

Abstract
Background and Purpose
Intracerebral hemorrhage (ICH) represents a devastating form of stroke for which there is no effective treatment. This pre-clinical study was designed to evaluate dimethyl fumarate (DMF), a substance recently approved for the treatment of multiple sclerosis, as therapy for in ICH. We hypothesized that DMF through activating the “master regulator” of cellular self-defense responses, transcription factor Nrf2, would act as effective treatment for ICH-mediated damage.
Methods
Male rats and mice, including Nrf2 knockouts were subjected to intracerebral injection of blood (to mimic ICH) and then treated with DMF. Neurological deficit, brain edema, gene induction profile and hematoma resolution were evaluated. Phagocytic functions of primary microglia in culture were used to study hematoma resolution.
Results
Treatment with DMF induced Nrf2-target genes, improved hematoma resolution, reduced brain edema and ultimately enhanced neurological recovery in rats and wild-type, but not Nrf2 knockout mice. Most importantly, the treatment of ICH with DMF showed a 24h window of therapeutic opportunity.
Conclusions
A clinically relevant dose of DMF demonstrates potent therapeutic efficacy and impressive 24h therapeutic window of opportunity. This study merits further evaluation of this compound as potential treatment for ICH in humans.
Keywords: Dimethyl fumarate, intracerebral hemorrhage, neuroprotection, inflammation, microglia phagocytosis, hematoma resolution
Introduction
Spontaneous intracerebral hemorrhage (ICH) is a devastating form of stroke with a high mortality and poor prognosis that affects an estimated 37,000-52,000 people in the United States annually1 and for which no effective therapy is currently available.2
At first, progressive accumulation of blood within the brain matter leads to increased intracranial pressure and cell/tissue damage that is primarily related to the mechanical injury.3, 4 In addition, it is considered that hematoma-derived toxic products (e.g. hemolysis products), oxidative stress and pro-inflammatory responses3, 5-7 in brain areas around the hematoma robustly aggravates the initial damage (a process often referred to as secondary brain injury) for many days following the ICH onset.3, 4, 8
We and others have recently identified that the transcription factor nuclear factor erythroid 2-related factor 2 (Nrf2), a master regulator of anti-oxidative and detoxification processes, plays a central role in inducing various defense processes which represent attractive targets in treating diverse aspects of the secondary injury after ICH.9-12
The objective of this study was to demonstrate that dimethyl fumarate (DMF), a substance that was recently approved for the treatment of multiple sclerosis (MS), by mechanisms involving mobilization of anti-inflammatory, anti-oxidative and immunomodulatory responses mediated by Nrf213, could also be effective in treating ICH.
Material and Methods
All animal studies followed the guidelines outlined in Guide for the Care and Use of Laboratory Animals from the National Institutes of Health and were approved by the Animal Welfare Committee of University of Texas Health Science Center at Houston. All studies were performed using randomization approach and all analyses were performed by the investigators blinded to treatment assignments.
Intracerebral hemorrhage (ICH) in rat and mice
ICH in rat and mouse was induced by intra-striatal injection of autologous blood as has been described previously. 4, 11, 14 Briefly, male Sprague-Dawley rats (250-350g), or Nrf2+/+ and Nrf2-/- mice 11 (both C57BL/6 background; 25-30g) under chloral hydrate anesthesia (0.35g/kg; i.p.) were immobilized onto a stereotaxic frame. A 1-mm-diameter burr hole was drilled in the skull and a 26-gauge stainless steel cannula was inserted for blood infusion (collected from femoral artery; 15μl/5min for mice, or 35μl/5min for rats) into the left corpus striatum. Stereotactic coordinates with respect to bregma were: SD rat, 0.5mm anterior, 2mm lateral, and 3.5mm deep; for mouse, 0.5mm anterior, 1.2mm lateral, and 2.5 mm deep. Core body temperature was maintained at 37±0.5°C during entire surgery and for 2h afterward.
Tissue harvesting
Animals were anesthetized with chloral hydrate (0.5g/kg; i.p.) and intracardially perfused with ice-cold PBS. The whole brains or the sub-dissected tissues representing hematoma-affected striatum were snap frozen by submersion in –80°C 2-methylbutane and stored in -80°C freezer prior to cryosectioning, RNA isolation, or protein analyses.
