

Abstract
Background and Purpose
Histone deacetylases (HDACs) 4 and 5 are abundantly expressed in the brain and have been implicated in the regulation of neurodegeneration. Under physiologic conditions, HDACs 4 and 5 are expressed in the cytoplasm of brain cells where they cannot directly access chromatin. In response to external stimuli, they can shuttle to the nucleus and regulate gene expression. However, the effect of stroke on nuclear shuttling of HDACs 4 and 5 remains unknown.
Methods
Using a rat model of middle cerebral artery occlusion (MCAO), we examined the subcellular localization of HDACs 4 and 5 in the peri-infarct cortex during brain repair after stroke.
Results
Stroke significantly increased nuclear HDAC4 immunoreactivity in neurons, but not in astrocytes or oligodendrocytes, of the peri-infarct cortex at 2, 7 and 14 days after MCAO. Neurons with nuclear HDAC4 immunoreactivity distributed across all layers of the peri-infarct cortex and were Ctip2+ excitatory and parvalbumin+ inhibitory neurons. These neurons were not TUNEL or BrdU positive. Furthermore, nuclear HDAC4 immunoreactivity was positively and significantly correlated with increased dendritic, axonal and myelin densities as determined by MAP-2, p-NFH and MBP, respectively. Unlike HDAC4, stroke did not alter nuclear localization of HDAC5.
Conclusions
Our data show that stroke induces nuclear shuttling of HDAC4 in neurons in the peri-infarct cortex, and that increased nuclear HDAC4 is strongly associated with neuronal remodeling but not neuronal cell death, suggesting a role for nuclear HDAC4 in promoting neuronal recovery after ischemic injury.
Keywords: Epigenetics, Stroke, Neuronal repair
Introduction
Stroke is a leading cause of morbidity and long-term disability in the United States and worldwide1. Therefore, a tremendous need exists for the development of new therapies for stroke. Stroke patients exhibit spontaneous recovery at the behavioral level within the first three months after ischemic injury 2. Studies from experimental stroke demonstrate that during stroke recovery, endogenous processes of neuronal remodeling such as axonal sprouting and formation of new cortical connections are induced in the area adjacent to the infarct border3-5. Clearly, endogenous mechanisms are not sufficient to restore full neurological function; however, pre-clinical data in rodents suggest that amplification of endogenous brain repair processes can lead to improved functional outcome after stroke6. Investigating the molecular mechanisms underlying neuronal remodeling after ischemic injury can help in identifying new targets for neurorestorative therapy for stroke.
Epigenetic post-translational modifications of histone proteins, such as lysine acetylation and deacetylation play a major role in the regulation of gene transcription in neurons7, 8. Histone deacetylases (HDACs) are a large family of enzymes that regulate histone acetylation levels by catalyzing the removal of acetyl moieties from lysine residues in histone tails. Histone deacetylation consequently leads to compaction of chromatin and gene repression9, 10.
HDACs are classified into four major classes (I-IV) based on homology to yeast enzymes11. Class II HDACs are mammalian Hda1 (histone deacetylase 1) like proteins which include HDACs 4, 5, 6, 7, 9 and 10. Based on their structure, class II HDACs are further subdivided into two subclasses: class IIa (4, 5, 7 and 9) and IIb (6 and 10)12, 13.
Two isoforms of class IIa HDACs (4 and 5) are abundantly expressed in the brain14, 15 and have been implicated in the regulation of neurodegeneration16-19, synaptic plasticity and memory formation20, 21. Constitutive HDAC4 knockout mice are not viable and die within 2 weeks of birth due to severe skeletal malformations22, while conditional loss of HDAC4 in the forebrain leads to impaired motor coordination, learning and memory20. Interestingly, conditional HDAC5 knockout mice do not display similar dysfunctions in learning and memory20, indicating that despite their similarity in structure, HDACs 4 and 5 play non-redundant roles in neurons.
HDACs 4 and 5 are predominantly expressed in the cytoplasm 15 where they cannot directly access chromatin, however, in response to external stimuli they can also shuttle to the nucleus23-25. For example, previously published studies showed that under physiologic conditions HDAC4 is localized to the cytoplasm of cerebellar granule neurons (CGNs), but shuttles to the nucleus when CGNs are treated with non-depolarizing media 16, 17.
