

Abstract
Hepatitis C virus (HCV) is a hepatotropic virus with a plus-strand RNA genome of ∼9.600 nt. Due to error-prone replication by its RNA-dependent RNA polymerase (RdRp) residing in nonstructural protein 5B (NS5B), HCV isolates are grouped into seven genotypes with several subtypes. By using whole-genome sequences of 106 HCV isolates and secondary structure alignments of the plus-strand genome and its minus-strand replication intermediate, we established refined secondary structures of the 5′ untranslated region (UTR), the cis-acting replication element (CRE) in NS5B, and the 3′ UTR. We propose an alternative structure in the 5′ UTR, conserved secondary structures of 5B stem–loop (SL)1 and 5BSL2, and four possible structures of the X-tail at the very 3′ end of the HCV genome. We predict several previously unknown long-range interactions, most importantly a possible circularization interaction between distinct elements in the 5′ and 3′ UTR, reminiscent of the cyclization elements of the related flaviviruses. Based on analogy to these viruses, we propose that the 5′–3′ UTR base-pairing in the HCV genome might play an important role in viral RNA replication. These results may have important implications for our understanding of the nature of the cis-acting RNA elements in the HCV genome and their possible role in regulating the mutually exclusive processes of viral RNA translation and replication.
Keywords: bioinformatics, HCV, RNA long-range interaction prediction, circularization, RNA secondary structure prediction
INTRODUCTION
Hepatitis C virus (HCV) is a hepatotropic plus-strand RNA virus causing acute or chronic liver disease that often leads to liver cirrhosis and hepatocellular carcinoma (Yamane et al. 2013; Zeisel et al. 2013). As the prototype member of the genus Hepacivirus within the family Flaviviridae, HCV is a representative of a large group of plus-strand RNA viruses that encode their own RNA-dependent RNA polymerases (RdRp). Since these enzymes lack 3′–5′-exonuclease proofreading activity, RNA replication of these viruses gives rise to a large number of progeny genomes with replication errors. Accordingly, a huge number of HCV isolates has been found worldwide, grouped into seven genotypes with several subtypes (Simmonds 2013; Jackowiak et al. 2014). For these reasons, HCV can be used as a model system to analyze the conservation of functional RNA sequences and secondary structures as well as long-range RNA–RNA interactions in a viral genome.
The single-stranded HCV genome of ∼9.6 kb is translated at the rough endoplasmatic reticulum in the cytosol. A single open reading frame (ORF) encodes for a single polyprotein that is co- and post-translationally processed by host and viral proteases into 10 cleavage products. These are classified as structural (core, envelope glycoproteins E1, E2, and p7) and nonstructural proteins (NS2, NS3, NS4A, NS4B, NS5A, and the RNA-dependent RNA polymerase [RdRp] residing in NS5B) (Lohmann 2013; Moradpour and Penin 2013).
The ORF encoding the polyprotein is flanked by highly structured 5′ and 3′ untranslated regions (UTRs), which are indispensable for translation and replication of the viral genome (Liu et al. 2009; Lohmann 2013). The 5′ UTR consists of four structured loop domains (SLI–SLIV), which have been extensively studied for genotypes 1 and 2 (Brown et al. 1992; Honda et al. 1999a; Friebe and Bartenschlager 2009; Lohmann 2013; Niepmann 2013). The SLI and SLII domains (Fig. 1) are essential for RNA replication (Friebe et al. 2001). This region contains two binding sites for the liver-specific microRNA-122 (miR-122) that stimulates HCV RNA translation and replication (Jopling et al. 2005; Henke et al. 2008) and enhances RNA stability (Shimakami et al. 2012; Sedano and Sarnow 2014) and translation (Henke et al. 2008).
FIGURE 1.
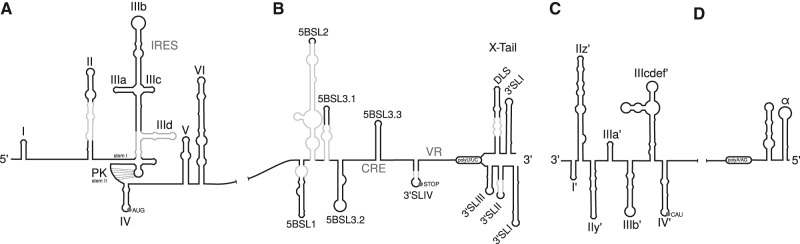
Overview of previously known (black) and novel (gray) RNA stem–loop structures of the HCV RNA genome. (A) 5′ UTR and core region SLV and SLVI of the viral plus-strand. (B) CRE region and 3′ UTR of the viral plus-strand. (C) 3′ End of the viral minus-strand. (D) 5′ End of the viral minus-strand.
Partially overlapping with these replication signals is the internal ribosomal entry site (IRES) which is formed by the domains SLII–SLIV along with the first 20–40 nt of the coding region (Friebe et al. 2001). This IRES confers cap-independent RNA translation (Brown et al. 1992; Honda et al. 1996).
Domains SLII, SLIIIa, d, e, and f as well as SLIV of the IRES are important for the recruitment of the ribosomal 40S subunit (Pestova et al. 1998; Spahn et al. 2001; Kieft et al. 2001, 2002). For this process the pseudoknot of the IRES has been shown to be essential (Wang et al. 1995; Berry et al. 2011). Additionally, the initiation factor eIF3 is recruited to the IRES mainly by determinants in domains SLIIIb and c (Buratti et al. 1998; Sizova et al. 1998; Kieft et al. 2002; Siridechadilok et al. 2005). Based on the crystal structure of the complete HCV IRES, a model has been proposed how the preinitiation complex might assemble to allow efficient RNA translation (Pérard et al. 2013).
The 3′ UTR is essential for viral minus-strand RNA synthesis (Yanagi et al. 1999; Friebe and Bartenschlager 2002; Yi and Lemon 2003; You and Rice 2008). It consists of a variable region (VR), a poly(U/UC) tract, and a conserved structure called 3′X or X-tail (Tanaka et al. 1995; Lohmann 2013). The variable region is poorly conserved among HCV isolates; this region is not essential, but enhances RNA replication (Friebe and Bartenschlager 2002). In the variable region resides the conserved stem–loop (SLIV) containing the stop codon of the polyprotein (Lohmann 2013). Conservation of a second stem–loop in the 3′ UTR among HCV isolates was not convincingly shown. The poly(U/UC) tract has a variable length, ranging between 30 and 90 nt (Lohmann 2013). The highly conserved X-tail can form two different conformations. One conformation consists of three experimentally verified stem–loop structures (SLI, SLII, SLIII, numbered in 3′ to 5′ direction with respect to the plus-strand) (Blight and Rice 1997) and is crucial for RNA replication (Friebe and Bartenschlager 2002). In an alternative conformation of the X-tail, only two stem–loops are formed. Here, SLI remains the same, but SLII and SLIII are merged to form a single stem–loop which includes a so-called dimerization linkage sequence (DLS) in its apical loop that was proposed to be necessary for viral RNA genome dimerization and possibly for genome encapsidation (Ivanyi-Nagy et al. 2006; Shetty et al. 2010).
It has been reported that the adjacent NS5B coding region might contain five structured stem–loops (Tuplin et al. 2002; Diviney et al. 2008) designated: 5BSL1 (also called SL9033), 5BSL2 (SL9132), 5BSL3.1 (SL9217), 5BSL3.2 (SL9266), and 5BSL3.3 (SL9324). Of these, 5BSL3.2 has been identified as a cis-acting replication element (CRE) that is important for viral replication (Lee et al. 2004; Friebe et al. 2005; Diviney et al. 2008). 5BSL3.2 is involved in several long-range interactions (LRIs). Its apical loop interacts with the SLII region of the X-tail, while its internal bulge can interact either with the IRES SLIIId apical loop (Romero-López and Berzal-Herranz 2009) or alternatively with a sequence called CRE 9110 which is located between 5BSL1 (SL9033) and 5BSL2 (SL9132); see Fig. 1 (Diviney et al. 2008).
The minus-strand of HCV is a replication intermediate that serves as template for the synthesis of progeny plus-strand RNAs. This minus-strand contains several CREs at its 3′ end, but owing to different G–U base-pairing and single-stranded regions, the secondary structures in this region (Smith et al. 2002) are very different from their complementary counterpart in the 5′ UTR of the plus-strand. In fact, the 3′ end of the minus-strand comprises several stem–loop structures, including stem–loops SL-I′, SL-IIz′, and SL-IIy′ that are crucial for RNA replication, and SL-III′ and SL-IV′ that contribute less to RNA replication efficiency (Friebe and Bartenschlager 2009).
Only little is known about the 5′ end of the minus-strand. Smith and Wu (2004) have shown that this part can form a mirror image of the structure of SLI of the X-tail, called SLα. The authors suggest that this structure protects the minus-strand from degradation by cytoplasmic exonucleases. However, thus far no structures corresponding to SLII and SLIII of the plus-strand could be identified in the minus-strand RNA (Smith and Wu 2004).
In this study, we present refined secondary structures of the 5′ UTR, the CRE-region in the NS5B RdRp coding region and of the 3′ UTR. We show alternative RNA secondary structures, flexible regions and several novel possible long-range interactions which are conserved among all HCV isolates analyzed. Based on all available HCV genomes, we hypothesize a 62-bp long 5′-UTR SLII/DLS interaction as the long-sought putative circularization sequence.
