

Abstract
RNA-regulatory factors bound to 3′ UTRs control translation and stability. Repression often is associated with poly(A) removal. The deadenylase CAF1 is a core component of the CCR4–NOT complex. Our prior studies established that CAF1 represses translation independent of deadenylation. We sought the mechanism of its deadenylation-independent repression in Xenopus oocytes. Our data reveal a chain of interacting proteins that links CAF1 to CCR4–NOT and to Xp54 and 4E-T. Association of CAF1 with NOT1, the major subunit of CCR4–NOT, is required for repression by CAF1 tethered to a reporter mRNA. Affinity purification-mass spectrometry and coimmunoprecipitation revealed that at least five members of the CCR4–NOT complex were recruited by CAF1. The recruitment of these proteins required NOT1, as did the ability of tethered CAF1 to repress translation. In turn, NOT1 was needed to recruit Xp54 and 4E-T. We examined the role of 4E-T in repression using mutations that disrupted either eIF4E-dependent or -independent mechanisms. Expression of a 4E-T truncation that still bound eIF4E alleviated repression by tethered CAF1, NOT1, and Xp54. In contrast, a mutant 4E-T that failed to bind eIF4E did not. Repression of global translation was affected only by the eIF4E-dependent mechanism. Reporters bearing IRES elements revealed that repression via tethered CAF1 and Xp54 is cap- and eIF4E-independent, but requires one or more of eIF4A, eIF4B, and eIF4G. We propose that RNA-binding proteins, and perhaps miRNAs, repress translation through an analogous chain of interactions that begin with the 3′ UTR-bound repressor and end with the noncanonical activity of 4E-T.
Keywords: CAF1, NOT1, CCR4–NOT, Xp54, 4E-T, translation
INTRODUCTION
mRNAs are tightly regulated from their first appearance to their ultimate destruction. Their proper control is critical in development, cell cycle progression, and cell proliferation (Gebauer and Hentze 2004; Molin and Puisieux 2005; Aslam et al. 2009; Chen and Shyu 2011). Repression and activation often correlate well with regulated changes in poly(A) tail length (Gebauer and Hentze 2004). Poly(A) lengthening typically stabilizes mRNAs, while shortening decreases stability and can cause translational repression (Chen and Shyu 2011). In addition, certain enzymes that remove the poly(A) tail repress translation independent of deadenylation (Cooke et al. 2010). Here, we sought to understand the basis of that deadenylation-independent activity.
Deadenylases are important in mRNA control and are recruited to target mRNAs by RNA-binding proteins and miRNAs (Yamashita et al. 2005; Goldstrohm et al. 2006; Garneau et al. 2007; Goldstrohm and Wickens 2008; Chen and Shyu 2011; Sandler et al. 2011). CAF1 is a member of the DEDD (Asp-Glu-Asp-Asp) deadenylase family, a class of enzymes named for the four residues required for catalysis (Wilusz et al. 2001; Goldstrohm and Wickens 2008; Suzuki et al. 2010). Vertebrates encode two CAF1 homologs, CAF1a and CAF1b, which are 76% identical in sequence. Both are highly conserved among higher eukaryotes (Bianchin et al. 2005).
CAF1 is a stable component of the CCR4–NOT complex. This large complex is composed of nine core subunits with roles in regulation of transcription, mRNA export, and mRNA decay (Lau et al. 2009; Collart and Panasenko 2012; Doidge et al. 2012; Miller and Reese 2012; Temme et al. 2014; Xu et al. 2014). The major component of CCR4–NOT is the 250 kDa NOT1 protein, which acts as a scaffold for the multisubunit complex (Nasertorabi et al. 2011; Petit et al. 2012; Bawankar et al. 2013). CAF1 binds directly to NOT1 through the MIF4G domain of NOT1 (Basquin et al. 2012). CAF1 is also associated with CCR4–NOT via an independent interaction with CCR4, another deadenylase subunit.
NOT1 interacts with many proteins, suggesting that it may act as a bridge to bring protein interactors into the CCR4–NOT complex. Specifically, GW182, an Argonaute-interacting protein in Drosophila (and its homolog TNRC6A-C in mammals), binds NOT1 to recruit factors to miRNA-bound mRNA targets to elicit translational repression (Braun et al. 2011; Chekulaeva et al. 2011; Fabian et al. 2011). AU-rich element (ARE) binding proteins, such as tristetraprolin (TTP) and its homologs, also bind directly to NOT1 to initiate deadenylation via the associated CAF1 (Sandler et al. 2011; Fabian et al. 2013). The CCR4–NOT complex is thus recruited to mRNAs by miRNAs and RNA-binding proteins (RBPs) to elicit translational repression or decay of the transcript (Braun et al. 2011; Chekulaeva et al. 2011; Fabian et al. 2011; Sandler et al. 2011; Wahle and Winkler 2013; Bhandari et al. 2014; Chen et al. 2014; Inada and Makino 2014; Mathys et al. 2014; Rouya et al. 2014; Temme et al. 2014).
CAF1 is unusual in that it represses translation independent of deadenylation. Xenopus CAF1a and CAF1b both possess this activity (Cooke et al. 2010), and this process is important for miRNA-mediated repression in somatic cells (Braun et al. 2011; Chekulaeva et al. 2011; Fabian et al. 2011). The mechanism through which this unexpected deadenylation-independent repression occurs is unknown. We hypothesized that the interaction between CAF1 and the CCR4–NOT complex might mediate other protein contacts important for repression.
We sought to determine how CAF1 elicits deadenylation-independent repression. To do so, we identified proteins associated with CAF1 and tested their functional roles using interaction-defective mutations. Our results reveal that NOT1 interaction with CAF1 is critical for repression and that the MIF4G domain of NOT1 recruits Xp54 and 4E-T. This repressive complex prevents translation in an eIF4E- and cap-independent manner.
RESULTS
CAF1 requires interaction with NOT1 to repress translation
To determine how CAF1 represses translation independent of deadenylation, we expressed mutant CAF1 proteins in Xenopus oocytes. We reasoned that CAF1 bound specific protein partners that exerted the repressive activity. To test this idea, we created mutations in Xenopus laevis CAF1b that were predicted to disrupt interaction with NOT1, CCR4, and TOB, each of which directly interacts with CAF1 (Fig. 1A; Horiuchi et al. 2009; Basquin et al. 2012). The known human CAF1a structure was used to guide mutagenesis (Horiuchi et al. 2009). We also mutated specific surface residues to alanine, reasoning that they too might mediate protein–protein contacts (Fig. 1A).
FIGURE 1.
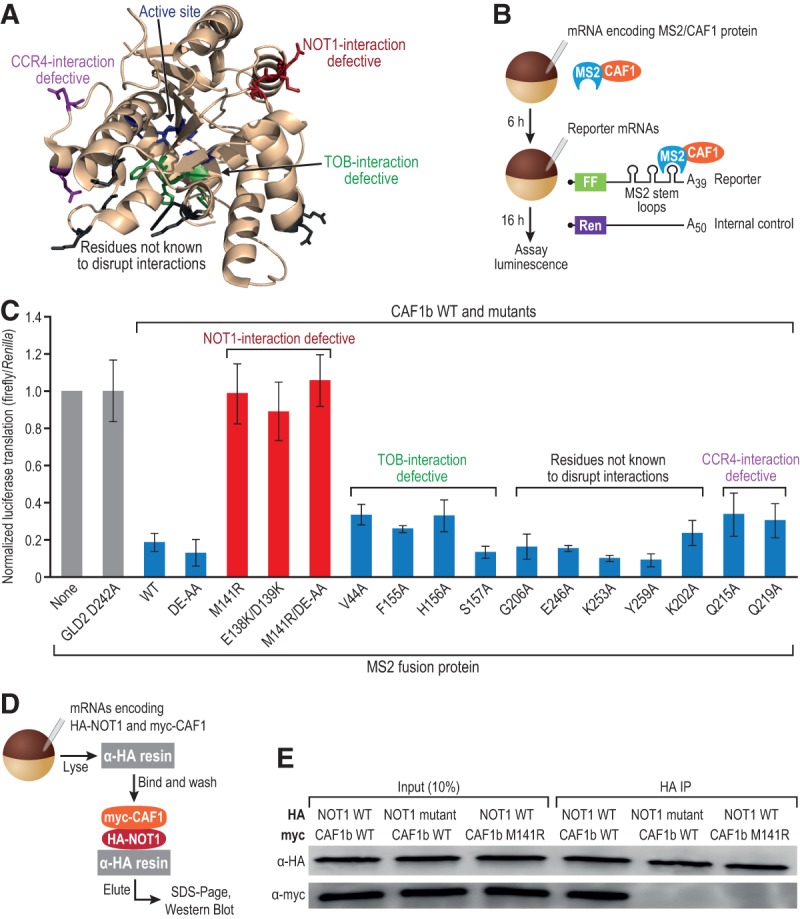
Mutational analysis of CAF1 proteins reveals requirement for NOT1 interaction. (A) Residues mutated are depicted on the human CAF1a structure (PDB 2D5R) and colored by the interaction disrupted. (B) Outline of the tethered function assay. (FF) firefly luciferase, (Ren) Renilla luciferase. (C) Results of tethered function assay, normalized to “no MS2 protein” control (none). Three independent experiments were performed, with four oocyte replicates each time. Error bars represent 1 SD. Student's two-tailed t-test was used to determine significant changes between CAF1 WT and mutants, and only NOT1-interaction-defective mutants were significantly (P < 0.005) different. (D) Outline of coimmunoprecipitation assay from Xenopus oocytes. (E) NOT1 interacts with wild-type CAF1 but not with the M141R mutant. A CAF1-interaction-defective mutant of NOT1 served as a negative control. Western blots depict a single representative experiment, and three biological replicates were conducted.
