

Abstract
Adoptive transfer of tumor-infiltrating lymphocytes (TILs) and genetically engineered T lymphocytes expressing chimeric antigen receptors (CARs) or conventional alpha/beta T-cell receptors (TCRs), collectively termed adoptive cell therapy (ACT), is an emerging novel strategy to treat cancer patients. Application of ACT has been constrained by the ability to isolate and expand functional tumor-reactive T cells. The transition of ACT from a promising experimental regimen to an established standard of care treatment relies largely on the establishment of safe, efficient, robust and cost-effective cell manufacturing protocols. The manufacture of cellular products under current good manufacturing practices (cGMPs) has a critical role in the process. Herein, we review current manufacturing methods for the large-scale production of clinical-grade TILs, virus-specific and genetically modified CAR or TCR transduced T cells in the context of phase I/II clinical trials as well as the regulatory pathway to get these complex personalized cellular products to the clinic.
INTRODUCTION
Adoptive cell therapy is an emerging therapeutic platform used to induce tumor regression or clearance of certain viral infections after organ transplantation or hematopoietic stem cell transplantation (HSCT). In addition to virus-specific T cells, two major T-cell sources can confer these therapeutic properties: (1) tumor-infiltrating lymphocytes (TILs) isolated, activated and expanded ex vivo; (2) peripheral blood T lymphocytes engineered to express conventional alpha/beta T-cell receptors (TCRs) or tumor-recognizing chimeric antigen receptors (CARs). Clinical cell doses of these autologous tumor-reactive lymphocytes can be manufactured and infused after suitable release testing.1–6
The generation of clinical-grade cellular products encompasses complex processes that are tightly regulated under current good manufacturing practices (cGMP) and requires adequate cell manufacturing facility, ancillary products and manufacturing processes to meet the Food and Drug Administration (FDA) guidelines.7 The end-of-process products characteristics such as safety, purity and potency need be carefully defined to meet the quality-control standards.8 The manufacturing process needs to be robust and reproducible as well as cost effective. Furthermore, autologous cell therapy products are unique, and the manufacturers must integrate the scientific knowledge defining the product with the FDA regulations. In fact, the regulatory guidelines need be tailored to each individual cell therapy product. In this review, we focus on the large-scale cGMP manufacturing of cells used in adoptive cell therapy including TILs, CAR- and TCR-expressing T cells and viral-specific CTLs.
T-CELL MANUFACTURING APPROACHES
Generation of a therapeutically suitable number of highly active antitumor T cells is a significant technical challenge, and remains critical for the application of adoptive cell therapy as a standard cancer therapy.
Manufacturing of TILs
Infusion of ex vivo-expanded TILs has proven to be a successful treatment regiment for refractory metastatic melanoma.9,10 The manufacture of tumor antigen-specific lymphocytes used in adoptive cell transfer is initiated from tumor fragments or single-cell enzymatic digests of resected tumor specimen. A microculture derived from a single tumor fragment or 106 viable cells derived from the single-cell enzymatic digestion are placed into one well of a 24-well plate with high dose interleukin-2 (IL-2). Growth medium is changed within 1 week; confluent wells are subsequently split into daughter wells and maintained as independent TIL cultures for generally 1–2 weeks. Cultures are subsequently fed twice per week and maintained at 0.8–1.6×106 mL−1 in flasks. A standard TIL culture typically generates 5 × 107 cells from each original well after 3 to 5 weeks of time. When tumor-reactive TIL cultures are expanded to the minimal requirement of 3 × 107 cells, independent TIL activity and specificity are determined by measuring interferon-gamma secretion by enzyme-linked immunosorbent assay post stimulation with tumor cells. Active individual TIL cultures are then expanded to therapeutic relevant numbers by using a rapid expansion protocol.11 During the rapid expansion phase, 106 TIL effector cells are combined with 2 × 108 irradiated, allogeneic healthy donor peripheral blood mononuclear cell (PBMC) feeder cells in presence of anti-CD3 OKT-3 monoclonal antibody (mAb) and high dose IL-2 in tissue culture flasks. Cell density is determined on day 6 of culture and thereafter to maintain a density of 106 mL−1 by splitting TIL cultures into flasks or culture bags. IL-2 (6000 U mL−1) is used throughout the process to promote cell expansion. Within 2 weeks of time since the start of the rapid expansion protocol, cells are harvested, washed, formulated and cryopreserved. The whole manufacture process takes ~6–8 weeks.12,13 Products meeting all quality-control tests are released for patient infusion (Figure 1a).
Figure 1.
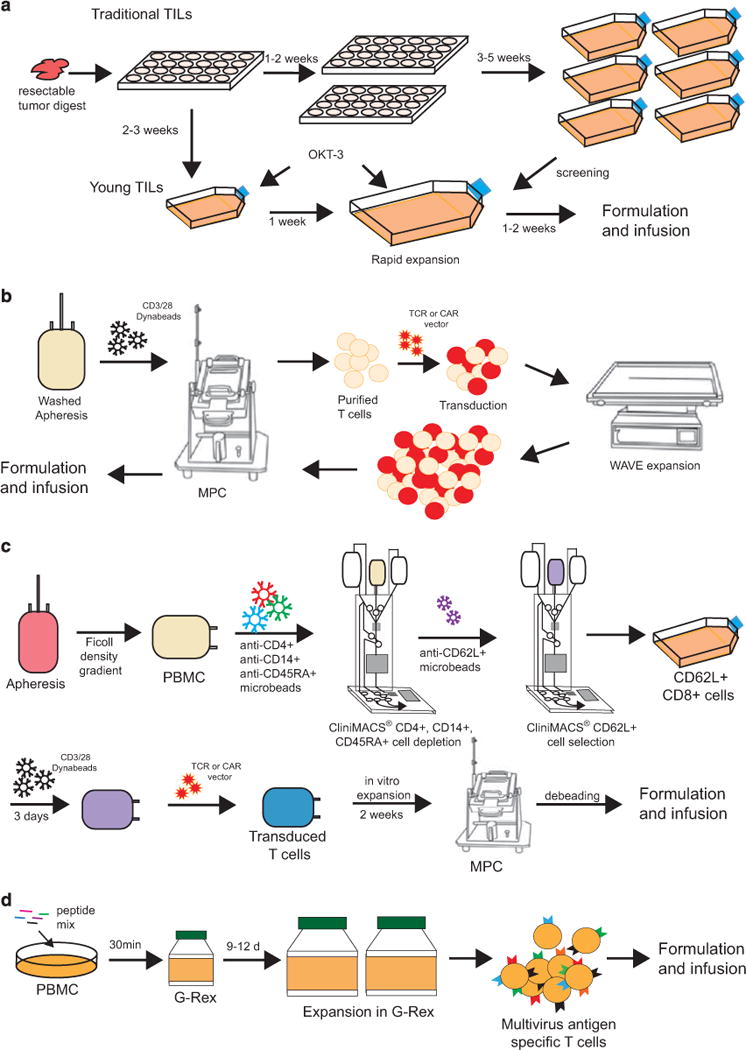
Schematic illustration of representative TIL, TCR/CAR-T, and EBV-CTL manufacturing platforms. (a) Traditional and young TIL manufacturing scheme. Single-cell digests of resected tumor are plated in 24-well plates as microcultures. For traditional TILs, the microcultures are passaged as independent cultures. Screened and selected TIL cultures are further expanded with a 2-week rapid expansion procedure using OKT-3 antibody. For young TILs, microcultures are pooled without screening. The pooled cells undergo the same 2-week rapid expansion procedure to reach the target dose. During the whole process, cells are maintained in culture with 6000 IU mL−1 of IL-2. (b) TCR/CAR-T manufacturing process. T cells are selected from washed apheresis product and activated by using CD3/28 Dynabeads and ClinExVivo magnetic particle concentrator (MPC). Activated T cells are transduced with TCR/CAR retroviral or lentiviral vectors, and transduced cells are expanded with WAVE bioreactor. CD3/28 magnetic Dynabeads are removed from the cells with MPC and end of the process cells are formulated for infusion. (c) CD8+ central memory TCR/CAR T-cell manufacturing process. PBMCs are first purified from apheresis product using Ficoll-Plaque gradient centrifugation, followed by CD4+, CD14+ and CD45RA+ cell depletion using anti-CD4, anti-CD14 and anti-CD45RA microbeads and CliniMACS. Collected cells undergo an additional CD62L positive selection procedure using anti-CD62L microbeads and CliniMACS. Selected CD8+CD62L+ cells are further activated with CD3/28 Dynabeads. Activated memory CD8+ cells are transduced with TCR/CAR vectors and expanded in vitro. Dynabeads are removed from the EOP cells using MCP before formulation. (d) Generation of multiviral antigen-specific T cells using G-Rex bioreactor. Donor PBMCs are pulsed with 15mer peptides mix spanning EBV, adenovirus, CMV, BK virus and human herpes virus antigen epitopes and transferred into G-Rex device. Multivirus antigen-specific T cells are expanded to high quantities in ~ 2 weeks of time in the presence of IL-4 and IL-7. CAR, chimeric antigen receptor; CTL, cytotoxic T lymphocytes; EBV, Epstein–Barr virus; IL, interleukin; PBMC, peripheral mononuclear blood cell; TCR, T-cell receptor; TIL, tumor-infiltrating lymphocyte.
