon response after viral infection. A recent study6 found a long (130–260 kb) haplotype called ‘N’ that overlaps this gene and that is broadly distributed across Eurasia. The haplotype is absent in sub-Saharan Africa, has a very ancient time to the most recent common ancestor (TMRCA) with other presentday human haplotypes (609 kya, assuming the faster mutation rate) and also has a very recent TMRCA with the Neanderthal genome (78 kya), suggesting it introgressed from this archaic hominin group. Though found throughout Eurasia at a frequency of ~5%, it is present at substantially elevated frequencies in Papuans (~54%). The authors used a neutrality test that controlled for demography to show this difference in frequency was likely due to positive selection acting on the region in Papuans. Though the study focused on STAT2 due to greater availability of sequence data in this gene, the introgressed N haplotype overlaps two other genes which could also have been the targets of selection: ERBB3 (coding for a peptide involved in cell growth and apoptosis68) or ESYT1 (coding for a transmembrane protein with a role in fibroblast differentation69, 70).
The highly polymorphic human leukocyte antigen (HLA) region in chromosome 6 also seems to show signatures of adaptive introgression, though in this case by balancing selection. The fact that the HLA region is highly prone to trans-specific polymorphisms makes it hard to distinguish adaptive introgression from balancing selection for different variants persisting over long time periods71. Various HLA haplotypes have been identified in Eurasians and Melanesians that carry functional variants that likely introgressed from archaic human groups.5 For example, a deeply divergent haplotype (B*73) with strong sequence homogeneity among its carriers is present at high frequencies in west Asia but absent or infrequent in the rest of the world. Simulations show that the global distribution of this haplotype is best explained by a model of introgression from an archaic source in west Asia.5 The Denisovan genome2 carries two variants (C*12:02 and C*15) that are in strong linkage disequilibrium with B*73, although the actual B*73 allele is absent in the Denisovan genome. These variants are also only found at high frequencies in west Asia. A separate analysis revealed a substantial Neanderthal HLA-A ancestry in modern non-African humans, with a wider geographic distribution than B*73 (ref. 5). In summary, unusual levels of LD and high archaic haplotype frequencies at the HLA locus in present-day humans seem to support the hypothesis of adaptive introgression.
Another HLA study examined an important amino acid motif in the family of antigen receptors in the HLA region (HLA-DPβ, allele DPB1*0401) that are required for allowing unmatched DRα and DPβ subunits to form a functional complex. The sequence closely matches the Neanderthal genome at the same locus72. However, a phylogenetic analysis of different variants of this motif in modern humans found that the putatively introgressed haplotype coalesces with other common modern human haplotypes before coalescing with the Neanderthal genome73. Furthermore, although the motif variant is present at 68% frequency across Europeans, it is also present in sub-Saharan Africans at 11% frequency. Finally, the divergence time between the haplotype of interest and the Neanderthal genome was estimated to be 2.2 Mya (assuming a mutation rate of 10−9 per bp per year), largely predating the modern human-Neanderthal population split time. Thus, it is more likely that the presence of this haplotype in Europeans is a consequence of ancient population structure in Africa73.
Other regions of the human genome show characteristic signatures of introgression, but the evidence for positive selection is weak or absent. For example, the OAS gene cluster (which helps to inhibit viral replication as part of the innate immune response) has been subject to two separate archaic introgression events: one into Non-Africans from Neanderthals (though the haplotype is absent in Papuans)74 and another one into Papuans from an extremely deep lineage (TMRCA with modern humans = 3.7 million years ago (Mya), assuming a mutation rate of 10−9 per bp per year)75. The haplotype for the latter matches best with the Denisova sequence across 90kb of high LD sequence, and has much higher sequence diversity than observed in other populations75. Although no evidence for positive selection in the region has been found,74, 75 the genetic distribution of the different haplotypes across continents is perhaps a signal of balancing selection74. The deep lineage could belong to another archaic hominin group that may have admixed with both Papuans and Denisovans, or with a common ancestor of the two populations75. In the same direction, but in a different study, Prüfer et al.4 recently found a genome-wide signal of “super-archaic” introgression in the Denisovan genome, likely due to an unsampled hominin group that diverged from modern humans before Denisovans and Neanderthals, and later interbred with Denisovans. However, the specific regions of the Denisovan genome that have super-archaic ancestry have yet to be identified, so it is unknown if the OAS gene cluster is one of them. Finally, evidence for adaptive introgression into Europeans from Neanderthals in the gene OAS2 was more recently confirmed using the CRF model.47
Pigmentation
In region p21.31 of chromosome 3, there is a 200kb haplotype of Neanderthal origin that has a high frequency (> 49%) in the East Asians sequenced as part of the 1000 Genomes Project7. The introgressed region shows very high LD and significantly high values of the iHS statistic49, which measures extended haplotype homozygosity and is a hallmark of a recent selective sweep. However, as mentioned before, it is unclear how the iHS score would be affected by admixture in the absence of selection. One of the most likely targets of selection is a nonsynonymous SNP in the gene HYAL2, involved in the cellular response to ultraviolet radiation. The SNP is absent in other non- African populations, so it appears to have been lost in the ancestors of Eurasians after migrating out of Africa, but was regained in East Asians via admixture with Neanderthals. The authors performed a bootstrapped phylogenetic analysis to support the shared ancestry of the haplotype with the Neanderthal sequence and obtained a significant p-value for the observed LD value compared to a null model without introgression. Its frequency distribution shows a weak latitudinal gradient, suggesting it was involved in the adaptive response to ultraviolet radiation as modern humans expanded throughout Asia7. A putative signal of adaptive introgression in East Asians in HYAL2 has also been identified using the CRF framework47.
