

Abstract
Bile acids (BAs) function as endocrine signaling molecules that activate multiple nuclear and membrane receptor signaling pathways to control fed-state metabolism. Since the detergent-like property of BAs causes liver damage at high concentrations, hepatic BA levels must be tightly regulated. BA homeostasis is regulated largely at the level of transcription by nuclear receptors, particularly the primary bile acid receptor, farnesoid X receptor (FXR), and small heterodimer partner (SHP) that inhibits BA synthesis by recruiting repressive histone-modifying enzymes. Although histone modifiers have been shown to regulate BA-responsive genes, their in vivo functions remain unclear. Here we show that lysine-specific histone demethylase1 (LSD1) is directly induced by BA-activated FXR, is recruited to BA synthetic genes, Cyp7a1 and Cyp8b1, and the BA uptake transporter gene, Ntcp, and removes a gene-activation mark, tri-methylated histone H3 lysine-4, leading to gene repression. LSD1 recruitment was dependent on SHP, and LSD1-mediated demethylation of H3K4-me3 was required for additional repressive histone modifications, H3K9/K14 deacetylation and H3K9 methylation. BA overload, feeding 0.5% cholic acid chow for 6 days, resulted in adaptive responses of altered expression of hepatic genes involved in BA synthesis, transport, and detoxification/conjugation. In contrast, adenoviral-mediated downregulation of hepatic LSD1 blunted these responses, which led to substantial increases in liver and serum BA levels, serum AST/ALT levels, and hepatic inflammation. This study identifies LSD1 as a novel histone-modifying enzyme in the orchestrated regulation mediated by the FXR and SHP that reduces hepatic BA levels and protects the liver against BA toxicity.
Keywords: SHP, epigenomic regulation, Cyp7a1, Ntcp, cholestasis
Introductory Statement
Bile acids (BAs) are amphipathic steroid molecules that are synthesized from cholesterol in liver (1, 2). BAs were long-considered to have a simple dietary role but are now recognized as key endocrine signaling molecules that activate multiple nuclear and membrane receptor-mediated signaling pathways to regulate fed-state metabolism and energy balance (2–5). When food enters the small intestine, bile acids in bile from the gall bladder are released and help solubilize and digest lipid nutrients. Over 95% of the BAs are returned to the liver by enterohepatic recycling, and the increased hepatic BA levels activate signaling pathways to reduce BA levels (1, 2, 4). Due to their detergent-like property, BAs are toxic to hepatocytes and, thus, hepatic BA levels must be tightly controlled through regulation of BA biosynthesis, metabolism, and transport (6–9). Deficiencies in these adaptive responses to high BA levels results in abnormal accumulation of BAs in the liver, resulting in liver damage and cholestatic disease.
BA homeostasis is regulated mainly at the level of transcription via the action of nuclear receptors, particularly by the primary BA receptor, Farnesoid X Receptor (FXR, NR1H4), its DNA binding partner, RXRα (NR2B1), and Small Heterodimer Partner (SHP, NR0B2) (2, 7). FXR/RXRα directly induces genes involved in the synthesis, transport, and detoxification of BAs or inhibits indirectly through the induction of SHP. RXRα also heterodimerizes with other BA-sensing nuclear receptors, PXR, CAR, and VDR, which contribute to regulation of hepatic genes involved in maintaining BA homeostasis. SHP coordinates epigenomic inhibition of the BA biosynthetic genes, Cyp7a1 and Cyp8b1 (10), by recruiting repressive chromatin-modifying enzymes (11–13). The role of chromatin modifiers in epigenomic regulation of BA synthetic genes has been shown (11–14), but the in vivo relevance remains largely unknown, although the in vivo role of MLL3/4 histone lysine methyltransferases in maintaining BA levels by regulating Bsep and Shp has been demonstrated (15, 16).
Transcriptional regulation in eukaryotes requires a coordinated interaction of chromatin-modifying coregulators and transcriptional factors, which results in histone modifications and gene activation or repression. While histone acetylation and deacetylation generally correlate with gene activation and gene repression, respectively, histone methylation leads to either gene activation or repression (17). Methylation of histone H3 at Lys-4 (H3K4) is associated with gene activation, whereas methylation at H3K9 and H3K27 is associated with gene repression. LSD1 (also KDM1A) is a lysine-specific histone demethylase that is associated with repressor proteins, such as, HDACs-containing coREST and SIRT1 deacetylase (18) and removes a gene-activating histone mark, mono-, di-, or tri-methylated H3K4 (17). Intriguingly, LSD1 was also shown to activate androgen receptor target genes by removing a gene-repression histone mark, methylated H3K9 (19). LSD1 was reported to regulate CYP7A1 in HepG2 cells but the in vivo role for LSD1 in regulation of hepatic BA levels has not been shown (20). In this study, we show that LSD1 functions in vivo as an important epigenomic regulator in the FXR/SHP pathway that functions to reduce hepatic BA levels and protect the liver against BA toxicity.
Experimental Procedures
Materials and reagents
Antibodies to SHP (sc-30169), GFP (sc-8334), actin (sc-1616), and HDAC1 (sc-7872), were purchased from Santa Cruz Biotechnology; to LSD1 (ab17721), Histone H3K4-me3 (ab8580), and F4/80 (ab6640) from Abcam; and to H3K9/K14-Ac (06-599), H3K9-me2 (07-030), and Prox1 (07-537) from Millipore. Prox1 siRNA was purchased from Dharmacon.
