

Background: Resveratrol and SRT1720 elicit beneficial metabolic effects, supposedly through activation of PGC-1α in skeletal muscle.
Results: Resveratrol/SRT1720 differentially affect transcriptional regulation in metabolic organs and modulates systemic parameters independently of muscle PGC-1α.
Conclusion: Skeletal muscle PGC-1α is largely dispensable for the systemic metabolic effects of resveratrol/SRT1720.
Significance: Identifying molecular targets of resveratrol and SRT1720 is important to understand their therapeutic effect.
Keywords: metabolic disease, metabolism, obesity, peroxisome proliferator-activated receptor gamma coactivator 1-alpha (PGC-1a) (PPARGC1A), resveratrol, skeletal muscle
Abstract
Resveratrol (RSV) and SRT1720 (SRT) elicit beneficial metabolic effects and are postulated to ameliorate obesity and related metabolic complications. The co-activator, peroxisome proliferator-activated receptor γ co-activator 1α (PGC-1α), has emerged as a major downstream effector responsible for metabolic remodeling of muscle and other metabolic tissues in response to RSV or SRT treatment. However, the requirement of PGC-1α in skeletal muscle for the systemic metabolic effects of these compounds has so far not been demonstrated. Using muscle-specific PGC-1α knock-out mice, we show that PGC-1α is necessary for transcriptional induction of mitochondrial genes in muscle with both RSV and SRT treatment. Surprisingly, the beneficial effects of SRT on glucose homeostasis and of both compounds on energy expenditure occur even in the absence of muscle PGC-1α. Moreover, RSV and SRT treatment elicit differential transcriptional effects on genes involved in lipid metabolism and mitochondrial biogenesis in liver and adipose tissue. These findings indicate that RSV and SRT do not induce analogous metabolic effects in vivo. Our results provide important insights into the mechanism, effects, and organ specificity of the caloric restriction mimetics RSV and SRT. These findings are important for the design of future therapeutic interventions aimed at ameliorating obesity and obesity-related metabolic dysfunction.
Introduction
In moderation, caloric restriction (CR)2 elicits various beneficial metabolic effects in humans (1). Thus, inducing a similar metabolic state through pharmacological interventions has been proposed as an approach to ameliorate obesity, type 2 diabetes, and other metabolic complications in patients. Resveratrol (RSV) is a naturally occurring compound that mimics both metabolic and transcriptional effects of CR in mice (2, 3). Moreover, RSV improves metabolic dysfunction and prolongs life span in high fat diet (HFD)-fed rodents (4, 5). The exact molecular mechanism of how RSV promotes these effects is not fully elucidated. AMP-activated protein kinase (AMPK) (6), sirtuin 1 (SIRT1) (7), and cAMP-degrading phosphodiesterases (8) are all proposed to be primary targets for this polyphenolic compound. It is, however, known that besides activation of these targets, RSV has additional properties and can act as an antioxidant, a phytoestrogen, and an anti-inflammatory agent (9). Thus, in the search for new, more specific pharmacological activators of SIRT1, several synthetic compounds have been identified, including SRT1720 (SRT) (10). SRT shows a higher efficacy and selectivity for activating SIRT1 compared with RSV (10) and mimics many of the transcriptional changes observed during both CR and RSV treatment in mice (3). SRT treatment can furthermore ameliorate metabolic defects in both HFD-fed (11) and aged mice (12), but as for RSV, it is controversial whether SRT directly activates SIRT1 to mediate these metabolic changes (13). However, for both compounds, peroxisome proliferator-activated receptor γ co-activator 1α (PGC-1α) has been proposed as the common downstream effector to modulate transcription of metabolic gene programs (5, 11, 14). This is regardless of the putative direct target of these compounds, because PGC-1α is activated by AMPK-mediated phosphorylation, SIRT1-induced deacetylation, and increased cAMP levels (15–17). In metabolic organs, PGC-1α is an important transcriptional co-activator that controls several integrated metabolic programs, such as mitochondrial biogenesis, oxidative phosphorylation (OXPHOS), and β-oxidation (18). These processes are furthermore postulated to be induced by CR (14) and upon administration of RSV and SRT in mice (5, 11). Accordingly, RSV and SRT treatment have been reported to activate PGC-1α and lead to metabolic remodeling in several organs, especially in skeletal muscle (5, 11). However, the requirement for muscle PGC-1α in mediating the effects of RSV and SRT in vivo has so far not been conclusively validated. Using mice with a skeletal muscle-specific deletion of PGC-1α, we now demonstrate that although PGC-1α is required for transcriptional induction of mitochondrial genes in muscle, this co-activator in muscle is dispensable for systemic metabolic effects of both RSV and SRT treatment, such as increased energy expenditure. Another important question is whether structurally distinct compounds, such as RSV and SRT, are able to induce similar effects in major metabolic organs. To answer this, we compared the effects elicited by RSV and SRT on metabolic processes in skeletal muscle, liver, and adipose tissue. Consequently, we could reveal several important analogous but also differential effects between RSV and SRT on transcriptional regulation of lipid metabolism and mitochondrial biogenesis.
Experimental Procedures
Animals and Diets
PGC-1α muscle-specific knock-out (MKO) mice were generated by crossing PGC-1αloxP/loxP mice with human α-skeletal actin-Cre transgenic mice, as described previously (19). Mice were housed in a conventional facility with a 12-h light/12-h dark cycle with free access to food and water. All experiments were performed in accordance with federal guidelines and were approved by the Kantonales Veterinäramt of Kanton Basel-Stadt. The HFD used for this study contains 60 kcal% fat derived mainly from lard (D12492, Research Diets Inc.), and either RSV (4 g/kg diet; Cayman Chemicals) or SRT1720 (1 g/kg diet; Sanofi Aventis) was added to the diet during production. Initially, mice were fed a HFD for 2 weeks, and then half of the groups were switched to a HFD containing either RSV for a period of 4 weeks (RSV-S), RSV for 13 weeks (RSV-L), or SRT1720 for 4 weeks (SRT). The respective control groups received a HFD without any additions for the duration of the study.
Metabolic Measurements
Body composition was measured using an EchoMRI-100TM analyzer (EchoMRI Medical Systems). An intraperitoneal glucose tolerance test was performed in overnight (16 h)-fasted mice. Mice were injected with a bolus of 2 g of glucose/kg body weight, and blood glucose was measured in tail vein blood at 0, 15, 30, 45, 60, 90, 120, and 180 min after injection using a handheld glucose meter (Accu-Chek, Roche Applied Science). To assess whole body metabolism through indirect calorimetry, mice were individually housed in metabolic chambers (CLAMS, Columbus Instruments) for 48 h after an initial acclimatization period. During the measurement period, oxygen consumption, food intake, and spontaneous activity were measured in 30-min intervals.
Blood Analysis
Blood glucose was measured in a tail vein blood sample from ad libitum-fed and overnight (16 h)-fasted mice using a hand-held glucose meter (Accu-Chek, Roche Applied Science). Whole tail-vein blood was collected in Microvette tubes (Sarstedt), and plasma was isolated. Levels of high density lipoprotein cholesterol (HDL-C) and low density lipoprotein cholesterol (LDL-C) in plasma were determined using an automated biochemical analyzer (Cobas c 111 analyzer; Roche Applied Science). Non-esterified free fatty acid levels in plasma were determined using a colorimetric commercial test (HR Series NEFA-HR(2); Wako Diagnostics) according to manufacturer's instructions. Insulin levels in plasma were determined using a colorimetric commercial test (Ultra-sensitive mouse insulin ELISA kit, Crystal Chem Inc.) according to the manufacturer's instructions.
Histology
Nicotinamide adenine dinucleotide (NADH) staining was performed on 10-μm cryo-sections from tibialis anterior by exposing the sections to NADH (Sigma) in the presence of nitro blue tetrazolium (Sigma). Fiber staining intensity was quantified for 100 fibers/section using ImageJ software on pictures taken from the same region of the muscle for all groups. Liver specimens were snap-frozen in 2-methyl butane precooled in liquid nitrogen. Tissues were cut into 5-μm-thick cryo-sections and stained with Oil Red O (Sigma) for lipid content. Oil Red O staining was evaluated by an expert pathologist. For studies of mitochondrial number, pieces of tibialis anterior muscle were fixed in 3% paraformaldehyde, 0.5% glutaraldehyde, further fixed in 1% osmium tetroxide, and embedded in Epon. 60–70-nm sections were cut, and pictures were taken with a Morgagni 268(D) transmission electron microscope (FEI). Quantification of the number of mitochondria in these pictures was performed using ImageJ software.
Cell Culture
C2C12 myoblasts were grown to confluence in Dulbecco's modified Eagle's medium (DMEM, Sigma) containing 10% fetal calf serum (FCS, Gibco) and 1% penicillin/streptomycin (Gibco). Confluent myoblasts were differentiated into myotubes through the addition of DMEM containing 2% horse serum (Gibco) and 1% penicillin/streptomycin. All experiments were conducted with myotubes that were differentiated for 4 days. PGC-1α was overexpressed by adenoviral transduction on day 3 of differentiation. Primary mouse myoblasts were isolated from 2–3-week-old male mice and cultured in DMEM containing 10% horse serum, 20% FCS, 1% chicken embryo extract, 2 mm Glutamax (Gibco), 5 ng/ml basic fibroblast growth factor, and 1% penicillin/streptomycin in plastic dishes coated with Matrigel (BD Biosciences). AML-12 mouse hepatocytes were cultured in DMEM/F-12 (1:1) (Gibco) containing 2 mm Glutamax, 10% FCS, 5 μg/ml insulin/selenium/transferrin (Gibco) and 40 ng/ml dexamethasone (Sigma). Cells were treated with vehicle (0.1% DMSO), RSV (50 μm; Cayman Chemicals) or SRT (10 μm; Sanofi Aventis) in serum-free DMEM or DMEM/F-12 containing 2 mm Glutamax for C2C12 cells and AML-12 cells, respectively. Cells were subsequently washed with ice-cold PBS and lysed using TRIzol reagent (Invitrogen) for RNA extraction or radioimmune precipitation assay buffer supplemented with protease inhibitors (cOmplete, Roche Applied Science) and phosphatase inhibitors (Sigma) for protein isolation.
RNA Extraction and RT-PCR
Snap-frozen gastrocnemius, liver, brown adipose tissue or white adipose tissue was homogenized, and total RNA was extracted using TRIzol reagent (Invitrogen). RNA concentration was adjusted, and 1 μg of total RNA was used for cDNA synthesis. Semiquantitative real-time PCR analysis was performed using Power SYBR Green master mix (Applied Biosystems) on a StepOnePlus real-time PCR system (Applied Biosystems). Relative expression levels for each gene of interest were calculated with the ΔΔCt method, using Eef2, Tbp, Polr2a, Hprt, or Rpl0 as normalization control. Primer sequences can be found in supplemental Table 1.