Promising clinical outcomes have been achieved using tumor-reactive TILs in combination with lymphodepletion.14 However, the extended duration of multiple microcultures and an individualized tumor recognition assay render the process time-consuming, complex and costly. To circumvent the limitations of such ‘standard’ method, Dudley and colleagues15 have simplified and standardized a process to culture ‘young’ TILs. ‘Young’ TIL cultures are made of bulk lymphocytes rather than individual microcultures, and the tumor recognition screening assay is eliminated from the process. Young TIL culture is initiated from enzymatic digestion of resected tumor specimen. Single-cell suspension is plated in individual wells of 24-well plates at 5 × 105 cells mL−1 in presence of 6000 IU mL−1 of IL-2. Five days after initiation, cells are fed and culture media is replaced every 2–3 days thereafter. By day 10 to 18, individual wells of cells are pooled and ~6–8 weeks.12,13 Products meeting all quality-control tests are released for patient infusion (Figure 1a).
Promising clinical outcomes have been achieved using tumor-reactive TILs in combination with lymphodepletion.14 However, the extended duration of multiple microcultures and an individualized tumor recognition assay render the process time-consuming, complex and costly. To circumvent the limitations of such ‘standard’ method, Dudley and colleagues15 have simplified and standardized a process to culture ‘young’ TILs. ‘Young’ TIL cultures are made of bulk lymphocytes rather than individual microcultures, and the tumor recognition screening assay is eliminated from the process. Young TIL culture is initiated from enzymatic digestion of resected tumor specimen. Single-cell suspension is plated in individual wells of 24-well plates at 5 × 105 cells mL−1 in presence of 6000 IU mL−1 of IL-2. Five days after initiation, cells are fed and culture media is replaced every 2–3 days thereafter. By day 10 to 18, individual wells of cells are pooled and ~6–8 weeks.12,13 Products meeting all quality-control tests are released for patient infusion (Figure 1a).
Promising clinical outcomes have been achieved using tumor-reactive TILs in combination with lymphodepletion.14 However, the extended duration of multiple microcultures and an individualized tumor recognition assay render the process time-consuming, complex and costly. To circumvent the limitations of such ‘standard’ method, Dudley and colleagues15 have simplified and standardized a process to culture ‘young’ TILs. ‘Young’ TIL cultures are made of bulk lymphocytes rather than individual microcultures, and the tumor recognition screening assay is eliminated from the process. Young TIL culture is initiated from enzymatic digestion of resected tumor specimen. Single-cell suspension is plated in individual wells of 24-well plates at 5 × 105 cells mL−1 in presence of 6000 IU mL−1 of IL-2. Five days after initiation, cells are fed and culture media is replaced every 2–3 days thereafter. By day 10 to 18, individual wells of cells are pooled and ~ 5×107 young TILs are obtained. Rapid expansion of young TILs is performed using the rapid 2-week expansion protocol as described above (Figure 1a). Continuous efforts are made to improve both standard and young TIL manufacturing processes to generate CD8+ T-cell-enriched culture.16 More recently, a process to generate epitope-specific TILs by stimulating patient PBMCs with clinical-grade peptides followed by sorting of antigen-specific T cells was published.17
Manufacturing of T cells genetically engineered to express an exogenous TCR or CAR
Despite the fact that TILs have been shown to mediate antitumor response in 50–70% of melanoma patients, TILs have only limited success in other types of cancers.18 Furthermore, the generation of TILs is not successful for all melanoma patients.10 To this end, the genetic modification of peripheral blood lymphocytes to endow these readily accessible cells with antitumor activity is an attractive approach. The power and promise of TCR and CAR-T therapy have been demonstrated by encouraging outcome in patients treated with NY-ESO-1 TCR19,20 and CD19-CAR T cells.21–24 Many ongoing clinical trials utilized genetically modified T cells, and numerous recent papers have reported their clinical success.25 The key requirement for this genetic modification methodology is the development of RNA vectors expressing TCRs and CARs. TCR can be cloned from the rare occurring patient tumor-reactive T-cell clones,26 from humanized murine models27,28 or using the phage display technology.29,30 The design of TCR and CAR has steadily improved over the past two decades.31–35 For CARs, tumor recognition is mediated by the single-chain variable fragment derived from a monoclonal antibody or humanized Fab. The rationale and strategy of TCR and CAR design and their evolution have been comprehensively reviewed elsewhere.36,37
The manufacture of T cells genetically engineered to express specific TCRs is initiated from Ficoll-purified PBMCs. T cells from PBMCs are activated with OKT-3 antibodies, transduced with a retroviral vector expressing a tumor antigen-specific TCR and cultured for ~ 2 weeks.38 For the CAR-T cells, large-scale transduction and expansion under cGMP has been established,39 and is also applicable to TCR-T cell manufacturing. The process is initiated from the selection and activation of T cells from patient apheresis products using Dynabeads CD3/CD28. CD3+CD28+ T cells are enriched using a magnetic particle concentrator, and are cultured at 106 mL−1. The activated T cells are transduced with retroviral vectors in RetroNectin-coated cell bags. The retroviral vector-transduced T cells are inoculated in a WAVE bioreactor on day 6 to day 8, and expanded with a continuous perfusion regime. By the end of the production run, the beads are removed with the same magnetic bead concentrator and the cells are formulated for infusion either fresh or frozen. The process takes ~ 2 weeks (Figure 1b). This semi-closed large-scale manufacturing platform successfully supports several ongoing clinical trials at MSKCC (NCT01416974, NCT01044069, NCT00466531, NCT01840566, NCT01860937, NCT01140373)21,23,39,40 and can be easily adapted for other clinical trials involving the transduction and expansion of autologous or donor T cells.
Other groups are focusing on defining which T-cell subsets are best suited for use in adoptive therapy to generate cell products enriched for these subsets.41 In animal models, T-cell transfer studies have shown that effector cells from TEM rapidly undergo apoptosis following adoptive transfer and do not persist beyond 7–14 days, whereas a subset of transferred CD8+ TE/CM can reacquire memory cell markers, and persist for years.42 Consequently, the authors developed a clinical CD8+ TCM purification, transduction and expansion platform that incorporates clinical scale polyclonal CD8+ TCM isolation from leukapheresis products, T-cell activation using anti-CD3/CD28 beads without exogenous feeder cells, lentiviral transduction and cell expansion in IL-2/IL-15.42 This process is performed with minimal open processing steps and reproducibly yields cryopreserved cell products in excess of 109 cells within 35 days (Figure 1c). This platform is currently being used to generate autologous CAR redirected CD19-specific CD8+ TE/CM for adoptive transfer following autologous HSCT for high-risk CD19+ non-Hodgkin lymphomas.42
Manufacturing of viral-specific T cells (G-Rex)
Adoptive transfer of viral antigen-specific T cells is a wellestablished procedure for effective treatment of transplantassociated viral infections and virus-related malignancies. Many laboratories have successfully generated and infused T cells specific for Epstein–Barr virus (EBV), cytomegalovirus and adenovirus using monocytes and EBV-transformed lymphoblastoid cells.43–46 Although therapeutic doses of trivirus-specific cytotoxic T lymphocytes can be generated, the original methodology requires 4 to 6 weeks of time for EBV–LCL generation and 4–8 weeks for CTL expansion.47,48 Recent process development has been reported for the generation of penta-viral-specific T cells for CMV, AdV, EBV, BK virus (BK) and human herpes virus 6 (HHP6) with dramatically reduced production complexity and time requirement. The process starts with incubation of 1.5 × 107 fresh PBMC and overlapping 15 amino acid peptide mix spanning EBV–LMP2, BZLF1, EBNA1; Adv-Penton, Hexon; CMV-pp65, IE-1; BKV-VP1, large T; HHV6-U11, U14 and U90. The cells are subsequently transferred to G-Rex bioreactors for continuous culture in presence of IL-4 and IL-7. Therapeutic doses can be achieved in ~10 days of time, in contrast to the 10-week period required with the traditional approach. Monovalent-, bivalent-, trivalent-, tetravalent- and pentavalent-specific T-cell products are efficiently generated with this method, whereas the range of antiviral activity is limited by the previous viral exposure of the donor T cells. The multiviral-specific T-cell lines generated using this method have been demonstrated to have up to a 94% response rate in post HSCT patient with viral infections49 (Figure 1d). Viral-specific T cells can be further genetically engineered to express TCRs and CARs to have a second specificity for tumor antigens.44,50–52 A GMP manufacture process has also been recently tested, in which the CliniMACS-purified viral-specific T cells were transduced with retroviral vectors, and expanded in vitro.53
To broaden the use of CAR-modified T cells that could provide a GVL effect after allo-HSCT without concomitant GVHD, Riddell et al.41 and others54 have proposed to combine the use of viral-specific T cells such as CMV- and EBV-specific CD8+ T cells to generate CAR expressing T cells derived from central memory T (TCM) cells as they are deemed capable of persisting long term.55 In one method, the CD45RA−CD8+ cell fraction is enriched by depletion of CD4+, CD14+ and CD45RA+ cells on the CliniMACS device using clinical-grade mAbs and paramagnetic beads.42 The CD62L+ cells are subsequently enriched by positive selection with a clinical-grade biotin-conjugated anti-CD62L mAb and anti-biotin microbeads. In brief, the enriched CD8+CD62L+ T cells are plated with either autologous γ-irradiated peptide-pulsed PBMCs or monocyte-derived dendritic cells in 50 IU mL−1 IL-2. On day 1 after stimulation, the T cells are exposed to lentiviral vector stocks encoding the CD19-CAR in presence of polybrene followed by spinoculation. After 8–10 days in culture, the cells are pooled and analyzed by flow cytometry after staining with virus-specific human leukocyte antigen tetramers. The transduced T cells are expanded in culture by plating with γ-irradiated LCLs and fed with 50 IU mL−1 IL-2. After 10–14 days of culture, cells are stained with virus-specific human leukocyte antigen tetramers and Abs specific for transduction markers. The virus-specific subset of transduced T cells is then purified using reversible class I MHC streptamers. The selected cells can also be transduced with a lentiviral vector CAR transgene modified to co-express a truncated version of the epidermal growth factor receptor that can be detected by biotinylated anti-EGFR (Erbitux) mAb.56–58
EXPRESSION VECTORS FOR GENETIC MODIFICATION OF T CELLS
Three main types of gene expression vectors are currently used in clinical applications for TCR and CAR delivery in T cells. They include gamma retroviral vectors, lentiviral vectors and transposons. We will focus herein on the large-scale manufacturing platforms for these critical reagents.