To test whether these mutations disrupted CAF1-mediated repression, wild-type and mutant forms of CAF1b were tethered to a reporter mRNA in X. laevis oocytes (Fig. 1B). Oocytes were injected with mRNAs encoding HA-tagged and MS2 coat protein-fused (HA-MS2) CAF1b proteins. After allowing time for the HA-MS2-CAF1 proteins to accumulate, the same oocytes were simultaneously injected with two reporter mRNAs. The first contained the firefly luciferase open reading frame linked to a 3′ UTR with binding sites for MS2 coat protein. The second contained the Renilla luciferase open reading frame lacking MS2 sites, which served as a negative control and was used for normalization.
CAF1 required interaction with NOT1 for repression (Fig. 1C). The mutations that disrupted interaction with NOT1 (M141R and E138K/D139K) both abrogated repression (Fig. 1C, red bars). M141R also disrupted repression in a deadenylation-defective CAF1 background (D40A, E42A; referred to as DE-AA), demonstrating that it affected the deadenylation-independent process. Mutations that disrupted interactions with CCR4 (Q215A, Q219A) or TOB (V44A, F155A, H156A, S157A) did not relieve repression. As expected, the negative control GLD2 (D242A) (Cooke et al. 2010) had no effect on translation, yielding the same level of luciferase as an oocyte expressing no MS2 protein (Fig. 1C, gray bars). Western blots with α-HA revealed that the MS2 fusion proteins were expressed (Supplemental Fig. S1). We confirmed that the interaction between CAF1 M141R and NOT1 had been disrupted by coimmunoprecipitation (co-IP), in which a NOT1 mutant defective for interaction with CAF1 served as a control (Fig. 1D,E). From these data, we inferred that NOT1 was required for the repression activity of CAF1 and next sought the partners of NOT1 that might be involved.
Mass spectrometry reveals novel CAF1 interactors
To identify proteins that interacted with CAF1 via NOT1 in oocytes, we used both candidate and proteomics approaches (Fig. 2A). In both cases, GST pull-downs were conducted with recombinant wild-type and mutant CAF1a and CAF1b proteins. Each was immobilized on glutathione magnetic resin, incubated with Xenopus oocyte lysate, and then washed to eliminate nonspecific interactions. GST protein served as a negative control. As expected, each GST protein was detected in equal amounts among the recovered proteins (Fig. 2B, top).
FIGURE 2.
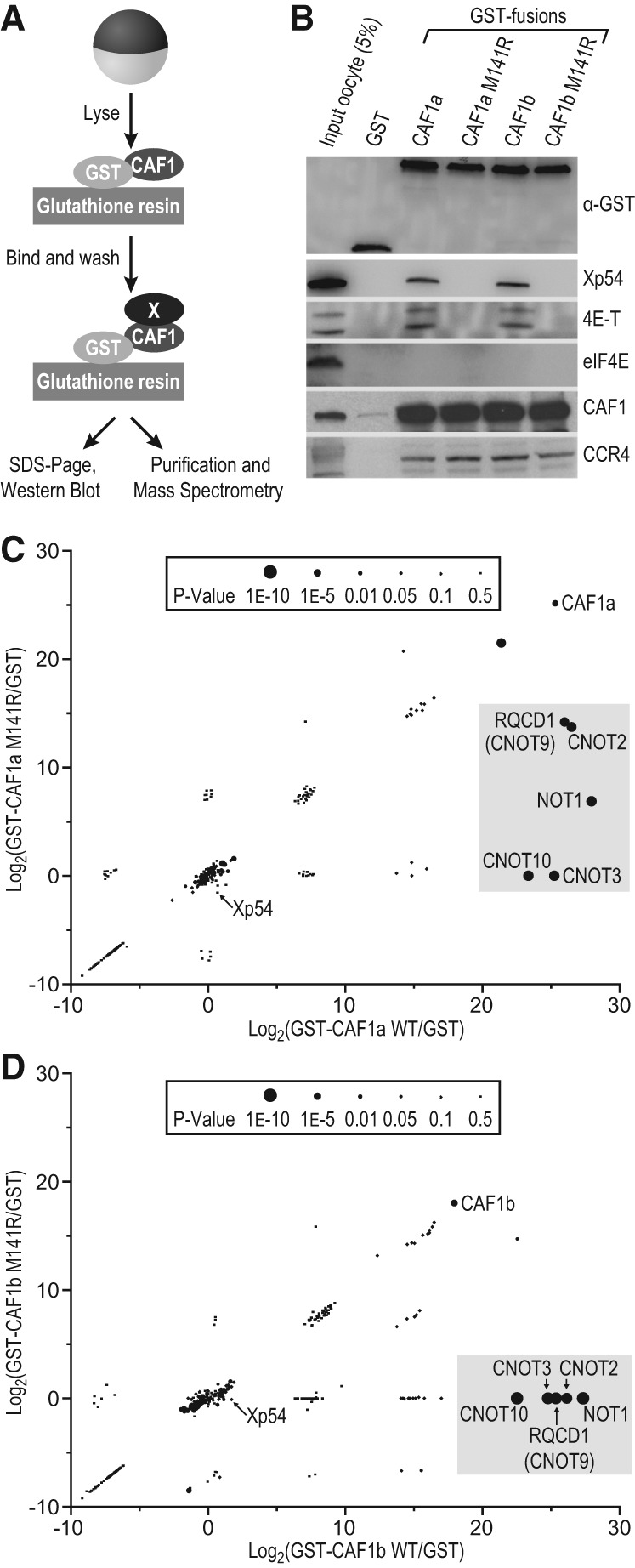
NOT1 mediates interactions between CAF1 and its protein partners. (A) Schematic representation of GST pull-down assay. (B) GST-CAF1 proteins are efficiently pulled down. Western blots with antibodies to known regulatory proteins were performed after GST pull-down to test for CAF1 interaction. Western blots depict a single representative experiment, and three biological replicates were conducted. (C) Graphical representation of mass spectrometry data from three biological replicates for GST-CAF1a wild type and mutant. P-values calculated using a two-tailed Student's t-test are indicated by dot size. Members of the CCR4–NOT complex (boxed) interact strongly with CAF1a WT but not the M141R mutant. Xp54 is also enriched specifically for CAF1a WT over the M141R mutant, but to a lesser extent. (D) Same as in C, but with CAF1b.
We found that interactions between CAF1 and Xp54, as well as multiple components of the CCR4–NOT complex, were facilitated by NOT1. Mass spectrometry was used to identify proteins enriched in the pull-downs with wild-type or M141R forms of CAF1. To quantify the mass spectrometry data, we measured enrichment in wild-type and mutant pull-down samples relative to the GST negative control. We then compared abundances of proteins in pull-downs performed with wild-type or mutant CAF1. Proteins that interacted specifically with the wild-type CAF1, and not M141R, fell into two groups, which were essentially the same whether CAF1a or CAF1b had been used as the affinity reagent (Fig. 2C,D; Supplemental Table S1). One group was enriched 20–30-fold relative to the GST negative control, with highly significant P-values (P < 1 × 10−5). The proteins in this group (highlighted in Fig. 2C,D) are all members of the CCR4–NOT complex and likely very stably associated with CAF1 and NOT1. A second group of interactors, including Xp54, exhibited twofold enrichment relative to GST with significant P-values (P < 0.05).
The candidate approach confirmed the results obtained by mass spectrometry. We tested interaction between the GST proteins and known RNA-regulatory factors by Western blot, using appropriate antibodies. Xp54 was also tested for interaction with CAF1, as it has recently been reported to directly contact the NOT1 MIF4G domain (Chen et al. 2014; Mathys et al. 2014; Rouya et al. 2014). Xp54 interacted with CAF1 in a manner that required the wild-type M141 allele (Fig. 2B). Since Xp54 associates with 4E-T (Minshall et al. 2007), we tested 4E-T interaction with CAF1. 4E-T also interacted with CAF1 dependent on the wild-type M141 allele (Fig. 2B). Disruption by the M141R mutation implied that CAF1 contacts both Xp54 and 4E-T via NOT1. Wild-type and mutant CAF1 proteins bound CCR4, implying that this interaction was independent of NOT1, as observed in other systems (Bai et al. 1999; Basquin et al. 2012). eIF4E, a protein that does not bind to CAF1 (Fabian et al. 2009), interacted with neither the wild-type nor mutant forms of CAF1.