Hematoma size measurement
Hematoma resolution was assessed by measuring the amount of hemoglobin (Hb) remaining in the hematoma–affected brain on d7 after ICH, as we detailed previously.14
We also used immunoblotting for Hb level determination. Brain homogenates from ICH-affected hemisphere were separated on SDS-PAGE, and processed for immunoblot. Rabbit anti-Hb antibody (Santa Cruz, sc-31332) followed by goat anti-rabbit Ig-HRP (Zymed) and ECL (Pierce, Rockford, IL) were employed to visualize Hb. Luminescence signal intensity was determined by analyses of optical density on X-ray film.
Dimethyl Fumarate or Sulforaphane treatment
For the animal experiments, 15mg/kg Dimethyl Fumarate15 (DMF, Sigma) dissolved in 10%DMSO was administered. In the SD rats, the DMF was injected intraperitoneally (i.p.) at 2h after ICH and then administered orally twice a day on d1, d2 and d3. In the mice experiment, the DMF was injected i.p. at 24h after ICH and then at d2 and d3. 10% DMSO in saline was used as the vehicle control.
For the cell culture phagocytosis experiments, 1-100μM DMF was directly applied into the culture medium at 16h before the cells were exposed to RBC. Sulforaphane, a prototypic Nrf2 activator, prepared in 10% DMSO, (SF, LKT Laboratories, Inc. S8044) was directly applied to the cell using the same conditions as for DMF. 0.5% DMSO (equal to the final concentration of DMSO in the experimental groups) in culture media was used as the vehicle control for SF and DMF.
Nrf2 knockout mice
Nrf2 knockout mice were constructed and characterized in Dr. Yuet Wai Kan's laboratory (Cardiovascular Research Institute, UCSF)16 and used in our earlier studies.11
Microglia culture
We isolated microglia using p1-p2 rat pups as we described.14 Briefly, the cells from brain tissue were seeded in 75cm2 TC flasks and cultured for 14d. The loosely adherent microglia were harvested, centrifuged and re-plated onto poly-L-lysine coated TC plates, with or without 12-mm diameter German-glass, at a density of 2∼5×105 cells/ml. 96% of all the cells were positive for CD11b.
Immunofluorescence
The immunohistochemistry for CD68 and RBC double labeling was performed using the procedure as we described.14 Briefly, the microglia cells grown on German-glass were fixed with 95% methanol containing 5% acetic acid for 10min at -20°C and incubated in mouse anti-CD68 (ED1, Serotec) and rabbit anti-rat RBC antibody (20R-RR012, Fitzgerald) overnight at 4°C. Goat anti-mouse IgG-Alexa Fluor 488 and goat anti-rabbit IgG-Alexa Fluor-546 (Invitrogen, USA) was used to visualize CD68-labeled microglia and the phagocytosed RBCs in the microglia. The nuclei were visualized with DAPI.
Image capture and cell counting
A Zeiss Axioskop-2 microscope equipped with CCD camera and operated by MetaMorph 7.4 software was used for image acquisition. The fluorescence-labeled cells were visualized using Ex/Em of 490/520 for Alexa 488, Ex/Em of 550/575nm for Alexa 546, and Ex/Em of 365/480 nm for DAPI.
RBC isolation and labeling for phagocytosis assay
The RBC were purified using density gradient centrifugation (BD Vacutainer® CPT™). After washing with PBS, the purified RBC were labeled with the fluorescent dye 5(6)-carboxyfluorescein diacetate (CFDA; Molecular Probes). The labeled RBCs were diluted to 109cells/ml and used as an indicator/target of phagocytosis, as we have previously reported.14
Phagocytosis in vitro
The phagocytosis assay was performed as we described earlier.14 At 2h after adding the CFDA-labeled RBC to the cultured microglia (20:1 ratio), we separated the unphagocytosed free floating RBC from phagocytes that are attached to the plastic by aspiration. The microglia containing engulfed RBC were lysed in distilled water and fluorescence intensity in the supernatant from the cell lysate was measured using a fluorometer with a 490/520nm filter set. The fluorescence intensity (OD) was referred as phagocytosis index.
In experiments with DMF or SF, we pre-incubated microglia in DMF or SF for 16h before adding the CFDA-labeled RBC, the targets of phagocytosis.
