

Abstract
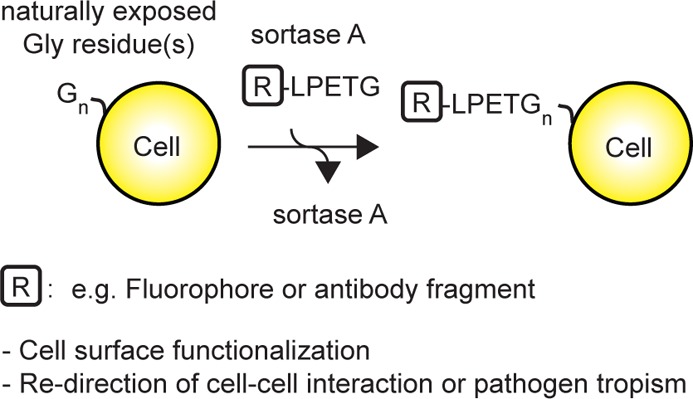
Surface display of engineered proteins has many useful applications. The expression of a synthetic chimeric antigen receptor composed of an extracellular tumor-specific antibody fragment linked to a cytosolic activating motif in engineered T cells is now considered a viable approach for the treatment of leukemias. The risk of de novo tumor development, inherent in the transfer of genetically engineered cells, calls for alternative approaches for the functionalization of the lymphocyte plasma membrane. We demonstrate the conjugation of LPXTG-tagged probes and LPXTG-bearing proteins to endogenous acceptors at the plasma membrane in a single step using sortase A. We successfully conjugated biotin probes not only to mouse hematopoietic cells but also to yeast cells, 293T cells, and Toxoplasma gondii. Installation of single domain antibodies on activated CD8 T cell redirects cell-specific cytotoxicity to cells that bear the relevant antigen. Likewise, conjugation of Toxoplasma gondii with single domain antibodies targets the pathogen to cells that express the antigen recognized by these single domain antibodies. This simple and robust enzymatic approach enables engineering of the plasma membrane for research or therapy under physiological reaction conditions that ensure the viability of the modified cells.
Engineering and functionalization of the eukaryotic cell surface has been achieved through genetic manipulation, covalent modification of glycans1−3 or lipids4,5 as well as by noncovalent modification using bifunctional small molecules6 or antibody moieties.7,8 These approaches enabled visualization of molecules otherwise refractory to genetic engineering (glycans and lipids),3−5 enhancement of antibody functions,6,9 or targeted lymphocyte engagement for therapeutic purposes.8,10 A clinically successful example of cell surface engineering is the viral transduction of human T cells with DNA encoding chimeric antigen receptors (CARs).11 CARs are composed of an extracellularly displayed targeting moiety specific for a tumor-associated antigen, connected to a cytoplasmic signaling domain that drives signal transduction, mimicking physiological receptor engagement. The binding of the target protein on a tumor cell via CAR receptors induces T cell activation, followed by tumor killing via T cell mediated cytoxicity.12 This approach has enjoyed clinical success in the treatment of leukemia.13 Genetic manipulation of cells for therapeutic purposes has drawbacks. Regardless of the vector used, genome modification entails the risk of lymphocyte transformation, and possibly even de novo tumor formation.14 Alternative approaches to functionalize cell surfaces that do not rely on genetic manipulation1,3−5 yet with desirable pharmacokinetic properties should therefore be explored. Direct chemical conjugation to cells of a targeting entity, such as an antibody—or a fragment derived from it—is not straightforward and requires reaction conditions that may be toxic to cells and that could affect the properties of the entity attached. Functional groups or proteins can also be coupled to lipids or other hydrophobic moieties to enable insertion into the plasma membrane,15−17 but the chemistry associated with lipid manipulation can be cumbersome and does not easily lend itself to general use.
Robust methods for covalent modification of cells should be fast, simple, compatible with standard tissue culture media and with most if not all cell types. The transpeptidase sortase A from Staphylococcus aureus conjugates peptides or proteins with (an) exposed N-terminal glycine(s) to a protein or peptide containing an LPTEG motif.18,19 As described below, we show that LPTEG-tagged probes and proteins can be conjugated using sortase A in a single step to glycines naturally exposed at the cell surface. We show that the conjugation of single domain antibodies to CD8 T cells and to Toxoplasma can redirect specific cytotoxicity and infection, respectively.