Thus, multiple components of the CCR4–NOT complex efficiently bound CAF1 via NOT1. Other proteins, including Xp54 and 4E-T, also were retained in a NOT1-dependent fashion, but were present in lower abundance. This may reflect their lower abundance in oocytes or the instability of their interactions (see Discussion). Xp54 and 4E-T are both known and conserved translational repressors. The former is an ATP-dependent RNA helicase and part of the decapping machinery (Coller et al. 2001; Fischer and Weis 2002; Minshall and Standart 2004), while the latter is an eIF4E-binding protein that is known to repress translation via two distinct mechanisms (Kamenska et al. 2014).
Coimmunoprecipitations confirm interactions in vivo
To confirm interactions detected by GST pull-down, we conducted coimmunoprecipitations from Xenopus oocytes. HA-tagged CAF1 mutant and wild-type proteins were expressed and immunoprecipitated (Fig. 3A), and the products were analyzed by Western blot.
FIGURE 3.
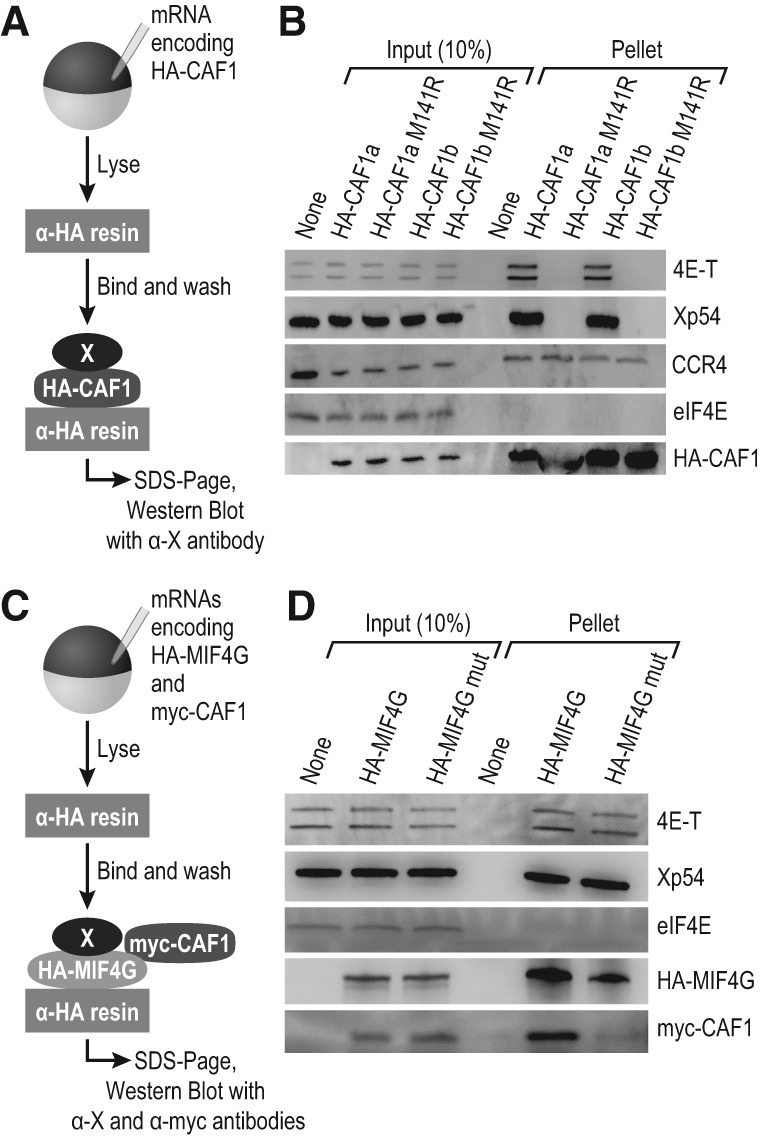
NOT1 MIF4G is required for CAF1 interactions in vivo. (A) Schematic of the coimmunoprecipitation assay with HA-CAF1 as the bait protein. (B) Western blots with antibodies to proteins that interact with CAF1 by GST pull-down were conducted to test interaction by co-IP. CCR4 is a positive control, and eIF4E is a negative control. Western blots depict a single representative experiment, and three biological replicates were conducted. (C) Schematic of the coimmunoprecipitation assay with HA-MIF4G (the NOT1 MIF4G domain) as the bait protein. Both HA-MIF4G WT and a CAF1-binding deficient mutant of the MIF4G domain (HA-MIF4G mut) were used. myc-CAF1 was coexpressed as a control. (D) Western blots were conducted to test whether 4E-T and Xp54 interact with the NOT1 MIF4G domain. eIF4E was used as a negative control, and myc-CAF1 also served as a control. Western blots depict a single representative experiment, and three biological replicates were conducted.
Both CAF1a and CAF1b required interaction with NOT1 to immunoprecipitate 4E-T and Xp54 (Fig. 3B). In contrast, CCR4 was immunoprecipitated by both wild-type and mutant CAF1a and CAF1b proteins, while eIF4E was not detected in any of the IPs. Probing with the α-HA antibody confirmed that the CAF1 proteins were immunoprecipitated (Fig. 3B, bottom).
Analogous IPs were conducted to determine whether the interactions via NOT1 were mediated by its MIF4G domain (Fig. 3C). We tested interactions of 4E-T and Xp54 with either a wild-type or CAF1-interaction-defective allele of the MIF4G domain (Basquin et al. 2012). Both 4E-T and Xp54 interacted with the MIF4G domain, independent of CAF1 interaction (Fig. 3D). eIF4E did not interact with the MIF4G domain. Wild-type and mutant MIF4G domains (detected with α-HA) were both immunoprecipitated, as expected.
These data demonstrate that the NOT1 MIF4G domain is responsible for recruitment of Xp54 and 4E-T.
NOT1 MIF4G domain mediates repression
To identify the role of the NOT1 MIF4G domain in translational repression in oocytes, we determined the repression activity of a series of NOT1 truncations. Fragments of NOT1, tagged with HA and MS2, were tethered to a reporter mRNA in Xenopus oocytes (Fig. 4A,B). CAF1-interaction-defective and Xp54-interaction-defective alleles (Basquin et al. 2012; Chen et al. 2014; Mathys et al. 2014; Rouya et al. 2014) of the NOT1 MIF4G domain were tested in parallel.
FIGURE 4.
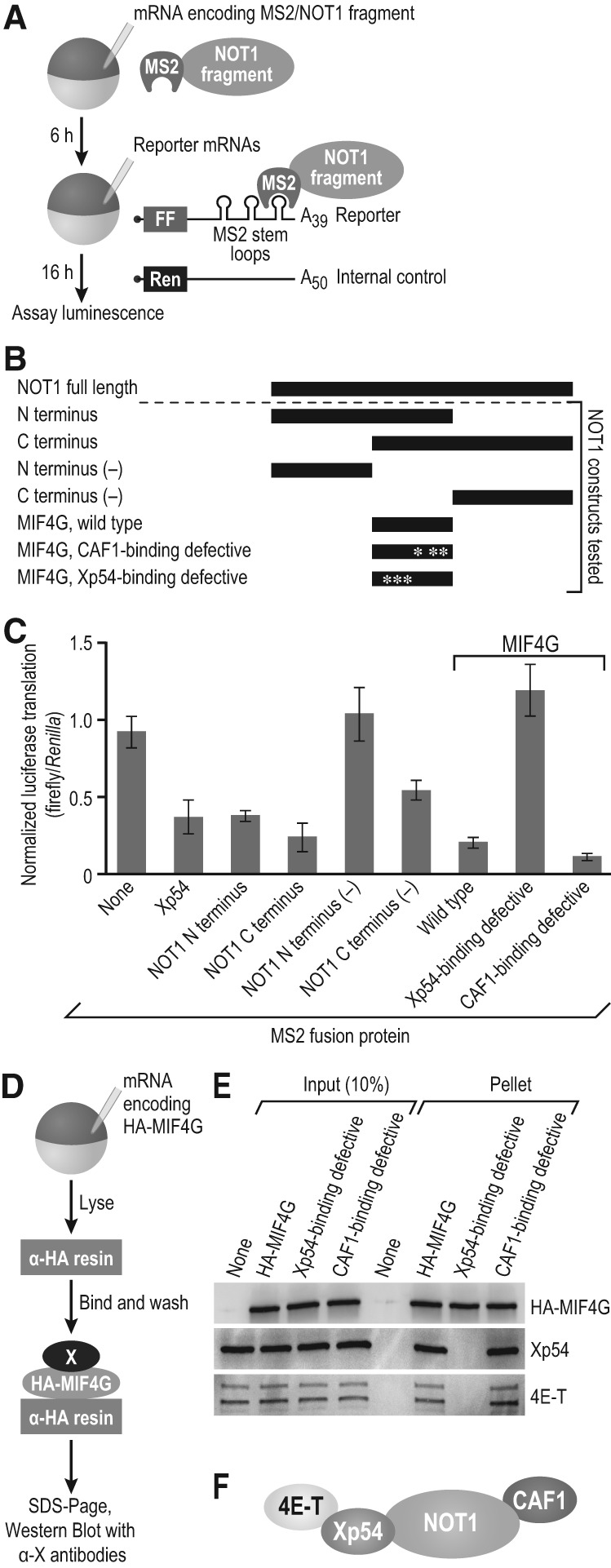
NOT1 MIF4G-Xp54 interaction is required for repression. (A) Schematic of tethered function assay with NOT1 fragments. (FF) firefly luciferase, (Ren) Renilla luciferase. (B) NOT1 fragments used in the tethered function assay. Mutations were made in the NOT1 MIF4G domain to obtain Xp54- and CAF1-binding defective constructs, as indicated by asterisks. Full-length NOT1 is depicted for scale. (C) Results of tethered function assay, normalized to “no MS2 protein” control (none). Xp54 was used as a control for repression. Three independent experiments were performed, with four oocyte replicates each time. Error bars represent 1 SD. Student's two-tailed t-test was used to compare the NOT1 amino terminus to amino terminus (−) and the wild-type MIF4G to the Xp54-binding defective MIF4G. Both were significantly (P < 0.005) different. (D) Schematic of the coimmunoprecipitation assay with HA-MIF4G (the NOT1 MIF4G domain) as the bait protein. HA-MIF4G WT, the CAF1-binding deficient mutant of the MIF4G domain, and the Xp54-binding deficient mutant were used. (E) Western blots were conducted to test whether 4E-T and Xp54 interact with the Xp54-binding deficient MIF4G domain. Western blots depict a single representative experiment, and three biological replicates were conducted. (F) Model of the interactions between NOT1, CAF1, Xp54, and 4E-T.