BNC2 seems to be a strong candidate for adaptive introgression, as shown in two genome-wide archaic ancestry analyses23, 47. Sankararaman et al.47 applied the CRF model to detect introgressed segments, and then inferred selection based on departures from a null model of neutrally introgressed alleles. Vernot and Akey23 also found the introgressed region using S*, then confirmed its ancestry by matching it with the Neanderthal genome, and finally inferred selection by observing that the region has high differentiation between Europeans and Asians, as measured by FST. A BNC2 SNP is associated with skin pigmentation76 and freckling in Europeans77, and the archaic haplotype is present at 70% frequency in Europeans, while it is absent in Asians. Interestingly both studies also found a strong adaptive introgression signal in a cluster of keratin genes on chromosome 12 in both Asians and Europeans23, 47.
Two neighboring genes (POU2F3 and TMEM136) have significant evidence for adaptive introgression in East Asians only, again based on the two genome-wide archaic ancestry analyses23, 47 (see above). POU2F3 is a transcription factor that mediates keratinocyte differentiation and proliferation, and the archaic haplotype is at 66% frequency in East Asians but almost absent in Europeans. TMEM136 codes for a transmembrane protein, but little information is available about its function.47
Ding et al.78 identified an introgressed haplotype of Neanderthal origin in Eurasians carrying a loss-of-function variant (Val92Met) in the gene MC1R, which encodes a melanocyte stimulating hormone receptor. This gene is known to affect hair color in mice79 and is associated with red hair, freckles and type I/II fair skin type in humans80, 81. The region, however, shows no significant departures from neutrality at the introgressed region in Europeans or East Asians, using either Tajima’s D52, Fu and Li’s test82, or iHS49, presumably because the frequency of the archaic haplotype only ranges from 5–22%. In addition, the lossof- function mutation (Val92Met) is not actually seen in the high-coverage Neanderthal genome4, despite being almost exclusively observed within haplotypes inferred to be introgressed from Neanderthals in Eurasian populations. The variant is also present in 3 African HapMap samples83, which weakens the argument for introgression into Eurasians, unless the variant was later introduced into Africans via admixture from Eurasians. Intriguingly, the same variant is found at very high frequencies in Taiwanese aborigines (60–70%), but lack of extensive sequence data at this locus has prevented formal rejection of neutrality at the putatively introgressed haplotype in these populations78.
Altitude
EPAS1 codes for a transcription factor that has a role in the response to hypoxia at high altitudes8. This gene was previously identified as being under positive selection in Tibetans in several studies58, 60, 84–88 but it did not fit with a simple model of selection from de novo mutation or from standing variation8. A set of 5 EPAS1 intronic mutations in a 32kb window are present in in the Denisova genome and at high frequency (~80%) in Tibetans, while absent in all of the individuals sequenced as part of the 1000 Genomes Project66 with the exception of two Han Chinese individuals.8 A haplotype network of present-day human and Denisovan haplotypes revealed that the Denisovan individual contained the closest-matching sequence to the introgressed haplotype. Their statistical evidence for introgression is based on highly significant D statistics, S* statistics (Supplementary information S1 (box), Box 1), and haplotype length. The unusually long haplotype length also rules out incomplete lineage sorting (Supplementary information S2 (box)) as a source of similarity to the Denisovan haplotype. Positive selection was supported by highly significant local differentiation based on the PBS58 statistic. Further, the putatively selected Tibetan variants in EPAS1 are significantly associated with hemoglobin concentrations, a phenotype that distinguishes Tibetans from lowland populations57. Denisovan ancestry throughout the genome of East Asian populations is very low4 (~0.2%), which suggests that selection was important in maintaining this specific haplotype at such high frequencies in Tibetans.
Metabolism
A recent study 67 proposed that Neanderthal alleles in lipid catabolism genes have been targets of recent positive natural selection in Europeans. The D statistic was used locally to detect regions of the genome supporting a gene tree in which a particular human population clustered with Neanderthals, to the exclusion of other human populations89. Lipid catabolism genes were significantly enriched for high D values (compared to the genome-wide expectation) in all European populations tested, but not in Asians or Africans. The authors then computed CMS scores54 (which serve to test for positive selection) along the genome, and these showed a significant positive correlation with the value of D for genes within the lipid catabolism gene set. However, it is possible that other factors (such as local effects of admixture or population structure) could have influenced the CMS score. The authors used phenotypic information to corroborate the result: they observed significantly diverged levels of lipid concentrations (by mass spectrometry) and lipid metabolic enzyme gene expression (by RNA-seq) in brain tissues of Europeans, when compared to Asian or African populations.
Solute carrier genes SLC16A11 and SLC16A13 are also implicated in lipid metabolic processes and have been found to harbor an introgressed haplotype. A GWAS study for Type 2 diabetes in thousands of Mexicans and Latin Americans revealed a novel association with the disease in a region that spanned these genes90. The strongest association was seen in a five SNP haplotype within SLC16A11, with four of the SNPs causing an amino acid change. The haplotype is at high frequency in Mexican populations, at lower frequency in Asians and very rare or absent in Europeans and Africans. Estimates of divergence time of the haplotype between Mexicans and Europeans of 799 kya (assuming a mutation rate of 10−9 per bp per year) suggested to the authors that the haplotype may have archaic origins. They found all of the 5 SNPs are present and homozygous in the high coverage Neanderthal sequence4. They provided two further lines of evidence for ancient admixture. Firstly, over an extended 73kb region around the five SNPs, those individuals with the 5 SNP haplotype have a TMRCA of 250 kya to Neanderthal compared to a TMRCA of 677 kya to Neanderthal for the individuals that do not have the five SNP haplotypes. Secondly, the length of the haplotype is significantly longer than would be expected from a split with Neanderthals unless a more recent admixture had occurred. As a functional assessment, they carried out a combination of in vitro experiments with the introgressed SLC16A11 sequence, determination of endogenous intracellular localization, and tissue specific expression, all pointing to a role for the protein in hepatic lipid metabolism. They propose that selection may be acting on the locus owing to the high frequency of the haplotype, but carried out no further analyses in this regard. We can speculate that the change in lipid metabolism caused by introgression may have conferred an evolutionary advantage to the recipient population, possibly in relation to an altered diet.