Results and Discussion
Engineering of the Cell Surface in Absence of Genetic Modification Using Sortase A
We and others have used sortase A from Gram-positive bacteria such as Staphylococcus aureus to conjugate modified probes onto the C-terminus of recombinant LPETG-tagged proteins, in a process referred to as “sortagging” (Figure 1a).20,21 The reaction proceeds as follows: sortase attacks the LPETG tag to cleave between T and G with concomitant formation of a covalent acyl-enzyme intermediate between sortase and the tagged protein.22,23 The covalent acyl-enzyme intermediate is resolved by a nucleophilic attack, using a peptide or protein that carries one or more exposed Gly residues at its NH2-terminus.20 This method can be applied to the modification of type II proteins on the surface of cells22,24 or on virus particles25 through the genetic insertion of a C-terminal sortase recognition tag. In a conceptually similar fashion, LPETG-tagged probes can be attached to the N terminus of NH2-G(n)-modified proteins (Figure 1b). This approach has been used to modify cells that display polyglycine peptides introduced genetically26 or chemically.27 In these cases, residual labeling was observed on unmodified cells, suggesting that exposed glycines might be naturally present on the surface of eukaryotic cells. These residues could therefore act as nucleophiles in the sortase reaction (Figure 1c).26,27 We incubated yeast cells, 293T cells, mouse splenocytes, or Toxoplasma gondii in the presence or absence of biotin-LPETG and sortase A (Figure 1d–g). We monitored conjugation of biotin-LPETG by SDS-PAGE, followed by immunoblotting using streptavidin HRP. We detected numerous streptavidin-reactive polypeptides in lysates of cells that had been incubated with both biotin-LPETG and sortase A (Figure 1d–g). Several endogenously biotinylated proteins28−32 could be detected in yeast lysate in absence of sortase and/or biotin probes (Figure 1d, lane 1 and 2). In silico analysis of yeast, Toxoplasma, human and mouse proteomes revealed a list of potentially modifiable proteins by virtue of the presence of a predicted N-terminal glycine after potential enzymatic removal of a signal peptide or the initiator methionine residue (Supporting Information Table 1). The complexity of this compendium of proteins reflects the expected differences in the protein composition expressed at the cell surface of the respective organisms.
Figure 1.

Sortagging of genetically unmanipulated cells. (a) Conjugation of G(n)-probe to the C-terminus of an LPETG-tagged protein using sortase A. (b) Conjugation of probe-LPETG to the N-terminus of a G(n)-tagged protein using sortase A. (c) Conjugation of LPETG-tagged probe or protein to naturally exposed N-terminal glycine residues at the surface of cells. (d–g) Saccharomyces cerevisiae, Toxoplasma gondii, human embryonic kidney HEK 293 T cells, or total mouse splenocytes from C57BL/6 mice were incubated for 1 h at room temperature (RT) with or without 500 μM of biotin-LPETG and with or without 20 μM of sortase A. Conjugation of the biotin-LPETG probe was analyzed by SDS PAGE followed by immunoblotting using streptavidin-HRP. *Endogenous biotinylated proteins. (h) Total splenocytes from C57BL/6 mice were incubated for 1 h at RT with 500 μM of biotin-LPETG in the presence (dark gray histograms) or absence (light gray histograms) of 20 μM of sortase A. Conjugation of biotin-LPETG was analyzed by flow cytometry using streptavidin–phycoerythrin (PE) together with antibodies specific for T cells (TCRβ), B cells (CD19), or erythrocytes (Ter119). (i) Erythrocyte-depleted splenocytes were incubated with 20 μM sortase A and 500 μM biotin-LPETG for the indicated times. Cells were washed and incubated with streptavidin-PE and analyzed by flow cytometry. Scatter plots show the mean fluorescence intensity of streptavidin-PE staining for each time point, normalized to maximum staining (120 min).
The mouse spleen is composed mainly of T cells, B cells and erythrocytes in addition to smaller numbers of myeloid cells. To investigate whether each subset was subject to modification, we monitored the installation of biotin-LPETG by flow cytometry, using fluorescently labeled streptavidin together with cell type-specific antibodies. Biotin-LPETG probes labeled T and B cells equally well and erythrocytes slightly less efficiently (Figure 1h). We measured by flow cytometry the kinetics with which biotin-LPETG was conjugated to erythrocyte-depleted splenocytes. Conjugation reached ∼30% of maximum after 5 min and ∼60% of maximum after 15 min (Figure 1i). Collectively, our data show that all cells tested were efficiently sortagged in a time frame compatible with biological experiments. Presumably most cells have naturally exposed glycines at their cell surface and will therefore be amenable to sortagging.