Nuclear Isolation and Immunoprecipitation
For nuclear isolation, liver was homogenized in hypotonic lysis buffer (HLB; 10 mm NaCl, 1.5 mm MgCl2, 20 mm HEPES, pH 7.4, 20% glycerol, 0.1% Triton X-100, 1 mm DTT) supplemented with protease inhibitors (cOmplete, Roche Applied Science), phosphatase inhibitors (Sigma), 10 mm nicotinamide A (Sigma), and 10 mm trichostatin A (Sigma), using a Dounce homogenizer. Intact nuclei were isolated through differential centrifugation at 880 × g for 3 min, and nuclear pellet was washed three times in HLB. Isolated nuclei were lysed using radioimmune precipitation assay buffer supplemented with protease inhibitors (cOmplete, Roche Applied Science), phosphatase inhibitors (Sigma), 10 mm nicotinamide A (Sigma), and 10 mm trichostatin A (Sigma). For immunoprecipitation, 500 μg of nuclear proteins were precleared with protein A beads (protein A-Sepharose 4B, Invitrogen). Precleared supernatant was incubated with 2 μg of pan-acetyl antibody (sc8649; Santa Cruz Biotechnology, Inc.) for 2 h, followed by overnight incubation with 50 μl of a 50% slurry of protein A beads. Immunoprecipitated proteins were denatured in 2× Laemmli sample buffer (Sigma) and prepared for immunoblotting.
Immunoblotting
Tissues were homogenized in radioimmune precipitation assay buffer supplemented with protease inhibitors (cOmplete, Roche Applied Science), phosphatase inhibitors (Sigma), 10 mm nicotinamide A (Sigma), and 10 mm trichostatin A (Sigma). Equal amounts of proteins were separated on SDS-PAGE under reducing conditions and transferred to a nitrocellulose membrane (Whatman). Proteins of interest were detected using the following antibodies: ACC (3662; Cell Signaling), phospho-ACC (3661; Cell Signaling), Mitoprofile (MS604; MitoSciences), AMPKα (2532; Cell Signaling), phospho-AMPKα Thr-172 (2531; Cell Signaling), eEF2 (2332; Cell Signaling), cAMP-response element-binding protein (CREB) (9197; Cell Signaling), phospho-CREB Ser-133 (9196; Cell Signaling), insulin receptor β (IRβ) (3025; Cell Signaling), phospho-IRβ Tyr-1146 (3021; Cell Signaling), Akt (9271; Cell Signaling), phospho-Akt Ser-473 (9271; Cell Signaling), phospho-Akt Thr-308 (4056; Cell Signaling), glycogen synthase kinase 3β (GSK3β) (9315; Cell Signaling), phospho-GSK3β Ser-9 (5558P; Cell Signaling), PGC-1 (AB3242; Millipore), polyclonal swine anti-rabbit immunoglobulin/HRP (P0399, Dako), polyclonal rabbit anti-mouse immunoglobulin/HRP (P0260, Dako). When applicable, densitometric analysis of immunoblots was performed on 6–7 individual samples using ImageJ software, and a representative selection is presented in each figure.
Hepatic Lipid Extraction
Liver tissue was homogenized in 1 ml of a 2:1 chloroform/methanol mixture and transferred to a glass tube together with 1 ml of distilled water. The sample was then vigorously mixed and centrifuged for 10 min at 2000 rpm. The upper aqueous phase was removed, and the lower organic phase was transferred to a new glass tube and dried under N2 at 50 °C. The sample was then resuspended in chloroform and added to a solid phase extraction column (UPTI-CLEAN NH2-S 100 mg/1 ml SPE columns, Interchim). Samples were eluted with chloroform, dried under N2 at 50 °C, and subsequently resuspended in chloroform containing 1% Triton X-100. Triglycerides were measured using a commercial enzymatic kit (TG PAP 150, BioMérieux) and normalized to the initial weight of the tissue used for extraction.
Statistical Analysis
All data are presented as means ± S.E. Unpaired Student's two-tailed t test was used to determine differences between groups.
Results
Increased Systemic Energy Expenditure with RSV and SRT Treatment Is Independent of Skeletal Muscle PGC-1α
To investigate the requirement of skeletal muscle PGC-1α for the metabolic effects of RSV and SRT, we used mice with a muscle-specific knock-out of PGC-1α (MKO) (19) and their floxed littermates with intact PGC-1α expression (CTRL). All mice were fed a HFD for 2 weeks and subsequently switched to a HFD containing either RSV (short term RSV treatment; RSV-S) or SRT for 4 consecutive weeks or a HFD containing RSV for 13 weeks (long term RSV treatment; RSV-L). All diets were administered ad libitum, and control groups were maintained on an unmodified HFD for a total of 6 weeks (RSV-S and SRT) or 15 weeks (RSV-L). In line with the suggested beneficial metabolic effects of RSV (2), RSV-treated mice displayed reduced body weight (Fig. 1A) and fat mass (Fig. 1B) at the end of the treatment period compared with HFD-fed mice. In contrast to the RSV-mediated effects, SRT treatment affected neither body weight (Fig. 1A) nor relative fat mass (Fig. 1B). The reduction in fat mass in RSV-treated mice could not be attributed to any changes in either food intake or spontaneous locomotor activity (data not shown). Importantly, based on their increased oxygen consumption, RSV-treated mice displayed an enhanced metabolic rate (Fig. 1C), which could explain the lean phenotype of these mice. SRT-treated mice also exhibited an enhanced metabolic rate (Fig. 1C), which, however, did not result in a reduced fat mass in this group (Fig. 1B). Surprisingly, the effects of RSV and SRT treatment on energy expenditure and fat mass were similar between PGC-1α MKO mice and control mice (Fig. 1, A–C). Hence, these data demonstrate that skeletal muscle PGC-1α is dispensable for the changes in body composition and whole body energy expenditure elicited by RSV and SRT. Skeletal muscle is the major site for insulin-induced glucose uptake and therefore an important organ in the regulation of whole body glucose homeostasis. Moreover, both RSV and SRT have been shown to improve glucose homeostasis in obese mice (3–5, 10–12). Hence, we investigated whether muscle PGC-1α is important to mediate the effects of RSV and SRT on glucose homeostasis in HFD-fed mice. Interestingly, with an intraperitoneal glucose tolerance test, we could detect that ablation of PGC-1α in skeletal muscle led to a basal improvement in glucose excursion rates (Fig. 1, D and E) and reduced fasted blood glucose levels (Fig. 1G) compared with control mice. This effect was especially prominent after 15 weeks of HFD feeding. RSV treatment led to a mild improvement of glucose excursion rates in control mice from the RSV-S group compared with HFD-fed mice (Fig. 1, D and E). However, this effect of RSV on glucose tolerance did not reach significance in control mice from the RSV-L group (Fig. 1, D and E). This trend toward an improved glucose homeostasis with RSV treatment could not be seen in PGC-1α MKO mice (Fig. 1, D and E). However, the mild effect elicited by RSV treatment could have been masked by the basal improvement of glucose tolerance in PGC-1α MKO mice compared with control mice (Fig. 1, D and E). Importantly, we could confirm that SRT treatment led to improved glucose excursion rates in HFD-fed mice (Fig. 1, D and E). This correlated with a reduction in both fed and fasted blood glucose levels in SRT-treated mice, which could not be observed with RSV administration (Fig. 1, F and G). Hence, SRT exerted a stronger effect on glucose homeostasis compared with RSV. Intriguingly, muscle-specific ablation of PGC-1α did not prevent the effects of SRT treatment on glucose homeostasis in HFD-fed mice (Fig. 1, D–G), indicating that skeletal muscle PGC-1α is dispensable for the effects of SRT on whole body glucose homeostasis.
FIGURE 1.

Systemic effects of RSV and SRT are independent of skeletal muscle PGC-1α. CTRL and PGC-1α MKO mice were fed a HFD for 2 weeks and subsequently switched to a HFD containing either RSV (4 g/kg diet) for 4 weeks (RSV-S) or 13 weeks (RSV-L) or SRT (1 g/kg diet) for 4 weeks. Control groups were maintained on an unmodified HFD for a total of 6 weeks (RSV-S and SRT) or 15 weeks (RSV-L). A, body weight at the end of the treatment period (n = 8–15/group). B, fat mass expressed as percentage of total body weight at the end of the treatment period (n = 5–9/group). C, basal energy expenditure as measured by 24-h average oxygen consumption (vO2) (n = 5–9/group). D, intraperitoneal glucose tolerance test. Mice were injected with a bolus of 2 g of glucose/kg body weight after a 16-h fasting period (n = 8–10/group). E, area under curve (in arbitrary units (a.u.)) for the intraperitoneal glucose tolerance test (n = 8–10/group). F and G, blood glucose levels measured in tail vein blood from ad libitum fed mice (n = 8–10/group) (F) or mice fasted for 16 h (n = 8–10/group) (G). Bars, means ± S.E. (error bars). Significant differences (p < 0.05) between CTRL and PGC-1α MKO mice are indicated (#) as well as significant differences between untreated and treated groups (*).