To our knowledge, no information is available about whether stroke affects the subcellular localization of class IIa HDACs. We hypothesized that ischemic conditions may stimulate nuclear shuttling of HDACs 4 and 5. Therefore, in the present study, using a rat model of focal cerebral ischemia, we investigated whether stroke induces nuclear shuttling of HDACs 4 and 5 in neurons in the peri-infarct brain. Furthermore, we also examined whether nuclear shuttling of HDACs is associated with apoptosis or promoted remodeling of neurons after ischemia.
Methods
All experimental procedures were approved by the Institutional Animal Care and Use Committee of Henry Ford Hospital.
Animal model
Adult male Wistar rats (270–300g, n=6/group) were subjected to permanent middle cerebral artery occlusion (MCAO) by advancing a 4–0 surgical nylon suture with an expanded tip26. Sham-operated rats were used for control. Rats were injected once daily with BrdU (please see Supplemental Methods) and sacrificed 2, 7 and 14 days after surgery.
Tissue preparation, Immunohistochemistry and TUNEL assay
The brains were fixed by transcardial perfusion with saline followed by 4% paraformaldehyde before being embedded in paraffin (please see Supplemental Methods for Immunhistochemistry and TUNEL assay).
Image acquisition and analysis
Laser-scanning confocal microscopy (Zeiss LSM 510 NLO, Carl Zeiss, Germany) was used for three dimensional imaging. For quantification, coronal sections were digitized under a 20× or 40× objective of an epifluorescence microscope (Axiophot, Carl Zeiss, Inc.) via a Micro Computer Imaging Device (MCID) system (Imaging Research Inc.). Six-eight fields of view were acquired from the peri-infarct cortex (up to 500 μm from the lesion border, Figure 1D). For comparison, additional fields of view from homologous areas were acquired from sham-operated rats and contralateral cortex. Immunoreactive cells were counted using NIH ImageJ software and immunoreactive areas were calculated using MCID imaging analysis system.
Figure 1. Distribution of HDAC4 within cortical neurons.

Confocal composite images and orthogonal views in panel A show that HDAC4 immunoreactivity in sham-operated rats was mainly localized to MAP-2+ cytoplasm (arrows) and dendrites (arrowheads) of cortical neurons. However, after MCAO, HDAC4 immunoreactivity was mainly detected in nuclei of MAP-2+ neurons in the peri-infarct cortex. Images from boxed area are orthogonal views of HDAC4 within cytoplasm and nuclei of MAP2+ neurons (A). Quantitative analysis of HDAC4/MAP-2 revealed the percentage of nuclear (B) and dendritic (C) HDAC4 immunoreactive neurons in the peri-infarct cortex at 2, 7 and 14 days after MCAO compared to sham. A schematic representation of a brain coronal section (D) shows that images were acquired from six layers of the peri-infarct cortex as outlined in the numbered boxes. Bar = 20μm.
Statistical analysis
One-way analysis of variance (ANOVA) with post hoc Bonferroni test was used for data analysis. Statistical significance was set at p<0.05. Pearson correlation coefficients were calculated among histological measurements and their correlation with nuclear HDAC4. All values are presented as mean ± SE for illustration.
Results
Stroke induces nuclear shuttling of HDAC4 in neurons
To examine whether stroke induces nuclear shuttling of HDAC4 in neurons during brain repair, rats were sacrificed 2, 7 and 14 days after MCAO and double immunohistochemistry was performed with antibodies specifically against HDAC4 along with MAP-2 and p-NFH. In the cortex of sham-operated rats, immunoreactivity of HDAC4 was mainly localized to MAP-2+ neuronal cytoplasm and dendrites (Figure 1A), but not pNFH+ axons (Supplemental Figure I). However, after MCAO, we found a robust increase in HDAC4 immunoreactivity in nuclei of neurons in the peri-infarct cortex (Figure 1A, D). Quantitative analysis of HDAC4/MAP-2 immunoreactive cells at multiple time points after MCAO revealed a temporal dynamic increase in the percentage of nuclear HDAC4+ neurons after stroke compared to sham (p<0.05, Figure 1B). Concomitantly, stroke-induced nuclear expression of HDAC4 was accompanied by a significant (p<0.05) decrease in HDAC4 immunoreactivity in neuronal dendrites over the same period (Figure 1C). We did not detect a significant increase in nuclear HDAC4 immunoreactivity in MAP-2+ neurons in contralateral homologeous areas of the cortex (31±5% at 2 days, 32±5% at 7 day and 27±2% at 14 days after MCAO vs. 27±3% in sham, p>0.05). Together, these data show that stroke induces nuclear shuttling of HDAC4 in neurons in the peri-infarct cortex.