RESULTS AND DISCUSSION
This work is based on computational predictions which were performed without applying prior knowledge about secondary structures of HCV (except the pseudoknot in the 5′ UTR). Obtained results were then compared with results from the literature. We are able to confirm and extend most of the previously described secondary structures of 5′ UTR, CRE and 3′ UTR for all HCV genotypes (Fig. 1). The results of our comprehensive secondary structure analysis, including novel possible long-range interactions, are summarized in Figure 7A below.
FIGURE 7.

Long-range interactions in 5′ UTR, CRE, VR, and X-tail. (A) Overview. (Gray) Known interactions (No. 13–17 from the literature, validated by our analysis for all examined isolates); (green) new interactions (from our calculations). Numbers indicate different interactions shown in detail in B. (B) Interactions for all available sequences labeled with numbers corresponding to A. Bottom numbers indicate number of sequences in the alignment used for the interaction. Interaction No. 1 can be extended for a possible circularization. (C) Possible circularization of the HCV RNA. The interaction of SLII and DLS of HCV plus-strand RNA can be extended to at least 62 bp in all available 19 isolates. (D) Corresponding interaction of minus-strand RNA.
We used a full-genome alignment of 106 isolates from all known genotypes with an average pairwise sequence identity (PSI) of 70.02%. Interestingly, the 5′-UTR alignment is based on a much higher sequence identity of 94.2%, showing the high functionality of this compact and essential region, including the IRES and the miR-122 binding sites. The 3′-UTR alignment (based on 19 isolates) has a PSI of 95.9%. This extremely high degree of conservation may reflect overlapping functions of the 3′-UTR sequences that do not allow for sequence variability like covariations in stem regions. Moreover, single mutations in the 3′ UTR can lead to losses of crucial binding sites (Niepmann 2013). The CRE-region is part of the coding region but remains with 73.6% PSI above the genome wide average, which may to be due to the multiple functions of this region.
5′-UTR-IRES region
For 5′-UTR secondary structure prediction we used 86 out of 106 isolates covering 60 out of 63 subtypes. The remaining 20 isolates comprise incomplete 5′ UTRs. The prediction based on structure conservation is very similar to previously proposed secondary structures (Honda et al. 1999a; Pang et al. 2011). However, there are some differences compared with previous studies (shown in Fig. 2A):
The predicted SLII is contradicting the structure of Honda et al. (1999a) and studies referring to it (Masante et al. 2008; Friebe and Bartenschlager 2009). However, our prediction is in agreement with the work of Zhao and Wimmer (2001) and Lukavsky et al. (2003) and with the SHAPE analysis of Pang et al. (2011) (Supplemental Fig. 1C).
The predicted apical stem of SLIIIb contains a shift of nucleotides compared with the data of Honda et al. (1999a). However, the unpaired nucleotides in the loop region which are part of the binding site for initiation factor eIF3 (Fraser and Doudna 2007) are identical.
We confirm the secondary structure of SLIIId as previously described and validated by experimental data (Supplemental Fig. 1A; Brown et al. 1992; Kolupaeva et al. 2000; Lukavsky et al. 2000; Odreman-Macchioli et al. 2000; Romero-López and Berzal-Herranz 2009; Pang et al. 2011; Hajdin et al. 2013). These secondary structures are additionally stabilized by an LRI introduced in Romero-López and Berzal-Herranz (2012) (No. 14 in Fig. 7, below).
FIGURE 2.

(A) Alignment based secondary structure prediction of the 5′ UTR of 86 isolates. The alignment was calculated by LocARNA, the consensus sequence and structure by RNAalifold. The sequence for SLI is contained in only nine isolate sequences and therefore appears here as white. SLII is different from that described in Honda et al. (1999a), but in agreement with Zhao and Wimmer (2001) and SHAPE analysis of Pang et al. (2011) (see Supplemental Fig. 1C). Here, we show an alternative secondary structure for SLIIId with a slightly better MFE and conservation compared with the known structure. (Blue sequences) Forbidden to pair by RNAalifold in order to build the pseudoknot that is essential for the recruitment of the ribosome and therefore for viral protein translation (Berry et al. 2011). (Gray) Secondary structure different from those proposed by Honda et al. (1999a). (B) Consensus sequences and secondary structures of the CRE, VR, and X-tail in two alternative foldings. CRE and VR based on 96 isolates, X-tail based on 19 isolates. Apical regions of 5BSL1, 5BSL2, and 5BSL3.1 show up as described previously (Diviney et al. 2008). Even though it seems that the apical part of the stems can vary between isolates, the consensus secondary structure leaves little variability (see dotplot in Fig. 3). SLIV is present in all tested isolates but appears white since the stem is variable (see supplement, STK file for variable region). (Gray) Differences in secondary structures of 5BSL1 (SL9033), and 5BSL2 (SL9033) compared with data of Diviney et al. (2008), of 5BSL3.1 (SL9132) compared with data of Diviney et al. (2008) and Romero-López et al. (2014), of SLIV (Kolykhalov et al. 1996), of SLII (Kolykhalov et al. 1996; Blight and Rice 1997; Ito and Lai 1997), and of DLS (Ivanyi-Nagy et al. 2006; Shetty et al. 2010; Romero-López et al. 2014). (Rectangle) Consensus of RNAalifold to establish 5BSL3.2, instead of the interaction in Figures 3 (oval) and 7, No. 15 (Diviney et al. 2008). A detailed explanation of the base pair color code is available in the Supplemental Material.
In addition, we propose an alternative secondary structure SLIIId* (Fig. 2A). This proposed secondary structure has a slightly better alignment minimum free energy (consisting of the average MFE and a covariance term, hereafter named MFE) and can be formed by all isolates tested. In this alternative structure, the position of SLIIId is shifted: The previously proposed apical loop region is base-pairing, whereas the region between SLIIId* and SLIIIe is base-pairing differently. We hypothesize the possibility of a coexistence of both structures in equilibria (Supplemental Fig. 1B).
Rep-Core
The secondary structure of the first 177 nt of the Core region is known to be involved in replication. The secondary structure yielded by our analysis (Supplemental Fig. 2C) is in agreement with the previously known structures SLIV, SLV, and SLVI (Wang et al. 2000; McMullan et al. 2007; Vassilaki et al. 2008).
CRE/Variable Region (VR)
The cis-acting replication element (CRE), located at the 3′ end of the NS5B coding region, plays an important role in RNA synthesis. The CRE-region consists of at least five structured stem–loops: 5BSL1 (SL9033), 5BSL2 (SL9132), 5BSL3.1 (SL9217), 5BSL3.2 (SL9266), and 5BSL3.3 (SL9324) (Tuplin et al. 2002; Lee et al. 2004; Friebe et al. 2005; Diviney et al. 2008). According to our analysis, all 96 isolates can form these five stem–loops (see Fig. 2B). Comparing the CRE/VR and X-tail region to previous literature, we obtain the following new features:
Here, we present for the first time a consensus secondary structure of 5BSL1, 5BLS2, and 5BLS3.1 based on 96 isolates. The three apical loop regions are identical to those proposed by Diviney et al. (2008); however, the stem regions differ significantly (see Fig. 2B, gray parts). In particular, the region shown as “SL9110” in Romero-López et al. (2014) that interacts with the internal loop in 5BSL3.2 appears not as a hairpin loop in our consensus calculations (Fig. 3, oval). In contrast, this sequence is located in the left basal region of 5BSL2. The basal regions of 5BLS1 and 5BSL2 may also assume alternative secondary structures or may be even single-stranded (Fig. 3); in the latter case, the sequence that interacts with 5BSL3.2 would be located in a single-stranded region between the core parts of 5BSL1 and 5BSL2.
We show that the NS5B stop codon is contained in a stem–loop (SLIV) in all available sequences (Supplemental Material, Stockholm file Dat. 1). This was unexpected due to the fact that the primary sequences downstream from the NS5B stop codon are highly variable among isolates, indicated by white colored base pairs in Figure 2B.
Several studies propose a second stem–loop downstream from the 3′SLIV (Kolykhalov et al. 1996; Ito and Lai 1997; Yi and Lemon 2003) but they did not provide structure probing data. From analyzing this region using secondary structure alignments, we were not able to find a suboptimal consensus structure that confirms this loop. However, when folding each sequence separately, an extremely variable stem–loop can be found in genotypes 1, 2, and 4, but rarely in genotypes 3 and 6 (Supplemental Material, Stockholm file Dat. 1).
FIGURE 3.