Mouse studies
Ten- to twelve-week old male FXR-KO or wild type (WT) C57BL/6J mice were fasted overnight and injected ip with corn oil or GW4064 (30 mg/kg) for 3 h, or fed 0.5% CA-chow for 6 h. For Ad-shRNA studies, mice were tail-vein injected with Ad-shLSD1 and 6 days later, mice were fed 0.5% CA chow for 6 h or 6 days. Ad-shLSD1 was generated using the published shRNA sequence (19) and SHP Ad-shRNA has been described (11). The adenoviral system, which selectively infects the liver (21), was used for liver-specific overexpression or downregulation of proteins or microRNAs (12, 22–24). Animal use and adenoviral protocols were approved by the Institutional Animal Care and Use and Biosafety Committees at the University of Illinois at Urbana-Champaign.
Liver histology and toxicity
Serum ALT/AST levels and BA levels were measured using colorimetric analysis kits purchased from Sigma and Trinity Biotech, respectively. Accumulated bile was detected by H&E staining.
Microscopy imaging studies
Paraffin-embedded liver sections were fixed with 4% paraformaldehyde. LSD1 or infiltrated macrophages were detected by IHC (Abcam, #Ab64261). Nuclei were stained with hematoxylin and samples were imaged with a Nanozoomer (Hamamatzu). Hepa1c1c7 cells were fixed and incubated with primary antibody, followed by secondary antibody conjugated with Alexa 488 and rhodamine, and images were taken by confocal microscopy (Zeiss, LSM700).
Luciferase reporter assay
Sequences containing the IR1 motifs in mouse Lsd1 were cloned into the pGL3 vector (Promega, Inc). IR1 motifs were mutated using site-directed mutagenesis (Agilent Technology). Cos-1 cells were transfected with plasmids, and 24 h later, treated with GW4064 (200 nM) or CDCA (50 μM) overnight and luciferase assays were done as previously described (11–13). The values for luciferase activities were normalized to β-gal activities.
Gel mobility shift assay
[γ-32P]ATP-labeled probe was incubated with partially purified flag-FXR and flag-RXR, 100 ng poly-dIdC, 20 μg BSA, and 1 μM GW4064 as described (22). Samples were pre-incubated with antibody or unlabeled oligo competitors for 10 min on ice before adding probe.
CoIP, GST pull-down, and ChIP
These assays were performed as previously described (11–13, 22). Briefly, in CoIP, mouse liver extracts were prepared in 50 mM Tris-HCl, pH. 8.0, 150 mM NaCl, 2 mM EDTA, 0.3% NP-40, and 10% glycerol. For GST pull-down, 1–2 μg of GST proteins were incubated with 35S-labeled proteins and bound proteins were detected by autoradiography. In ChIP, chromatin was immunoprecipitated with 1 μg of antisera at 4°C overnight. The immune complex was washed, the bound chromatin was eluted, and DNA was purified for q-PCR.
q-RTPCR
Total RNA was isolated using Trizol (Invitrogen), cDNA was synthesized using a kit (Promega), q-RTPCR was performed with a Step One Plus Thermal Cycler (Perkin Elmer), and the amount of mRNA was normalized to that of 36B4 mRNA.
Results
Pharmacologically activated FXR directly binds to LSD1 IR1 motifs
FXR binding peaks were detected within the Lsd1 gene by mouse liver ChIP-seq (25) (Fig. S1A). An inverted repeat1 (IR1) motif, known to bind FXR, was detected at a major FXR binding peak in the 15th intron, and 15 additional IR1 motifs were present within the Lsd1 gene or 50 Kb to either side (Fig. 1A, S1B). Occupancy of FXR at these sites, analyzed by ChIP assay, was increased prominently by treatment with the FXR agonist, GW4064, at IR1 motifs #6 and #7 (Fig. 1B) in introns 15 and 9, respectively, suggesting that FXR may directly regulate expression of LSD1 in liver.
Fig. 1. Pharmacologically activated FXR directly binds to IR1 motifs in the Lsd1 gene.

(A) IR1 motifs detected within the 50 kb Lsd1 gene and 50 kb of flanking regions. (B) ChIP: Mice were treated with GW4064 or vehicle for 3 h, livers were pooled from 3 mice, and FXR occupancy to the IR1 motifs was detected by ChIP. (C, D) Gel mobility shift assays were performed using partially purified flag-FXR and flag-RXR, and a probe containing the IR motif #6, in presence or absence of antibody (C) or competitor oligonucleotides (D) as indicated. (E, F) Cos-1 cells were transfected with a luc-reporters containing Lsd1 IR1 motif #6 (E) or #7 (F) and expression plasmids as indicated, and 36 h later, the cells were treated with GW4064 (200 nM) or CDCA (50 μM) overnight and reporter assays were performed. The mean and SEM (n=3) are plotted. Consistent results were observed in 2 independent triplicate reporter assays.
Binding of FXR to the Lsd1 IR1 motifs was examined by gel-shift analysis. Addition of partially purified flag-FXR and flag-RXRα (Fig. S2) resulted in the formation of a FXR/RXR/DNA complex with motif #6 as probe (Fig. 1C, lane, 4–6), which was supershifted by addition of M2 (flag peptide) or FXR antibodies, but not control Gal4 DBD antibody (lanes, 7–9), and mutation of the IR1 motif completely abolished the binding (lane 10). Gel-shift assays using mouse liver nuclear extracts were consistent with binding of endogenous FXR and RXRα to the probe (Fig. S3). Addition of unlabeled oligonucleotides with sequences of IR1 motif #6 (Fig 1D, left panel, lanes 3,4) or a positive control SHP IR1 motif (lanes 7,8) effectively competed for the binding, while mutation of IR1 #6 motif had only modest effects (lanes 5,6). Similarly, FXR/RXR bound the IR1 #7 probe, and wild type (WT), but not mutated IR1 #7 oligonucleotides effectively competed for the binding (Fig. 1D, right panel). These results indicate that FXR directly binds to IR1 motifs #6 and #7 in the intron regions of the Lsd1 gene.
