

Abstract
The 3′ untranslated regions (3′ UTRs) of mRNAs regulate transcripts by serving as binding sites for regulatory factors, including microRNAs and RNA binding proteins. Binding of such trans-acting factors can control the rates of mRNA translation, decay, and other aspects of mRNA biology. To better understand the role of 3′ UTRs in gene regulation, we performed a detailed analysis of a model mammalian 3′ UTR, that of Hmga2, with the principal goals of identifying the complete set of regulatory elements within a single 3′ UTR, and determining the extent to which elements interact with and affect one another. Hmga2 is an oncogene whose overexpression in cancers often stems from mutations that remove 3′-UTR regulatory sequences. We used reporter assays in cultured cells to generate maps of cis-regulatory information across the Hmga2 3′ UTR at different resolutions, ranging from 50 to 400 nt. We found many previously unidentified regulatory sites, a large number of which were up-regulating. Importantly, the overall location and impact of regulatory sites was conserved between different species (mouse, human, and chicken). By systematically comparing the regulatory impact of 3′-UTR segments of different sizes we were able to determine that the majority of regulatory sequences function independently; only a very small number of segments showed evidence of any interactions. However, we discovered a novel interaction whereby terminal 3′-UTR sequences induced internal up-regulating elements to convert to repressive elements. By fully characterizing one 3′ UTR, we hope to better understand the principles of 3′-UTR-mediated gene regulation.
Keywords: 3′ UTR, Hmga2, microRNA, RNA-binding protein, regulatory sequence
INTRODUCTION
Precisely controlling the amount of protein made from each gene is a fundamental cellular process; while much of this regulation derives from transcriptional control, it is increasingly clear that regulation acting upon the mature transcript also plays a crucial role. The sequences underpinning post-transcriptional regulation are most often located within the transcripts 5′ and 3′ untranslated regions (UTRs). These cis-regulatory sequence elements within UTRs serve as binding sites for trans-factors such as RNA binding proteins (RBPs) and noncoding RNAs (for review, see Barrett et al. 2012). One relatively well-understood paradigm for post-transcriptional control is that of microRNAs (miRNAs), a class of ∼22-nt small RNAs, which act in concert with the proteins of the RNA-induced silencing complex as trans-regulators of mRNAs. The effective binding sites for miRNAs are most often located within the 3′ UTR, and recruitment of a miRNA to a transcript results in transcript destabilization and translational repression (for review, see Bartel 2009). While miRNAs are, perhaps, the most prevalent example of mammalian post-transcriptional regulation, a wide variety of other mechanisms exist. Hundreds of RBPs have been identified based on the presence of predicted RNA binding domains (Cook et al. 2010), but only a modest subset has been studied. Most post-transcriptional regulatory RBPs that have been studied have binding sites within 3′ UTRs (e.g., Hafner et al. 2010; Yoon et al. 2014). The 3′ UTRs of mammalian genomes, in particular, are typically both well conserved (compared with other regions of the genome; Siepel et al. 2005; Xie et al. 2005) and extensive in length (averaging 1.2 kb, versus 1.5 kb for coding sequences). Moreover, recent studies have indicated that 3′ UTRs are densely bound by proteins (Baltz et al. 2012), many of which are likely to have regulatory roles. It seems the 3′ UTR serves as a switchboard that combines complex inputs leading to proper post-transcriptional regulation, a similar role to that of enhancers in transcriptional regulation.
While many, and likely most, 3′-UTR regulatory elements are uncharacterized, an increasingly large number have been identified. The majority of elements are between ∼6 and 12-nt long and are recognized by trans-factors by virtue of their primary sequence (Xie et al. 2005). Longer elements have also been described, whose identity and function derives from both structure and sequence (Goodarzi et al. 2012). Both site-types tend to be degenerate and therefore difficult to predict from examining the primary 3′-UTR sequence. However, some informative sequence motifs corresponding to cis-regulatory elements have been characterized genome-wide, including miRNA target sites, Pumilio Response elements, and binding sites for a small number of other regulatory proteins (Fox et al. 2005; Friedman et al. 2009). It is, nevertheless, far from clear how many cis-regulatory elements exist in a typical 3′ UTR. Few 3′ UTRs have been systematically examined with the goal of identifying all of their regulatory sequences. However, studies that have begun to address this question suggest that 3′ UTRs might contain a large number of regulatory sequences; for example, mutational analysis of the Caenorhabditis elegans cog-1 3′ UTR shows that many parts of the 3′ UTR contribute to the post-transcriptional regulation of the cog-1 transcript (Didiano and Hobert 2008). Systematic studies of the regulatory landscape of 3′ UTRs to reveal the numbers and impacts of regulatory elements within them should enhance our understanding of the roles of 3′ UTRs in post-transcriptional biology.
The vast majority of investigations into 3′-UTR-mediated regulation has focused on isolated, individual elements within a 3′ UTR and have not determined how collections of elements might interact with one another. However, there exist a handful of examples of 3′ UTRs containing combinations of cis-regulatory elements that function cooperatively: Closely spaced binding sites for miRNAs can function synergistically, enhancing their regulatory impact (Grimson et al. 2007; Sætrom et al. 2007; Broderick et al. 2011), and the binding of RBPs has been reported to both activate adjacent cryptic miRNA binding sites (Kim et al. 2009, 2012; Kedde et al. 2010; Miles et al. 2012) and mask otherwise functional sites (Bhattacharyya et al. 2006; Kedde et al. 2007; Léveillé et al. 2011; Kundu, et al. 2012). For example, two studies demonstrated that the binding of human Pumilio proteins within a 3′ UTR can induce a conformational change in the RNA structure. This structural change allows the silencing machinery access to miRNA target sites that were previously hidden within hairpins (Kedde et al. 2010; Miles et al. 2012) and thus enhances miRNA repression. Alternatively, the binding of RBM38 (Léveillé et al. 2011) or Dnd1 (Kedde et al. 2007) within certain 3′ UTRs inhibits miRNA-mediated repression of mRNAs by inhibiting the binding of the silencing machinery, an example of an inhibitory interaction (or negative synergism). Importantly, many such interactions mediate major regulatory control of the underlying transcripts with important biological consequences.
These, and a handful of other examples, illustrate the potential importance of complex interactions between 3′-UTR regulatory elements—but are such interactions rare or are they commonplace? At one extreme, if the impact of 3′ UTRs on expression can, typically, be derived from the independent contribution of individual elements, then a systematic approach to describe the role 3′ UTRs play in gene expression is relatively straightforward. Alternatively, if post-transcriptional regulation encoded within 3′ UTRs typically entails complex interactions between elements, then a genome-wide understanding of 3′-UTR-mediated regulation represents a nontrivial problem. In this study, we have systematically identified regulatory sequences within a single 3′ UTR, that of Hmga2, and developed a new approach to methodically determine the extent to which identified regulatory elements function independently or synergistically.
We selected the Hmga2 transcript as a case study because it is strongly repressed post-transcriptionally through sites within the long (2.9 kb) and highly conserved 3′ UTR (Borrmann et al. 2001). Normal expression of Hmga2, which encodes a nonhistone chromatin protein, is limited to embryonic tissues, and overexpression strongly correlates with poor prognoses for cancer patients. Moreover, Hmga2 has been identified as a driver for metastasis (Morishita et al. 2013). One key component to the repression of Hmga2 in normal nonembryonic tissues is the tumor suppressing miRNA let-7, which has seven target sites within the 3′ UTR. Loss of post-transcriptional regulation, and specifically loss of let-7 targeting (Lee and Dutta 2007; Mayr et al. 2007), leads to overexpression of Hmga2 and oncogenic transformation. Interestingly, the RNA binding protein IGF2BP3 has recently been shown to protect the Hmga2 transcript from let-7-mediated repression in embryonic tissues and cancer cells (Jønson et al. 2014). While let-7 repression and IGF2BP3 sequestration are clearly important, these alone do not explain the extensive conservation of the Hmga2 3′ UTR. Thus, it seems likely that additional regulatory sequences exist within what is already a relatively well-studied 3′ UTR.
A handful of 3′ UTRs have been examined systematically by truncation analysis with the goal of identifying important sequences. Most such studies were performed at low resolution (e.g., 400 nt), giving limited information about specific sequences (Borrmann et al. 2001; Khaziapoul et al. 2012; Diab et al. 2013; Melanson et al. 2013). Two recent studies have taken a more comprehensive look at 3′ UTRs. Wirsing et al. (2011) first performed a low-resolution truncation analysis, and then performed higher resolution mapping of selected 3′-UTR fragments. A high-resolution map of regulatory sequences within the CXCL2 3′ UTR has been generated by analyzing a large collection of point mutations within the 3′ UTR, though only measuring mRNA steady-state levels (Zhao et al. 2014). Although this study represents one of the most thorough investigations of a single 3′ UTR, focusing only on elements that control mRNA levels is not optimal, as elements that alter translation will be missed; moreover, elements robust to single point mutations would not have been detected, which may constitute a relatively large fraction of 3′ UTR elements. We have taken a different approach, mapping the Hmga2 3′ UTR to high-resolution (50 nt) using reporter assays that monitor total protein output. Importantly, we also measured the regulatory impact of larger overlapping fragments (100, 200, and 400 nt) and used comparisons between different sized 3′-UTR fragments to determine whether combinations of regulatory elements are interacting synergistically. We found a large number of previously unidentified regulatory segments, many of which confer up-regulation. Importantly, our data suggest that elements within the 3′ UTR largely function independently of one another. We did, however, observe an exception to this rule, whereby distal sequences within the 3′ UTR induce otherwise positive regulatory elements to function as repressive elements.
