

Abstract
Background
Cork oak (Quercus suber L.) has a natural distribution across western Mediterranean regions and is a keystone forest tree species in these ecosystems. The fruiting phase is especially critical for its regeneration but the molecular mechanisms underlying the biochemical and physiological changes during cork oak acorn development are poorly understood. In this study, the transcriptome of the cork oak acorn, including the seed, was characterized in five stages of development, from early development to acorn maturation, to identify the dominant processes in each stage and reveal transcripts with important functions in gene expression regulation and response to water.
Results
A total of 80,357 expressed sequence tags (ESTs) were de novo assembled from RNA-Seq libraries representative of the several acorn developmental stages. Approximately 7.6 % of the total number of transcripts present in Q. suber transcriptome was identified as acorn specific. The analysis of expression profiles during development returned 2,285 differentially expressed (DE) transcripts, which were clustered into six groups. The stage of development corresponding to the mature acorn exhibited an expression profile markedly different from other stages. Approximately 22 % of the DE transcripts putatively code for transcription factors (TF) or transcriptional regulators, and were found almost equally distributed among the several expression profile clusters, highlighting their major roles in controlling the whole developmental process. On the other hand, carbohydrate metabolism, the biological pathway most represented during acorn development, was especially prevalent in mid to late stages as evidenced by enrichment analysis. We further show that genes related to response to water, water deprivation and transport were mostly represented during the early (S2) and the last stage (S8) of acorn development, when tolerance to water desiccation is possibly critical for acorn viability.
Conclusions
To our knowledge this work represents the first report of acorn development transcriptomics in oaks. The obtained results provide novel insights into the developmental biology of cork oak acorns, highlighting transcripts putatively involved in the regulation of the gene expression program and in specific processes likely essential for adaptation. It is expected that this knowledge can be transferred to other oak species of great ecological value.
Electronic supplementary material
The online version of this article (doi:10.1186/s12870-015-0534-1) contains supplementary material, which is available to authorized users.
Keywords: Quercus suber, Fruit, Seed, Transcriptomics, Transcription factor, Transcriptional regulators, Response to water, Carbohydrate metabolism
Background
Seed protection and dispersal are the main functions of the fruit. Fruit initiation and development play a crucial role in plant adaptation, and successful fruiting strategies are important drivers of colonization of new niches. Fruit and seed set are generally characterized by extensive cell division and coordinated development of maternal and filial tissues, while growth and maturation stages are characterized by cell expansion and accumulation of storage products, mainly proteins, starch and oils [1]. The transcriptomic and proteomic analyses of genetic networks operating during specific processes of fruit and seed development have revealed the involvement of a wide range of molecular players including enzymes, regulatory proteins as well as hormonal signals. Molecular studies of fruit development have been mostly conducted in fleshy fruits such as tomato [2–7], grape [8–10], blueberry [11, 12], sweet orange [13] or melon [14], due to their importance for human consumption. Genes related to fruit ripening have been extensively studied in tomato, grape and sweet orange [10, 13, 15, 16] and genes specifically expressed in fruits have been identified in apple [17] and date palm [18]. In addition, Arabidopsis has proven very informative because its silique is a dehiscent fruit characteristic of the legumes and thus represents another exceptionally important fruit type in terms of human and animal food.
Much less attention has been paid to other types of fruits that although not generally used for human consumption, have huge ecological importance. The Fagaceae family comprises more than one thousand species half of which belong to the Quercus genus, commonly known as oaks. The oaks produce an indehiscent fruit, usually termed acorn, and are characteristically adapted to extremely variable habitats being widely distributed throughout the northern hemisphere in an almost continuous pattern.
Cork oak (Quercus suber L.), native to the western Mediterranean and north Africa regions [19] characterized by hot and dry summers, has been considered a keystone forest tree species in the ecosystems where it grows [20, 21]. The species is mostly recognized for producing cork, which is removed from adult trees at regular intervals of at least 9 years, sustaining highly profitable cork industries [22]. Cork oak has an unusual fruiting strategy as it is the only known oak species with annual and biennial acorns on the same tree [23–27]. Other features such as bigger acorn size have been related to drought tolerance [28–30] and higher seed germination ability [29–32] and thus may strongly impact the capacity for species natural regeneration, the most common way of cork oak propagation [33]. Seed development and germination are critical for the successful maintenance of the cork oak growing regions [34–36].
During development, the acorn undergoes many biochemical and physiological changes which likely confer the ability to survive the severe drought periods and high temperatures. The few studies that have been conducted in oak acorns have focused on morphological, physiological and phenological aspects [24, 29, 37] and a few reports exist on aspects of the male and female flower development [33, 38–40] and flower/fruit anatomy [41]. Although some transcriptomic and genomic studies have also been published in oak species [42–44], it was only recently that the transcriptome of cork oak has started to be analysed in multiple tissues, developmental stages and physiological conditions [45]. In this context, and to gain knowledge on the molecular mechanisms underlying the development of cork oak acorn and to identify transcripts putatively related with adaptive traits, we have analysed the dynamics of the transcriptome of acorns along five stages of development, defined according to morphological characters, from early fruiting stages to fruit and seed maturation. Our approach identified genes with potentially relevant roles during acorn development focusing specially on transcripts coding for putative transcription factors and transcription regulators or transcripts associated to water related processes including response, transport and deprivation.
Results
Categorization of cork oak acorns into different developmental stages
Although the fruits are often defined as seed-bearing structures formed from a mature ovary, many structures that might be defined as fruit are in fact composed of different tissue types [46]. Other definitions have been proposed, such as the one by Van der Pijl [47] that considers the fruit as the dispersal unit. In this work we use the term acorn for simplicity, referring in fact to all the tissues enclosed by the pericarp, including the seed. It should be pointed out that at maturity most of the acorn mass consists of seed tissues, mainly cotyledons. Cork oak acorns were collected from late June to November in order to cover all developmental stages, from early development to full maturation. A staging system was established based on several morphological aspects (Fig. 1 and Table 1). Since the dimensions of the acorns were variable in the same collection date among trees in different locations, additional features were used to establish developmental classes. These included the presence of a visible endosperm, multiple embryos or a dominant embryo within the developing seed, covering of the acorn by the cupule and colour of the pericarp (Table 1). Accordingly, eight stages of acorn development were established (S1–S8, Fig. 1a, b). In the first stage (S1), fertilization of the ovules may have occurred already but in most cases the endosperm was not yet visible. In the S2 stage multiple fertilised ovules were visible, however only one continued to grow becoming dominant and causing abortion of the other ovules (S3). During S4 and S5, the embryo continues to develop and in the remaining stages (S6, S7) further enlargement of the cotyledons takes place, with full maturation being reached in S8.
Fig. 1.

Developmental stages established for the cork oak acorn. a Cork oak fruits collected at different developmental stages (S1-S8). The scale bar corresponds to 1 mm in S1 to S3 and to 5 mm in S4 to S8. b Cork oak fruits at stages S3-S5 after removal of the cupule (above), or cupule and pericarp (below) exposing the seed, and acorn measurement parameters (S7) used for acorn staging. D, maximum diameter of the acorn; Pi, portion of the acorn outside the cupule; P, acorn portion covered by the cupule. The scale bar corresponds to 1 mm in S3 to S5 and to 5 mm in S7
Table 1.
Criteria used for categorizing the cork oak acorn into different developmental stages. Representation of each stage in the normalized (N) and non-normalized (nN) cDNA libraries. Embryo tissues isolated from acorns belonging to stages S3-S5 and S8 were classified as EM3-EM5 and EM8
| Developmental stage | Max Ø with cupule (mm) | Max Ø without cupule (mm) | Isolation of embryos | Other observations; library type |
|---|---|---|---|---|
| S1 | 2 - 3 | nd | no | no endosperm visible; N and nN |
| S2 | 3 - 5 | nd | no | multiple fertilized ovules, some aborted; N and nN |
| S3 | 5 - 8 | nd | yes (EM3) | dominant embryo; N and nN |
| S4 | 8 - 12 | nd | yes (EM4) | acorn completely covered by the cupule; N and nN |
| S5 | 12 - 17 | 7 - 11 | yes (EM5) | acorn already visible out of the cupule; N and nN |
| S6 | 17 - 20 | 11 - 16 | no | ~1/2 of the acorn out of the cupule |
| S7 | >20 | >16 | no | Green acorn mostly out of the cupule; N |
| S8 | >20 | >16 | yes (EM8) | Brown acorn; N and nN |
nd: not determined
Sequencing and assembly of the cork oak acorn transcriptome
The sequencing of the five non-normalized libraries corresponding to samples from stages S1, S2, S3 + S4, S5 and S8 aimed at gene expression analysis during acorn development. In addition, two normalized libraries prepared from RNA of cork oak acorns or from isolated embryos were sequenced to favour the detection of rare transcripts and thereby facilitate the assembly. After pre-processing, 2,088,335 high-quality sequences were retained and used in the assembly and mapping steps. The final average length of the reads was 215 and 400 bp for the normalized and non-normalized libraries, respectively (Table 2).
Table 2.
Read statistics from libraries of cork oak acorn and embryos before and after pre-processing. Embryo tissues isolated from acorns belonging to stages S3-S5 and S8 were termed EM3-EM5 and EM8, respectively
| Non-normalized cDNA libraries | Normalized cDNA libraries | |||||||
|---|---|---|---|---|---|---|---|---|
| S1 | S2 | S3 + S4 | S5 | S8 | S1 to S8 | EM3 to EM5 + EM8 | ||
| Raw reads | Total | 111,703 | 373,962 | 200,862 | 302,253 | 102,250 | 738,266 | 705,151 |
| Range (bp) | 51-1,200 | 52-1,200 | 47-1,200 | 50-1,200 | 55-1,200 | 50-1,201 | 52-1,201 | |
| Mode (bp) | 520 | 521 | 519 | 522 | 519 | 516 | 516 | |
| Mean (bp) | 503.7 | 515.8 | 510.7 | 524.8 | 513 | 538.5 | 538.7 | |
| Size (Mbp) | 47.5 | 162.5 | 86.1 | 133.6 | 44.3 | 314.7 | 298.8 | |
| Valid reads | Total | 100,081 | 332,674 | 179,588 | 267,171 | 88,261 | 572,665 | 547,895 |
| Range (bp) | 40-568 | 40-579 | 40-571 | 40-565 | 40-546 | 40-576 | 40-616 | |
| Mode (bp) | 209 | 213 | 220 | 217 | 214 | 409 | 380 | |
| Mean (bp) | 254.1 | 255.2 | 253.6 | 253.5 | 258.1 | 304.3 | 289.2 | |
| Size (Mbp) | 27.6 | 92.7 | 49.5 | 74.4 | 25.5 | 201.0 | 183.0 | |
The seven libraries were assembled by MIRA and Newbler (Table 3). MIRA assembly contained 104,862 contigs, 52.2 % of which were longer than 500 bp. Newbler assembly contained 33,034 contigs, 79.6 % of which were longer than 500 bp. The merging of the MIRA and the Newbler assembly using CAP3 resulted in 80,357 contigs that were deposited in ENA (the accession number of the de novo transcriptome is [ENA: HABZ01000000] and the accession numbers of the contigs are [ENA: HABZ01000001–HABZ01080357]). 62.5 % of these contigs were longer than 500 bp. The assembled transcripts were classified as complete, terminal, internal or novel by comparison with the complete plant proteins in UniprotKB database (Table 3 and Additional file 1). 23,840 contigs did not have any homologous sequence in the tested database (Complete plant Uniprot proteins). However, it was possible to predict a clear ORF for 4,658 of them, and they were classified as novel.
Table 3.
De novo transcriptome assemblies and classification of the assembled cork oak transcripts
| MIRA | Newbler 2.6 | Final (Merged) | |
|---|---|---|---|
| Contigs | 104,862 | 33,034 | 80,357 |
| Contigs > 500 bp | 54,764 (52.2 %) | 26,313 (79.6 %) | 50,197 (62.5 %) |
| Contigs < 200 bp | 6,306 (6 %) | 481 (1.5 %) | 4,510a |
| Contigs with homologous in Uniprot | 73,103 (69.7 %) | 26,951 (81.6 %) | 56,517 (70.3 %) |
| Unique Uniprot IDs | 28,154 (38.5 %) | 16,047 (48.6 %) | 24,474 (43.3 %) |
| Complete contigs | 16,149 (15.4 %) | 12,953 (39.2 %) | 19,146 (23.8 %) |
| C-terminus contigs | 15,818 (15.1 %) | 5,342 (16.2 %) | 11,410 (14.2 %) |
| N-terminus contigs | 15,368 (14.7 %) | 4,064 (12.3 %) | 10,108 (12.6 %) |
| Internal contigs | 25,432 (24.2 %) | 4,575 (13.8 %) | 15,509 (19.3 %) |
| Misassembled contigs | 336 (0.32 %) | 17 (0.05 %) | 344 (0.43 %) |
| Contigs without UNIPROT homologous | 31,759 (30.3 %) | 6,083 (18.40 %) | 23,840 (29.6 %) |
| Novel genes | 5,388 (5.14 %) | 1,320 (4 %) | 4,658 (5.8 %) |
| Complete ORF | 2,758 (2.6 %) | 664 (2.01 %) | 2,318 (2.9 %) |
| Partial ORF | 2,630 (2.5 %) | 656 (1.99 %) | 2,340 (2.9 %) |
| Unknown contigs | 26,346 (25.1 %) | 4,757 (14.4 %) | 19,163 (23.8 %) |
| Unknown contigs < 200 bp | 5,471 (5.2 %) | 407 (1.2 %) | 0a |
| Putative ncRNAs | 25 (0.02 %) | 6 (0.02 %) | 19 (0.02 %) |
| Reads mappedb | 2,020,921 (96.8 %) | 1,909,842 (91.5 %) | 2,009,759 (96.2 %) |
| Unique mapped reads | 1,703,996 (84.3 %) | 1,549,803 (74,2 %) | 1,491,131 (71.4 %) |
| Duplicated mapped reads | 273,619 (13.1 %) | 270,027 (14.1 %) | 367,486 (17.6 %) |
| Mapping more than two times | 43,306 (2.1 %) | 90,012 (4.7 %) | 151,142 (7.2 %) |
| Average coverage | 7.8 | 14.2 | 8 |
aContigs shorter than 200 bp were filtered out before analyzing
bThe percentage of reads that mapped over a possible total of 2,088,230 reads
Completeness of the Q. suber transcriptome by comparison with other Fagaceae
The proteins in the Q. suber assembly were compared to the proteins from other four Quercus spp., two Castanea spp. and a Fagus sp. For this purpose, we obtained the assembled transcriptomes from the Fagaceae project (www.fagaceae.org) or NCBI (for Q. robur and Q. petraea). The transcripts in each transcriptome were classified as complete, terminal, internal or novel by comparison with the complete plant proteins in UniprotKB database, as we had previously done for Q. suber (Additional file 2). Our Q. suber assembly had the higher number of complete proteins (19,146) and the second higher number of total proteins (56,517).
On average, 94.2 % of the proteins from any of the tested species could be found in our de novo assembled Q. suber transcriptome when it was used as the target database (Additional file 3). On the other hand, when the Q. suber proteins were used as query, we found that between 60 and 80 % of the queries aligned to each of the other transcriptomes, and the higher ratios corresponded to the more complete Castanea mollisina and Castanea dentata transcriptomes. Of all the queries, only contig20020 was not found in any other transcriptome.