To examine whether these Lsd1 IR1 motifs are functional, WT and mutated IR1 #6 and IR1 #7 sequences were inserted into luciferase reporter plasmids (Fig. 1E, F). Expression of FXR increased luciferase reporter activity in Cos-1 cells treated with either GW4064 or a natural FXR agonist, chenodeoxycholic acid (CDCA), and mutation of the IR1 sequence attenuated the increase (Fig. 1E, F). These results demonstrate that FXR directly binds to these IR1 motifs and that they are functional.
Pharmacological and physiological activation of FXR signaling increased hepatic expression of LSD1
To determine whether FXR acts as an in vivo physiological regulator of LSD1 expression, we examined the effects of GW4064 treatment, CA feeding, or feeding/fasting on expression of hepatic LSD1 in WT and FXR-KO mice. Treatment of WT mice with GW4064 increased LSD1 mRNA and pre-mRNA levels nearly 5-fold, indicating that gene expression was increased, but no increase was detected in FXR-KO mice (Fig. 2A). LSD1 protein levels detected by IB (Fig. 2B) or IHC (Fig. S4) were also increased after GW4064 treatment in WT mice, but not in FXR-KO mice. Similar effects on expression of Lsd1 were observed in mice fed with 0.5% cholic acid (CA)-supplemented chow (Fig. 2C) and in fed compared to fasted mice (Fig. 2D). These results demonstrate that pharmacological or physiological activation of FXR signaling induces hepatic expression of Lsd1 in an FXR-dependent manner.
Fig. 2. Activation of FXR signaling increased hepatic expression of LSD1 in an FXR-specific manner.
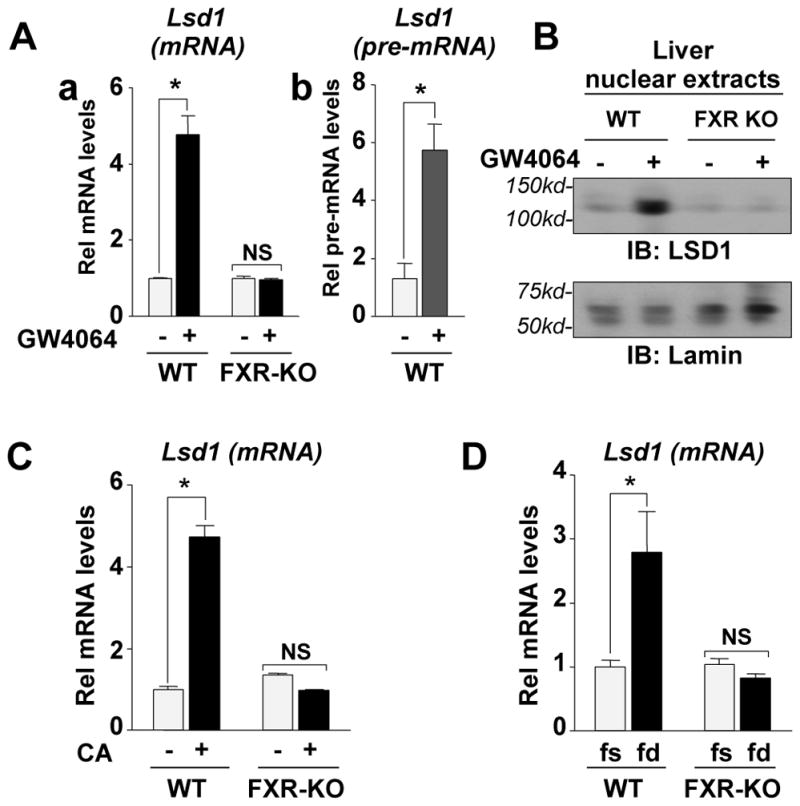
(A) WT or FXR-KO mice were treated with GW4064 or vehicle for 3 h and livers were collected for q-RTPCR to measure Lsd1 mRNA (left) or pre-mRNA (right) levels. (B) Expression of hepatic nuclear LSD1 was detected by IB in mice that had been treated with GW4064 for 3 h. (C, D) Mice were fed normal chow or 0.5% CA supplemented chow for 6 h (C) or were fasted overnight and further fasted or refed for 6 h (D). Livers were collected for q-RTPCR. The mean and SEM are plotted and statistical significance was measured using Student’s t-test. *p<0.05 and NS statistically not significant, n=3.
LSD1 directly interacts with SHP and enhances gene repression by SHP
Since SHP-mediated the inhibition of the expression of Cyp7a1 and Cyp8b1 by recruitment of the histone-modifying enzymes, HDACs and G9a (12), we asked if FXR-induced LSD1, which removes a histone activation mark (17), contributes to SHP-mediated epigenomic repression. First, the interaction of LSD1 with SHP was studied after 3 h of CA feeding when LSD1 levels are unchanged (Fig. 3A-a) so that interaction independent of induction could be examined. LSD1 interaction with SHP was increased about 4-fold (Fig. 3A-b, IgG control in Fig. 3A-c) in liver after CA feeding. In GST-pull down assays, LSD1 directly interacted with the N-terminal domain of SHP (Fig. 3B-a) and conversely, SHP interacted with the catalytic domain of LSD1 (Fig. 3B-b). Immunofluorescence studies also showed that LSD1 co-localized with SHP after CDCA treatment (Fig. S5). These studies suggest that LSD1 interaction with SHP is increased upon BA signaling.
Fig. 3. LSD1 directly interacts with SHP and enhances its repression activity.
