

Abstract
Motivation: The study of RNA virus populations is a challenging task. Each population of RNA virus is composed of a collection of different, yet related genomes often referred to as mutant spectra or quasispecies. Virologists using deep sequencing technologies face major obstacles when studying virus population dynamics, both experimentally and in natural settings due to the relatively high error rates of these technologies and the lack of high performance pipelines. In order to overcome these hurdles we developed a computational pipeline, termed ViVan (Viral Variance Analysis). ViVan is a complete pipeline facilitating the identification, characterization and comparison of sequence variance in deep sequenced virus populations.
Results: Applying ViVan on deep sequenced data obtained from samples that were previously characterized by more classical approaches, we uncovered novel and potentially crucial aspects of virus populations. With our experimental work, we illustrate how ViVan can be used for studies ranging from the more practical, detection of resistant mutations and effects of antiviral treatments, to the more theoretical temporal characterization of the population in evolutionary studies.
Availability and implementation: Freely available on the web at http://www.vivanbioinfo.org
Contact: nshomron@post.tau.ac.il
Supplementary information: Supplementary data are available at Bioinformatics online.
1 Introduction
The study of RNA virus populations is a challenging task. Each population of RNA virus is composed of a collection of different, yet related genomes often referred to as mutant spectra or quasispecies (Lauring and Andino, 2010). This genotypic diversity is the result of the high mutation rate of RNA viruses, which surpasses that of DNA organisms by orders of magnitude (Sanjuán et al., 2010). This intrinsic high error-rate is largely attributed to the viral RNA dependent RNA polymerases that replicate their genomes (Steinhauer and Holland, 1987). Recent studies demonstrate that this high-error rate is essential for viral adaptation and survival (Ahlquist, 2002; Crotty et al., 2001) and can have significant implications in vaccine and antiviral efficacy (Love et al., 2010; Woo and Reifman, 2012). Changes in the rate at which these errors occur may affect virus infectivity, fitness and pathogenesis (Coffey et al., 2011; Crotty and Andino, 2002; Gnädig et al., 2012; Graham et al., 2012). Moreover, it was shown that the diversity within an RNA virus population enhances tissue tropism and dissemination, possibly by improving adaptability and mutual support (Vignuzzi et al., 2005). These studies suggest that characterizing RNA virus populations as a whole, rather than focusing on dominant viral haplotypes (e.g. sequences shared by a significant amount of members in the population) or consensus sequence is far more informative to study pathogenesis. Currently, RNA viruses are genetically characterized through a variety of techniques. The most basic and common method involves classic Sanger sequencing of cDNA produced by RT-PCR of total viral RNA (i.e. consensus sequencing). Although informative, the consensus sequence is the genetic average of all variants within the virus population and the limitations of this method include its low resolution and ability to detect only highly abundant variants. To increase sensitivity and obtain a small, representative sample of viral variants in a given population by Sanger sequencing, individual viruses can be isolated and amplified (biological clones) (Vignuzzi et al., 2005) or RT-PCR amplicons of the whole population can be subcloned into individual plasmids (molecular clones) (Levi et al., 2010; Sanjuán et al., 2010). An additional method is Single Genome Amplification in which viral RNA is extracted from a sample and copied into cDNA, which in turn is subjected to limiting dilution and PCR amplification. Thus the obtained PCR products are the result of the amplification of one single molecule of cDNA. These PCR products are then sequenced directly without cloning. Nevertheless, these three methods are time intensive and tedious. Furthermore, the limited coverage that can be sampled (generally <100 clones and 100 K nucleotides per population) is far from the range of variants (105–109) typically found in virus samples. Newer high-throughput sequencing (HTS) technologies have expedited research in microbiology in