RESULTS
High-resolution mapping reveals many discrete regulatory sequence elements within the Hmga2 3′ UTR
To begin to understand the regulatory sequences present within the Hmga2 3′ UTR, we generated a set of luciferase reporter constructs, each containing ∼100-nt fragments (100mers) of the mouse 3′ UTR, such that the complete 2.97-kb sequence was represented in the set (Fig. 1A). Experiments in which reporter activities are compared with each other can be used to calculate the relative impact on gene expression for each reporter. They cannot, however, be used to infer the absolute impact on gene expression, because there is no way to establish a baseline. To address this problem, we reasoned that 3′ UTRs comprising randomized sequences are unlikely to contain any sequence elements that could impact gene expression, and are therefore ideal inert controls with which to establish a baseline for expression. We generated three different reporter constructs, each containing a different random 100-nt sequence (random 100mers) as their 3′ UTR. Importantly, there were no significant differences in reporter activity between the three random 100mer constructs, nor between them and a no-3′-UTR control (Fig. 1B), indicating that none contained sequences impacting gene expression in our assay and confirming their usefulness as inert controls. Although the random-sequence controls are equivalent in their expression to a no-3′-UTR control for 3′ UTRs of this size, such size-matched random-sequence constructs have the benefit of controlling for the influence of 3′ UTR size per se on gene expression (Tanguay and Gallie 1996; Hogg and Goff 2010; Nicholson et al. 2010), a consideration that is more important for larger 3′-UTR fragments (Fig. 2A). This approach allowed us to establish the baseline for all of our reporter experiments, thus enabling us to calculate the absolute impact on gene expression for all 3′-UTR fragments assayed.
FIGURE 1.

Regulatory sequences in vertebrate Hmga2 3′ UTRs. (A) Reporter assays measuring regulatory impact of tiled 100-nt fragments (100mers) of the mouse Hmga2 3′ UTR. Histogram indicates log2 fold change conferred by 3′-UTR 100mers, relative to random-sequence 100mer reporters (B); significance assessed with Bonferroni-corrected Wilcoxon rank-sum tests (n > 12; P < 0.05, 1 × 10−3, and 1 × 10−5 significance thresholds indicated by *, **, and ***, respectively; error bars indicate the third largest and third smallest values among 12 replicates, approximating 68% of the data as a nonparametric analog of one standard deviation; for measurements with more than 12 replicates, values were selected so that error bars also approximate 68% of the data). The x-axis shows the approximate coordinates of each 3′-UTR sequence; the positions of let-7 miRNA target sites are indicated with blue arrowheads. (B) Reporter assays measuring regulatory impact of a no-3′-UTR control (No) and 100mer 3′-UTR fragments of random sequence (A–C). Three different control sequences mediate regulatory effects equivalent to one another and to the no-3′-UTR control (n = 9; P > 0.05, Wilcoxon rank-sum test). (C) Mutated let-7 sites abrogate the repression of Hmga2 100mer 3′-UTR fragments. Reporter assays comparing the regulatory impact of 100mer fragments containing predicted let-7 target sites (sites numbered as in A) to otherwise identical fragments in which the let-7 target sites were disrupted (n = 9; ** indicates P < 1 × 10−3, Wilcoxon rank-sum test). (D) Let-7 mediated repression of the full-length Hmga2 3′ UTR. As in C, comparing full-length Hmga2 3′ UTR constructs (normalized to a no-3′ UTR control) with all let-7 target sites intact (WT) or disrupted (m7) (n = 9, P < 1 × 10−3, Wilcoxon rank-sum test). (E,F) Reporter assays measuring regulatory impact of successive 100-nt fragments of human (E) and chicken (F) HMGA2 3′-UTR sequences. Boundaries for human and chicken 3′-UTR fragments are orthologous to mouse coordinates; otherwise as described in A (n = 9). (G) Conservation of regulatory sequence impact. Heat maps (top) illustrate reporter data from A,D,E (color key on right); the positions of let-7 miRNA target sites are indicated with blue arrowheads, as in A. Table (bottom) contains correlation coefficients (Spearman and Pearson) and P values comparing regulation of orthologous 100-nt fragments tiled across the 3′ UTR. (H) Nucleotide diversity (X-axis) is not well-correlated with divergence in regulatory impact of different 3′-UTR fragments from human, mouse, and chicken (Pearson R = 0.21, P > 0.08). (I) Impact of Hmga2 3′-UTR regulatory sequences compared in different cell types. Reporter assays (data depicted as heat maps, as described above) comparing regulatory impact of mouse 100mer 3′-UTR reporters performed in indicated cell lines. Each pair-wise comparison between reporter data is summarized with Spearman and Pearson correlation coefficients along with P values. (J) Regulatory effect of full-length Hmga2 3′ UTRs compared in different cell types. Full-length Hmga2 3′-UTR constructs (normalized to a no-3′-UTR control) with all let-7 target sites intact (WT) or disrupted (m7) in different cell types (n = 9; * and ** indicate P < 0.05 and P < 1 × 10−3, respectively, Wilcoxon rank-sum test).
FIGURE 2.
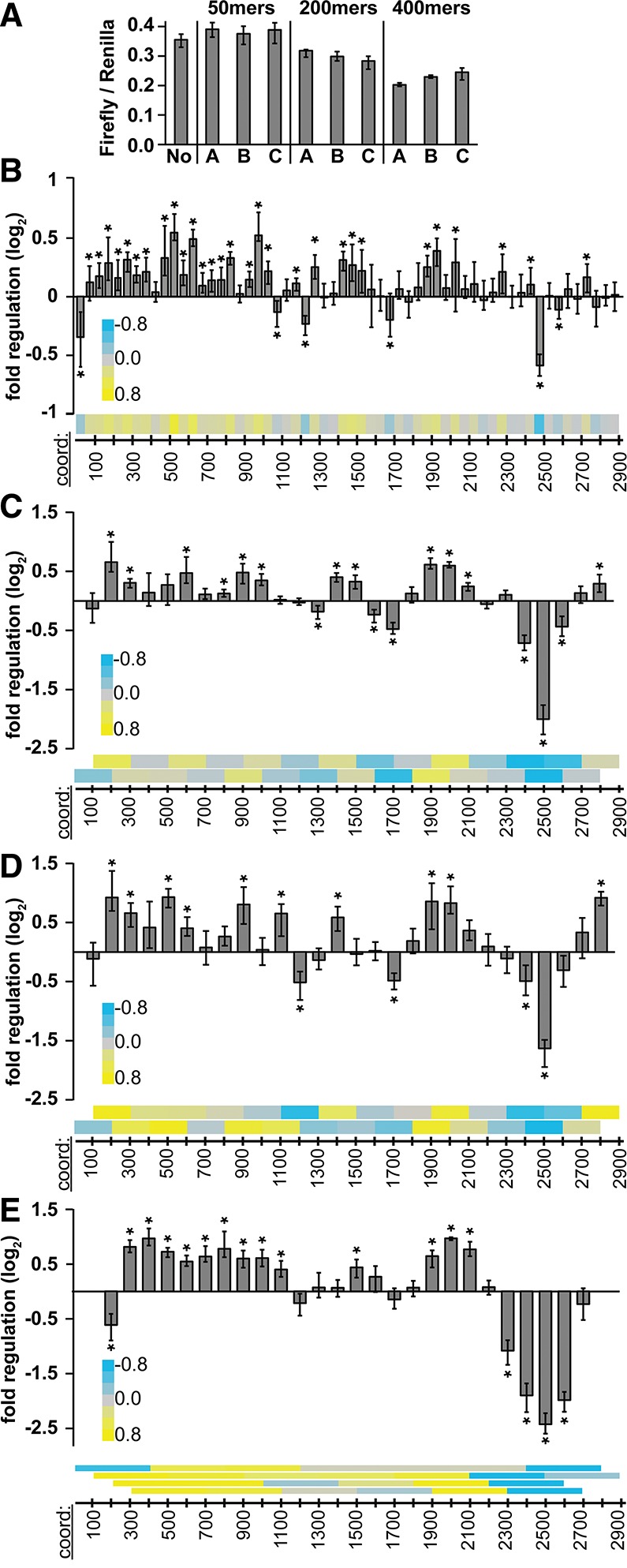
Regulatory sequences in the mouse Hmga2 3′ UTR at different size resolutions. (A) Reporter assays measuring regulatory impact of 50-, 200-, and 400-nt (n = 9) 3′-UTR fragments of random sequence, otherwise as described in Figure 1B. (B–E) Reporter assays measuring regulatory impact of tiled 50mer fragments (n > 12) of mouse Hmga2 3′-UTR sequences (B); 200mer fragments tiled at 100-nt intervals (C,D, mouse and human sequences, respectively); and 400mer mouse 3′-UTR fragments tiled at 100-nt intervals (E). Heatmaps (bottom of each panel) show same data while illustrating tiling strategy (each bar is centered over the middle of the corresponding square in the heatmap). Significance of regulation determined with Bonferroni-corrected Wilcoxon rank-sum tests (n = 9; * indicates P < 0.05), otherwise as described in Figure 1A.
We first assayed our set of mouse Hmga2 100mer reporters in A549 cells (Fig. 1A). As expected, the fragments containing previously identified let-7 miRNA target sites corresponded well to those that significantly down-regulated reporter expression. We confirmed that let-7 was responsible for the repression observed by comparing reporters with and without intact target sites (Fig. 1C). We also measured the total impact of let-7 repression of the full-length Hmga2 3′ UTR by comparing a wild-type full-length construct to a 3′ UTR construct that has all seven let-7 target sites disrupted (m7) (Fig. 1D). Let-7 repression of the full-length matches well with expectation based on the 100mer data. In addition to let-7 containing fragments, only two other fragments contained active repressive elements, the identities of which were not readily apparent. The presence of such elements, however, is not unexpected, as the Hmga2 3′ UTR is considered a strongly repressive sequence (Fig. 1D,J; Borrmann et al. 2001). We observed that many fragments significantly up-regulated reporter expression, a somewhat surprising result given the presence of counteracting repressive elements.