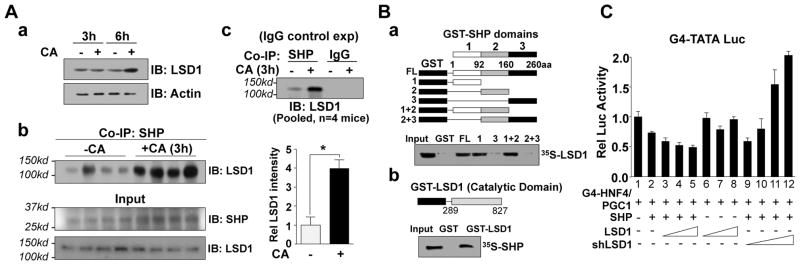
(A) CoIP: (A-a) Mice were fed CA chow for 3 h or 6 h, extracts were prepared from livers pooled from 4 mice and immunoblots (IB) were performed. (A-b) Mice were fed CA chow for 3 h and CoIP assays were performed. In the quantitative analysis, the band intensity for the control (-CA) group = 1. The mean and SEM are plotted and statistical significance was measured using Student’s t-test. *p <0.05 (n= 4). (A-c) CoIP assay with an IgG control. (B) GST-pull down: Schematic diagrams of fragments of SHP and LSD1 in GST fusion proteins are shown above the autoradiograms. LSD or SHP was 35S-labeled using the in vitro TNT system and interaction was detected by autoradiography. (C) Cos-1 cells were transfected with a Gal4-TATA-luc reporter and expression plasmids as indicated, and 36 h later, the cells were treated with CDCA overnight and reporter assays were performed. The mean and SEM (n=3) are plotted.
We next examined whether LSD1 enhances SHP-mediated repression by reporter assays. Expression of SHP decreased transactivation mediated by Gal4-HNF4/PGC-1α, and expression of LSD1 enhanced the decrease (Fig. 3C, lanes 2–5), but had little effect if SHP was not expressed (lane, 6–8). Remarkably, downregulation of LSD1 reversed the SHP repression and dramatically increased luciferase activity, (lanes 9–12). Similar results were observed with a native BA-responsive CYP7A1 promoter-reporter when activated by CDCA treatment (Fig. S6). These results suggest that LSD1 directly interacts with SHP and enhances SHP repression.
LSD1 is recruited to BA synthetic genes in a SHP-dependent manner and removes a gene activation histone mark, H3K4-me3
We next determined the effects of CDCA treatment on the occupancy of LSD1 and SHP and on H3K4-me3 levels at the SHP-target BA synthetic genes, CYP7A1 and CYP8B1. Occupancy of LSD1 and SHP was increased by CDCA treatment of HepG2 cells, while H3K4-me3 levels were reduced (Fig. 4A) and these effects were reversed and inhibition of these genes were compromised in SHP-downregulated cells (Fig. 4B, Fig. S7). Further, the decrease in H3K4-me3 levels was reversed by downregulation of LSD1 (Fig. 4C, Fig. S8). Since SHP-dependent H3K9/K14 deacetylation and H3K9 methylation also have been shown to occur at the CYP7A1 promoter (12), we further examined effects of LSD1 downregulation on these histone modifications and occupancy of SHP. Treatment with CDCA decreased H3K9/14-Ac levels and increased H3K9-me2 levels and these effects were reversed in LSD1-downregulated cells (Fig. 4C), whereas SHP occupancy was not changed (Fig. S8). Consistent with the changes in histone modifications, the expression of these biosynthetic genes was inhibited by CDCA treatment and this inhibition was reversed by downregulation of LSD1 (Fig. 4D). These results suggest that LSD1 represses CYP7A1 and CYP8B1 by erasing a gene activation histone mark, H3K4-me3, and that this step is important for the following decreased H3K9/14 acetylation and increased H3K9 methylation.
Fig. 4. LSD1 is recruited to CYP7A1 and CYP8B1 genes in a SHP-dependent manner and demethylates H3K4-me3, which is required for additional repressive histone modifications in HepG2 cells.

(A) Effects of CDCA treatment on occupancy of LSD1 and SHP and histone H3K4-me3 levels at the CYP7A1 and CYP8B1 promoters as detected by ChIP. (B) Effects of downregulation of SHP on occupancy of LSD1 and histone H3K4-me3 levels. SHP levels detected by IB are shown at the left. (C) Effects of downregulation of LSD1 on histone H3K4-me3, H3K9-me2, and H3K9/14-Ac levels. Reduced LSD1 levels detected by IB are shown at the left. (D) Effects of downregulation of LSD1 on mRNA levels of CYP7A1 and CYP8B1. The mean and SEM (n=3) are plotted and statistical significance was measured using Student’s t-test. *p<0.05.
LSD1-mediated epigenomic repression of BA synthetic and uptake genes occurs in vivo
To determine whether the LSD1-mediated demethylation of histone H3K4-me3 also occurred in vivo, the effects of downregulation of LSD1 in liver were examined on Cyp7a1 and Cyp8b1, as well as a SHP-target BA uptake transporter gene, Ntcp, that plays a crucial role in regulating hepatic BA levels (26) (Fig. 5A). Adenoviral-mediated downregulation of LSD1 led to reduced hepatic nuclear LSD1 protein levels (Fig. 5B, IHC data in Fig. S9) and reduced Lsd1, but not Shp, mRNA levels (Fig. S10). In mice fed CA chow for a short time, 6 h, occupancies of LSD1 and SHP were increased and H3K4-me3 levels were decreased, and the effects on H3K4-me3, but not SHP, were reversed by downregulation of LSD1 (Fig. 5C). CA feeding resulted in inhibition of expression of Cyp7a1, Cyp8b1, and Ntcp, and strikingly, the inhibition of Ntcp was largely reversed by downregulation of LSD1, while the inhibition of Cyp7a1 and Cyp8b1 was only partially reversed (Fig. 5D). These results indicate that in vivo pathways independent of LSD1 contribute to inhibition of expression of Cyp7a1 and Cyp8b1 genes by BA. Nevertheless, downregulation of LSD1 increased the basal expression of these genes and partially reversed the inhibition by BAs, suggesting that LSD1 is an important in vivo regulator of these genes.