RNA isolation and reverse transcription-polymerase chain reaction (RT-PCR)
The ipsilateral corpus striatum (hematoma and peri-hematoma areas) was dissected on ice, snap-frozen and processed for mRNA extraction using Trizol-Reagents. RT-PCR analyses were done as we described.14, 17 We used glyceraldehyde-3-phosphate dehydrogenase (GAPDH) gene as an internal standard. The sequences of primers are listed in Table 1 (supplement). Measurements of the gene products were normalized to the optical density of GAPDH bands. The results were calculated as percentage change over the control (naïve animal, in vivo).
Neurological Deficits Score (NDS)
All behavioral tests were conducted in a quiet and low-lit room by an experimenter blinded with respect to the treatment groups. The NDS was determined by a battery of behavioral tests, including Postural Flexing, Circling test, Footfault, Forelimb Placing and Wire, as we have previously described.11 All animals were pre-tested and then evaluated behaviorally at time points as indicated.
Brain Edema
The brain edema was measured using the wet-weight/dry-weight method.18 Briefly, the brains were removed without perfusion and the ICH-affected left brain hemispheres were dissected. A brain coronal section (4mm-thick) at 2mm anterior and 2mm posterior to the blood injection site was excised. The tissue weight was determined before and after drying in a 95°C oven for 48h. The brain edema was expressed as percent of water content as: (wet weight – dry weight)/wet weight X100.
Statistics
All results are expressed as mean±SEM. For the in vitro experiments, we pooled the samples from three culture wells and repeated the experiments three times. We performed statistical analyses using the GraphPad and InStat programs. One-way analysis of variance (ANOVA) followed by Newman-Keuls post-test was used for multiple group comparisons. Non-paired t-test was used when two groups were compared.
Results
DMF reduced functional deficit and brain edema in rats after ICH
Our initial objective was to determine whether DMF, at doses that are recognized to ameliorate dysfunction in animal model of MS15, reduced neurological deficit and brain edema caused by ICH. The rats were treated with DMF at 2h after ICH and then behaviorally tested at day 1 and day 3, followed by brain edema determination. This study demonstrated that DMF significantly ameliorates the neurological deficit at both day 1 and day 3 after ICH (Fig 1A) and that this functional improvement corresponded with robust reduction in brain edema in DMF treated rats (Fig 1B).
Figure 1.

(A) Neurological Deficit Score (NDS) in SD rats on d1 and d3 after ICH; *p≤0.05, vs. vehicle control and **p≤0.05, vs all other groups. The DMF was dissolved in 10% DMSO at 15mg/kg and injected, i.p., at 2h after ICH, and then orally on d1 and d2. (B) The brain edema on d3 after ICH is expressed as percent of water content in the ICH-affected brain hemisphere; *p≤0.05 vs. vehicle. Number of animals per each group is indicated above the graph.
DMF induces expression of gene profile suggesting activation of Nrf2 and anti-inflammatory activities
By working under the assumption that DMF protects brain by activating Nrf2, we first decided to establish that therapeutic doses of DMF could indeed induce the expression of Nrf2 gene targets in ICH-affected brain tissue. To make sure that the DMF-mediated gene induction in fact represents Nrf2 activation, we contrasted the gene expression pattern between Nrf2-KO and the wild-type mice. Mice were subjected to ICH, treated with DMF and the gene profile was assessed at 48h. Our study established that DMF was indeed able to upregulate the prototypic Nrf2-responsive genes including heme oxygenase-1 (HO-1), CD36, catalase or quinolone oxidoreductase-1 (NQO1), as well as Nrf2 itself in the ICH-affected brain (Fig 2). Among many genes with potential roles in ICH, we found that DMF induced the expression of haptoglobin (Hp) (key hemoglobin/Hb detoxifying protein18) and also CD163 (Hb-Hp scavenger receptor)(Fig 2). The upregulation of all the above genes by DMF was reduced (or even neutralized) in the Nrf2-KO mice, stressing the underlying role of Nrf2 in the induction process. In addition, we found that DMF was also effective in inhibiting the expression of pro-inflammatory mediators, as probed with iNOS and IL-1β expression, while increasing the expression of anti-inflammatory IL-10 (Fig 2). Interestingly, DMF-mediated responses toward the expression of inflammatory genes were similar in WT and Nrf2-deficiency mice, suggesting a Nrf2 independent mechanism of DMF in this process.