To investigate whether the novel regulatory elements within the Hmga2 3′ UTR are conserved, we performed an equivalent study of both the human and chicken HMGA2 3′ UTRs (83% and 76% identical to mouse, respectively), in which we generated and characterized a complete series of 100mer fragments in a luciferase reporter system (Fig. 1E–G). When generating the human and chicken reporters, the 100mer coordinates were selected based on an alignment of the three 3′ UTRs, allowing us to ensure that comparisons between species were specific to orthologous regions of each 3′ UTR. Importantly, because of the high pair-wise identity, we could confidently identify orthologous positions between the 3′ UTRs. We found significant correlations in regulatory impact of equivalent 3′-UTR fragments between the three species (Fig. 1G), indicating conservation of regulatory sequences. The extent to which regulatory impact deviated between species was not strongly correlated to the sequence divergence (nucleotide diversity) of the corresponding 100mer fragments (Fig. 1H), perhaps suggesting the existence of relatively small conserved regulatory elements that are responsible for the regulatory effect of each fragment. Notably, both human and chicken HMGA2 3′ UTRs also contained multiple positive regulatory sites, providing additional support for their biological importance within the Hmga2 3′ UTR.
To gain additional perspective on the regulatory potential of the Hmga2 3′ UTR in a variety of different trans-factor environments, we repeated our experiments, testing mouse 100mers in a series of different cell lines: mouse 3T3 and F9 cells and human HeLa cells. Although we found instances of cell-type specific differences in regulation, the overall regulatory landscape was extremely consistent between different cell lines (Fig. 1I). The largest exception to this trend was found in F9 cells, a mouse testicular teratoma cell line that should not express let-7 (Zovoilis et al. 2008). Indeed, when we compared the full-length wild-type and let-7 disrupted (m7) reporters in these cell lines, the F9 cells were the only environment where there was not a substantial difference in expression between the constructs (Fig. 1J). Importantly, we selected a range of different types of cells, thus, the majority of the regulatory events detected here are relatively robust to differences in cellular environment.
Map of regulatory sequences within the Hmga2 3′ UTR at different nucleotide resolutions
The regulatory capacity of a 3′ UTR cannot be determined by analyzing 100-nt fragments alone, in part because many bona fide regulatory elements are significantly smaller than 100 nucleotides, and also because some elements may require larger surrounding sequence context to recapitulate regulatory impact found in their native context. To address these concerns, we generated three additional sets of reporters, each of different sizes, derived from the mouse 3′ UTR. As before, we generated and assayed size-matched random-sequence control constructs; as expected, random-sequence reporters of the same size impacted reporter expression equivalently (Fig. 2A), allowing us to establish an appropriate size-matched baseline for each of the three different sets of reporters. Our observation that longer random-sequence controls were more repressive than shorter ones implies a sequence-independent repressive effect with increasing 3′-UTR length; a result consistent with previous observations (Hogg and Goff 2010).
We first generated a 50mer fragment set, which was tiled in nonoverlapping ∼50-nt windows across the entire mouse 3′ UTR (Fig. 2B), maintaining the same boundaries used previously to generate the 100mer reporters (Fig. 1A). Within this high-resolution regulatory map, very few 50mers show repressive effects (6 of 58), and most of these (5 of 6) contain let-7 target sites, consistent with the 100-nt data set. A large number of fragments (29 of 58) mediate significant positive regulatory effects, again consistent with our analysis of the 100mer reporters. Together, these data indicate that the Hmga2 3′ UTR contains at least 35 discrete regulatory elements. It is worth mentioning that the identification of specific regulatory elements within the Hmga2 3′ UTR is greatly facilitated by narrowing the resolution from 100 to 50 nt (e.g., Fig. 6, below).
FIGURE 6.
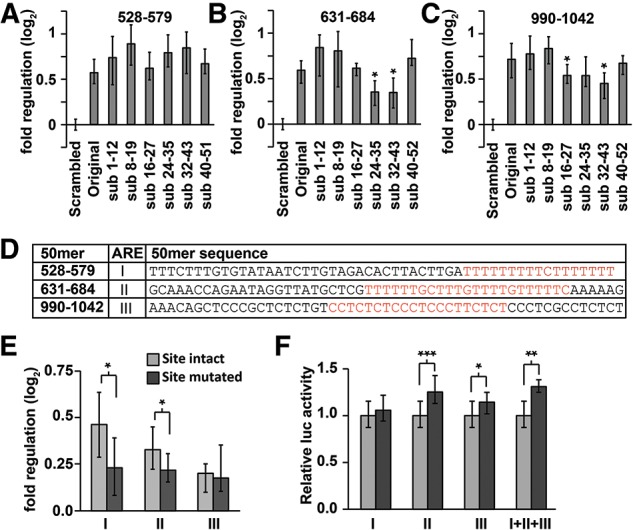
Fine-resolution mapping of positive regulatory sequence elements within the Hmga2 3′ UTR. (A–C) Scanning mutagenesis identifies regulatory sites within 631–684 and 990–1042 50mers (B,C, respectively), but not within 528–579 50mer (A). Reporter assays comparing the original endogenous-sequence 50mers to substitution mutant 50mers, relative to size-matched inert random-sequence controls. Significant reductions (Wilcoxon rank-sum tests) in positive regulatory impact are signified with an asterisk (n = 9). (D) Sequence of three strongest positive 50mers with candidate sequence elements highlighted in red. ARE sequence elements are denoted I, II, and III. (E,F) Reporter assays determining the effect of a targeted deletion of candidate elements from 200mers (E) or full-length Hmga2 (F). Reporter activities of 200mers are shown relative to size-matched random-sequence inert controls; full-length reporters with the targeted deletion are shown relative to an intact Hmga2 3′-UTR reporter. Significant effects on reporter expression were determined using Wilcoxon rank-sum tests (n ≥ 12; P < 0.05, 1 × 10−3, and 1 × 10−5 significance thresholds indicated by *, **, and ***, respectively).
We next generated a 200mer fragment set, which was tiled in overlapping ∼100-nt windows across the entire 3′ UTR using the same boundaries as the original set of 100mers. We generated 200mer sets for both the mouse (Fig. 2C) and human (Fig. 2D) sequences. Multiple Hmga2 200mer fragments also show significant up-regulatory potential and the overall pattern is highly similar between the mouse and human (Pearson correlation coefficient, R = 0.88, P < 1 × 10−6). Finally, we generated an overlapping 400mer fragment set for the mouse 3′ UTR, tiled in ∼100-nt offsets using the same boundaries as both the 100 and 200mer sets (Fig. 2E). Together, these data sets confirm the presence of multiple positive regulatory sequences within the Hmga2 3′ UTR.
It is worth noting that despite the presence of multiple positive sites, the complete Hmga2 3′ UTRs of all three species examined are repressive, when compared with a no-3′ UTR control (22-, 12-, and 29-fold repressed for mouse, human, and chicken, respectively). Moreover, the mouse Hmga2 3′ UTR remains repressive even when let-7 target sites have been disrupted (Fig. 1J). While some, perhaps the majority, of this repression may derive from the 3′ UTRs large size (top 15% for 3′ UTR length in humans), it may also indicate that the regulatory impact of the full-length 3′ UTR is not simply the sum of its component elements.
Nonadditive interactions between neighboring sequence elements are rare within the Hmga2 3′ UTR
Though rare examples of sequence element interactions have been identified, it is not clear how commonly this occurs in mammalian 3′ UTRs. A major motivation for this study was, therefore, to systematically examine a single 3′ UTR for evidence of nonadditive interactions between different regulatory elements. Since the Hmga2 3′ UTR is both long and contains many regulatory sequences (as we have shown above), it is an ideal subject to test how often distinct sequence elements interact. One approach to this question is to compare the regulatory impact of each 3′ UTR fragment to that predicted by the corresponding pairs of smaller fragments. Toward this goal, we modeled the expected regulation of a particular 3′ UTR fragment as the product (log additive) of the regulatory impact of constituent smaller fragments (Fig. 3A). This model assumes that sites act autonomously of one another, each contributing independently to the cumulative regulation observed when sites are combined within a 3′ UTR. Notably, this model recapitulates well experimental measurements of the regulatory impact of multiple miRNA target sites (Grimson et al. 2007). If the observed and the modeled values correlate strongly, this indicates that most sites within the Hmga2 3′ UTR function independently of each other. On the other hand, a failure to correlate could indicate the presence of interacting sequence elements, either synergistic or inhibitory.
FIGURE 3.