Fig. 5. LSD1-mediated epigenomic regulation of BA synthetic and uptake genes also occurs in mouse liver in response to CA feeding.

(A) Experimental outline. (B) Effects of downregulation of LSD1 on hepatic nuclear levels of LSD1 detected by IB. (C, D) Effects of downregulation of LSD1 on occupancy of SHP and LSD1, and H3K4-me3 levels at Cyp7a1, Cyp8b1, and Ntcp promoters (C), and mRNA levels of these genes (D).
LSD1 protects from liver toxicity in responses to a chronic BA overload
We next examined the role of LSD1 in vivo in the response to a chronic BA overload, 6 days of feeding a CA diet (Fig. 6A). Injection with Ad-shLSD1 resulted in substantial decreases in hepatic LSD1 protein levels (Fig. 6B, Fig. S11) and Lsd1, but not Shp, mRNA levels (Fig. S11). CA feeding for 6 days increased the weight of gall bladders and volume of bile, BA pool size, liver and serum BA levels (Fig. 6C); and indicators of liver toxicity, serum AST and ALT levels (Fig. 6D). Consistent with increased liver toxicity, pathological histological changes in the liver were observed; including loosened liver structure, degeneration of individual hepatocytes, and abnormally accumulated bile (Fig. 6E). Consistent with findings that increased hepatic BA levels causes liver injury and inflammatory responses (26, 27), increased macrophage infiltration was evident in LSD1-downregulated mice challenged with a high BA load (Fig. 6F). These results indicate that downregulation of LSD1 increased hepatic BA levels and liver toxicity in mice that were challenged with a dietary BA overload.
Fig. 6. In vivo downregulation of LSD1 leads to impaired adaptive responses to a high BA load, resulting in increased hepatic BA levels, liver toxicity, and inflammation.

(A) Experimental outline: Mice were injected with Ad-shRNA for LSD1 or control virus and 6 days later, the injected mice were fed normal chow or 0.5% CA chow for 6 days, and tissues and serum were collected. (B) Effects of downregulation of LSD1 in mice fed normal chow or 0.5% CA chow for 6 days on hepatic LSD1 protein levels detected by IB or IHC. (C, D) Effects of downregulation of LSD1 on gall bladder and BA pool sizes and liver and serum BA levels (C), on serum ALT/AST (D), and on liver histology detected by H&E staining (E). Abnormally accumulated bile is indicated by the arrows (E). (F) Effects of downregulation of LSD1 on infiltration of macrophages detected by IHC. (E, F) Mice were fed normal chow (-CA) or CA chow for 6 days (+CA). Arrows indicate infiltrated macrophages (F). The mean and SEM (n=3) are plotted and statistical significance was measured using the Student’s t-test. *p<0.05, **p<0.01 and NS statistically not significant.
LSD1 plays a role in adaptive transcriptional responses to a BA overload
Finally, we examined the role of LSD1 in the transcriptional responses to a BA overload, feeding CA for 6 days. In liver ChIP assays, the CA feeding resulted in increased occupancy of SHP and LSD1 and decreased H3K4-me3 levels at Cyp7a1, Cyp8b1, and Ntcp promoters (Fig. 7A). Downregulation of LSD1 had little effect on occupancy of SHP, but largely blocked the decrease in H3K4-me3 levels (Fig. 7A), indicating the importance of LSD1 in demethylation of H3K4-me3. In gene expression studies, the CA feeding had variable effects on hepatic BA-responsive genes. Expression of the BA synthetic genes was strongly inhibited by BA overload and downregulation of LSD1 increased basal expression by 2- to 3-fold and a strong response to CA was still retained although the levels after the BA load were several-fold higher than in the control Ad-E mice (Fig. 7B). Expression of the BA import transporters was inhibited in response to a BA overload and downregulation of LSD1 increased the basal levels of expression and largely reversed the inhibition, particularly expression of Ntcp (Fig. 7C). For BA export transporter genes, BA overload had modest effects on expression, except for an increase in Ostβ expression, and downregulation of LSD1 also had modest effects (Fig. 7C, Fig. S12, and Model in Fig. 7D). Expression of the detoxification and conjugation genes were modestly increased by the CA feeding and downregulation of LSD1 reversed the response so that expression was inhibited (Fig. 7E). Thus, downregulation of LSD1 contributes to increased liver BA levels by increased BA synthesis, increased BA hepatic uptake, decreased BA export, and decreased metabolism of BAs, which contribute to an increase in hepatic bile acids resulting in liver toxicity and inflammation. Consistent these changes, basal expression and expression after BA overload of inflammatory genes were increased by downregulation of LSD1 (Fig. 7F). Further, activated JNK and ERK levels were increased in mice fed CA chow for 6 days (Fig. S13). These results indicate that downregulation of LSD1 led to impaired transcriptional responses to a high BA load and the net result is substantial increases in liver and serum BA levels and liver toxicity (Fig. 6) based on increased serum AST/ALT levels and inflammation.
Fig. 7. In vivo downregulation of LSD1 leads to impaired transcriptional responses to a high BA load.