Stroke induces HDAC4 shuttling across cortical layers in interneurons and pyramidal neurons
The cerebral cortex is composed of six layers that are mainly composed of pyramidal neurons (excitatory) and interneurons (inhibitory)27. We used HDAC4/MAP-2 double immunohistochemistry to examine whether nuclear shuttling of HDAC4 after stroke exhibits a specific laminar distribution (Figure 2A, Supplemental Figure II). We found that in the normal brain, HDAC4 immunoreactivity was predominantly in the cytoplasm of neurons in both superficial (L1-4) and deep (L5-6) cortical layers. The number of nuclear HDAC4 immunoreactive neurons in the normal cortex was low and evenly distributed throughout the superficial and deep cortical layers. Starting 2 days after stroke, we found a significant increase in nuclear HDAC4 immunoreactive neurons in deep cortical layers (L5-6) compared to sham control (Figure 2A, Supplemental Figure II). The percentage of nuclear HDAC4 immunoreactive neurons in deep cortical layers continued to increase until 2 weeks after MCAO (Figure 2A, Supplemental Figure II). On the other hand, nuclear HDAC4 immunoreactivity in superficial cortical layers was delayed and significantly increased only at 7 and 14 days after MCAO (Figure 2A, Supplemental Figure II). Together, these data show that stroke-induced nuclear HDAC4 shuttling first starts in the deep cortical layers and then it expands to superficial layers.
Figure 2. Distribution of HDAC4 across cortical layers in neurons.
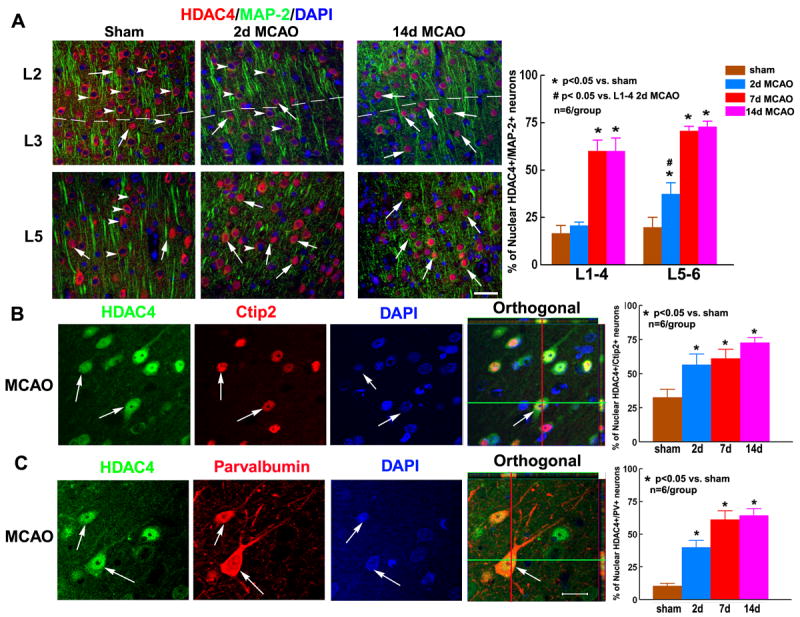
In superficial (L2-3) and deep (L5-6) cortical layers of sham-operated animals, HDAC4 immunoreactivity was predominantly detected in the cytoplasm of MAP-2+ neurons (A, sham, arrowheads for cytoplasmic HDAC4, arrows for nuclear HDAC4). At 2 days after stroke, nuclear HDAC4 was increased in neurons of deep (A, L5-6), but not superficial (A, L2-3) cortical layers. Only at 7 and 14 days after MCAO, nuclear HDAC4 was also detected in superficial layers (A, L2-3). Confocal composite images and quantitative data in panels B-C show that stroke increased nuclear HDAC4 immunoreactivity in Ctip2+ pyramidal neurons (B, arrows) and parvalbumin+ interneurons (C, arrows). Orthogonal views of HDAC4 within nuclei of Ctip2+ and parvalbumin+ neurons are also shown (orthogonal in B and C, arrow). Bar = 20μm.
To examine whether nuclear HDAC4 shuttling is induced in a specific neuronal population after stroke, we performed double immunohistochemistry for HDAC4 with Ctip2 (pyramidal neuron marker) and parvalbumin (interneuron marker). We found that stroke significantly increased nuclear HDAC4 immunoreactivity in both Ctip2+ pyramidal cells (Figure 2B) and parvalbumin+ interneurons (Figure 2C). These data suggest that stroke induces nuclear HDAC4 shuttling in both excitatory and inhibitory neurons of the peri-infarct cortex.