PGC-1α Is Required for Induction of Mitochondrial Gene Transcription but Not for Enhanced Oxidative Capacity in Skeletal Muscle of RSV- or SRT-treated Mice
Because the systemic effects of RSV and SRT were unaffected by the ablation of PGC-1α in muscle, we were interested in how these compounds would affect skeletal muscle metabolism in our mice. Because RSV and SRT have been postulated to influence mitochondrial function and biogenesis via activation of PGC-1α (14), we specifically focused on these parameters in muscle. As previously shown, ablation of PGC-1α resulted in a reduced oxidative capacity in muscle (20). This was evident in our mice by the reduced transcript levels of several mitochondrial genes, namely cytochrome c (Cytc), cytochrome c oxidase subunit 5b (Cox5b), citrate synthase (Cs), and NADH dehydrogenase (ubiquinone) 1β subcomplex 8 (Ndufb8) in muscle (Fig. 2A). In all groups, we could also detect a mild compensatory increase in PGC-1β expression when PGC-1α was deleted in skeletal muscle (Fig. 2A). PGC-1α MKO mice furthermore displayed reduced levels of proteins involved in mitochondrial OXPHOS (Fig. 2B). The RSV-S group displayed no induction of mRNA expression of mitochondrial genes (Fig. 2A) or OXPHOS protein content (Fig. 2B) in skeletal muscle. Additionally, short term RSV treatment resulted in a mild decrease in fibers with a high NADH activity in CTRL mice (Fig. 2C). In the RSV-L group, however, we could detect a trend toward increased PGC-1α gene transcription (Fig. 2A) and a mild induction of several mitochondrial PGC-1α target genes (Cytc, Cox5b, and Cs) (Fig. 2A). Importantly, the effects of RSV treatment on mitochondrial gene transcription were dependent on PGC-1α, because the induction of these genes was abolished in PGC-1α MKO mice (Fig. 2A). However, RSV treatment had no effect on either OXPHOS protein levels (Fig. 2B) or oxidative enzymatic staining in muscle (Fig. 2C). SRT treatment led to increased mRNA (Fig. 2A) and protein levels (Fig. 2B) of Ndfub8 in skeletal muscle. Apart from Ndufb8, no other mitochondrial gene tested was found to be induced by SRT treatment (Fig. 2A). Importantly, the effect on Ndufb8 transcription was dependent on PGC-1α, because both induction of mRNA (Fig. 2A) and protein levels (Fig. 2B) of Ndufb8 with SRT treatment were blunted in PGC-1α MKO mice. Despite the mild effect on mitochondrial gene transcription, SRT treatment resulted in an enhanced oxidative capacity in muscle. This was indicated by a higher percentage of dark blue fibers in skeletal muscle stained for NADH activity (Fig. 2C). Surprisingly, this effect also occurred in PGC-1α MKO mice (Fig. 2C), implying that this effect of SRT treatment was independent of muscle PGC-1α. Increased oxidative capacity with SRT treatment could be the consequence of a higher mitochondrial number in skeletal muscle. We therefore quantified mitochondria in electron microscopy pictures from skeletal muscle and could detect an increased number of intramyofibrillar mitochondria with SRT treatment in both genotypes, although this did not reach statistical significance for PGC-1α MKO mice (Fig. 2D). Hence, the increase in oxidative capacity with SRT treatment might be a consequence of a higher mitochondrial number in skeletal muscle.
FIGURE 2.

PGC-1α is required for the transcriptional induction of mitochondrial genes in skeletal muscle. A, mRNA levels of PGC-1α/β and mitochondrial genes in SKM relative to Polr2a (RSV-S and RSV-L) or Hprt (SRT) (n = 5–8/group). B, representative immunoblots of mitochondrial proteins in SKM (n = 6/group). C, representative pictures of NADH stainings of SKM sections. Bar graphs show quantification of the intensity of 100 muscle fibers per section (n = 3/group). D, representative electron micrographs from SKM. The bar graph shows average mitochondrial number per image normalized to HFD CTRL and expressed in arbitrary units (a.u.) (n = 3/group). Bars, means ± S.E. (error bars). Significant differences (p < 0.05) between CTRL and PGC-1α MKO mice are indicated (#) as well as significant differences between untreated and treated groups (*). ATP5A, ATP synthase, H+-transporting, mitochondrial F1 complex, α subunit 1; UQCRC2, ubiquinol cytochrome c reductase core protein 2; SDHB, succinate dehydrogenase complex, subunit B; eEF2, eukaryotic elongation factor 2.
RSV and SRT treatment have been shown to affect the transcription of several processes in skeletal muscle besides mitochondrial function (e.g. glycolysis, β-oxidation, and myosin heavy chain isoforms) (5, 11). However, in our experimental cohorts, neither RSV nor SRT had a strong effect on the transcriptional induction of these processes in muscle (Fig. 3, A–C). We observed a PGC-1α-independent induction of uncoupling protein 3 (Ucp3) mRNA expression (Fig. 3A) in RSV-S mice, whereas other genes involved in fatty acid β-oxidation, such as medium-chain acyl-CoA dehydrogenase (Mcad), carnitine palmitoyltransferase 1B (Cpt1b), and pyruvate dehydrogenase kinase isozyme 4 (Pdk4), were not significantly altered (Fig. 3A). Interestingly, the increase in Ucp3 expression was a transient effect of RSV treatment, because this effect was no longer present in the RSV-L group (Fig. 3B). In contrast, a mild PGC-1α-dependent induction of myosin heavy chain-2a (MyHCIIA) and -2b (MyHCIIB) mRNA expression was observed in RSV-L mice (Fig. 3B), which was not present in RSV-S mice. SRT treatment led to the induction of hexokinase 2 (Hk2) and Mcad (Fig. 3C) in muscle, whereas mRNA expression of other genes involved in glycolysis, β-oxidation, and MyHC subtypes were unchanged (Fig. 3C). In the PGC-1α MKO group, induction of Hk2 and Mcad by SRT did not reach statistical significance (Fig. 3C). We furthermore assessed AMPKα phosphorylation in muscle as readout of RSV- and SRT-induced signaling. In line with the mild transcriptional effects of these compounds, we could detect no significant induction of AMPKα phosphorylation (Thr-172) in muscle from either RSV- or SRT-treated mice (Fig. 3D). Because systemic compensatory effects could potentially have masked the effects of RSV and SRT in muscle in vivo, we further tested the transcriptional response of these compounds in vitro. Interestingly, C2C12 myotubes treated with either RSV or SRT displayed a significant reduction in PGC-1α transcript levels and unaltered or slightly reduced transcription of known PGC-1α target genes, such as Cytc, Cox5b, Cs, Ndufb8, Ucp3, Mcad, or Hk2 (Fig. 3E). Pdk4 transcription was, however, induced by RSV treatment (Fig. 3E). To determine whether the lack of effect with RSV and SRT treatment could be attributed to the low basal expression of PGC-1α in C2C12 myotubes, we also performed this experiment with either C2C12 myotubes overexpressing PGC-1α (565 ± 62-fold overexpression; data not shown) (Fig. 3F) or in primary mouse myotubes (Fig. 3G). However, apart from a boosted PGC-1α expression with SRT treatment in PGC-1α-overexpressing myotubes (Fig. 3F), neither compound led to any prominent transcriptional effect on the above mentioned genes in either setup (Fig. 3, F and G). Conclusively, the minor transcriptional effects of RSV and SRT in cultured myotubes mirror our in vivo findings (Fig. 3, A–C). This prompted us to investigate AMPK signaling in RSV- and SRT-treated myotubes. In line with other studies (11), SRT did not affect the phosphorylation status of AMPKα (Thr-172) in C2C12 myotubes (Fig. 3H). Surprisingly, RSV-treated myotubes displayed decreased AMPKα phosphorylation (Thr-172) (Fig. 3H). In conclusion, our in vitro data support our in vivo findings that RSV and SRT elicit only minor effects on transcription of mitochondrial and metabolic gene programs in skeletal muscle. Moreover, we show that we cannot enhance the transcriptional effect of RSV and SRT in muscle cells by concomitant overexpression of PGC-1α.
FIGURE 3.

RSV and SRT treatment elicit minor transcriptional effects in skeletal muscle cells. A–C, mRNA levels of genes involved in glucose uptake and glycolysis, myosin heavy chain (MyHC) subtypes, and fatty acid β-oxidation in SKM relative to Polr2a (RSV-S and RSV-L) or Hprt (SRT) (n = 5–8/group). D, representative immunoblots of AMPKα total protein and AMPKα phosphorylation (Thr-172) in SKM (n = 6/group). E–G, mRNA levels of the indicated genes normalized to vehicle group (dashed line) in C2C12 myotubes (E), C2C12 myotubes overexpressing (OE) PGC-1α through adenoviral transduction (F), or primary mouse myotubes (G). All were treated for 24 h with either 50 μm RSV or 10 μm SRT, relative to Tbp. H, immunoblots of AMPKα total protein and AMPKα phosphorylation (Thr-172) in C2C12 mouse myotubes treated for 24 h with either 50 μm RSV or 10 μm SRT1720. Bars, means ± S.E. (error bars). Significant differences (p < 0.05) between CTRL and PGC-1α MKO mice are indicated (#) as well as significant differences between untreated and treated groups (*). PKM1, pyruvate kinase muscle isozyme 1.
RSV Improves Mitochondrial Biogenesis and Reduces Inflammation in White Adipose Tissue
Because RSV and SRT exerted only a mild effect on skeletal muscle metabolism and because PGC-1α MKO mice displayed a similar systemic response to both RSV and SRT treatment as control mice, this suggests only a minor role for skeletal muscle PGC-1α in the regulation of systemic metabolism by these compounds. We therefore continued to analyze the effects of RSV and SRT treatment in other major metabolic organs, such as brown and white adipose tissue (BAT and WAT, respectively) as well as liver. To avoid confounding metabolic cross-talk due to ablation of PGC-1α in muscle, the following experiments were performed in mice with intact PGC-1α expression in skeletal muscle.
BAT is an important organ for regulation of both energy expenditure and glucose homeostasis (21). Hence, we were interested in elucidating whether RSV and SRT would exert a more prominent effect in this organ, which could explain the systemic metabolic effects in our mice. Intriguingly, neither RSV nor SRT affected the transcription of PGC-1α, PGC-1β, or mitochondrial genes in BAT (Fig. 4, A–C). Moreover, whereas the RSV-S group displayed a significant increase in type II iodothyronine deiodinase (Dio2) (Fig. 4A), neither compound led to an induction of uncoupling protein 1 (Ucp1) or peroxisome proliferator-activated receptor γ (Pparγ) isoforms (Fig. 4, A–C) in BAT. Because neither compound affected Ucp1 expression in BAT, we also decided to investigate the thermogenic gene expression in WAT. However, we could detect no induction of Ucp1, Dio2, cell death-inducing DFFA-like effector a (Cidea) or PR domain-containing 16 (Prdm16) expression in WAT with either RSV or SRT (Fig. 4D). The unaltered transcription of key thermogenic genes in BAT and the lack of Ucp1 induction in WAT indicate that the increased energy expenditure seen with both RSV and SRT treatment (Fig. 1C) is probably not a direct consequence of either enhanced BAT thermogenesis or WAT browning. Additionally, we measured the expression of genes important for glucose uptake and glycolysis in BAT. Intriguingly, we could detect a significant induction of genes involved in glucose uptake and glycolysis (glucose transporter 1 (Glut1), Hk2, and phosphofructokinase 1 (Pfkm)) in BAT with SRT treatment (Fig. 4C). This could not be seen in RSV-treated mice (Fig. 4, A and B). Because BAT plays a fundamental role in the regulation of systemic glucose homeostasis (21), the transcriptional induction of genes involved in glucose uptake and glycolysis in this tissue could contribute to the beneficial effect of SRT treatment on glucose homeostasis in our mice.