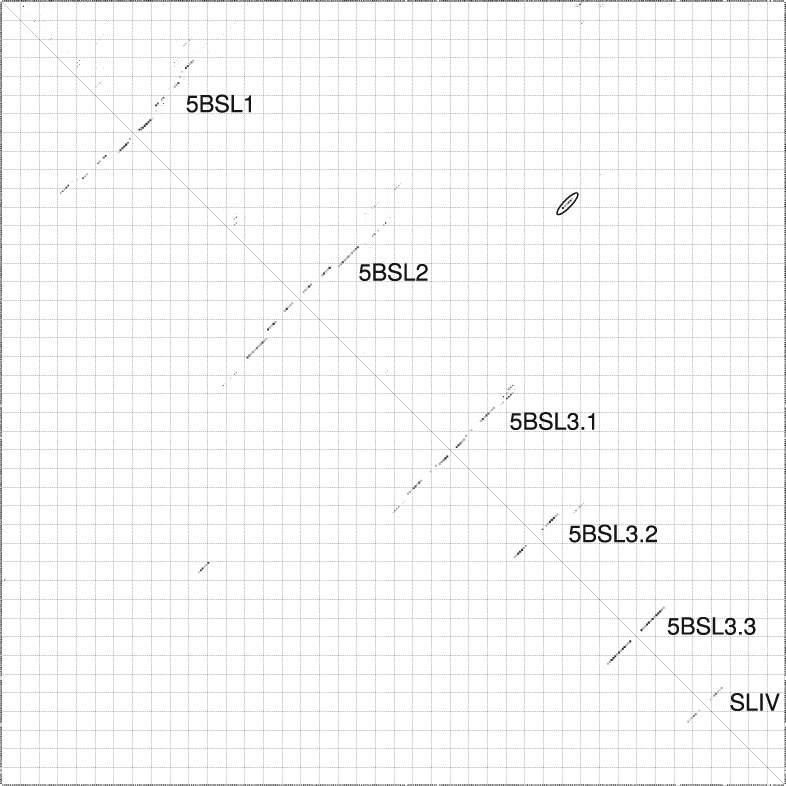
RNAalifold base-pairing probability matrix of the CRE and VR region, based on 96 isolates. The upper half of the matrix shows all base pair probabilities <10−6. The lower half shows the base pair probabilities of the MFE structure. (Oval) The interaction of the internal bulge of 5BSL3.2 with basal region of 5BSL2 (Fig. 7, No. 15), described previously in Diviney et al. (2008) and Tuplin et al. (2012). Dotplot and structure of CRE, VR, and X-tail region (19 isolates) without constraints are available in the Supplemental Material (Supplemental Fig. 3A,B).
Thus, we can only speculate that formation of this putative stem–loop may not be of general functional importance.
X-tail
According to the dotplot of the X-tail region based on analysis of the available 19 isolates from 13 subtypes (Fig. 4), we find up to four possible secondary structures of the X-tail (Fig. 4A,B are in agreement with the SHAPE analyses of Romero-López et al. 2014), which demonstrates a high flexibility of the very conserved sequences in particular in the region of 3′SLII and 3′SLIII which can form a variety of different secondary structures. These structure variants which are compatible with all considered sequences include:
Formation of the terminal 3′SLI and the known upstream stem–loop that includes the dimer linkage sequence (DLS) (Fig. 4A; Ivanyi-Nagy et al. 2006; Shetty et al. 2010).
The commonly accepted stem–loop structures 3′SLI, 3′SLII, and 3′SLIII (Fig. 4B; Blight and Rice 1997; Friebe and Bartenschlager 2002). However, in our analysis the apical part of 3′SLII is predicted different from Friebe et al. (2005). Taking into account the known long-range interaction of 5BSL3.2 with 3′SLII, we consider that the entire apical region may be locally unpaired.
Another, previously unknown, predicted secondary structure corresponds to the SLI and an alternative stem–loop containing the DLS (Fig. 4C).
Finally, another alternative structure can be obtained that contains SLI, SLIII, and an alternative version of SLII that is shifted by 4 nt compared with the previously known version of SLII (Fig. 4D).
FIGURE 4.
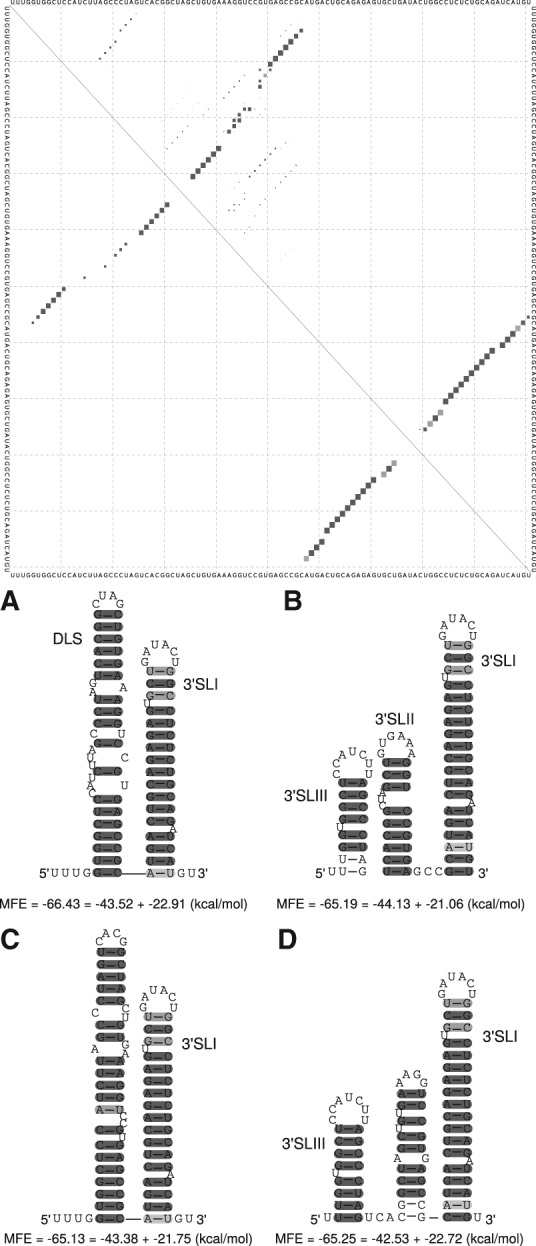
Alignment based secondary structures of the X-tail using sequences of 19 isolates. (Top) RNAalifold base-pairing probability matrix of the X-tail region. (A) Consensus secondary structure without applying any constraints. (B) Consensus secondary structure using constraints that force the postulated structure of Blight and Rice (1997). (C,D) Previously unknown secondary structures of the X-tail. All alternative structures contain SLI but show shifted hairpins compared with DLS or SLII.
5′ End of minus-strand
Our prediction for the 5′ end of the minus-strand (based on 19 isolates) returns an overall structure similar to the complementary plus-strand (Fig. 5A), consistent with previous studies (Smith and Wu 2004; Ivanyi-Nagy et al. 2006), see Supplemental Figure 5 for dotplot, although structures mirroring 3′SLII and 3′SLIII were previously not identified by structural probing (Smith and Wu 2004; Ivanyi-Nagy et al. 2006). We forced RNAalifold to calculate this structure, resulting in an only slightly higher MFE (Fig. 5B, −58.18 kcal/mol). Interestingly, the probing data of Ivanyi-Nagy et al. (2006) also do not contradict the latter structure. Additionally, we were able to detect a possible new secondary structure (similarly proposed in Smith and Wu 2004), displayed in Figure 5C, with a MFE of −60.13 kcal/mol. In conclusion, different structures seem possible for the 5′ end of the minus-strand.
FIGURE 5.
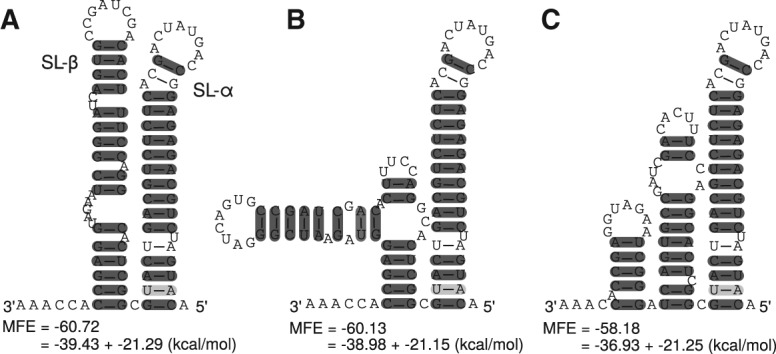
Alignment based secondary structure of the 5′ end of the minus-strand with 19 isolates. (A) Without constraints; (B) with constraints aiming at a similar structure like the mirror plus-strand; (C) suboptimal structure according to dotplot (Supplemental Fig. 5).
3′ End of minus-strand
Our analysis comprising 86 isolates returns a consensus MFE structure with eight stem–loop structures (Fig. 6A). This secondary structure contains the SLI′, SLIIz′, SLIIy′, SLIIIa′, SLIIIb′ (Smith et al. 2002; Dutkiewicz et al. 2008; Mahias et al. 2010) and SLIV′ (Smith et al. 2002) which are already known from experimental data. However, there is no unique structure between SLIIIb′ and SLIV′ (Fig. 6A).
FIGURE 6.

Alignment based secondary structure of the 3′ end of the minus-strand with 86 isolates. (A) RNAalifold base-pairing probability matrix. The upper half of the matrix shows all base pair probabilities <10−6. The lower half shows the base pair probabilities of the MFE structure. (B) Consensus secondary structure without constraints. (C) Consensus secondary structure with constraints: The last 3 nt at the very 3′ end were forced to be unpaired (blue). The resulting structure mirrors the secondary structure of the domain SLIII from RNA plus strand. (D) Consensus secondary structure with constraints forcing the postulated structure of Smith et al. (2002). (E) Consensus secondary structure with constraints forcing the postulated structure of Dutkiewicz et al. (2008). (F) Consensus secondary structure with constraints forcing the postulated structure of Schuster et al. (2002). (Dots) Nucleotides base-pairing with an extended version of the 3′ end; for details see Schuster et al. (2002). (Connected rectangles) Further interactions calculated by LocARNA. White base-pairings are derived from isolates being sequenced only partially.