Functional annotation of the Q. suber transcriptome
All 80,357 transcripts were compared with the NCBI non-redundant (nr) protein database using Blastx with an E-value of 1e-10, which resulted in 53,670 (66.8 %) sequences with a significant alignment (Additional file 4). From the total number of transcripts, 19,757 (24.6 %) transcripts had the best match to Vitis vinifera sequences, followed by 9,329 (11.6 %); 8,636 (10.7 %) and 5,324 (6.6 %) transcripts that matched to Ricinus communis, Populus trichocarpa and Glycine max sequences, respectively. Similar results have been obtained in the transcriptome annotation for other plant species and linked to conserved biochemical, morphological and developmental characteristics [12]. The number of alignments obtained among the Fagaceae family was very low: 122, 101, 97 and 75 sequences matched with sequences of Castanea sativa, Fagus sylvatica, Quercus suber and Castanea mollissima, respectively (Additional file 4A). This is mainly due to the limited amount of data available at the GenBank database for non-sequenced species. Most of the alignments showed a similitude between 75 and 90 % (Additional file 4B). Only about 7.6 % of the total number of transcripts present in Q. suber transcriptome was found to be fruit or seed specific but, according to our analysis of the conserved motifs and structures in the sequences, the majority of these transcripts are unknown (Additional file 1).
50,228 (62.5 %) transcripts were annotated with at least one Gene Ontology (GO) term (Additional file 5). There was a direct relation between the sequence length and percentage of annotated sequences and over 75 % of the sequences longer than 1 Kb could be annotated (Additional file 4C).
49,945 (52.15 %) Q. suber transcripts had a homologous in the A. thaliana genome (Blastx E-value < 1e-10). Each transcript was annotated with the GO terms of its Arabidopsis homologous gene. Additionally, each A. thaliana gene was annotated with its NCBI COGs, if any exists, and this annotation was also associated backwards to the original Q. suber transcript (Additional file 6). In order to compare our de novo transcriptome and identify COGs specific to Q. suber, a similar approach was followed for Q. petraea, Q. robur and the cork oak ESTs database (CODB). 44,300 (55.1 %); 59,572 (74.1 %) and 51,916 (64.6 %) transcripts from Q. petraea, Q. robur and CODB were homologous to genes from the A. thaliana genome, respectively. The distribution of protein clusters is summarized in a Venn diagram (Additional file 7). 2,254 (72.5 %) of a total of 3,110 COGs were present in all the species and 222 COGs were specific to Q. suber. Of these 222 COGs, 12.2 % were involved in replication, recombination and repair, 6.3 % in RNA processing and modification, 5.9 % in translation, ribosomal structure and biogenesis, 5.4 % in cell cycle control, cell division and chromosome partition, 5 % in post-translational modifications, protein turnover and chaperones and 5 % in transcription. Finally, 20.8 % of the 222 COGs were unknown or poorly characterized (Additional file 6: File S3 and Additional file 8).
Pathway analysis during cork oak acorn development
15,612 (19.4 %) sequences were annotated according to their homology with known enzymes that belonged to 140 pathways (KEGG level-3 pathways) and all 14 KEGG groups of related pathways (KEGG level-2 pathways) (Additional file 9). The carbohydrate metabolism was the group most represented, which also includes several of the more represented pathways, such as starch and sucrose, glycolysis and gluconeogenesis, amino sugar and nucleotide sugar, pyruvate, and galactose metabolic pathways. The second most represented group was amino acid metabolism, which includes phenylalanine metabolism. When the number of different enzymes is considered, the more relevant pathways were glycine, serine and threonine; arginine and proline and cysteine and methionine pathways. The third most represented group was lipid metabolism followed by energy metabolism. Among the most represented pathways were also purine and pyrimidine metabolism, methane metabolism, and phenylpropanoid biosynthesis (Additional file 9).
The reads from the five non-normalized libraries were mapped to the transcripts in the final assembly to quantify the expression in each stage. The number of mapped reads of the transcripts belonging to the same pathway was summed up to determine the expression of each pathway on time (Additional file 9). The normalized expression values for the level 2 pathways were represented in a heatmap (Fig. 2). While the immune system was the most up-regulated pathway in the first acorn developmental stage (S1), followed by metabolism of other aminoacids and secondary metabolites, in middle stages of development (S2 to S5) up-regulation of carbohydrate, nucleotide, glycan biosynthesis and energy metabolism was observed. Signal transduction pathways were up-regulated only in S2, while amino acid metabolism and translation were specifically up-regulated in S3S4. S8 exhibited an expression profile markedly different from other developmental stages where lipid metabolism and metabolism of cofactors and vitamins were specifically up-regulated.
Fig. 2.
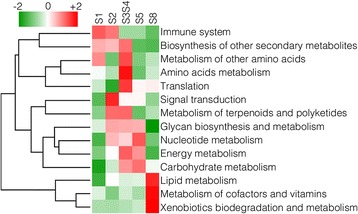
Heatmap of the expression levels of the KEGG level 2 pathways. The expression levels were normalized in Z scores, with signals from red (higher expression) to green (lower expression)
Differentially expressed genes (DEGs) during cork oak acorn development
From the mapping of the reads of the five non-normalized libraries to the assembled transcriptome, 58,839 genes were identified as expressed during any of the developmental stages, 7,824 transcripts (13.3 %) were expressed in all the stages and 22,802 (38.8 %) were specific to one stage. The total number of transcripts present in each stage was 23,104 (39.3 %); 37,501 (63.7 %); 30,035 (51 %); 33,367 (56.7 %) and 17,310 (29.4 %), respectively from S1 to S8 (Additional file 10).
Of the 58,839 transcripts expressed during acorn development, 2,285 (3.9 %) were considered DE (Additional file 11). From those 710 (31.1 %), 475 (20.8 %), 685 (30 %) and 1,078 (47.2 %) transcripts were DE between stages S2 and S1, S3S4 and S2, S5 and S3S4, and S8 and S5, respectively. 568 transcripts (24.9 %) were DE in more than one stage transition (Fig. 3). From the DEGs only 30 (~1.3 %) were found as acorn specific, with 5 transcripts in stage S1, 14 in stage S3S4, 7 in stage S5 and 4 in the last stage of the acorn development (S8). However, the majority of these transcripts are of unknown function (Additional file 11).
Fig. 3.
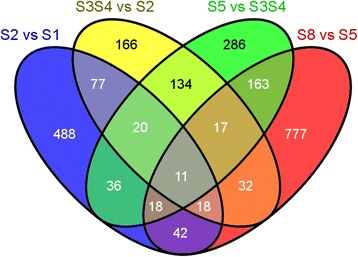
Venn diagram illustrating the number of transcripts differentially expressed between two consecutive stages of development
An enrichment analysis by F-fisher test (FDR < 0.05) comparing the set of 2,285 DEGs versus the complete transcriptome evidenced 466 over-represented GO terms (Additional file 12). One third of the DEGs were involved in responses to abiotic stimulus, one fifth in carbohydrate catabolism, and other fifth in the catabolism and generation of energetic compounds. GO terms related with transport process, such as water and auxin polar transport, or development and growth were also significantly represented (Additional file 13).
DEGs were clustered in six groups according to their expression profile on time (Fig. 4 and Additional file 11). Since each cluster contains genes with a peak of expression in specific stage(s) of development, an enrichment analysis (FDR < 0.01) of the genes in each cluster versus the complete transcriptome evidenced the dominant processes in those stages (Additional file 12). Eight GO terms were over-represented at S1 (cluster A) including response to stimulus, response to osmotic and salt stress, and hexose transmembrane transport. Forty-one GO terms were over-represented at S2 (cluster B), including response to stress, to water, to water deprivation, to osmotic and to salt stresses, as well as water transport, and transmembrane transport. At S3S4 stages (cluster C) 73 GO terms were over-represented, including the previous terms related with response, and also regulation of meristem growth and of meristem development, among others. Thirty-one GO terms were over-represented at S5 (cluster D), and 68 GO terms at S5 and S8 (cluster E), including glycogen synthesis and metabolism, carbohydrates (glucose, hexose, glucan) metabolism, as well as starch synthesis and xylem development. Fifty GO terms were over-expressed at S8 (cluster F), including chitin binding and metabolism and aminoglycan, amino sugar, glucosamine and polysaccharide catabolism.
Fig. 4.
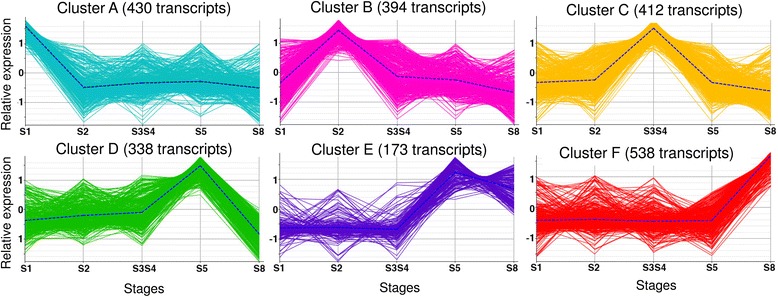
Clustering analysis of differentially expressed genes (DEGs) according to their expression profiles
Genes related to response to water, water deprivation or water transport
From the total DEG, 211 (9.23 %) were related to water response, deprivation and transport, and distributed across all the developmental stages but over-represented in S2, followed by the last developmental stage (S8) (Additional file 14). A shortlist of these transcripts is presented in Table 4, including those that have a known Arabidopsis homolog, are specific of a single cluster and have a level of expression at a given stage higher than 85 % compared with its expression in all stages. Fulfilling these criteria and mostly expressed in the first stage of acorn development (S1-cluster A) we found an homolog of the ABC transporter family, MULTIDRUG RESISTANCE P-GLYCOPROTEINS (MDR4/PGP4/ABCB4) as well as an homolog of the UBIQUITIN-CONJUGATING ENZYME 32 (UBC32). In cluster B, with a peak of expression in stage S2, an homolog of the Arabidopsis β-AMYLASE 1 (BAM1) was identified. In the subsequent stages of acorn development (S3S4, cluster C) we found a transcript with homology to the RESPONSIVE TO DESICCATION 22 (RD22). Specific to cluster D we found homologs of the ALPHA-GLUCAN PHOSPHORYLASE 1 (PHS1), EARLY RESPONSIVE TO DEHYDRATION 5 (ERD5) and the ABA INSENSITIVE 3 (ABI3). With an expression profile that fits in cluster E, was a member of the EARLY RESPONSIVE TO DEHYDRATION, an homolog of the ERD15. Transcripts putatively encoding for members of Late Embryogenesis Abundant protein family, such as LATE EMBRYOGENESIS ABUNDANT 4–5 (LEA4–5), DEHYDRIN XERO 1 (XERO1) and DROUGHT-INDUCED 21 (DI21), for LIPID TRANSFER PROTEIN 3 (LTP3), for URIDINE DIPHOSPHATE GLYCOSYLTRANSFERASE 74E2 (UGT74E2) and for METALLOTHIONEIN 3 (MT3) were almost uniquely expressed in the last stage of the acorn development (Table 4 and Additional file 14).
Table 4.
Shortlist of differentially expressed transcripts annotated as involved in response to water. Transcripts with a known Arabidopsis homolog were selected from a total of 211 DEGs in this category, based on their specificity to a single cluster and higher expression level in a given stage as determined by a stage expression factor (SEFa) higher than 0.85. To have transcripts specific of cluster D in the short list, the stage expression factor considered in this case was 0.57. Expression in each stage is represented as normalized counts
| Cluster | Transcript name | At Locus | At homologous | Annotation | counts | SEFa | ||||
|---|---|---|---|---|---|---|---|---|---|---|
| S1 | S2 | S3S4 | S5 | S8 | ||||||
| A | Contig16112 | AT2G47000 | MDR4, PGP4, ABCB4 | Multidrug resistance 4, P-glycoprotein 4, ATP binding cassette subfamily B4 | 19.8 | 0 | 0.6 | 0.4 | 0 | 0.95 |
| A | Qs-dev_rep_c84235 | AT3G17000 | UBC32 | Ubiquitin-conjugating enzyme 32 | 25 | 0 | 1.2 | 0 | 1.2 | 0.91 |
| B | Contig18768 | AT3G23920 | BAM1 | β-Amylase 1 | 1.2 | 29.6 | 0.6 | 1.2 | 2.2 | 0.85 |
| Qs-dev_rep_c76744 | 0.4 | 54.2 | 0.2 | 8.4 | 0.8 | 0.85 | ||||
| C | Qs-dev_rep_c77033 | AT5G25610 | RD22 | Responsive to desiccation 22 | 0.4 | 0 | 34.4 | 0 | 3.6 | 0.90 |
| D | Contig17288 | AT3G30775 | ERD5, PRODH, ATPOX, ATPDH, PRO1, PDH1 | Methylenetetrahydrofolate reductase family protein | 0 | 0 | 0 | 22 | 15 | 0.59 |
| D | Qs-dev_rep_c76415 | AT3G29320 | PHS1 | Glycosyl transferase | 0.4 | 0.6 | 0.2 | 25.4 | 16.6 | 0.59 |
| D | Contig19318 | AT3G24650 | ABI3 | AP2/B3-like transcriptional factor family protein | 0 | 0 | 7.2 | 66.8 | 42.2 | 0.57 |
| E | Contig19981 | AT2G41430 | ERD15, LSR1, CID1 | Dehydration-induced protein (ERD15) | 0 | 2.2 | 0.6 | 25.8 | 37.2 | 0.96 |
| F | Contig18471 | AT5G59320 | LTP3 | Lipid transfer protein 3 | 0 | 0.2 | 0 | 5.6 | 38.6 | 0.87 |
| F | Contig16935 | AT4G15910 | DI21 | Late embryogenesis abundant protein Lea5; drought-induced 21 | 1.2 | 0 | 3.6 | 18.8 | 179.2 | 0.88 |
| Qs-dev_c42419 | 0 | 0 | 0 | 0 | 16.4 | 1 | ||||
| F | Contig18808 | AT5G06760 | LEA4-5 | Late embryogenesis abundant 4-5 | 1.2 | 2.2 | 0.6 | 0.4 | 53 | 0.92 |
| F | Contig17917 | AT3G50980 | XERO1 | Dehydrin xero1 | 3 | 0.8 | 0.6 | 3.4 | 50.8 | 0.87 |
| F | Contig19993 | AT1G05680 | UGT74E2 | Uridine diphosphate glycosyltransferase 74E2 | 0 | 0.8 | 0 | 4.2 | 33.8 | 0.87 |
| F | Qs-dev_rep_c73281 | AT3G15353 | MT3 | Metallothionein 3 | 0.4 | 3 | 0.2 | 4.6 | 75 | 0.90 |
| Qs-dev_rep_c79475 | 0.4 | 0 | 0.2 | 0.8 | 18.8 | 0.93 | ||||
a
Transcriptional regulators differentially expressed during acorn development
Transcription factors have important roles in gene expression due to their ability to bind specific DNA sequences and control transcription by acting as transcriptional activators or repressors. Out of 2,285 DEGs during acorn development a total of 498 (21.8 %) were annotated as TFs or transcriptional regulators (Additional file 15). These transcripts were almost equally distributed among the different clusters, but slightly up-regulated in early development, representing approximately 22.5 and 20.7 % of the transcripts in clusters A and B, respectively, and less expressed in the late stages of acorn development representing 7.4 and 15.9 % of the total DE TFs in clusters E and F, respectively.