FIGURE 4.

RSV and SRT elicit differential transcriptional effects in white and brown adipose tissue. A–C, mRNA levels of PGC-1α/β and mitochondrial genes, genes involved in adaptive thermogenesis and glycolysis/glucose uptake in BAT, relative to Polr2a (RSV-S and RSV-L) or eEF2 (SRT) (n = 5–8/group). D, mRNA levels of genes involved in adaptive thermogenesis in WAT, relative to Polr2a (RSV-S and RSV-L) or Hprt (SRT) (n = 5–8/group). E, mRNA levels of PGC-1α/β and mitochondrial genes in WAT, relative to Polr2a (RSV-S and RSV-L) or Hprt (SRT) (n = 5–8/group). Shown are representative immunoblots of PGC-1α and mitochondrial proteins (F) and AMPKα total protein and AMPKα phosphorylation (Thr-172) (G) in WAT (n = 6/group). Error bars, means ± S.E. *, significant differences (p < 0.05) between untreated and treated groups.
Metabolic dysfunction in WAT plays an important role in the development of obesity and metabolic syndrome. Hence, we were interested in the effects of RSV and SRT on WAT metabolism. Intriguingly, in contrast to the effects seen in skeletal muscle and BAT, RSV-S mice displayed a significant increase in PGC-1α transcript levels in WAT (Fig. 4E). RSV-L mice showed no significant induction of PGC-1α mRNA levels (Fig. 4E), but PGC-1α protein levels for this time point were significantly increased (Fig. 4F). In line with the higher levels of PGC-1α, RSV treatment resulted in a significant induction of mitochondrial genes (Uqcrc2 and succinate dehydrogenase complex, subunit A (Sdha)) at both time points (Fig. 4E), whereas only RSV-L mice displayed enhanced mitochondrial protein levels of SDHB (Fig. 4F). SRT treatment resulted in a trend toward increased PGC-1α transcription in WAT (Fig. 4E) but had no further effect on mitochondrial transcript (Fig. 4E) or protein levels (Fig. 4F). Moreover, neither compound resulted in significantly enhanced AMPKα phosphorylation (Thr-172) in WAT (Fig. 4G). These data suggest that increased AMPK activity is most likely not the main mechanism by which RSV treatment increases mitochondrial transcript and protein levels in WAT. Another possible explanation for the increased mitochondrial gene transcription is through down-regulation of activating transcription factor 3 (Atf3), a transcriptional repressor known to reduce transcription of PGC-1α and mitochondrial genes in WAT (22). Interestingly, we could detect a significant reduction in Atf3 transcript levels in WAT of RSV-L mice and a trend (p = 0.06) toward diminished Atf3 transcription in RSV-S mice (Fig. 5A). To substantiate these findings, we also measured the expression of adiponectin (Adipoq), a gene known to be repressed by ATF3 in adipocytes (23). In line with the reduced Atf3 mRNA levels, expression of adiponectin was significantly increased specifically in RSV-treated mice (Fig. 5A), supporting a reduced transcriptional repression by ATF3. In SRT-treated mice, however, no alterations in either ATF3 or adiponectin expression were observed (Fig. 5A). These findings correlate well with the unchanged mitochondrial gene transcription with SRT treatment (Fig. 4E) and suggest that repression of ATF3 is mediated specifically by RSV. To gain more insight into the mechanisms that underlie the repression of Atf3 expression by RSV, we assessed the activity of CREB, because increased CREB activity has been associated with increased Atf3 transcription in WAT during obesity (24). In line with the reduced transcript levels of Atf3 in RSV-treated mice, we could also detect reduced levels of CREB phosphorylation (Ser-133) in RSV-treated but not SRT-treated mice (Fig. 5B), corresponding to a reduced activity of this transcription factor with RSV administration. Besides various beneficial systemic effects of adiponectin on whole body metabolism, this hormone also possesses anti-inflammatory properties (25). We therefore measured gene expression of inflammatory markers in WAT of our mice. In line with the repression of Atf3 and induction of adiponectin, inflammatory gene transcription (tumor necrosis factor α (Tnf), monocyte chemotactic protein-1 (Mcp-1), and cluster of differentiation 68 (Cd68)) was decreased in RSV-treated mice (Fig. 5C). However, SRT treatment did not result in any changes in proinflammatory gene expression.
FIGURE 5.

RSV treatment affects lipid metabolism in white adipose tissue. A, mRNA levels of Atf3 and Adipoq in WAT, relative to Polr2a (RSV-S and RSV-L) or Hprt (SRT) (n = 5–8/group). B, representative immunoblots of CREB total protein and CREB phosphorylation (Ser-133) in WAT (n = 6/group). C and D, mRNA levels of genes associated with inflammation (C) and lipogenesis (D) in WAT, relative to Polr2a (RSV-S and RSV-L) or Hprt (SRT) (n = 5–8/group). E, representative immunoblots of IRβ, Akt, and GSK3β as well as their respective phosphorylations in WAT (n = 6/group). F, insulin levels in plasma from ad libitum-fed and 16-h-fasted mice (n = 5–8/group). G, mRNA levels of genes involved in lipolysis and glycerol 3-phosphate generation in WAT, relative to Polr2a (RSV-S and RSV-L) or Hprt (SRT) (n = 5–8/group). H, non-esterified fatty acid levels in plasma from ad libitum-fed and 16-h-fasted mice (n = 5–8/group). Bars, means ± S.E. (error bars). *, significant differences (p < 0.05) between untreated and treated groups.
Long Term RSV Treatment Leads to an Induction of Genes Involved in Lipogenesis and Lipolysis in WAT
Next, we were interested whether RSV or SRT would affect lipid handling in WAT. Interestingly, RSV-L mice displayed a robust induction of a number of lipogenic genes (sterol regulatory element-binding protein-1c (Srebp1c), acetyl-CoA carboxylase 1 (Acc1), stearoyl-CoA desaturase-1 (Scd1), fatty acid synthase (Fasn)) as well as genes involved in fatty acid re-esterification (glycerophosphate acyltransferase (Gpat) and diglyceride acyltransferase 1 (Dgat1)) in WAT (Fig. 5D). In contrast, neither RSV-S nor SRT groups displayed any alterations in lipogenic gene transcription, with the exception of reduced Fasn transcript levels in RSV-S mice (Fig. 5D). This suggests that the effects on lipogenic gene transcription are more prominent with long term (13-week) as compared with short term (4-week) RSV treatment. To assess whether increased insulin signaling in WAT from RSV-L mice could explain the induction of lipogenic genes specifically in this group, we analyzed insulin signaling in WAT. Interestingly, RSV-L mice displayed increased protein levels and autophosphorylation of IRβ (Tyr-1146), Akt (Ser-473), and the Akt target GSK3β (Ser-9) in WAT (Fig. 5E). These findings indicate an enhanced insulin signaling, which could provide an explanation for the increased transcription of lipogenic genes observed in the RSV-L group. However, enhanced insulin signaling in WAT could not be attributed to alterations in circulating insulin levels, because RSV-L mice showed no significant alterations in plasma insulin concentration in either fed or fasted states (Fig. 5F). In contrast to the RSV-L group, RSV-S mice displayed no alterations in either IRβ (Tyr-1146), Akt (Ser-473 and Thr-308), and GSK3β (Ser-9) phosphorylation in WAT (Fig. 5E). This finding highlights another difference between short and long term RSV administration. SRT-treated mice furthermore displayed no obvious differences in either insulin signaling (Fig. 5E) or plasma insulin levels (Fig. 5F) compared with control mice. Increased lipogenesis and fatty acid re-esterification could be indicative of an increased WAT mass. However, in contrast to the elevation of lipogenic gene expression, short and long term RSV treatment led to a reduced fat mass (Fig. 1B). Hence, we speculated that not only lipogenesis, but also other aspects of triglyceride handling might be affected in WAT of RSV-treated mice. Indeed, in addition to the transcriptional activation of lipogenic processes in the RSV-L group, we also found an induction of genes involved in lipolysis (adipose triglyceride lipase (Atgl) and hormone-sensitive lipase (Hsl)) (Fig. 5G). However, despite the induction of lipolytic genes with RSV treatment, no alterations in plasma non-esterified fatty acid levels in the RSV-L group were observed (Fig. 5H). These data, together with the concurrent induction of lipolysis and fatty acid re-esterification, would point toward an increased fatty acid recycling in the RSV-L mice. Because the availability of glycerol 3-phosphate is essential for fatty acid re-esterification, we investigated transcriptional regulation of genes involved in glycerol 3-phosphate synthesis. Interestingly, we could observe an induction of genes involved in two processes important for glycerol 3-phosphate generation, namely glucose uptake/glycolysis (glucose transporter type 4 (Glut4) and Hk2) and glyceroneogenesis (phosphoenolpyruvate carboxykinase (Pepck)) in the RSV-L group (Fig. 5G). No differences in these genes could be detected for the RSV-S and SRT groups, apart from a significant up-regulation of Hk2 in SRT-treated mice (Fig. 5G).
SRT Induces Transcription of PGC-1α Target Genes in Liver
Because RSV and SRT treatment elicited distinct mitochondrial and metabolic alterations in adipose tissue, we were also interested in how these compounds would affect liver metabolism. In contrast to the effect in WAT, RSV treatment at either time point led to a reduced transcription of PGC-1α but did not further affect transcription of mitochondrial genes in the liver (Fig. 6A). Conversely, SRT treatment increased the transcription of several mitochondrial genes (Cox5b, Cytc, Cs, and Uqcrc2) (Fig. 6A) in the liver, along with a small significant increase in UQCRC2 and ATP synthase subunit α5 (ATP5A) protein levels (Fig. 6B). Moreover, SRT treatment led to a small elevation in PGC-1α protein levels (Fig. 6B). Because several PGC-1α target genes involved in mitochondrial OXPHOS were induced in the liver of SRT-treated mice, this indicates that SRT treatment resulted in an increased transcriptional activity of PGC-1α. Because SRT is an activator of SIRT1 (10), which can deacetylate and thereby activate PGC-1α (17), we next determined the acetylation status of PGC-1α in the liver of control and SRT-treated animals. However, we could detect no difference in PGC-1α acetylation status between HFD-fed control mice and SRT-treated mice (Fig. 6C). Interestingly, SRT-treated mice displayed a strong trend toward increased hepatic AMPKα phosphorylation (Thr-172) with SRT treatment (Fig. 6D), correlating with an increased phosphorylation of the AMPK-sensitive phosphorylation site (Ser-79) on ACC (Fig. 6D). Because AMPK can phosphorylate and thereby increase the transcriptional activity of PGC-1α (16), AMPK activation upon SRT treatment could account for the increased hepatic PGC-1α activity in SRT-treated mice. Importantly, AMPKα phosphorylation (Thr-172) was not induced in the liver of RSV-treated mice at either time point (Fig. 6E). SRT treatment furthermore induced several known PGC-1α target genes involved in mitochondrial fatty acid oxidation (Mcad and long-chain acyl-CoA dehydrogenase (Lcad)), cellular and mitochondrial fatty acid import (carnitine-acylcarnitine translocase (Cact), lipoprotein lipase (LPL), and cluster of differentiation 36 (Cd36)), ketogenesis (acetyl-CoA acetyltransferase 1 (Acat1), 3-hydroxymethyl-3-methylglutaryl-CoA lyase (Hmgcl), and 3-hydroxy-3-methylglutaryl-CoA synthase 2 (Hmgcs2)) and gluconeogenesis (Pepck and glucose-6-phosphatase (G6pc)) (Fig. 6F). Intriguingly, the induction of PGC-1α-regulated gene programs in liver could not be detected in RSV-treated mice (Fig. 6F), indicating that this effect is elicited specifically by SRT administration.