Redirection of T Cell Cytotoxicity through Surface Conjugation of Single Domain Antibodies
Expression of tumor-specific chimeric antigen receptors at the surface of CD8 T cells is emerging as practical approach to eradicate leukemic cells.13 To investigate whether LPETG-tagged single domain antibodies (VHH) could be attached to the surface of activated CD8 T cells by means of sortagging, we incubated cytotoxic OTI CD8 T cells with increasing concentrations of a GFP-specific (“enhancer”)33 or a mouse class II MHC-specific (“VHH7”)34 single domain antibody in the presence or absence of sortase A. To monitor installation of VHHs as well as assess their specificity in a cell-bound format, we measured the ability of cells to bind GFP by flow cytometry. Only cells incubated with enhancer-LPETG and sortase A bound recombinant GFP in a dose-dependent fashion (Figure 2a). The concentration of VHHs used for the reaction is proportional to the number of VHHs installed. We estimated the number of VHHs installed per cell by measuring the number of bound GFP molecules by SDS-PAGE and immunoblotting, using a solution of GFP of known concentration as standard (Figure 2b). Sortagging of T cells in the presence of 500 μM of VHHs and sortase A resulted in the conjugation of ∼1 million VHHs per cell. To investigate whether two different probes could be installed on lymphocytes, we incubated erythrocytes-cell depleted splenocytes with or without enhancer-LPETG for 60 min, followed by the addition of biotin-LPETG to the reaction for 15 min (Figure 2c). Cells incubated with enhancer-LPETG prior to biotin-LPETG had similar amounts of surface-conjugated biotin compared to cells incubated with biotin-LPTEG alone (Figure 2c). These data suggest that sortagging of VHHs to cells only minimally affect subsequent conjugation of biotin-LPETG. The smaller LPETG-tagged probes may have more ready access to surface-displayed nucleophiles left unoccupied by larger LPETG-tagged proteins.
Figure 2.

Installation of VHHs on mouse lymphocytes. (a) In vitro activated CD8 T cells from OTI RAG–/– mice were incubated for 1 h at RT with or without 500, 50, or 5 μM of enhancer-LPETG or VHH7-LPETG and with or without 20 μM of sortase A. Control or sortagged cells were incubated with purified GFP. Binding of GFP was analyzed by flow cytometry. (b) Control or sortagged cells were incubated with purified GFP. The amount of bound GFP was estimated by analysis of cell lysates by SDS-PAGE and immunoblotting against GFP and comparing the resultant signal to a GFP standard (right lanes). (c) Erythrocyte-depleted splenocytes were incubated with or without 500 μM enhancer-LPETG and 20 μM sortase A. After 60 min, 500 μM biotin-LPETG was added to reactions where indicated for a further 15 min. Dot plots show the binding of APC-conjugated streptavidin and GFP by sortagged cells after washing. (d) In vitro activated CD8 T cells from OTI RAG–/– mice were incubated for 1 h at RT with or without 500, 50, or 5 μM of enhancer-LPETG or VHH7-LPETG and 20 μM of sortase A. Sortagged cells were incubated with splenocytes from WT mice for 20 h. Histograms show the percentage of propidium iodide-negative CD4 and CD19 cells, compared to cells incubated with control activated OTI CD8 T cells. Error bars: standard deviation (n = 3). (e) CAR T cells are genetically engineered to express a synthetic receptor composed of an extracellular single-chain variable fragment and one or several cytoplasmic activating motifs that mediate signal transduction and T cell activation upon antigen binding. (f) Sortase-mediated conjugation of VHHs on activated T cells affords redirection of cytotoxicity toward cells expressing the targeted antigen.
We next addressed whether the conjugation of VHHs to activated OTI T cells enables cell-specific killing toward cells that bear the antigen for which the VHH is specific. Co-incubation of preactivated OTI T cells sortagged using the anti-class-II MHC VHH7 with mouse splenocytes resulted in specific killing of class II MHC-positive B cells but not class II MHC negative CD4 T cells (Figure 2d). Specific cytotoxicity showed a direct correlation with the number of conjugated VHHs. VHHs can thus be efficiently sortagged onto T cells with full retention of the VHH’s antigen binding capacity. The sortagging reaction preserved cytotoxic functions and allowed redirected killing based on VHH specificity, in a fashion analogous to, but conceptually distinct from CAR T cells (Figure 2e and f). We have installed 2 different probes on the surface of lymphocytes (Figure 2c), and there is no reason to assume that two is the limit to the number of different proteins that can be attached to T cells in this manner. Our approach opens new possibilities in the functionalization of (T) lymphocytes with cytotoxic and targeting moieties to achieve enhanced and specific tumor killing.
This enzymatic approach has several key advantages over current methods. First, because this method does not rely on genetic manipulation of the lymphocyte, it reduces genetic hazards and simplifies the process of functionalization of virtually any cell type. Second, the extent of modification can be controlled, and last, the temporary nature of these modifications, dictated by the lifespan of individual activated lymphocytes, may help to prevent undesired chronic response against healthy cells that express the targeted antigen.