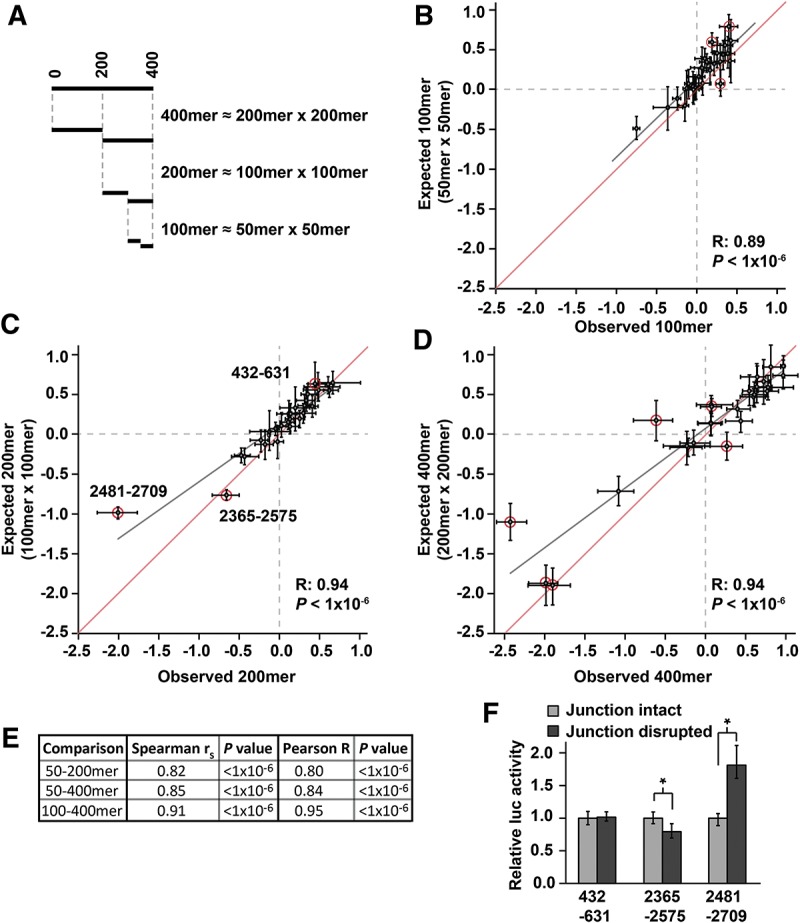
3′-UTR regulatory sequences impact gene expression independently of one another within Hmga2. (A) Depiction of regulatory impact of different 3′-UTR fragments, assuming independent action of regulatory elements. The cumulative effect of regulatory elements within a 3′-UTR fragment is modeled as the product of the regulatory impacts of constituent smaller segments. (B–D) The observed regulatory impact (x-axis) for each 100mer, 200mer, and 400mer fragment compared with the prediction (y-axis) based on the 50mer, 100mer, and 200mer data set, respectively, for the mouse Hmga2 3′ UTR. The gray line represents a Pearson best-fit regression between x- and y-values; for comparison, the red line shows a y = x line. Error bars represent the middle 68% of the data as a nonparametric equivalent to one standard deviation. 3′-UTR fragments that deviate significantly (see Materials and Methods) from the regression are circled in red. (E) Comparison of the observed regulatory impact of 200mer and 400mer 3′-UTR fragments to the prediction using values observed for 50mers (for 200mers and 400mers) and 100mers (for 400mers). Each comparison is summarized with correlation coefficients (Spearman and Pearson) and P values. (F) Sequences located at the junction of adjacent 100mer fragments alter the regulatory behavior of two of the three 200mer fragments whose activity is not well predicted by constituent 100mers (circled red in C). For each 200mer fragment, mutations were introduced at the junction between the two corresponding 100mers. The activity of each mutant derivative was normalized to the original 200mer reporter constructs. Significant difference between intact and disrupted junction constructs was determined with a Wilcoxon rank-sum test (n = 9; * indicates P < 0.05).
We implemented the approach described above at three different resolutions, comparing observed to expected reporter data for 100mer, 200mer, and 400mer fragments using, respectively, 50mer, 100mer, and 200mer reporter data to generate expected values. Strikingly, we found strong and significant correlations between observed and expected values at all three resolutions (Fig. 3B–D, for mouse 100mers, 200mers, and 400mers, respectively). Indeed, these correlations were almost as strong as simulated correlations for perfectly independent sites that take into account the degree of experimental noise intrinsic to the experiments (Rmax = 0.95, 0.99, and 0.99 for 100mers, 200mers, and 400mers, respectively).
As a more strenuous test of the model of site independence, we tested whether observed 50mer reporter data predicted 200mer and 400mer data, and whether observed 100mer data predicted 400mer data. In all three cases, we observed significant (P < 1 × 10−6) and pronounced (R > 0.80) correlations between the directly measured regulatory impact of large fragments and that derived from measurements of multiple constituent fragments (Fig. 3E). These results illustrate that cis-element interactions over short to mid-range distances (up to 400 nt) are rare in Hmga2, and that the 3′-UTR functions as the sum of many independent elements. Additionally, these results indicate that our approach to mapping regulatory sequences within 3′ UTRs is appropriate, as the vast majority of 3′-UTR fragments we examined consistently recapitulate the behavior of larger portions of the 3′ UTR. This final point is most striking when considering that 50mer reporter data captures well (R = 0.80, P < 1 × 10−6) the regulatory impact measured for 400-nt portions of the Hmga2 3′ UTR.
We did observe a small number of fragments with a statistically significant (P < 0.01), but usually small, discrepancy between observed and expected measurements (3 of 29 100mers, 3 of 28 200mers, and 6 of 26 400mers). Such exceptions likely derive from two possible sources. Firstly, they may represent bona fide cases in which two (or more) regulatory elements in adjacent fragments interact with either positive or negative synergism. Secondly, they may arise from technical limitations of our approach. For example, a functional regulatory element located in the middle of a 200mer might be divided between each of the constituent 100mers, and therefore no longer functional in either. To differentiate between these possibilities, we mutated sequences at the center of the deviating 200mers (where the two 100mers would meet) and examined the effect on reporter expression (Fig. 3F). For two of the three deviating 200mers, this mutational analysis indicated the likely existence of a single regulatory element within the 200mer that is disrupted in both constituent 100mers (these results also likely explain certain of the discrepancies between expected and observed 400mers). In contrast, the remaining 200mer may contain a pair of interacting elements. In summary, only one of 28 200mer fragments (of which almost all contain detectable regulatory sequences) showed evidence indicating nonadditive interactions between regulatory elements. Taken together, these data indicate that synergistic interactions between regulatory elements rarely occur within this 3′ UTR, at least within the size resolutions we examined.
The PIM1 3′ UTR contains multiple regulatory elements, which largely function independently
Fundamentally, our comprehensive study of the Hmga2 3′ UTR identified many discrete regulatory sequence elements with minimal nonadditive interactions between neighboring elements. We tested whether these results extend to an additional 3′ UTR by creating a similar data set for the PIM1 3′ UTR, which is relatively well conserved (Wang et al. 2001) but has not been systematically characterized with respect to post-transcriptional biology. As before, we generated both 100- and 200-nt sets of reporters, tiled at 100-nt intervals across the complete PIM1 3′ UTR. Reminiscent of our results with Hmga2, most (8 of 13 100mers and 10 of 12 200mers) of the 3′-UTR fragments mediated significant regulation (Fig. 4A,B). Notably, the regulatory impact of 200mers was again well recapitulated by constituent 100mers (Fig. 4C). Also, as with Hmga2, the full-length PIM1 3′ UTR is repressive (compared with a no-3′-UTR control; Fig. 4D) despite the presence of multiple positive sites, although the extent to which the repression can be attributed to the size of the PIM1 3′ UTR could not be determined. Along with our investigations of regulatory sequences within the Hmga2 3′ UTR, these results imply that the regulatory sequence elements found in 3′ UTRs typically function independently of one another.
FIGURE 4.
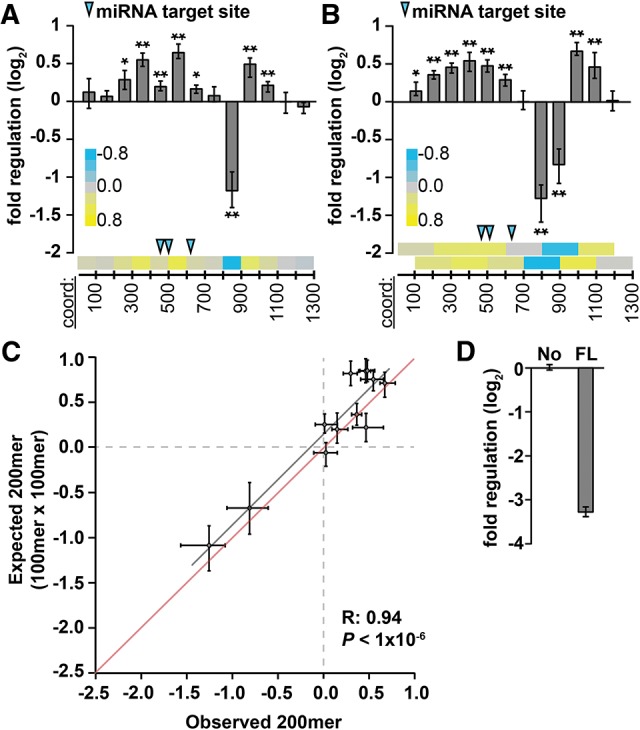
Regulatory sequences impact gene expression independently of one another within the PIM1 3′ UTR. (A,B) Reporter assays measuring regulatory impact of 100- and 200-nt 3′-UTR fragments (A,B, respectively) tiled across the PIM1 3′ UTR at 100-nt intervals (n = 9; P < 0.05 and 1 × 10−3 indicated by * and **, respectively, Bonferroni-corrected Wilcoxon rank-sum tests). Heatmaps (bottom of each panel) show same data while illustrating tiling strategy; blue arrows indicate location of target sites for miRNAs expressed in A549 cells. (C) The measured regulatory impact of each 200mer fragment (observed, x-axis) modeled as the product of the regulatory impact of constituent 100mer fragments (expected, y-axis), for mouse PIM1 3′-UTR sequences, otherwise as described in Figure 3C. (D) The full-length PIM1 3′ UTR (FL) is repressive (normalized to a no-3′ UTR control [No], n = 9).
A role for HuR in mediating regulation within the Hmga2 3′ UTR
When examining the up-regulating Hmga2 50mer sequences, we noted that many contained U-rich sequences consistent with a possible function as AU-rich elements (AREs). AREs interact with multiple different trans-factors and their presence within a transcript can mediate both positive and negative post-transcriptional regulatory effects (for review, see Barreau et al. 2005). HuR is an established ARE-binding protein and is one of the few known to confer positive regulation (Fan and Steitz 1998; Peng et al. 1998). Publicly available PAR-CLIP data (Kishore et al. 2011) indicate several HuR binding sites within the Hmga2 3′ UTR. Moreover, all such sites fall within fragments we identified as containing positive regulatory elements (Fig. 5A). To test whether HuR is required for the observed positive regulation of the Hmga2 3′-UTR fragments, we used RNAi (using lentiviral-delivered shRNAs) to knock down HuR in A549 cells. The efficacy of HuR knockdown was assessed by qRT-PCR (Fig. 5B), identifying two different shRNAs that effectively target HuR. We then performed reporter assays in the HuR knockdown cells to determine the effect on Hmga2 3′-UTR reporter constructs. Inert shRNAs targeting GFP and LacZ served as negative controls for RNAi.