Gamma-retroviral vector
Gamma-retroviral vectors were the first viral vectors used for clinical application.59 They are still used as gene-transfer vehicles in about 20% of the current clinical trials.60 The wide usage of gamma retroviral vectors is due to their broad cell tropism, efficient integration and stable gene expression in target cells. In addition, they can be consistently manufactured at relatively low cost. Many stable packaging cell lines, such as PA317,61 PG1362 and 293GP,63 have been developed. We share with several groups the combinatorial use of the SFG vector and PG13 packaging cell line.23,44,64–66 The clonal selection and expansion of high-titer-producer cells can yield the desired stable gamma retroviral cell clone. Subsequently, a master cell bank of the stable packaging cell clone can be generated and qualified. Large-scale manufacturing protocols have been reported from different laboratories. The manufacturing process starts from the expansion of stable producer cells in roller bottles,67,68 10-layer cell factories64 or bioreactors69 (Figure 2a). Gamma-retroviral vectors cannot be harvested longer than three consecutive days due to the relative short half-life of gamma retroviral vectors. The harvests are pooled at the end of the production run and filtered using a step-filtration step to efficiently remove packaging cell contaminants from the vector stocks.64,67 The vector stocks are aliquoted and frozen in vials or cryobags. In the case of gamma retroviral vectors with a self-inactivating design, vector manufacturing relies on transient transfection-based techniques similar to lentiviral vector production.70
Figure 2.
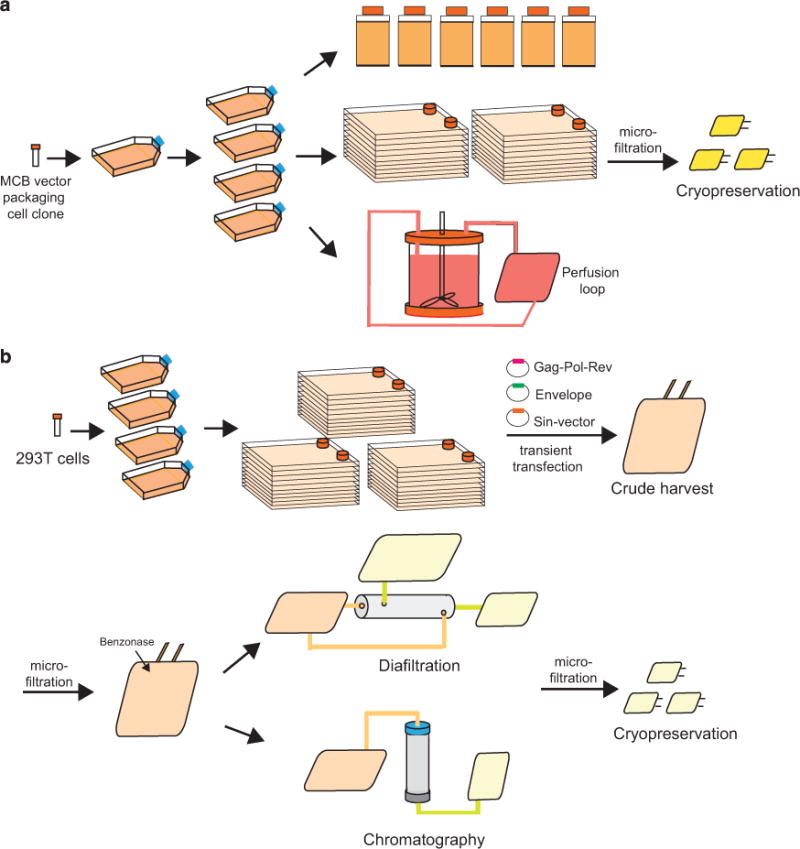
Retroviral and lentiviral vectors’ manufacturing platform. (a) Generation of retroviral vector. High titer-producer cells from master cell bank are thawed and expanded in T flasks. Cells are further expanded in either roller bottles, cell factories or bioreactors. Vector stocks are harvested in the optimized harvesting window, filtered to removed contaminants and cryopreserved for biosafety testing before release for clinical use. (b) Manufacturing of lentiviral vector using transient transfection in 10-layer cell factories. 293T cells or derivatives are expanded to large quantity to inoculate multiple 10-layer cell factories. Cells are transiently transfected with packaging, envelope, and SIN-vector plasmids. Crude vector stocks are harvested and filtered. Benzonase is added to the crude harvests to remove plasmid contaminants. Vector stocks need to be further purified and concentrated using diafiltration or chromatography methodologies. Purified and concentrated vector stocks are cryopreserved for biosafety testing to be qualified for clinical use. MCB, master cell bank; SIN, self-inactivating.