FIGURE 6.

RSV and SRT treatment elicit differential effects on metabolic processes in liver. A, mRNA levels of PGC-1α/β and mitochondrial genes in liver, relative to Rpl0 (RSV-S) or Polr2a (RSV-L, SRT) (n = 5–8/group). B, representative immunoblots of PGC-1α and mitochondrial proteins in liver. Bar graphs show quantification of PGC-1α and mitochondrial protein content relative to eEF2 (n = 6/group). C, immunoprecipitation of acetylated lysines (Ac-K) from hepatic nuclear extracts, followed by immunoblotting against PGC-1α. For the input, an immunoblot against PGC-1α from hepatic nuclear extracts is shown (n = 3/group). D and E, representative immunoblots of AMPKα total protein and AMPKα phosphorylation (Thr-172) as well as ACC total protein and ACC phosphorylation (Ser-79) in liver (n = 6/group). F and G, mRNA levels of genes involved in ketogenesis, gluconeogenesis fatty acid β-oxidation, and fatty acid uptake (F) and lipogenesis (G) in liver, relative to Rpl0 (RSV-S) or Polr2a (RSV-L and SRT) (n = 5–8/group). H, representative immunoblot of IRβ, Akt, and GSK3β as well as their respective phosphorylations (n = 6/group). Bars, means ± S.E. (error bars). *, significant differences (p < 0.05) between untreated and treated groups.
RSV and SRT Differentially Regulate Hepatic Lipogenic Gene Transcription
Due to the observed metabolic remodeling in liver by SRT, we also measured the expression of genes important for hepatic lipogenesis in our mice. SRT treatment led to a transcriptional induction of the lipogenic transcription factor Srebp1c as well as several of its target genes involved in lipogenesis (Acc2, Scd1, and Fasn) and fatty acid re-esterification (Gpat) (Fig. 6G). In line with the transcriptional induction of Acc2, protein levels of ACC were also up-regulated in liver from SRT-treated mice (Fig. 6D). Because insulin is a known regulator of Srebp1c transcription and processing (26), we investigated whether increased hepatic insulin signaling could account for the increased lipogenic gene transcription upon SRT treatment. In the SRT group, Akt phosphorylation (Thr-308) was increased, whereas phosphorylation of IRβ (Tyr-1146) and phosphorylation of the Akt target GSK3β (Ser-9) were reduced (Fig. 6H). Moreover, as mentioned earlier, circulating insulin levels were unchanged in SRT-treated mice (Fig. 5F). Collectively, these findings indicate no direct link between hepatic insulin signaling and the increased transcriptional activation of lipogenic genes seen with SRT treatment. Interestingly, RSV resulted in an opposite regulation of lipogenic gene transcription compared with SRT in liver. RSV treatment led to a significant reduction in Scd1 and Acc2 transcript (Fig. 6G) and ACC protein (Fig. 6E) levels in liver at both time points. Moreover, the RSV-S group displayed a significant reduction of genes involved in lipogenesis (Fasn) and fatty acid re-esterification (Gpat) as well as a statistically non-significant trend toward reduced Srebp1c transcription for both time points (Fig. 6G). Interestingly, whereas lipogenic gene transcription in the liver was reduced with RSV treatment, the same lipogenic genes were up-regulated in WAT of the RSV-L group (Fig. 5D) at the same time point. RSV thus exerted opposing effects on lipogenic gene transcription in WAT compared with liver. Additionally, SRT did not influence lipogenic gene transcription in WAT (Fig. 5D), whereas a strong effect was observed in liver (Fig. 6G). We next investigated whether the strikingly opposite transcriptional responses between RSV and SRT in liver would affect hepatic triglyceride content. In the RSV-S group, a trend (p = 0.056) toward reduced triglyceride content was observed (Fig. 7, A and B). Surprisingly, this effect was not present in the RSV-L group (Fig. 7, A and B), although lipogenic gene transcription was still reduced at this time point (Fig. 6G). Despite the increased transcription of lipogenic genes with SRT treatment, this did not further affect hepatic triglyceride content (Fig. 7, A and B). We next studied the effect of RSV and SRT treatment on hepatic cholesterol biosynthesis. RSV treatment did not affect transcription of genes involved in cholesterol biosynthesis and esterification (acyl-CoA:cholesterol acyltransferase 2 (Acat2), farnesyl-diphosphate farnesyltransferase 1 (Fdft1), and 7-dehydrocholesterol reductase (Dhcr7)) in the liver at either time point (Fig. 7C). Nevertheless, RSV-S mice exhibited reduced levels of LDL-C in plasma (Fig. 7D), whereas HDL-C (Fig. 7D) levels remained unaltered. Moreover, the reduction in circulating LDL-C levels was most likely a transient effect of RSV treatment, because no differences in either LDL or HDL cholesterol were observed in the RSV-L group (Fig. 7D). Conversely, SRT-treated mice displayed increased transcription of genes involved in cholesterol biosynthesis and esterification (Acat2, Fdft1, and Dhcr7) (Fig. 7C). In line with this, circulating levels of LDL-C and HDL-C were significantly increased in plasma of SRT-treated mice (Fig. 7D). Because RSV and SRT elicited distinct effects on hepatic metabolism, we were interested in whether this was due to differential effects of these compounds or whether it could be attributed to compensatory systemic effects in vivo. To address this question, we treated AML-12 cells, a mouse hepatocyte cell line, with either RSV or SRT. In contrast to the effects seen in vivo, PGC-1α expression was induced with both RSV (Fig. 7E) and SRT treatment (Fig. 7F) in AML-12 cells, whereas only SRT treatment led to an induction of PGC-1β (Fig. 7F). Moreover, RSV and SRT treatment led to a reduction in Hmgcs2 transcription in AML-12 cells, which indicates that this gene is a common target for both RSV and SRT in vitro (Fig. 7, E and F). Interestingly, Hmgcs2 transcription was either unaffected with RSV or induced with SRT treatment in liver in vivo (Fig. 6F). Moreover, Cact transcription was elevated by RSV treatment in vitro (Fig. 7E), whereas this only occurred in SRT-treated mice in vivo (Fig. 6F). These differential transcriptional effects observed in vitro as compared with in vivo could stem from systemic compensatory effects in vivo. Hence, this observation further highlights the importance of studying the therapeutic effects of these compounds in vivo. Nonetheless, in line with our in vivo findings, SRT treatment resulted in a transcriptional induction of genes involved in lipogenesis (Srebp1c, Scd1, and Fasn) and cholesterol synthesis (Fdft1 and Dhcr7) in AML-12 cells (Fig. 7F), whereas these genes were either unaltered or down-regulated with RSV treatment (Fig. 7E). Hence, the differential effects of RSV and SRT on lipogenic gene transcription in liver in vivo are most likely not caused by systemic compensatory mechanisms. Moreover, the distinct transcriptional profiles of RSV and SRT in cultured hepatocytes (Fig. 7, E and F) demonstrate that these compounds do not elicit analogous effects on hepatic metabolism. We next measured the expression of genes involved in lipogenesis and cholesterol synthesis in C2C12 myotubes to determine whether the SRT-mediated effects on lipogenic gene transcription were liver cell-specific. Intriguingly, whereas SRT treatment led to a reduction in PGC-1α and mitochondrial gene expression in C2C12 myotubes (Fig. 3E), SRT treatment boosted transcription of lipogenic genes (Scd1, Fas, and Acc1) also in muscle cells (Fig. 7H). In contrast to the effects in liver (Fig. 7C) and cultured hepatocytes (Fig. 7F), SRT did not significantly induce genes involved in cholesterol synthesis in muscle cells (Fig. 7H). Furthermore, in analogy with our findings in liver (Figs. 6G and 7C) and cultured hepatocytes (Fig. 7E), RSV treatment did not lead to the induction of genes involved in either lipogenesis or cholesterol synthesis in C2C12 myotubes (Fig. 7G). Hence, we demonstrate that SRT is a potent transcriptional activator of lipogenic gene transcription in both liver and muscle cells. Moreover, we also show that RSV and SRT elicit differential transcriptional effects in hepatocytes, both in vivo and in vitro.
FIGURE 7.

SRT treatment affects lipid homeostasis. A, triglyceride (TG) content in liver expressed in arbitrary units (a.u.), relative to HFD-fed groups (n = 5–8/group). B, representative pictures of Oil Red O staining in liver sections (n = 5/group). C, mRNA levels of genes involved in cholesterol biogenesis in liver, relative to Rpl0 (RSV-S) or Polr2a (RSV-L and SRT) (n = 5–8/group). D, plasma LDL and HDL cholesterol levels (n = 8–10/group). E and F, mRNA levels of the indicated genes in AML-12 mouse hepatocytes treated for 6 h with either 50 μm RSV (E) or 10 μm SRT1720 (F), relative to Tbp (RSV) or Rpl0 (SRT). G and H, mRNA levels of the indicated genes in C2C12 mouse myotubes treated for 24 h with either 50 μm RSV (G) or 10 μm SRT1720 (H), relative to Tbp. Bars, means ± S.E. (error bars). *, significant differences (p < 0.05) between untreated and treated groups.