FIGURE 5.
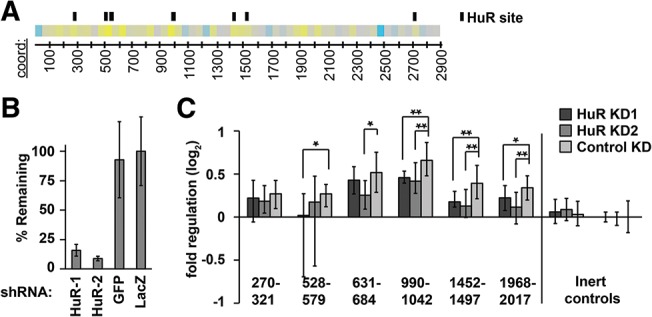
A role for HuR in regulation of Hmga2. (A) HuR binding sites correspond to up-regulating 50mers. Location of HuR binding sites (black boxes) according to available PAR-CLIP data (Kishore et al. 2011) displayed parallel to a heatmap representation of the Hmga2 3′-UTR 50mer reporter data. (B) Evaluation of HuR knockdown. A549 cells were infected with two different shRNA hairpins targeting the HuR mRNA, and the effect on HuR transcript levels determined with qRT-PCR; two different hairpins (targeting GFP and LacZ) were used as negative controls (n = 3). Error bars indicate one standard deviation. (C) Reporter assays of selected Hmga2 50mer reporters, and one random 50mer control reporter, in HuR knockdown cells. Reporter expression, normalized to a second inert random-sequence control (right-most data), was compared between HuR knockdown cells and cells infected with control shRNAs (targeting GFP). Significant changes in reporter expression were determined using Wilcoxon rank-sum tests (n = 12; P < 0.05 and 1 × 10−3 indicated by * and **, respectively).
We found consistent evidence that HuR is required for the positive regulatory impact of multiple 50mer reporter fragments (Fig. 5C). For three different 50mer fragments, both HuR knockdown experiments resulted in significant reductions in positive regulatory impact. For the remaining three 50mer fragments, HuR knockdown with either shRNA consistently impaired the positive regulatory impact of the reporters, but only two of the experiments were statistically significant. Overall, these results suggest a role for HuR in regulating the Hmga2 transcript. Surprisingly, however, the results did not extend to a full-length Hmga2 3′-UTR reporter, as it was only minimally affected by HuR knockdown (data not shown; see Discussion).
Mapping positive regulatory sequence elements within the Hmga2 3′ UTR
To further examine the positive regulatory impact conferred by fragments of the Hmga2 3′ UTR, we performed a scanning mutagenesis analysis of the three 50mer fragments with the strongest positive impact on reporter expression. We first generated a series of reporter constructs in which we replaced 12 nt of endogenous 3′-UTR sequence with 12 nt of inert sequence (derived from our random-sequence controls), with the 12-nt window tiled at 8-nt intervals across each of the three 50mers. We then assayed the reporters, expecting to identify a subset whose ability to mediate increased reporter activity was compromised, thereby identifying the specific nucleotides comprising the positive regulatory elements within these Hmga2 fragments. This strategy was successful for two of the three 50mer reporters (Fig. 6B,C), identifying nucleotides 660–671 and 1009–1029 as the specific sequences required for positive regulation. For the remaining 50mer the results were less clear, possibly indicating the existence of multiple separate regulatory elements (Fig. 6A).
The sequences identified by the scanning mutagenesis as candidate positive regulatory elements coincided well with U-rich or CU-rich sequences (consistent with AREs) (Fig. 6D). Indeed, even the 50mer refractory to this approach (Fig. 6A) contains a similar pyrimidine-rich sequence (Fig. 6D). To determine if U-rich sequences are responsible for these up-regulating effects, we deleted the candidate AREs from 3′-UTR reporters containing either the full-length Hmga2 3′ UTR or 200mer fragments containing the U-rich sequences (Fig. 6D–F). Experiments performed with 200mer fragments were consistent with the mutational analyses, supporting a role for pyrimidine-rich tracts as positive regulatory sequences (Fig. 6E). Surprisingly, removal of poly-pyrimidine tracts within the full-length Hmga2 had an effect of the same magnitude but in the opposite direction (Fig. 6F), suggesting that the same sequences can also function as negative regulatory elements, but only when located within the complete 3′ UTR. Thus, while it was clear that these sites regulate expression, the exact nature of this regulation is remarkably context dependent.
Terminal sequences within the Hmga2 3′ UTR induce a functional switch in candidate AREs from positive to negative regulatory elements
As individual AREs are known to bind many different proteins (for review, see Barreau et al. 2005), it is conceivable that events elsewhere in the 3′ UTR can modulate which ARE-binding protein stably associates with AREs within that same 3′ UTR, resulting in a switch in function. We therefore hypothesized that sites elsewhere in the Hmga2 3′ UTR were responsible for the functional switching of the candidate AREs responsible for up-regulation within 200mer fragments to down-regulation in the full-length Hmga2 3′ UTR (Fig. 6). To identify the region responsible, we generated a truncation series of the full-length Hmga2 3′ UTR, and compared this series with a parallel truncation series in which the ARE at nucleotides 660–671 (ARE II) was deleted. These experiments revealed two regions of the 3′ UTR that alter the behavior of the distal ARE: nucleotides 6–431 and 2481–2855 (Fig. 7A). In 3′-UTR reporters lacking either region, the ARE is rendered inert. Furthermore, we found that for 3′-UTR reporters lacking both regions, the same ARE functions as a positive regulatory element. Any additional truncation had no effect on the function of the ARE sequence. This pattern was recapitulated when tested in the mouse 3T3 cell line (data not shown). Together, these results indicate that distal sequences within the 3′-UTR control the function of at least one ARE located hundreds of nucleotides distant.
FIGURE 7.

Mapping regions involved in switching ARE function. (A) Truncation analysis reveals regions necessary for switching. The effect on reporter expression of ARE II deletion (shown on right) was determined in a set of truncation reporter constructs (illustrated on left with approximate coordinates shown at top). For each pair of truncation constructs, significant differences between otherwise wild-type and ARE II-delete reporters were determined with Wilcoxon rank-sum tests (n = 9; P < 0.05 and 1 × 10−3 indicated by * and **, respectively). The location of ARE II is marked in green and regions that affect ARE II function are marked in pink. (B) ARE II deletion has an equivalent effect on reporter expression in both a wild-type and a let-7-disrupted full-length Hmga2 reporter (Wilcoxon rank-sum tests; n = 9, P > 0.2). (C) Impact of terminal deletions on the overall regulatory effect of the Hmga2 3′ UTR. Full-length Hmga2 3′ UTR (m7) is compared with terminal truncation mutants (all constructs normalized to a no-3′ UTR control) (n = 9). (D) A model showing two possible mechanisms for the ARE II switching phenotype. On the left, a localization model depicts a trans-factor (in blue) binding to localization signals within the terminal regions, leading to a change in the transcripts local environment, thereby altering the set of trans-factors available to bind ARE II. On the right, a protein–protein interaction model depicts a trans-factor (in blue) interacting with sequences within the terminal regions of the 3′ UTR; binding of this factor governs the selection of regulatory proteins bound to ARE II.
Notably, the 3′-UTR regions that affect the distant ARE both contain highly effective let-7 target sites (Fig. 1A; Mayr et al. 2007), suggesting that let-7 targeting might play a role in this phenomenon. To address this possibility, we deleted ARE II from a full-length Hmga2 3′-UTR construct that had all seven let-7 target sites disrupted. Orthogonally, we also examined our truncation reporters (Fig. 7A) in F9 cells, which do not express let-7. If let-7 targeting is necessary for the ARE to convert to a repressive element, this conversion should occur in neither F9 cells nor in reporters containing disrupted let-7 target sites. Instead, our data clearly indicates that loss of let-7 targeting (through loss of either the target site or the miRNA; Fig. 7B and data not shown, respectively) had no effect on the functional switch in the ARE. Thus, the Hmga2 3′ UTR contains unknown terminal regulatory elements whose function appears to render ineffective internal positive regulatory elements, converting them to repressive elements.
To investigate whether terminal regulatory elements induce a more global switching of otherwise positive regulatory elements within the Hmga2 3′ UTR, we directly compared the full-length sequence to reporters containing terminal deletions. In particular, we were interested in whether such experiments could explain the apparent discrepancy between the repressiveness of the full-length 3′ UTR and the prevalence of positive regulatory elements detected in 50–400-nt reporters. To better enable comparisons between the different reporters, each of which contain different numbers of let-7 target sites, we tested only reporters in which the let-7 target sites were disrupted (m7). Although a reporter containing nucleotides 432–2480 of the Hmga2 3′-UTR-mediated regulation equivalent to that of the full-length reporter, the activity of a reporter containing nucleotides 432–1645 was significantly and substantially increased (P < 1 × 10−4; ∼2.4-fold), compared with the full length (Fig. 7C). These results support a model whereby the deleted sequences normally act as control sequences involved in distal element switching. However, it is important to note that such experiments are unable to distinguish between regulation conferred by specific regulatory elements, and the effect of 3′-UTR size, per se (Fig. 2A; Hogg and Goff 2010; Nicholson et al. 2010). In particular, the results highlight sequences within nucleotides 6–431 and 1645–2844 as important for the complete repressive effect observed for the full-length 3′ UTR.