Taken together, nuclear shuttling of HDAC4 after stroke is induced in different neuronal populations across all cortical layers, suggesting that nuclear shuttling of HDAC4 play a major role in regulating neuronal response to ischemia.
HDAC4, but not HDAC5, shuttles to neuronal nuclei after stroke
It has been reported that both HDACs 4 and 5 possess the ability to translocate between the cytoplasm and nucleus23, 28. Therefore, we investigated whether stroke also induces nuclear shuttling of HDAC5 in neurons. In sham-operated rats, HDAC5/MAP-2 and HDAC5/p-NFH double immunohistochemistry showed that the subcellular localization of HDAC5 in cortical neurons was similar to HDAC4; HDAC5 immunoreactivity was also mainly localized to MAP-2+ cytoplasm and dendrites of neurons (Figure 3A) and not pNFH+ axons (Supplemental Figure I). However, in contrast to HDAC4, stroke did not significantly increase nuclear HDAC5 in neurons in the peri-infarct cortex across cortical layers at 2, 7 or 14 days after MCAO (Fig. 3A). These data suggest that stroke only induces nuclear shuttling of HDAC4 in neurons in the peri-infarct cortex.
Figure 3. Distribution of HDAC5 in cortical neurons and of HDAC4 in glial cells.
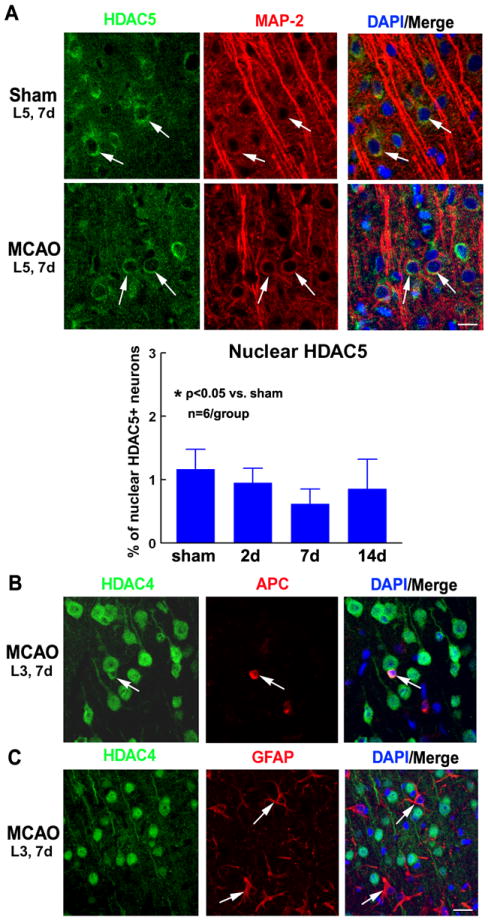
In sham-operated and ischemic rats (2, 7 and 14 days after MCAO), HDAC5 immunoreactivity was predominantly detected in the cytoplasm and dendrites of MAP-2+ neurons (A). A bar graph shows quantitative data of the percentage of neurons with nuclear HDAC5 immunoreactivity (A). In glial cells of the peri-infarct cortex, HDAC4 immunoreactivity was detected only in the cytoplasm of some APC+ oligodendrocytes (B, arrows) and not detected at all in GFAP+ astrocytes (C, arrows). Representative images were acquired from L5, 7d after sham surgery or MCAO (A), L3, 7d after MCAO (B-C). Bar = 20μm.
Stroke-induced nuclear shuttling of HDAC4 is specific to neurons
Previously, we found that stroke induces diverse changes in the expression profiles of individual class I and II HDACs in oligodendrocytes 29. Using double immunohistochemistry for HDAC4 with APC (oligodendrocytes marker) and GFAP (astrocytes marker), we also examined whether stroke induces nuclear shuttling of HDAC4 in glial cells. We did not detect nuclear HDAC4 immunoreactivity in APC+ oligodendrocytes or GFAP+ astrocytes in the peri-infarct cortex (Figure 3B-C, 7 days after MCAO). These data indicate that stroke-induced nuclear shuttling of HDAC4 is specific to neurons and does not occur in glial cells of the ischemic brain.