Uncharacterized function
For some adaptive introgression candidates, it is less clear what the selective pressure may have been. One example is the dystrophin gene (DMD), which was identified by Zietkiewicz et al.91 as having an unusually diverged 8kb X-linked haplotype, B006, and confirmed by both Sankararaman et al.47 and Yotova et al.92 to have come from Neanderthals. The B006 haplotype is found in all non-African human populations at low but considerable frequencies (7% in Middle Eastern individuals, 12.9% in Europeans, 4.1% in Asians, 17.6% in Native Americans) and is extremely rare in sub- Saharan African populations (average 0.7% frequency). Therefore, it may have undergone weak positive selection in non-Africans, but there is no formal evidence in favor of this hypothesis yet. The DMD gene encodes a protein that is an important structural component of skeletal muscle, although the confirmed archaic SNP is non-coding (intronic), and its functional effect remains uncertain.
Future studies
While many individual loci have been proposed to be adaptively introgressed, the total extent and relative contribution of adaptive introgression in human evolution remains unknown. Some of the recently reported candidate adaptive introgression regions have limited or no clear evidence for positive selection. Furthermore, in certain cases, ILS or ancestral structure has not been definitively ruled out. This requires more extensive analyses: for example, by simulating data under alternative scenarios that do not include selection and/or introgression to derive more rigorous null distributions of the statistics being used to investigate hypotheses regarding adaptive introgression.
A further caveat is that the true donor archaic population of an introgressed haplotype may be confounded by ILS in the population ancestral to multiple archaic (but not modern) human groups and/or gene flow between archaic populations. It may be difficult to distinguish between Neanderthal and Denisovan admixture in genomic regions where these populations are not highly differentiated.
While we have focused here on admixture between modern and archaic humans, there is well-documented evidence for admixture between different groups of modern humans in recent93, 94, as well as in ancient times42, 95. This type of genetic exchange between modern humans has already been proposed to have facilitated adaptation to harsh environments96, 97. However, the extent of adaptive gene flow among modern human populations remains an open question. For example, a recent study of nearly 30,000 African Americans found no evidence for adaptive gene flow in this recently admixed population98. As most genetic variation is shared among modern human populations, there is less opportunity for gene flow to introduce new genetic variation that was previously absent in the recipient population.
Interestingly, there is also genetic evidence for admixture between different archaic groups. The sequencing of the Neanderthal and Denisovan genomes at high coverage revealed signals of admixture from eastern Neanderthals into Denisovans, as well as into Denisovans from an unknown ‘super-archaic’ group that diverged from present-day humans before Neanderthals and Denisovans3, 4 (see also Online Resources for earlier proposals of the super-archaic admixture model using the lower coverage draft Neanderthal and Denisovan genomes). The introgressed regions for most of these events have yet to be identified, but once found, they may reveal important adaptations that were shared between archaic humans. For example, Neanderthal introgression in Denisovans has been identified at loci that include the CRISP cluster of genes, known to have a role in sperm maturation and egg fertilization, as well as the HLA region4.
All instances of putative archaic adaptive introgression published so far have been found by either: a) detecting signatures of selection in regions previously known to be introgressed, b) detecting signatures of introgression in regions known to be selected, or c) observing archaic haplotypes in regions identified in GWAS studies. No method yet exists that can jointly infer the probability of admixture and selection in a particular region. One approach could be to develop informative summary statistics or combinations of summary statistics (like D, S*, the number of uniquely shared sites and Fst-based statistics) under different models of introgression and adaptation. These could be incorporated into a likelihood framework for adaptive introgression to scan the genome in search for this signal. To truly be able to discern archaic introgression from other forces, this method should also require that the length of archaic segments be consistent with archaic introgression (Figure 1), under a realistic demographic scenario. Such a model-oriented approach could potentially allow for estimation of informative parameters, like selection coefficients and times of introgression.
Conclusions
The biological processes that appear to have been affected by adaptive introgression in humans closely mirror the processes that are typically found to be targeted by positive selection in humans and in other organisms. It appears that this process has provided a rich reservoir of new genetic variation that has allowed humans to adapt rapidly to a variety of new environmental conditions. The studies reviewed here suggest that adaptive introgression should be considered an important mode of selection in human population genetics, and can provide fascinating insights into the evolution of our species.
However, rigorously identifying regions affected by adaptive introgression in humans is not trivial: care should be taken to distinguish ancestral polymorphism from true introgression, and to use appropriate null models that include introgression when testing for selection. The advent of larger numbers and a broader range of human genome sequence data, both modern and archaic, in the near future may allow us to further disentangle the details of the process of adaptive introgression in humans, ultimately advancing our understanding of the genetic basis of phenotypic differences among human populations.
Supplementary Material
Key Points.
Recent genomic analysis of sequence data from archaic humans has detected evidence of gene flow from the genomes of archaic to modern humans. Several studies have identified DNA segments within the genomes of modern humans showing strong signatures of both introgression and positive selection.
We review statistical analysis methods developed for the detection of surviving archaic human DNA segments, as well as those used to ascertain that these introgressed segments show signatures of positive selection.