Figure 2.

DMF-induced gene expression pattern in wild-type (WT) and Nrf2 knockout (Nrf2-KO) mice in brains at 48h after ICH, determined with RT-PCR, suggest activation of Nrf2 and amelioration of inflammatory responses with DMF. DMF (15mg/kg) was administered i.p. at 2h, 24h and 40h after ICH. Except for inducible genes (HO-1, iNOS, IL-10) expression where the baseline mRNA levels are negligible and the relative densitometrical values (OD) are used to indicate alteration in mRNA, all the other gene expression changes are presented as fold changes over the WT Naïve control. *p≤0.05. (n=3-4)
DMF stimulates phagocytic activities of microglia toward RBC and improves hematoma resolution
Our earlier research suggests that the effective cleanup process conducted by microglia/macrophages after ICH, including in response to the Nrf2 activator sulforaphane (SF), may improve hematoma resolution and improve post-ICH recovery.10 Thus, in a proof of concept experiment, we now demonstrated that DMF (similar to SF) activates microglia in culture toward more effective engulfment of RBC (Fig 3). We next employed the ICH model in rat and showed that treatment with DMF that reduced neurological dysfunction (Fig-4A) led to improved hematoma resolution, as measured by comparing the amount of brain's residual hemoglobin using an immunoblot (Fig 4B& D; p<0.05) or Drabkin's reagent (Fig 4C; p=0.057) at days 7 after ICH.
Figure 3.
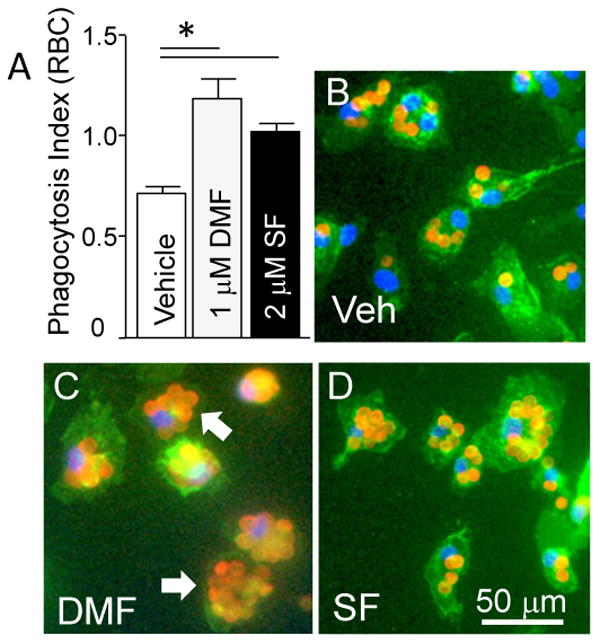
(A) Phagocytosis Index indicating the efficacy of microglia in engulfment of RBC in presence of DMF (1μM) and sulforaphane (SF; 2μM)(n=3 independent experiments). *p≤0.05, vs. vehicle control group. (B,C&D) Representative photomicrograph of microglia (Green; Phalloidin-FITC) in culture that phagocytosed RBC (Red; immunolabeled for RBC) in presence of vehicle (B), DMF (C) and SF (D). The nuclei are labeled with DAPI (blue). The arrow points at microglial cells that internalized RBC.
Figure 4.

(A) Neurological Deficit Score (NDS) determined in rats on d7 after ICH (n=5); DMF at 15mg/kg was injected, i.p., at 2h after ICH and then orally for 3 days. *p≤0.05 vs. vehicle. To establish the hematoma resolution, the hemorrhage size on d7 after ICH was quantified by measuring the remnant Hb in the hematoma-affected brain tissue by Western blot (B and D – representative immunoblot) and by Drabkin's reagent (C).The data are expressed as mean±SEM (n = 5).*p≤0.05.