A list of selected transcripts that have a known Arabidopsis homolog and are annotated as transcription factors or transcriptional regulators is presented in Table 5, including those that are specific of a single cluster and are also stage-specific or belong to TF families with well characterized roles in plant development. We found homologs of MYB DOMAIN PROTEIN 36 (MYB36) and AUXIN RESPONSE FACTOR 4 (ARF4) specifically expressed in stage S1 (cluster A) and a member of the MYB-related family, homolog of the PEROXIDASE 72 (PRX72) specifically present in stage S3S4 (cluster C). Also other transcripts were identified which expression was restricted to the late stages of development, such as a homolog of the FAR1-RELATED SEQUENCE 4 (FRS4) in stage S8 (cluster F). Interesting genes from well-known families of TFs or transcriptional regulators such as NAC, bHLH, class II KNOTTED1-like homeobox and OLEOSIN are also represented during the cork oak acorn development. Up-regulation in the early stages of development (S1 and S2) was observed for class II KNOTTED1-like homeobox genes. During the late stages of the acorn development transcripts putatively coding for OLEOSIN (OLEO) were up-regulated. Transcripts coding for NAC and bHLH transcription factor families were found DE across all the studied developmental stages (Table 5 and Additional file 15).
Table 5.
Shortlist of differentially expressed transcripts putatively coding for transcription factors and transcriptional regulators. Transcripts with a known Arabidopsis homolog were selected according the following criteria: transcript is either specific of a single cluster and is stage-specific or belongs to TF families with well characterized roles in plant development
| Cluster | Transcript name | Transcription factor | Domain /Family | At Locus | At homologous | Annotation | counts | SEFa | ||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| S1 | S2 | S3S4 | S5 | S8 | ||||||||
| A | Contig20395 | Vitis_vinifera_ GSVIVP00000629001 | MYB_DNA binding/MYB | AT5G57620 | MYB36 | MYB domain protein 36 | 24.2 | 0 | 0 | 0 | 0 | 1 |
| A | Contig14583 | Vitis_vinifera_ GSVIVP00024505001 | B3/ARF | AT5G60450 | ARF4 | Auxin response factor 4 | 21.6 | 0 | 0 | 0 | 0 | 1 |
| C | Contig20996 | Oryza_sativa_subsp._Indica_OsIBCD010288 | MYB_DNA binding/MYB-related | AT5G66390 | PRX72 | Peroxidase superfamily protein | 0 | 0 | 16.2 | 0 | 0 | 1 |
| F | Contig14504 | Oryza_sativa_subsp._japonica_LOC_Os11g45530.1 | FAR1/FAR1 | AT1G76320 | FRS4, CPD25 | FAR1-related sequence 4 | 0 | 0 | 0 | 0 | 23.2 | 1 |
| A | Contig23988 | Carica_papaya_emv.TU.supercontig_187.28 | KNOX1/HB | AT5G25220 | KNAT3 | KNOTTED1-like homeobox gene 3 | 35.6 | 0.8 | 3.8 | 11.6 | 13.8 | 0.54 |
| B | Contig17627 | Vitis_vinifera_ GSVIVP00005632001 | AT5G11060 | KNAT4 | KNOTTED1-like homeobox gene 4 | 1.2 | 34 | 27.4 | 5.6 | 2 | 0.48 | |
| E | Contig24728 | Carica_papaya_emv.TU.supercontig_996.1 | AP2/AP2-AREBP DUF260/LOB | AT4G25140 | OLEO1 | Oleosin 1 | 0 | 0 | 3 | 69.2 | 58.5 | 0.98 |
| E | Contig20473 | Vitis_vinifera_ GSVIVP00016010001 | 0 | 0 | 8.2 | 428.2 | 192.4 | 0.99 | ||||
| E | Contig23293 | Vitis_vinifera_ GSVIVP00016010001 | DUF260/LOB | AT3G27660 | OLEO4 | Oleosin 4 | 0 | 0 | 0 | 71 | 25.8 | 1 |
| F | Qs-dev_rep_c103605 | Vitis_vinifera_ GSVIVP00016010001 | 0.4 | 0 | 0 | 9.2 | 39.6 | 0.80 | ||||
| F | Qs-dev_c70574 | Vitis_vinifera_ GSVIVP00016010001 | AT5G40420 | OLEO2 | Oleosin 2 | 0.2 | 0 | 0 | 2.2 | 49.8 | 0.95 | |
| B | Contig1681 | Carica_papaya_evm.TU.supercontig_3.229 | HLH/bHLH | AT5G08130 | BIM1 | 0 | 22.8 | 0 | 1.2 | 8.8 | 0.70 | |
| D | Contig25637 | Vitis_vinifera_GSVIVP00008627001 | AT1G01260 | JAM2 | Jasmonate associated MYC2 like 2 | 5.2 | 16.6 | 7.4 | 21.6 | 0 | 0.43 | |
| E | Contig18945 | Populus_trichocarpa_180347 | AT5G41315 | GL3, MYC6.2 | Glabra 3, Glabrous 3, MYC6.2 | 0 | 4.6 | 0.6 | 24.6 | 23.8 | 0.90 | |
| A | Contig4505 | Populus_trichocarpa_218417 | NAM/NAC | AT1G56010 | NAC1 | NAC domain containing protein 1 | 34.2 | 2.2 | 0.6 | 0 | 0 | 0.92 |
| A | Contig23086 | Populus_trichocarpa_714847 | AT1G34190 | NAC017, ANAC017 | NAC domain containing protein 17 | 20.2 | 0 | 0 | 11 | 4.6 | 0.56 | |
| B | Contig23085 | 0 | 27.8 | 22.8 | 18.4 | 7 | 0.37 | |||||
| A | Contig16452 | Vitis_vinifera_ GSVIVP00027622001 | AT4G27410 | RD26, ANAC072 | Responsive to desiccation 26 | 608.8 | 184.2 | 138.6 | 32 | 11 | 0.62 | |
| B | Contig25368 | Vitis_vinifera_ GSVIVP00000471001 | AT4G35580 | NTL9 | NAC transcription factor-like 9 | 18.2 | 42 | 8.2 | 14.8 | 0 | 0.50 | |
| D | Contig26025 | Vitis_vinifera_ GSVIVP00027621001 | AT3G15510 | NAC2 | NAC domain containing protein 2 | 1.2 | 1.6 | 12 | 19.8 | 0 | 0.57 | |
a
Validation of the differential expression profiles by RT-qPCR
Several genes were selected to validate the data obtained by sequencing with 454 Technology (Table 6). Twenty DEGs related to water responses, seven of which also annotated as TFs, were chosen for the validation of expression profiles by reverse transcription quantitative real-time PCR (RT-qPCR). Two transcripts belong to cluster A, six to cluster B, three to C, four transcripts belong to D and five to F. Correlation between the gene expression levels and the profiles obtained by 454 technology was demonstrated by Pearson correlation (Fig. 5) with most of the genes showing strong or moderately strong correlation [48]. In addition, these results also validate that the transcript assemblies are correct for the sequences tested and support the robustness of the transcriptome assembly performed in this work.
Table 6.
Primers used in RT-qPCR for validation of the expression profile obtained by 454 sequencing.
| Clustera | Gene abbreviationb | Gene description | Transcript name | At Locus | Primer sequences (forward / reverse) |
|---|---|---|---|---|---|
| A | MRP10, ABCC14 | ABC transporter C family member 14 | Contig3296 | AT3G62700 | GCTGCCTTTGCCCCACACT/ TGGAAGAGCCTTGAACGCTGCC |
| A | CPK10, CDPK1 | Calcium-dependent protein kinase | Contig4133 | AT1G18890 | GCTCACCTCCCTCGCACAACT/ AGGCGCGAGGTGGCGATAAT |
| B | ASPG1 | Protein aspartic protease in guard cell | Contig19387 | AT3G18490 | ATGCAGCTCGCTCGACGTGT/ TGTCGTGTCCGCAACCCAGA |
| B | ABF2 | Abscisic acid responsive elements-binding factor 2 | Contig22999 | AT1G45249 | CGGCCTGCTTTTTCCGCAACT/ AGTCAGCTGCAAGGTCACGAGC |
| B | AVP1 | Pyrophosphate-energized vacuolar membrane proton pump | Contig 8793 | AT1G15690 | CGCACTTGAGAACGACGCT/ TGCGCGGTCGTCGGAATCAT |
| B | HSP90-7 | Chaperone protein htpG family protein | Qs-dev_rep_c72408 | AT4G24190 | CAGCATCAGCTTCAGCCTCGGT/ AGCGGCTTCACGCCTTCTGA |
| B | BAM1, BMY7, TR-BAMY | β-amylase 1 | Contig 25491 | AT3G23920 | AACGCGAGTTTGCAGGCGCT/ CGACGTTTCCGCCGCATTGA |
| B | GolS2 | Galactinol synthase 2 | Contig 17943 | AT1G56600 | TGACGCCATAACATGGCCAGCA/ TTGCCAGCAGTGCCCTGACA |
| C | ALDH7B4 | Aldehyde dehydrogenase 7B4 | Contig 9124 | AT1G54100 | AGGGTGCTCCAACGACTCCA/ ACGCACAGCTAGGCCGATG |
| C | OPR3 | Oxophytodienoate-reductase 3 | Contig 16616 | AT2G06050 | TTGGTCTCTAACACAGCCGCCG/ CACGGCCGACATGCCATA |
| C | PAL1 | Phenylalanine ammonia-lyase 1 | Contig 9321 | AT2G37040 | TGCTAACTGGCCGCCCCAAT/ GCCAGAACCAACAGCTGTGCCA |
| D | ABI3, SIS10 | Abscisic acid insensitive 3 | Contig 19318 | AT3G24650 | GCAGTGGCCGTGGTGCAAT/ ATGGCGAGGCAAAGGCGGTT |
| D | PHS1 | Glycosyl transferase, family 35 | Contig 10143 | AT3G29320 | TGGCTTGAGATGGGCAACCCT/ TTTGGTGTGCTCCCCGGCAT |
| D | SIR | Sulfite reductase | Contig 19883 | AT5G04590 | TGCAATGGCATGCCCAGCCT/ ACTGGGGTTTCCACCAAGCCA |
| D | PIP2B | Aquaporin PIP2-4 | Contig 24062 | AT2G37170 | GCTTGGGCCTTTGGTGGCA/ TGGCTCCACCACCGTACTTGCT |
| F | COR414-TM1 | Cold-regulated 314 inner membrane 1 | Contig 19349 | AT1G29395 | TGGTGAGCTTGAGCTGCCGT/ TGCAAGCTGCCTGCTCGTGA |
| F | DI21 | Late embryogenesis abundant protein | Contig 16865 | AT4G15910 | AGCGTGCTCTCACCTTGTGGT/ TGCGGCTGCATCACATGCCT |
| F | JMT | Jasmonic acid carboxyl methyltransf. | Contig 17776 | AT1G19640 | TGGCTATCTTCGCCGCCCTT/ TGCCGTGGTTTGCACCTACTTCG |
| F | GSTU25 | Glutathione S-transferase TAU 25 | Contig 17981 | AT1G17180 | ACCACCTCGTCCGCCATTGT/ AGCCTGACCGACTCCATGGCAA |
| F | LTP3 | Lipid transfer protein 3 | Contig 19993 | AT5G59320 | TGCCCAGGCCACCATAACATGC/ TGTGGTCTTGGCCATGCCGTT |
a According to their expression profiles each transcript belongs to a different cluster
b Corresponds to the homologous in Arabidopsis thaliana
Fig. 5.
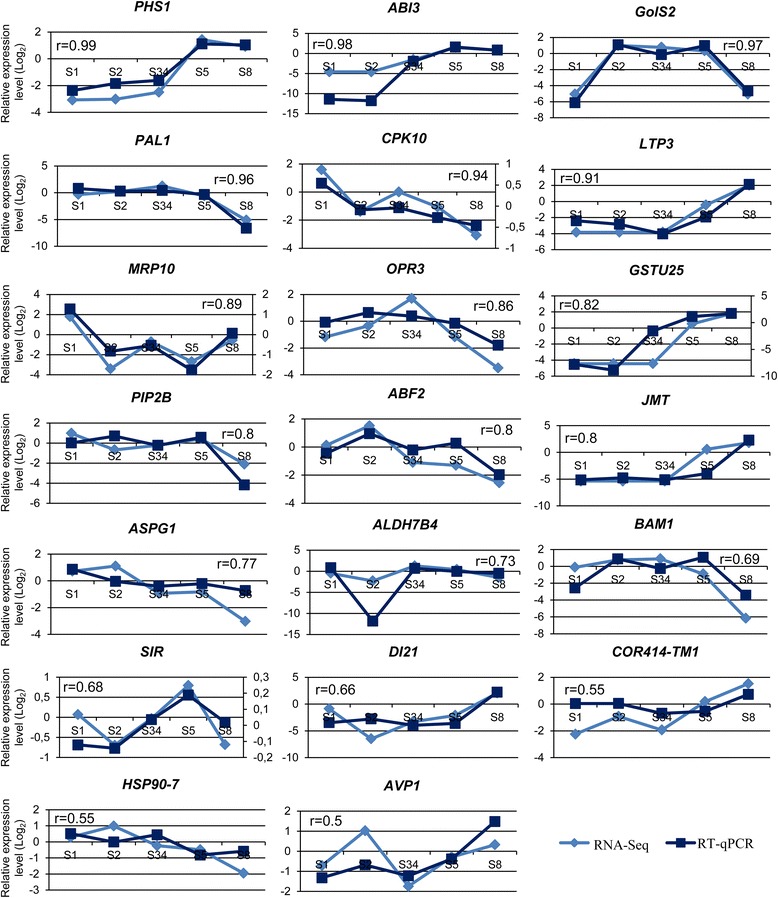
Validation of the RNA-Seq transcript profiles. Comparison of transcripts expression patterns from RNA-Seq data and from reverse transcription quantitative real-time PCR (RT-qPCR). In the y-axis it is represented the Log2 of the relative expression level in each developmental stage and the five acorn developmental stages are represented in the x-axis. The numbers above the graphics correspond to the values obtained with the Pearson correlation
Discussion
Based on available genomic resources and NGS technologies we provide here the first overview of the dynamics of the transcriptome along different stages of the acorn development in a Fagaceae species. The analysis of our data highlighted specific genes and processes relevant to better understand the molecular mechanisms involved in cork oak acorn development, and it is expected that this knowledge can be transferable to other oak species of great ecological value. The studied stages of development were established according to morphological criteria and to previously described reports on cork oak reproductive features [33] in order to cover the whole fruit and seed developmental process.
A set of 2,285 differentially expressed genes were identified with roles in a range of biological processes. We then focused our analysis on groups of transcripts with putative functions in transcriptional regulation and traits likely relevant to seed survival and dispersal, including mechanisms related to water response, water transport and water deprivation.
A de novo transcriptome of cork oak acorn
A de novo transcriptome assembly with the data here generated allowed us to identify the transcripts expressed during the acorn developmental process, some of which classified as novel. This de novo assembly facilitated the mapping of reads in unique positions since it was not necessary to allow mismatches between reads and reference. In fact, we discarded the marginal number of reads mapping in several positions. Assemblers of 454 transcriptome data have been systematically compared using real and simulated datasets. In such reviews, Newbler [49] and MIRA [50] outperformed other assemblers [51–54]. Newbler usually assembles longer contigs that often cover more than the 80 % of the reference sequences. MIRA joins reads in a more conservative way than Newbler, which prevents chimeric contigs and generates bigger assemblies using more bases and containing higher number of contigs, but some of them are redundant. Kumar et al. [52] proposed an assembly strategy that was used for the de novo assembly of pyrosequencing data from chickpea [53], by merging individual assemblies using a traditional Overlap-Layout-Consensus assembler, such as CAP3 [55]. Merged datasets aligned better to reference datasets and were more consistent in the total span and number and size of contigs than individual assemblies. In our case, the number of complete contigs (19,146) was higher in the merged assembly than in the individual ones, while the percentage of C-terminal and N-terminal contigs was smaller in the merged assembly than in any of the original assemblies (Table 3). This supports that several contigs from the same transcript were merged. When compared with other Fagaceae transcriptomes (Additional file 2), we report the highest number of complete proteins and different unique Uniprot IDs, which evidenced the advantages of this strategy.