Stroke-induced HDAC4 nuclear shuttling in neurons is not associated with apoptosis
Previous studies presented contradictory evidence regarding whether nuclear HDAC4 shuttling has a positive or deleterious effect on neuronal survival16, 17. We thus, examined whether nuclear HDAC4 immunoreactive neurons in the ischemic brain are primed to die. Using immunohistochemistry for HDAC4 with TUNEL assay for identification of DNA fragmentation, we found that nuclear HDAC4+ cells in the peri-infarct cortex did not exhibit TUNEL staining (Figure 4A, 7 days after MCAO), indicating that nuclear HDAC4 immunoreactive neurons are not apoptotic cells. Additionally, we examined whether nuclear HDAC4+ cells re-enter the cell cycle using double immunofluorescent staining with antibodies against HDAC4 and BrdU. We found that nuclear HDAC4 immunoreactive cells were not BrdU+ (Figure 4B, 7 days after MCAO), suggesting that nuclear HDAC4 immunoreactive neurons do not re-enter an abortive cell cycle that could have led to apoptosis30. Together, these data show that nuclear HDAC4 shuttling under ischemic conditions in vivo is not associated with neuronal apoptosis or abortive cell cycle re-entry.
Figure 4. Correlation of nuclear HDAC4 with MAP-2, pNFH and MBP.

Cells with nuclear HDAC4 immunoreactivity were not TUNEL+ (A, arrows) or BrdU+ (B, arrows). Furthermore, nuclear HDAC4 immunoreactivity after stroke was significantly correlated to an increase in density of MAP-2 (C), p-NFH (D) and MBP (D) immunoreactivity. Representative images were acquired from L5, 7d after MCAO (A, B) and L5, 14d after MCAO (C). Data used for correlation analyses in panels C-D were acquired from 2, 7 and 14d after MCAO. Bar = 20μm.
Nuclear shuttling of HDAC4 is correlated with increased dendritic, axonal and myelin densities after stroke
Neuronal remodeling such as rewiring neuronal circuitry and re-myelination occurs in peri-infarct region during stroke recovery3, 31. We thus, examined whether increased nuclear HDAC4 immunoreactivity after stroke is associated with increased neuronal remodeling, by performing double immunofluorescent staining for HDAC4/MAP-2, HDAC4/pNFH and HDAC4/MBP at 2, 7 and 14 days after MCAO. Correlation analysis of the data showed that nuclear HDAC4 immunoreactivity was significantly and positively correlated with MAP-2 immunoreactivity (an index of dendrites, r = 0.71, p<0.05, Figure 4C) in the peri-infarct cortex. Additionally, positive and significant (p<0.05) correlations were detected between nuclear HDAC4 and pNFH+ density (an index of axons, r = 0.69, Figure 4D), as well as MBP+ density (an index of myelin, r = 0.68, Figure 4D). Altogether, our data showed that stroke-induced nuclear shuttling of HDAC4 in neurons in the peri-infarct cortex was strongly associated with improved indices of neuronal remodeling including axonal, dendritic and myelin densities, suggesting a positive role for nuclear HDAC4 shuttling in promoting neuronal recovery after stroke.
Discussion
In the present study, we show for the first time that stroke induces nuclear shuttling of HDAC4 in neurons in the peri-infarct cortex and that nuclear HDAC4 is strongly associated with improved neuronal remodeling and not neuronal cell death, suggesting a role for nuclear HDAC4 in promoting brain repair after ischemic injury.
Class IIa HDACs are mainly expressed in the cytoplasm in physiological conditions, but are also able to shuttle into the nucleus. In the nucleus, HDACs can access histone proteins and act as epigenetic regulators15, 23-25. Interestingly, in addition to their known cytoplasmic and nuclear localizations, we also found that HDACs 4 and 5 are expressed in dendrites of neurons in the normal and ischemic cortex. This finding suggests a function for HDACs in dendritic outgrowth in addition to gene regulation.
Synaptic activity in hippocampal and cerebellar granular neurons is known to regulate the intracellular shuttling of class IIa HDACs23, 32. Whether ischemic stimuli can similarly affect the subcellular localization of HDACs has not been previously explored. The present study shows that stroke induces nuclear shuttling of HDAC4 in neurons in the peri-infarct sensorimotor cortex. The translocation of HDAC4 from the cytoplasm to nuclei induced by stroke is specific because nuclear translocation of HDAC5 was not detected, although both HDACs are closely similar in structure (70% homology)11. Moreover, along with increased nuclear HDAC4 shuttling after stroke, we detected a significant decrease in dendritic HDAC4 immunoreactivity. Thus, our data strongly suggest that cerebral ischemia affects the subcellular localization of HDAC4 leading to increased trafficking of HDAC4 from dendrites and cytoplasm into the nucleus. While nuclear HDAC4 continued to gradually increase up to 14 days after MCAO, dendritic HDAC4 decreased from 19% in sham to 6-8% at 2 days after MCAO and remained stable afterwards. This finding suggests that trafficking of HDAC4 from dendrites to the nucleus is most robust in early stages after stroke. Possibly, a minimal level of dendritic HDAC4 is required to maintain dendritic integrity and function. Later on, at 7 and 14 days after MCAO, the increase in nuclear HDAC4 most likely results from increased shuttling of HDAC4 from the cytoplasm to the nucleus.