Cell-Specific Targeting of Toxoplasma gondii
The modification of CD8 T cells through sortagging does not obviously interfere with cytotoxic functions (Figure 2d). We decided to extend these methods to the manipulation of other cell–cell interactions. To investigate whether modified T. gondii tachyzoites would still be able to invade host cells, we sortagged parasites with TAMRA-modified LPETG peptides and incubated them together with human fibroblasts. Sortagged parasites visualized by fluorescence microscopy were perfectly capable of invading fibroblasts (Figure 3a and Supporting Information Movie 1). The image shows an intracellular parasite with a distinctly labeled plasma membrane (yellow arrow) and a parasite in the process of invasion characterized by a bright arrow pointed at the constricted moving junction (white arrows).35 To address whether T. gondii could be targeted to specific cells, we sortagged parasites with biotin, biotin plus enhancer or VHH7, followed by incubation with WT splenocytes. Sortagging of VHH7 to T. gondii not only resulted in a dramatic increase of B cells targeted by the parasite together with a significant decrease of binding to non-B cells (Figure 3b) but also enhanced the percentages of B cells lysed upon infection (Figure 3c). The possibility of targeting genetically engineered cytolytic pathogens to cancer cells has been explored by others.36,37 In principle, the approach described here lends itself to similar strategies without the need of genome modifications: this approach is compatible not only with the majority of Gram-positive bacteria that already use sortase enzymes but also with parasites (Figure 3) and possibly with viruses.
Figure 3.
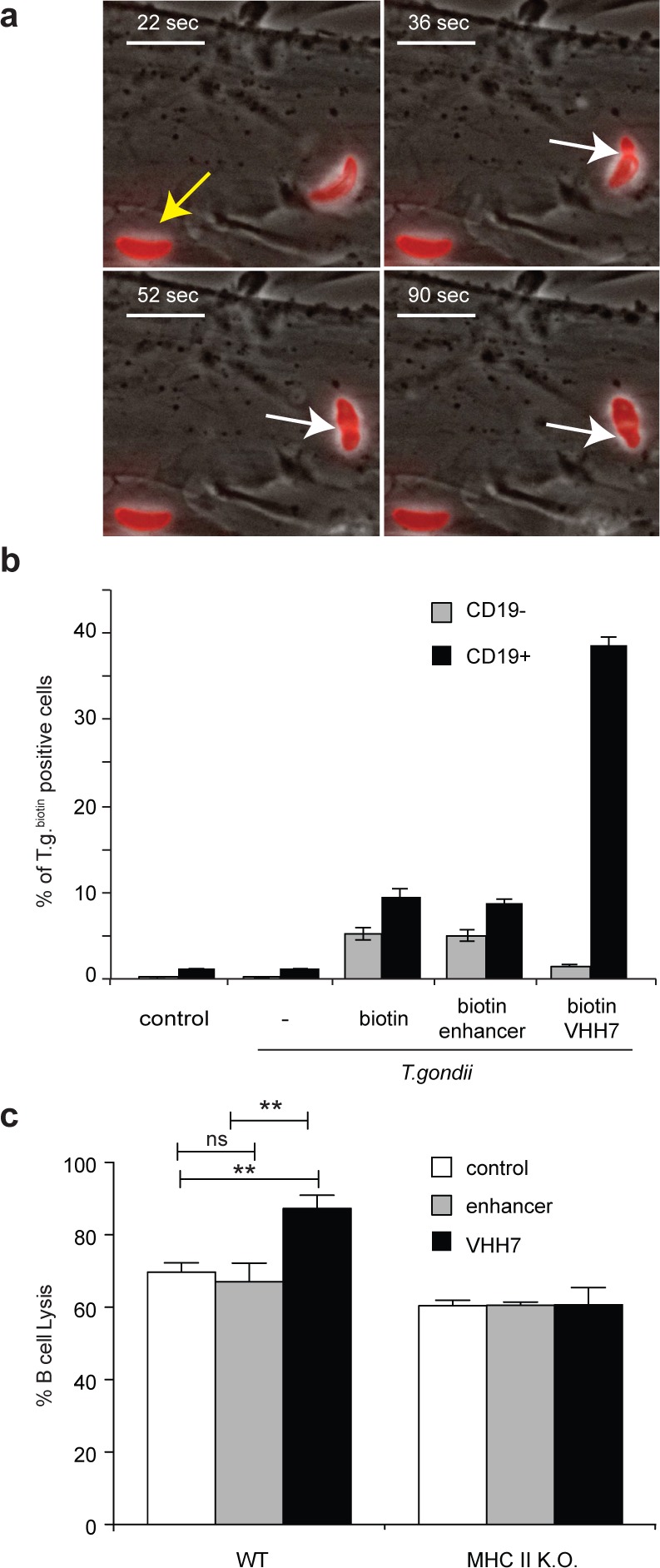
Toxoplasma gondii sortagging. (a) Toxoplasma gondii tachyzoites were incubated with 500 μM TAMRA-LPETG and 20 μM sortase A for 15 min. Parasites were then washed and incubated with human foreskin fibroblasts. Images show the juxtaposition of bright and fluorescent fields. Yellow arrow: intracellular parasite. White arrows: invading parasite. Scale bar: 10 μm. (b) Toxoplasma gondii tachyzoites were incubated with or without 50 μM enhancer- or VHH7-LPETG and 20 μM sortase A. After 20 min, biotin-LPETG was added for 15 min. Parasites were then washed and incubated with red-cell depleted splenocytes for 1 h at a multiplicity of infection of 5. Cells were then washed and stained with a CD19-specific antibody and fluorescently labeled streptavidin. The histogram shows the percentage of sortagged Toxoplasma gondii (T.g.) positive cells within CD19 negative or positive populations. Error bars: standard deviation (n = 3). (c) Purified B cells from WT or class II MHC knock out (k.o.) mice were incubated together with control T. gondii or T. gondii sortagged with enhancer or VHH7 at a multiplicity of infection of 5. Fifteen hours after infection, cell lysis was measured and normalized to uninfected (0%) and detergent-lysed B cells (100%). Error bars: standard deviation (n = 3). **p < 0.01 at Student t test.