The NOT1 MIF4G domain was sufficient for repression, and that activity was dependent on its interaction with Xp54 (Fig. 4C). Wild-type and CAF1-interaction-defective alleles of the MIF4G domain possessed similar activities, suggesting that CAF1 was dispensable for repression. This is consistent with data obtained in human and Drosophila cells (Chen et al. 2014; Mathys et al. 2014; Rouya et al. 2014). The amino terminus of NOT1 repressed only when it included the MIF4G domain, while the carboxy terminus retained some repressive activity even without the MIF4G domain. This is likely due to the fact that the carboxy terminus contains a domain necessary for interaction with CNOT9. NOT1 interaction with CNOT9 contributes to, but is not required for, repression in other systems (Chen et al. 2014; Mathys et al. 2014). Xp54 (Fig. 4C), used as a control, repressed translation relative to an oocyte expressing no MS2 tagged protein as expected (none, Fig. 4C). Expression of the appropriate proteins was confirmed by Western blot (Supplemental Fig. S2); the amino- and carboxy-terminal fragments lacking the MIF4G domain were less abundant than the other fragments, relative to actin.
The Xp54-binding defective mutation in the NOT1 MIF4G did indeed disrupt recruitment of Xp54, as judged by coimmunoprecipitation (co-IP) (Fig. 4D,E). Similarly, 4E-T interacted with NOT1 in an Xp54-dependent manner. From these data, we propose that Xp54 directly interacts with NOT1 and is a bridge that links NOT1 and 4E-T (Fig. 4F).
4E-T is responsible for repression in an eIF4E-independent manner
Xp54 interacts with the protein 4E-T (Minshall et al. 2007), which represses translation through two distinct mechanisms that can be differentiated by mutational analysis (Kamenska et al. 2014). Expression of wild-type 4E-T represses translation of cellular mRNAs (Kamenska et al. 2014). 4E-T mutations (YLL-AAA) that disrupt binding to eIF4E lack that activity (Fig. 5A). 4E-T mutants that possess only the amino-terminal region (aa 1–180) bind eIF4E and are general translational repressors; importantly, they do not further repress mRNA to which they are tethered via the 3′ UTR (Kamenska et al. 2014). Thus, 4E-T can repress translation via a canonical eIF4E-dependent mechanism (Fig. 5A, “Mechanism 1”) and through a distinct eIF4E-independent mechanism (Fig. 5A, “Mechanism 2”).
FIGURE 5.
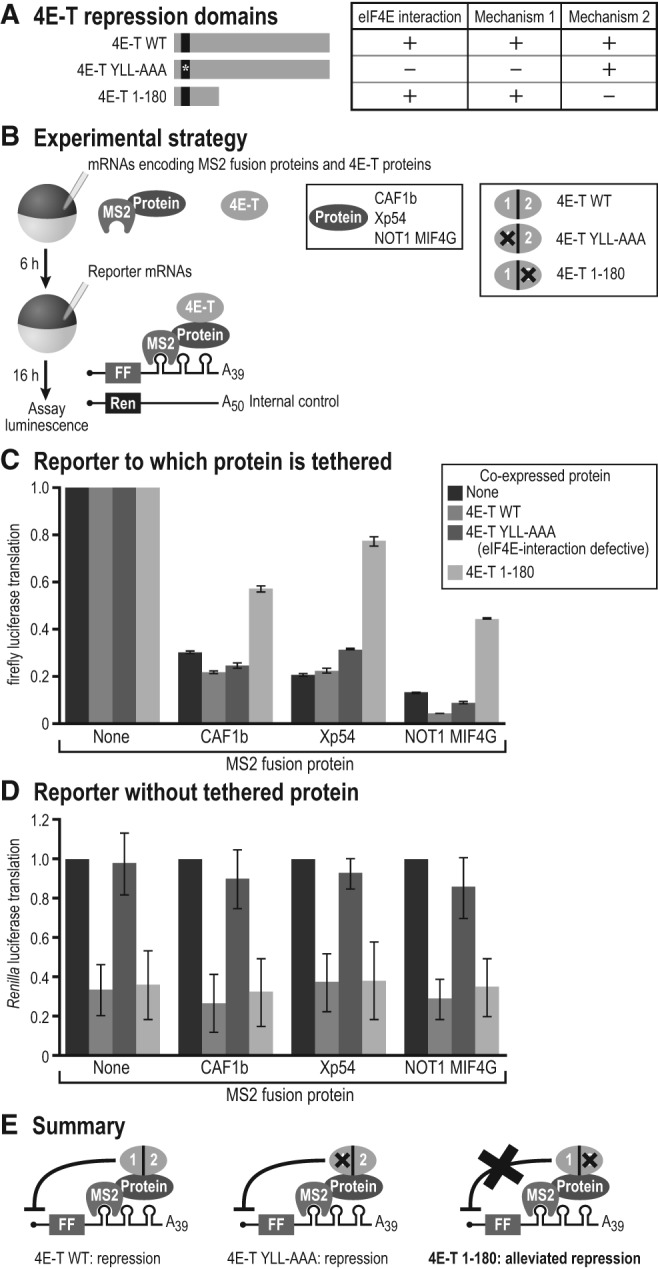
4E-T mediates repression in an eIF4E-independent manner. (A) 4E-T protein depicted to show eIF4E-binding motif (black box) and mutants used for modified tethered function assay. Table is used to show the different repression mechanisms of 4E-T (“Mechanism 1,” eIF4E-dependent and “Mechanism 2,” eIF4E-independent). (B) Schematic of modified tethered function assay with coexpressed 4E-T proteins. (FF) firefly luciferase, (Ren) Renilla luciferase. 4E-T represents the wild-type and mutant 4E-T constructs expressed, with 1 and 2 indicating the repression mechanisms from panel A. “Protein” represents CAF1b, Xp54, or NOT1 MIF4G. (C) Results of tethered function assay, normalized to “no MS2 protein” control (none). Black bars indicate no coexpressed 4E-T protein, gray shaded bars indicate wild-type 4E-T protein, the eIF4E-interaction-defective allele of 4E-T (YLL-AAA), and the 4E-T 1–180 truncation. Three independent experiments were performed, with four oocyte replicates each time. Error bars represent 1 SD. Student's two-tailed t-test was used to compare the 4E-T 1–180 data to the 4E-T WT data. For CAF1b and Xp54, the difference was highly significant (P < 0.005), while the NOT1 MIF4G domain had a slightly less significant difference of P < 0.05. (D) Graph of Renilla luciferase translation from the same experiment to show effects of the 4E-T coexpressed proteins on general translation. (E) Schematics depicting the summary of the tethered function assay data. “Protein” represents CAF1b, Xp54, and NOT1 MIF4G.
Since our data indicated that Xp54 was required for repression in oocytes, and Xp54 interacts with 4E-T, we sought to determine the role of 4E-T in repression. We used a modified tethered function assay, in which HA-MS2 fusions of CAF1b, Xp54, and the NOT1 MIF4G domain were coexpressed with untethered HA-tagged 4E-T proteins (Fig. 5B). We tested both previously identified 4E-T mutants (YLL-AAA and 1–180) (Kamenska et al. 2014) and wild-type 4E-T. This strategy, diagrammed in Figure 5B, enabled us to examine the relative contributions of eIF4E-dependent and -independent mechanisms (Kamenska et al. 2014). Since 4E-T associates with Xp54, NOT1, and CAF1b (Figs. 2–4; Minshall et al. 2007), we reasoned that over-expressed 4E-T proteins would interact with tethered Xp54, NOT1, and CAF1b proteins. If the 4E-T interaction with eIF4E were required for repression by tethered CAF1b, Xp54, and NOT1, then expression of the YLL-AAA mutant would abolish that repression activity. However, if repression were elicited via the eIF4E-independent mechanism, then the YLL-AAA mutant would still repress, while the 4E-T 1–180 truncation would abrogate repression of the bound reporter mRNA.