We previously reported that stroke induces diverse changes in the expression profiles of class I and II HDACs in oligodendrocytes 29. However, the present study demonstrated that stroke-induced nuclear shuttling of HDAC4 is neuronal specific and is not induced in oligodendrocytes or astrocytes. In addition, examination of the laminar distribution of nuclear HDAC4 shuttling after stroke revealed that nuclear HDAC4 shuttling starts in deep cortical layers and then expands to superficial layers. We also found that nuclear HDAC4 shuttling is not specific to one neuronal population and is induced in both excitatory and inhibitory neurons, indicating that nuclear HDAC4 may play a major role in regulating neuronal response to ischemic injury.
The role of nuclear HDAC4 in neuronal cell death and survival remains debated in the literature33. The controversy largely stems from contradictory reports on the effect of HDAC4 shuttling in cerebellar granule neurons (CGNs). Bolger and Yao16 reported that in response to low potassium or excitatory glutamate conditions that induce CGNs death, HDAC4 shuttled from the cytoplasm to the nucleus. Nuclear HDAC4 then repressed the activity of MEF2 (myocyte enhancer factor 2), a known pro-survival transcription factor and promoted neuronal apoptosis. On the other hand, Majdzaeh et al17 reported that forced expression of HDAC4 in CGNs protected them against low potassium induced apoptosis. The protective effect of HDAC4 was shown to occur in the nucleus and mediated by preventing abortive cell-cycle reentry through inhibition of CDK1 (cyclin-dependent kinase 1). These reports may have yielded contradictory results due to heavily relying on in vitro experiments33. Therefore, in the current study we used an in vivo model for ischemia to examine whether endogenous stroke-induced nuclear HDAC4 shuttling is associated with cell death or not.
Using double immunohistochemistry, we found lack of co-localization of nuclear HDAC4 immunoreactive neurons with TUNEL assay and BrdU labeling, suggesting that stroke-induced nuclear shuttling of HDAC4 does not trigger apoptotic cell death or abortive cell cycle re-entry. Furthermore, nuclear HDAC4 shuttling persists for at least two weeks after stroke and is positively correlated with endogenous processes of neuronal repair after ischemia. Surviving cortical neurons in the peri-infarct cortex experience axonal sprouting, dendritic outgrowth and re-myelination3, 4, 34 which may contribute to spontaneous functional recovery following stroke. Indeed, our data show that nuclear HDAC4 is strongly and positively correlated with improved indices of neuronal remodeling including dendritic, axonal and myelin as determined by MAP-2, p-NFH and MBP densities, respectively. These findings highlight that further studies are warranted to show a causative relationship between nuclear HDAC4 shuttling and improved neurological outcome after stroke.
In summary, our data show that stroke induces nuclear shuttling of HDAC4 in neurons in the peri-infarct cortex and that nuclear HDAC4 is strongly associated with improved neuronal remodeling and not neuronal cell death, suggesting a possible positive role for nuclear shuttling of HDAC4 in promoting neuronal recovery after ischemic injury. Further studies to investigate the effects of specific blocking of neuronal HDAC4 in the ischemic brain would advance our understanding of the mechanisms by which HDAC4 possibly promotes neuronal remodeling after stroke.
Supplementary Material
Acknowledgments
The authors wish to thank Qinge Lu and Sutapa Santra for technical assistance.
Sources of Funding
This work was supported by NIH grants RO1 NS088656 (MC) and RO1 NS075156 (ZGZ). The content is solely the responsibility of the authors and does not necessarily represent the official view of the NIH.