Regardless of the utility of sortagging to endow lymphocytes with novel recognition specificities for therapeutic applications, the presence of endogenous nucleophiles also provides a mild enzymatic method to install any entity of choice on the surface of a living cell without the need for genetic modification or the use of harsh chemical conditions. Identification at the molecular level of the substrates to which the label is attached may not be required, and the ease of surface modification under mild and physiological conditions compares favorably with methods that employ radioisotopes or those requiring reaction conditions that ensure reactivity of chemical tags with primary amines or thiols at the cell surface. Sortase-based modifications can be performed in protein-containing buffers and at pH values that would preclude the use of standard chemical labeling strategies.
Methods
Mice
C57BL/6, class II MHC deficient mice and OTI RAG deficient mice38 were purchased from Jackson laboratory, bred in the animal facility of the Whitehead Institute for Biomedical Research (Cambridge, MA) and maintained according to protocols approved by the MIT Committee on Animal Care.
Cell Culture
Erythrocytes cells were depleted from total mouse splenocytes using red blood cell lysis buffer (Sigma, cat. R7757). Mouse lymphocytes were cultured in RPMI 1640 (cat. 11875; Gibco) supplemented with 10% (v/v) inactivated FCS (Gibco), 0.0002% β-mercaptoethanol final (Sigma, cat. M7522), penicillin 50 units per liter–streptomycin 50 mg per liter (Sigma, cat. P4333), 1 mM sodium pyruvate final (Gibco, cat. 11360) nonessential amino acids (Life Technologies, cat. 11140) and 2 mM glutamine (US Biological, cat. G7120). HEK 293T cells were cultured in Dulbecco’s Modified Eagle’s Medium supplemented with 10% (v/v) inactivated FBS (Gibco). Saccharomyces cerevisiae strain W303 was cultured in YPD medium. Toxoplasma gondii (RH strain) tachyzoites were grown in human foreskin fibroblasts (HFF) cultured in Dulbecco’s Modified Eagles Medium (DMEM; Invitrogen) supplemented with 10% tetracycline-free FBS (HyClone), 2 mM glutamine, 10 mM HEPES (pH 7.5), and 20 μg/mL gentamicin.
Antibodies and Reagents
Anti-PGK (clone 22C5D8, Invitrogen), antimouse/human actin (clone Ab-5, BD biosciences), anti-Toxoplasma gondii actin,39 horseradish peroxidase-conjugated goat antirabbit Ig (Southern Biotech, cat. 4041-05), horseradish peroxidase-conjugated antimouse Ig (GE Healthcare, cat. NXA931), anti-TCRβ (clone H57, BD Pharmingen), anti-CD4 (clone GK1.5, ebiosciences), anti-CD19 (clone 1D4, BD Pharmingen), anti-TER119 (clone TER-119, BD Pharmingen), allophycocyanin-conjugated streptavidin (ebiosciences, cat. number 17-4317), phycoerythrin-conjugated streptavidin (Southern Biotech, cat. number 7100-09S). Propidium iodide (Sigma-Aldrich, cat. number P4864). B cell isolation kit (Miltenyi, cat. 130-090-862). For the experiments described here, we used a modified form of sortase A from Staphylococcus aureus that lacks the first 59 residues, and with the following mutations: E105 K/E108A40 and P94R/D160N/D165A/K190E/K196T:24 Sortase A was produced and purified as described elsewhere.20 Enhancer33 and VHH734 were produced and purified as described elsewhere.34 Biotin-LPETG (Biotin- aminohexanoic acid-LPETGG) was produced by the MIT biopolymer facility through standard solid phase peptide synthesis. TAMRA-LPETG was produced as described elsewhere.20
Cell Sortagging
Unless described otherwise, reactions were performed at room temperature (RT) for 1 h in PBS or in HHE buffer (Hanks Balanced Salt Solution, 1 mM EDTA, 25 mM HEPES pH 7). Sortase A was used at a final concentration of 20–40 μM and LPETG substrates at 500 μM unless indicated otherwise. Mouse splenocytes or lymphocytes were used at 20–100 million cells per milliliter, HEK 293T cells at 20 million per milliliter, Toxoplasma gondii at 200 to 400 million per milliliter, yeast at 6 OD 280 units per milliliter. For biochemical analysis, enzymatically modified cells were washed twice with PBS after sortagging and lysed in 1× reducing Laemmli sample buffer.