We group the reported examples of adaptive introgression by putative selective pressures, including pathogens, temperatures, altitude and diet. Candidate genes showing evidence for adaptive introgression have functional annotations suggesting roles in immune function, pigmentation, response to high altitude and metabolism.
While recent studies have identified several well-supported examples of adaptive introgression in humans, we still lack a framework that jointly models the effects of introgression and positive selection. Studies of the synergistic effect of these two forces will lead to a better characterization of adaptive introgression and of its relative importance in human adaptation.
Acknowledgments
E.H.S. is supported by startup funds from the University of California Merced. R.N and S..S are supported by the US National Institutes of Health (R01HG003229-09, K99 GM111744). F.R. is supported by the US National Institutes of Health grant to Montgomery Slatkin (R01-GM40282). We thank Fergal Casey for useful discussions and help with Box 1. We also thank Eric Durand for help with Supplementary information S1 (box).
Glossary
- Admixture
Genetic exchange between individuals from two populations that were isolated in the past
- Archaic introgression
The introduction of genetic material into the ancestors of an extant population (e.g. East Asians) from an archaic population that is currently extinct (e.g. Neanderthals), via admixture
- Archaic humans
A broad category of human populations that diverged from present-day humans 550–765 kya4 (assuming a mutation rate of 0.5*10−9 per bp per year) before present-day human populations started diverging from each other (86–130 kya, assuming the same mutation rate4) and that are now extinct. This includes the Neanderthal and Denisovan populations
- Modern humans
Present-day humans and their recent ancestors, up to the time at which they diverged from their most closely related archaic human groups, the Neanderthals and Denisovans
- Linkage disequilibrium
A non-random association of alleles in different loci along the same chromosome, due to low recombination rate, population structure, and/or selection
- Uniquely shared site
A site containing a high-frequency derived allele in a particular population that is also present in a distantly related population, but is absent or at low frequencies in other populations that are more closely related to the first population. Such a site serves as necessary, but not sufficient, evidence for adaptive introgression from the distantly related population
- Hidden Markov Model (HMM)
A statistical modeling method used to infer hidden states from observed data along an ordered sequence, wherein each hidden variable is independent of all other hidden variables, conditional on knowing the state of the immediately previous hidden variable
- Conditional Random Field (CRF)
A statistical modeling method that is similar to an HMM, but that also allows contextual information (regional data not directly contiguous to a site in a sequence) to provide information about the state of a hidden variable
- Haplotype
A sequence of contiguous alleles that are closely linked and that tend to be inherited together as a single unit
- Balancing selection
Selection that favors the maintenance of variability in a population, which can prevent any single allele from reaching fixation. Examples include frequency-dependent selection and heterozygous advantage (overdominance)
- Positive directional selection
Selection that favors a specific allele over others. The allele may consequently rise to high frequency or fixation. Hitchhiking of neutral alleles tightly linked to the favored allele leaves a known genetic footprint in the genome, sometimes allowing detection of positive selection at a particular locus
- D-statistics
Summary statistics based on differential sharing of derived alleles among different pairs of populations. When applied on a genome-wide scale, they can be used to detect significant deviations from a strict population tree with no admixture or migration
- S* statistic
Summary statistic based on patterns of linkage disequilibrium that can be used to detect introgressed haplotypes
- TMRCA
Time in generations back into the past until two copies of an allele or two haplotypes find their most recent common ancestor (MRCA). This is often an unknown parameter that can be estimated from genetic data
- Incomplete lineage sorting (ILS)
ILS occurs when two or more lineages from different populations or species share a common ancestor more recently than their respective MRCA within populations, causing discordance between the population tree and a gene-tree
- Ancestral structure
A demographic scenario in which an ancestral population is not homogenously mixing. For example, some sub-populations might exchange more migrants with certain other sub-populations than with the rest because of geography or mate choice
- Ancestral polymorphism
Used to describe the existence of more than one allele at a site or haplotype in the ancestor of two populations, before they diverged from each other
- Site-frequency spectrum
A vector of size n-1 whose ith entry is the number of polymorphic sites at which a derived (i.e. non-ancestral) allele is present in i copies in a sample of size n from a population. It can be used as a summary statistic for demographic inference
- Human mutation rate
The rate (per base-pair) at which mutations appear in the genome sequence of an individual at each generation or year. Currently, the exact value of this rate in humans is a topic of debate, with most estimates ranging from a value of 0.5*10−9 per bp per year to a value of 10−9 per bp per year
- Out of Africa model
A model of recent human evolution positing that all present day humans had a recent origin in Africa, and then expanded across the world, replacing other archaic groups
- Coalescence
An event in the past at which two genetic lineages sampled in the present find their most recent common ancestor, at a specific locus in the genome
- Emission functions
Functions that relate the hidden variables to the observed data in the CRF framework
- Negative selection
Selection that acts to prune away deleterious variants from the genome
- Hybrid sterility
Reduced viability or fertility of offspring from a mating between individuals from populations or species that diverged a long time ago, often due to incompatible mutations that occurred in each daughter population after they separated from each other
Biographies
Fernando Racimo is a graduate student at the Department of Integrative Biology at University of California Berkeley. He is interested in human paleogenomics and works on developing methods that use archaic genomes to infer demographic models and detect signals of positive selection in modern humans.
Sriram Sankararaman is a postdoctoral fellow at the Department of Genetics, Harvard Medical School. He completed his PhD in Computer Science at the University of California Berkeley. As a postdoc he has developed methods to estimate archaic ancestry and admixture times in modern humans. His interests lie at the interface of computational biology, statistical genomics and statistical machine learning.
Rasmus Nielsen is a Professor at the Department of Integrative Biology and the Department of Statistics at University of California Berkeley. The Nielsen lab’s research encompasses population genetics, statistical genetics, medical genetics, evolutionary biology and phylogenetics.