The experimental outline is as described in the Fig. 6 legend. (A) Effects of downregulation of hepatic LSD1 on protein occupancy and histone modification at Cyp7a1, Cyp8b1, and Ntcp promoters. (B, C, E, F) Effects of downregulation of LSD1 on expression of the indicated genes detected by q-RTPCR. The mean and SEM (n=3) are plotted and statistical significance was measured using Student’s t-test. *p<0.05, **p<0.01and NS statistically not significant. (D) Expected changes in liver BA flow in LSD1-downregulated mice challenged with a BA overload based on transcriptional changes are illustrated.
Discussion
This study identifies LSD1 as an in vivo histone-modifying enzyme as a component of epigenomic regulation orchestrated by the FXR/SHP pathway that reduces hepatic BA levels and protects the liver against BA toxicity. BA-activated FXR directly binds to and induces hepatic expression of LSD1 as well as SHP. Induced LSD1 interacts with SHP, is recruited to BA biosynthetic genes, Cyp7a1 and Cyp8b1, and a BA uptake gene, Ntcp, and removes a gene activation histone mark, histone H3K4-me3, resulting in epigenomic repression of these genes. The epigenomic repression of these genes by LSD1 inhibits BA synthesis and blocks BA entry into hepatocytes, contributing to the homeostatic reduction of hepatic BA levels in response to elevated BA levels.
The functional role of the nuclear receptors, SHP, FXR, and RXRα, in regulating hepatic BA levels and protecting the liver against injury is well established. BA-induced toxicity in response to a BA load was increased in SHP-null mice (28) and similarly, RXRα-deficient mice exhibited aggravated liver injury (26). Selective activation of intestinal FXR protected mice against cholestasis by inducing FGF15 (29). Abnormally high BA levels and juvenile onset of cholestasis were detected in FXR/SHP double KO mice (30). Interestingly, LSD1 is associated with the FXR/SHP regulatory pathway in two ways: FXR/RXRα induces LSD1 and LSD1-mediated demethylation of H3K4-me3 enhances repression by SHP. BA signaling also increased LSD1 interaction with SHP. Although the mechanism is unknown, BA-induced phosphorylation of SHP by PKCζ is important for increased interaction of SHP with HDACs and G9a (23) and may underlie the increased interaction with SHP.
LSD1-mediated epigenomic regulation of Cyp7a1, Cyp8b1, and Ntcp, provides a molecular mechanism for the feedback inhibition of BA synthesis and uptake regulated by the FXR/SHP pathway. We have shown previously that hepatic BA signaling resulted in SHP-dependent repressive histone modifications, H3K9/14 deacetylation by HDACs and H3K9 methylation by G9a (12). These epigenomic events were impaired when LSD1 was downregulated, suggesting that LSD1-mediated H3K4-me3 demethylation is upstream of these histone modifications. Notably, recruitment of LSD1 and the subsequent H3K4 demethylation were dependent on SHP. In addition to SHP, other proteins, such as, Prox1 may be important for recruitment of LSD1 (20). We observed that downregulation of Prox1 partially inhibited recruitment of LSD1 and HDAC1 to the CYP7A1 promoter, whereas downregulation of SHP completely inhibited the recruitment, suggesting both SHP and Prox1 contribute to recruitment of LSD1 and HDAC1 but SHP is likely the dominant factor in LSD1 recruitment (Fig. S14).
The current study reveals differences between the effects of downregulation of LSD1 on expression of Cyp7a1 and Cyp8b1 in vitro and in vivo. Changes in H3K4-me3 levels were similar in both systems, but BA inhibition of these genes was completely reversed by downregulation of LSD1 in HepG2 cells, while only partial reversed in vivo. These results indicate that in vivo pathways independent on LSD1 contribute to inhibition of expression of these genes in response to CA and partially override the epigenomic regulation by LSD1. The intestinal FGF15/19 is a possible pathway that has emerged as a key regulator of BA levels (31). Although both, FXR/SHP and FGF15/19, pathways are important for reducing liver BA levels under physiological conditions, they may act at different times. The BA-activated FXR/SHP pathway should predominate at relatively earlier times when liver BA levels are elevated after recycling from enterohepatic circulation, but the intestinal FGF15/19 signal would be more important at relatively late postprandial periods. As liver BA levels must be tightly regulated, these BA-induced pathways likely ensure tight regulation of BA levels.
Our study demonstrates that LSD1 is protective against toxicity due to a BA overload by effects on basal expression and CA responsiveness. Downregulation of LSD1 at physiological levels of bile acids results in elevated “basal” levels of BA synthetic, detoxification, and uptake transport genes, while responsiveness to a BA overload differs for these categories of genes. Cyp7a1 and Cyp8b1, as noted, respond similarly to BA overload when LSD1 is downregulated, but because of the elevated basal levels, are still expressed at higher levels than in the BA overloaded control mice. Notably, CA responsiveness for the liver uptake genes, in particular Ntcp, is blunted so the elevated basal levels are sustained. For detoxification genes, CA responsiveness is reversed so that even though basal levels are increased, expression of these genes is decreased by a BA overload. The combined effects of LSD1 downregulation on these genes involved in BA synthesis, transport, and detoxification primarily contribute to the increased hepatic BA levels and toxicity upon BA overload.
The transcriptional regulatory mechanisms that lead to the different responsiveness of BA-responsive genes are likely to be complex. After lengthy CA feeding, secondary and indirect regulatory changes may be in play depending on the combination of factors that regulate a specific gene. For example, under pathological conditions like liver cholestasis, aberrant signaling pathways, such as, stress-induced ERK and JNK (32) or inflammatory pathways (26, 27), modulate the FXR/SHP pathway as well as intestinal FGF15/19 signaling pathway and may affect CA responsiveness. However, mechanisms for aberrant regulation are largely unknown and warrant future investigation.