general, including virology, due to their high throughput sequence data production (Capobianchi et al., 2013; Didelot et al., 2012; Radford et al., 2012; Shomron, 2013). The ultra-deep coverage afforded by HTS technologies provides obvious improvements to the characterization of RNA viruses. However, most HTS tools in virology are designed for virus discovery, de novo assembly of unknown viral genomes, and the characterization of viral biodiversity found in different organs, organisms or environments (virome) (Delwart, 2013; Foulongne et al., 2012; Hurwitz and Sullivan, 2013; Roux et al., 2012; Yin et al., 2012). More recently, researchers have begun characterizing the intra-host and intra-strain virus diversity and population dynamics for a variety of viruses of clinical or agricultural relevance. For example, some studies addressed the temporal evolution of variants, the pre-existing presence of drug resistant mutants and the dynamics and emergence of escape mutations in the presence of various types of pressure (Archer et al., 2012b; Barzon et al., 2011; Beerenwinkel and Zagordi, 2011; Bull et al., 2011; Escobar-Gutiérrez et al., 2012; Grad et al., 2014; Love et al., 2010; Martínez et al., 2012; Selleri et al., 2012; Töpfer et al., 2013; Willerth et al., 2010; Wright et al., 2011). However, virologists using HTS technologies for the purpose of rare variant detection, face major obstacles when studying virus population dynamics, both experimentally and in natural settings due to the relatively high error rates of these technologies (Archer et al., 2012a; Watson et al., 2013; Wright et al., 2011). In order to overcome these hurdles several methods have been proposed (Acevedo et al., 2013; Eriksson et al., 2008; Flaherty et al., 2012; Ghedin et al., 2012; Jabara et al., 2011; Kinde et al., 2011; Macalalad et al., 2012; Mangul et al., 2014; Schmitt et al., 2012; Watson et al., 2013; Wilm et al., 2012; Wright et al., 2011; Wu et al., 2014; Zagordi et al., 2011). These methods can be split into two main categories. The first increases variant detection fidelity by modulating the library preparation step. Schmitt et al. tag and compare both strands of a DNA segment, efficiently pinpointing strand-discordant variants as errors. Jabara et al., Kinde et al., Mangul et al. and Wu et al. use unique identifiers for each sequenced template, facilitating the identification of PCR and sequencing introduced errors and biases. Acevedo et al. used circularized genomic RNA fragments to generate tandem repeats which originate from a single individual within the viral population. Variants that appear in every copy of a set of repeats are then considered true variants. The second category includes methods that utilize post-sequencing parameters in order to identify PCR and sequencing introduced errors. Ghedin et al. utilize the calculated overall baseline error rates while Flaherty et al. and Wilm et al. calculate these rates per sequenced base. Watson et al. apply a two step quality based filtration prior to variant calling and Macalalad et al. used covariation (i.e. phasing) between variants and an expectation maximization-based quality recalibration. Methods which aim to identify the genomes of individual haplotypes in the population (Eriksson et al., 2008; Zagordi et al., 2011) utilize clustering of overlapping reads and haplotype reconstruction in order to correct for sequencing errors. The performance of these methods depends upon the length of sequence reads with long reads enabling the reconstruction of entire viral genome sequences. Some methods (Ghedin et al., 2012; McElroy et al., 2013; Wilm et al., 2012) also include an additional test for strand bias (Guo et al., 2012) in order to further reduce false positive calls. Although these second category methods demonstrate very high specificity (positions without variance are correctly identified as such), sensitivity (pinpointing true variant positions) remains the major limiting factor (Wilm et al., 2012). All of the aforementioned tools focus on accurate variant detection and do not facilitate any downstream analysis and interpretation of the detected variants. Moreover, although the end-users for the aforementioned tools and methods are, for the most part, virologists studying highly diverse viral populations, these tools require the use of unix command line and other computational proficiencies which are not common in the field.