Emilia Huerta-Sánchez is an Assistant Professor at the School of Natural Sciences at University of California Merced. She was previously a postdoc in the Departments of Statistics and Integrative Biology at the University of California Berkeley. Her lab focuses on modeling and characterizing the effects of natural selection and demography in natural populations. For more details visit https://www.stat.berkeley.edu/~emiliahs/
References
- 1.Green RE, et al. A draft sequence of the Neandertal genome. Science. 2010;328:710–22. doi: 10.1126/science.1188021. The draft sequence of a Neanderthal genome. Analysis of this sequence provided the first estimates of the proportion of archaic admixture in present-day human genomes. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Reich D, et al. Genetic history of an archaic hominin group from Denisova Cave in Siberia. Nature. 2010;468:1053–60. doi: 10.1038/nature09710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Meyer M, et al. A high-coverage genome sequence from an archaic Denisovan individual. Science. 2012;338:222–6. doi: 10.1126/science.1224344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Prüfer K, et al. The complete genome sequence of a Neanderthal from the Altai Mountains. Nature. 2014;505:43–9. doi: 10.1038/nature12886. Release of the first high coverage sequence of a Neanderthal genome. Analysis of this sequence allowed for the identification of Neanderthal segments in present-day humans. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Abi-Rached L, et al. The shaping of modern human immune systems by multiregional admixture with archaic humans. Science. 2011;334:89–94. doi: 10.1126/science.1209202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Mendez FL, Watkins JC, Hammer MF. A haplotype at STAT2 Introgressed from neanderthals and serves as a candidate of positive selection in Papua New Guinea. Am J Hum Genet. 2012;91:265–74. doi: 10.1016/j.ajhg.2012.06.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ding Q, Hu Y, Xu S, Wang J, Jin L. Neanderthal introgression at chromosome 3p21.31 was under positive natural selection in East Asians. Mol Biol Evol. 2014;31:683–95. doi: 10.1093/molbev/mst260. [DOI] [PubMed] [Google Scholar]
- 8.Huerta-Sánchez E, et al. Altitude adaptation in Tibetans caused by introgression of Denisovan-like DNA. Nature. 2014;512:194–7. doi: 10.1038/nature13408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Wolpoff MH, Wu X, Thorne AG. In: The Origins of Modern Humans: A World Survey of the Fossil Evidence. Smith FH, Spencer F, editors. Liss; New York: 1984. pp. 411–483. [Google Scholar]
- 10.Stringer CB, Andrews P. Genetic and fossil evidence for the origin of modern humans. Science. 1988;239:1263–8. doi: 10.1126/science.3125610. [DOI] [PubMed] [Google Scholar]
- 11.Bräuer G. The Afro-European sapiens hypothesis and hominid evolution in East Asia during the late Middle and Upper Pleistocene. Cour Forschungsinst Senckenb. 1984;69:145–165. [Google Scholar]
- 12.Smith FH, Janković I, Karavanić I. The assimilation model, modern human origins in Europe, and the extinction of Neandertals. Quaternary International. 2005;137:7–19. [Google Scholar]
- 13.Plagnol V, Wall JD. Possible ancestral structure in human populations. PLoS Genet. 2006;2:e105. doi: 10.1371/journal.pgen.0020105. A novel statistic algorithm, S*, is developed here in order to find candidate linked SNPs within haplotypes that may have been introduced by admixture with archaic humans. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Wall JD, Lohmueller KE, Plagnol V. Detecting ancient admixture and estimating demographic parameters in multiple human populations. Mol Biol Evol. 2009;26:1823–7. doi: 10.1093/molbev/msp096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Higham T, et al. The timing and spatiotemporal patterning of Neanderthal disappearance. Nature. 2014;512:306–9. doi: 10.1038/nature13621. [DOI] [PubMed] [Google Scholar]
- 16.Castellano S, et al. Patterns of coding variation in the complete exomes of three Neandertals. Proc Natl Acad Sci U S A. 2014;111:6666–71. doi: 10.1073/pnas.1405138111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Arnold ML, Martin NH. Adaptation by introgression. J Biol. 2009;8:82. doi: 10.1186/jbiol176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Rieseberg LH. Evolution: replacing genes and traits through hybridization. Curr Biol. 2009;19:R119–22. doi: 10.1016/j.cub.2008.12.016. [DOI] [PubMed] [Google Scholar]
- 19.Hedrick PW. Adaptive introgression in animals: examples and comparison to new mutation and standing variation as sources of adaptive variation. Mol Ecol. 2013;22:4606–18. doi: 10.1111/mec.12415. An introductory review of adaptive introgression in non-human species, providing a comparison with other types of selection and a list of many specific examples. [DOI] [PubMed] [Google Scholar]
- 20.Hawks J, Cochran G, Harpending HC, Lahn BT. A genetic legacy from archaic Homo. Trends Genet. 2008;24:19–23. doi: 10.1016/j.tig.2007.10.003. [DOI] [PubMed] [Google Scholar]
- 21.Hawks J, Cochran G. Dynamics of adaptive introgression from archaic to modern humans. PaleoAnthropology. 2006;101–115 A visionary review that proposed adaptive introgression as a mode of adaptation in humans, before the availability of archaic genome sequences. [Google Scholar]
- 22.Wall JD, et al. Higher levels of neanderthal ancestry in East Asians than in Europeans. Genetics. 2013;194:199–209. doi: 10.1534/genetics.112.148213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Vernot B, Akey JM. Resurrecting surviving Neandertal lineages from modern human genomes. Science. 2014;343:1017–21. doi: 10.1126/science.1245938. A key paper estimating the sum total of Neandertal segments in a large sample of present-day human genomes using an LD-based statistic, S*, and identifying segments that may be targets of positive selection. [DOI] [PubMed] [Google Scholar]