Of the 4E-T constructs tested, only 4E-T 1–180 alleviated repression by the tethered proteins (Fig. 5C). It interfered with repression by tethered CAF1b, Xp54, or the NOT1 MIF4G domain. Neither the wild-type nor the eIF4E-interaction-defective variant interfered with repression by any of the three tethered proteins tested, relative to control oocytes lacking any tethered protein. Thus, repression by tethered CAF1, Xp54, and NOT1 occurred via the eIF4E-independent activity of 4E-T. Expression of the proteins was confirmed by Western blot (Supplemental Fig. S3).
To ensure that the effect of 4E-T on repression by the tethered proteins was due to the eIF4E-independent mechanism, we examined the effects of the 4E-T proteins on general translation by analysis of Renilla luciferase levels (Fig. 5D). This reporter lacks MS2 sites, and so is not subject to the effects of the MS2 fusion proteins. As seen in human cells (Kamenska et al. 2014), wild-type 4E-T and the 1–180 truncation reduced general translation, while the YLL-AAA mutant did not. These findings demonstrate that the 4E-T YLL-AAA protein is functional, but its ability to bind eIF4E is dispensable for repression mediated through 3′ UTR-bound proteins.
Taken together, our studies revealed two mechanisms of repression by 4E-T in oocytes. This is consistent with previous experiments conducted in human cells (Kamenska et al. 2014). The first mechanism was independent of the eIF4E interaction and acted specifically on the firefly reporter to which the proteins were tethered (Fig. 5C, “Mechanism 1”). The second was dependent on the eIF4E interaction and affected general translation, as monitored by the Renilla reporter (Fig. 5D, “Mechanism 2”). These results, summarized in Figure 5E, demonstrate that 4E-T mediates repression by the CCR4–NOT complex in oocytes, and does so in a manner independent of its interaction with eIF4E.
Repression by CAF1 and Xp54 is independent of the 5′ cap and eIF4E
Since our data implied that 4E-T acted independently of its interaction with eIF4E, we probed the role of the cap and translation initiation factors in repression by CAF1. To do so, we used reporters bearing different IRES regions, with varying initiation factor requirements (Fig. 6A,B).
FIGURE 6.
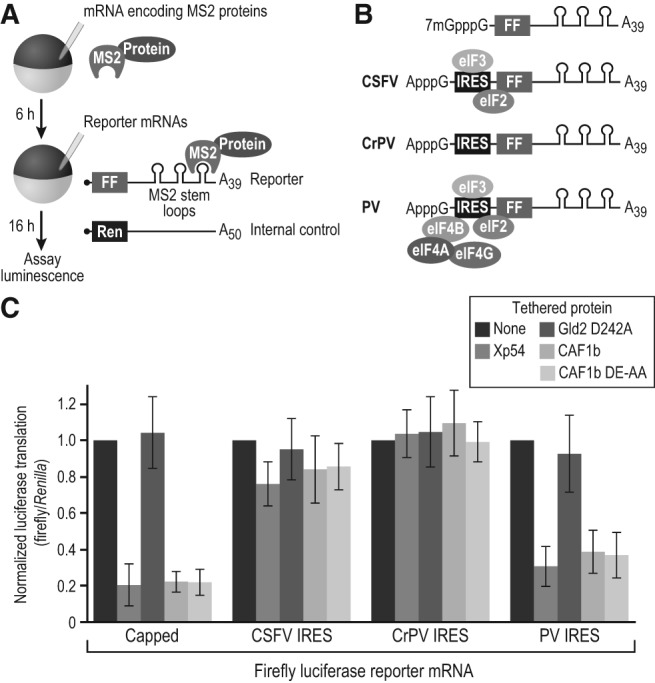
Xp54 and CAF1 repress translation independent of eIF4E and the 5′ cap. (A) Schematic of the tethered function assay. (FF) firefly luciferase, (Ren) Renilla luciferase. “Protein” represents GLD2-D242A (negative control), CAF1b, CAF1b DE-AA, or Xp54. (B) Schematic of the firefly luciferase reporters used. The IRES translation factor requirements are depicted. (C) Results of tethered function assay, normalized to “no MS2 protein” control (none, black bars). Three independent experiments were performed, with four oocyte replicates each time. Error bars represent 1 SD. Student's two-tailed t-test was used to compare the Xp54, CAF1b, and CAF1b DE-AA data to the no MS2 protein data for each reporter. For CAF1b, CAF1b DE-AA, and Xp54, the difference was highly significant (P < 0.005) only for the capped and PV IRES reporters.
CAF1b and Xp54 were tethered to a firefly reporter mRNA possessing either a 7-methyl GpppG cap or one of three IRESes, each with different translation factor requirements (Fig. 6B). An ApppG cap was used to protect the IRES reporters from degradation. As a positive control, we used our standard 5′ UTR with a 7mGpppG cap. The Renilla luciferase reporter served as a negative control, as it contained no tethering sites. Expression of the tethered proteins was confirmed by Western blot (Supplemental Fig. S4).
Xp54 and both wild-type and deadenylation-defective (DE-AA) CAF1b failed to repress via the Classical Swine Fever Virus (CSFV) or Cricket Paralysis Virus (CrPV) IRESes (Fig. 6C). Strikingly, they did repress RNAs bearing the Poliovirus (PV) IRES. As expected, the negative control GLD2 (D242A) had no effect on translation. The ability of CAF1 and Xp54 to repress the PV IRES reporter suggests that the cap structure is dispensable for repression. CSFV and PV IRESes share a requirement for eIF3 and eIF2. They differ in that the PV IRES also recruits eIF4G and therefore eIF4A and eIF4B, but not eIF4E (Pestova et al. 1996; Kieft 2008; Plank and Kieft 2012). This suggests that one or more of eIF4G, eIF4A, and eIF4B are required for repression by CAF1 and Xp54.
DISCUSSION
Our efforts to understand the mechanism of deadenylation-independent repression by CAF1 in oocytes led to a chain of protein–protein interactions that link tethered CAF1 first to NOT1, then Xp54, and ultimately to 4E-T and an eIF4E-independent mechanism (Fig. 7A). The significance of each interaction has been validated using interaction-defective mutations. Tethered CAF1 represses only if it is capable of interaction with NOT1, the scaffolding component of the CCR4–NOT complex. Similarly, tethered NOT1 represses in an Xp54-dependent fashion (Fig. 4C). 4E-T is responsible for repression by CAF1, NOT1, and Xp54 (Fig. 5B), and this repression is both eIF4E- and cap-independent (Fig. 6).
FIGURE 7.
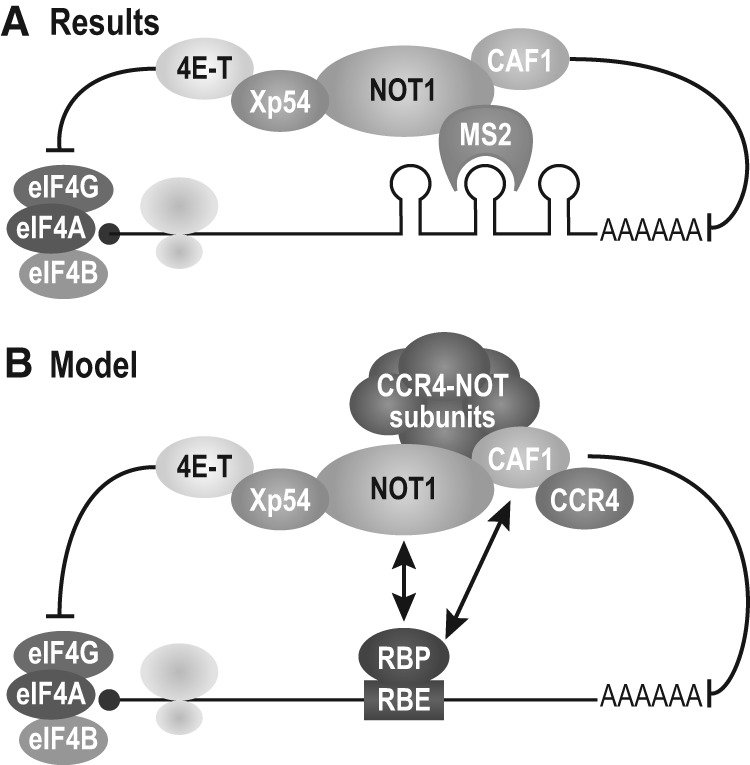
The NOT1 MIF4G domain recruits Xp54 and 4E-T to mediate eIF4E- and cap-independent repression by CAF1. (A) Model of repression when a tethered repressor is on the 3′ end of a reporter mRNA. (B) We propose that repression may occur in a similar manner by RNA-binding proteins that recruit the CAF1/NOT1/Xp54/4E-T complex when bound to target mRNAs.