To release the vector stocks for clinical use, a series of biosafety testing is required, which include but are not limited to sterility on end-of-process cells (EOP) and final product vector stocks (FP), mycoplasma testing (EOP and FP), general safety (FP), transmission electron microscopy (EM) on bulk vector stocks, in vitro adventitious virus testing (FP) and GalV replication-competent retrovirus (GalV RCR; EOP and FP).39 The production of gamma retroviral vectors in serum-free media or media containing serum replacement is highly desirable for clinical trials beyond phase I but remains a challenge.71,72 Gamma-retroviral vectors have been shown to be safe in patients who received T cells genetically modified to express LNGF-R, HSV-TK, neomycin, adenosine deaminase or an anti-HIV-1 tat ribozyme. After up to 10 years follow-up, these patients have not developed any evidence of T-cell clonal expansion.73–76
Lentiviral vector
Lentiviral vectors have been successfully utilized to engineer hematopoietic stem cells for the treatment of adrenoleukodystropy,77 beta-thalassemia,78,79 Wiskott–Aldrich syndrome80 and metachromatic leukodystropy81 as well as CAR T cells for hematologic diseases.22,56,82 Similar to gamma retroviral vectors, lentiviral vectors mediate efficient gene transfer and high level of transgene expression. The commonly used VSV-G pseudo envelop also endows broad tropism. Compared with gamma retroviral vectors, lentiviral vectors display several favorable features such as the ability to transduce nondividing cells83–85 and relative safer chromosome integration profile;86 it should be noted that gamma retroviral vectors have not been reported to be genotoxic in terminally differentiated cells such as T lymphocytes.73–76 Significant hurdles in production and purification processes to obtain sufficient quantities of GMP grade lentiviral vector stocks for phase I clinical trials and beyond need to be overcome. Stable producer cell lines are difficult to generate and are not widely available for lentiviral vector production.87,88 The commonly used manufacturing platforms for the third- and fourth-generation packaging systems are based on transient transfection of three or four independent plasmids encoding gag-pol-rev, the self-inactivating transfer vector and the pseudo envelope. For the fourth-generation packaging system, the rev gene can also be encoded on a separate plasmid. HEK293 cell and its derivatives such as 293T,89 293E90 are the principle cell lines used for lentiviral vector production. The calcium phosphate precipitation method is traditionally used for transfection. Another cost-effective compound, polyethylenimine, has also been qualified and used in recent years91,92 as well as flow electroporation.93 Other lipidbased methods are still too expensive to be used in a large-scale manufacturing setting. For large-scale lentiviral vector production, HEK293-derived cells are expanded in large quantity. The method of culture expansion is a critical component for generating vector stocks with high titer and yield. The available scalable expansion systems include the cell factory system, the HYPERFlask, microcarriers and bioreactors.70,94,95 The downstream processes for lentiviral vector production aims at removing cell and plasmid contaminants, concentrating vector particles to achieve high titer vector stocks while maintaining vector potency. These are challenging tasks that typically encompass the following steps: (1) Vector stocks harvesting. Owing to the nature of transient transfection, crude lentiviral vector stocks can be harvested for 2 days. Generally, the titer of the vector stocks beyond 2-day harvest is too low to be used; (2) Clarification. This step is to eliminate producer cells and cell debris from the crude harvest. It can be achieved by centrifugation or dead-end filtration. Microfiltration is needed to achieve greater clarification for downstream ultrafiltration or chromatography; (3) Nucleic acid digestion. Plasmid DNAs used for transfection are the major source of DNA contaminants. Cellular DNA and RNA may also be released during cell culture. Nucleic acids need to be removed to meet safety requirements and decrease sample viscosity, a major cause of column clotting. Benzonase is commonly used for this purpose; (4) Concentration and purification. Ultracentrifugation is the most widely used method for lentiviral vector concentration in a research setting. Ultrafiltration and chromatography are the preferred methods for manufacturing under cGMP. Although different filtration modes and devices are available for ultrafiltration, tangential-flow filtration is the most widely used method for its effectiveness and better yield. Chromatography is another preferred method for GMP manufacturing. A number of chromatography methods, including anion exchange chromatography, affinity chromatography and size exclusion chromatography96 have been reported for the purification of lentiviral vector particles; (5) Sterile filtration and storage. Membrane filtration through 0.22 μm pores is the last step in the generation of clinicalgrade lentiviral vector (Figure 2b). Vectors are packaged and stored in −80°C and a series of quality-control assays are performed before release for clinical use (Table 1). Similar to gamma retroviral vectors, the production in serum-free media is desirable for clinical trials beyond phase I but remains challenging.97 The production of lentiviral vectors has been comprehensively reviewed by Schweizer and Merten.98
Table 1.
Quality-control assays for clinical-grade retroviral and lentiviral vectors
| Testing | Example assays | Criteria | |
|---|---|---|---|
| Purity | Lentiviral vector | ||
| Total proteins (ng mL−1) | ELISA | Report results | |
| Bovine serum albumin (ng mL−1) | ELISA | Report results | |
| Benzonase (ng mL−1) | ELISA | Report results or <100 ng mL−1 | |
| Plasmid DNA (copies per 100 ng) | VSVg qPCR optional or serial washes | Below detection or decrease over time | |
| Host cell specific DNA (copies per 100 ng) | qPCR | Report results | |
| SV40 LTA and E1A qPCR | qPCR | Below detection | |
| Safety | Retroviral and lentiviral vector | ||
| Sterility | USP, No growth for 14 days culture on Vero indicator, PTC | No growth for 14 days | |
| Mycoplasma | Negative | ||
| Endotoxin/pyrogen | LAL | <10 EU mL−1 | |
| In vitro Adventitious agents | Assay on MRC5, Vero and A549 cells | Negative | |
| Retroviral vector | |||
| RCR | Marker-rescue cell culture assay | No RCR detected | |
| General safety (first lot) | Current USP | Absence of adverse agents | |
| Lentiviral vector | |||
| RCL | Co-culture on C8166 cells with amplification and indicator phases | No RCL detected | |
| Potency | Retroviral and lentiviral vector | ||
| Infectious viral particles | Gene transfer/expression assay in cell line of choice | Report results | |
| Retroviral vector | |||
| Total viral particles | EM | Only type C retroviral like particles detected | |
| Lentiviral vector | |||
| Total viral particles | p24 ELISA, qRT-PCR | Report value | |
| Others | Retroviral and lentiviral vector | ||
| Physicochemical characteristics | pH (optional) | 6.9–7.8 | |
| Appearance | Opaque |
Abbreviations: ELISA, enzyme-linked immunosorbent assay; LAL, limulus amebocyte lysate; qPCR, quantitative PCR; qRT-PCR, quantitative reverse transcription PCR; RCL, replication competent lentivirus; RCR, replication competent retrovirus; USP, U.S. pharmacopeial convention.
Sleeping beauty transposon/transposase system
Transposon/transposase is a relatively new expression system in the gene therapy field. It is a nonviral, plasmid-based methodology. The transposon/transposase system is derived from fish and has been adapted for gene therapy. The sleeping beauty (SB) system consists of two DNA plasmids: one plasmid is the transposon that encodes the gene of interest, such as CAR or TCR; the second plasmid expresses the transposase that enables the insertion of the transgene into TA dinucleotide repeats. The SB transposon/transposase have been used to produce genetically modified CAR-T cells for phase I/II clinical trial,99,100 in which SB transposon/transposase are introduced into T cells by electroporation. Transfected T cells are subsequently expanded on artificial antigen-presenting cells.101 The advantages of using the SB system are that the clinical-grade plasmids are much simpler to produce, and the cost effectiveness due to lesser safety testing requirements when compared to cell products genetically modified with gamma retroviral or lentiviral vectors (Table 2).
Table 2.
Example of Quality-control assays for clinical-grade SB plasmid
| Testing | Example assays | Criteria | |
|---|---|---|---|
| Purity | Identity | Restriction mapping and agarose gel | Expected band size |
| Sequencing | Sanger, high through put | Confirm the original coding sequence | |
| Concentration | absorbance at 260nm | 2.0 ± 0.2 mg mL−1 | |
| A260/A280 absorbance | assay name | 1.8 ± 0.2 | |
| Plasmid Form | HPLC | >90% supercoiled | |
| Safety | Sterility Test | USP, no growth within 14 days | No growth for 14 days |
| Bacterial endotoxin | Kinetic LAL test | <50 EU mg−1 | |
| E. coli host protein | ELISA | <0.3% | |
| E. coli RNA | HPLC | <10% | |
| Others | Appearance | Observation | Clear, colorless, liquid |
Abbreviations: E. coli, Escherichia coli; ELISA, enzyme-linked immunosorbent assay; HPLC, high-performance liquid chromatography; SB, Sleeping Beauty.
PROCESS VALIDATION
The bench-to-bedside transition for innovative adaptive cell therapy requires carefully designed scale-up and validation processes. Process validation is required to establish scientific evidence that a process is capable of consistently delivering quality products. FDA issued new guidelines for process validation in 2011.102 Process validation can be broken down into the following three stages.
Process design stage
Process design is based on the knowledge gained through process development and scale-up activities, including those gained from research laboratories, process engineering, pilot and small-scale studies. The goal of this stage is to design a process suitable for routine manufacturing procedures. Early process design experiments do not need to be performed under cGMP; however, maintaining detailed records of reagents and procedures is highly advisable. During this stage, maintaining the right balance between process complexity and practicality is important to ensure broad downstream application. The selection of reagents that allows freedom to operate can be a challenge as these therapies demonstrate promising outcomes.
Process qualification stage
During this stage, the process design is evaluated to determine whether it is performing in the intended manner. This stage includes two elements: (1) Facility design as well as equipment and utility qualification; and (2) process performance qualification. Current GMP-compliant procedures must be followed at this stage.103 Equipment and utility qualification can either be performed under individual plans or as part of an overall project plan. The quality-control unit must review and approve the qualification plan and report. The process performance qualification combines the actual facility, utilities, equipment and trained personnel with the manufacturing process. A written protocol specifying the manufacturing conditions, controls, testing and expected outcome is essential at this stage. In most cases, process performance qualifications have a higher level of sampling, additional testing and greater scrutiny. A performance analysis report should be prepared in a timely fashion post completion of the process. The successful execution of process performance qualification is a major step in the product life cycle.
Continued process verification
This stage of validation ensures that the manufacturing process remains in a state of control. The equipment and facility qualification status must be maintained through routine monitoring, maintenance and calibration procedures.
Data generated during processes related to product quality need to be collected and analyzed in a timely fashion by qualified individuals. These results help the manufacturers gain deeper understanding of the source of variability, the presence and degree of variation and the impact of these variations on the process and product. A change /optimization of the process may be warranted based on the data collected.
Documentation at all stages of process validation life cycle is essential for effective communication in the complicated, lengthy, and multidisciplinary process of cell manufacturing. Quality is built in the product, not solely tested in the final product. An open and ongoing dialogue between manufacturing team and quality assurance/quality control teams is the key for establishing a successful manufacturing platform.