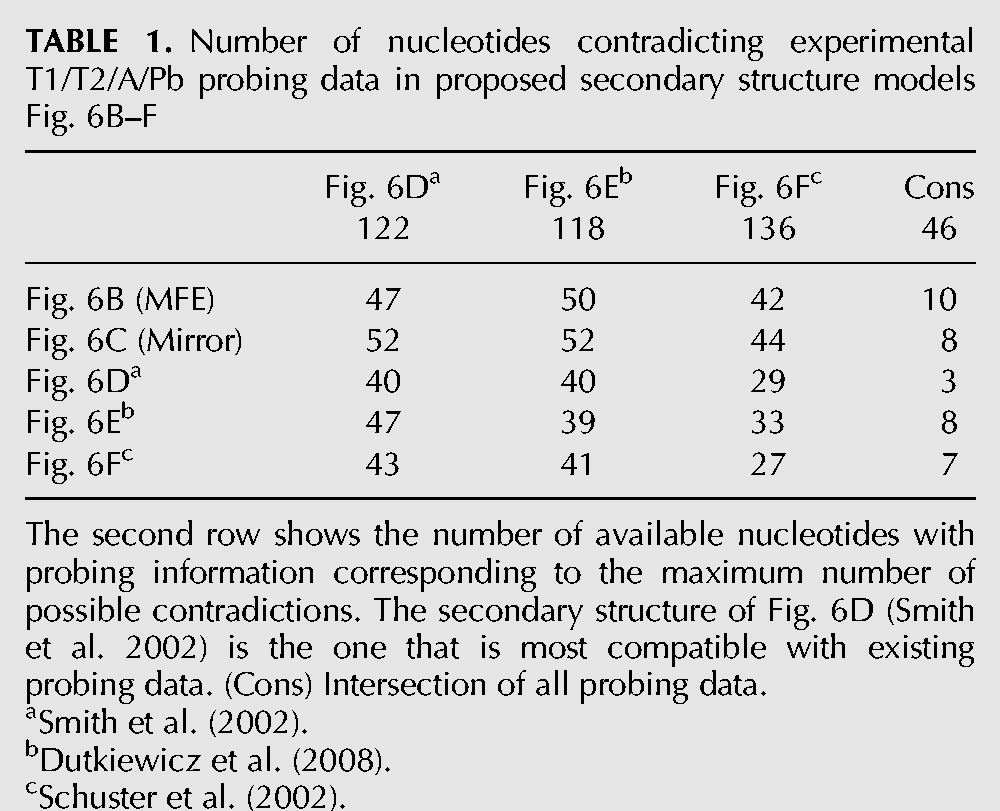
The following secondary structures appear with a similar MFE: Figure 6B shows the consensus structure with best MFE for all available isolates. In order to make this secondary structure comparable with those known from the literature, in a separate approach we disallowed only the last 3 nt to bind. Surprisingly, this approach returned a secondary structure of the domain SLIII (Fig. 6C) that mirrors the upper regions of the SLIII in the plus strand, with a similar MFE score. When forcing the experimentally verified secondary structures from Schuster et al. (2002), Smith et al. (2002), and Dutkiewicz et al. 2008), we obtained the structures shown in Figure 6D–F which are also very conserved but have MFEs considerably worse than that of the calculated optimal structure (Fig. 6B).
When we compare the structures from Figure 6B–F with available probing data from different sources (Table 1), the secondary structure of Figure 6D (Smith et al. 2002) appears most accurate among the proposed probing data. We suppose that (in analogy to RNAs from other organisms, e.g., bacteria) not a rigid structure is present in solution, but dynamic changes between closely related structures can occur in solution even in one molecule (Narberhaus 2010).
TABLE 1.
Number of nucleotides contradicting experimental T1/T2/A/Pb probing data in proposed secondary structure models Fig. 6B–F
Long-range interactions
When searching for long-range interactions in 5′ UTR, Rep-Core, CRE, VR, and X-tail, we could verify several known long-range interactions in every HCV genome analyzed:
The interaction of SLIIId–5BSL3.2 (No. 14 in Fig. 7A,B, obtained in 104 out of 104 sequences) (Romero-López and Berzal-Herranz 2012) possibly blocks HCV IRES-dependent translation and therefore may have a critical role in the viral replication cycle (Romero-López and Berzal-Herranz 2009; 2012). Furthermore, we were able to extend this interaction (Supplemental Fig. 6).
The interaction of the sequence called CRE “9110” with the internal bulge of 5BSL3.2 (No. 15 in Fig. 7A,B; Tuplin et al. 2012; Romero-López et al. 2014) is consistent in 106 out of 106 isolates and was verified for isolate Con1 (genotype 1b) by SHAPE analysis (Tuplin et al. 2012).
The interaction of the apical loop of 5BSL3.2 and 3′SLII (No. 16 in Fig. 7A,B, consistent in 19 out of 19 isolates) (Friebe et al. 2005; Liu et al. 2009) is necessary for replication (Friebe et al. 2005) and was speculated to be involved in a control switch (Romero-López et al. 2014).
The interaction of the SLVI (Core) and the single-stranded region between 5′-UTR SLI and SLII (No. 17 in Fig. 7, in 77 out of 77 isolates) has been described before in several studies (Honda et al. 1999b; Wang et al. 2000; Kim et al. 2003; Beguiristain et al. 2005). The interaction of 5′ UTR and Rep-Core is displayed in Supplemental Figure 2A. Apart from this interaction the 5′ UTR and Rep-Core seem to be independent.
In addition to the above known interactions, we identified several novel possible long-range interactions, described in the following sections.
Collapse of 3′-UTR structure
When folding the entire CRE/3′-UTR region we obtain a conserved interaction of 7 nt of the basal 5BSL2 and of the basal 3′SLII (No. 8 in Fig. 7A,B). This new interaction results in an additional hairpin directly adjacent to the 3′SLIII that is equivalent to the apical part of the DLS. This finding reinforces the assumption that the secondary structures are in a dynamic equilibrium and may provide a “switch” between translation and replication (Ivanyi-Nagy et al. 2006; Shetty et al. 2010; Romero-López et al. 2014). This switch might even be initiated by miRNA-122, binding to the downstream seven interacting nucleotides of 5BSL2 (Nasheri et al. 2011).
The interacting 3′SLIV
When folding the complete CRE/VR/X-Tail region, we obtain a strong conserved long-range interaction (No. 10 in Fig. 7A,B) between the region basal to 5BSL2 (called region “9110” in Romero-López et al. 2014) and the apical loop of the 3′SLIV containing the NS5B stop codon. This interaction would compete interaction No. 15. Furthermore, we can show a possible interaction of 3′SLIV to 5BSL1 (No. 9 in Fig. 7A,B) that would compete with interaction No. 10. Thus, these interactions may perhaps dynamically change during translation of the genomic RNA.
Other long-range interactions
In addition to the interactions described above, we were able to find about 20 more possible interactions (Supplemental Fig. 7), of which the most reliable eight are also presented in Figure 7 (No. 2–No. 7 and No. 11–No. 12). Interestingly, some conserved hairpins show no direct interaction with the remaining regions of the HCV genome investigated in this study, but they might possibly be sites for protein interactions (e.g., 5BSL3.1, 3′SLI), whereas other parts of the genome seem highly reactive to the examined genomic regions (e.g., SLII, 3′SLIV).
Possible circularization of HCV
The most interesting long-range interaction of this HCV study is the genome circularization between 5′-UTR SLII and 3′-UTR DLS. We initially identified a conserved interaction between the apical loops of SLII and DLS (No. 1 in Fig. 7A,B). This initial interaction can be extended to include 62 interacting base pairs (Fig. 7C). For such a possible circularization interaction, SLI and SLII of the 5′ UTR would need to unfold and interact with the completely unfolded X-tail region.
Viral genome circularization has been described in several RNA plus-strand viruses to be important for translation, transcription and viral replication (Edgil and Harris 2006). Such a genome circularization might be also involved in 5′–3′-end communication of the HCV RNA genome to facilitate RNA translation (Song et al. 2006) and/or RNA replication (Isken et al. 2007). Importantly, since both processes are mutually exclusive, genome circularization might be one mechanism to regulate both processes in a temporal manner. Accordingly, we propose that also in case of HCV this genome circularization might be involved in RNA replication. In a similar way, also both ends of the minus-strand are predicted to form such an extended long-range circularization interaction (Fig. 7D), that could be involved in the regulation of plus-strand RNA synthesis initiation.