We identified two groups of CAF1 interactors through GST pull-down and mass spectrometry. The first group comprised CCR4–NOT subunits and was highly enriched for wild-type CAF1, suggestive of a stoichiometric and stable interaction. Xp54 was identified among a less enriched group of proteins that likely interact with lower stoichiometry with CAF1 and NOT1. Other 3′ UTR-binding proteins likely make contacts to CAF1 and CCR4–NOT but were not detected due to their low abundance or the transience of their interactions. RNA-binding proteins known to mediate repression of specific families of mRNAs in oocytes may enter and leave the complex to control translation of their targets during oogenesis, oocyte maturation, and early embryonic development.
We observe two mechanisms of translational repression by 4E-T (Fig. 5). General translation, as monitored by reporter mRNAs without MS2 sites, is repressed by 4E-T via interaction with eIF4E. Importantly, repression due to 3′ UTR-bound factors acts via the CCR4–NOT complex through an eIF4E-independent mechanism. The molecular basis of this activity of 4E-T has not yet been determined.
The use of multiple IRES reporters demonstrates that repression via tethered CAF1 requires one or more of eIF4A, eIF4B, and eIF4G. Our IRES results are similar to those obtained in studies of miRNA-mediated repression (Meijer et al. 2013). In human cells, let-7 miRNA repressed mRNAs bearing the EMCV IRES, but not those of HCV or CrPV IRES (Meijer et al. 2013). These findings suggested that miRNA-mediated repression required subunits of eIF4F other than eIF4E, and were consistent with the conclusion that eIF4A2 was critical (Meijer et al. 2013). The factors required for repression via miRNAs and RNA-binding proteins overlap. It will be of interest to determine the extent to which their mechanisms diverge.
During early development, RNA-binding proteins repress and activate translation of specific mRNAs by binding to regulatory elements in their 3′ UTRs. While many of these proteins recruit the CCR4–NOT complex, they do so by interacting with different subunits. For example, PUF proteins in the fly embryo bind NOT4 via interaction with NANOS1 (Kadyrova et al. 2007), while yeast PUF proteins bind directly to the CAF1 homolog POP2 (Goldstrohm et al. 2006). Similarly, the fly proteins NANOS2 and NANOS3 interact with CAF1 and NOT1, respectively (Suzuki et al. 2014), but TTP in mammalian cells binds NOT1 directly (Sandler et al. 2011; Fabian et al. 2013). Human TOB1 binds to CAF1 to recruit CCR4–NOT to the RNA-binding protein CPEB3 (Hosoda et al. 2011). We propose that these diverse modes of recruitment of the CCR4–NOT complex allow for independent contacts that elicit regulation of mRNAs (Fig. 7B). For instance, if an interaction between one CCR4–NOT subunit and a 3′ UTR-bound protein were disrupted, different RNA-binding proteins would remain bound to CCR4–NOT via the other subunits. Thus, various RNA-binding proteins can use CCR4–NOT to deadenylate or repress their target mRNAs. Depletion of 4E-T increases the half-life of ARE-containing mRNAs (Ferraiuolo et al. 2005), suggesting that ARE-binding proteins such as TTP do indeed exploit 4E-T.
Our work suggests a model in which RNA-binding proteins associated with the 3′ UTR elicit repression via a network of interactions that lead to recruitment of 4E-T and its eIF4E-independent mechanism of action (Fig. 7B). miRNA-mediated repression in human and Drosophila cells similarly requires recruitment of NOT1, CAF1, and the Xp54 homolog DDX6, but via Ago and GW182 (TNRC6A-C) (Chen et al. 2014; Mathys et al. 2014; Rouya et al. 2014). In oocytes, Ago is not present and miRNA-mediated repression does not occur (Lund et al. 2011). Thus we propose that the action of regulatory 3′ UTR-binding proteins in the oocyte is through the chain of protein–protein interactions in Figure 7B, without Ago or miRNAs. The balance among modes of recruitment appears to vary in oocytes and somatic cells, though the chain of connectivity is similar. It will be of interest to determine whether multiple 3′ UTR-binding proteins that act during early development recruit identical complexes.
MATERIALS AND METHODS
Plasmid construction and mutational analysis
pCS2+3HAMS2 containing CAF1, Xp54, and GLD2-D242A proteins have been described previously (Cooke et al. 2010). CAF1 mutants in all plasmids were generated using site-directed mutagenesis. For protein purifications, pGEX-6P-1 plasmids containing CAF1 proteins cloned into the SalI/NotI sites were used. NOT1 fragments and mutants (amino-terminus: amino acids 1–1317; carboxy-terminus: amino acids 1093–2376; MIF4G domain: amino acids 1093–1317; amino terminus (−): amino acids 1–1092; carboxy terminus (−): amino acids 1318–2376; CAF1-binding defective: P1209Y, P1257Y, V1251R; Xp54 binding-defective: E1142R, N1144A, F1145A) were amplified from the human NOT1 cDNA clone (Thermo Scientific, Accession: BC040523.1, MGC: 23019, IMAGE: 5266600) and cloned into the XhoI/SnaBI sites of pCS2 + 3HAMS2 for tethered function assays or the same sites in pCS2 + 3HA or pCS2 + 9xmyc for co-IPs. Full-length NOT1 was cloned the same way, but did not express in oocytes so was not used for experiments. pCS2 + 9xmyc was constructed by amplifying the 9xmyc tag from the pCDNA3 + 9xmyc plasmid into the BamHI and StuI sites of pCS2 + 3HA, replacing the 3 HA tags.
Reporter mRNA plasmids for tethered function assays (pLG-MS2 and pCSFV-Luc-MS2 for firefly and pSP65-ren for Renilla) have been described previously (Dickson et al. 2001; Kwak et al. 2004). pPV-Luc-MS2 and pEJ4 with MS2 were kindly provided by Nicola Gray. All reporter mRNAs possessed a poly(A) tail.
4E-T was amplified from the X. laevis eIF4ENIF1 cDNA clone (GE Healthcare, MGC:80355, IMAGE:5074419) and cloned into the pCS2 + 3HA vector using NcoI/XbaI sites. The YLL-AAA (Y28A, L33A, L34A) mutant was generated using site-directed mutagenesis, and the 1–180 truncation was amplified from the full-length cDNA clone and put into pCS2 + 3HA with NcoI/XbaI.
In vitro transcription and RNA preparation
Plasmids for protein expression were linearized with NotI (Fermentas) and plasmids for luciferase reporter mRNAs were linearized with BamHI (pLG-MS2) or SalI (pSP65-ren). Linearized plasmids were transcribed using either AmpliScribe SP6 high yield transcription or T7-Flash transcription kits (CellScript). m7G(5′)ppp(5′)G cap analog (New England Biolabs) was added into the transcription reactions to cap the mRNAs. After transcription and DNase I treatment, mRNAs were purified using the Fermentas RNA purification kit.
Oocyte injections
Oocyte injections were performed as described previously (Gray et al. 2000; Kwak et al. 2004).
Tethered function assays
Tethered function assays were conducted as described previously (Cooke et al. 2010). Briefly, Stage VI oocytes were injected with 50 nL of 400 ng/µL capped mRNAs for MS2 or HA protein expression. Six hours later, the same oocytes were injected again with 50 nL of 30:10 ng/µL (firefly:Renilla) reporter mRNA mix. After 16 h, oocytes were collected, lysed, and assayed using the Promega Dual Luciferase Assay kit.
Protein purification
Protein expression plasmids were transformed into BL21-CodonPlus(DE3)-RIL cells and grown in LB + ampicillin/2% glucose medium at 37°C until OD600 ∼0.8. Protein expression was induced with 0.5 mM isopropyl-1-thio-β-D-galactopyranoside overnight at 16°C. Cells were resuspended in lysis buffer (20 mM HEPES-KOH, pH 7.4, 0.5 M NaCl, 5 mM DTT, 0.02% Tween-20, with protease inhibitors [EDTA-free, Roche]), and lysed using a Cell Disrupter (2.2kW TS Series, Constant Systems Ltd.). Cleared lysate was incubated with prewashed glutathione magnetic resin (Novagen GST•Mag) for 2 h at 4°C. The resin was washed three times with lysis buffer, and a final wash was conducted with 1× PBS with protease inhibitors (EDTA-free, Roche). The protein-bound resin was resuspended in 1× PBS with protease inhibitors and 30% glycerol. Protein concentrations were determined by Bradford assays, and proteins were aliquoted and flash frozen prior to storage at −80°C.
GST pull-down assays
Oocytes were lysed with a pestle in TNMEN buffer with 0.05% Igepal (50 mM Tris–HCl pH 8.0, 1 mM EDTA, 2 mM MgCl2, 150 mM NaCl) with protease inhibitors (EDTA-free, Roche). Oocyte lysate was cleared and RNase A/T1 (Promega) was added to digest RNA. Cleared lysate was incubated with single aliquots of GST or GST-CAF1 proteins immobilized on glutathione magnetic resin after the proteins were washed with TNMEN + 0.05% Igepal and protease inhibitors to remove glycerol. The lysate and proteins were incubated at 4°C for 2 h. GST proteins were washed twice with TNMEN with 0.01% Igepal, followed by two more washes with TNMEN with 0.01% deoxycholate. GST proteins and bound partners were eluted from the resin in 50 mM glutathione in 20 mM Tris pH 8 and 0.01% deoxycholate. Elution was performed twice at room temperature for 45 min. Eluted proteins were used for Western blot analysis (performed as described previously) (Cooke et al. 2010) and mass spectrometry.