Pathway analysis revealed that carbohydrate metabolism was the group (KEGG level-2 pathway) most represented in the transcriptome of developing cork oak acorns, especially in the middle stages of development. The enrichment analysis performed in the different clusters evidenced also the timing when a specific metabolic process appears prevalent. Using this approach, carbohydrates metabolism and starch synthesis, were found over-represented in the transcriptome of acorns at late stages of development, both S5 and S8. However, specific processes like hexose transmembrane transport were found over-represented in early stages of acorn development, where actively dividing cells contribute to a rapid growth of the fruit. In general, hexoses favour cell division and expansion, whereas sucrose favours differentiation and maturation [56]. This is also supported by the analysis of DEGs. For instance during the middle stages of acorn development, several up-regulated DEGs homologous to SUCROSE SYNTHASES were identified which are putatively involved in the synthesis of UDP-glucose and ADP-glucose linked to cellulose and starch biosynthesis [57]. These include SUCROSE SYNTHASE 3 (SUS3), and SUCROSE SYNTHASE 4 (SUS4). This is consistent with an active synthesis of cellulose and starch during these developmental stages, possibly related to the mobilization of sucrose into pathways involved in structural and storage functions. One fifth of the identified DEGs are related to carbohydrate metabolism and some of these transcripts, involved in water response or transcriptional regulation, are discussed below.
Response to water across acorn development
At complete maturity cork oak acorns contain a large and fleshy embryo with high water content. The natural shedding of cork oak acorns coincides with complete maturity and acorns left on the ground after shedding will either germinate or lose their viability as a result of desiccation [24]. Increased tolerance to desiccation may thus represent an important factor in cork oak regeneration success and the identification of transcripts related to response to water, water deprivation or water transport may prove relevant for highlighting genes with adaptive roles. In agreement with previous reports in Arabidopsis [58, 59], the DEGs annotated as being related to water responses during acorn development are not fruit specific. A high number of DEGs in this category were identified in the cluster of transcripts with a higher expression at the last stage of acorn development corresponding to maturity, probably reflecting some reduction in water content at this stage, but also during the early stages. The early stage of fruit and seed development is one of the most sensitive periods of the plant life cycle to abiotic stresses such as drought [60]. Therefore, it is not surprising that transcripts involved in response to water stress are up regulated at these stages as to ensure the protection of offspring against conditions of low water availability common during the summer.
Among the transcripts strongly related to drought stress that were identified in early development are several homologs of the RD family, including RD22 and other members not listed in Table 4 such as RD19, RD21, RD26 and RD29B. RD22 and RD26, were previously described to respond to dehydration [61–64] and used as drought-induced marker genes in different plant species [65–69]. RD22 is expressed in the early and middle stages of Arabidopsis seed development [62] and in our transcriptome analysis it is also highly expressed in the middle stages of the acorn development (S3S4, cluster C). During the early stage of the acorn development (cluster A), a putative homolog of MDR4 annotated as being involved in response to water deprivation was also highly expressed. MDR4 belongs to the large family of the ABC (ATP binding cassete) transporter superfamily and has been mainly characterized for its role in the basipetal redirection of auxin from the root tip [70, 71]. With the same expression profile (cluster A) was a putative homolog of UBC32, highly induced under drought stress [72]. Recently, functional studies in tomato revealed that UBC32 is involved in the regulation of fruit ripening [73] and, it is possible that it plays a similar function in cork oak acorn development. Carbohydrate metabolism-related transcripts with homologies to β-AMYLASE genes, which are described as contributing to osmoprotection during desiccation [74], were also found differentially expressed during acorn development. While some of the amylase transcripts, e.g. homologous to the A. thaliana chloroplast β-AMYLASEs BAM1/BAMY7, were up-regulated from the early to middle stages, other transcripts with homology to β-AMYLASEs BAM5 or BAM6 were up-regulated in later stages up to S8, being almost absent in earlier stages. The significance of these divergent expression patterns is not currently clear. It is possible that different roles related for instance to hormone signalling and/or acquisition of desiccation tolerance involving the accumulation of soluble sugars through starch degradation [75] are played by these enzymes during seed development.
Specific to cluster D are two transcripts putatively related to abscisic acid (ABA) signalling/responses, homologs of PHS1 and ABI3. PHS1 is a negative regulator of ABA signalling involved in the regulation of stomatal aperture [76]. Its expression pattern under different stresses vary significantly among species but it has been associated to drought responses [77]. Like in barley, we detected low expression of this transcript in early acorn development and its putative involvement in the regulation of starch synthesis during endosperm development [77, 78] may explain its up-regulation in later stages of seed development (S5). With the same expression profile is a homolog of a well characterized gene related to acquisition of tolerance to seed desiccation, ABI3, putatively encoding a TF expressed in seeds that mediates ABA responses. In Arabidopsis, ABI3 is required during seed maturation for the accumulation of seed storage proteins, dormancy onset and maintenance and for the acquisition of seed desiccation tolerance [79, 80]. Our results seem in agreement with a tight regulation of the mechanisms controlled by ABI3 during cork oak seed development towards maturation, such as ABA responsiveness and synthesis and accumulation of proline in order to increase stress tolerance in the embryo [81]. The expression profile of ABI3 seems consistent with alterations in ABA content as described for the recalcitrant seeds (desiccation-sensitive) of Quercus robur [82, 83] and corresponding ABA-induced processes along development. Like PHS1 and ABI3, an homolog of the EARLY RESPONSIVE TO DEHYDRATION 15 (ERD15) is up-regulated in stage S5 (cluster E). ERD genes are rapidly activated by dehydration and the encoded proteins constitute the first line of defense against drought stress [84, 85]. In Arabidopsis ERD15 is a common regulator of the ABA response and salicylic acid (SA)-dependent pathway [86] encoding a protein with functions in stress responses, including drought [87].
During the last stage of acorn development (S8, cluster F) several transcripts protrude from the list of DEGs annotated as related to water responses. A homolog of LTP3, known in Arabidopsis to be highly expressed in mature siliques and induced by drought [88, 89], was specific to the late stages of acorn development. The Arabidopsis LTP3 positively regulates the plant response to drought stress through the transcriptional activation of the cuticular wax biosynthesis to avoid dehydration [88, 89]. Also well represented in cluster F are the LATE EMBRYOGENESIS ABUNDANT (LEA), the protein family to which DI21 belongs, as well as XERO1 and LEA4–5. This protein family plays major roles in desiccation tolerance [90] and is characteristically accumulated in the last stages of seed development [91] as also observed in our data. The Arabidopsis XERO1 was only detected in seeds whereas DI21 was detected ubiquitously [92]. LEA4-5 highly accumulates during late Arabidopsis embryogenesis and in dry seeds, and upon constitutive expression it confers tolerance to severe drought [91]. In oil palm LEA4 is solely expressed in the mesocarp, and similarly to what we have obtained in the cork oak acorns, it is expressed in the late stages of fruit development, being probably involved in plant adaptation and stress (drought) responsive pathway [93]. Also DI21 is known to be up-regulated under abiotic stresses and XERO1 is thought to protect cellular components from dehydration stress [92]. Accumulation of LEA proteins during seed maturation and in response to altered water status was previously observed in oak species [82, 94, 95]. However, their effect in increasing drought tolerance in oaks is not as extensive as in non-recalcitrant seeds [83]. Another worth mentioning transcript putatively related to desiccation tolerance and up-regulated in the last stage of the acorn development is a homolog of the UGT74E2, encoding a hydrogen peroxide–responsive UDP-glucosyltransferase. Ectopic expression of UGT74E2 was associated to changes in plant architecture and to an increased tolerance to drought and salt stresses [96]. Increased tolerance to abiotic stresses, such as drought, was also verified in transgenic tobacco plants overexpressing the cotton type 3 metallothionein (GhMT3a) [97], a homolog of which was also identified up-regulated in late acorn development. Interestingly, many members of this group are associated with fruit ripening [98–101]. Like in the cork oak acorns, also in apple and kiwifruit MT3 was barely detected in young fruits but accumulated later with fruit development and fruit ripening [98, 100] suggesting a role in the ripening process, as well as in setting drought tolerance in this phase of development.
Transcriptional regulators during acorn development
Transcriptional regulators are crucial for plant developmental processes through their function in the regulation of gene expression, and fruit and seed development is no exception. In Arabidopsis which, like cork oak, has dry fruits, it has been revealed that the core and extended genetic network controlling fruit development consists entirely of interactions among transcription factors [46]. In fact, we found a significant number of differentially expressed transcripts coding for putative transcription factors and other transcriptional regulators along the acorn developmental stages covered by our analysis. However, our results support a slightly more prominent role of these transcripts in early development (S1 and S2) becoming less represented towards acorn maturity. In fact, it is during this stage that the transcription machinery seems more active as evidenced by the higher number of expressed transcripts.
Genes of the auxin response factor family (ARFs) are key regulators of auxin-modulated gene expression and can either activate or repress transcription of auxin-responsive genes [102]. A putative ortholog of ARF4 was found specifically expressed in cluster A, suggesting an important role in the initial phase of the acorn development. In tomato, ARF4 was described to repress the expression of auxin-responsive genes, playing a key role in the control of sugar metabolism during fruit development, through the regulation of photosynthetic activity as well as chlorophyll and starch accumulation [103]. Moreover, it was found highly expressed in the pericarp tissues of immature fruit and then undergoing a marked decline at the onset of ripening associated with the increase in sugar content accumulation [103]. It is possible that the transcript identified in our data has similar functions in the cork oak acorn development. Interestingly, ARF4 is a putative target gene of ta-siRNAs produced from cleavage of the ta-siRNA locus 3 (TAS3) directed by miR390 which was reported as differentially expressed in cork oak tissues [104].
Two putative class II KNOTTED1-like homeobox (KNOX2) genes were also up-regulated in the early stages of acorn development, one in S1 (KNAT3) and the other one in S2 (KNAT4). KNAT3 was previously reported to have a role in seed development, specifically in embryo sac development and during megagametogenesis [105]. More recently, KNOX2 TFs were shown to have a critical role in establishing an alternation of generations in land plants by preventing the haploid-specific body plan from developing in the diploid plant body [106], which appears consistent with the up-regulated expression of cork oak putative KNOX2 genes in early acorn developmental stages.
One putative MYB and one putative MYB-related transcripts, homologs of the Arabidopsis MYB36 and PRX72, respectively, were specifically detected in the early and mid-stages of acorn development (cluster A and C, respectively). MYB transcription factors have diverse functions in plants including development, secondary metabolism, hormone signal transduction, disease resistance and abiotic stress tolerance [107, 108]. MYB36 belongs to the R2R3-MYB gene family [109], which members have been described as the primary regulators of fruit flavonoid biosynthesis [110], among other functions [111]. Within the major flavonoid compounds present in flowers and fruits, the proanthocyanidins are astringent compounds that can offer protection during the early stages of fruit development against herbivory and pathogen attack [112, 113]. The specific presence of this transcript in the first stage of fruit development here analysed would be consistent with such a function in cork oak acorns, however, other roles should not be excluded. Our study also showed that the cork oak putative homolog to AtPRX72 was expressed in a specific stage of acorn developmental, S3S4 (cluster C). This may be related to the significant increase in the size of the acorn from this point onwards, and therefore increased cell wall expansion processes, but it can be also due to an increase in hardiness of the fruit, suggesting that this gene may function in the lignification process in cork oak acorn, consistent with previously described roles [114–117].
Transcripts identified as specifically expressed in the last stages of development, included a putative transcription factor gene of the FAR1 family, similar to the FRS4, which is a homolog of FHY3. Several studies report that FHY3 and FAR1 are required for regulating various aspects of plant processes, such as far-red-mediated seedling de-etiolation, the circadian clock, chloroplast division, and chlorophyll biosynthesis [118–124]. Tang et al. [125] have recently reported that knock-out mutants of FHY3 and/or FAR1 have reduced sensitivity to ABA-mediated inhibition of seed germination and seedling growth, lose water faster, and are less tolerant to drought stress than are wild type plants. Given the significance of drought tolerance traits for the successful cork oak natural regeneration process, it is tempting to consider that the putative homolog of FRS4 in the cork oak mature acorn may be involved in conferring tolerance to water stress conditions. However, other roles are also possible [126].
Another family of proteins described as transcriptional regulators (http://plntfdb.bio.uni-potsdam.de/), the OLEOSINS (OLEO1 and OLEO4) were also found specifically up-regulated in the last stages of the acorn development (S5 and S8). This is in accordance with the reported function of these plant specific genes in the control of oil body structure and accumulation of seed reserves, affecting seed germination and embryo phenotypes [127, 128]. Recent work in different species showed an increase in their accumulation during seed development [129–131].
Several members of two major families of TFs were present in all stages of the acorn development, bHLH and NAC. As an example of a putative bHLH TF gene, a homolog of the Arabidopsis bHLH GLABRA3 (GL3), was up-regulated during the late stages of the acorn development (S5 and S8- cluster E) when the acorn pericarp starts to become visible out of the cupule and turning brown. If the function of this transcript is conserved, then it is tempting to speculate that its up-regulation during late acorn development is related to the regulation of anthocyanin biosynthesis [132, 133] that occurs during this phase, and that it may play an important function in seed dispersal by attracting herbivores. Putative NAC family members were also found along different stages of the acorn development. This family of TFs plays important roles in responses to plant biotic and abiotic stress [134, 135] but also in developmental processes such as seed and embryo development [135, 136]. Our data revealed three different up-regulated transcripts coding for the NAC protein family at the S1 developmental stage, two at S2 and S5 and one in both S5 and S8. One of these transcripts up-regulated in S1, RD26, has been mentioned above in relation to response to water [61, 63].
Conclusions
In summary, our analysis allowed to cluster transcripts differentially expressed along acorn development in different profiles showing up-regulation in specific stages of development. While the DE transcripts putatively coding for transcriptional regulators associated to several biological processes were found up-regulated in given developmental stages throughout the whole acorn developmental process, other transcripts involved in specific processes such as response to water or carbohydrate metabolism were over-represented in particular stages. Future functional analysis of genes of interest identified in this work will be important to devise successful strategies for regeneration and breeding of this important species. This is especially important when considering the climate changes predicted for the Mediterranean region in the near future [137, 138] and the fact that drought is the main selection agent in Mediterranean ecosystems [21, 28]. Additionally, this dataset significantly contributes to increase the still scarce information on cork oak genomics providing tools for further molecular dissection of cork oak biology.
Methods
Plant material
Acorns were collected between mid June and late November 2009 from cork oak trees growing in private properties at six different locations in the South and Centre of Portugal: Quinta da Serra (Vila Nogueira de Azeitão), Alter do Chão, São Brás de Alportel, Monchique, Calhariz (Santarém) and Abrantes, after obtaining the required permissions. Samples were collected in accordance with the Portuguese legislation (Decree-laws n.° 169/2001 and n.° 155/2004) for cork oak. The term acorn is here used to refer the whole structure consisting of the pericarp and all the tissues enclosed by the pericarp including the seed. Acorns in the early stages of development (S1 to S4, Fig. 1a), were collected from 1 to 8 trees in each location and were immediately frozen in liquid nitrogen. To obtain the samples at S5 developmental stage and subsequent stages, branches bearing the acorns were kept at 4 ° C for up to 24 h before collecting the acorns and freezing in liquid nitrogen. In S8 stage samples, approximately 1/3 of the acorn part opposite to the embryo radicle was removed to minimize the presence of polysaccharides that could compromise the purity of isolated RNA. Additionally, some acorns from stage S3, S4, S5 and S8 were opened for embryo isolation (Fig. 1b); embryos from S8 stage were isolated by excising the embryo axis but excluding most cotyledonal tissue. In each collection, acorns and isolated embryos were carefully observed for evaluation and documentation of the developmental stage.