In Vitro Cytotoxicity Assay
Pooled lymph nods cells and erythrocytes-depleted splenocytes from OTI rag deficient mice were seeded in complete RPMI at 2 million cells per milliliter in 24 well plate previously coated with 2 μg/mL anti-CD3 (clone 17A2) and 2 μg/mL anti-CD28 (clone 37–51). After 72 h, cells were washed twice with PBS and sortagged for 1 h in PBS as described above and the figure legends. Cells were then washed once with PBS and once with complete RPMI. 1–2 × 105 sortagged or control cells were incubated together with 2 × 105 WT splenocytes in complete RPMI in U bottom 96 well plates. After 16 to 20 h cells the percentage of living propidium iodide negative CD4 positive and CD19 positive cells were measured by flow cytometry using fluorescently labeled antibodies specific for CD4 and CD19 together with propidium iodide (final concentration 1 μg/mL).
Toxoplasma gondii Invasion Assay
Toxoplasma gondii tachyzoites were incubated with 500 μM TAMRA-LPETG and 20 μM sortase A at RT for 15 min in HHE buffer. Parasites were then washed and incubated with HFF. Parasite targeting to class II MHC positive cells: Toxoplasma gondii tachyzoites were incubated with or without 50 μM enhancer- or VHH7-LPETG and 20 μM sortase A at RT in HHE buffer. After 20 min, biotin-LPETG was added for 15 min. Parasites were then washed and incubated with red-cell depleted splenocytes for 1 h at a multiplicity of infection of 5. Cells were then washed and stained with a CD19-specific antibody and fluorescently labeled streptavidin. Cells were fixed (BD Cytofix/Cytoperm, cat. 554722) prior to analysis by flow cytometry. B cells lysis assay: Toxoplasma gondii tachyzoites were incubated with or without 50 μM enhancer- or VHH7-LPETG and 20 μM sortase A at RT in HHE buffer for 15 min. After washing T. gondii was incubated together with 0.5 million magnetic beads-purified splenic B cells from WT or class II MHC k.o. at a multiplicity of infection of 5 in 100 μL or complete RPMI in 96 flat bottom well plates. After 15 h supernatants were harvested and cell lysis was measured using CytoTox 96 Non-Radioactive Cytotoxicity Assay kit (Promega, cat. G1781) according to manufacturer’s instructions.
Acknowledgments
This work was funded by a R21 grant from the National Health Institute (CA184568). L.K.S. acknowledges the Swiss Foundation for Grants in Biology and Medicine (SFGBM). Toxoplasma work was supported in part by National Institutes of Health (NIH) grant 1DP5OD017892 to S.L.
Supporting Information Available
Table 1, Movie 1, and additional methods. This material is available free of charge via the Internet at http://pubs.acs.org.
The authors declare the following competing financial interest(s): Hidde L. Ploegh is a founder of 121 Bio.
Funding Statement
National Institutes of Health, United States
Supplementary Material
References
- Mahal L. K.; Yarema K. J.; Bertozzi C. R. (1997) Engineering chemical reactivity on cell surfaces through oligosaccharide biosynthesis. Science 276, 1125–1128. [DOI] [PubMed] [Google Scholar]
- Prescher J. A.; Dube D. H.; Bertozzi C. R. (2004) Chemical remodelling of cell surfaces in living animals. Nature 430, 873–877. [DOI] [PubMed] [Google Scholar]
- Chang P. V.; Bertozzi C. R. (2012) Imaging beyond the proteome. Chem. Commun. 48, 8864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jao C. Y.; Roth M.; Welti R.; Salic A. (2009) Metabolic labeling and direct imaging of choline phospholipids in vivo. Proc. Natl. Acad. Sci. U.S.A. 106, 15332–15337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neef A. B.; Schultz C. (2009) Selective fluorescence labeling of lipids in living cells. Angew. Chem., Int. Ed. Engl. 48, 1498–1500. [DOI] [PubMed] [Google Scholar]
- McEnaney P. J.; Parker C. G.; Zhang A. X.; Spiegel D. A. (2012) Antibody-recruiting molecules: An emerging paradigm for engaging immune function in treating human disease. ACS Chem. Biol. 7, 1139–1151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mølhøj M.; Crommer S.; Brischwein K.; Rau D.; Sriskandarajah M.; Hoffmann P.; Kufer P.; Hofmeister R.; Baeuerle P. A. (2007) CD19-/CD3-bispecific antibody of the BiTE class is far superior to tandem diabody with respect to redirected tumor cell lysis. Mol. Immunol. 44, 1935–1943. [DOI] [PubMed] [Google Scholar]