Coimmunoprecipitations
Oocytes were injected with 50 nL of 400 ng/µL capped mRNAs expressing the appropriate HA-tagged or myc-tagged proteins. After proteins were allowed to accumulate for 6 h, oocytes of each condition were collected and lysed as for GST pull-downs. Cleared lysate (with RNase A/TI added) was incubated with HA or myc magnetic resin (Pierce α-HA magnetic beads or MBLI α-myc tag mAb-magnetic beads). The lysate and resin were incubated at 4°C for 2 h. The resin was washed four times with TNMEN with 0.01% Igepal. SDS-PAGE loading dye was added to the resin to elute proteins prior to analysis by Western blot (performed as described previously) (Cooke et al. 2010).
Mass spectrometry
Eluates were brought to 8 M urea, 50 mM Tris (pH 8.0), 50 mM sodium chloride, and 1× protease inhibitor (Roche). Disulfide bonds were reduced with 5 mM dithiothreitol, alkylated with 15 mM iodoacetamide, and the alkylating reaction was quenched with 5 mM dithiothreitol. Samples were diluted to 1.5 M urea and digested with trypsin (Promega) at a 1:100 enzyme-to-protein ratio. Samples were acidified to pH 2 using trifluoroacetic acid and centrifuged to remove detergents. Peptides were desalted using C18 columns (SepPak, Waters), dried under vacuum, and resuspended in 0.2% formic acid. LC-MS/MS was performed on a Thermo Q Exactive coupled to a nanoAcquity UPLC (Waters). Mobile phase A comprised water, 0.2% formic acid, and 5% DMSO and mobile phase B comprised acetonitrile and 0.2% formic acid. Peptides were separated over a 100 min gradient on a 75-μm inner diameter fused silica capillary packed with 5 μm diameter, 130 Å pore size Bridged Ethylene Hybrid C18 particles (Waters) heated to 60°C. Eluting peptides were converted to gas phase ions by electrospray ionization. The mass spectrometer method consisted of an MS1 survey scan followed by MS2 scans of the 20 most abundant precursors. The isolation width was set to 2 m/z, and peptides were subjected to HCD with normalized collision energy set to 25. The MS2 AGC target was set at 1e5 with a maximum injection time of 60 msec. Precursors with a charge state of less than two were rejected, and dynamic exclusion was set to 20 sec.
Mass spectrometry data analysis
Raw data from three biological replicates were analyzed using MaxQuant software (Cox and Mann 2008). Spectra were searched using the Andromeda search engine (Cox et al. 2011) against a Xenopus reference proteome obtained from UniProt (http://www.uniprot.org/). Search parameters include the following: allowance of up to two missed cleavages, oxidation of methionine as a variable modification, and carbamidomethylation of cysteine as a fixed modification. Precursor ion mass tolerance was set to 4.5 ppm, and product ions were allowed a 20 ppm mass tolerance. Peptide and protein identifications were filtered to a 1% false discovery rate using a target-decoy method (Nesvizhskii and Aebersold 2005). Label-free quantification of proteins was done using the MaxLFQ algorithm (Cox et al. 2014). Parameters for quantification include the following: Minimum ratio count was set to 2, Fast LFQ was selected, minimum number of neighbors was set to 3, and average number of neighbors was set to 6.
Antibodies
The following antibodies were diluted 1:1000 in 5% milk in 1× TBST: α-HA-11 Clone 16B12 (Covance), α-c-myc (Sigma C3956), α-DDX6 (A300-460A, Bethyl Laboratories), α-eIF4ENIF1 (ab6034, Abcam), α-eIF4E C46H6 (2067S, Cell Signaling Technologies). α-Actin antibody (MAB1501, Millipore) was diluted 1:10,000 in 5% milk in 1× TBST. Secondary antibodies, α-Goat IgG (KPL, 14-13-06), α-rabbit IgG (KPL, 074-1506), α-mouse IgG (KPL, 474-1806) were diluted 1:20,000 in 5% milk in 1× TBST.
SUPPLEMENTAL MATERIAL
Supplemental material is available for this article.
Supplementary Material
ACKNOWLEDGMENTS
We thank Michael Sheets, Alex Hebert, and members of the Wickens laboratory for helpful discussion. We also thank Nicola Gray and Amy Cooke for plasmids and Laura Vanderploeg for help preparing figures. This work was supported by National Institutes of Health (NIH) grants GM50942 (M.W.), GM31892 (M.W.), and GM80148 (J.J.C.).
Footnotes
Article published online ahead of print. Article and publication date are at http://www.rnajournal.org/cgi/doi/10.1261/rna.051565.115.
REFERENCES
- Aslam A, Mittal S, Koch F, Andrau JC, Winkler GS. 2009. The Ccr4-NOT deadenylase subunits CNOT7 and CNOT8 have overlapping roles and modulate cell proliferation. Mol Biol Cell 20: 3840–3850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bai Y, Salvadore C, Chiang YC, Collart MA, Liu HY, Denis CL. 1999. The CCR4 and CAF1 proteins of the CCR4-NOT complex are physically and functionally separated from NOT2, NOT4, and NOT5. Mol Cell Biol 19: 6642–6651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Basquin J, Roudko VV, Rode M, Basquin C, Seraphin B, Conti E. 2012. Architecture of the nuclease module of the yeast Ccr4-NOTcomplex: the NOT1-Caf1-Ccr4 interaction. Mol Cell 48: 207–218. [DOI] [PubMed] [Google Scholar]
- Bawankar P, Loh B, Wohlbold L, Schmidt S, Izaurralde E. 2013. NOT10 and C2orf29/NOT11 form a conserved module of the CCR4-NOT complex that docks onto the NOT1 N-terminal domain. RNA Biol 10: 228–244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhandari D, Raisch T, Weichenrieder O, Jonas S, Izaurralde E. 2014. Structural basis for the Nanos-mediated recruitment of the CCR4-NOT complex and translational repression. Genes Dev 28: 888–901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bianchin C, Mauxion F, Sentis S, Séraphin B, Corbo L. 2005. Conservation of the deadenylase activity of proteins of the Caf1 family in human. RNA 11: 487–494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Braun JE, Huntzinger E, Fauser M, Izaurralde E. 2011. GW182 proteins directly recruit cytoplasmic deadenylase complexes to miRNA targets. Mol Cell 44: 120–133. [DOI] [PubMed] [Google Scholar]
- Chekulaeva M, Mathys H, Zipprich JT, Attig J, Colic M, Parker R, Filipowicz W. 2011. miRNA repression involves GW182-mediated recruitment of CCR4-NOT through conserved W-containing motifs. Nat Struct Mol Biol 18: 1218–1226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen CY, Shyu AB. 2011. Mechanisms of deadenylation-dependent decay. Wiley Interdiscip Rev RNA 2: 167–183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Y, Boland A, Kuzuoğlu-Öztürk D, Bawankar P, Loh B, Chang CT, Weichenrieder O, Izaurralde E. 2014. A DDX6-CNOT1 complex and W-binding pockets in CNOT9 reveal direct links between miRNA target recognition and silencing. Mol Cell 54: 737–750. [DOI] [PubMed] [Google Scholar]
- Collart MA, Panasenko OO. 2012. The Ccr4–not complex. Gene 492: 42–53. [DOI] [PubMed] [Google Scholar]
- Coller JM, Tucker M, Sheth U, Valencia-Sanchez MA, Parker R. 2001. The DEAD box helicase, Dhh1p, functions in mRNA decapping and interacts with both the decapping and deadenylase complexes. RNA 7: 1717–1727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cooke A, Prigge A, Wickens M. 2010. Translational repression by deadenylases. J Biol Chem 285: 28506–28513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cox J, Mann M. 2008. MaxQuant enables high peptide identification rates, individualized p.p.b.-range mass accuracies and proteome-wide protein quantification. Nat Biotechnol 26: 1367–1372. [DOI] [PubMed] [Google Scholar]
- Cox J, Neuhauser N, Michalski A, Scheltema RA, Olsen JV, Mann M. 2011. Andromeda: a peptide search engine integrated into the MaxQuant environment. J Proteome Res 10: 1794–1805. [DOI] [PubMed] [Google Scholar]
- Cox J, Hein MY, Luber CA, Paron I, Nagaraj N, Mann M. 2014. Accurate proteome-wide label-free quantification by delayed normalization and maximal peptide ratio extraction, termed MaxLFQ. Mol Cell Proteomics 13: 2513–2526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dickson KS, Thompson SR, Gray NK, Wickens M. 2001. Poly(A) polymerase and the regulation of cytoplasmic polyadenylation. J Biol Chem 276: 41810–41816. [DOI] [PubMed] [Google Scholar]
- Doidge R, Mittal S, Aslam A, Winkler GS. 2012. Deadenylation of cytoplasmic mRNA by the mammalian Ccr4-Not complex. Biochem Soc Trans 40: 896–901. [DOI] [PubMed] [Google Scholar]