Genome circularizations are known to be necessary for the RNA replication in Human immunodeficiency virus (HIV) and in Poliovirus (Herold and Andino 2001; Ooms et al. 2007; Beerens and Kjems 2010). Also in the related members of the Flaviviridae family, which include West Nile virus (WNV), yellow fever virus (YFV) and dengue virus (DENV), such a genome circularization has been described to be essential for RNA replication (You et al. 2001; Corver et al. 2003; Alvarez et al. 2005; Zhang et al. 2008; Friebe et al. 2011). Moreover, in the case of DENV this RNA circularization has been observed by atomic force microscopy in the absence of viral or cellular proteins, demonstrating that such a RNA circularization can be formed solely by RNA base-pairing (Alvarez et al. 2005). Nevertheless, cellular and/or viral proteins could facilitate this process. For example, the helicase NS3 could unwind the stem–loops structures to facilitate this circularization. Several cellular RNA binding proteins have been identified by proteomics that could be involved in this process and in different steps of HCV replication. For instance, 26 host proteins were found to specifically interact with the IRES (Lu et al. 2004) and more than 70 cellular factors were identified to interact with the 3′ UTR of the plus-strand (Harris et al. 2006). Accordingly, it has been speculated that a RNA circularization could be a general replication mechanism for all plus-strand RNA viruses (Herold and Andino 2001).
CONCLUSION
Using a comprehensive bioinformatical approach, we confirmed several important previously experimentally proven RNA secondary structures and interactions. Moreover, we provide strong evidence for new and alternative, yet unknown structures and interactions based on their conservation in all seven genotypes, suggesting possible biological functions. Among the newly identified interactions, in particular the possible circularization of the plus-strand and of the minus-strand RNA may have important biological implications for the initiation of RNA synthesis on the viral RNA genome and its replication intermediate.
From 950 downloaded isolate sequences we selected the two longest per group and retrieved 86 sequences containing a nearly complete 5′-UTR sequence and 19 genomes with a complete 3′ UTR. Although in the future we need more 5′/3′ ends of HCV sequenced, we applied no constraints to the foldings to identify possible RNA secondary structures in an unbiased approach (an exception was a constraint applied to the plus-strand 5′-UTR sequence which forces formation of the pseudoknot; nucleotides marked blue in Fig. 2A).
Among the newly identified secondary structures and interactions, some may have biological relevance, such as the alternative structure of SLIIId* in the newly proposed structure of the 5′ UTR (Fig. 2A), in addition to the known structure (Supplemental Fig. 1A). Some cryo-electron microscopy analyses do not provide sufficient resolution to unambiguously identify the known SLIIId structure (Spahn et al. 2001; Boehringer et al. 2005; Siridechadilok et al. 2005). However, experimental data strongly argue for the classical known SLIIId structure to be present at least when the IRES RNA is in solution (Brown et al. 1992; Odreman-Macchioli et al. 2000; Romero-López and Berzal-Herranz 2009; Pang et al. 2011; Hajdin et al. 2013), and nuclease footprints in the apical loop of the classical version of SLIIId disappear upon binding of the small 40S ribosomal subunit (Kolupaeva et al. 2000; Lukavsky et al. 2000). Moreover, the classical version of SLIIId is strongly supported by data showing a four-way junction including SLIII, SLIIIe, and SLIIIf (Berry et al. 2011; Yamamoto et al. 2014). Thus, we can only speculate if the newly identified highly conserved alternative SLIIId* structure has a biological function. For example, refolding of the IRES to the newly proposed SLIIId* structure could favor dissociation of the IRES from the ribosome to allow genome RNA encapsidation into new viral particles. Our analysis of the 5′ UTR also clearly confirms (in both shown 5′-UTR structures) that the SLII sequence is optimized to assume the conformation experimentally validated by Zhao and Wimmer (2001).
Our study suggests that other regions of the HCV genome may also be very dynamic. The 3′ UTR of the plus-strand as well as the minus-strand 5′- and 3′-ends display different structures with each very close MFEs. Thus, these structures may exist in equilibrium in the HCV RNA and adopt different structures easily to serve different functions in the HCV life cycle. The SLII of the 3′ UTR (Fig. 4B) was reported to interact with the 5BSL3.2 in a kissing loop interaction that has a role in replication; mutations disabling this interaction impair HCV replication that can be restored by complementary mutations restoring base-pairing (Friebe et al. 2005). The alternative structure (Fig. 4A) exposes the DLS sequence that is suspected to have a role in genome dimerization and perhaps also encapsidation (Ivanyi-Nagy et al. 2006; Shetty et al. 2010; Romero-López et al. 2014). However, we find two other very highly conserved structures (Fig. 4C,D) with stabilities comparable with the two known structures. We do not know a function of these structures, but their high degree of conservation and stability suggests that they may have a biological role. Currently, we can only speculate if the different conserved structures found for the 5′ end of the minus-strand (Fig. 5) just result from the constraints shaping plus-strand sequences, or if these minus-strand structures may have genuine functions.
The alternative proposed structures of the 3′ end of the minus-strand (Fig. 6) have two different features. On one hand, they have in common that the terminal stem–loops SL-I′ and SL-IIz′ essentially form in all proposed models, underlining the importance of these sequences as shown in various experimental approaches (Astier-Gin et al. 2005; Friebe and Bartenschlager 2009; Mahias et al. 2010). On the other hand, the conservation of the sequences upstream of SL-IIz′ allows varying conserved secondary structures. Besides our consensus structure model (Fig. 6B) we found that their structure is also well compatible with the model of Smith et al. (2002). Thus, both the consensus structure prediction and the experimental results suggest that these structures are of minor importance for replication.
The variety of known and newly identified long-range interactions (LRIs) shown in Figure 7 suggests that the interactions between different regions of the HCV RNA may be very dynamic. These LRIs may be classified into four groups. First, there are interactions within the 5′ region (already known: No. 13, new: 11 and 12). Among these, interaction No. 13 is known to be involved in dynamic changes in IRES structure that augment entry of the HCV sequence containing the AUG start codon into the entry channel of the 40S ribosomal subunit (Filbin and Kieft 2011). Interaction No. 17 was reported to be involved in repressing HCV translation. This interaction can be relieved in the presence of microRNA-122 which by that can indirectly stimulate translation (Díaz-Toledano et al. 2009; Goergen and Niepmann 2012). In contrast, a possible biological role of the proposed interactions No. 11 and 12 is not yet known. Second, the interactions within in the NS5B CRE region (already known: No. 15; new: 9 and 10) might stabilize the local aggregation of secondary structures. These interactions could be involved, e.g., in exposing the apical loop of 5BSL3.2 for binding of the NS5B polymerase (Lee et al. 2004). Third, there are interactions between the 3′-terminal NS5B coding region and the 3′ UTR (already known: No. 16; new: 8). We do not know what the biological role of these interactions is, but they might transfer the NS5B polymerase trapped by 5BSL3.2 to the 3′ UTR, the actual site of minus-strand initiation.
The fourth class is long-range interactions between the two genome ends. These interactions involve one already known interaction between the IRES SLIIId and the 5BSL3.2 CRE element (No. 14) and several newly identified possible interactions between the sequences in the NS5B coding region and the IRES (No. 3, 4, 6, and 7), as well as interactions between 3′ UTR and 5′ UTR (No. 1, 2, and 5). In addition to the one known interaction, we identified in total seven new interactions between the two genome ends. Some of these interactions are mutually exclusive, like interaction 1 versus 2 plus 5, since they depend on two different conformations of the 3′ UTR. Nevertheless, these hybridizations suggest intense and perhaps dynamic interactions between 3′-terminal and 5′-terminal sequences of the viral genome which might serve different functions in the replication cycle.
Surprisingly, the interaction between the IRES SLII and the 3′ UTR DLS (No. 1) could be extended up to a 62 nt long base-pairing, resulting in extensive interaction between both genome ends (Fig. 7C). A corresponding interaction could be also shown for the minus-strand (Fig. 7D), although slightly different due to different G–U base-pairing. Such a genome circularization of the plus-strand can be hypothesized to have an important role in the initiation of minus-strand RNA synthesis and/or regulation of a switch from RNA translation to genome replication. These results strongly support earlier reports which show that the HCV genome ends interact with each other (Isken et al. 2007) to facilitate RNA replication. This circularization may be promoted by proteins, in particular by RNA helicases (Ariumi et al. 2007; Isken et al. 2007; Jangra et al. 2010) that might be involved in resolving and forming base-pairing interactions between RNA secondary structures at the genome ends. Furthermore, other studies showed that the 3′ UTR stimulates RNA translation (Song et al. 2006 and references therein), and a recent study suggested that the 3′ UTR is also associated with the ribosomal 40S subunit (Bai et al. 2013). Currently, we can only speculate what occurs first, if binding of both genome ends to the ribosome facilitates genome end hybridization, or if hybridization of the 3′X region to those regions of the 5′ UTR which are not directly involved in ribosome binding facilitates stimulation of translation by the 3′ UTR and paves the way for minus-strand initiation by genome circularization.
In this context, it is important to note that the circularization of the plus-strand ends results in a situation in which the very 3′ end of the plus-strand, which was assumed to be hidden in the stable 3′-SLI structure, now is presented to the viral replicase with a free single-stranded conformation (see Fig. 7C). In analogy to studies with the related Dengue Virus (Alvarez et al. 2005, 2008), we propose that this circularization may have an important role in HCV replication.
Taken together, our results propose several new RNA–RNA interactions in the HCV RNA, leading to the concept of highly dynamic local and long-range interactions in the viral RNA genome that may serve multiple functions in the viral life cycle.