DMF reduces neurological deficit after ICH in wild-type, but not Nrf2 knockout, mouse with 24h therapeutic window
In this experiment, WT and Nrf2-KO mice were subjected to ICH and then treatment with DMF starting at 24h after the onset of ICH. Despite this marked delay with the intervention, DMF significantly improved the neurological performance of animals, as judged based on the composite neurological score (postural flexing, forward placing, wire hanging, footfault, and circling tests) (Fig 5), as well as based on individual test performance (Fig I Supplement), as assessed on day 3 after ICH. In addition, we repeated this experiment, using separate group of animals, and this time established that the therapeutic benefit of DMF lasts for at least 7 days (Fig II Supplement). Importantly, the beneficial effect of DMF was lost in Nrf2-KO mice, again, suggesting an important function of Nrf2 in the DMF-mediated therapy. Finally, Nrf2-KO mice as compared to WT mice showed less neurological improvement over time, again implicating endogenous Nrf2 as critical component of self-protection in this insult.
Figure 5.

Time course changes in grand NDS in WT and in Nrf2-KO mice on d1, d3 after ICH. The DMF at 15 mg/kg was injected i.p, at 24h after ICH and then on d2, and d3 after ICH. *p≤0.05 vs. indicated groups. Number of animals per group is indicated above the graph.
Discussion
This study shows for the first time that DMF potently attenuates neurological deficit and improves hematoma resolution in a mouse and rat model of intracerebral hemorrhage, even when administered as late as 24h after the onset of ICH. The beneficial effect of DMF was lost in the Nrf2-KO animals, suggesting that its therapeutic effect is through activating Nrf2. The treatment of WT, but not Nrf2-KO mice with DMF indeed induced brain expression of many signature Nrf2 genes and also selected Nrf2-target genes of the detoxification-cleanup system (e.g. CD36, CD163, and haptoglobin). Furthermore, DMF reduced expression of genes associated with pro-inflammatory responses. In vitro, DMF activated the pro-phagocytic function of microglia, suggesting that DMF-mediated hematoma resolution may in part involve improved effectiveness of phagocytosis mediated by microglia/macrophages.
DMF has been extensively studied in animal models of MS15 and recently was approved by FDA as a possible treatment for people with relapsing-remitting MS.19, 20 Based on mechanistic studies with cell culture and experimental autoimmune encephalomyelitis, an animal model of MS, it was proposed that the underlying therapeutic and immunomodulatory mechanism of action of DMF, at a cellular level, is activation of Nrf2.15
Nrf2 is a pleiotropic transcription factor and a key genomic homeostatic regulator that, through antioxidant response elements (ARE) within the regulatory region of many target genes, coordinates anti-oxidative and detoxification processes allowing all types of cells in the neurovascular unit to adapt to detrimental conditions caused by intracellular or extracellular stress.11, 21, 22 We and others have proposed that Nrf2 by its pleiotropic mechanism of action resulting in reduction of oxidative damage, reduction of inflammation, blood product detoxification and improved microglia/macrophages-mediated cleanup process may represent an excellent target for the treatment of ICH.9-12 In agreement with this assumption, we now demonstrate that DMF, at clinically relevant doses, benefits the brain by improving the anti-oxidative milieu of the brain through enhancement of HO-1, catalase or NQO1 genes expression. DMF enhanced expression of CD36, a scavenger receptor that plays a prominent role in the phagocyte-mediated hematoma cleanup process.14 In addition, DMF was effective in amplifying the expression of mRNA for haptoglobin (Hp) and CD163 in ICH-affected brain. Hp, by forming virtually irreversible complexes with hemoglobin (Hb), is known to neutralize the cytotoxicity of Hb (including after ICH)18, while CD163 serves as a scavenger receptor on microglia/macrophages for endocytosis of Hp-Hb complexes, a process that is necessary for the permanent removal of Hb from brain parenchyma.23, 24 Finally, DMF reduced expression of IL-1β and iNOS, two prototypic pro-inflammatory products, and upregulated the expression of anti-inflammatory IL-10. In contrast to the gene products mentioned earlier that required Nrf2 for their induction, DMF-mediated change in IL-1β, iNOS or IL-10 could be detected in both Nrf2-proficient and Nrf2-deficient mice. This may suggest that the regulation of inflammation (at least the components measured in this study) by DMF may not directly involve Nrf2 activation. As such, since Nrf2 proficiency was a prerequisite for the therapeutic/neurological benefit with DMF (Fig 5), it could furthermore be concluded that the mechanisms underlying the therapeutic effect of DMF could include enhanced anti-oxidative mechanisms, neutralization of hemolytic products and cleanup, and are likely not directly based on the regulation of inflammatory responses.