RELEASE TESTING AND CERTIFICATE OF ANALYSIS
An appropriate set of practical and scientifically defendable release criteria is essential to guarantee the products’ integrity, consistency and efficacy. The underlying principle for release criteria is to provide adequate testing to ensure the product identity, purity, safety and potency. The cellular identity of T-cell products is commonly characterized by cell surface marker expression detected by flow cytometry analysis. It can also include more defined cell subset composition42 or residual tumor contamination. The interpretation of purity here means lack of endotoxin or other potential harmful materials contaminating the product during manufacturing. Safety of the product requires that it is sterile and free of mycoplasma contamination and of RCR or RCL. Cellular products need to be viable (generally ⩾ 70%), and genetically modified cellular products may need to reach a minimum transduction efficiency as a potency criteria.
Sterility is a fundamental test required for the release of cellular products. Standard sterility tests described in 21CFR610.2 for bacterial and fungal contamination requires 14 days of incubation. Bactec automated-based method are also being considered and can be validated for cultured cell products.104,105 When only short time intervals are foreseeable between completion of manufacturing and product release, Gram staining can be used in combination with sterility results on in process samples collected 24 and 48 h before formulation. ‘Points to Consider’ is the method recommended by the FDA for mycoplasma testing for all ex vivo cultures. Although there are other commercially available PCR-based kits to detect this contaminant, these methods are not approved by the regulatory agency. They may be used if they are properly validated during the process validation. A rapid release assay for endotoxin has been developed using the Endosafe PTS endotoxin device, which takes about 20 min and is approved by the FDA.106 Viability assessment of cells is a routine requirement that can be done by various methods, including trypan blue exclusion, 7-aminoactinomycin D staining coupled with flow cytometric analysis, and acridine orange and propidium iodide staining followed by automatic cell counting. Other required product-specific assays should be established earlier on in the process development phase and approved by FDA under the investigational new drug application.
The release of the cell product for infusion is handled through the issuance of a certificate of analysis (C of A). The C of A summarizes the characteristics of the product and the tests performed. The C of A also details the release specifications and results of each test including the method used, assay sensitivity and acceptable range of results. Example of released tests used for CAR-T cells were previously published by our group39 and by others.82,101
CONCLUSION
Treating cancers by harnessing the power of the immune system holds great promise for future cancer therapies. Cumulative evidence shows that adoptive T-cell therapy is an effective treatment for various tumors, including melanoma, hematological cancers and some viral infections post organ transplantation and HSCT. Yet, breakthroughs are still awaited in the field of solid tumors. TILs and the genetically modified TCR and CAR transduced T cells as therapeutic modalities are progressing toward a more mature stage. Although autologous cell therapy poses unprecedented challenges in terms of manufacturing and distribution for commercialization purposes, TCR- and CAR-transduced T cells recently became part of the portfolio of biotechnology and large pharmaceutical companies. Multiple partnerships between academic centers and industry have been established.107 As a result, improved and semi-automated manufacturing platforms are likely to be developed that will allow wide dissemination of these promising therapies and will encourage novel research approaches. Adoptive T-cell therapies are poised to become part of the standard of care treatments for patients with cancer.
Acknowledgments
We thank Dr Michel Sadelain for critically reviewing the manuscript. This work is supported by NCI P30 CA08748, P50 CA086438.
Footnotes
CONFLICT OF INTEREST
IR is a scientific cofounder of and a consultant for Juno Therapeutics. XW declares no conflict of interest.
References
- 1.Rosenberg SA, Restifo NP, Yang JC, Morgan RA, Dudley ME. Adoptive cell transfer: a clinical path to effective cancer immunotherapy. Nat Rev Cancer. 2008;8:299–308. doi: 10.1038/nrc2355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Restifo NP, Dudley ME, Rosenberg SA. Adoptive immunotherapy for cancer: harnessing the T cell response. Nat Rev Immunol. 2012;12:269–281. doi: 10.1038/nri3191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Humphries C. Adoptive cell therapy: honing that killer instinct. Nature. 2013;504:S13–S15. doi: 10.1038/504S13a. [DOI] [PubMed] [Google Scholar]
- 4.Maus MV, Fraietta JA, Levine BL, Kalos M, Zhao Y, June CH. Adoptive immunotherapy for cancer or viruses. Annu Rev Immunol. 2014;32:189–225. doi: 10.1146/annurev-immunol-032713-120136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Yee C. The use of endogenous T cells for adoptive transfer. Immunol Rev. 2014;257:250–263. doi: 10.1111/imr.12134. [DOI] [PubMed] [Google Scholar]
- 6.Davila ML, Brentjens R, Wang X, Riviere I, Sadelain M. How do CARs work?: Early insights from recent clinical studies targeting CD19. Oncoimmunology. 2012;1:1577–1583. doi: 10.4161/onci.22524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.FDA. Guidance for industry: current good manufacturing practice for blood and blood components: (1). Quarantine and disposition of units from prior collections from donors with repeatedly reactive screening test for antibody to hepatitis C virus (anti-HCV); (2) Supplemental testing, and the notification of consignees and blood recipients of donor test results for anti-HCV; availability—FDA Notice. Fed Regist. 1998;63:56198–56199. [PubMed] [Google Scholar]
- 8.Gee AP. Product release assays. Cytotherapy. 1999;1:485–491. doi: 10.1080/0032472031000141309. [DOI] [PubMed] [Google Scholar]
- 9.Dudley ME, Wunderlich JR, Robbins PF, Yang JC, Hwu P, Schwartzentruber DJ, et al. Cancer regression and autoimmunity in patients after clonal repopulation with antitumor lymphocytes. Science. 2002;298:850–854. doi: 10.1126/science.1076514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Dudley ME, Yang JC, Sherry R, Hughes MS, Royal R, Kammula U, et al. Adoptive cell therapy for patients with metastatic melanoma: evaluation of intensive myeloablative chemoradiation preparative regimens. J Clin Oncol. 2008;26:5233–5239. doi: 10.1200/JCO.2008.16.5449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Riddell SR, Greenberg PD. The use of anti-CD3 and anti-CD28 monoclonal antibodies to clone and expand human antigen-specific T cells. J Immunol Methods. 1990;128:189–201. doi: 10.1016/0022-1759(90)90210-m. [DOI] [PubMed] [Google Scholar]
- 12.Topalian SL, Muul LM, Solomon D, Rosenberg SA. Expansion of human tumor infiltrating lymphocytes for use in immunotherapy trials. J Immunol Methods. 1987;102:127–141. doi: 10.1016/s0022-1759(87)80018-2. [DOI] [PubMed] [Google Scholar]