MATERIALS AND METHODS
Alignment generation
We downloaded 950 HCV genomes from NCBI (17.01.2014, Nucleotides, Search term: (Hepatitis C virus[porgn:__txid11103] AND complete genome[Title])) and the HCV database (v. 2008 [Kuiken et al. 2005] http://hcv.lanl.gov/content/sequence/NEWALIGN/align.html), see Supplemental Material (“Initial data”). Sequences which occur in both data sets were only included once. Nucleotides “Y,” “S,” “W,” “R” have been replaced by corresponding valid nucleotides. An alignment with MAFFT –auto (v.6.8 [Katoh et al. 2002]) was performed and a phylogenetic tree was built with Geneious (v.6.1 [Kearse et al. 2012]) Neighbor-Joining method Tamura-Nei (Tamura and Nei 1993). Based on this tree and on the annotations from NCBI and the HCV database the data set was reduced to two of the longest genomes from each subtype if available (see Supplemental Material, “Selected data”). With the reduced data set of 106 sequences from 65 subtypes, we performed a second MAFFT –maxiterate 1000 –localpair alignment. This alignment was used to define the ranges of 5′ UTR, Rep-Core, CRE, 3′ UTR and protein coding regions and is used in the following (see Supplemental Material, “Selected data”).
Alignment based secondary structures
For 5′ UTR, Rep-Core, CRE, and 3′ UTR we calculated a secondary structure based alignment with LocARNA (v.1.7.2 [Will et al. 2012]). We compared the quality of the secondary structure alignment from LocARNA, DAFS (Sato et al. 2012), and Lara (Bauer et al. 2007) based on the CRE-region. Although all programs present nearly the same results, we decided to use LocARNA, because it has a lower running time and refining of the CRE-region alignment performed best (see Supplemental Fig. 8).
We define the combined 5′-UTR-IRES region of the plus-strand (referred to as “5′ UTR” hereafter) to range from MAFFT alignment position 0–358 (based on 86 isolates, see Supplemental Material, “Selected data”). The latter position refers to 12 nt downstream from the start codon of the polyprotein coding region to allow the formation of SLIV (Honda et al. 1999a; Niepmann 2013). The Core region part known to be needed for replication (Rep-Core) is defined from position 330–520 of the MAFFT alignment (based on 106 isolates). For interaction of 5′ UTR and Rep-Core we used positions 1–520 (based on 77 isolates). The 3′ end of minus-strand is defined from 0–366 to form known possible hairpins (Smith et al. 2002; Dutkiewicz et al. 2008). We used 86 sequences for the above analyses since 20 sequences lack most of the 5′ UTR. The CRE ranges from MAFFT alignment position 9399–9775, and its structure is calculated together with that of the variable region of the 3′ UTR (9399–9820) followed by a poly-U region and the remaining X-tail, from 10,000–10,101. For the latter calculation we included all available sequences covering the complete region (96 sequences for CRE/VR and 19 sequences for X-tail). We performed alignments for the whole region (9399–10,101) as well as for CRE/VR and X-tail separately. Positions for the 5′ end of the minus-strand refer to the plus-strand. The consensus structures are calculated with RNAalifold -r -p –color –MEA (v.2.0.7 [Lorenz et al. 2011]) and -C when constraints were used, as described in the main text. Each minimum free energy we use herein is the sum of the average minimum free energy of all sequences in the given alignment plus the covariance term as used in RNAalifold.
Long-range interactions
For each of the calculated secondary structures (Fig. 2A, Supplemental Figs. 1, 2B, 4A,B) all alignment regions containing at least three consecutive nonpairing nucleotides, added with two flanking nucleotides, were concatenated to a new alignment, separated with “NNN.” For each of the artificial alignments 5′ UTR/CRE, 5′ UTR/X-Tail A and 5′-UTR/X-Tail B a base pair probability matrix (Supplemental Fig. 7) was calculated. Based on this matrix we picked the most probable interactions, recalculated the selected interactions with LocARNA and filtered the results manually from dotplots of, e.g., the 5′ UTR (Supplemental Fig. 1B) and CRE (Supplemental Fig. 3B,D). We used the manual approach for LRI identification, as alignment based secondary predictions tools alone—such as RNAalifold, RNAaliduplex (Lorenz et al. 2011), LocARNA, or PETcofold (Seemann et al. 2011)—are not able to predict all of the previously known LRIs from the full defined regions (5′ UTR, CRE, 3′ UTR).
The z-score, based on RNAalifold MFE was calculated as
whereas X is the MFE of the corresponding interaction, μ is the mean MFE and σ the corresponding standard deviation of the 1000 randomly dinucleotide shuffled alignments (generated by multiperm) (Anandam et al. 2009).
SUPPLEMENTAL MATERIAL
Supplemental material is available for this article at http://www.rna.uni-jena.de/supplements/hcv/.
ACKNOWLEDGMENTS
This work was in part funded by Deutsche Forscheungsgemeinschaft (DFG) projects MA-5082/1, NI-604/2-2, SPP 1596, IRTG 1384 and the Carl-Zeiss-Stiftung.
Footnotes
Article published online ahead of print. Article and publication date are at http://www.rnajournal.org/cgi/doi/10.1261/rna.049338.114.
REFERENCES
- Alvarez DE, Lodeiro MF, Ludueña SJ, Pietrasanta LI, Gamarnik AV. 2005. Long-range RNA-RNA interactions circularize the dengue virus genome. J Virol 79: 6631–6643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alvarez DE, Filomatori CV, Gamarnik AV. 2008. Functional analysis of dengue virus cyclization sequences located at the 5′ and 3′UTRs. Virology 375: 223–235. [DOI] [PubMed] [Google Scholar]
- Anandam P, Torarinsson E, Ruzzo WL. 2009. Multiperm: shuffling multiple sequence alignments while approximately preserving dinucleotide frequencies. Bioinformatics 25: 668–669. [DOI] [PubMed] [Google Scholar]
- Ariumi Y, Kuroki M, Abe K, Dansako H, Ikeda M, Wakita T, Kato N. 2007. DDX3 DEAD-box RNA helicase is required for hepatitis C virus RNA replication. J Virol 81: 13922–13926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Astier-Gin T, Bellecave P, Litvak S, Ventura M. 2005. Template requirements and binding of hepatitis C virus NS5B polymerase during in vitro RNA synthesis from the 3′-end of virus minus-strand RNA. FEBS J 272: 3872–3886. [DOI] [PubMed] [Google Scholar]
- Bai Y, Zhou K, Doudna JA. 2013. Hepatitis C virus 3′UTR regulates viral translation through direct interactions with the host translation machinery. Nucleic Acids Res 41: 7861–7874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bauer M, Klau GW, Reinert K. 2007. Accurate multiple sequence-structure alignment of RNA sequences using combinatorial optimization. BMC Bioinformatics 8: 271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beerens N, Kjems J. 2010. Circularization of the HIV-1 genome facilitates strand transfer during reverse transcription. RNA 16: 1226–1235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beguiristain N, Robertson HD, Gómez J. 2005. RNase III cleavage demonstrates a long range RNA: RNA duplex element flanking the hepatitis C virus internal ribosome entry site. Nucleic Acids Res 33: 5250–5261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berry KE, Waghray S, Mortimer SA, Bai Y, Doudna JA. 2011. Crystal structure of the HCV IRES central domain reveals strategy for start-codon positioning. Structure 19: 1456–1466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blight KJ, Rice CM. 1997. Secondary structure determination of the conserved 98-base sequence at the 3′ terminus of hepatitis C virus genome RNA. J Virol 71: 7345–7352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boehringer D, Thermann R, Ostareck-Lederer A, Lewis JD, Stark H. 2005. Structure of the hepatitis C virus IRES bound to the human 80S ribosome: remodeling of the HCV IRES. Structure 13: 1695–1706. [DOI] [PubMed] [Google Scholar]
- Brown EA, Zhang H, Ping LH, Lemon SM. 1992. Secondary structure of the 5′ nontranslated regions of hepatitis C virus and pestivirus genomic RNAs. Nucleic Acids Res 20: 5041–5045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buratti E, Tisminetzky S, Zotti M, Baralle FE. 1998. Functional analysis of the interaction between HCV 5′UTR and putative subunits of eukaryotic translation initiation factor eIF3. Nucleic Acids Res 26: 3179–3187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corver J, Lenches E, Smith K, Robison RA, Sando T, Strauss EG, Strauss JH. 2003. Fine mapping of a cis-acting sequence element in yellow fever virus RNA that is required for RNA replication and cyclization. J Virol 77: 2265–2270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Díaz-Toledano R, Ariza-Mateos A, Birk A, Martínez-García B, Gómez J. 2009. In vitro characterization of a miR-122-sensitive double-helical switch element in the 5′ region of hepatitis C virus RNA. Nucleic Acids Res 37: 5498–5510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diviney S, Tuplin A, Struthers M, Armstrong V, Elliott RM, Simmonds P, Evans DJ. 2008. A hepatitis C virus cis-acting replication element forms a long-range RNA-RNA interaction with upstream RNA sequences in NS5B. J Virol 82: 9008–9022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dutkiewicz M, Swiatkowska A, Figlerowicz M, Ciesiołka J. 2008. Structural domains of the 3′-terminal sequence of the hepatitis C virus replicative strand. Biochemistry 47: 12197–12207. [DOI] [PubMed] [Google Scholar]