In order to better integrate and utilize HTS technology in viral population studies, we developed a computational pipeline and web server, termed ViVan (Viral Variance Analysis). ViVan is a complete pipeline to facilitate the identification, characterization and comparison of sequence variance in deep sequenced virus populations (Fig. 1). ViVan performs per-sample allele rate analysis, translates the detected changes into amino acid changes, compares and outputs several informative metrics regarding each analyzed sample. Applying ViVan on deep sequenced data obtained from samples that were previously characterized by more classical approaches, we achieved superior sensitivity and uncovered novel and potentially crucial aspects of virus populations, which could not have been identified otherwise and in much shorter time scales. ViVan efficiently identified low-frequency minority variants across the entire viral genome and changes in population diversity that correlate with in vitro and in vivo data of previously described mutator and antimutator strains of RNA viruses. Additionally, it accurately determined dosage-dependent and drug-specific alterations in mutation frequency associated with mutagenic, antiviral compounds. More generally, we monitored temporal changes occurring within RNA virus populations during experimental evolution and pinpointed unique variable positions in viral genomes found in specific host environments that are indicative of positive selection and adaptation. Overall, we show that ViVan allows rapid characterization of the genetic composition and population dynamics of rapidly evolving viral mutant spectra.
Fig. 1.

Schematic of ViVan pipeline workflow. The analysis starts with raw sequence reads output by deep sequencing of a virus population sample. First, these raw reads undergo quality trimming where low quality bases are removed from both ends of the read. Second, these quality reads are aligned against a user-supplied reference sequence and a pileup is produced for each position. The pileup output is then analyzed, true variants are identified, variant frequencies are modified and confidence intervals calculated. From these modified significant variants, an assortment of variation metrics is produced, including information regarding the predicted amino acid change in each protein, the variation rates across the viral genome, transition/transversion rates and specific nucleotide change tables. Additionally, once variant frequencies have been calculated, a consensus sequence is produced, utilizing the major allele in each position. This modified consensus sequence can then be used for the alignment of the initial quality reads, hence improving overall alignment and accuracy. Once the analysis is done for each virus sequence sample, a comparison is performed between groups of samples in order to pinpoint both common and unique variants in each group
2 Methods
2.1 Performance validation
Validation data-sets were generated and published by Wilm et al. (2012). LoFreq (Wilm et al., 2012) version 0.6.1 was run on the data using default parameters. Variants with a P-value lower than 0.05 were marked as positive calls, regardless of the strand-bias P-value. VPhaser2 (Yang et al.), was ran on the data using default parameters. In order to maximize sensitivity, all the variants found in the raw output file (without strand-bias or FDR corrections) were considered as positive. In the first data set, Pileup was included in the comparison and every detected non-reference allele was considered as a true variant.
2.2 Reproducibility confirmation
In order to test the hypothesis that the frequencies of variants detected only by one replicate (non-reproducible variant; NRV) are higher than expected in the other un-detected samples, for each NRV we counted the number of times its frequency is higher than the other non-reference, non-variant alleles in each replicate. If the NRV frequency is found to be higher in >3 of the replicates (P < 0.05) we determine false negative as the reason of non-reproducibility.
2.3 Measuring diversity of viral fidelity variants
The production and generation of DS libraries of passage 3 low fidelity variants is described previously (Gnädig et al., 2012). The raw data were reanalyzed for this article. (For additional details see Supplementary Data 3).
2.4 Statistical methods
Correlation significance between samples was calculated using Pearson’s correlation. An unpaired Student t-test was used for comparison between samples (temporal accumulation of variations).
2.5 ViVan pipeline
Our ViVan pipeline is composed of both established HTS data analysis tools and in-house Python scripts. The entire process, from raw sequencing data to analysis output can be divided into several steps: (i) quality control, (ii) alignment and Pileup, (iii) variant frequency collection and filtering, (iv) variant annotation, (v) per-sample metrics and supplements and (vi) group comparison. (For additional details see Supplementary Data 4)
2.6 Variant sites identification with allele frequency and confidence interval estimation from deep sequence data
We have a sample with minor allele frequency (MAF) f at a genomic locus of interest. The sequencer generates reads covering that locus. The ith read is denoted yi and is either 0 or 1, where 0 is a non-mutant allele and 1 is the mutant allele. Each read is associated with a quality score pi indicating the probability that it is incorrect. The probability of observing a minor allele is given by:
and the probability of observing a reference allele is:
Because each yi is in fact a Bernoulli variable, we can write down the log of the likelihood function as a sum of log-likelihood functions:
The optimum of the log-likelihood function has no closed form expression, but the function can be effectively optimized numerically to find its maximizer, denoted , which is the maximum likelihood estimator of the MAF f in the sample. We can then construct a 1 − α confidence set using Wilks’ theorem:
where is the 1 − α percentile of the chi-square distribution with one degree of freedom. This confidence set is consistent when the estimator is not on the boundary, and conservative when it is.