- 13.Dudley ME, Wunderlich JR, Shelton TE, Even J, Rosenberg SA. Generation of tumor-infiltrating lymphocyte cultures for use in adoptive transfer therapy for melanoma patients. J Immunother. 2003;26:332–342. doi: 10.1097/00002371-200307000-00005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Rosenberg SA. Raising the bar: the curative potential of human cancer immunotherapy. Sci Transl Med. 2012;4:127ps8. doi: 10.1126/scitranslmed.3003634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Tran KQ, Zhou J, Durflinger KH, Langhan MM, Shelton TE, Wunderlich JR, et al. Minimally cultured tumor-infiltrating lymphocytes display optimal characteristics for adoptive cell therapy. J Immunother. 2008;31:742–751. doi: 10.1097/CJI.0b013e31818403d5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Dudley ME, Gross CA, Langhan MM, Garcia MR, Sherry RM, Yang JC, et al. CD8+ enriched ‘young’ tumor infiltrating lymphocytes can mediate regression of metastatic melanoma. Clin Cancer Res. 2010;16:6122–6131. doi: 10.1158/1078-0432.CCR-10-1297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Labarriere N, Fortun A, Bellec A, Khammari A, Dreno B, Saiagh S, et al. A full GMP process to select and amplify epitope-specific T lymphocytes for adoptive immunotherapy of metastatic melanoma. Clin Dev Immunol. 2013;2013:932318. doi: 10.1155/2013/932318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hinrichs CS, Rosenberg SA. Exploiting the curative potential of adoptive T-cell therapy for cancer. Immunol Rev. 2014;257:56–71. doi: 10.1111/imr.12132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Robbins PF, Morgan RA, Feldman SA, Yang JC, Sherry RM, Dudley ME, et al. Tumor regression in patients with metastatic synovial cell sarcoma and melanoma using genetically engineered lymphocytes reactive with NY-ESO-1. J Clin Oncol. 2011;29:917–924. doi: 10.1200/JCO.2010.32.2537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hunder NN, Wallen H, Cao J, Hendricks DW, Reilly JZ, Rodmyre R, et al. Treatment of metastatic melanoma with autologous CD4+ T cells against NY-ESO-1. N Engl J Med. 2008;358:2698–2703. doi: 10.1056/NEJMoa0800251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Brentjens RJ, Davila ML, Riviere I, Park J, Wang X, Cowell LG, et al. CD19-targeted T cells rapidly induce molecular remissions in adults with chemotherapy-refractory acute lymphoblastic leukemia. Sci Transl Med. 2013;5:177ra38. doi: 10.1126/scitranslmed.3005930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Grupp SA, Kalos M, Barrett D, Aplenc R, Porter DL, Rheingold SR, et al. Chimeric antigen receptor-modified T cells for acute lymphoid leukemia. N Engl J Med. 2013;368:1509–1518. doi: 10.1056/NEJMoa1215134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Davila ML, Riviere I, Wang X, Bartido S, Park J, Curran K, et al. Efficacy and toxicity management of 19-28z CAR T cell therapy in B cell acute lymphoblastic leukemia. Sci Transl Med. 2014;6:224ra25. doi: 10.1126/scitranslmed.3008226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kochenderfer JN, Dudley ME, Kassim SH, Somerville RP, Carpenter RO, Stetler-Stevenson M, et al. Chemotherapy-refractory diffuse large B-cell lymphoma and indolent B-cell malignancies can be effectively treated with autologous T cells expressing an anti-CD19 chimeric antigen receptor. J Clin Oncol. 2014 doi: 10.1200/JCO.2014.56.2025. (e-pub ahead of print) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Aranda F, Vacchelli E, Obrist F, Eggermont A, Galon J, Herve Fridman W, et al. Trial watch:adoptive cell transfer for anticancer immunotherapy. Oncoimmunology. 2014;3:e28344. doi: 10.4161/onci.28344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Johnson LA, Heemskerk B, Powell DJ, Jr, Cohen CJ, Morgan RA, Dudley ME, et al. Gene transfer of tumor-reactive TCR confers both high avidity and tumor reactivity to nonreactive peripheral blood mononuclear cells and tumor-infiltrating lymphocytes. J Immunol. 2006;177:6548–6559. doi: 10.4049/jimmunol.177.9.6548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Parkhurst MR, Joo J, Riley JP, Yu Z, Li Y, Robbins PF, et al. Characterization of genetically modified T-cell receptors that recognize the CEA:691-699 peptide in the context of HLA-A2.1 on human colorectal cancer cells. Clin Cancer Res. 2009;15:169–180. doi: 10.1158/1078-0432.CCR-08-1638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Cohen CJ, Zheng Z, Bray R, Zhao Y, Sherman LA, Rosenberg SA, et al. Recognition of fresh human tumor by human peripheral blood lymphocytes transduced with a bicistronic retroviral vector encoding a murine anti-p53 TCR. J Immunol. 2005;175:5799–5808. doi: 10.4049/jimmunol.175.9.5799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Li Y, Moysey R, Molloy PE, Vuidepot AL, Mahon T, Baston E, et al. Directed evolution of human T-cell receptors with picomolar affinities by phage display. Nat Biotechnol. 2005;23:349–354. doi: 10.1038/nbt1070. [DOI] [PubMed] [Google Scholar]
- 30.Varela-Rohena A, Molloy PE, Dunn SM, Li Y, Suhoski MM, Carroll RG, et al. Control of HIV-1 immune escape by CD8 T cells expressing enhanced T-cell receptor. Nat Med. 2008;14:1390–1395. doi: 10.1038/nm.1779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Baum C, Schambach A, Bohne J, Galla M. Retrovirus vectors: toward the plentivirus? Mol Ther. 2006;13:1050–1063. doi: 10.1016/j.ymthe.2006.03.007. [DOI] [PubMed] [Google Scholar]
- 32.Robbins PF, Li YF, El-Gamil M, Zhao Y, Wargo JA, Zheng Z, et al. Single and dual amino acid substitutions in TCR CDRs can enhance antigen-specific T cell functions. J Immunol. 2008;180:6116–6131. doi: 10.4049/jimmunol.180.9.6116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Cohen CJ, Zhao Y, Zheng Z, Rosenberg SA, Morgan RA. Enhanced antitumor activity of murine-human hybrid T-cell receptor (TCR) in human lymphocytes is associated with improved pairing and TCR/CD3 stability. Cancer Res. 2006;66:8878–8886. doi: 10.1158/0008-5472.CAN-06-1450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Cohen CJ, Li YF, El-Gamil M, Robbins PF, Rosenberg SA, Morgan RA. Enhanced antitumor activity of T cells engineered to express T-cell receptors with a second disulfide bond. Cancer Res. 2007;67:3898–3903. doi: 10.1158/0008-5472.CAN-06-3986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Voss RH, Willemsen RA, Kuball J, Grabowski M, Engel R, Intan RS, et al. Molecular design of the Calphabeta interface favors specific pairing of introduced TCRalphabeta in human T cells. J Immunol. 2008;180:391–401. doi: 10.4049/jimmunol.180.1.391. [DOI] [PubMed] [Google Scholar]
- 36.Suerth JD, Schambach A, Baum C. Genetic modification of lymphocytes by retrovirus-based vectors. Curr Opin Immunol. 2012;24:598–608. doi: 10.1016/j.coi.2012.08.007. [DOI] [PubMed] [Google Scholar]
- 37.Sadelain M, Brentjens R, Riviere I. The basic principles of chimeric antigen receptor design. Cancer Discov. 2013;3:388–398. doi: 10.1158/2159-8290.CD-12-0548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Morgan RA, Dudley ME, Wunderlich JR, Hughes MS, Yang JC, Sherry RM, et al. Cancer regression in patients after transfer of genetically engineered lymphocytes. Science. 2006;314:126–129. doi: 10.1126/science.1129003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Hollyman D, Stefanski J, Przybylowski M, Bartido S, Borquez-Ojeda O, Taylor C, et al. Manufacturing validation of biologically functional T cells targeted to CD19 antigen for autologous adoptive cell therapy. J Immunother. 2009;32:169–180. doi: 10.1097/CJI.0b013e318194a6e8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Brentjens RJ, Riviere I, Park JH, Davila ML, Wang X, Stefanski J, et al. Safety and persistence of adoptively transferred autologous CD19-targeted T cells in patients with relapsed or chemotherapy refractory B-cell leukemias. Blood. 2011;118:4817–4828. doi: 10.1182/blood-2011-04-348540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Riddell SR, Sommermeyer D, Berger C, Liu LS, Balakrishnan A, Salter A, et al. Adoptive therapy with chimeric antigen receptor-modified T cells of defined subset composition. Cancer J. 2014;20:141–144. doi: 10.1097/PPO.0000000000000036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Wang X, Naranjo A, Brown CE, Bautista C, Wong CW, Chang WC, et al. Phenotypic and functional attributes of lentivirus-modified CD19-specific human CD8+ central memory T cells manufactured at clinical scale. J Immunother. 2012;35:689–701. doi: 10.1097/CJI.0b013e318270dec7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Koehne G, Smith KM, Ferguson TL, Williams RY, Heller G, Pamer EG, et al. Quantitation, selection, and functional characterization of Epstein-Barr virus-specific and alloreactive T cells detected by intracellular interferon-gamma production and growth of cytotoxic precursors. Blood. 2002;99:1730–1740. doi: 10.1182/blood.v99.5.1730. [DOI] [PubMed] [Google Scholar]
- 44.Pule MA, Savoldo B, Myers GD, Rossig C, Russell HV, Dotti G, et al. Virus-specific T cells engineered to coexpress tumor-specific receptors: persistence and antitumor activity in individuals with neuroblastoma. Nat Med. 2008;14:1264–1270. doi: 10.1038/nm.1882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Doubrovina E, Oflaz-Sozmen B, Prockop SE, Kernan NA, Abramson S, Teruya-Feldstein J, et al. Adoptive immunotherapy with unselected or EBV-specific T cells for biopsy-proven EBV+ lymphomas after allogeneic hematopoietic cell transplantation. Blood. 2012;119:2644–2656. doi: 10.1182/blood-2011-08-371971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Heslop HE, Slobod KS, Pule MA, Hale GA, Rousseau A, Smith CA, et al. Long-term outcome of EBV-specific T-cell infusions to prevent or treat EBV-related lymphoproliferative disease in transplant recipients. Blood. 2010;115:925–935. doi: 10.1182/blood-2009-08-239186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Rooney CM, Smith CA, Ng CY, Loftin S, Li C, Krance RA, et al. Use of gene-modified virus-specific T lymphocytes to control Epstein-Barr-virus-related lymphoproliferation. Lancet. 1995;345:9–13. doi: 10.1016/s0140-6736(95)91150-2. [DOI] [PubMed] [Google Scholar]