Discussion
Caloric restriction mediates several beneficial metabolic effects in organisms ranging from yeast to rodents and humans (1). As a result, the concept of using pharmacological CR mimetics, such as RSV and SRT, to ameliorate metabolic disorders has received a lot of attention in recent years (1, 27). In line with other studies (3–5, 10–12), we can demonstrate that RSV and SRT treatment elicit beneficial metabolic effects in HFD-fed mice, such as an increased metabolic rate, improved glucose homeostasis, and reduced adiposity. However, in contrast to previous hypotheses implying muscle PGC-1α as the common molecular effector of RSV and SRT, we now show that skeletal muscle PGC-1α is dispensable for the improved metabolic rate of these compounds and the improved glucose homeostasis seen with SRT treatment. Interestingly, PGC-1α was required for the induction of mitochondrial gene transcription in muscle with RSV and SRT treatment, whereas it was dispensable for the induction of non-transcriptional changes, such as increased oxidative capacity and enhanced mitochondrial number, in skeletal muscle with SRT treatment. Importantly, a similar observation was made in a study by Finley et al. (28), where the authors investigated the requirement for skeletal muscle PGC-1α for the systemic metabolic effects of CR. Analogous to our data, this study showed that during CR, PGC-1α was required for the enhanced transcription of mitochondrial genes in muscle, whereas basal oxygen consumption increased to the same extent even in the absence of skeletal muscle PGC-1α (28). These data and our findings collectively indicate that enhanced oxidative capacity in muscle during CR or SRT treatment is dissociated from the induction of mitochondrial gene transcription seen in this context and would thus be independent of muscle PGC-1α. However, these and our results were obtained in sedentary animals and therefore do not preclude a more prominent role for muscle PGC-1α in the regulation of the metabolic phenotype of physically active mice in which muscle contributes more significantly to the global metabolic rate. It is also important to note that PGC-1β levels in skeletal muscle were not reduced in the PGC-1α MKO-model used in our study and in the study by Finley et al. (28). Because PGC-1β is a known deacetylation target of SIRT1 (29), a caveat of our study would be that PGC-1β could, to a certain extent, compensate for the loss of PGC-1α in skeletal muscle. Moreover, PGC-1β could be responsible for mediating the effects of RSV and SRT also in other organs, a subject that will be important to investigate in future studies.
Despite the structural difference between RSV and SRT, the effects of these compounds are considered to be analogous and ultimately mediated through the same downstream signaling pathways. For example, the transcriptional response to these two compounds overlaps in liver (12) and is similar to the hepatic transcriptional profile during CR (2, 3). However, this overlap is small in comparison with the total number of altered genes (12), suggesting that many effects of RSV and SRT on metabolic processes in the liver are in fact unique. In line with this, we have identified several hepatic metabolic processes that are induced at a transcriptional level both in vivo and in vitro by SRT treatment but not by RSV treatment. Hence, these effects are most likely due to a direct differential effect of these compounds and not due to compensatory systemic effects in vivo. This is evident for the transcriptional regulation of lipogenic genes, which were induced specifically by SRT treatment in both hepatocytes and muscle cells. Moreover, SRT, but not RSV, enhanced transcription of genes involved in cholesterol metabolism, fatty acid β-oxidation, ketogenesis, and gluconeogenesis in liver. Collectively, these findings suggest that RSV and SRT elicit distinct effects on hepatic metabolism. We could also detect a small difference between RSV and SRT treatment on hepatic triglyceride content in vivo. In agreement with other studies (4, 30, 31), we observed that short term RSV treatment resulted in a mild protection against hepatic steatosis, whereas this could not be seen for SRT treatment at the same time point. This lack of effect is in contrast to the reduced hepatic lipid content reported with SRT treatment in other studies (11, 12, 32). However, at least in one of these studies (11), the SRT-linked reduction in hepatic lipid accumulation was only observed at a dose that was 5 times higher than that used in our study. Interestingly, Qiang et al. (33) could show that mice carrying an extra copy of the Sirt1 gene develop hepatic steatosis and display increased circulating cholesterol levels when fed a HFD. In analogy with these findings, the transcriptional activation of cholesterol biosynthesis in liver by SRT treatment also correlated with increased circulating levels of HDL and LDL cholesterol in our study. Importantly, increased cholesterol synthesis and transcriptional induction of lipogenesis might constitute a caveat for using SRT as a treatment in obese patients. In contrast, RSV had a more beneficial effect on whole body lipid parameters in our mice, because we could demonstrate a trend toward reduced hepatic lipid accumulation as well as significantly lower LDL-C levels in the RSV-S group. Additionally, reduced hepatic steatosis and LDL-C levels with RSV treatment has also been reported in human patients (34–36).
Differential effects between RSV and SRT treatment could also be observed in adipose tissue. Whereas SRT did not affect adiposity in HFD-fed mice, RSV treatment resulted in a reduced fat mass at both time points. This correlated with a gene expression profile in RSV-treated mice indicating improved adipose tissue health, which could not be detected in SRT-treated mice. Both RSV-S and RSV-L groups displayed increased transcription of mitochondrial genes, enhanced adiponectin expression, and reduced adipose tissue inflammation. These effects of RSV have also been observed in other studies with both rodents (37–39) and non-human primates (40). In search of a mechanism for how RSV mediates these effects, we could show that RSV treatment resulted in a reduced phosphorylation of CREB in WAT, correlating with a reduced transcription of ATF3. CREB is a positive regulator of Atf3 transcription during obesity (24), and ATF3 in turn has been shown to inhibit transcription of GLUT4 (24), adiponectin (23), and mitochondrial genes (22). Interestingly, mice expressing a dominant negative form of CREB in WAT (24) display similar phenotypic changes as seen with RSV treatment in our study, including reduced inflammation, improved GLUT4 expression, and improved whole body glucose tolerance. Moreover, a transcription factor analysis performed in WAT from humans receiving RSV treatment predicted reduced CREB1 activity with RSV administration (41). These studies are in agreement with our findings and give credence to the theory that the repression of CREB-ATF3 axis by RSV treatment in WAT might also be relevant in humans. Surprisingly, although the above mentioned effects were detected in both RSV-S and RSV-L groups, we could detect enhanced insulin signaling and transcriptional induction of lipogenesis and lipolysis in WAT only in the RSV-L group. These findings demonstrate a differential response to RSV administration in WAT, depending on the duration of the treatment. This is an important point to consider for future studies and for the use of RSV in a clinical setting. Moreover, due to the fact that a long term SRT treatment was not investigated in our current study, we cannot exclude the possibility that long term SRT treatment could promote similar effects on lipogenic gene transcription in WAT as shown for RSV, in particular because SRT treatment induced lipogenic gene transcription in both liver and muscle cells in our study. Finally, to provide an explanation for the increased energy expenditure seen with RSV and SRT treatment, we investigated thermogenic gene expression in both WAT and BAT. Although neither compound had significant effects on transcriptional regulation of adipose tissue thermogenesis, SRT treatment led to the transcriptional induction of glycolytic genes in BAT. Considering the fundamental role of BAT in the regulation of glucose homeostasis (21), this could provide an explanation for the improved glucose tolerance in SRT-treated mice.
The efficacy of RSV in increasing life span and health span in different species is somewhat controversial (2, 42, 43). Moreover, the main molecular target for RSV in vivo is not well established (44), and recent studies in particular have questioned the effects of RSV in skeletal muscle (45). In line with our findings, Higashida et al. (45) could not detect induction of mitochondrial biogenesis in skeletal muscle with RSV treatment. Additionally, nor could Ringholm et al. (46) detect any strong effect of RSV treatment on oxidative protein or mitochondrial DNA content in their study. Interestingly, these studies (45, 46) as well as our own used a similar concentration of RSV (4 g/kg diet) as was used in a study by Lagouge et al. (5). Although the study by Lagouge et al. (5) could observe an increased mitochondrial oxidative capacity in skeletal muscle with RSV treatment, this finding could not be replicated in our study. However, one important difference between the Lagouge study (5) and our study is the choice of HFD. Whereas the diet our mice were fed is lard-based (D12492, Research Diet) and thus rich in long-chain fatty acids, the HFD used by Lagouge et al. (5) (D12327, Research Diet) contains mainly coconut oil, which primarily consists of medium-chain fatty acids. HFDs rich in medium-chain fatty acids have been shown to induce mitochondrial metabolism and oxidative capacity in skeletal muscle (47, 48), whereas HFDs containing long-chain fatty acid have a more detrimental effect on adiposity and glucose tolerance in mice than medium-chain fatty acid-based diets (47). Hence, the different composition of the HFDs might have influenced the basal metabolic state of skeletal muscle and, as a consequence, the response to RSV. Higashida et al. (45) speculate that the lack of effect of RSV treatment in their study could be due to the low bioavailability of the compound in blood. However, using the same dose of RSV, we and others could clearly demonstrate systemic metabolic effects in mice, metabolic adaptation of muscle as well as modulation of signaling pathways in both liver and adipose tissue. Taken together, these findings indicate that the effects of RSV on skeletal muscle metabolism and function seem to be complex and highly dependent on the specific experimental context.
In summary, we show that PGC-1α is required for the transcriptional effects of both RSV and SRT on mitochondrial gene expression in skeletal muscle, whereas muscle PGC-1α is dispensable for the systemic metabolic effects elicited by RSV and SRT, such as the enhanced metabolic rate. Importantly, treatment with RSV and SRT resulted in more prominent metabolic changes in adipose tissue and liver as compared with skeletal muscle, at least in the sedentary HFD-fed mice used in this study. It might therefore be of importance to adopt a broader, less muscle-centric view on the metabolic effects of RSV and SRT treatment, in particular in the context of a potential therapy of sedentary, obese patients. Furthermore, the comparison of short and long term administration of RSV highlights some potential caveats for the design of such a therapy; for example, increased lipogenesis in WAT of the long term RSV-treated animals could indicate that administration of this compound in patients should be time-limited. Intriguingly, despite apparently similar systemic effects of RSV and SRT, these compounds exert differential effects on key metabolic organs, including skeletal muscle, liver, WAT, and BAT (Fig. 8A). Hence, our findings reveal important insights into the specific effects of these structurally distinct compounds. Importantly, increased hepatic lipogenesis and LDL-C levels should be considered when designing SRT-based therapeutic approaches. Whereas both in vivo and in vitro experiments clearly demonstrate differential transcriptional effects of RSV and SRT treatment in hepatocytes, it is currently unclear whether this is due to activation of different molecular targets and signaling pathways or due to non-SIRT1-mediated effects of RSV (e.g. its phytoestrogenic, anti-inflammatory, or antioxidant properties). For WAT and liver, it is currently unclear whether these differences between RSV and SRT could be further caused by variable bioavailability in these tissues. In either case, it is of importance to consider the differential effects of RSV and SRT treatment for future study design and even more significantly for the use of these compounds in a clinical context.
FIGURE 8.