- 24.Reich D, et al. Denisova admixture and the first modern human dispersals into Southeast Asia and Oceania. Am J Hum Genet. 2011;89:516–28. doi: 10.1016/j.ajhg.2011.09.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Skoglund P, Jakobsson M. Archaic human ancestry in East Asia. Proc Natl Acad Sci U S A. 2011;108:18301–6. doi: 10.1073/pnas.1108181108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hammer MF, Woerner AE, Mendez FL, Watkins JC, Wall JD. Genetic evidence for archaic admixture in Africa. Proc Natl Acad Sci U S A. 2011;108:15123–8. doi: 10.1073/pnas.1109300108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lachance J, et al. Evolutionary history and adaptation from high-coverage whole-genome sequences of diverse African hunter-gatherers. Cell. 2012;150:457–69. doi: 10.1016/j.cell.2012.07.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Veeramah KR, Hammer MF. The impact of whole-genome sequencing on the reconstruction of human population history. Nat Rev Genet. 2014;15:149–62. doi: 10.1038/nrg3625. [DOI] [PubMed] [Google Scholar]
- 29.Currat M, Excoffier L. Strong reproductive isolation between humans and Neanderthals inferred from observed patterns of introgression. Proc Natl Acad Sci U S A. 2011;108:15129–34. doi: 10.1073/pnas.1107450108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Eriksson A, Manica A. Effect of ancient population structure on the degree of polymorphism shared between modern human populations and ancient hominins. Proc Natl Acad Sci U S A. 2012;109:13956–60. doi: 10.1073/pnas.1200567109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Liang M, Nielsen R. The Lengths of Admixture Tracts. Genetics. 2014;197:953–967. doi: 10.1534/genetics.114.162362. This work presents important theoretical results on the expected length of admixture tracts and how they can be used to identify particular models of admixture. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Pool JE, Nielsen R. Inference of historical changes in migration rate from the lengths of migrant tracts. Genetics. 2009;181:711–9. doi: 10.1534/genetics.108.098095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Gravel S. Population genetics models of local ancestry. Genetics. 2012;191:607–19. doi: 10.1534/genetics.112.139808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Fu Q, et al. Genome sequence of a 45,000-year-old modern human from western Siberia. Nature. 2014;514:445–9. doi: 10.1038/nature13810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Seguin-Orlando A, et al. Genomic structure in Europeans dating back at least 36,200 years. Science. 2014;346:1113–1118. doi: 10.1126/science.aaa0114. [DOI] [PubMed] [Google Scholar]
- 36.Sankararaman S, Patterson N, Li H, Pääbo S, Reich D. The date of interbreeding between Neandertals and modern humans. PLoS Genet. 2012;8:e1002947. doi: 10.1371/journal.pgen.1002947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Gronau I, Hubisz MJ, Gulko B, Danko CG, Siepel A. Bayesian inference of ancient human demography from individual genome sequences. Nat Genet. 2011;43:1031–4. doi: 10.1038/ng.937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Gravel S, et al. Demographic history and rare allele sharing among human populations. Proc Natl Acad Sci U S A. 2011;108:11983–8. doi: 10.1073/pnas.1019276108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Harris K, Nielsen R. Inferring demographic history from a spectrum of shared haplotype lengths. PLoS Genet. 2013;9:e1003521. doi: 10.1371/journal.pgen.1003521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Yang MA, Malaspinas AS, Durand EY, Slatkin M. Ancient structure in Africa unlikely to explain Neanderthal and non-African genetic similarity. Mol Biol Evol. 2012;29:2987–95. doi: 10.1093/molbev/mss117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Lohse K, Frantz LA. Neandertal admixture in Eurasia confirmed by maximum-likelihood analysis of three genomes. Genetics. 2014;196:1241–51. doi: 10.1534/genetics.114.162396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Patterson N, et al. Ancient admixture in human history. Genetics. 2012;192:1065–93. doi: 10.1534/genetics.112.145037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Durand EY, Patterson N, Reich D, Slatkin M. Testing for ancient admixture between closely related populations. Molecular biology and evolution. 2011;28:2239–2252. doi: 10.1093/molbev/msr048. A detailed analysis of Patterson’s D statistic to test for admixture with archaic humans. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Martin SH, Davey JW, Jiggins CD. Evaluating the use of ABBA–BABA statistics to locate introgressed loci. Molecular biology and evolution. 2014;32:244–257. doi: 10.1093/molbev/msu269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Wall JD. Detecting ancient admixture in humans using sequence polymorphism data. Genetics. 2000;154:1271–1279. doi: 10.1093/genetics/154.3.1271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Viterbi AJ. Error bounds for convolutional codes and an asymptotically optimum decoding algorithm. Information Theory, IEEE Transactions on. 1967;13:260–269. [Google Scholar]