Footnotes
Disclosures
None
References
- 1.Go AS, Mozaffarian D, Roger VL, Benjamin EJ, Berry JD, Blaha MJ, et al. Heart disease and stroke statistics--2014 update: A report from the american heart association. Circulation. 2014;129:e28–e292. doi: 10.1161/01.cir.0000441139.02102.80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Nakayama H, Jorgensen HS, Raaschou HO, Olsen TS. Recovery of upper extremity function in stroke patients: The copenhagen stroke study. Archives of physical medicine and rehabilitation. 1994;75:394–398. doi: 10.1016/0003-9993(94)90161-9. [DOI] [PubMed] [Google Scholar]
- 3.Liu Z, Zhang RL, Li Y, Cui Y, Chopp M. Remodeling of the corticospinal innervation and spontaneous behavioral recovery after ischemic stroke in adult mice. Stroke; a journal of cerebral circulation. 2009;40:2546–2551. doi: 10.1161/STROKEAHA.109.547265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Carmichael ST, Wei L, Rovainen CM, Woolsey TA. New patterns of intracortical projections after focal cortical stroke. Neurobiology of disease. 2001;8:910–922. doi: 10.1006/nbdi.2001.0425. [DOI] [PubMed] [Google Scholar]
- 5.Dancause N, Barbay S, Frost SB, Plautz EJ, Chen D, Zoubina EV, et al. Extensive cortical rewiring after brain injury. The Journal of neuroscience : the official journal of the Society for Neuroscience. 2005;25:10167–10179. doi: 10.1523/JNEUROSCI.3256-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Chen J, Venkat P, Zacharek A, Chopp M. Neurorestorative therapy for stroke. Frontiers in human neuroscience. 2014;8:382. doi: 10.3389/fnhum.2014.00382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Berger SL. The complex language of chromatin regulation during transcription. Nature. 2007;447:407–412. doi: 10.1038/nature05915. [DOI] [PubMed] [Google Scholar]
- 8.Borrelli E, Nestler EJ, Allis CD, Sassone-Corsi P. Decoding the epigenetic language of neuronal plasticity. Neuron. 2008;60:961–974. doi: 10.1016/j.neuron.2008.10.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kouzarides T. Chromatin modifications and their function. Cell. 2007;128:693–705. doi: 10.1016/j.cell.2007.02.005. [DOI] [PubMed] [Google Scholar]
- 10.Jenuwein T, Allis CD. Translating the histone code. Science. 2001;293:1074–1080. doi: 10.1126/science.1063127. [DOI] [PubMed] [Google Scholar]
- 11.de Ruijter AJ, van Gennip AH, Caron HN, Kemp S, van Kuilenburg AB. Histone deacetylases (hdacs): Characterization of the classical hdac family. The Biochemical journal. 2003;370:737–749. doi: 10.1042/BJ20021321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Verdin E, Dequiedt F, Kasler HG. Class ii histone deacetylases: Versatile regulators. Trends in genetics : TIG. 2003;19:286–293. doi: 10.1016/S0168-9525(03)00073-8. [DOI] [PubMed] [Google Scholar]
- 13.Yang XJ, Gregoire S. Class ii histone deacetylases: From sequence to function, regulation, and clinical implication. Molecular and cellular biology. 2005;25:2873–2884. doi: 10.1128/MCB.25.8.2873-2884.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Broide RS, Redwine JM, Aftahi N, Young W, Bloom FE, Winrow CJ. Distribution of histone deacetylases 1-11 in the rat brain. J Mol Neurosci. 2007;31:47–58. doi: 10.1007/BF02686117. [DOI] [PubMed] [Google Scholar]
- 15.Darcy MJ, Calvin K, Cavnar K, Ouimet CC. Regional and subcellular distribution of hdac4 in mouse brain. The Journal of comparative neurology. 2010;518:722–740. doi: 10.1002/cne.22241. [DOI] [PubMed] [Google Scholar]
- 16.Bolger TA, Yao TP. Intracellular trafficking of histone deacetylase 4 regulates neuronal cell death. The Journal of neuroscience : the official journal of the Society for Neuroscience. 2005;25:9544–9553. doi: 10.1523/JNEUROSCI.1826-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Majdzadeh N, Wang L, Morrison BE, Bassel-Duby R, Olson EN, D’Mello SR. Hdac4 inhibits cell-cycle progression and protects neurons from cell death. Developmental neurobiology. 2008;68:1076–1092. doi: 10.1002/dneu.20637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Chen B, Cepko CL. Hdac4 regulates neuronal survival in normal and diseased retinas. Science. 