- Edgil D, Harris E. 2006. End-to-end communication in the modulation of translation by mammalian RNA viruses. Virus Res 119: 43–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Filbin ME, Kieft JS. 2011. HCV IRES domain IIb affects the configuration of coding RNA in the 40S subunit's decoding groove. RNA 17: 1258–1273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fraser CS, Doudna JA. 2007. Structural and mechanistic insights into hepatitis C viral translation initiation. Nat Rev Microbiol 5: 29–38. [DOI] [PubMed] [Google Scholar]
- Friebe P, Bartenschlager R. 2002. Genetic analysis of sequences in the 3′ nontranslated region of hepatitis C virus that are important for RNA replication. J Virol 76: 5326–5338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Friebe P, Bartenschlager R. 2009. Role of RNA structures in genome terminal sequences of the hepatitis C virus for replication and assembly. J Virol 83: 11989–11995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Friebe P, Lohmann V, Krieger N, Bartenschlager R. 2001. Sequences in the 5′ nontranslated region of hepatitis C virus required for RNA replication. J Virol 75: 12047–12057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Friebe P, Boudet J, Simorre JP, Bartenschlager R. 2005. Kissing-loop interaction in the 3′ end of the hepatitis C virus genome essential for RNA replication. J Virol 79: 380–392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Friebe P, Shi PY, Harris E. 2011. The 5′ and 3′ downstream AUG region elements are required for mosquito-borne flavivirus RNA replication. J Virol 85: 1900–1905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goergen D, Niepmann M. 2012. Stimulation of Hepatitis C Virus RNA translation by microRNA-122 occurs under different conditions in vivo and in vitro. Virus Res 167: 343–352. [DOI] [PubMed] [Google Scholar]
- Hajdin CE, Bellaousov S, Huggins W, Leonard CW, Mathews DH, Weeks KM. 2013. Accurate SHAPE-directed RNA secondary structure modeling, including pseudoknots. Proc Natl Acad Sci 110: 5498–5503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harris D, Zhang Z, Chaubey B, Pandey VN. 2006. Identification of cellular factors associated with the 3′-nontranslated region of the hepatitis C virus genome. Mol Cell Proteomics 5: 1006–1018. [DOI] [PubMed] [Google Scholar]
- Henke JI, Goergen D, Zheng J, Song Y, Schüttler CG, Fehr C, Jünemann C, Niepmann M. 2008. microRNA-122 stimulates translation of hepatitis C virus RNA. EMBO J 27: 3300–3310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herold J, Andino R. 2001. Poliovirus RNA replication requires genome circularization through a protein-protein bridge. Mol Cell 7: 581–591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Honda M, Brown EA, Lemon SM. 1996. Stability of a stem-loop involving the initiator AUG controls the efficiency of internal initiation of translation on hepatitis C virus RNA. RNA 2: 955–968. [PMC free article] [PubMed] [Google Scholar]
- Honda M, Beard MR, Ping LH, Lemon SM. 1999a. A phylogenetically conserved stem-loop structure at the 5′ border of the internal ribosome entry site of hepatitis C virus is required for cap-independent viral translation. J Virol 73: 1165–1174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Honda M, Rijnbrand R, Abell G, Kim D, Lemon SM. 1999b. Natural variation in translational activities of the 5′ nontranslated RNAs of hepatitis C virus genotypes 1a and 1b: evidence for a long-range RNA-RNA interaction outside of the internal ribosomal entry site. J Virol 73: 4941–4951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Isken O, Baroth M, Grassmann CW, Weinlich S, Ostareck DH, Ostareck-Lederer A, Behrens SE. 2007. Nuclear factors are involved in hepatitis C virus RNA replication. RNA 13: 1675–1692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ito T, Lai MM. 1997. Determination of the secondary structure of and cellular protein binding to the 3′-untranslated region of the hepatitis C virus RNA genome. J Virol 71: 8698–8706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ivanyi-Nagy R, Kanevsky I, Gabus C, Lavergne JP, Ficheux D, Penin F, Fossé P, Darlix JL. 2006. Analysis of hepatitis C virus RNA dimerization and core–RNA interactions. Nucleic Acids Res 34: 2618–2633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jackowiak P, Kuls K, Budzko L, Mania A, Figlerowicz M, Figlerowicz M. 2014. Phylogeny and molecular evolution of the hepatitis C virus. Infect Genet Evol 21: 67–82. [DOI] [PubMed] [Google Scholar]
- Jangra RK, Yi M, Lemon SM. 2010. DDX6 (Rck/p54) is required for efficient hepatitis C virus replication but not for internal ribosome entry site-directed translation. J Virol 84: 6810–6824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jopling CL, Yi M, Lancaster AM, Lemon SM, Sarnow P. 2005. Modulation of hepatitis C virus RNA abundance by a liver-specific microRNA. Science 309: 1577–1581. [DOI] [PubMed] [Google Scholar]
- Katoh K, Misawa K, Kuma K, Miyata T. 2002. MAFFT: a novel method for rapid multiple sequence alignment based on fast Fourier transform. Nucleic Acids Res 30: 3059–3066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kearse M, Moir R, Wilson A, Stones-Havas S, Cheung M, Sturrock S, Buxton S, Cooper A, Markowitz S, Duran C, et al. 2012. Geneious Basic: an integrated and extendable desktop software platform for the organization and analysis of sequence data. Bioinformatics 28: 1647–1649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kieft JS, Zhou K, Jubin R, Doudna JA. 2001. Mechanism of ribosome recruitment by hepatitis C IRES RNA. RNA 7: 194–206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kieft JS, Zhou K, Grech A, Jubin R, Doudna JA. 2002. Crystal structure of an RNA tertiary domain essential to HCV IRES-mediated translation initiation. Nat Struct Biol 9: 370–374. [DOI] [PubMed] [Google Scholar]
- Kim YK, Lee SH, Kim CS, Seol SK, Jang SK. 2003. Long-range RNA–RNA interaction between the 5′ nontranslated region and the core-coding sequences of hepatitis C virus modulates the IRES-dependent translation. RNA 9: 599–606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kolupaeva VG, Pestova TV, Hellen CU. 2000. An enzymatic footprinting analysis of the interaction of 40S ribosomal subunits with the internal ribosomal entry site of hepatitis C virus. J Virol 74: 6242–6250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kolykhalov AA, Feinstone SM, Rice CM. 1996. Identification of a highly conserved sequence element at the 3′ terminus of hepatitis C virus genome RNA. J Virol 70: 3363–3371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuiken C, Yusim K, Boykin L, Richardson R. 2005. The Los Alamos hepatitis C sequence database. Bioinformatics 21: 379–384. [DOI] [PubMed] [Google Scholar]
- Lee H, Shin H, Wimmer E, Paul AV. 2004. cis-acting RNA signals in the NS5B C-terminal coding sequence of the hepatitis C virus genome. J Virol 78: 10865–10877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Y, Wimmer E, Paul AV. 2009. Cis-acting RNA elements in human and animal plus-strand RNA viruses. Biochim Biophys Acta 1789: 495–517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lohmann V. 2013. Hepatitis C virus RNA replication, Vol. 369 of Current topics in microbiology and immunology. Springer, Berlin, Heidelberg. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lorenz R, Bernhart SH, Höner zu Siederdissen C, Tafer H, Flamm C, Stadler PF, Hofacker IL. 2011. ViennaRNA Package 2.0. Algorithms Mol Biol 6: 26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu H, Li W, Noble WS, Payan D, Anderson DC. 2004. Riboproteomics of the hepatitis C virus internal ribosomal entry site. J Proteome Res 3: 949–957. [DOI] [PubMed] [Google Scholar]
- Lukavsky PJ, Otto GA, Lancaster AM, Sarnow P, Puglisi JD. 2000. Structures of two RNA domains essential for hepatitis C virus internal ribosome entry site function. Nat Struct Biol 7: 1105–1110. [DOI] [PubMed] [Google Scholar]
- Lukavsky PJ, Kim I, Otto GA, Puglisi JD. 2003. Structure of HCV IRES domain II determined by NMR. Nat Struct Biol 10: 1033–1038. [DOI] [PubMed] [Google Scholar]
- Mahias K, Ahmed-El-Sayed N, Masante C, Bitard J, Staedel C, Darfeuille F, Ventura M, Astier-Gin T. 2010. Identification of a structural element of the hepatitis C virus minus strand RNA involved in the initiation of RNA synthesis. Nucleic Acids Res 38: 4079–4091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Masante C, Mahias K, Lourenço S, Dumas E, Cahour A, Trimoulet P, Fleury H, Astier-Gin T, Ventura M. 2008. Seven nucleotide changes characteristic of the hepatitis C virus genotype 3 5′ untranslated region: correlation with reduced in vitro replication. J Gen Virol 89: 212–221. [DOI] [PubMed] [Google Scholar]
- McMullan LK, Grakoui A, Evans MJ, Mihalik K, Puig M, Branch AD, Feinstone SM, Rice CM. 2007. Evidence for a functional RNA element in the hepatitis C virus core gene. Proc Natl Acad Sci 104: 2879–2884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moradpour D, Penin F. 2013. Hepatitis C virus proteins: from structure to function. Curr Top Microbiol Immunol 369: 113–142. [DOI] [PubMed] [Google Scholar]