RNA extraction
Frozen samples were first ground in a mill (M 20 Universal mill, Ika) previously cooled with liquid nitrogen and then transferred to a cooled mortar and reduced to a fine powder. Total RNA was isolated following a protocol described by Reid et al. with some minor modifications [139]. RNA isolation was performed separately by developmental stage, date of collection and mother tree.
Total RNA was purified using the RNeasy MinElute Cleanup kit (Qiagen) with on-column DNase I treatment (Qiagen RNase-Free DNase Set) and only samples with A260/280 > 1.8 were used for further steps. RNA integrity was assessed in 1 % (w/v) agarose gels after ethidium bromide staining and for a rigorous assessment of RNA quality, the RNA samples were run on a RNA Pico6000 chip in Agilent 2100 Bioanalyzer RNA (Agilent). Additionally, each sample was quantified by fluorescence with the Quant-iT Ribogreen RNA Assay kit (Invitrogen).
Preparation of cDNA libraries and RNA-Seq
Two normalized and 5 non-normalized cDNA libraries were prepared. Normalized libraries were prepared with RNA isolated from acorn tissues or from isolated embryos, collected from a total of 5 and 3 trees, respectively. In each library, a pool of 2 μg total RNA containing equal amounts of RNA extracted from the several acorn (from S1 to S5, S7 and S8) or embryo developmental stages (from S3 to S5 and S8) was prepared. Double stranded cDNA was obtained using SMART technology [140] and the normalization was performed with the Duplex-Specific Nuclease (DSN) technology [141].
The 5 non-normalized cDNA libraries corresponded to different acorn developmental stages (S1, S2, S3 + S4, S5 and S8) collected from 7 different trees. For each library, a pool of total RNA was prepared containing 50 μg of total RNA. MicroPoly(A)Purist kit (Ambion) was used to isolate mRNA from each total RNA pool and 200 ng of mRNA were fragmented and used as template for double stranded cDNA production using cDNA Synthesis System Kit (Roche) followed by adaptor ligation.
Pyrosequencing of the normalized and non-normalized libraries was performed in the Titanium GS-FLX (454-Roche) at Biocant (Cantanhede, Portugal).
The data were deposited in the European Nucleotide Archive (ENA) under the accession number PRJEB6178/ERP005652. For each non-normalized library the accession numbers are the following: [ENA: ERX455655 for S1, ENA: ERX455656 for S2, ENA: ERX455657 for S3S4, ENA: ERX455658 for S5 and ENA: ERX455659 for S8]. For the normalized libraries [ENA: ERX455660 and ENA: ERX455661] are the accession numbers of the acorn and isolated embryo libraries, respectively.
Reads pre-processing, de novo assembly and transcriptome annotation
The full workflow is schematized in the Additional file 16. First, the raw reads were filtered by SeqTrimNext [142] to remove adapter sequences and low quality/complexity sequences, which included fragments (window of 15 nts) with a quality value lower than 20, more than an 80 % of indeterminations, or 75 % of polyA or polyT sequences. Fragments with an E-value < 1e-10 and 85 % identity to contaminants such as plastids, mitochondria, ribosome and virus/bacteria sequences, were trimmed. Final sequences shorter than 40 nts were also excluded. RNA-Seq data was de novo assembled using MIRA version 3.4.0 [50] and Newbler version 2.6 [49]. MIRA was executed with the default 454 settings and without clipping steps. Newbler was executed with the default parameters. The individual assemblies were merged with CAP3 with default options and an identity threshold of 95 %.
Transcripts were compared with the Uniprot and Trembl databases using NCBI Blastx with an E-value of 1e-6. Only full-length plant proteins were included in the target database. “Full Lengther Next” scripts (https://rubygems.org/gems/full_lengther_next) were used to compare the aligned regions in query and target, in order to determine the right translation frame and classify the transcripts as complete, internal or terminal. These translated proteins constitute the Q. suber proteome used in subsequent comparisons. For those transcripts without any alignment, the program runs an Open Reading Frame (ORF) prediction step. Novel ORFs with a result higher than 0.7 (default threshold) were annotated as novel transcripts. The conserved motifs and structures in the sequences of these novel transcripts were identified by comparison against the motifs databases in EBI Interpro (http://www.ebi.ac.uk/interpro/interproscan.html). Transcripts were compared with the NCBI non-redundant (nr) and Arabidopsis TAIR protein databases using NCBI Blastx with an E-value of 1e-10. Results were imported in Blast2GO [143] to annotate the gene ontology terms, enzymatic protein codes and KEGG pathways. The reads of each transcript belonging to the same pathway were summed up. For each pathway, the number of reads in each stage was transformed in Z-scores, clustered, and plotted in a heatmap using Mayday [144]. Gene ontology terms and NCBI COGs (Clusters of Orthologous Groups of proteins) associated to each Arabidopsis gene were downloaded from TAIR (www.arabidopsis.org) and NCBI (ftp://ftp.ncbi.nih.gov/pub/COG/KOG/), and associated back to the original Q. suber transcript.
The transcriptomes of other Fagaceae species were downloaded from NCBI and Fagaceae Genomics Project (www.fagaceae.org). The proteome for each of them was built in a similar way as for Q. suber by comparison with the Uniprot and Trembl full plant proteins using the Full Lengther Next scripts. Proteomes were compared by pairs using NCBI Blastp. Proteins in a query species were considered as having an orthologous in a target species if they shared both a minimal identity and coverage of 70 %.
An alignment of the reads obtained from 19 normalized cork oak cDNA libraries prepared from different tissues and made available by [45], against the reads obtained from the normalized libraries described in this work, was performed to identify the transcripts specifically expressed in fruit and seed tissues. The de novo transcripts without aligned reads were considered as fruit and seed specific.
Expression analysis and differentially expressed gene (DEG) clustering
The CLC Genomics Workbench (http://www.clcbio.com), version 6.7.2, was used to quantify the expression of the RNA-Seq data in four steps. Firstly, the reads from each of the five non-normalized libraries were aligned to the 80,357 contigs in the Q. suber assembly using the default scoring values and ignoring reads not uniquely mapping. Secondly, the number of aligned reads in each library was normalized by quartile normalization to take into account the different total number of reads per library. Third, a statistical analysis that compares the expected versus observed proportions of mapped reads by Kal’s z-test [145] implemented in CLC software between consecutive acorn developmental stages was used to identify the differentially expressed genes (DEGs). Finally, p-values were False Discovery Rate (FDR) corrected. Transcripts with a FDR value lower than 0.01 were considered as differentially expressed (DE).
DEGs were divided in 6 clusters according to the normalized number of aligned reads in each stage by Neural Gas clustering implemented in Mayday [144] based on Euclidian correlation between expression values. Neural Gas is an alternative to K-means clustering that resulted in clusters with a better balance of members. The list of transcripts in each cluster was used in Blast2GO to identify the enriched GO terms. Blast2GO enrichment analysis was based on a F-fisher test (FDR < 0.05). The relation among GO terms was assigned using REVIGO with the Resvik algorithm option [146] and R treemap library. DEGs annotated as related to response to water, including water transport and water deprivation were identified. We used the Plant Transcription Factor database (PLNTFDB, http://plntfdb.bio.uni-potsdam.de/) as reference to identify the TFs and other transcriptional regulators in our transcriptome. The database contains close to 30 K protein sequences of experimentally-identified elements from diverse plant species, and their classification in families according to their protein domains by HMM methods. The transcript sequences of the DEGs were aligned to the PLNTFDB using Blastx and a minimum E-value of 1e-10. We considered any transcript with a result under that threshold as a TF/transcriptional regulator, and annotated it within the family of the homologous with a lower E-value.
Quantitative RT-PCR analysis
Reverse transcription quantitative real-time PCR (RT-qPCR) of a set of 20 DEGs was carried out to validate the expression profile obtained by 454 sequencing (Table 6). RNA samples were first treated with TURBO DNase (Ambion) and afterwards all cDNAs were synthesized from 1.5 μg of total RNA using the Transcriptor High Fidelity cDNA Synthesis Kit (Roche) with the anchored-oligo(dT)18 primers. Specific primers were designed using Geneious software [147]. Quantitative real-time PCR experiments were then performed in LightCycler 480 (Roche) using SYBR Green I Master (Roche) and 96-well plates. For the genes tested, 3 biological replicates were used and the reaction mixtures were performed in a final volume of 16 μl containing 8 μl of 2× SYBR Green I Master, 400 nM of each primer and 1.5 μl of cDNA as template. The amplification program was the same for all genes tested: 95 ° C for 10 min, 45 cycles of 10 s at 95 ° C, 20 s at 60 ° C and 10 s at 72 ° C, except for β-AMYLASE 1 (BAM1), GALACTINOL SYNTHASE 2 (GolS2) and CALCIUM DEPENDENT PROTEIN KINASE 10 (CPK10) for which the annealing temperature was 62 ° C. A calibrator sample was used in each plate to normalize the values obtained and the potential differences among plates. Normalization was carried out with two reference genes ACTIN (ACT) and CLATHRIN ADAPTOR COMPLEXES (CAC) [139]. Normalized relative quantities were obtained through the formula , where E is the efficiency of the amplification for each primer pair in each tissue, f the number of reference genes used to normalize the data, goi and ref are the gene of interest and the reference gene, respectively, and ΔCt is the Ct of the calibrator minus the Ct value of the sample in test [148–150]. The data obtained from the RNA-Seq experiment and the RT-qPCR were compared. From the RNA-Seq a logarithmic ratio of base 2 between the counts of a gene in each developmental stage and the mean counts of the same gene in all developmental stages were made. A similar approach was followed for the data obtained by RT-qPCR by doing a logarithmic ratio of base 2 between the normalized quantities of the gene of interest in each developmental stage and the mean normalized quantities of the same gene in all developmental stages in analysis. For genes where the RNA-Seq values were zero in some of the developmental stages, a value of 1 was added to all the RNA-Seq results of those genes to avoid dividing by zero [151].
Availability of supporting data
The data sets supporting the results of this article are available in the European Nucleotide Archive (ENA) under the accession number PRJEB6178, http://www.ebi.ac.uk/ena/data/view/PRJEB6178. The reads libraries are also available in the ENA repository, http://www.ebi.ac.uk/ena/data/view/ERX455655-ERX455661. The cork oak transcriptome assembly performed in this work is directly available in ENA, http://www.ebi.ac.uk/ena/data/view/HABZ01000000, as well as each of the individual contigs, http://www.ebi.ac.uk/ena/data/view/HABZ01000001-HABZ01080357.
Acknowledgements
This project was funded by “Fundação para a Ciência e a Tecnologia” (FCT) through projects PEst-OE/EQB/LA0004/2011 and Research unit GREEN-it “Bioresources for Sustainability” (UID/Multi/04551/2013), SOBREIRO/0029/2009, IF/01168/2013, the doctoral fellowship to Andreia Miguel (SFRH/BD/44474/2008) and the post-doctoral fellowship to Liliana Marum (BPD/47679/2008).
Abbreviations
- DE
Differentially expressed
- DEGs
Differentially expressed genes
- ESTs
Expressed sequence tags
- TF
Transcription factor.
Additional files
Quercus suber transcript classification and predicted function. Annotation and classification of the assembled transcripts using Full Lengther Next scripts and functional annotation using the Uniprot database. Annotation and characterization (against the motifs databases from EBI Interpro) of the transcripts specific to the cork oak acorn.
Comparison of the transcriptomes of several Fagaceae tree species with full length plant proteins in UniprotKB. Transcriptomes were retrieved from the UniprotKB database.
Identification of shared proteins among some members of the Fagaceae family. Sequences from a query species (rows) with homologous in a different target species (columns). Proteins were considered homologous between the query and target species if they share a minimal identity and coverage of 70 %.
Quality assessment of the gene annotation. (A) Species with the best BLASTX alignment of each query sequence. (B) Values of similitude from Blastx alignments. (C) Number of gene ontology (GO) terms per sequence length.
Functional annotation of Quercus suber transcripts. Annotation was based on the homologous in NCBI and Arabidopsis (TAIR) databases using Blastx with an E-value of 1e-10 and results were submitted in Blast2GO to annotate gene ontology (GO) terms.
Classification of Orthologous Groups (COG). Analysis of the COGs present in data retrieved from Q. suber, CODB (Cork oak Database), Q. petraea and Q. robur and association to each Arabidopsis gene.
Distribution of Clusters of Orthologs Groups (COGs) between different species of the Quercus genus: Q. suber, Q. petraea and Q. robur and the data from the Cork Oak Database (CODB).
Biological domain of the clusters of Orthologous Groups of proteins (COGs) specific to Q. suber.
Number of cork oak transcripts in KEGG level 2 and 3 pathways. Expression of each pathway was quantified as the sum of the counts (mapped reads) of all the transcripts belonging to it.
Venn diagram showing the number of transcripts expressed in the different acorn developmental stages.
Identification of the genes differentially expressed during acorn development by Z-test. Results of the clustering, fold-change and functional annotation (NCBI, TAIR, UNIPROT) of the differentially expressed genes (DEGs) in consecutive stages of acorn development. Annotation of the DEGs specific to the cork oak acorn.
Enrichment analysis of the differentially expressed genes (DEGs). Comparison of the DEGs with the complete transcriptome evidencing the gene ontology (GO) terms over-represented in all the clusters and separately in each cluster.
Treemap of the GO terms from the DEGs. The area of each cell is proportional to the number of genes annotated with that term.
Differentially expressed genes related to response to water, water deprivation or water transport. Cluster annotation, Uniprot locus, association with Arabidopis homologs, normalized counts in each acorn developmental stage, fold-change between consecutive stages, stage expression factor (SEF) and contigs specific to each cluster.
Differentially expressed genes annotated as transcription factors and transcriptional regulators. Cluster annotation, Uniprot locus, association with Arabidopis homologs, normalized counts in each acorn developmental stage, fold-change between consecutive stages, stage expression factor (SEF) and contigs specific to each cluster.
Workflow of the 454 sequencing data analyses. After sequencing, the raw reads obtained were pre-processed to remove non desired sequences, including low quality sequences and contaminants. The clean reads from all libraries were then assembled using two different assemblers and then joined using CAP3. The obtained contigs that were then associated to gene ontology (GO) terms and searched for transcription factors. After mapping the differentially expressed genes (DEGs) along cork oak acorn development were identified.
Footnotes
Andreia Miguel and José de Vega-Bartol contributed equally to this work.
Competing interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Authors’ contributions
This study was conceived by CMM, MCV and JL and directed by CMM. Collection and identification of field material was performed by AM, LM, MCV, JL and CMM. Sample preparation and nucleic acid isolation were performed by AM and TS. RT-qPCR was performed by AM and LM. Data analysis and biological interpretation of results were conducted by AM, JVB, IC and CMM. AM, JVB and CMM wrote the paper. All authors read and approved the final manuscript.
Contributor Information
Andreia Miguel, Email: andreiamiguel@itqb.unl.pt.
José de Vega-Bartol, Email: devegabartol@itqb.unl.pt.
Liliana Marum, Email: marum@itqb.unl.pt.
Inês Chaves, Email: ichaves@itqb.unl.pt.