DISCUSSION
Prior to this study, the most widely studied aspect of HMGA2 regulation was the post-transcriptional repression of the Hmga2 transcript by let-7 (Lee and Dutta 2007; Mayr et al. 2007). Notably, however, let-7 was responsible for only ∼15% of the total regulatory effects observed in our study. In fact, our findings illustrate that many previously uncharacterized regions, distributed throughout the Hmga2 3′ UTR, have significant regulatory impact—most of comparable magnitude to the let-7 target sites. The majority of 3′-UTR fragments mediated similar effects when compared between mouse, human, and chicken sequences, a level of conservation likely indicating important biological roles. Moreover, the regulatory map of the 3′ UTR was remarkably robust to different cellular environments, suggesting that the corresponding trans-factors are broadly expressed.
In the study that originally determined that Hmga2 expression was regulated by sequences within its 3′ UTR, Borrmann et al. (2001) characterized it using a strategy based on successive ∼500-nt truncations. Their analysis revealed two negative and a single positive regulatory region, which, while informative, gives an incomplete depiction of the complexity of post-transcriptional regulation of Hmga2. Given the relatively small size of many 3′-UTR regulatory elements and the absence of reliable predictive tools, high-resolution mapping is one of the few appropriate approaches to defining a near-complete set of regulatory sequences within a 3′ UTR. Here, by using this approach, we identify at least 35 distinct regulatory elements within the Hmga2 3′ UTR, a density of ∼12 elements per 1000 nt of 3′-UTR sequence. Given the biological importance of post-transcriptional control of HMGA2 by let-7 (Lee and Dutta 2007; Mayr et al. 2007), these newly identified cis-regulatory regions may also play important roles in development and oncogenesis, through their control of HMGA2 levels.
The extent to which discrete cis-regulatory elements in 3′ UTRs act synergistically (either positively or negatively) is a major unanswered question in post-transcriptional biology. Here, we attempted to answer this question using a methodical search for evidence of synergism within the Hmga2 and PIM1 3′ UTRs. Although our comprehensive analysis was limited to two 3′ UTRs, in both cases our results indicate that such interactions, at least over the distance ranges we examined (up to 400 nt), are not a typical paradigm for cis-elements within 3′ UTRs. Our finding that most sites act independently, if generalizable, will greatly simplify endeavors toward a full understanding of the impact of 3′ UTRs on gene expression. However, our data also indicate that despite an absence of synergistic interactions over short to medium distances, the behavior of sites in the full-length 3′ UTR is not necessarily fully recapitulated by behavior of sites within smaller fragments.
In many cases, synergism can derive from local effects, whereby binding of a trans-factor at one site affects the affinity of a second trans-factor to a neighboring site. This can be accomplished through changes in RNA conformation (Kedde et al. 2010) or through physical interactions between trans-factors, resulting either in increased affinity to sites (Jing et al. 2005) or blocking of a second site through steric hindrance (Kedde et al. 2007). These types of interactions are discoverable by our approach, although we may not capture every instance. Another, perhaps more rare, mode of synergism derives from long-distance interactions. An example would be a site at one end of a 3′ UTR that controls transcript localization (for example to processing bodies); such a change in localization can readily alter the set of trans-factors available to the transcript and thereby alter the function of a cis-element anywhere within a 3′ UTR. Our primary approach is not well suited to identify such interactions, as the maximum fragment size we examined was 400 nt. It is clear that long-range interactions do occur; indeed IGF2BP3 control of the HMGA2 transcript is one such example (Jønson et al. 2014), as is our discovery of distal control of AREs within the Hmga2 3′ UTR. Systematic identification of such interactions requires both knowledge of the regulatory impact of discrete pieces of a 3′ UTR, together with experiments that investigate the roles of identified elements within the complete native 3′ UTR. It remains to be seen how frequent such complex interactions are. It is worth mentioning that even if such interactions are rare, their impact on regulation can be profound, and may even be relatively common in a small subset of 3′ UTRs whose post-transcriptional regulation is particularly critical.
Although the primary focus of this study was to characterize cis-regulatory elements in the Hmga2 3′ UTR, we also showed that disrupting the expression (by RNAi) of the positive regulatory RBP HuR had a significant effect on multiple individual fragments of the Hmga2 3′ UTR. Surprisingly, we were unable to see the same effect on the full-length 3′ UTR. While this may indicate that HuR has a minimal role in Hmga2 regulation, there are other possible explanations. The expression of RBPs that bind AREs (ARE-BPs), including HuR, are known to be extensively interconnected, such that inhibition of one ARE-BP results in complex changes in the levels of other ARE-BPs (Pullmann et al. 2007). For example, repression of HuR led to at least twofold changes in the protein levels of three other ARE-BPs, with KSRP and TIA-1 levels going down and AUF1 going up. Thus, knockdown of HuR may yield interpretable results when focusing on isolated, discrete 3′-UTR fragments that contain only AREs with high affinity to HuR, but results may not be so clear for a 3′-UTR sequence containing multiple AREs, some of which can bind other ARE-BPs. A recent study identified another RBP that has an important role in HMGA2 regulation: Jønson et al. (2014) demonstrated that IGF2BP3 protects let-7 targeted transcripts, and HMGA2 in particular, from miRNA repression by sequestration into “IGF2BP3 granules.” Unfortunately, we were unable to address this aspect of Hmga2 biology, as in our hands, shRNA-mediated inhibition of IGF2BP3 had no effect on full-length Hmga2 3′-UTR reporters (data not shown). This has two likely explanations: It is possible that a stronger knockdown is needed to see an effect, or, it may be that IGF2BP3 does not regulate Hmga2 in A549 cells.
Although our results indicate that interactions between 3′-UTR elements are rare, we did discover one striking exception within Hmga2. In isolation, our data clearly indicate that AREs within the Hmga2 3′ UTR are positive regulatory elements, but in the context of the full-length sequence, the same AREs became repressive elements. We demonstrated that terminal sequences within the 3′ UTR are responsible for the conversion of at least one of the internal AREs from activating to repressive sequence elements. We also showed that this effect was fully independent of let-7 targeting. The most likely explanation for this switch in behavior is a change in the identity of the ARE-BP associated with the site, from HuR to an ARE-BP that mediates repression. We have considered three possible mechanisms that could be driving this interaction. The first model, and the one we favor, is that sub-cellular localization of the Hmga2 transcript is controlled by sequences within the terminal regions of the 3′ UTR; altered localization, in turn, changes the RBPs available for binding to Hmga2 (Fig. 7D, left). The second model posits that physical interactions between trans-factors bound at terminal sequences within the 3′ UTR and ARE-binding proteins control the identity of the specific ARE-BP that binds to the central sites (Fig. 7D, right). The final model is that the native function of the ARE elements are determined by the structural conformation of the Hmga2 3′ UTR, which requires the sequences at both termini. This final possibility seems least likely for two reasons: We can find no plausible predicted RNA structures supporting this model, and terminal truncations at only one end inactivated the internal ARE but did not induce switching. Determining which of these mechanisms is responsible will require further study. While we only demonstrated the switching effect for the three strongest positive regulatory sites (and only mapped the interacting region for one of them), it is possible, indeed likely, that this applies to many of the remaining sites we identified as positive regulatory elements. If true, this may explain the seemingly contradictory identification of multiple positive sites that would counteract the negative effect of let-7 targeting, as well as the apparent contrast between the regulatory effect of the full-length 3′ UTR and the pieces that comprise it. It is easy to imagine how such switchable AREs could contribute to regulation of Hmga2. In adult tissues, when repression of Hmga2 is essential for normal cellular function, the AREs function as repressive sites, along with let-7 target sites. However, in situations where Hmga2 is highly expressed, such as certain embryonic tissues, the same AREs could act as positive regulators of Hmga2 expression.
In summary, our study has three major findings. First, the Hmga2 3′ UTR is crowded with regulatory sequences, many of which were previously unidentified. Second, despite the density of regulatory sequences, synergistic interactions over short to medium distances are exceedingly rare; we identified and validated only one such example, and found that all other adjacent regulatory sequences largely functioned independently. These findings extended to a second 3′ UTR, indicating the discoveries may be generally applicable. If true, the lack of widespread interactions means that useful information can be gathered by studying sequence elements in isolation with limited surrounding sequence context. However, it is clear that complex interactions between elements do occur and can have pronounced effects on the role of a 3′ UTR. This is particularly clear in light of our third discovery: A long-range interaction within Hmga2 reconfigured multiple positive elements to repressive sequence elements. That synergistic interactions appear to be rare within 3′ UTRs suggests that those evolved to contain them likely correspond to genes whose post-transcriptional control is of particular biological importance. The interactions identified to date have commonly involved genes with important roles in cell-cycle control (Kedde et al. 2010), development (Kedde et al. 2007), and cancer (Miles et al. 2012), processes that are generally under very complex control. Though preferences in research topics may account for some of this apparent enrichment, it would be interesting to see if an unbiased search for element interactions would find interesting patterns in which types of genes evolves interacting 3′-UTR elements.
MATERIALS AND METHODS
3′-UTR reporter constructs
Forward and reverse PCR primers were designed at ∼100-nt intervals spanning the entire Hmga2 (mouse, human, and chicken) and PIM1 (human) 3′ UTRs (see Supplemental Tables S1,S2, respectively). Each primer also contained SpeI/NheI (forward) or NotI (reverse) restriction sites to use for cloning. PCR amplicons of ∼100 and ∼200 (Hmga2 and PIM1) and 400 bp (Hmga2 only) were generated using these primers, creating amplicons tiled at ∼100-nt intervals across each 3′ UTR. To generate the 50-nt insert sequences, oligos were designed so that they could anneal to one another, and after primer extension create double-stranded molecules tailed with appropriate restriction sites (see Supplemental Table S3). Each 3′ UTR segment was then inserted downstream from the firefly luciferase coding sequence and upstream of the SV40 late poly(A) signal of Promega's pmirGLO Dual-Luciferase miRNA Target Expression vector, using the NheI and NotI vector restriction sites. The insert sequences and cloning junctions of all resulting constructs were fully sequence-validated.