- Baeuerle P. A.; Reinhardt C. (2009) Bispecific T-cell engaging antibodies for cancer therapy. Cancer Res. 69, 4941–4944. [DOI] [PubMed] [Google Scholar]
- Parker C. G.; Domaoal R. A.; Anderson K. S.; Spiegel D. A. (2009) An antibody-recruiting small molecule that targets HIV gp120. J. Am. Chem. Soc. 131, 16392–16394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mack M.; Riethmüller G.; Kufer P. (1995) A small bispecific antibody construct expressed as a functional single-chain molecule with high tumor cell cytotoxicity. Proc. Natl. Acad. Sci. U.S.A. 92, 7021–7025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sadelain M.; Brentjens R.; Rivière I. (2009) The promise and potential pitfalls of chimeric antigen receptors. Curr. Opin. Immunol. 21, 215–223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sadelain M.; Brentjens R.; Rivière I. (2013) The basic principles of chimeric antigen receptor design. Cancer Discovery 3, 388–398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grupp S. A.; Kalos M.; Barrett D.; Aplenc R.; Porter D. L.; Rheingold S. R.; Teachey D. T.; Chew A.; Hauck B.; Wright J. F.; Milone M. C.; Levine B. L.; June C. H. (2013) Chimeric antigen receptor-modified T cells for acute lymphoid leukemia. N. Engl. J. Med. 368, 1509–1518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hacein-Bey-Abina S.; Von Kalle C.; Schmidt M.; McCormack M. P.; Wulffraat N.; Leboulch P.; Lim A.; Osborne C. S.; Pawliuk R.; Morillon E.; Sorensen R.; Forster A.; Fraser P.; Cohen J. I.; de Saint Basile G.; Alexander I.; Wintergerst U.; Frebourg T.; Aurias A.; Stoppa-Lyonnet D.; Romana S.; Radford-Weiss I.; Gross F.; Valensi F.; Delabesse E.; Macintyre E.; Sigaux F.; Soulier J.; Leiva L. E.; Wissler M.; Prinz C.; Rabbitts T. H.; Le Deist F.; Fischer A.; Cavazzana-Calvo M. (2003) LMO2-associated clonal T cell proliferation in two patients after gene therapy for SCID-X1. Science 302, 415–419. [DOI] [PubMed] [Google Scholar]
- Tomita U.; Yamaguchi S.; Maeda Y.; Chujo K.; Minamihata K.; Nagamune T. (2013) Protein cell-surface display through in situ enzymatic modification of proteins with a poly(ethylene glycol)-lipid. Biotechnol. Bioeng. 110, 2785–2789. [DOI] [PubMed] [Google Scholar]
- Tatsumi K.; Ohashi K.; Teramura Y.; Utoh R.; Kanegae K.; Watanabe N.; Mukobata S.; Nakayama M.; Iwata H.; Okano T. (2012) The non-invasive cell surface modification of hepatocytes with PEG-lipid derivatives. Biomaterials 33, 821–828. [DOI] [PubMed] [Google Scholar]
- Antos J. M.; Miller G. M.; Grotenbreg G. M.; Ploegh H. L. (2008) Lipid modification of proteins through sortase-catalyzed transpeptidation. J. Am. Chem. Soc. 130, 16338–16343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ton-That H.; Liu G.; Mazmanian S. K.; Faull K. F.; Schneewind O. (1999) Purification and characterization of sortase, the transpeptidase that cleaves surface proteins of Staphylococcus aureus at the LPXTG motif. Proc. Natl. Acad. Sci. U.S.A. 96, 12424–12429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Navarre W. W.; Schneewind O. (1999) Surface proteins of gram-positive bacteria and mechanisms of their targeting to the cell wall envelope. Microbiol. Mol. Biol. Rev. 63, 174–229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guimaraes C. P.; Witte M. D.; Theile C. S.; Bozkurt G.; Kundrat L.; Blom A. E. M.; Ploegh H. L. (2013) Site-specific C-terminal and internal loop labeling of proteins using sortase-mediated reactions. Nat. Protoc. 8, 1787–1799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsukiji S.; Nagamune T. (2009) Sortase-mediated ligation: A gift from Gram-positive bacteria to protein engineering. ChemBioChem 10, 787–798. [DOI] [PubMed] [Google Scholar]
- Popp M. W.; Antos J. M.; Grotenbreg G. M.; Spooner E.; Ploegh H. L. (2007) Sortagging: A versatile method for protein labeling. Nat. Chem. Biol. 3, 707–708. [DOI] [PubMed] [Google Scholar]
- Aulabaugh A.; Ding W.; Kapoor B.; Tabei K.; Alksne L.; Dushin R.; Zatz T.; Ellestad G.; Huang X. (2007) Development of an HPLC assay for Staphylococcus aureus sortase: Evidence for the formation of the kinetically competent acyl enzyme intermediate. Anal. Biochem. 360, 14–22. [DOI] [PubMed] [Google Scholar]