A P-value can be similarly derived using Wilks’ theorem:
where is the cumulative distribution function of the chi-square distribution with one degree of freedom.
This analysis is performed for every non-reference allele in every position with sufficient coverage (set at the start of the analysis). The variation rate at position i is defined as the proportion (F) of significant non-reference alleles (k) and is denoted Vi
The region-wide variation rate is the averaged variation rate across all covered positions in the region (denoted n).
3 Results
3.1 Performance validation
Minority variants have been shown to be associated with various viral features, such as viral evolution, pathogenesis and the outcome of drug treatment (Nájera et al., 1995; Vignuzzi et al., 2005). To determine how accurately our method detects low-frequency variants in a viral population, and to demonstrate how it compares with currently available methods, we utilized published data generated by Wilm et al. (2012). We selected three data sets: (i) simulated dengue virus (DENV) population composed of 10 different in-silico generated haplotypes in varying rates (0.1–50%) and varying coverage levels (50×, 100×, 500×, 1000×, 5000×, 10 000×), (ii) simulated DENV population using six sequenced clinical samples sub-sampled at various rates. (iii) six library replicates of DENV2 TSV01 viruses. On each data set, we compared ViVan’s performance with LoFreq (Wilm et al., 2012) and VPhaser2 (Yang et al., 2013). As the number of true negatives (non-variant alleles) greatly surpasses the number of true positives (resulting in high specificity values across methods), we chose to focus on positive predictive values (PPVs; Rate of true variants out of the total identified variants) instead of specificity for performance assessment. The first data set was used to test performance as a function of sequencing coverage. In this data set, we also tested SAMtools pileup’s (Li et al., 2009) performance in order to mark the upper limit of sensitivity and to demonstrate the importance of variant significance testing. We demonstrated that ViVan presents a significantly higher sensitivity across coverage levels compared with LoFreq and VPhaser2 (P < 0.005; Fig. 2A and B), identifying 90 ± 5% of the variants with frequencies <0.2% (64 and 0% in LoFreq and VPhaser2, respectively). The PPV cost of such an increase in sensitivity was found to be minor and significant only in low coverage levels (<500×; P < 0.01) with a PPV of 0.968 ± 0.028 (specificity of 0.99995 ± 0.00004) in 50× coverage. Calling variants using pileup, resulted in the highest sensitivity across coverage levels, but with a much higher cost in PPV. pileup’s PPV ranged from 0.376 ± 0.022 in 50× to 0.066 ± 0.00003 in 10 000× (93.4% of detected variants are false). This emphasizes the need for computational variant assessment methods such as those employed by ViVan, LoFreq and VPhaser2 in order to greatly reduce the number of false positive calls. The second data set, which includes real sequencing data, was used to demonstrate ViVan’s performance in the context of coverage and quality biases (Fig. 2C). In this set, variants identified in positions that differ from the reference in any of the six virus strains composing the simulated population were considered true variants (Table 1.)
Fig. 2.

Performance validation of ViVan using data sets compiled by Wilm et al. (A,B) Simulated sequencing of an in-silico generated virus population with varying coverage levels. Performance is demonstrated using sensitivity and the F1-score which incorporates both sensitivity and PPV (2*(Sensitivity*PPV)/(Sensitivity + PPV)). As coverage increases, low frequency variants are detected and sensitivity levels rise across methods. ViVan demonstrates high sensitivity and specificity across coverage levels with only a minor cost in PPV, maintaining an overall F1-score higher than the other methods. SAMtools pileup’s decline in F1-score is the result of a decrease in PPV as coverage increases. (C) Simulated virus population, using sub-sampling of clinical samples with known variant locations. Comparing true positive variant calls between methods, ViVan demonstrated the highest sensitivity, identifying all but one of the variants detected by the other methods, and detecting 24 additional low-rate variants. (D) Reproducibility analysis using six libraries replicates of the same virus population. Reproducibility was defined as % of variants detected in more than one replicate. ViVan demonstrates high reproducibility in the higher frequency levels (>0.5%) which decreases as frequencies drop. This decrease in reproducibility is due to detection of extremely low frequency variants by ViVan only in some of the replicates and may be alleviated by sufficient coverage
Table 1.