Tatiana Santo, Email: ts.tatianasanto@gmail.com.
José Leitão, Email: jleitao@ualg.pt.
Maria Carolina Varela, Email: carolina.varela@iniav.pt.
Célia M. Miguel, Phone: +351 214469627, Email: cmiguel@itqb.unl.pt
References
- 1.Ruan YL, Patrick JW, Bouzayen M, Osorio S, Fernie AR. Molecular regulation of seed and fruit set. Trends Plant Sci. 2012;17:656–65. doi: 10.1016/j.tplants.2012.06.005. [DOI] [PubMed] [Google Scholar]
- 2.Alba R, Payton P, Fei Z, McQuinn R, Debbie P, Martin GB, Tanksley SD, Giovannoni JJ. Transcriptome and selected metabolite analyses reveal multiple points of ethylene control during tomato fruit development. Plant Cell. 2005;17:2954–65. doi: 10.1105/tpc.105.036053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Rocco M, D’Ambrosio C, Arena S, Mirelle F, Scaloni A, Marra M. Proteomic analysis of tomato fruits from two ecotypes during ripening. Proteomics. 2006;6:3781–91. doi: 10.1002/pmic.200600128. [DOI] [PubMed] [Google Scholar]
- 4.Mintz-Oron S, Mandel T, Rogachev I, Feldberg L, Lotan O, Yativ M, Wang Z, Jetter R, Venger I, Adato A, Aharoni A. Gene expression and metabolism in tomato fruit surface tissues. Plant Physiol. 2008;147:823–51. doi: 10.1104/pp.108.116004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Vriezen WH, Feron R, Maretto F, Keijman J, Mariani C. Changes in tomato ovary transcriptome demonstrate complex hormonal regulation of fruit set. New Phytol. 2008;177:60–76. doi: 10.1111/j.1469-8137.2007.02254.x. [DOI] [PubMed] [Google Scholar]
- 6.Karlova R, Rosin FM, Busscher-Lange J, Parapunova V, Phuc TD, Fernie AR, Fraser PD, Baxter C, Angenent GC, Maagd RA. Transcriptome and metabolite profiling show that APETALA2a is a major regulator of tomato fruit ripening. Plant Cell. 2011;23:923–41. doi: 10.1105/tpc.110.081273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Osorio S, Alba R, Damasceno CMB, Lopez-Casado G, Lohse M, Zanor MI, Tohge T, Usadel B, Rose JKC, Fei Z, Giovannoni JJ, Fernie AR. Systems biology of tomato fruit development: Combined transcript, protein, and metabolite analysis of tomato transcription factor (nor, rin) and ethylene receptor (Nr) mutants reveals novel regulatory interactions. Plant Physiol. 2011;157:405–25. doi: 10.1104/pp.111.175463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Goes F, Iandolino A, Al-kayal F, Bohlmann MC, Cushman MA, Lim H, Ergul A, Figueroa R, Kabuloglu EK, Osborne C, Rowe J, Tattersall E, Leslie A, Xu J, Baek J, Cramer GR, Cushman JC, Cook DR. Characterizing the grape transcriptome. Analysis of Expressed Sequence Tags from multiple Vitis species and development of a compendium of gene expression during berry development. Plant Physiol. 2005;139:574–97. doi: 10.1104/pp.105.065748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Peng FY, Reid KE, Liao N, Schlosser J, Lijavetzky D, Holt R, Martínez Zapater JM, Jones S, Marra M, Bohlmann J, Lund ST. Generation of ESTs in Vitis vinifera wine grape (Cabernet Sauvignon) and table grape (Muscat Hamburg) and discovery of new candidate genes with potential roles in berry development. Gene. 2007;402:40–50. doi: 10.1016/j.gene.2007.07.016. [DOI] [PubMed] [Google Scholar]
- 10.Sweetman C, Wong DC, Ford CM, Drew DP. Transcriptome analysis at four developmental stages of grape berry (Vitis vinifera cv. Shiraz) provides insights into regulated and coordinated gene expression. BMC Genomics. 2012;13:691. doi: 10.1186/1471-2164-13-691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Rowland LJ, Alkharouf N, Darwish O, Ogden EL, Polashock JJ, Bassil NV, Main D. Generation and analysis of blueberry transcriptome sequences from leaves, developing fruit, and flower buds from cold acclimation through deacclimation. BMC Plant Biol. 2012;12:46. doi: 10.1186/1471-2229-12-46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Gupta V, Estrada AD, Blakley I, Reid R, Patel K, Meyer MD, et al. RNA-Seq analysis and annotation of a draft blueberry genome assembly identifies candidate genes involved in fruit ripening, biosynthesis of bioactive compounds, and stage-specific alternative splicing. Gigascience. 2015;4. [DOI] [PMC free article] [PubMed]
- 13.Yu K, Xu Q, Da X, Guo F, Ding Y, Deng X. Transcriptome changes during fruit development and ripening of sweet orange (Citrus sinensis) BMC Genomics. 2012;13:10. doi: 10.1186/1471-2164-13-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Clepet C, Joobeur T, Zheng Y, Jublot D, Huang M, Truniger V, Boualem A, Hernandez-Gonzalez ME, Dolcet-Sanjuan R, Portnoy V, Mascarell-Creus A, Caño-Delgado AI, Katzir N, Bendahmane A, Giovannoni JJ, Aranda MA, Garcia-Mas J, Fei Z. Analysis of expressed sequence tags generated from full-length enriched cDNA libraries of melon. BMC Genomics. 2011;12:252. doi: 10.1186/1471-2164-12-252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Moore S, Vrebalov J, Payton P, Giovannoni J. Use of genomics tools to isolate key ripening genes and analyse fruit maturation in tomato. J Exp Bot. 2002;53:2023–30. doi: 10.1093/jxb/erf057. [DOI] [PubMed] [Google Scholar]
- 16.Fei Z, Tang X, Alba RM, White JA, Ronning CM, Martin GB, Tanksley SD, Giovannoni JJ. Comprehensive EST analysis of tomato and comparative genomics of fruit ripening. Plant J. 2004;40:47–59. doi: 10.1111/j.1365-313X.2004.02188.x. [DOI] [PubMed] [Google Scholar]
- 17.Park S, Sugimoto N, Larson MD, Beaudry R, Nocker SV. Identification of genes with potential roles in apple fruit development and biochemistry through large-scale statistical analysis of Expressed Sequence Tags. Plant Physiol. 2006;141:811–24. doi: 10.1104/pp.106.080994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Zhang G, Pan L, Yin Y, Liu W, Huang D, Zhang T, Wang L, Xin C, Lin Q, Sun G, Abdullah MMB, Zhang X, Hu S, Al-Mssallem IS, Yu J. Large-scale collection and annotation of gene models for date palm (Phoenix dactylifera, L.) Plant Mol Biol. 2012;79:521–36. doi: 10.1007/s11103-012-9924-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Bugalho MN, Caldeira MC, Pereira JS, Aronson J, Pausas JG. Mediterranean cork oak savannas require human use to sustain biodiversity and ecosystem services. Front Ecol Environ. 2011;9:278–86. [Google Scholar]
- 20.Aronson J, Pereira JS, Pausas JG. Cork Oak Woodlands on the Edge: Ecology, Adaptive Management, and Restoration. Washington DC: Island Press; 2009. pp. 11–21. [Google Scholar]
- 21.Ramírez-Valiente J, Lorenzo Z, Soto A, Valladares F, Gil L, Aranda I. Elucidating the role of genetic drift and natural selection in cork oak differentiation regarding drought tolerance. Mol Ecol. 2009;18:3803–15. doi: 10.1111/j.1365-294X.2009.04317.x. [DOI] [PubMed] [Google Scholar]
- 22.Silva SP, Sabino MA, Fernandes EM, Correlo VM, Boesel LF, Reis RL. Cork: properties, capabilities and applications. Int Mater Rev. 2005;50:345–65. [Google Scholar]
- 23.Corti R. Ricerche sul ciclo riproduttivo di specie del genere Quercus della Flora Italiana. II. Contributo alla biologia e allla sistematica di Q. suber L. e in particolare della forma a sviluppo biennale della ghianda. Ann Acad Ital Sci For. 1955;4:55–133. [Google Scholar]
- 24.Merouani H, Apolinário L, Almeida M, Pereira J. Morphological and physiological maturation of acorns of cork oak (Quercus suber L.) Seed Sci Technol. 2003;31:111–24. [Google Scholar]
- 25.Díaz-Fernández PM, Climent J, Gil L. Biennial acorn maturation and its relationship with flowering phenology in Iberian populations of Quercus suber. Trees. 2004;18:615–21. [Google Scholar]
- 26.Vinagre PR, Santos L, Nóbrega F, Varela MC: Estudos comparativos entre as duas primeiras frutificações do sobreiro: bastão e lande. In Sociedade Portuguesa de Ciências Florestais (ed) Proceedings of the V Congresso Nacional Florestal, 16-19 May 2005 (Insituto Politécnico de Viseu, Portugal).
- 27.Pons J, Pausas JG. The coexistence of acorns with different maturation patterns explains acorn production variability in cork oak. Oecologia. 2012;169:723–31. doi: 10.1007/s00442-011-2244-1. [DOI] [PubMed] [Google Scholar]
- 28.Ramírez-Valiente JA, Valladares F, Gil L, Aranda I. Population differences in juvenile survival under increasing drought are mediated by seed size in cork oak (Quercus suber L.) For Ecol Manage. 2009;257:1676–83. [Google Scholar]
- 29.Bonito A, Varone L, Gratani L. Relationship between acorn size and seedling morphological and physiological traits of Quercus ilex L. from different climates. Photosynthetica. 2011;49:75–86. [Google Scholar]
- 30.Moles AT, Westoby M. Seedling survival and seed size: a synthesis of the literature. J Ecol. 2004;92:372–83. [Google Scholar]
- 31.Leishman MR, Wright IJ, Moles AT, Westoby M: The evolutionary ecology of seed size. In Fenner M (ed) Seeds: the ecology of regeneration in plant communities. CABI Publishing, Wallingford, UK; 2000:31–58.
- 32.Gómez JM. Bigger is not always better: conflicting selective pressures on seed size in Quercus ilex. Evolution. 2004;58:71–80. doi: 10.1111/j.0014-3820.2004.tb01574.x. [DOI] [PubMed] [Google Scholar]
- 33.Boavida LC, Varela C, Feijó JA. Sexual reproduction in the cork oak (Quercus suber L.). I. The progamic phase. Sex Plant Reprod. 1999;11:347–53. [Google Scholar]
- 34.Belletti P: Seed and seedling management in cork oak. In Management and long term storage of cork-oak acorns. The research of European Community DIVAPRA, Turin; 2001:1–9.
- 35.Belletti P, Monteleone I, Cartarasa M: Cork-oak and stone-oak seed management aimed at implementing nursery production of seedlings. In Nursery production and stand establishment of broad-leaves to promote sustainable forest management. Edited by Ciccarese L, Lucci S, Mattsson A, APAT – Italy`s agency for the protection of the environment and for technical services, Rome; 2004:21–24.
- 36.Pausas JG, Marañón T, Caldeira M, Pons J: Natural Regeneration. In Cork Oak Woodlands on the Edge. Volume 1. Edited by Aronson J, Pereira JS, Pausas JG. Island Press, Washington DC; 2009:115–125.