In conclusion, this study identifies a novel function for LSD1 in the FXR/SHP pathway that controls hepatic BA levels. The data are consistent with a model in which FXR-induced LSD1 is recruited to direct SHP-target BA synthetic and uptake transport genes, which results in the removal of the activating H3K4-me3 and permits epigenomic repression of these genes by additional repressive histone modifications. Epigenomic regulation by histone-modifying enzymes is a promising area for development of potential therapies for treating human diseases (32). In this regard, small molecules that specifically modulate LSD1 function may be promising agents for the treatment of BA-related hepatobiliary diseases, including cholestatic liver disease.
Supplementary Material
Acknowledgments
Financial Support
This study was supported by National Institutes of Health Grants, DK062777 and DK095842 to J.K.
We are grateful to Ronald Schule for pCMX-flag-LSD1 and pSUPER si LSD1, Tadashi Baba for pGEX-LSD1, and Johan Auwerx for pGEX-GST SHP constructs.
List of Abbreviations
- BA
bile acid
- FXR
Farnesoid X Receptor
- SHP
Small Heterodimer Partner
- LSD1
Lysine specific demethylase1
- RXRα
Retinoid X Receptorα
- IR1
inverted repeat1
- FXRE
FXR response element
- CA
cholic acid
- CDCA
chenodeoxycholic acid
- ChIP
chromatin immunoprecipitation
- ChIP-seq
ChIP-sequencing
- H3K4-me3
tri-methylated histone 3 on lysine 4
- H3K4/K19-Ac
acetylated histone 3 on lysine 9 and 14
- H3K9-me2/3
di- or tri-methylated histone 3 on lysine 9
- Cyp
cytochrome P450
- HDAC1
histone deacetylase 1
- Prox1
Prospero homeobox 1
- IB
immunoblotting
- IHC
immunohistochemistry
Footnotes
Author Contribution
YK, SF, and JK designed research; YK, SF, SB, and SS performed experiments; YK, SF, SB, SS, BK, and JK analyzed data; and YK, BK, JK wrote the paper.
Conflict of Interest
The authors declare no conflict of interest.
References
- 1.Russell DW. The enzymes, regulation, and genetics of bile acid synthesis. Annu Rev Biochem. 2003;72:137–174. doi: 10.1146/annurev.biochem.72.121801.161712. [DOI] [PubMed] [Google Scholar]
- 2.Chiang JY. Bile acids: Regulation of synthesis. J Lipid Res. 2009;50:1955–1966. doi: 10.1194/jlr.R900010-JLR200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.de Aguiar Vallim TQ, Tarling EJ, Edwards PA. Pleiotropic roles of bile acids in metabolism. Cell Metab. 2013;17:657–669. doi: 10.1016/j.cmet.2013.03.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Lefebvre P, Cariou B, Lien F, Kuipers F, Staels B. Role of bile acids and bile acid receptors in metabolic regulation. Physiol Rev. 2009;89:147–191. doi: 10.1152/physrev.00010.2008. [DOI] [PubMed] [Google Scholar]
- 5.Schaap FG, Trauner M, Jansen PL. Bile acid receptors as targets for drug development. Nat Rev Gastroenterol Hepatol. 2014;11:55–67. doi: 10.1038/nrgastro.2013.151. [DOI] [PubMed] [Google Scholar]
- 6.Trauner M, Boyer JL. Bile salt transporters: Molecular characterization, function, and regulation. Physiol Rev. 2003;83:633–671. doi: 10.1152/physrev.00027.2002. [DOI] [PubMed] [Google Scholar]
- 7.Wagner M, Zollner G, Trauner M. Nuclear receptors in liver disease. Hepatology. 2011;53:1023–1034. doi: 10.1002/hep.24148. [DOI] [PubMed] [Google Scholar]
- 8.Kosters A, Karpen SJ. Bile acid transporters in health and disease. Xenobiotica. 2008;38:1043–1071. doi: 10.1080/00498250802040584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Dawson PA, Lan T, Rao A. Bile acid transporters. J Lipid Res. 2009;50:2340–2357. doi: 10.1194/jlr.R900012-JLR200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Smith Z, Ryerson D, Kemper JK. Epigenomic regulation of bile acid metabolism: Emerging role of transcriptional cofactors. Mol Cell Endocrinol. 2012;368:59–70. doi: 10.1016/j.mce.2012.04.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kemper J, Kim H, Miao J, Bhalla S, Bae Y. Role of a mSin3A-swi/snf chromatin remodeling complex in the feedback repression of bile acid biosynthesis by SHP. Mol Cell Biol. 2004;24:7707–7719. doi: 10.1128/MCB.24.17.7707-7719.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Fang S, Miao J, Xiang L, Ponugoti B, Treuter E, Kemper JK. Coordinated recruitment of histone methyltransferase G9a and other chromatin-modifying enzymes in SHP-mediated regulation of hepatic bile acid metabolism. Mol Cell Biol. 2007;27:1407–1424. doi: 10.1128/MCB.00944-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Miao J, Fang S, Lee J, Comstock C, Knudsen KE, Kemper JK. Functional specificities of brm and brg-1 swi/snf ATPases in the feedback regulation of hepatic bile acid biosynthesis. Mol Cell Biol. 2009;29:6170–6181. doi: 10.1128/MCB.00825-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Mitro N, Godio C, De Fabiani E, Scotti E, Galmozzi A, Gilardi F, Caruso D, et al. Insights in the regulation of cholesterol 7alpha-hydroxylase gene reveal a target for modulating bile acid synthesis. Hepatology. 2007;46:885–897. doi: 10.1002/hep.21819. [DOI] [PubMed] [Google Scholar]