One unique aspect of the ICH-mediated damage is chemical insult caused by lysis of red blood cells within hematoma. This process doesn't start until 1-3 days after ICH6 and escalates over time leading to buildup of toxic levels of intraparenchymal Hb, heme, and iron, ultimately inflicting oxidative stress, cellular damage, BBB disruption and edema.3, 5 We have recently proposed that the progressive nature of the chemical insult may be combated with a therapy that may pre-condition cells around the hematoma, so that they acquire resistance to forthcoming chemical insult.9 We also suggested that since chemical insult starts 1-3 days after the ictus, this approach may show a particularly long therapeutic window. We now demonstrate that DMF can ameliorate ICH-mediated injury with a therapeutic window of at least 24h. We suggest that DMF may represent a viable treatment for ICH.
Supplementary Material
Acknowledgments
Source of Funding: Supported by National Institute of Health, National Institute of Neurological Disorders and Stroke, grants NS060768 and NS064109.
Footnotes
Disclosure: NONE
References
- 1.Broderick JP, Adams HP, Jr, Barsan W, Feinberg W, Feldmann E, Grotta J, et al. Guidelines for the management of spontaneous intracerebral hemorrhage: A statement for healthcare professionals from a special writing group of the stroke council, american heart association. Stroke; a journal of cerebral circulation. 1999;30:905–915. doi: 10.1161/01.str.30.4.905. [DOI] [PubMed] [Google Scholar]
- 2.Qureshi AI, Mendelow AD, Hanley DF. Intracerebral haemorrhage. Lancet. 2009;373:1632–1644. doi: 10.1016/S0140-6736(09)60371-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Keep RF, Hua Y, Xi G. Intracerebral haemorrhage: Mechanisms of injury and therapeutic targets. Lancet neurology. 2012;11:720–731. doi: 10.1016/S1474-4422(12)70104-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Felberg RA, Grotta JC, Shirzadi AL, Strong R, Narayana P, Hill-Felberg SJ, et al. Cell death in experimental intracerebral hemorrhage: The “black hole” model of hemorrhagic damage. Annals of neurology. 2002;51:517–524. doi: 10.1002/ana.10160. [DOI] [PubMed] [Google Scholar]
- 5.Aronowski J, Zhao X. Molecular pathophysiology of cerebral hemorrhage: Secondary brain injury. Stroke; a journal of cerebral circulation. 2011;42:1781–1786. doi: 10.1161/STROKEAHA.110.596718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Wagner KR, Sharp FR, Ardizzone TD, Lu A, Clark JF. Heme and iron metabolism: Role in cerebral hemorrhage. Journal of cerebral blood flow and metabolism : official journal of the International Society of Cerebral Blood Flow and Metabolism. 2003;23:629–652. doi: 10.1097/01.WCB.0000073905.87928.6D. [DOI] [PubMed] [Google Scholar]
- 7.Wang J. Preclinical and clinical research on inflammation after intracerebral hemorrhage. Progress in neurobiology. 2010;92:463–477. doi: 10.1016/j.pneurobio.2010.08.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Aronowski J, Hall CE. New horizons for primary intracerebral hemorrhage treatment: Experience from preclinical studies. Neurological research. 2005;27:268–279. doi: 10.1179/016164105X25225. [DOI] [PubMed] [Google Scholar]
- 9.Zhao X, Aronowski J. Nrf2 to pre-condition the brain against injury caused by products of hemolysis after ich. Translational stroke research. 2013;4:71–75. doi: 10.1007/s12975-012-0245-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Zhao X, Sun G, Ting SM, Song S, Zhang J, Edwards NJ, et al. Cleaning up after ich: The role of nrf2 in modulating microglia function and hematoma clearance. Journal of neurochemistry. 2015;133:144–152. doi: 10.1111/jnc.12974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Zhao X, Sun G, Zhang J, Strong R, Dash PK, Kan YW, et al. Transcription factor nrf2 protects the brain from damage produced by intracerebral hemorrhage. Stroke; a journal of cerebral circulation. 2007;38:3280–3286. doi: 10.1161/STROKEAHA.107.486506. [DOI] [PubMed] [Google Scholar]