- 37.Elena-Rosello J, Rio J, Valdecantos JG, Santamaria I. Ecological aspects of the floral phenology of the cork-oak (Q. suber. L): why do annual and biennial biotypes appear? Ann For Sci. 1993;50:114–21. [Google Scholar]
- 38.Boavida LC, Silva JP, Feijó JA. Sexual reproduction in the cork oak (Quercus suber L). II. Crossing intra- and interspecific barriers. Sex Plant Reprod. 2001;14:143–52. [Google Scholar]
- 39.Varela M, Valdiviesso T. Phenological phases of Quercus suber L. flowering. For Genet. 1996;3:93–102. [Google Scholar]
- 40.Rocheta M, Sobral R, Magalhães J, Amorim MI, Ribeiro T, Pinheiro M, Egas C, Morais-Cecílio L, Costa MMR. Comparative transcriptomic analysis of male and female flowers of monoecious Quercus suber. Front Plant Sci. 2014;5:1–16. doi: 10.3389/fpls.2014.00599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Borgardt S, Nixon K. A comparative flower and fruit anatomical study of Quercus acutissima, a biennial-fruiting oak from the Cerris group (Fagaceae) Am J Bot. 2003;90:1567–84. doi: 10.3732/ajb.90.11.1567. [DOI] [PubMed] [Google Scholar]
- 42.Ueno S, Le Provost G, Léger V, Klopp C, Noirot C, Frigerio J-M, Salin F, Salse J, Abrouk M, Murat F, Brendel O, Derory J, Abadie P, Léger P, Cabane C, Barré A, de Daruvar A, Couloux A, Wincker P, Reviron M-P, Kremer A, Plomion C. Bioinformatic analysis of ESTs collected by Sanger and pyrosequencing methods for a keystone forest tree species: oak. BMC Genomics. 2010;11:650. doi: 10.1186/1471-2164-11-650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Ueno S, Klopp C, Leplé JC, Derory J, Noirot C, Léger V, Prince E, Kremer A, Plomion C, Le Provost G. Transcriptional profiling of bud dormancy induction and release in oak by next-generation sequencing. BMC Genomics. 2013;14:236. doi: 10.1186/1471-2164-14-236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Rampant PF, Lesur I, Boussardon C, Bitton F, Martin-Magniette M-L, Bodénès C, Le Provost G, Bergès H, Fluch S, Kremer A, Plomion C. Analysis of BAC end sequences in oak, a keystone forest tree species, providing insight into the composition of its genome. BMC Genomics. 2011;12:292. doi: 10.1186/1471-2164-12-292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Pereira-Leal JB, Abreu IA, Alabaça CS, Almeida MH, Almeida P, Almeida T, Amorim MI, Araújo S, Azevedo H, Badia A, Batista D, Bohn A, Capote T, Carrasquinho I, Chaves I, Coelho AC, Costa MM, Costa R, Cravador A, Egas C, Faro C, Fortes AM, Fortunato AS, Gaspar MJ, Gonçalves S, Graça J, Horta M, Inácio V, Leitão JM, Lino-Neto T, et al. A comprehensive assessment of the transcriptome of cork oak (Quercus suber) through EST sequencing. BMC Genomics. 2014;15:371. doi: 10.1186/1471-2164-15-371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Seymour GB, Østergaard L, Chapman NH, Knapp S, Martin C. Fruit development and ripening. Annu Rev Plant Biol. 2013;64:219–41. doi: 10.1146/annurev-arplant-050312-120057. [DOI] [PubMed] [Google Scholar]
- 47.Van der Pijl L. Principles of Dispersal in Higher Plants. Volume 26. Berlin Heidelberg: Springer; 1982. [Google Scholar]
- 48.Chan YH. Biostatistics 104: Correlational Analysis. Singapore Med J. 2003;44:614–9. [PubMed] [Google Scholar]
- 49.Margulies M, Egholm M, Altman WE, Attiya S, Bader JS, Bemben LA, Berka J, Braverman MS, Chen Y-J, Chen Z, Dewell SB, Du L, Fierro JM, Gomes XV, Godwin BC, He W, Helgesen S, Ho CH, Ho CH, Irzyk GP, Jando SC, Alenquer MLI, Jarvie TP, Jirage KB, Kim J-B, Knight JR, Lanza JR, Leamon JH, Lefkowitz SM, Lei M, et al. Genome sequencing in microfabricated high-density picolitre reactors. Nature. 2005;437:376–80. doi: 10.1038/nature03959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Chevreux B, Pfisterer T, Drescher B, Driesel AJ, Müller WEG, Wetter T, Suhai S. Using the miraEST assembler for reliable and automated mRNA transcript assembly and SNP detection in sequenced ESTs. Genome Res. 2004;14:1147–59. doi: 10.1101/gr.1917404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Weber APM, Weber KL, Carr K, Wilkerson C, Ohlrogge JB. Sampling the Arabidopsis transcriptome with massively parallel pyrosequencing. Plant Physiol. 2007;144:32–42. doi: 10.1104/pp.107.096677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Kumar S, Blaxter ML. Comparing de novo assemblers for 454 transcriptome data. BMC Genomics. 2010;11:571. doi: 10.1186/1471-2164-11-571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Garg R, Patel RK, Jhanwar S, Priya P, Bhattacharjee A, Yadav G, Bhatia S, Chattopadhyay D, Tyagi AK, Jain M. Gene discovery and tissue-specific transcriptome analysis in chickpea with massively parallel pyrosequencing and web resource development. Plant Physiol. 2011;156:1661–78. doi: 10.1104/pp.111.178616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Mundry M, Bornberg-Bauer E, Sammeth M, Feulner PGD. Evaluating characteristics of de novo assembly software on 454 transcriptome data: a simulation approach. PLoS One. 2012;7 doi: 10.1371/journal.pone.0031410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Huang X, Madan A. CAP3: A DNA sequence assembly program. Genome Res. 1999;9:868–77. doi: 10.1101/gr.9.9.868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Koch K. Sucrose metabolism: regulatory mechanisms and pivotal roles in sugar sensing and plant development. Curr Opin Plant Biol. 2004;7:235–46. doi: 10.1016/j.pbi.2004.03.014. [DOI] [PubMed] [Google Scholar]
- 57.Baroja-Fernández E, Muñoz FJ, Li J, Bahaji A, Almagro G, Montero M, Etxeberria E, Hidalgo M, Sesma MT, Pozueta-Romero J. Sucrose synthase activity in the sus1/sus2/sus3/sus4 Arabidopsis mutant is sufficient to support normal cellulose and starch production. Proc Natl Acad Sci U S A. 2012;109:321–6. doi: 10.1073/pnas.1117099109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Bray EA. Genes commonly regulated by water-deficit stress in Arabidopsis thaliana. J Exp Bot. 2004;55:2331–41. doi: 10.1093/jxb/erh270. [DOI] [PubMed] [Google Scholar]
- 59.Le BH, Cheng C, Bui AQ, Wagmaister JA, Henry KF, Pelletier J, Kwong L, Belmonte M, Kirkbride R, Horvath S, Drews GN, Fischer RL, Okamuro JK, Harada JJ, Goldberg RB. Global analysis of gene activity during Arabidopsis seed development and identification of seed-specific transcription factors. Proc Natl Acad Sci U S A. 2010;107:8063–70. doi: 10.1073/pnas.1003530107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Barnabás B, Jäger K, Fehér A. The effect of drought and heat stress on reproductive processes in cereals. Plant, Cell Environ. 2008;31:11–38. doi: 10.1111/j.1365-3040.2007.01727.x. [DOI] [PubMed] [Google Scholar]
- 61.Fujita M, Fujita Y, Maruyama K, Seki M, Hiratsu K, Ohme-Takagi M, Tran L-SP, Yamaguchi-Shinozaki K, Shinozaki K. A dehydration-induced NAC protein, RD26, is involved in a novel ABA-dependent stress-signaling pathway. Plant J. 2004;39:863–76. doi: 10.1111/j.1365-313X.2004.02171.x. [DOI] [PubMed] [Google Scholar]
- 62.Yamaguchi-Shinozaki K, Shinozaki K. The plant hormone abscisic acid mediates the drought-induced expression but not the seed-specific expression of rd22, a gene responsive to dehydration stress in Arabidopsis thaliana. MGG Mol Gen Genet. 1993;238:17–25. doi: 10.1007/BF00279525. [DOI] [PubMed] [Google Scholar]
- 63.Tran LP, Nakashima K, Sakuma Y, Simpson SD, Fujita Y, Maruyama K, Fujita M, Seki M, Shinozaki K, Yamaguchi-shinozaki K. Isolation and functional analysis of Arabidopsis stress-inducible NAC transcription factors that bind to a drought-responsive cis-element in the early responsive to dehydration stress 1 Promoter. Plant Cell. 2004;16:2481–98. doi: 10.1105/tpc.104.022699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Singh AK, Sharma V, Pal AK, Acharya V, Ahuja PS. Genome-wide organization and expression profiling of the NAC transcription factor family in potato (Solanum tuberosum L.) DNA Res. 2013;20:403–23. doi: 10.1093/dnares/dst019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Xu H, Li Y, Yan Y, Wang K, Gao Y, Hu Y. Genome-scale identification of soybean BURP domain-containing genes and their expression under stress treatments. BMC Plant Biol. 2010;10:197. doi: 10.1186/1471-2229-10-197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Park MY, Chung MS, Koh HS, Lee DJ, Ahn SJ, Kim CS. Isolation and functional characterization of the Arabidopsis salt-tolerance 32 (AtSAT32) gene associated with salt tolerance and ABA signaling. Physiol Plant. 2009;135:426–35. doi: 10.1111/j.1399-3054.2008.01202.x. [DOI] [PubMed] [Google Scholar]
- 67.Song H, Zhao R, Fan P, Wang X, Chen X, Li Y. Overexpression of AtHsp90.2, AtHsp90.5 and AtHsp90.7 in Arabidopsis thaliana enhances plant sensitivity to salt and drought stresses. Planta. 2009;229:955–64. doi: 10.1007/s00425-008-0886-y. [DOI] [PubMed] [Google Scholar]
- 68.Kim EY, Seo YS, Park KY, Kim SJ, Kim WT. Overexpression of CaDSR6 increases tolerance to drought and salt stresses in transgenic Arabidopsis plants. Gene. 2014;552:146–54. doi: 10.1016/j.gene.2014.09.028. [DOI] [PubMed] [Google Scholar]
- 69.Matus JT, Aquea F, Espinoza C, Vega A, Cavallini E, Santo SD, Cañón P, de la Guardia AR-H, Serrano J, Tornielli GB, Arce-Johnson P. Inspection of the grapevine BURP superfamily highlights an expansion of RD22 genes with distinctive expression features in berry development and ABA-mediated stress responses. PLoS One. 2014;9 doi: 10.1371/journal.pone.0110372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Terasaka K, Blakeslee JJ, Titapiwatanakun B, Peer WA, Bandyopadhyay A, Makam SN, Lee OR, Richards EL, Murphy AS, Sato F, Yazaki K. PGP4, an ATP binding cassette P-glycoprotein, catalyzes auxin transport in Arabidopsis thaliana roots. Plant Cell. 2005;17:2922–39. doi: 10.1105/tpc.105.035816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Tripathi V, Syed N, Laxmi A, Chattopadhyay D. Role of CIPK6 in root growth and auxin transport. Plant Signal Behav. 2009;4:663–5. doi: 10.4161/psb.4.7.9002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Cui F, Liu L, Zhao Q, Zhang Z, Li Q, Lin B, Wu Y, Tang S, Xie Q. Arabidopsis ubiquitin conjugase UBC32 is an ERAD component that functions in brassinosteroid-mediated salt stress tolerance. Plant Cell. 2012;24:233–44. doi: 10.1105/tpc.111.093062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Wang Y, Wang W, Cai J, Zhang Y, Qin G, Tian S. Tomato nuclear proteome reveals the involvement of specific E2 ubiquitin-conjugating enzymes in fruit ripening. Genome Biol. 2014;15. [DOI] [PMC free article] [PubMed]
- 74.Kaplan F, Guy CL. β-Amylase induction and the protective role of maltose during temperature shock. Plant Physiol. 2004;135:1674–84. doi: 10.1104/pp.104.040808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Moore JP, Le NT, Brandt WF, Driouich A, Farrant JM. Towards a systems-based understanding of plant desiccation tolerance. Trends Plant Sci. 2009;14:110–7. doi: 10.1016/j.tplants.2008.11.007. [DOI] [PubMed] [Google Scholar]
- 76.Quettier AL, Bertrand C, Habricot Y, Miginiac E, Agnes C, Jeannette E, Maldiney R. The phs1-3 mutation in a putative dual-specificity protein tyrosine phosphatase gene provokes hypersensitive responses to abscisic acid in Arabidopsis thaliana. Plant J. 2006;47:711–9. doi: 10.1111/j.1365-313X.2006.02823.x. [DOI] [PubMed] [Google Scholar]
- 77.Ma J, Jiang QT, Zhang XW, Lan XJ, Pu ZE, Wei YM, Liu C, Lu ZX, Zheng YL. Structure and expression of barley starch phosphorylase genes. Planta. 2013;238:1081–93. doi: 10.1007/s00425-013-1953-6. [DOI] [PubMed] [Google Scholar]
- 78.Satoh H, Shibahara K, Tokunaga T, Nishi A, Tasaki M, Hwang S-K, Okita TW, Kaneko N, Fujita N, Yoshida M, Hosaka Y, Sato A, Utsumi Y, Ohdan T, Nakamura Y. Mutation of the plastidial alpha-glucan phosphorylase gene in rice affects the synthesis and structure of starch in the endosperm. Plant Cell. 2008;20:1833–49. doi: 10.1105/tpc.107.054007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Parcy F, Valon C, Raynal M, Gaubier-Comella P, Delseny M, Giraudat J. Regulation of gene expression programs during Arabidopsis seed development: roles of the ABI3 locus and of endogenous abscisic acid. Plant Cell. 1994;6:1567–82. doi: 10.1105/tpc.6.11.1567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Liu X, Zhang H, Zhao Y, Feng Z, Li Q, Yang H-Q, Luan S, Li J, He Z-H. Auxin controls seed dormancy through stimulation of abscisic acid signaling by inducing ARF-mediated ABI3 activation in Arabidopsis. Proc Natl Acad Sci U S A. 2013;110:15485–90. doi: 10.1073/pnas.1304651110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Hare PD, Cress WA, van Staden J. Proline synthesis and degradation: a model system for elucidating stress-related signal transduction. J Exp Bot. 1999;50:413–34. [Google Scholar]
- 82.Prewein C, Endemann M, Reinöhl V, Salaj J, Sunderlikova V, Wilhelm E. Physiological and morphological characteristics during development of pedunculate oak (Quercus robur L.) zygotic embryos. Trees. 2005;20:53–60. [Google Scholar]
- 83.Finch-Savage WE, Clay HA. Evidence that ethylene, light and abscisic acid interact to inhibit germination in the recalcitrant seeds of Quercus robur L. J Exp Bot. 1994;45:1295–9. [Google Scholar]
- 84.Kiyosue T, Yamaguchi-Shinozaki K, Shinozaki K. Cloning of cDNAs for genes that are early-responsive to dehydration stress (ERDs) in Arabidopsis thaliana L.: identification of three ERDs as HSP cognate genes. Plant Mol Biol. 1994;25:791–8. doi: 10.1007/BF00028874. [DOI] [PubMed] [Google Scholar]
- 85.Alves MS, Fietto LG: Functional Diversity of Early Responsive to Dehydration (ERD) Genes in Soybean. In A Comprehensive Survey of International Soybean Research - Genetics, Physiology, Agronomy and Nitrogen Relationships. Edited by E. J.; 2013:475–487.