- 48.Smith CA, Ng CY, Heslop HE, Holladay MS, Richardson S, Turner EV, et al. Production of genetically modified Epstein-Barr virus-specific cytotoxic T cells for adoptive transfer to patients at high risk of EBV-associated lymphoproliferative disease. J Hematother. 1995;4:73–79. doi: 10.1089/scd.1.1995.4.73. [DOI] [PubMed] [Google Scholar]
- 49.Papadopoulou A, Gerdemann U, Katari UL, Tzannou I, Liu H, Martinez C, et al. Activity of broad-spectrum T cells as treatment for AdV, EBV, CMV, BKV, and HHV6 infections after HSCT. Sci Transl Med. 2014;6:242ra83. doi: 10.1126/scitranslmed.3008825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Rossig C, Bollard CM, Nuchtern JG, Rooney CM, Brenner MK. Epstein-Barr virus-specific human T lymphocytes expressing antitumor chimeric T-cell receptors: potential for improved immunotherapy. Blood. 2002;99:2009–2016. doi: 10.1182/blood.v99.6.2009. [DOI] [PubMed] [Google Scholar]
- 51.Heemskerk MH, Hoogeboom M, Hagedoorn R, Kester MG, Willemze R, Falkenburg JH. Reprogramming of virus-specific T cells into leukemia-reactive T cells using T cell receptor gene transfer. J Exp Med. 2004;199:885–894. doi: 10.1084/jem.20031110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Savoldo B, Rooney CM, Di Stasi A, Abken H, Hombach A, Foster AE, et al. Epstein Barr virus specific cytotoxic T lymphocytes expressing the anti-CD30zeta artificial chimeric T-cell receptor for immunotherapy of Hodgkin disease. Blood. 2007;110:2620–2630. doi: 10.1182/blood-2006-11-059139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.van Loenen MM, de Boer R, van Liempt E, Meij P, Jedema I, Falkenburg JH, et al. A Good Manufacturing Practice procedure to engineer donor virus-specific T cells into potent anti-leukemic effector cells. Haematologica. 2014;99:759–768. doi: 10.3324/haematol.2013.093690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Louis CU, Savoldo B, Dotti G, Pule M, Yvon E, Myers GD, et al. Antitumor activity and long-term fate of chimeric antigen receptor-positive T cells in patients with neuroblastoma. Blood. 2011;118:6050–6056. doi: 10.1182/blood-2011-05-354449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Wang X, Berger C, Wong CW, Forman SJ, Riddell SR, Jensen MC. Engraftment of human central memory-derived effector CD8+ T cells in immunodeficient mice. Blood. 2011;117:1888–1898. doi: 10.1182/blood-2010-10-310599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Wang X, Chang WC, Wong CW, Colcher D, Sherman M, Ostberg JR, et al. A transgene-encoded cell surface polypeptide for selection, in vivo tracking, and ablation of engineered cells. Blood. 2011;118:1255–1263. doi: 10.1182/blood-2011-02-337360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Neudorfer J, Schmidt B, Huster KM, Anderl F, Schiemann M, Holzapfel G, et al. Reversible HLA multimers (Streptamers) for the isolation of human cytotoxic T lymphocytes functionally active against tumor- and virus-derived antigens. J Immunol Methods. 2007;320:119–131. doi: 10.1016/j.jim.2007.01.001. [DOI] [PubMed] [Google Scholar]
- 58.Terakura S, Yamamoto TN, Gardner RA, Turtle CJ, Jensen MC, Riddell SR. Generation of CD19-chimeric antigen receptor modified CD8+ T cells derived from virus-specific central memory T cells. Blood. 2012;119:72–82. doi: 10.1182/blood-2011-07-366419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Rosenberg SA, Aebersold P, Cornetta K, Kasid A, Morgan RA, Moen R, et al. Gene transfer into humans–immunotherapy of patients with advanced melanoma, using tumor-infiltrating lymphocytes modified by retroviral gene transduction. N Engl J Med. 1990;323:570–578. doi: 10.1056/NEJM199008303230904. [DOI] [PubMed] [Google Scholar]
- 60.Deichmann A, Schmidt M. Biosafety considerations using gamma-retroviral vectors in gene therapy. Curr Gene Therapy. 2013;13:469–477. doi: 10.2174/15665232113136660004. [DOI] [PubMed] [Google Scholar]
- 61.Miller AD, Buttimore C. Redesign of retrovirus packaging cell lines to avoid recombination leading to helper virus production. Mol Cell Biol. 1986;6:2895–2902. doi: 10.1128/mcb.6.8.2895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Miller AD, Garcia JV, von Suhr N, Lynch CM, Wilson C, Eiden MV. Construction and properties of retrovirus packaging cells based on gibbon ape leukemia virus. J Virol. 1991;65:2220–2224. doi: 10.1128/jvi.65.5.2220-2224.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Ghani K, Cottin S, Kamen A, Caruso M. Generation of a high-titer packaging cell line for the production of retroviral vectors in suspension and serum-free media. Gene Ther. 2007;14:1705–1711. doi: 10.1038/sj.gt.3303039. [DOI] [PubMed] [Google Scholar]
- 64.Przybylowski M, Hakakha A, Stefanski J, Hodges J, Sadelain M, Riviere I. Production scale-up and validation of packaging cell clearance of clinical-grade retroviral vector stocks produced in cell factories. Gene Ther. 2006;13:95–100. doi: 10.1038/sj.gt.3302648. [DOI] [PubMed] [Google Scholar]
- 65.Gallardo HF, Tan C, Ory D, Sadelain M. Recombinant retroviruses pseudotyped with the vesicular stomatitis virus G glycoprotein mediate both stable gene transfer and pseudotransduction in human peripheral blood lymphocytes. Blood. 1997;90:952–957. [PubMed] [Google Scholar]
- 66.Savoldo B, Ramos CA, Liu E, Mims MP, Keating MJ, Carrum G, et al. CD28 costimulation improves expansion and persistence of chimeric antigen receptor-modified T cells in lymphoma patients. J Clin Invest. 2011;121:1822–1826. doi: 10.1172/JCI46110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Reeves L, Cornetta K. Clinical retroviral vector production: step filtration using clinically approved filters improves titers. Gene Ther. 2000;7:1993–1998. doi: 10.1038/sj.gt.3301328. [DOI] [PubMed] [Google Scholar]
- 68.Feldman SA, Goff SL, Xu H, Black MA, Kochenderfer JN, Johnson LA, et al. Rapid production of clinical-grade gammaretroviral vectors in expanded surface roller bottles using a ‘modified’ step-filtration process for clearance of packaging cells. Hum Gene Ther. 2011;22:107–115. doi: 10.1089/hum.2010.064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Merten OW, Cruz PE, Rochette C, Geny-Fiamma C, Bouquet C, Goncalves D, et al. Comparison of different bioreactor systems for the production of high titer retroviral vectors. Biotechnol Prog. 2001;17:326–335. doi: 10.1021/bp000162z. [DOI] [PubMed] [Google Scholar]
- 70.van der Loo JC, Swaney WP, Grassman E, Terwilliger A, Higashimoto T, Schambach A, et al. Scale-up and manufacturing of clinical-grade self-inactivating gamma-retroviral vectors by transient transfection. Gene Ther. 2012;19:246–254. doi: 10.1038/gt.2011.102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Rodrigues AF, Carmo M, Alves PM, Coroadinha AS. Retroviral vector production under serum deprivation: The role of lipids. Biotechnol Bioeng. 2009;104:1171–1181. doi: 10.1002/bit.22499. [DOI] [PubMed] [Google Scholar]
- 72.Ghani K, Wang X, de Campos-Lima PO, Olszewska M, Kamen A, Riviere I, et al. Efficient human hematopoietic cell transduction using RD114- and GALV-pseudotyped retroviral vectors produced in suspension and serum-free media. Hum Gene Ther. 2009;20:966–974. doi: 10.1089/hum.2009.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Bonini C, Grez M, Traversari C, Ciceri F, Marktel S, Ferrari G, et al. Safety of retroviral gene marking with a truncated NGF receptor. Nat Med. 2003;9:367–369. doi: 10.1038/nm0403-367. [DOI] [PubMed] [Google Scholar]
- 74.Brenner MK, Heslop HE. Is retroviral gene marking too dangerous to use? Cytotherapy. 2003;5:190–193. doi: 10.1080/14653240310001307. [DOI] [PubMed] [Google Scholar]
- 75.Macpherson JL, Boyd MP, Arndt AJ, Todd AV, Fanning GC, Ely JA, et al. Long-term survival and concomitant gene expression of ribozyme-transduced CD4+ T-lymphocytes in HIV-infected patients. J Gene Med. 2005;7:552–564. doi: 10.1002/jgm.705. [DOI] [PubMed] [Google Scholar]