- 12.Wang J, Fields J, Zhao C, Langer J, Thimmulappa RK, Kensler TW, et al. Role of nrf2 in protection against intracerebral hemorrhage injury in mice. Free radical biology & medicine. 2007;43:408–414. doi: 10.1016/j.freeradbiomed.2007.04.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Scannevin RH, Chollate S, Jung MY, Shackett M, Patel H, Bista P, et al. Fumarates promote cytoprotection of central nervous system cells against oxidative stress via the nuclear factor (erythroid-derived 2)-like 2 pathway. The Journal of pharmacology and experimental therapeutics. 2012;341:274–284. doi: 10.1124/jpet.111.190132. [DOI] [PubMed] [Google Scholar]
- 14.Zhao X, Sun G, Zhang J, Strong R, Song W, Gonzales N, et al. Hematoma resolution as a target for intracerebral hemorrhage treatment: Role for peroxisome proliferator-activated receptor gamma in microglia/macrophages. Annals of neurology. 2007;61:352–362. doi: 10.1002/ana.21097. [DOI] [PubMed] [Google Scholar]
- 15.Linker RA, Lee DH, Ryan S, van Dam AM, Conrad R, Bista P, et al. Fumaric acid esters exert neuroprotective effects in neuroinflammation via activation of the nrf2 antioxidant pathway. Brain: a journal of neurology. 2011;134:678–692. doi: 10.1093/brain/awq386. [DOI] [PubMed] [Google Scholar]
- 16.Chan K, Lu R, Chang JC, Kan YW. Nrf2, a member of the nfe2 family of transcription factors, is not essential for murine erythropoiesis, growth, and development. Proceedings of the National Academy of Sciences of the United States of America. 1996;93:13943–13948. doi: 10.1073/pnas.93.24.13943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Zhao X, Strong R, Zhang J, Sun G, Tsien JZ, Cui Z, et al. Neuronal ppargamma deficiency increases susceptibility to brain damage after cerebral ischemia. The Journal of neuroscience: the official journal of the Society for Neuroscience. 2009;29:6186–6195. doi: 10.1523/JNEUROSCI.5857-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Zhao X, Song S, Sun G, Strong R, Zhang J, Grotta JC, et al. Neuroprotective role of haptoglobin after intracerebral hemorrhage. The Journal of neuroscience : the official journal of the Society for Neuroscience. 2009;29:15819–15827. doi: 10.1523/JNEUROSCI.3776-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Gold R, Kappos L, Arnold DL, Bar-Or A, Giovannoni G, Selmaj K, et al. Placebo-controlled phase 3 study of oral bg-12 for relapsing multiple sclerosis. The New England journal of medicine. 2012;367:1098–1107. doi: 10.1056/NEJMoa1114287. [DOI] [PubMed] [Google Scholar]
- 20.Arnold DL, Gold R, Kappos L, Bar-Or A, Giovannoni G, Selmaj K, et al. Effects of delayed-release dimethyl fumarate on mri measures in the phase 3 define study. Journal of neurology. 2014;261:1794–1802. doi: 10.1007/s00415-014-7412-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Moi P, Chan K, Asunis I, Cao A, Kan YW. Isolation of nf-e2-related factor 2 (nrf2), a nf-e2-like basic leucine zipper transcriptional activator that binds to the tandem nf-e2/ap1 repeat of the beta-globin locus control region. Proceedings of the National Academy of Sciences of the United States of America. 1994;91:9926–9930. doi: 10.1073/pnas.91.21.9926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.van Muiswinkel FL, Kuiperij HB. The nrf2-are signalling pathway: Promising drug target to combat oxidative stress in neurodegenerative disorders. Curr Drug Targets CNS Neurol Disord. 2005;4:267–281. doi: 10.2174/1568007054038238. [DOI] [PubMed] [Google Scholar]
- 23.Zhao X, Grotta J, Gonzales N, Aronowski J. Hematoma resolution as a therapeutic target: The role of microglia/macrophages. Stroke; a journal of cerebral circulation. 2009;40:S92–94. doi: 10.1161/STROKEAHA.108.533158. [DOI] [PubMed] [Google Scholar]
- 24.Zuwala-Jagiello J. Haemoglobin scavenger receptor: Function in relation to disease. Acta Biochim Pol. 2006;53:257–268. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.