- 86.Kariola T, Brader G, Helenius E, Li J, Heino P, Palva ET. EARLY RESPONSIVE TO DEHYDRATION 15, a negative regulator of abscisic acid responses in Arabidopsis. Plant Physiol. 2006;142:1559–73. doi: 10.1104/pp.106.086223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Aalto MK, Helenius E, Kariola T, Pennanen V, Heino P, Hõrak H, Puzõrjova I, Kollist H, Palva ET. ERD15-An attenuator of plant ABA responses and stomatal aperture. Plant Sci. 2012;182:19–28. doi: 10.1016/j.plantsci.2011.08.009. [DOI] [PubMed] [Google Scholar]
- 88.Seo PJ, Lee SB, Suh MC, Park M-J, Go YS, Park C-M. The MYB96 transcription factor regulates cuticular wax biosynthesis under drought conditions in Arabidopsis. Plant Cell. 2011;23:1138–52. doi: 10.1105/tpc.111.083485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Guo L, Yang H, Zhang X, Yang S. Lipid transfer protein 3 as a target of MYB96 mediates freezing and drought stress in Arabidopsis. J Exp Bot. 2013;64:1755–67. doi: 10.1093/jxb/ert040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Candat A, Paszkiewicz G, Neveu M, Gautier R, Logan DC, Avelange-Macherel M-H, Macherel D. The ubiquitous distribution of Late Embryogenesis Abundant proteins across cell compartments in Arabidopsis offers tailored protection against abiotic stress. Plant Cell. 2014;26:1–20. doi: 10.1105/tpc.114.127316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Olvera-Carrillo Y, Campos F, Reyes JL, Garciarrubio A, Covarrubias AA. Functional analysis of the group 4 late embryogenesis abundant proteins reveals their relevance in the adaptive response during water deficit in Arabidopsis. Plant Physiol. 2010;154:373–90. doi: 10.1104/pp.110.158964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Hundertmark M, Hincha DK. LEA (late embryogenesis abundant) proteins and their encoding genes in Arabidopsis thaliana. BMC Genomics. 2008;9:118. doi: 10.1186/1471-2164-9-118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Hualkasin W, Thongin W, Petsean K, Phongdara A, Nakkaew A. Molecular cloning and characterization of the late embryogenesis abundant group 4 (EgLEA4) gene from oil palm (Elaeis guineensis Jacq) Songklanakarin J Sci Technol. 2013;35:275–85. [Google Scholar]
- 94.Sunderlíková V, Wilhelm E. High accumulation of legumin and Lea-like mRNAs during maturation is associated with increased conversion frequency of somatic embryos from pedunculate oak (Quercus robur L.) Protoplasma. 2002;220:97–103. doi: 10.1007/s00709-002-0025-8. [DOI] [PubMed] [Google Scholar]
- 95.Sunderlíková V, Salaj J, Kopecky D, Salaj T, Wilhem E, Matusíková I. Dehydrin genes and their expression in recalcitrant oak (Quercus robur) embryos. Plant Cell Rep. 2009;28:1011–21. doi: 10.1007/s00299-009-0710-6. [DOI] [PubMed] [Google Scholar]
- 96.Tognetti VB, Van Aken O, Morreel K, Vandenbroucke K, van de Cotte B, De Clercq I, Chiwocha S, Fenske R, Prinsen E, Boerjan W, Genty B, Stubbs KA, Inzé D, Van Breusegem F. Perturbation of indole-3-butyric acid homeostasis by the UDP-glucosyltransferase UGT74E2 modulates Arabidopsis architecture and water stress tolerance. Plant Cell. 2010;22:2660–79. doi: 10.1105/tpc.109.071316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Xue T, Li X, Zhu W, Wu C, Yang G, Zheng C. Cotton metallothionein GhMT3a, a reactive oxygen species scavenger, increased tolerance against abiotic stress in transgenic tobacco and yeast. J Exp Bot. 2009;60:339–49. doi: 10.1093/jxb/ern291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Reid SJ, Ross GS. Up-regulation of two cDNA clones encoding metallothionein-like proteins in apple fruit during cool storage. Physiol Plant. 1997;100:183–9. [Google Scholar]
- 99.Abdullah SNA, Cheah SC, Murphy DJ. Isolation and characterisation of two divergent type 3 metallothioneins from oil palm, Elaeis guineensis. Plant Physiol Biochem. 2002;40:255–63. [Google Scholar]
- 100.Ledger SE, Gardner RC. Cloning and characterization of five cDNAs for genes differentially expressed during fruit development of kiwifruit (Actinidia deliciosa var. deliciosa) Plant Mol Biol. 1994;25:877–86. doi: 10.1007/BF00028882. [DOI] [PubMed] [Google Scholar]
- 101.Dabrowska G, Mierek-Adamska A, Goc A. Plant metallothioneins: Putative functions identified by promoter analysis in silico. Acta Biol Cracoviensia Ser Bot. 2012;54:109–20. [Google Scholar]
- 102.Liscum E, Reed JW. Genetics of Aux / IAA and ARF action in plant growth and development. Plant Mol Biol. 2002;49:387–400. [PubMed] [Google Scholar]
- 103.Sagar M, Chervin C, Mila I, Hao Y, Roustan J-P, Benichou M, Gibon Y, Biais B, Maury P, Latché A, Pech J-C, Bouzayen M, Zouine M. SlARF4, an auxin response factor involved in the control of sugar metabolism during tomato fruit development. Plant Physiol. 2013;161:1362–74. doi: 10.1104/pp.113.213843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Chaves I, Lin YC, Pinto-Ricardo C, Van de Peer Y, Miguel C. miRNA profiling in leaf and cork tissues of Quercus suber reveals novel miRNAs and tissue-specific expression patterns. Tree Genet Genomes. 2014;10:721–37. [Google Scholar]
- 105.Pagnussat GC, Yu H-J, Sundaresan V. Cell-fate switch of synergid to egg cell in Arabidopsis eostre mutant embryo sacs arises from misexpression of the BEL1-like homeodomain gene BLH1. Plant Cell. 2007;19:3578–92. doi: 10.1105/tpc.107.054890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Sakakibara K, Ando S, Yip HK, Tamada Y, Hiwatashi Y, Murata T, Deguchi H, Hasebe M, Bowman JL. KNOX2 genes regulate the haploid-to-diploid morphological transition in land plants. Science. 2013;339:1067–70. doi: 10.1126/science.1230082. [DOI] [PubMed] [Google Scholar]
- 107.Allan AC, Hellens RP, Laing WA. MYB transcription factors that colour our fruit. Trends Plant Sci. 2008;13:99–102. doi: 10.1016/j.tplants.2007.11.012. [DOI] [PubMed] [Google Scholar]
- 108.Cominelli E, Tonelli C. A new role for plant R2R3-MYB transcription factors in cell cycle regulation. Cell Res. 2009;19:1231–2. doi: 10.1038/cr.2009.123. [DOI] [PubMed] [Google Scholar]
- 109.Vimolmangkang S, Han Y, Wei G, Korban SS. An apple MYB transcription factor, MdMYB3, is involved in regulation of anthocyanin biosynthesis and flower development. BMC Plant Biol. 2013;13:176. doi: 10.1186/1471-2229-13-176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Zoratti L, Karppinen K, Luengo Escobar A, HÃggman H, Jaakola L. Light-controlled flavonoid biosynthesis in fruits. Front Plant Sci. 2014;5:1–16. doi: 10.3389/fpls.2014.00534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Stracke R, Werber M, Weisshaar B. The R2R3-MYB gene family in Arabidopsis thaliana. Curr Opin Plant Biol. 2001;4:447–56. doi: 10.1016/s1369-5266(00)00199-0. [DOI] [PubMed] [Google Scholar]
- 112.Koes R, Verweij W, Quattrocchio F. Flavonoids: a colorful model for the regulation and evolution of biochemical pathways. Trends Plant Sci. 2005;10:236–42. doi: 10.1016/j.tplants.2005.03.002. [DOI] [PubMed] [Google Scholar]
- 113.Bogs J, Jaffé FW, Takos AM, Walker AR, Robinson SP. The grapevine transcription factor VvMYBPA1 regulates proanthocyanidin synthesis during fruit development. Plant Physiol. 2007;143:1347–61. doi: 10.1104/pp.106.093203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Armengaud P, Breitling R, Amtmann A. The potassium-dependent transcriptome of Arabidopsis reveals a prominent role of jasmonic acid in nutrient signaling. Plant Physiol. 2004;136:2556–76. doi: 10.1104/pp.104.046482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Valério L, De Meyer M, Penel C, Dunand C. Expression analysis of the Arabidopsis peroxidase multigenic family. Phytochemistry. 2004;65:1331–42. doi: 10.1016/j.phytochem.2004.04.017. [DOI] [PubMed] [Google Scholar]
- 116.Irshad M, Canut H, Borderies G, Pont-Lezica R, Jamet E. A new picture of cell wall protein dynamics in elongating cells of Arabidopsis thaliana: confirmed actors and newcomers. BMC Plant Biol. 2008;8:94. doi: 10.1186/1471-2229-8-94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Herrero J, Fernández-Pérez F, Yebra T, Novo-Uzal E, Pomar F, Pedreño MÁ, Cuello J, Guéra A, Esteban-Carrasco A, Zapata JM. Bioinformatic and functional characterization of the basic peroxidase 72 from Arabidopsis thaliana involved in lignin biosynthesis. Planta. 2013;237:1599–612. doi: 10.1007/s00425-013-1865-5. [DOI] [PubMed] [Google Scholar]
- 118.Hudson M, Ringli C, Boylan MT, Quail PH. The FAR1 locus encodes a novel nuclear protein specific to The FAR1 locus encodes a novel nuclear protein specific to phytochrome A signaling. Genes Dev. 1999;13:2017–27. doi: 10.1101/gad.13.15.2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Wang H, Xing WD. Arabidopsis FHY3 defines a key phytochrome A signaling component directly interacting with its homologous partner FAR1. EMBO J. 2002;21:1339–49. doi: 10.1093/emboj/21.6.1339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Allen T, Koustenis A, Theodorou G, Somers DE, Kay SA, Whitelam GC, Devlin PF. Arabidopsis FHY3 specifically gates phytochrome signaling to the circadian clock. Plant Cell. 2006;18:2506–16. doi: 10.1105/tpc.105.037358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Lin R, Ding L, Casola C, Ripoll DR, Feschotte C, Wang H. Transposase-Derived transcription factors regulate light signaling in Arabidopsis. Science. 2008;318:1302–5. doi: 10.1126/science.1146281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Li G, Siddiqui H, Teng Y, Lin R, Wan X, Li J, Lau O-S, Ouyang X, Dai M, Wan J, Devlin PF, Deng XW, Wang H. Coordinated transcriptional regulation underlying the circadian clock in Arabidopsis. Nat Cell Biol. 2011;3:616–22. doi: 10.1038/ncb2219. [DOI] [PubMed] [Google Scholar]
- 123.Ouyang X, Li J, Li G, Li B, Chen B, Shen H, Huang X, Mo X, Wan X, Lin R, Li S, Wang H, Deng XW. Genome-Wide binding site analysis of FAR-RED ELONGATED HYPOCOTYL3 reveals its novel function in Arabidopsis development. Plant Cell. 2011;23:2514–35. doi: 10.1105/tpc.111.085126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Tang W, Wang W, Chen D, Ji Q, Jing Y, Wang H, Lin R. Transposase-derived proteins FHY3/FAR1 nteract with PHYTOCHROME-INTERACTING FACTOR1 to regulate chlorophyll biosynthesis by modulating HEMB1 during deetiolation in Arabidopsis. Plant Cell. 2012;24:1984–2000. doi: 10.1105/tpc.112.097022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Tang W, Ji Q, Huang Y, Jiang Z, Bao M, Wang H, Lin R. FAR-RED ELONGATED HYPOCOTYL3 and FAR-RED IMPAIRED RESPONSE1 transcription factors integrate light and abscisic acid signaling in Arabidopsis. Plant Physiol. 2013;163:857–66. doi: 10.1104/pp.113.224386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Gao Y, Liu H, An C, Shi Y, Liu X, Yuan W, Zhang B, Yang J, Yu C, Gao H. Arabidopsis FRS4/CPD25 and FHY3/CPD45 work cooperatively to promote the expression of the chloroplast division gene ARC5 and chloroplast division. Plant J. 2013;75:795–807. doi: 10.1111/tpj.12240. [DOI] [PubMed] [Google Scholar]
- 127.Siloto RMP, Findlay K, Lopez-villalobos A, Yeung EC, Nykiforuk CL, Moloney MM. The accumulation of Oleosins determines the size of seed oilbodies in Arabidopsis. Plant Cell. 2006;18:1961–74. doi: 10.1105/tpc.106.041269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Miquel M, Trigui G, D’Andréa S, Kelemen Z, Baud S, Berger A, Deruyffelaere C, Trubuil A, Lepiniec L, Dubreucq B. Specialization of oleosins in OB dynamics during seed development in Arabidopsis thaliana seeds. Plant Physiol. 2014;164:1866–78. doi: 10.1104/pp.113.233262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Li DD, Fan YM. Cloning, characterisation, and expression analysis of an oleosin gene in coconut (Cocos nucifera L.) pulp. J Hortic Sci Biotechnol. 2009;84:483–8. [Google Scholar]
- 130.Popluechai S, Froissard M, Jolivet P, Breviario D, Gatehouse AMR, O’Donnell AG, Chardot T, Kohli A. Jatropha curcas oil body proteome and oleosins: L-form JcOle3 as a potential phylogenetic marker. Plant Physiol Biochem. 2011;49:352–6. doi: 10.1016/j.plaphy.2010.12.003. [DOI] [PubMed] [Google Scholar]
- 131.Cao H, Zhang L, Tan X, Long H, Shockey JM. Identification, classification and differential expression of Oleosin genes in Tung Tree (Vernicia fordii) PLoS One. 2014;9 doi: 10.1371/journal.pone.0088409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Zhang F, Gonzalez A, Zhao M, Payne CT, Lloyd A. A network of redundant bHLH proteins functions in all TTG1-dependent pathways of Arabidopsis. Development. 2003;130:4859–69. doi: 10.1242/dev.00681. [DOI] [PubMed] [Google Scholar]
- 133.Feller A, Machemer K, Braun EL, Grotewold E. Evolutionary and comparative analysis of MYB and bHLH plant transcription factors. Plant J. 2011;66:94–116. doi: 10.1111/j.1365-313X.2010.04459.x. [DOI] [PubMed] [Google Scholar]
- 134.Nakashima K, Takasaki H, Mizoi J, Shinozaki K, Yamaguchi-Shinozaki K. NAC transcription factors in plant abiotic stress responses. Biochim Biophys Acta. 1819;2012:97–103. doi: 10.1016/j.bbagrm.2011.10.005. [DOI] [PubMed] [Google Scholar]
- 135.Nuruzzaman M, Sharoni AM, Kikuchi S. Roles of NAC transcription factors in the regulation of biotic and abiotic stress responses in plants. Front Microbiol. 2013;4:248. doi: 10.3389/fmicb.2013.00248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Olsen AN, Ernst HA, Lo LL, Skriver K. NAC transcription factors: structurally distinct, functionally diverse. Trends Plant Sci. 2005;10:79–87. doi: 10.1016/j.tplants.2004.12.010. [DOI] [PubMed] [Google Scholar]
- 137.Christensen JH, Hewitson B, Busuioc A, Chen A, Gao X, Held I, Jones R, Kolli RK, Kwon WT, Laprise R, Magaña Rueda V, Mearns L, Menéndez CG, Räisänen J, Rinke A, Sarr A, Whetton P. Regional climate projections. In: Fu C, Giorgi F, editors. Climate change 2007: The Physical Science Basis. Contribution of Working Group I to the Fourth Assessment Report of the Intergovernmental Panel on Climate Change. Cambridge: University Press; 2007. pp. 872–9. [Google Scholar]
- 138.Giorgi F, Lionello P. Climate change projections for the Mediterranean region. Glob Planet Change. 2008;63:90–104. [Google Scholar]
- 139.Marum L, Miguel A, Ricardo CP, Miguel C. Reference gene selection for quantitative real-time PCR normalization in Quercus suber. PLoS One. 2012;7:1–10. doi: 10.1371/journal.pone.0035113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Zhu YY, Machleder EM, Chenchik A, Li R, Siebert PD. Reverse transcriptase template switching: a SMART approach for full-length cDNA library construction. Biotechniques. 2001;30:892–7. doi: 10.2144/01304pf02. [DOI] [PubMed] [Google Scholar]
- 141.Zhulidov PA, Bogdanova EA, Shcheglov AS, Vagner LL, Khaspekov GL, Kozhemyako VB, Matz MV, Meleshkevitch E, Moroz LL, Lukyanov SA, Shagin DA. Simple cDNA normalization using kamchatka crab duplex-specific nuclease. Nucleic Acids Res. 2004;32 doi: 10.1093/nar/gnh031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Falgueras J, Lara AJ, Fernández-Pozo N, Cantón FR, Pérez-Trabado G, Claros MG. SeqTrim: a high-throughput pipeline for pre-processing any type of sequence read. BMC Bioinformatics. 2010;11:38. doi: 10.1186/1471-2105-11-38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Conesa A, Götz S, García-Gómez JM, Terol J, Talón M, Robles M. Blast2GO: a universal tool for annotation, visualization and analysis in functional genomics research. Bioinformatics. 2005;21:3674–6. doi: 10.1093/bioinformatics/bti610. [DOI] [PubMed] [Google Scholar]
- 144.Battke F, Symons S, Nieselt K. Mayday–integrative analytics for expression data. BMC Bioinformatics. 2010;11:121. doi: 10.1186/1471-2105-11-121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Kal A, van Zonneveld A, Benes V, van den Berg M, Koerkamp M, Albermann K, Strack N, Ruijter J, Richter A, Dujon B, Ansorge W, Tabak H. Dynamics of gene expression revealed by comparison of serial analysis of gene expression transcript profiles from yeast grown on two different carbon sources. Mol Biol Cell. 1999;10:1859–72. doi: 10.1091/mbc.10.6.1859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Supek F, Bošnjak M, Škunca N, Šmuc T. REVIGO summarizes and visualizes long lists of gene ontology terms. PLoS One. 2011;6 doi: 10.1371/journal.pone.0021800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Kearse M, Moir R, Wilson A, Stones-Havas S, Cheung M, Sturrock S, Buxton S, Cooper A, Markowitz S, Duran C, Thierer T, Ashton B, Meintjes P, Drummond A. Geneious Basic: an integrated and extendable desktop software platform for the organization and analysis of sequence data. Bioinformatics. 2012;28:1647–9. doi: 10.1093/bioinformatics/bts199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Pfaffl MW. A new mathematical model for relative quantification in real-time RT-PCR. Nucleic Acids Res. 2001;29 doi: 10.1093/nar/29.9.e45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2^(-Delta Delta CT) Method. Methods. 2001;25:402–8. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
- 150.Hellemans J, Mortier G, De Paepe A, Speleman F, Vandesompele J. qBase relative quantification framework and software for management and automated analysis of real-time quantitative PCR data. Genome Biol. 2007;8:R19. doi: 10.1186/gb-2007-8-2-r19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Stewart R, Rascón CA, Tian S, Nie J, Barry C, Chu L-F, Ardalani H, Wagner RJ, Probasco MD, Bolin JM, Leng N, Sengupta S, Volkmer M, Habermann B, Tanaka EM, Thomson JA, Dewey CN. Comparative RNA-seq analysis in the unsequenced axolotl: the oncogene burst highlights early gene expression in the blastema. PLoS Comput Biol. 2013;9:1–15. doi: 10.1371/journal.pcbi.1002936. [DOI] [PMC free article] [PubMed] [Google Scholar]