- Chen I.; Dorr B. M.; Liu D. R. (2011) A general strategy for the evolution of bond-forming enzymes using yeast display. Proc. Natl. Acad. Sci. U.S.A. 108, 11399–11404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Popp M. W.-L.; Karssemeijer R. A.; Ploegh H. L. (2012) Chemoenzymatic site-specific labeling of influenza glycoproteins as a tool to observe virus budding in real time. PLoS Pathog. 8, e1002604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi J.; Kundrat L.; Pishesha N.; Bilate A.; Theile C.; Maruyama T.; Dougan S. K.; Ploegh H. L.; Lodish H. F. (2014) Engineered red blood cells as carriers for systemic delivery of a wide array of functional probes. Proc. Natl. Acad. Sci. U.S.A. 111, 10131–10136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ta H. T.; Prabhu S.; Leitner E.; Jia F.; von Elverfeldt D.; Jackson K. E.; Heidt T.; Nair A. K. N.; Pearce H.; von zur Muhlen C.; Wang X.; Peter K.; Hagemeyer C. E. (2011) Enzymatic single-chain antibody tagging: A universal approach to targeted molecular imaging and cell homing in cardiovascular disease. Circ. Res. 109, 365–373. [DOI] [PubMed] [Google Scholar]
- van Werven F. J.; Timmers H. T. M. (2006) The use of biotin tagging in Saccharomyces cerevisiae improves the sensitivity of chromatin immunoprecipitation. Nucleic Acids Res. 34, e33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brewster N. K.; Val D. L.; Walker M. E.; Wallace J. C. (1994) Regulation of pyruvate carboxylase isozyme (PYC1, PYC2) gene expression in Saccharomyces cerevisiae during fermentative and nonfermentative growth. Arch. Biochem. Biophys. 311, 62–71. [DOI] [PubMed] [Google Scholar]
- Hoja U.; Marthol S.; Hofmann J.; Stegner S.; Schulz R.; Meier S.; Greiner E.; Schweizer E. (2004) HFA1 encoding an organelle-specific acetyl-CoA carboxylase controls mitochondrial fatty acid synthesis in Saccharomyces cerevisiae. J. Biol. Chem. 279, 21779–21786. [DOI] [PubMed] [Google Scholar]
- Kim H. S.; Hoja U.; Stolz J.; Sauer G.; Schweizer E. (2004) Identification of the tRNA-binding protein Arc1p as a novel target of in vivo biotinylation in Saccharomyces cerevisiae. J. Biol. Chem. 279, 42445–42452. [DOI] [PubMed] [Google Scholar]
- Hasslacher M.; Ivessa A. S.; Paltauf F.; Kohlwein S. D. (1993) Acetyl-CoA carboxylase from yeast is an essential enzyme and is regulated by factors that control phospholipid metabolism. J. Biol. Chem. 268, 10946–10952. [PubMed] [Google Scholar]
- Kirchhofer A.; Helma J.; Schmidthals K.; Frauer C.; Cui S.; Karcher A.; Pellis M.; Muyldermans S.; Casas-Delucchi C. S.; Cardoso M. C.; Leonhardt H.; Hopfner K.-P.; Rothbauer U. (2010) Modulation of protein properties in living cells using nanobodies. Nat. Struct. Mol. Biol. 17, 133–138. [DOI] [PubMed] [Google Scholar]
- Witte M. D.; Cragnolini J. J.; Dougan S. K.; Yoder N. C.; Popp M. W.; Ploegh H. L. (2012) Preparation of unnatural N-to-N and C-to-C protein fusions. Proc. Natl. Acad. Sci. U.S.A. 109, 11993–11998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Håkansson S.; Morisaki H.; Heuser J.; Sibley L. D. (1999) Time-lapse video microscopy of gliding motility in Toxoplasma gondii reveals a novel, biphasic mechanism of cell locomotion. Mol. Biol. Cell 10, 3539–3547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Massa P. E.; Paniccia A.; Monegal A.; de Marco A.; Rescigno M. (2013) Salmonella engineered to express CD20-targeting antibodies and a drug-converting enzyme can eradicate human lymphomas. Blood 122, 705–714. [DOI] [PubMed] [Google Scholar]
- Dang L. H.; Bettegowda C.; Huso D. L.; Kinzler K. W.; Vogelstein B. (2001) Combination bacteriolytic therapy for the treatment of experimental tumors. Proc. Natl. Acad. Sci. U.S.A. 98, 15155–15160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hogquist K. A.; Jameson S. C.; Heath W. R.; Howard J. L.; Bevan M. J.; Carbone F. R. (1994) T cell receptor antagonist peptides induce positive selection. Cell 76, 17–27. [DOI] [PubMed] [Google Scholar]
- Dobrowolski J. M.; Carruthers V. B.; Sibley L. D. (1997) Participation of myosin in gliding motility and host cell invasion by Toxoplasma gondii. Mol. Microbiol. 26, 163–173. [DOI] [PubMed] [Google Scholar]
- Hirakawa H.; Ishikawa S.; Nagamune T. (2012) Design of Ca2+-independent Staphylococcus aureus sortase A mutants. Biotechnol. Bioeng. 109, 2955–2961. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.