Random-sequence and no-3′-UTR control reporter constructs
All luciferase reporter experiments were performed using a panel of size-matched reporter constructs in which the sequence of the 3′ UTR was randomly generated. The sequence of the random 100mer 3′ UTRs were generated to mimic the base composition of the Hmga2 3′ UTR (A:29%, U:30%, C:23%, G:18%). These percentages represent the nucleotide ratios in the full-length Hmga2 3′ UTR, and are close to the average composition of all 3′ UTRs (A:27%, U:29%, C:22%, G:22%). Three different random 100mer sequences were generated, and oligonucleotides designed to generate the corresponding double-stranded DNA suitable for cloning into pmirGLO.
Three different 50mer control sequences were derived from 100mer control sequences, and were cloned using oligonucleotide extension, as described above. Three different 200mer control sequences were made by combining different random 100mer controls using overlap extension PCR. Two different random 400mer sequences were designed as described above for random 100mers and one was designed as a combination of two neutral 200mers. All three random 400mers were synthesized as gBlocks (IDT), which were inserted into pmirGLO. See Supplemental Table S4 for random sequences. The no-3′ UTR control was generated by digesting the pmirGLO vector with NheI and NotI and using T4 DNA polymerase to create blunt ends, which were then ligated.
Mutated 3′-UTR reporters
let-7 mutants (Fig. 1C,D,J): 100mer reporters containing disrupted let-7 target sites were PCR amplified from an Hmga2 3′-UTR construct in which all let-7 binding sites were disrupted (Mayr et al. 2007); amplicons were cloned into the pmirGLO vector, as described above. See Supplemental Table S5 for primers.
Junction mutants (Fig. 3F): For fragments with coordinates 432–631 and 2365–2575, 10 nt of 3′ UTR sequence were removed from the region where the two corresponding 100mers would meet and replaced with scrambled versions of the sequences. For 2481–2709, the let-7 target site that is located at the junction between the two corresponding 100mers was disrupted by altering three nucleotides within the seed region. Mutants were generated using overlap extension PCR using the primers in Supplemental Table S6. The altered versions were inserted into pmirGLO, as described above.
Scanning mutagenesis (Fig. 6A–C): For each of the three positive 50mers we generated a set of mutants by replacing 12 nt of endogenous 3′-UTR sequence with 12 nt of inert 3′-UTR sequence (derived from one of our random-sequence controls), with the 12-nt window tiled at 8-nt intervals across each of the three 50mers. Mutant 50mer constructs were generated by oligo annealing and primer extension, as described above for endogenous 50mer sequences (see Supplemental Table S7 for primers).
U-rich sequence deletions (Fig. 6E–F): Candidate U-rich elements were deleted from the full-length Hmga2 (or let-7 disrupted Hmga2) reporter using overlap extension PCR (see Supplemental Table S8 for primers); the resulting amplicons were inserted into pmirGLO, as described above. 200mer reporters containing the same mutations were PCR amplified from the mutated full-length Hmga2 reporters (using primers from Supplemental Table S1) and cloned as described above.
Truncation analysis (Fig. 7A–C): Truncated Hmga2 3′-UTR reporters were generated by PCR amplifying the indicated regions (Fig. 7A) from wild-type, ARE II disrupted and let-7-disrupted full-length Hmga2 3′ UTR reporters using primers from Supplemental Table S1. The resulting amplicons were inserted into pmirGLO, as described above.
The inserts and cloning junctions of all mutated constructs were fully sequence-validated.
Dual-Luciferase reporter assays
Cells were seeded 24 h pre-transfection at densities appropriate for each cell line (70,000 cells/well for A549, 52,500 cells/well for HeLa, 50,000 cells/well for F9 and 3T3 cells) in 24-well plates. A549 and HeLa cells were transfected with Lipofectamine 2000 (Invitrogen); F9 and 3T3 cells were transfected with Lipofectamine LTX and Plus reagent (Invitrogen), following the manufacturer's protocols, and using 2.5–50 ng of pmirGLO-derived reporter plasmids, with the amount determined by the identity of the cell lines, to account for differences in transfection efficiency, and/or inherent differences in expression from the plasmid. Carrier DNA (pUC19) was included to increase transfection efficiency (100–140 ng/well). Cells were harvested 30 h post-transfection by removing the media, washing once with 1× PBS, and frozen at −80°C. Luciferase assays were performed using the Promega Dual-Luciferase Reporter Assay kit and a Veritas Microplate Luminometer (Turner Biosystems) according to manufacturer's protocols. The resulting Firefly values were first normalized to the Renilla values for each individual well, thus controlling for transfection efficiency. Normalized Firefly values were then scaled relative to the geometric mean of normalized firefly values for the appropriate baseline control (i.e., the random-sequence controls for most experiments). The resulting data was plotted as log2 values. The error bars were estimated as the nonparametric equivalent to one standard deviation (∼68% of the data is within the error bars). To determine significant differences between different reporters, a Wilcoxon rank-sum two-sample test was used. Multiple comparison corrections (Bonferroni) were used when appropriate (as indicated).
Quantifying sequence divergence and regulatory impact divergence between mouse, human, and chicken Hmga2 3′ UTRs
Sequence divergence was calculated as previously described (Nei and Li 1979), counting gaps as sequence differences. Divergence in regulatory impact was calculated as the absolute value of the deviation from the mean regulatory impact (across species), summed across all three species.
Calculating expected regulatory potential
We used a Monte Carlo sampling strategy to estimate an expected regulatory impact for a 3′-UTR fragment from reporter values corresponding to two constituent fragments (Figs. 3, 4). We used all individual normalized luciferase values (as described above) for each of the two constituent fragments and randomly sampled one from each distribution and multiplied them. This procedure was repeated 100,000 times, generating a distribution of simulated values, which represent the expected values assuming no regulatory interactions between the two constituent fragments (i.e., the model outlined in Fig. 3A). This distribution of simulated values was then treated equivalently to the distributions of all other reporter data (as described above).
Identifying significant outliers from observed-modeled regressions
We used a Monte Carlo sampling strategy to identify significant deviations in expected regulatory impact (y-axis values in Fig. 3). We simulated individual y-axis values by assuming that the true value lied precisely on the regression line, and then used the experimentally determined variations in reporter data to generate the simulated reporter data for that y-axis value. This procedure was repeated 100,000 times for each reporter fragment, generating a distribution of simulated values, which were used to estimate the probability that the actual value was a significant outlier. This same approach was also used to estimate the maximum possible correlations between observed and expected values for the data shown in Fig. 3.
shRNA knockdown experiments
shRNA hairpin plasmids (The RNAi Consortium, see Supplemental Table S9 for hairpin reference numbers) were used to generate shRNA virus according to TRC protocols (Root et al. 2006). Media supernatant with virus was harvested on day 2 and 3 post transfection and pooled.
A549 cells were plated at 80,000 cells/mL into 6-well plates (three wells per infection) and infected with shRNA virus 24 h later in polybrene media (DMEM 10% FBS, 8 µg/mL polybrene [Sigma]) at an MOI of 4. Cells were selected with puromycin media (DMEM 10% FBS and 3 µg/mL puromycin [Sigma]) and expanded to 75 cm3 flasks. On day 4 post infection, cells were plated for transfection and luciferase assays (as above). Cells were also lysed in TRIzol for RNA isolation, following the manufacturer's recommended protocol (Life Technologies).
qRT-PCR
RNA isolated from shRNA knock down cells was treated with recombinant DNase I (Roche) for 20 min, and then phenol chloroform extracted. cDNA was generated using Thermo Scientific RevertAid Reverse transcriptase and an oligo(dT)18 primer at 42°C for 60 min; the reaction was heat inactivated at 70°C for 10 min. qPCR reactions were performed using Taq polymerase and SYBR Green (Life Technologies) as the detection agent and using GAPDH as a house-keeping gene to which to normalize. For qPCR primers used see Supplemental Table S10. Each qPCR reaction was done in triplicate, and performed on at least two biological replicate samples.
SUPPLEMENTAL MATERIAL
Supplemental material is available for this article.
Supplementary Material
ACKNOWLEDGMENTS
This work was supported by a Research Scholar Grant from the American Cancer Society to A.G. (RSG-13-057-01-RMC), and National Institutes of Health (NIH) grant 1R01GM105668 to A.G. We thank Rae Wilson and Nu Tang for assistance with reporter experiments, Alfred Simkin for assistance with quantifying sequence divergence, and Christine Mayr for full-length wild-type and let-7 disrupted mouse Hmga2 3′ UTRs. We thank Jen Grenier and members of the Grimson laboratory for helpful discussions and suggestions.
Footnotes
Article published online ahead of print. Article and publication date are at http://www.rnajournal.org/cgi/doi/10.1261/rna.051177.115.
Freely available online through the RNA Open Access option.