- 47.Sankararaman S, et al. The genomic landscape of Neanderthal ancestry in present-day humans. Nature. 2014;507:354–7. doi: 10.1038/nature12961. A framework that combines different DNA sequence features to identify Neandertal segments in present-day humans, and to identify which of those segments may have been positively selected. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Moorjani P, et al. The history of African gene flow into Southern Europeans, Levantines, and Jews. PLoS Genet. 2011;7:e1001373. doi: 10.1371/journal.pgen.1001373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Voight BF, Kudaravalli S, Wen X, Pritchard JK. A map of recent positive selection in the human genome. PLoS Biol. 2006;4:e72. doi: 10.1371/journal.pbio.0040072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Sabeti PC, et al. Detecting recent positive selection in the human genome from haplotype structure. Nature. 2002;419:832–7. doi: 10.1038/nature01140. [DOI] [PubMed] [Google Scholar]
- 51.Sabeti PC, et al. Genome-wide detection and characterization of positive selection in human populations. Nature. 2007;449:913–8. doi: 10.1038/nature06250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Tajima F. Statistical method for testing the neutral mutation hypothesis by DNA polymorphism. Genetics. 1989;123:585–595. doi: 10.1093/genetics/123.3.585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Fay JC, Wu CI. Hitchhiking under positive Darwinian selection. Genetics. 2000;155:1405–13. doi: 10.1093/genetics/155.3.1405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Grossman SR, et al. A composite of multiple signals distinguishes causal variants in regions of positive selection. Science. 2010;327:883–6. doi: 10.1126/science.1183863. [DOI] [PubMed] [Google Scholar]
- 55.Barreiro LB, Laval G, Quach H, Patin E, Quintana-Murci L. Natural selection has driven population differentiation in modern humans. Nat Genet. 2008;40:340–5. doi: 10.1038/ng.78. [DOI] [PubMed] [Google Scholar]
- 56.Lewontin RC, Krakauer J. Distribution of gene frequency as a test of the theory of the selective neutrality of polymorphisms. Genetics. 1973;74:175–95. doi: 10.1093/genetics/74.1.175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Shriver MD, et al. The genomic distribution of population substructure in four populations using 8,525 autosomal SNPs. Hum Genomics. 2004;1:274–86. doi: 10.1186/1479-7364-1-4-274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Yi X, et al. Sequencing of 50 human exomes reveals adaptation to high altitude. Science. 2010;329:75–8. doi: 10.1126/science.1190371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Chakraborty R. Gene admixture in human populations: models and predictions. Yearbook of Physical Anthropology. 1986;29:1–43. [Google Scholar]
- 60.Beall CM, et al. Natural selection on EPAS1 (HIF2alpha) associated with low hemoglobin concentration in Tibetan highlanders. Proc Natl Acad Sci U S A. 2010;107:11459–64. doi: 10.1073/pnas.1002443107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Gould SJ, Lewontin RC. The spandrels of San Marco and the Panglossian paradigm: a critique of the adaptationist programme. Proc R Soc Lond B Biol Sci. 1979;205:581–98. doi: 10.1098/rspb.1979.0086. [DOI] [PubMed] [Google Scholar]
- 62.Hardy J, et al. Evidence suggesting that Homo neanderthalensis contributed the H2 MAPT haplotype to Homo sapiens. Biochem Soc Trans. 2005;33:582–5. doi: 10.1042/BST0330582. [DOI] [PubMed] [Google Scholar]
- 63.Evans PD, Mekel-Bobrov N, Vallender EJ, Hudson RR, Lahn BT. Evidence that the adaptive allele of the brain size gene microcephalin introgressed into Homo sapiens from an archaic Homo lineage. Proc Natl Acad Sci U S A. 2006;103:18178–83. doi: 10.1073/pnas.0606966103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Setó-Salvia N, et al. Using the neanderthal and denisova genetic data to understand the common MAPT 17q21 inversion in modern humans. Hum Biol. 2012;84:633–40. doi: 10.3378/027.084.0605. [DOI] [PubMed] [Google Scholar]
- 65.Ségurel L, Wyman MJ, Przeworski M. Determinants of mutation rate variation in the human germline. Annu Rev Genomics Hum Genet. 2014;15:47–70. doi: 10.1146/annurev-genom-031714-125740. [DOI] [PubMed] [Google Scholar]
- 66.Abecasis GR, et al. An integrated map of genetic variation from 1,092 human genomes. Nature. 2012;491:56–65. doi: 10.1038/nature11632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Khrameeva EE, et al. Neanderthal ancestry drives evolution of lipid catabolism in contemporary Europeans. Nat Commun. 2014;5:3584. doi: 10.1038/ncomms4584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Baselga J, Swain SM. Novel anticancer targets: revisiting ERBB2 and discovering ERBB3. Nat Rev Cancer. 2009;9:463–75. doi: 10.1038/nrc2656. [DOI] [PubMed] [Google Scholar]
- 69.Lalioti V, et al. The atypical kinase Cdk5 is activated by insulin, regulates the association between GLUT4 and E-Syt1, and modulates glucose transport in 3T3-L1 adipocytes. Proc Natl Acad Sci U S A. 2009;106:4249–53. doi: 10.1073/pnas.0900218106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Min SW, Chang WP, Südhof TC. E-Syts, a family of membranous Ca2+-sensor proteins with multiple C2 domains. Proc Natl Acad Sci U S A. 2007;104:3823–8. doi: 10.1073/pnas.0611725104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.DeGiorgio M, Lohmueller KE, Nielsen R. A model-based approach for identifying signatures of ancient balancing selection in genetic data. PLoS Genet. 2014;10:e1004561. doi: 10.1371/journal.pgen.1004561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Temme S, et al. A novel family of human leukocyte antigen class II receptors may have its origin in archaic human species. J Biol Chem. 2014;289:639–53. doi: 10.1074/jbc.M113.515767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Ding Q, Hu Y, Jin L. Non-Neanderthal origin of the HLA-DPB1*0401. J Biol Chem. 2014;289:10252. doi: 10.1074/jbc.L114.547505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Mendez FL, Watkins JC, Hammer MF. Neandertal origin of genetic variation at the cluster of OAS immunity genes. Mol Biol Evol. 2013;30:798–801. doi: 10.1093/molbev/mst004. [DOI] [PubMed] [Google Scholar]