- 15.Ananthanarayanan M, Li Y, Surapureddi S, Balasubramaniyan N, Ahn J, Goldstein JA, Suchy FJ. Histone H3K4 trimethylation by MLL3 as part of ASCOM complex is critical for NR activation of bile acid transporter genes and is downregulated in cholestasis. Am J Physiol Gastrointest Liver Physiol. 2011;300:G771–81. doi: 10.1152/ajpgi.00499.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kim DH, Kim J, Lee JW. Requirement for MLL3 in p53 regulation of hepatic expression of small heterodimer partner and bile acid homeostasis. Mol Endocrinol. 2011;25:2076–2083. doi: 10.1210/me.2011-1198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kouzarides T. Chromatin modifications and their function. Cell. 2007;128:693–705. doi: 10.1016/j.cell.2007.02.005. [DOI] [PubMed] [Google Scholar]
- 18.Mulligan P, Yang F, Di Stefano L, Ji JY, Ouyang J, Nishikawa JL, Toiber D, et al. A SIRT1-LSD1 corepressor complex regulates notch target gene expression and development. Mol Cell. 2011;42:689–699. doi: 10.1016/j.molcel.2011.04.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Metzger E, Wissmann M, Yin N, Muller JM, Schneider R, Peters AH, Gunther T, et al. LSD1 demethylates repressive histone marks to promote androgen-receptor-dependent transcription. Nature. 2005;437:436–439. doi: 10.1038/nature04020. [DOI] [PubMed] [Google Scholar]
- 20.Ouyang H, Qin Y, Liu Y, Xie Y, Liu J. Prox1 directly interacts with LSD1 and recruits the LSD1/NuRD complex to epigenetically co-repress CYP7A1 transcription. PLoS One. 2013;8:e62192. doi: 10.1371/journal.pone.0062192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Connelly S, Mech C. Delivery of adenoviral DNA to mouse liver. Methods Mol Biol. 2004;246:37–52. doi: 10.1385/1-59259-650-9:37. [DOI] [PubMed] [Google Scholar]
- 22.Kemper JK, Xiao Z, Ponugoti B, Miao J, Fang S, Kanamaluru D, Tsang S, et al. FXR acetylation is normally dynamically regulated by p300 and SIRT1 but constitutively elevated in metabolic disease states. Cell Metabolism. 2009;10:392–404. doi: 10.1016/j.cmet.2009.09.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Seok S, Kanamaluru D, Xiao Z, Ryerson D, Choi SE, Suino-Powell K, Xu HE, et al. Bile acid signal-induced phosphorylation of small heterodimer partner by protein kinase czeta is critical for epigenomic regulation of liver metabolic genes. J Biol Chem. 2013;288:23252–23263. doi: 10.1074/jbc.M113.452037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Fu T, Choi SE, Kim DH, Seok S, Suino-Powell KM, Xu HE, Kemper JK. Aberrantly elevated microRNA-34a in obesity attenuates hepatic responses to FGF19 by targeting a membrane coreceptor beta-klotho. Proc Natl Acad Sci U S A. 2012;109:16137–16142. doi: 10.1073/pnas.1205951109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lee J, Seok SM, Yu P, Kim K, Smith Z, Rivas-Astroza M, Zhong S, et al. Genomic analysis of hepatic farnesoid X receptor (FXR) binding sites reveals altered binding in obesity and direct gene repression by FXR. Hepatol. 2012;56:108–117. doi: 10.1002/hep.25609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kosters A, Felix JC, Desai MS, Karpen SJ. Impaired bile acid handling and aggravated liver injury in mice expressing a hepatocyte-specific RXRalpha variant lacking the DNA-binding domain. J Hepatol. 2014;60:362–369. doi: 10.1016/j.jhep.2013.09.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Allen K, Jaeschke H, Copple BL. Bile acids induce inflammatory genes in hepatocytes: A novel mechanism of inflammation during obstructive cholestasis. Am J Pathol. 2011;178:175–186. doi: 10.1016/j.ajpath.2010.11.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Park YJ, Qatanani M, Chua SS, LaRey JL, Johnson SA, Watanabe M, Moore DD, et al. Loss of orphan receptor small heterodimer partner sensitizes mice to liver injury from obstructive cholestasis. Hepatology. 2008;47:1578–1586. doi: 10.1002/hep.22196. [DOI] [PubMed] [Google Scholar]
- 29.Modica S, Petruzzelli M, Bellafante E, Murzilli S, Salvatore L, Celli N, Di Tullio G, et al. Selective activation of nuclear bile acid receptor FXR in the intestine protects mice against cholestasis. Gastroenterology. 2012;142:355–65. e1–4. doi: 10.1053/j.gastro.2011.10.028. [DOI] [PubMed] [Google Scholar]
- 30.Anakk S, Watanabe M, Ochsner SA, McKenna NJ, Finegold MJ, Moore DD. Combined deletion of fxr and shp in mice induces Cyp17a1 and results in juvenile onset cholestasis. J Clin Invest. 2011;121:86–95. doi: 10.1172/JCI42846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Inagaki T, Choi M, Moschetta A, Peng L, Cummins CL, McDonald JG, Luo G, et al. Fibroblast growth factor 15 functions as an enterohepatic signal to regulate bile acid homeostasis. Cell Metab. 2005;2:217–225. doi: 10.1016/j.cmet.2005.09.001. [DOI] [PubMed] [Google Scholar]
- 32.Haberland M, Montgomery RL, Olson EN. The many roles of histone deacetylases in development and physiology: Implications for disease and therapy. Nat Rev Genet. 2009;10:32–42. doi: 10.1038/nrg2485. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.