REFERENCES
- Baltz AG, Munschauer M, Schwanhäusser B, Vasile A, Murakawa Y, Schueler M, Youngs N, Penfold-Brown D, Drew K, Milek M, et al. 2012. The mRNA-bound proteome and its global occupancy profile on protein-coding transcripts. Mol Cell 46: 674–690. [DOI] [PubMed] [Google Scholar]
- Barreau C, Paillard L, Osborne HB. 2005. AU-rich elements and associated factors: Are there unifying principles? Nucleic Acids Res 33: 7138–7150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barrett LW, Fletcher S, Wilton SD. 2012. Regulation of eukaryotic gene expression by the untranslated gene regions and other non-coding elements. Cell Mol Life Sci 69: 3613–3634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bartel DP. 2009. MicroRNAs: target recognition and regulatory functions. Cell 136: 215–233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhattacharyya SN, Habermacher R, Martine U, Closs EI, Filipowicz W. 2006. Relief of microRNA-mediated translational repression in human cells subjected to stress. Cell 125: 1111–1124. [DOI] [PubMed] [Google Scholar]
- Borrmann L, Wilkening S, Bullerdiek J. 2001. The expression of HMGA genes is regulated by their 3′UTR. Oncogene 20: 4537–4541. [DOI] [PubMed] [Google Scholar]
- Broderick JA, Salomon WE, Ryder SP, Aronin N, Zamore PD. 2011. Argonaute protein identity and pairing geometry determine cooperativity in mammalian RNA silencing. RNA 17: 1858–1869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cook KB, Kazan H, Zuberi K, Morris Q, Hughes TR. 2010. RBPDB: a database of RNA-binding specificities. Nucleic Acids Res 39: D301–D308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diab T, Hanoun N, Bureau C, Christol C, Buscail L, Cordelier P, Torrisani J. 2013. The role of the 3′ untranslated region in the post-transcriptional regulation of KLF6 gene expression in hepatocellular carcinoma. Cancers (Basel) 6: 28–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Didiano D, Hobert O. 2008. Molecular architecture of a miRNA-regulated 3′ UTR. RNA 14: 1297–1317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fan XC, Steitz JA. 1998. Overexpression of HuR, a nuclear–cytoplasmic shuttling protein, increases the in vivo stability of ARE-containing mRNAs. EMBO J 17: 3448–3460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fox M, Urano J, Reijo Pera RA. 2005. Identification and characterization of RNA sequences to which human PUMILIO-2 (PUM2) and deleted in Azoospermia-like (DAZL) bind. Genomics 85: 92–105. [DOI] [PubMed] [Google Scholar]
- Friedman RC, Farh KK-H, Burge CB, Bartel DP. 2009. Most mammalian mRNAs are conserved targets of microRNAs. Genome Res 19: 92–105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goodarzi H, Najafabadi HS, Oikonomou P, Greco TM, Fish L, Salavati R, Cristea IM, Tavazoie S. 2012. Systematic discovery of structural elements governing stability of mammalian messenger RNAs. Nature 485: 264–268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grimson A, Farh KK-H, Johnston WK, Garrett-Engele P, Lim LP, Bartel DP. 2007. MicroRNA targeting specificity in mammals: determinants beyond seed pairing. Mol Cell 27: 91–105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hafner M, Landthaler M, Burger L, Khorshid M, Hausser J, Berninger P, Rothballer A, Ascano M Jr, Jungkamp A-C, Munschauer M, et al. 2010. Transcriptome-wide identification of RNA-binding protein and microRNA target sites by PAR-CLIP. Cell 141: 129–141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hogg JR, Goff SP. 2010. Upf1 senses 3′UTR length to potentiate mRNA decay. Cell 143: 379–389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jing Q, Huang S, Guth S, Zarubin T, Motoyama A, Chen J, Di Padova F, Lin S-C, Gram H, Han J. 2005. Involvement of microRNA in AU-rich element-mediated mRNA instability. Cell 120: 623–634. [DOI] [PubMed] [Google Scholar]
- Jønson L, Christiansen J, Hansen TVO, Vikeså J, Yamamoto Y, Nielsen FC. 2014. IMP3 RNP safe houses prevent miRNA-directed HMGA2 mRNA decay in cancer and development. Cell Rep 7: 539–551. [DOI] [PubMed] [Google Scholar]
- Kedde M, Strasser MJ, Boldajipour B, Oude Vrielink JA, Slanchev K, le Sage C, Nagel R, Voorhoeve PM, van Duijse J, Ørom UA, et al. 2007. RNA-binding protein Dnd1 inhibits microRNA access to target mRNA. Cell 131: 1273–1286. [DOI] [PubMed] [Google Scholar]
- Kedde M, van Kouwenhove M, Zwart W, Oude Vrielink JAF, Elkon R, Agami R. 2010. A Pumilio-induced RNA structure switch in p27-3′ UTR controls miR-221 and miR-222 accessibility. Nat Cell Biol 12: 1014–1020. [DOI] [PubMed] [Google Scholar]
- Khaziapoul S, Pearson MJ, Pryme IF, Stern B, Hesketh JE. 2012. CUG binding protein 1 binds to a specific region within the human albumin 3′ untranslated region. Biochem Biophys Res Commun 426: 539–543. [DOI] [PubMed] [Google Scholar]
- Kim HH, Kuwano Y, Srikantan S, Lee EK, Martindale JL, Gorospe M. 2009. HuR recruits let-7/RISC to repress c-Myc expression. Genes Dev 23: 1743–1748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim BC, Lee HC, Lee J-J, Choi C-M, Kim D-K, Lee JC, Ko Y-G, Lee J-S. 2012. Wig1 prevents cellular senescence by regulating p21 mRNA decay through control of RISC recruitment. EMBO J 31: 4289–4303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kishore S, Jaskiewicz L, Burger L, Hausser J, Khorshid M, Zavolan M. 2011. A quantitative analysis of CLIP methods for identifying binding sites of RNA-binding proteins. Nat Methods 8: 559–564. [DOI] [PubMed] [Google Scholar]
- Kundu P, Fabian MR, Sonenberg N, Bhattacharyya SN, Filipowicz W. 2012. HuR protein attenuates miRNA-mediated repression by promoting miRISC dissociation from the target RNA. Nucleic Acids Res 40: 5088–5100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee YS, Dutta A. 2007. The tumor suppressor microRNA let-7 represses the HMGA2 oncogene. Genes Dev 21: 1025–1030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Léveillé N, Elkon R, Davalos V, Manoharan V, Hollingworth D, Oude Vrielink J, le Sage C, Melo CA, Horlings HM, Wesseling J, et al. 2011. Selective inhibition of microRNA accessibility by RBM38 is required for p53 activity. Nat Commun 2: 513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mayr C, Hemann MT, Bartel DP. 2007. Disrupting the pairing between let-7 and Hmga2 enhances oncogenic transformation. Science 315: 1576–1579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Melanson BD, Cabrita MA, Bose R, Hamill JD, Pan E, Brochu C, Marcellus KA, Zhao TT, Holcik M, McKay BC. 2013. A novel cis-acting element from the 3′UTR of DNA damage-binding protein 2 mRNA links transcriptional and post-transcriptional regulation of gene expression. Nucleic Acids Res 41: 5692–5703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miles WO, Tschöp K, Herr A, Ji J-Y, Dyson NJ. 2012. Pumilio facilitates miRNA regulation of the E2F3 oncogene. Genes Dev 26: 356–368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morishita A, Zaidi MR, Mitoro A, Sankarasharma D, Szabolcs M, Okada Y, D'Armiento J, Chada K. 2013. HMGA2 is a driver of tumor metastasis. Cancer Res 73: 4289–4299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nei M, Li WH. 1979. Mathematical model for studying genetic variation in terms of restriction endonucleases. Proc Natl Acad Sci 76: 5269–5273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nicholson P, Yepiskoposyan H, Metze S, Orozco RZ, Kleinschmidt N, Mühlemann O. 2010. Nonsense-mediated mRNA decay in human cells: mechanistic insights, functions beyond quality control and the double-life of NMD factors. Cell Mol Life Sci 5: 677–700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peng SS-Y, Chen C-YA, Xu N, Shyu A-B. 1998. RNA stabilization by the AU-rich element binding protein, HuR, an ELAV protein. EMBO J 17: 3461–3470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pullmann R Jr, Kim HH, Abdelmohsen K, Lal A, Martindale JL, Yang X, Gorospe M. 2007. Analysis of turnover and translation regulatory RNA-binding protein expression through binding to cognate mRNAs. Mol Cell Biol 27: 6265–6278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Root DE, Hacohen N, Hahn WC, Lander ES, Sabatini DM. 2006. Genome-scale loss-of-function screening with a lentiviral RNAi library. Nat Methods 3: 715–719. [DOI] [PubMed] [Google Scholar]
- Sætrom P, Heale BSE, Snøve O Jr, Aagaard L, Alluin J, Rossi JJ. 2007. Distance constraints between microRNA target sites dictate efficacy and cooperativity. Nucleic Acids Res 35: 2333–2342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siepel A, Bejerano G, Pedersen JS, Hinrichs AS, Hou M, Rosenbloom K, Clawson H, Spieth J, Hillier LW, Richards S, et al. 2005. Evolutionarily conserved elements in vertebrate, insect, worm, and yeast genomes. Genome Res 15: 1034–1050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanguay RL, Gallie DR. 1996. Translational efficiency is regulated by the length of the 3′ untranslated region. Mol Cell Biol 16: 146–156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Z, Petersen K, Weaver MS, Magnuson NS. 2001. cDNA cloning, sequencing and characterization of bovine pim-1. Vet Immunol Immunopathol 78: 177–195. [DOI] [PubMed] [Google Scholar]
- Wirsing A, Senkel S, Klein-Hitpass L, Ryffel GU. 2011. A systematic analysis of the 3′UTR of HNF4A mRNA reveals an interplay of regulatory elements including miRNA target sites. PLoS One 6: e27438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie X, Lu J, Kulbokas EJ, Golub TR, Mootha V, Lindblad-Toh K, Lander ES, Kellis M. 2005. Systematic discovery of regulatory motifs in human promoters and 3′ UTRs by comparison of several mammals. Nature 434: 338–345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoon J-H, De S, Srikantan S, Abdelmohsen K, Grammatikakis I, Kim J, Kim KM, Noh JH, White EJF, Martindale JL, et al. 2014. PAR-CLIP analysis uncovers AUF1 impact on target RNA fate and genome integrity. Nat Commun 5: 5248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao W, Pollack JL, Blagev DP, Zaitlen N, McManus MT, Erle DJ. 2014. Massively parallel functional annotation of 3′ untranslated regions. Nat Biotechnol 32: 387–391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zovoilis A, Nolte J, Drusenheimer N, Zechner U, Hada H, Guan K, Hasenfuss G, Nayernia K, Engel W. 2008. Multipotent adult germline stem cells and embryonic stem cells have similar microRNA profiles. Mol Hum Reprod 14: 521–529. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.