- Fabian MR, Mathonnet G, Sundermeier T, Mathys H, Zipprich JT, Svitkin YV, Rivas F, Jinek M, Wohlschlegel J, Doudna JA, et al. 2009. Mammalian miRNA RISC recruits CAF1 and PABP to affect PABP-dependent deadenylation. Mol Cell 35: 868–880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fabian MR, Cieplak MK, Frank F, Morita M, Green J, Srikumar T, Nagar B, Yamamoto T, Raught B, Duchaine TF, et al. 2011. miRNA-mediated deadenylation is orchestrated by GW182 through two conserved motifs that interact with CCR4-NOT. Nat Struct Mol Biol 18: 1211–1217. [DOI] [PubMed] [Google Scholar]
- Fabian MR, Frank F, Rouya C, Siddiqui N, Lai WS, Karetnikov A, Blackshear PJ, Nagar B, Sonenberg N. 2013. Structural basis for the recruitment of the human CCR4-NOT deadenylase complex by tristetraprolin. Nat Struct Mol Biol 20: 735–739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferraiuolo MA, Basak S, Dostie J, Murray EL, Schoenberg DR, Sonenberg N. 2005. A role for the eIF4E-binding protein 4E-T in P-body formation and mRNA decay. J Cell Biol 170: 913–924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fischer N, Weis K. 2002. The DEAD box protein Dhh1 stimulates the decapping enzyme Dcp1. EMBO J 21: 2788–2797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garneau NL, Wilusz J, Wilusz CJ. 2007. The highways and byways of mRNA decay. Nat Rev Mol Cell Biol 8: 113–126. [DOI] [PubMed] [Google Scholar]
- Gebauer F, Hentze MW. 2004. Molecular mechanisms of translational control. Nat Rev Mol Cell Biol 5: 827–835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldstrohm AC, Wickens M. 2008. Multifunctional deadenylase complexes diversify mRNA control. Nat Rev Mol Cell Biol 9: 337–344. [DOI] [PubMed] [Google Scholar]
- Goldstrohm AC, Hook BA, Seay DJ, Wickens M. 2006. PUF proteins bind Pop2p to regulate messenger RNAs. Nat Struct Mol Biol 13: 533–539. [DOI] [PubMed] [Google Scholar]
- Gray NK, Coller JM, Dickson KS, Wickens M. 2000. Multiple portions of poly(A)-binding protein stimulate translation in vivo. EMBO J 19: 4723–4733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horiuchi M, Takeuchi K, Noda N, Muroya N, Suzuki T, Nakamura T, Kawamura-Tsuzuku J, Takahasi K, Yamamoto T, Inagaki F. 2009. Structural basis for the antiproliferative activity of the Tob-hCaf1 complex. J Biol Chem 284: 13244–13255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hosoda N, Funakoshi Y, Hirasawa M, Yamagishi R, Asano Y, Miyagawa R, Ogami K, Tsujimoto M, Hoshino S. 2011. Anti-proliferative protein Tob negatively regulates CPEB3 target by recruiting Caf1 deadenylase. EMBO J 30: 1311–1323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inada T, Makino S. 2014. Novel roles of the multi-functional CCR4-NOT complex in post-transcriptional regulation. Front Genet 5: 135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kadyrova LY, Habara Y, Lee TH, Wharton RP. 2007. Translational control of maternal Cyclin B mRNA by Nanos in the Drosophila germline. Development 134: 1519–1527. [DOI] [PubMed] [Google Scholar]
- Kamenska A, Lu WT, Kubacka D, Broomhead H, Minshall N, Bushell M, Standart N. 2014. Human 4E-T represses translation of bound mRNAs and enhances microRNA-mediated silencing. Nucleic Acids Res 42: 3298–3313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kieft JS. 2008. Viral IRES RNA structures and ribosome interactions. Trends Biochem Sci 33: 274–283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kwak JE, Wang L, Ballantyne S, Kimble J, Wickens M. 2004. Mammalian GLD-2 homologs are poly(A) polymerases. Proc Natl Acad Sci 101: 4407–4412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lau NC, Kolkman A, van Schaik FM, Mulder KW, Pijnappel WW, Heck AJ, Timmers HT. 2009. Human Ccr4-Not complexes contain variable deadenylase subunits. Biochem J 422: 443–453. [DOI] [PubMed] [Google Scholar]
- Lund E, Sheets MD, Imboden SB, Dahlberg JE. 2011. Limiting Ago protein restricts RNAi and microRNA biogenesis during early development in Xenopus laevis. Genes Dev 25: 1121–1131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mathys H, Basquin J, Ozgur S, Czarnocki-Cieciura M, Bonneau F, Aartse A, Dziembowski A, Nowotny M, Conti E, Filipowicz W. 2014. Structural and biochemical insights to the role of the CCR4-NOT complex and DDX6 ATPase in microRNA repression. Mol Cell 54: 751–765. [DOI] [PubMed] [Google Scholar]
- Meijer HA, Kong YW, Lu WT, Wilczynska A, Spriggs RV, Robinson SW, Godfrey JD, Willis AE, Bushell M. 2013. Translational repression and eIF4A2 activity are critical for microRNA-mediated gene regulation. Science 340: 82–85. [DOI] [PubMed] [Google Scholar]
- Miller JE, Reese JC. 2012. Ccr4-Not complex: the control freak of eukaryotic cells. Crit Rev Biochem Mol Biol 47: 315–333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Minshall N, Standart N. 2004. The active form of Xp54 RNA helicase in translational repression is an RNA-mediated oligomer. Nucleic Acids Res 32: 1325–1334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Minshall N, Reiter MH, Weil D, Standart N. 2007. CPEB interacts with an ovary-specific eIF4E and 4E-T in early Xenopus oocytes. J Biol Chem 282: 37389–37401. [DOI] [PubMed] [Google Scholar]
- Molin L, Puisieux A. 2005. C. elegans homologue of the Caf1 gene, which encodes a subunit of the CCR4-NOT complex, is essential for embryonic and larval development and for meiotic progression. Gene 358: 73–81. [DOI] [PubMed] [Google Scholar]
- Nasertorabi F, Batisse C, Diepholz M, Suck D, Bottcher B. 2011. Insights into the structure of the CCR4-NOT complex by electron microscopy. FEBS Lett 585: 2182–2186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nesvizhskii AI, Aebersold R. 2005. Interpretation of shotgun proteomic data: the protein inference problem. Mol Cell Proteomics 4: 1419–1440. [DOI] [PubMed] [Google Scholar]
- Pestova TV, Hellen CU, Shatsky IN. 1996. Canonical eukaryotic initiation factors determine initiation of translation by internal ribosomal entry. Mol Cell Biol 16: 6859–6869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petit AP, Wohlbold L, Bawankar P, Huntzinger E, Schmidt S, Izaurralde E, Weichenrieder O. 2012. The structural basis for the interaction between the CAF1 nuclease and the NOT1 scaffold of the human CCR4-NOT deadenylase complex. Nucleic Acids Res 40: 11058–11072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Plank TD, Kieft JS. 2012. The structures of nonprotein-coding RNAs that drive internal ribosome entry site function. Wiley Interdiscip Rev RNA 3: 195–212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rouya C, Siddiqui N, Morita M, Duchaine TF, Fabian MR, Sonenberg N. 2014. Human DDX6 effects miRNA-mediated gene silencing via direct binding to CNOT1. RNA 20: 1398–1409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sandler H, Kreth J, Timmers HT, Stoecklin G. 2011. Not1 mediates recruitment of the deadenylase Caf1 to mRNAs targeted for degradation by tristetraprolin. Nucleic Acids Res 39: 4373–4386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suzuki A, Igarashi K, Aisaki K, Kanno J, Saga Y. 2010. NANOS2 interacts with the CCR4-NOT deadenylation complex and leads to suppression of specific RNAs. Proc Natl Acad Sci 107: 3594–3599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suzuki A, Niimi Y, Saga Y. 2014. Interaction of NANOS2 and NANOS3 with different components of the CNOT complex may contribute to the functional differences in mouse male germ cells. Biol Open 3: 1207–1216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Temme C, Simonelig M, Wahle E. 2014. Deadenylation of mRNA by the CCR4-NOT complex in Drosophila: molecular and developmental aspects. Front Genet 5: 143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wahle E, Winkler GS. 2013. RNA decay machines: deadenylation by the Ccr4-not and Pan2-Pan3 complexes. Biochim Biophys Acta 1829: 561–570. [DOI] [PubMed] [Google Scholar]
- Wilusz CJ, Wormington M, Peltz SW. 2001. The cap-to-tail guide to mRNA turnover. Nat Rev Mol Cell Biol 2: 237–246. [DOI] [PubMed] [Google Scholar]
- Xu K, Bai Y, Zhang A, Zhang Q, Bartlam MG. 2014. Insights into the structure and architecture of the CCR4-NOT complex. Front Genet 5: 137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamashita A, Chang TC, Yamashita Y, Zhu W, Zhong Z, Chen CY, Shyu AB. 2005. Concerted action of poly(A) nucleases and decapping enzyme in mammalian mRNA turnover. Nat Struct Mol Biol 12: 1054–1063. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.