- 76.Muul LM, Tuschong LM, Soenen SL, Jagadeesh GJ, Ramsey WJ, Long Z, et al. Persistence and expression of the adenosine deaminase gene for 12 years and immune reaction to gene transfer components: long-term results of the first clinical gene therapy trial. Blood. 2003;101:2563–2569. doi: 10.1182/blood-2002-09-2800. [DOI] [PubMed] [Google Scholar]
- 77.Cartier N, Hacein-Bey-Abina S, Bartholomae CC, Veres G, Schmidt M, Kutschera I, et al. Hematopoietic stem cell gene therapy with a lentiviral vector in X-linked adrenoleukodystrophy. Science. 2009;326:818–823. doi: 10.1126/science.1171242. [DOI] [PubMed] [Google Scholar]
- 78.Cavazzana-Calvo M, Payen E, Negre O, Wang G, Hehir K, Fusil F, et al. Transfusion independence and HMGA2 activation after gene therapy of human beta-thalassaemia. Nature. 2010;467:318–322. doi: 10.1038/nature09328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Boulad F, Wang X, Qu J, Taylor C, Ferro L, Karponi G, et al. Safe mobilization of CD34+ cells in adults with beta-thalassemia and validation of effective globin gene transfer for clinical investigation. Blood. 2014;123:1483–1486. doi: 10.1182/blood-2013-06-507178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Aiuti A, Biasco L, Scaramuzza S, Ferrua F, Cicalese MP, Baricordi C, et al. Lentiviral hematopoietic stem cell gene therapy in patients with Wiskott-Aldrich syndrome. Science. 2013;341:1233151. doi: 10.1126/science.1233151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Biffi A, Montini E, Lorioli L, Cesani M, Fumagalli F, Plati T, et al. Lentiviral hematopoietic stem cell gene therapy benefits metachromatic leukodystrophy. Science. 2013;341:1233158. doi: 10.1126/science.1233158. [DOI] [PubMed] [Google Scholar]
- 82.Kalos M, Levine BL, Porter DL, Katz S, Grupp SA, Bagg A, et al. T cells with chimeric antigen receptors have potent antitumor effects and can establish memory in patients with advanced leukemia. Sci Transl Med. 2011;3:95ra73. doi: 10.1126/scitranslmed.3002842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Naldini L, Blomer U, Gallay P, Ory D, Mulligan R, Gage FH, et al. In vivo gene delivery and stable transduction of nondividing cells by a lentiviral vector. Science. 1996;272:263–267. doi: 10.1126/science.272.5259.263. [DOI] [PubMed] [Google Scholar]
- 84.Reiser J, Harmison G, Kluepfel-Stahl S, Brady RO, Karlsson S, Schubert M. Transduction of nondividing cells using pseudotyped defective high-titer HIV type 1 particles. Proc Natl Acad Sci USA. 1996;93:15266–15271. doi: 10.1073/pnas.93.26.15266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Cavalieri S, Cazzaniga S, Geuna M, Magnani Z, Bordignon C, Naldini L, et al. Human T lymphocytes transduced by lentiviral vectors in the absence of TCR activation maintain an intact immune competence. Blood. 2003;102:497–505. doi: 10.1182/blood-2003-01-0297. [DOI] [PubMed] [Google Scholar]
- 86.Wang GP, Levine BL, Binder GK, Berry CC, Malani N, McGarrity G, et al. Analysis of lentiviral vector integration in HIV+ study subjects receiving autologous infusions of gene modified CD4+ T cells. Mol Ther. 2009;17:844–850. doi: 10.1038/mt.2009.16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Ni Y, Sun S, Oparaocha I, Humeau L, Davis B, Cohen R, et al. Generation of a packaging cell line for prolonged large-scale production of high-titer HIV-1-based lentiviral vector. J Gene Med. 2005;7:818–834. doi: 10.1002/jgm.726. [DOI] [PubMed] [Google Scholar]
- 88.Broussau S, Jabbour N, Lachapelle G, Durocher Y, Tom R, Transfiguracion J, et al. Inducible packaging cells for large-scale production of lentiviral vectors in serum-free suspension culture. Mol Ther. 2008;16:500–507. doi: 10.1038/sj.mt.6300383. [DOI] [PubMed] [Google Scholar]
- 89.Pear WS, Nolan GP, Scott ML, Baltimore D. Production of high-titer helper-free retroviruses by transient transfection. Proc Natl Acad Sci USA. 1993;90:8392–8396. doi: 10.1073/pnas.90.18.8392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Pham PL, Kamen A, Durocher Y. Large-scale transfection of mammalian cells for the fast production of recombinant protein. Mol Biotechnol. 2006;34:225–237. doi: 10.1385/MB:34:2:225. [DOI] [PubMed] [Google Scholar]
- 91.Toledo JR, Prieto Y, Oramas N, Sanchez O. Polyethylenimine-based transfection method as a simple and effective way to produce recombinant lentiviral vectors. Appl Biochem Biotechnol. 2009;157:538–544. doi: 10.1007/s12010-008-8381-2. [DOI] [PubMed] [Google Scholar]
- 92.Ansorge S, Lanthier S, Transfiguracion J, Durocher Y, Henry O, Kamen A. Development of a scalable process for high-yield lentiviral vector production by transient transfection of HEK293 suspension cultures. J Gene Med. 2009;11:868–876. doi: 10.1002/jgm.1370. [DOI] [PubMed] [Google Scholar]
- 93.Witting SR, Li LH, Jasti A, Allen C, Cornetta K, Brady J, et al. Efficient large volume lentiviral vector production using flow electroporation. Hum Gene Therapy. 2012;23:243–249. doi: 10.1089/hum.2011.088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Kutner RH, Puthli S, Marino MP, Reiser J. Simplified production and concentration of HIV-1-based lentiviral vectors using HYPERFlask vessels and anion exchange membrane chromatography. BMC Biotechnol. 2009;9:10. doi: 10.1186/1472-6750-9-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Ausubel LJ, Hall C, Sharma A, Shakeley R, Lopez P, Quezada V, et al. Production of CGMP-Grade Lentiviral Vectors. Bioprocess Int. 2012;10:32–43. [PMC free article] [PubMed] [Google Scholar]
- 96.Segura MM, Mangion M, Gaillet B, Garnier A. New developments in lentiviral vector design, production and purification. Expert Opin Biol Ther. 2013;13:987–1011. doi: 10.1517/14712598.2013.779249. [DOI] [PubMed] [Google Scholar]
- 97.Segura MM, Garnier A, Durocher Y, Ansorge S, Kamen A. New protocol for lentiviral vector mass production. Methods Mol Biol. 2010;614:39–52. doi: 10.1007/978-1-60761-533-0_2. [DOI] [PubMed] [Google Scholar]
- 98.Schweizer M, Merten OW. Large-scale production means for the manufacturing of lentiviral vectors. Curr Gene Therapy. 2010;10:474–486. doi: 10.2174/156652310793797748. [DOI] [PubMed] [Google Scholar]
- 99.Kebriaei P, Huls H, Jena B, Munsell M, Jackson R, Lee DA, et al. Infusing CD19-directed T cells to augment disease control in patients undergoing autologous hematopoietic stem-cell transplantation for advanced B-lymphoid malignancies. Hum Gene Ther. 2012;23:444–450. doi: 10.1089/hum.2011.167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Huls MH, Figliola MJ, Dawson MJ, Olivares S, Kebriaei P, Shpall EJ, et al. Clinical application of Sleeping Beauty and artificial antigen presenting cells to genetically modify T cells from peripheral and umbilical cord blood. J Vis Exp. 2013:e50070. doi: 10.3791/50070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Singh H, Figliola MJ, Dawson MJ, Olivares S, Zhang L, Yang G, et al. Manufacture of clinical-grade CD19-specific T cells stably expressing chimeric antigen receptor using Sleeping Beauty system and artificial antigen presenting cells. PloS One. 2013;8:e64138. doi: 10.1371/journal.pone.0064138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.FDA Process Validation: General Principle and Practices. 2011 [Google Scholar]
- 103.FDA. Guidance for Industry: CGMP for Phase 1 Investigational Drugs. 2008 [Google Scholar]
- 104.Khuu HM, Patel N, Carter CS, Murray PR, Read EJ. Sterility testing of cell therapy products: parallel comparison of automated methods with a CFR-compliant method. Transfusion. 2006;46:2071–2082. doi: 10.1111/j.1537-2995.2006.01041.x. [DOI] [PubMed] [Google Scholar]
- 105.Hocquet D, Sauget M, Roussel S, Malugani C, Pouthier F, Morel P, et al. Validation of an automated blood culture system for sterility testing of cell therapy products. Cytotherapy. 2014;16:692–698. doi: 10.1016/j.jcyt.2013.09.005. [DOI] [PubMed] [Google Scholar]
- 106.Gee AP, Sumstad D, Stanson J, Watson P, Proctor J, Kadidlo D, et al. A multicenter comparison study between the Endosafe PTS rapid-release testing system and traditional methods for detecting endotoxin in cell-therapy products. Cytotherapy. 2008;10:427–435. doi: 10.1080/14653240802075476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Bouchie A, Allison M, Webb S, DeFrancesco L. Nature Biotechnology’s academic spinouts of 2013. Nat Biotech. 2014;32:229–238. doi: 10.1038/nbt.2846. [DOI] [PubMed] [Google Scholar]