2009;323:256–259. doi: 10.1126/science.1166226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Li J, Chen J, Ricupero CL, Hart RP, Schwartz MS, Kusnecov A, et al. Nuclear accumulation of hdac4 in atm deficiency promotes neurodegeneration in ataxia telangiectasia. Nature medicine. 2012;18:783–790. doi: 10.1038/nm.2709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kim MS, Akhtar MW, Adachi M, Mahgoub M, Bassel-Duby R, Kavalali ET, et al. An essential role for histone deacetylase 4 in synaptic plasticity and memory formation. The Journal of neuroscience : the official journal of the Society for Neuroscience. 2012;32:10879–10886. doi: 10.1523/JNEUROSCI.2089-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Sando R, 3rd, Gounko N, Pieraut S, Liao L, Yates J, 3rd, Maximov A. Hdac4 governs a transcriptional program essential for synaptic plasticity and memory. Cell. 2012;151:821–834. doi: 10.1016/j.cell.2012.09.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Vega RB, Matsuda K, Oh J, Barbosa AC, Yang X, Meadows E, et al. Histone deacetylase 4 controls chondrocyte hypertrophy during skeletogenesis. Cell. 2004;119:555–566. doi: 10.1016/j.cell.2004.10.024. [DOI] [PubMed] [Google Scholar]
- 23.Grozinger CM, Schreiber SL. Regulation of histone deacetylase 4 and 5 and transcriptional activity by 14-3-3-dependent cellular localization. Proceedings of the National Academy of Sciences of the United States of America. 2000;97:7835–7840. doi: 10.1073/pnas.140199597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Wang AH, Kruhlak MJ, Wu J, Bertos NR, Vezmar M, Posner BI, et al. Regulation of histone deacetylase 4 by binding of 14-3-3 proteins. Molecular and cellular biology. 2000;20:6904–6912. doi: 10.1128/mcb.20.18.6904-6912.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Zhao X, Ito A, Kane CD, Liao TS, Bolger TA, Lemrow SM, et al. The modular nature of histone deacetylase hdac4 confers phosphorylation-dependent intracellular trafficking. The Journal of biological chemistry. 2001;276:35042–35048. doi: 10.1074/jbc.M105086200. [DOI] [PubMed] [Google Scholar]
- 26.Chen H, Chopp M, Zhang ZG, Garcia JH. The effect of hypothermia on transient middle cerebral artery occlusion in the rat. Journal of cerebral blood flow and metabolism : official journal of the International Society of Cerebral Blood Flow and Metabolism. 1992;12:621–628. doi: 10.1038/jcbfm.1992.86. [DOI] [PubMed] [Google Scholar]
- 27.Shipp S. Structure and function of the cerebral cortex. Current biology : CB. 2007;17:R443–449. doi: 10.1016/j.cub.2007.03.044. [DOI] [PubMed] [Google Scholar]
- 28.Wang AH, Yang XJ. Histone deacetylase 4 possesses intrinsic nuclear import and export signals. Molecular and cellular biology. 2001;21:5992–6005. doi: 10.1128/MCB.21.17.5992-6005.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kassis H, Chopp M, Liu XS, Shehadah A, Roberts C, Zhang ZG. Histone deacetylase expression in white matter oligodendrocytes after stroke. Neurochemistry international. 2014;77:17–23. doi: 10.1016/j.neuint.2014.03.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Bauer S, Patterson PH. The cell cycle-apoptosis connection revisited in the adult brain. The Journal of cell biology. 2005;171:641–650. doi: 10.1083/jcb.200505072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Gensert JM, Goldman JE. Endogenous progenitors remyelinate demyelinated axons in the adult cns. Neuron. 1997;19:197–203. doi: 10.1016/s0896-6273(00)80359-1. [DOI] [PubMed] [Google Scholar]
- 32.Chawla S, Vanhoutte P, Arnold FJ, Huang CL, Bading H. Neuronal activity-dependent nucleocytoplasmic shuttling of hdac4 and hdac5. Journal of neurochemistry. 2003;85:151–159. doi: 10.1046/j.1471-4159.2003.01648.x. [DOI] [PubMed] [Google Scholar]
- 33.Majdzadeh N, Morrison BE, D’Mello SR. Class iia hdacs in the regulation of neurodegeneration. Frontiers in bioscience : a journal and virtual library. 2008;13:1072–1082. doi: 10.2741/2745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ueno Y, Chopp M, Zhang L, Buller B, Liu Z, Lehman NL, et al. Axonal outgrowth and dendritic plasticity in the cortical peri-infarct area after experimental stroke. Stroke; a journal of cerebral circulation. 2012;43:2221–2228. doi: 10.1161/STROKEAHA.111.646224. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.