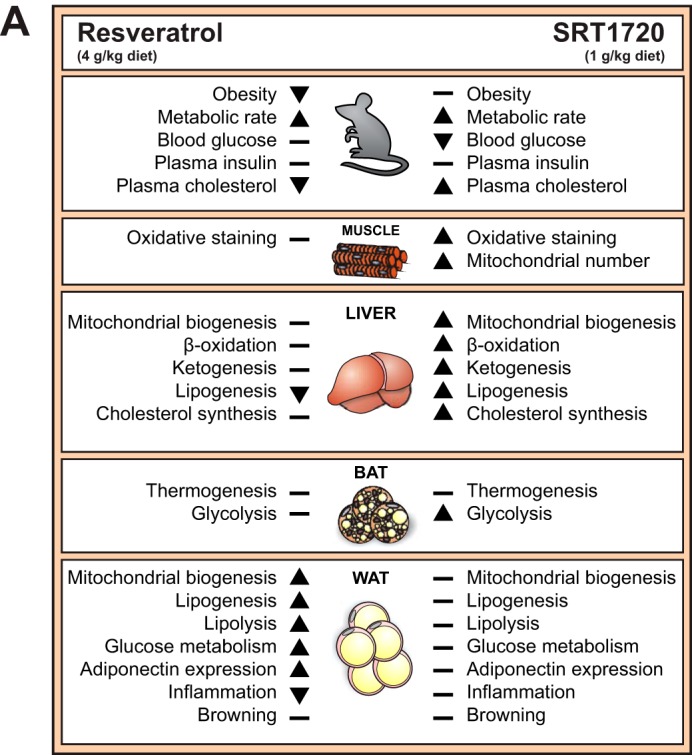
RSV and SRT elicit differential effects in key metabolic organs. A, schematic representation of the effects of resveratrol and SRT1720 on whole body level as well as relevant gene programs affected in skeletal muscle, liver, BAT, and WAT.
Supplementary Material
Acknowledgments
We thank Dr. Aimo Kannt (Sanofi-Aventis) for providing the SRT1720 compound used in this study. We also acknowledge the great technical assistance of Vesna Olivieri (Center for Microscopy (ZMB), University of Basel).
Author Contributions—K. S. and C. H. designed the research; K. S., S. S., V. A., B. C., L. Q., and L. T. performed research and analyzed data; K. S. and C. H. wrote the paper.
This project was supported by the ERC Consolidator Grant 616830-MUSCLE_NET, the Swiss National Science Foundation, SystemsX.ch, the Swiss Society for Research on Muscle Diseases (SSEM), the Neuromuscular Research Association Basel (NeRAB), the Gebert-Rüf Foundation “Rare Diseases” Program, the “Novartis Stiftung für medizinisch-biologische Forschung,” the University of Basel, and the Biozentrum. The authors declare that they have no conflicts of interest with the contents of this article.

This article contains supplemental Table 1.
- CR
- caloric restriction
- BAT
- brown adipose tissue
- CTRL
- control
- HDL-C
- high density lipoprotein cholesterol
- HFD
- high fat diet
- LDL-C
- low density lipoprotein cholesterol
- MKO
- PGC-1α muscle-knockout
- OXPHOS
- oxidative phosphorylation
- RSV
- resveratrol
- RSV-L
- long term resveratrol treatment
- RSV-S
- short term resveratrol treatment
- SRT
- SRT1720
- WAT
- white adipose tissue
- AMPK
- AMP-activated protein kinase
- SIRT1
- sirtuin 1
- PGC-1α
- peroxisome proliferator-activated receptor γ co-activator 1α
- IRβ
- insulin receptor β
- GSK3β
- glycogen synthase kinase 3β
- SKM
- skeletal muscle
- CREB
- cAMP-response element-binding protein.
References
- 1. Speakman J. R., Mitchell S. E. (2011) Caloric restriction. Mol. Aspects Med. 32, 159–221 [DOI] [PubMed] [Google Scholar]
- 2. Pearson K. J., Baur J. A., Lewis K. N., Peshkin L., Price N. L., Labinskyy N., Swindell W. R., Kamara D., Minor R. K., Perez E., Jamieson H. A., Zhang Y., Dunn S. R., Sharma K., Pleshko N., Woollett L. A., Csiszar A., Ikeno Y., Le Couteur D., Elliott P. J., Becker K. G., Navas P., Ingram D. K., Wolf N. S., Ungvari Z., Sinclair D. A., de Cabo R. (2008) Resveratrol delays age-related deterioration and mimics transcriptional aspects of dietary restriction without extending life span. Cell Metab. 8, 157–168 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Smith J. J., Kenney R. D., Gagne D. J., Frushour B. P., Ladd W., Galonek H. L., Israelian K., Song J., Razvadauskaite G., Lynch A. V., Carney D. P., Johnson R. J., Lavu S., Iffland A., Elliott P. J., Lambert P. D., Elliston K. O., Jirousek M. R., Milne J. C., Boss O. (2009) Small molecule activators of SIRT1 replicate signaling pathways triggered by calorie restriction in vivo. BMC Syst. Biol. 3, 31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Baur J. A., Pearson K. J., Price N. L., Jamieson H. A., Lerin C., Kalra A., Prabhu V. V., Allard J. S., Lopez-Lluch G., Lewis K., Pistell P. J., Poosala S., Becker K. G., Boss O., Gwinn D., Wang M., Ramaswamy S., Fishbein K. W., Spencer R. G., Lakatta E. G., Le Couteur D., Shaw R. J., Navas P., Puigserver P., Ingram D. K., de Cabo R., Sinclair D. A. (2006) Resveratrol improves health and survival of mice on a high-calorie diet. Nature 444, 337–342 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Lagouge M., Argmann C., Gerhart-Hines Z., Meziane H., Lerin C., Daussin F., Messadeq N., Milne J., Lambert P., Elliott P., Geny B., Laakso M., Puigserver P., Auwerx J. (2006) Resveratrol improves mitochondrial function and protects against metabolic disease by activating SIRT1 and PGC-1α. Cell 127, 1109–1122 [DOI] [PubMed] [Google Scholar]
- 6. Um J. H., Park S. J., Kang H., Yang S., Foretz M., McBurney M. W., Kim M. K., Viollet B., Chung J. H. (2010) AMP-activated protein kinase-deficient mice are resistant to the metabolic effects of resveratrol. Diabetes 59, 554–563 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Price N. L., Gomes A. P., Ling A. J., Duarte F. V., Martin-Montalvo A., North B. J., Agarwal B., Ye L., Ramadori G., Teodoro J. S., Hubbard B. P., Varela A. T., Davis J. G., Varamini B., Hafner A., Moaddel R., Rolo A. P., Coppari R., Palmeira C. M., de Cabo R., Baur J. A., Sinclair D. A. (2012) SIRT1 is required for AMPK activation and the beneficial effects of resveratrol on mitochondrial function. Cell Metab. 15, 675–690 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Park S. J., Ahmad F., Philp A., Baar K., Williams T., Luo H., Ke H., Rehmann H., Taussig R., Brown A. L., Kim M. K., Beaven M. A., Burgin A. B., Manganiello V., Chung J. H. (2012) Resveratrol ameliorates aging-related metabolic phenotypes by inhibiting cAMP phosphodiesterases. Cell 148, 421–433 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Baur J. A. (2010) Biochemical effects of SIRT1 activators. Biochim. Biophys. Acta 1804, 1626–1634 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Milne J. C., Lambert P. D., Schenk S., Carney D. P., Smith J. J., Gagne D. J., Jin L., Boss O., Perni R. B., Vu C. B., Bemis J. E., Xie R., Disch J. S., Ng P. Y., Nunes J. J., Lynch A. V., Yang H., Galonek H., Israelian K., Choy W., Iffland A., Lavu S., Medvedik O., Sinclair D. A., Olefsky J. M., Jirousek M. R., Elliott P. J., Westphal C. H. (2007) Small molecule activators of SIRT1 as therapeutics for the treatment of type 2 diabetes. Nature 450, 712–716 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Feige J. N., Lagouge M., Canto C., Strehle A., Houten S. M., Milne J. C., Lambert P. D., Mataki C., Elliott P. J., Auwerx J. (2008) Specific SIRT1 activation mimics low energy levels and protects against diet-induced metabolic disorders by enhancing fat oxidation. Cell Metab. 8, 347–358 [DOI] [PubMed] [Google Scholar]
- 12. Minor R. K., Baur J. A., Gomes A. P., Ward T. M., Csiszar A., Mercken E. M., Abdelmohsen K., Shin Y. K., Canto C., Scheibye-Knudsen M., Krawczyk M., Irusta P. M., Martín-Montalvo A., Hubbard B. P., Zhang Y., Lehrmann E., White A. A., Price N. L., Swindell W. R., Pearson K. J., Becker K. G., Bohr V. A., Gorospe M., Egan J. M., Talan M. I., Auwerx J., Westphal C. H., Ellis J. L., Ungvari Z., Vlasuk G. P., Elliott P. J., Sinclair D. A., de Cabo R. (2011) SRT1720 improves survival and healthspan of obese mice. Sci. Rep. 1, 70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Pacholec M., Bleasdale J. E., Chrunyk B., Cunningham D., Flynn D., Garofalo R. S., Griffith D., Griffor M., Loulakis P., Pabst B., Qiu X., Stockman B., Thanabal V., Varghese A., Ward J., Withka J., Ahn K. (2010) SRT1720, SRT2183, SRT1460, and resveratrol are not direct activators of SIRT1. J. Biol. Chem. 285, 8340–8351 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Martin-Montalvo A., de Cabo R. (2013) Mitochondrial metabolic reprogramming induced by calorie restriction. Antioxid. Redox Signal. 19, 310–320 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Herzig S., Long F., Jhala U. S., Hedrick S., Quinn R., Bauer A., Rudolph D., Schutz G., Yoon C., Puigserver P., Spiegelman B., Montminy M. (2001) CREB regulates hepatic gluconeogenesis through the coactivator PGC-1. Nature 413, 179–183 [DOI] [PubMed] [Google Scholar]
- 16. Jäger S., Handschin C., St-Pierre J., Spiegelman B. M. (2007) AMP-activated protein kinase (AMPK) action in skeletal muscle via direct phosphorylation of PGC-1α. Proc. Natl. Acad. Sci. U.S.A. 104, 12017–12022 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Rodgers J. T., Lerin C., Haas W., Gygi S. P., Spiegelman B. M., Puigserver P. (2005) Nutrient control of glucose homeostasis through a complex of PGC-1α and SIRT1. Nature 434, 113–118 [DOI] [PubMed] [Google Scholar]
- 18. Handschin C. (2009) The biology of PGC-1α and its therapeutic potential. Trends Pharmacol. Sci. 30, 322–329 [DOI] [PubMed] [Google Scholar]