- Narberhaus F. 2010. Translational control of bacterial heat shock and virulence genes by temperature-sensing mRNAs. RNA Biol 7: 84–89. [DOI] [PubMed] [Google Scholar]
- Nasheri N, Singaravelu R, Goodmurphy M, Lyn RK, Pezacki JP. 2011. Competing roles of microRNA-122 recognition elements in hepatitis C virus RNA. Virology 410: 336–344. [DOI] [PubMed] [Google Scholar]
- Niepmann M. 2013. Hepatitis C virus RNA translation. Curr Top Microbiol Immunol 369: 143–166. [DOI] [PubMed] [Google Scholar]
- Odreman-Macchioli FE, Tisminetzky SG, Zotti M, Baralle FE, Buratti E. 2000. Influence of correct secondary and tertiary RNA folding on the binding of cellular factors to the HCV IRES. Nucleic Acids Res 28: 875–885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ooms M, Abbink TE, Pham C, Berkhout B. 2007. Circularization of the HIV-1 RNA genome. Nucleic Acids Res 35: 5253–5261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pang PS, Elazar M, Pham EA, Glenn JS. 2011. Simplified RNA secondary structure mapping by automation of SHAPE data analysis. Nucleic Acids Res 39: e151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pérard J, Leyrat C, Baudin F, Drouet E, Jamin M. 2013. Structure of the full-length HCV IRES in solution. Nat Commun 4: 1612. [DOI] [PubMed] [Google Scholar]
- Pestova TV, Shatsky IN, Fletcher SP, Jackson RJ, Hellen CU. 1998. A prokaryotic-like mode of cytoplasmic eukaryotic ribosome binding to the initiation codon during internal translation initiation of hepatitis C and classical swine fever virus RNAs. Genes Dev 12: 67–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Romero-López C, Berzal-Herranz A. 2009. A long-range RNA–RNA interaction between the 5′ and 3′ ends of the HCV genome. RNA 15: 1740–1752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Romero-López C, Berzal-Herranz A. 2012. The functional RNA domain 5BSL3.2 within the NS5B coding sequence influences hepatitis C virus IRES-mediated translation. Cell Mol Life Sci 69: 103–113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Romero-López C, Barroso-Deljesus A, García-Sacristán A, Briones C, Berzal-Herranz A. 2014. End-to-end crosstalk within the hepatitis C virus genome mediates the conformational switch of the 3′X-tail region. Nucleic Acids Res 42: 567–582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sato K, Kato Y, Akutsu T, Asai K, Sakakibara Y. 2012. DAFS: simultaneous aligning and folding of RNA sequences via dual decomposition. Bioinformatics 28: 3218–3224. [DOI] [PubMed] [Google Scholar]
- Schuster C, Isel C, Imbert I, Ehresmann C, Marquet R, Kieny MP. 2002. Secondary structure of the 3′ terminus of hepatitis C virus minus-strand RNA. J Virol 76: 8058–8068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sedano CD, Sarnow P. 2014. Hepatitis C virus subverts liver-specific miR-122 to protect the viral genome from exoribonuclease Xrn2. Cell Host Microbe 16: 257–264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seemann SE, Richter AS, Gesell T, Backofen R, Gorodkin J. 2011. PETcofold: predicting conserved interactions and structures of two multiple alignments of RNA sequences. Bioinformatics 27: 211–219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shetty S, Kim S, Shimakami T, Lemon SM, Mihailescu MR. 2010. Hepatitis C virus genomic RNA dimerization is mediated via a kissing complex intermediate. RNA 16: 913–925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shimakami T, Yamane D, Jangra RK, Kempf BJ, Spaniel C, Barton DJ, Lemon SM. 2012. Stabilization of hepatitis C virus RNA by an Ago2-miR-122 complex. Proc Natl Acad Sci 109: 941–946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simmonds P. 2013. The origin of hepatitis C virus. Curr Top Microbiol Immunol 369: 1–15. [DOI] [PubMed] [Google Scholar]
- Siridechadilok B, Fraser CS, Hall RJ, Doudna JA, Nogales E. 2005. Structural roles for human translation factor eIF3 in initiation of protein synthesis. Science 310: 1513–1515. [DOI] [PubMed] [Google Scholar]
- Sizova DV, Kolupaeva VG, Pestova TV, Shatsky IN, Hellen CU. 1998. Specific interaction of eukaryotic translation initiation factor 3 with the 5′ nontranslated regions of hepatitis C virus and classical swine fever virus RNAs. J Virol 72: 4775–4782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith RM, Wu GY. 2004. Secondary structure and hybridization accessibility of the hepatitis C virus negative strand RNA 5′-terminus. J Viral Hepat 11: 115–123. [DOI] [PubMed] [Google Scholar]
- Smith RM, Walton CM, Wu CH, Wu GY. 2002. Secondary structure and hybridization accessibility of hepatitis C virus 3′-terminal sequences. J Virol 76: 9563–9574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song Y, Friebe P, Tzima E, Jünemann C, Bartenschlager R, Niepmann M. 2006. The hepatitis C virus RNA 3′-untranslated region strongly enhances translation directed by the internal ribosome entry site. J Virol 80: 11579–11588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spahn CM, Kieft JS, Grassucci RA, Penczek PA, Zhou K, Doudna JA, Frank J. 2001. Hepatitis C virus IRES RNA-induced changes in the conformation of the 40s ribosomal subunit. Science 291: 1959–1962. [DOI] [PubMed] [Google Scholar]
- Tamura K, Nei M. 1993. Estimation of the number of nucleotide substitutions in the control region of mitochondrial DNA in humans and chimpanzees. Mol Biol Evol 10: 512–526. [DOI] [PubMed] [Google Scholar]
- Tanaka T, Kato N, Cho MJ, Shimotohno K. 1995. A novel sequence found at the 3′ terminus of hepatitis C virus genome. Biochem Biophys Res Commun 215: 744–749. [DOI] [PubMed] [Google Scholar]
- Tuplin A, Wood J, Evans DJ, Patel AH, Simmonds P. 2002. Thermodynamic and phylogenetic prediction of RNA secondary structures in the coding region of hepatitis C virus. RNA 8: 824–841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tuplin A, Struthers M, Simmonds P, Evans DJ. 2012. A twist in the tail: SHAPE mapping of long-range interactions and structural rearrangements of RNA elements involved in HCV replication. Nucleic Acids Res 40: 6908–6921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vassilaki N, Friebe P, Meuleman P, Kallis S, Kaul A, Paranhos-Baccalà G, Leroux-Roels G, Mavromara P, Bartenschlager R. 2008. Role of the hepatitis C virus core+1 open reading frame and core cis-acting RNA elements in viral RNA translation and replication. J Virol 82: 11503–11515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang C, Le SY, Ali N, Siddiqui A. 1995. An RNA pseudoknot is an essential structural element of the internal ribosome entry site located within the hepatitis C virus 5′ noncoding region. RNA 1: 526–537. [PMC free article] [PubMed] [Google Scholar]
- Wang TH, Rijnbrand RC, Lemon SM. 2000. Core protein-coding sequence, but not core protein, modulates the efficiency of cap-independent translation directed by the internal ribosome entry site of hepatitis C virus. J Virol 74: 11347–11358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Will S, Joshi T, Hofacker IL, Stadler PF, Backofen R. 2012. LocARNA-P: accurate boundary prediction and improved detection of structural RNAs. RNA 18: 900–914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamamoto H, Unbehaun A, Loerke J, Behrmann E, Collier M, Bürger J, Mielke T, Spahn CM. 2014. Structure of the mammalian 80S initiation complex with initiation factor 5B on HCV-IRES RNA. Nat Struct Mol Biol 21: 721–727. [DOI] [PubMed] [Google Scholar]
- Yamane D, McGivern DR, Masaki T, Lemon SM. 2013. Liver injury and disease pathogenesis in chronic hepatitis C. Curr Top Microbiol Immunol 369: 263–288. [DOI] [PubMed] [Google Scholar]
- Yanagi M, St Claire M, Emerson SU, Purcell RH, Bukh J. 1999. In vivo analysis of the 3′ untranslated region of the hepatitis C virus after in vitro mutagenesis of an infectious cDNA clone. Proc Natl Acad Sci 96: 2291–2295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yi M, Lemon SM. 2003. 3′ Nontranslated RNA signals required for replication of hepatitis C virus RNA. J Virol 77: 3557–3568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- You S, Rice CM. 2008. 3′ RNA elements in hepatitis C virus replication: kissing partners and long poly(U). J Virol 82: 184–195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- You S, Falgout B, Markoff L, Padmanabhan R. 2001. In vitro RNA synthesis from exogenous dengue viral RNA templates requires long range interactions between 5′- and 3′-terminal regions that influence RNA structure. J Biol Chem 276: 15581–15591. [DOI] [PubMed] [Google Scholar]
- Zeisel MB, Felmlee DJ, Baumert TF. 2013. Hepatitis C virus entry. Curr Top Microbiol Immunol 369: 87–112. [DOI] [PubMed] [Google Scholar]
- Zhang B, Dong H, Stein DA, Shi PY. 2008. Co-selection of West Nile virus nucleotides that confer resistance to an antisense oligomer while maintaining long-distance RNA/RNA base pairings. Virology 382: 98–106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao WD, Wimmer E. 2001. Genetic analysis of a poliovirus/hepatitis C virus chimera: new structure for domain II of the internal ribosomal entry site of hepatitis C virus. J Virol 75: 3719–3730. [DOI] [PMC free article] [PubMed] [Google Scholar]