- 75.Mendez FL, Watkins JC, Hammer MF. Global genetic variation at OAS1 provides evidence of archaic admixture in Melanesian populations. Mol Biol Evol. 2012;29:1513–20. doi: 10.1093/molbev/msr301. [DOI] [PubMed] [Google Scholar]
- 76.Jacobs LC, et al. Comprehensive candidate gene study highlights UGT1A and BNC2 as new genes determining continuous skin color variation in Europeans. Hum Genet. 2013;132:147–58. doi: 10.1007/s00439-012-1232-9. [DOI] [PubMed] [Google Scholar]
- 77.Eriksson N, et al. Web-based, participant-driven studies yield novel genetic associations for common traits. PLoS Genet. 2010;6:e1000993. doi: 10.1371/journal.pgen.1000993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Ding Q, et al. Neanderthal origin of the haplotypes carrying the functional variant Val92Met in the MC1R in modern humans. Mol Biol Evol. 2014;31:1994–2003. doi: 10.1093/molbev/msu180. [DOI] [PubMed] [Google Scholar]
- 79.Nachman MW, Hoekstra HE, D’Agostino SL. The genetic basis of adaptive melanism in pocket mice. Proc Natl Acad Sci U S A. 2003;100:5268–73. doi: 10.1073/pnas.0431157100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Valverde P, et al. The Asp84Glu variant of the melanocortin 1 receptor (MC1R) is associated with melanoma. Hum Mol Genet. 1996;5:1663–6. doi: 10.1093/hmg/5.10.1663. [DOI] [PubMed] [Google Scholar]
- 81.Valverde P, Healy E, Jackson I, Rees JL, Thody AJ. Variants of the melanocyte-stimulating hormone receptor gene are associated with red hair and fair skin in humans. Nat Genet. 1995;11:328–30. doi: 10.1038/ng1195-328. [DOI] [PubMed] [Google Scholar]
- 82.Fu YX, Li WH. Statistical tests of neutrality of mutations. Genetics. 1993;133:693–709. doi: 10.1093/genetics/133.3.693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Altshuler DM, et al. Integrating common and rare genetic variation in diverse human populations. Nature. 2010;467:52–8. doi: 10.1038/nature09298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Bigham A, et al. Identifying signatures of natural selection in Tibetan and Andean populations using dense genome scan data. PLoS Genet. 2010;6:e1001116. doi: 10.1371/journal.pgen.1001116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Simonson TS, et al. Genetic evidence for high-altitude adaptation in Tibet. Science. 2010;329:72–5. doi: 10.1126/science.1189406. [DOI] [PubMed] [Google Scholar]
- 86.Peng Y, et al. Genetic variations in Tibetan populations and high-altitude adaptation at the Himalayas. Mol Biol Evol. 2011;28:1075–81. doi: 10.1093/molbev/msq290. [DOI] [PubMed] [Google Scholar]
- 87.Xu S, et al. A genome-wide search for signals of high-altitude adaptation in Tibetans. Mol Biol Evol. 2011;28:1003–11. doi: 10.1093/molbev/msq277. [DOI] [PubMed] [Google Scholar]
- 88.Wang B, et al. On the origin of Tibetans and their genetic basis in adapting high-altitude environments. PLoS One. 2011;6:e17002. doi: 10.1371/journal.pone.0017002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Abecasis GR, et al. A map of human genome variation from population-scale sequencing. Nature. 2010;467:1061–73. doi: 10.1038/nature09534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Williams AL, et al. Sequence variants in SLC16A11 are a common risk factor for type 2 diabetes in Mexico. Nature. 2014;506:97–101. doi: 10.1038/nature12828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Zietkiewicz E, et al. Haplotypes in the dystrophin DNA segment point to a mosaic origin of modern human diversity. Am J Hum Genet. 2003;73:994–1015. doi: 10.1086/378777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Yotova V, et al. An X-linked haplotype of Neandertal origin is present among all non-African populations. Mol Biol Evol. 2011;28:1957–62. doi: 10.1093/molbev/msr024. [DOI] [PubMed] [Google Scholar]
- 93.Moorjani P, et al. Genetic evidence for recent population mixture in India. Am J Hum Genet. 2013;93:422–38. doi: 10.1016/j.ajhg.2013.07.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Hellenthal G, et al. A genetic atlas of human admixture history. Science. 2014;343:747–51. doi: 10.1126/science.1243518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Lazaridis I, et al. Ancient human genomes suggest three ancestral populations for present-day Europeans. Nature. 2014;513:409–13. doi: 10.1038/nature13673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Huerta-Sánchez E, et al. Genetic signatures reveal high-altitude adaptation in a set of ethiopian populations. Mol Biol Evol. 2013;30:1877–88. doi: 10.1093/molbev/mst089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Jeong C, et al. Admixture facilitates genetic adaptations to high altitude in Tibet. Nat Commun. 2014;5:3281. doi: 10.1038/ncomms4281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Bhatia G, et al. Genome-wide Scan of 29,141 African Americans Finds No Evidence of Directional Selection since Admixture. Am J Hum Genet. 2014;95:437–44. doi: 10.1016/j.ajhg.2014.08.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
Online Resources
- Hawks J. John Hawks Weblog. 2011 ( http://johnhawks.net/weblog/reviews/denisova/denisova-mcph1-2011.html.
- Waddell PJ, Tan X. New g%AIC, g%AICc, g%BIC, and Power Divergence Fit Statistics Expose Mating between Modern Humans, Neanderthals and other Archaics. arXiv. 2012 ( http://arxiv.org/abs/1212.6820)
- Waddell PJ. Happy New Year Homo erectus? More evidence for interbreeding with archaics predating the modern human/Neanderthal split. 2013 arXiv. http://arxiv.org/abs/1312.7749.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.