- 19. Pérez-Schindler J., Summermatter S., Santos G., Zorzato F., Handschin C. (2013) The transcriptional coactivator PGC-1α is dispensable for chronic overload-induced skeletal muscle hypertrophy and metabolic remodeling. Proc. Natl. Acad. Sci. U.S.A. 110, 20314–20319 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Handschin C., Choi C. S., Chin S., Kim S., Kawamori D., Kurpad A. J., Neubauer N., Hu J., Mootha V. K., Kim Y. B., Kulkarni R. N., Shulman G. I., Spiegelman B. M. (2007) Abnormal glucose homeostasis in skeletal muscle-specific PGC-1α knockout mice reveals skeletal muscle-pancreatic beta cell cross-talk. J. Clin. Invest. 117, 3463–3474 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Townsend K. L., Tseng Y. H. (2014) Brown fat fuel utilization and thermogenesis. Trends Endocrinol. Metab. 25, 168–177 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Jang M. K., Son Y., Jung M. H. (2013) ATF3 plays a role in adipocyte hypoxia-mediated mitochondria dysfunction in obesity. Biochem. Biophys. Res. Commun. 431, 421–427 [DOI] [PubMed] [Google Scholar]
- 23. Kim H. B., Kong M., Kim T. M., Suh Y. H., Kim W. H., Lim J. H., Song J. H., Jung M. H. (2006) NFATc4 and ATF3 negatively regulate adiponectin gene expression in 3T3-L1 adipocytes. Diabetes 55, 1342–1352 [DOI] [PubMed] [Google Scholar]
- 24. Qi L., Saberi M., Zmuda E., Wang Y., Altarejos J., Zhang X., Dentin R., Hedrick S., Bandyopadhyay G., Hai T., Olefsky J., Montminy M. (2009) Adipocyte CREB promotes insulin resistance in obesity. Cell Metab. 9, 277–286 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Tilg H., Wolf A. M. (2005) Adiponectin: a key fat-derived molecule regulating inflammation. Expert Opin. Ther. Targets 9, 245–251 [DOI] [PubMed] [Google Scholar]
- 26. Horton J. D., Goldstein J. L., Brown M. S. (2002) SREBPs: activators of the complete program of cholesterol and fatty acid synthesis in the liver. J. Clin. Invest. 109, 1125–1131 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Ingram D. K., Zhu M., Mamczarz J., Zou S., Lane M. A., Roth G. S., deCabo R. (2006) Calorie restriction mimetics: an emerging research field. Aging Cell 5, 97–108 [DOI] [PubMed] [Google Scholar]
- 28. Finley L. W., Lee J., Souza A., Desquiret-Dumas V., Bullock K., Rowe G. C., Procaccio V., Clish C. B., Arany Z., Haigis M. C. (2012) Skeletal muscle transcriptional coactivator PGC-1α mediates mitochondrial, but not metabolic, changes during calorie restriction. Proc. Natl. Acad. Sci. U.S.A. 109, 2931–2936 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Kelly T. J., Lerin C., Haas W., Gygi S. P., Puigserver P. (2009) GCN5-mediated transcriptional control of the metabolic coactivator PGC-1β through lysine acetylation. J. Biol. Chem. 284, 19945–19952 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Gómez-Zorita S., Fernández-Quintela A., Macarulla M. T., Aguirre L., Hijona E., Bujanda L., Milagro F., Martínez J. A., Portillo M. P. (2012) Resveratrol attenuates steatosis in obese Zucker rats by decreasing fatty acid availability and reducing oxidative stress. Br. J. Nutr. 107, 202–210 [DOI] [PubMed] [Google Scholar]
- 31. Jin S. H., Yang J. H., Shin B. Y., Seo K., Shin S. M., Cho I. J., Ki S. H. (2013) Resveratrol inhibits LXRα-dependent hepatic lipogenesis through novel antioxidant Sestrin2 gene induction. Toxicol. Appl. Pharmacol. 271, 95–105 [DOI] [PubMed] [Google Scholar]
- 32. Yamazaki Y., Usui I., Kanatani Y., Matsuya Y., Tsuneyama K., Fujisaka S., Bukhari A., Suzuki H., Senda S., Imanishi S., Hirata K., Ishiki M., Hayashi R., Urakaze M., Nemoto H., Kobayashi M., Tobe K. (2009) Treatment with SRT1720, a SIRT1 activator, ameliorates fatty liver with reduced expression of lipogenic enzymes in MSG mice. Am. J. Physiol. Endocrinol. Metab. 297, E1179–E1186 [DOI] [PubMed] [Google Scholar]
- 33. Qiang L., Lin H. V., Kim-Muller J. Y., Welch C. L., Gu W., Accili D. (2011) Proatherogenic abnormalities of lipid metabolism in SirT1 transgenic mice are mediated through Creb deacetylation. Cell Metab. 14, 758–767 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Bhatt J. K., Thomas S., Nanjan M. J. (2012) Resveratrol supplementation improves glycemic control in type 2 diabetes mellitus. Nutr. Res. 32, 537–541 [DOI] [PubMed] [Google Scholar]
- 35. Timmers S., Konings E., Bilet L., Houtkooper R. H., van de Weijer T., Goossens G. H., Hoeks J., van der Krieken S., Ryu D., Kersten S., Moonen-Kornips E., Hesselink M. K., Kunz I., Schrauwen-Hinderling V. B., Blaak E. E., Auwerx J., Schrauwen P. (2011) Calorie restriction-like effects of 30 days of resveratrol supplementation on energy metabolism and metabolic profile in obese humans. Cell Metab. 14, 612–622 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Tomé-Carneiro J., Gonzálvez M., Larrosa M., Garcia-Almagro F. J., Avilés-Plaza F., Parra S., Yáñez-Gascón M. J., Ruiz-Ros J. A., García-Conesa M. T., Tomás-Barberán F. A., Espín J. C. (2012) Consumption of a grape extract supplement containing resveratrol decreases oxidized LDL and ApoB in patients undergoing primary prevention of cardiovascular disease: a triple-blind, 6-month follow-up, placebo-controlled, randomized trial. Mol. Nutr. Food Res. 56, 810–821 [DOI] [PubMed] [Google Scholar]
- 37. Beaudoin M. S., Snook L. A., Arkell A. M., Simpson J. A., Holloway G. P., Wright D. C. (2013) Resveratrol supplementation improves white adipose tissue function in a depot-specific manner in Zucker diabetic fatty rats. Am. J. Physiol. Regul. Integr. Comp. Physiol. 305, R542–R551 [DOI] [PubMed] [Google Scholar]
- 38. Gómez-Zorita S., Fernández-Quintela A., Lasa A., Hijona E., Bujanda L., Portillo M. P. (2013) Effects of resveratrol on obesity-related inflammation markers in adipose tissue of genetically obese rats. Nutrition 29, 1374–1380 [DOI] [PubMed] [Google Scholar]
- 39. Rivera L., Morón R., Zarzuelo A., Galisteo M. (2009) Long-term resveratrol administration reduces metabolic disturbances and lowers blood pressure in obese Zucker rats. Biochem. Pharmacol. 77, 1053–1063 [DOI] [PubMed] [Google Scholar]
- 40. Jimenez-Gomez Y., Mattison J. A., Pearson K. J., Martin-Montalvo A., Palacios H. H., Sossong A. M., Ward T. M., Younts C. M., Lewis K., Allard J. S., Longo D. L., Belman J. P., Malagon M. M., Navas P., Sanghvi M., Moaddel R., Tilmont E. M., Herbert R. L., Morrell C. H., Egan J. M., Baur J. A., Ferrucci L., Bogan J. S., Bernier M., de Cabo R. (2013) Resveratrol improves adipose insulin signaling and reduces the inflammatory response in adipose tissue of rhesus monkeys on high-fat, high-sugar diet. Cell Metab. 18, 533–545 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Konings E., Timmers S., Boekschoten M. V., Goossens G. H., Jocken J. W., Afman L. A., Muller M., Schrauwen P., Mariman E. C., Blaak E. E. (2013) The effects of 30 days resveratrol supplementation on adipose tissue morphology and gene expression patterns in obese men. Int. J. Obes. (Lond.) 38, 470–473 [DOI] [PubMed] [Google Scholar]
- 42. Hector K. L., Lagisz M., Nakagawa S. (2012) The effect of resveratrol on longevity across species: a meta-analysis. Biol. Lett. 8, 790–793 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Miller R. A., Harrison D. E., Astle C. M., Baur J. A., Boyd A. R., de Cabo R., Fernandez E., Flurkey K., Javors M. A., Nelson J. F., Orihuela C. J., Pletcher S., Sharp Z. D., Sinclair D., Starnes J. W., Wilkinson J. E., Nadon N. L., Strong R. (2011) Rapamycin, but not resveratrol or simvastatin, extends life span of genetically heterogeneous mice. J. Gerontol. A Biol. Sci. Med. Sci. 66, 191–201 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Kulkarni S. S., Cantó C. (2015) The molecular targets of resveratrol. Biochim. Biophys. Acta 1852, 1114–1123 [DOI] [PubMed] [Google Scholar]
- 45. Higashida K., Kim S. H., Jung S. R., Asaka M., Holloszy J. O., Han D. H. (2013) Effects of resveratrol and SIRT1 on PGC-1α activity and mitochondrial biogenesis: a reevaluation. PLoS Biol. 11, e1001603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Ringholm S., Olesen J., Pedersen J. T., Brandt C. T., Halling J. F., Hellsten Y., Prats C., Pilegaard H. (2013) Effect of lifelong resveratrol supplementation and exercise training on skeletal muscle oxidative capacity in aging mice; impact of PGC-1α. Exp. Gerontol. 48, 1311–1318 [DOI] [PubMed] [Google Scholar]
- 47. Montgomery M. K., Osborne B., Brown S. H., Small L., Mitchell T. W., Cooney G. J., Turner N. (2013) Contrasting metabolic effects of medium- versus long-chain fatty acids in skeletal muscle. J. Lipid Res. 54, 3322–3333 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Turner N., Hariharan K., TidAng J., Frangioudakis G., Beale S. M., Wright L. E., Zeng X. Y., Leslie S. J., Li J. Y., Kraegen E. W., Cooney G. J., Ye J. M. (2009) Enhancement of muscle mitochondrial oxidative capacity and alterations in insulin action are lipid species dependent: potent tissue-specific effects of medium-chain fatty acids. Diabetes 58, 2547–2554 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.