Performance comparison on simulated virus population:
| ViVan | LoFreq | VPhaser2 | SAMTools pileup | |
|---|---|---|---|---|
| True positives | 231 | 219 | 196 | 244 |
| False positives | 4 | 1 | 2 | 1399 |
| True negatives | 31918 | 31921 | 31920 | 30523 |
| False negatives | 13 | 25 | 48 | 0 |
| sensitivity | 0.947 | 0.898 | 0.803 | 1.000 |
| specificity | 1.000 | 1.000 | 1.000 | 0.956 |
| PPV | 0.983 | 0.995 | 0.990 | 0.149 |
| NPV | 0.999 | 0.998 | 0.995 | 1.000 |
| F1-Score | 0.965 | 0.944 | 0.887 | 0.259 |
Simulated DENV population using six sequenced clinical samples sub-sampled at various rates was used to demonstrate ViVan’s performance in the context of real sequencing data
Because the coverage in this data set was low (100×) ViVan’s PPV was the lowest. However, ViVan demonstrated the highest sensitivity out of the three methods, identifying all but one of the variants found in the other. This dataset also highlights the need for sequencing-error aware methods in order to reduce false positive calls such as the ones produced by naïve pileup.
Because the coverage in this data set was low (100×) ViVan’s PPV was the lowest (0.983, 0.995 and 0.99 for ViVan, LoFreq and VPhaser2, respectively). However, ViVan demonstrated the highest sensitivity out of the three methods (0.947, 0.828 and 0.803 for ViVan, LoFreq and VPhaser2, respectively), identifying all but one of the variants found in the other methods (n = 208), and detecting 24 additional true, low-frequency variants at the cost of four false positive variants. As we demonstrate using the first data-set, a coverage increase should compare ViVan’s PPV to the other methods’ while maintaining higher sensitivity.
The third and final data set, composed of six technical replicates of the same sequenced DENV population, was used to test ViVan’s reproducibility. Reproducibility was calculated as the percentage of variants found in more than one replicate, out of the total number of detected variants. When limiting the minimal variant rate for detection, we see that ViVan demonstrates the highest reproducibility (>91.7%) among methods in variants with frequencies >0.5%. Lower variant frequencies thresholds resulted in lower reproducibility across tools, with ViVan detecting the highest number of variants but at the cost of lower reproducibility (Fig. 2D). We suspected that this decrease in reproducibility is due to coverage limitation and in fact represents false negative calls in extreme low frequency variants in the disagreeing replicates in oppose to false positive calls in the replicate with detected variant. We therefore reviewed the allele frequencies in the variants not reproduced and found that the majority of them (93/133) did demonstrate a higher frequency than expected in the disagreeing replicates (P < 0.05; For more details see Methods section), suggesting that they are in fact true variants. Simply put, ViVan managed to detect extreme low coverage variants in some of the replicates, which, given sufficient coverage, would have been reproduced in the other samples as well. This demonstrates that ViVan maintains high reproducibility (88.4%) even in low variant rates, given sufficient coverage (Fig. 2D).
3.2 Identifying differences in population diversity and mutagen effects
Although HTS could be a less labor-intensive approach to quantifying diversity than more classic methods involving Sanger sequencing or biochemical incorporation assays, the relatively high error rate of the technology might prove to be too great to distinguish between subtle differences in the genetic diversity of populations. To test whether ViVan can detect such differences, we deep sequenced wild type CVB3 virus and two well-characterized low fidelity mutator strains, I230F and F232Y, that were shown by classical assays to generate 3-fold more mutations than wild type (Gnädig et al., 2012). By molecular cloning, wild type virus was found to have a diversity measure of 0.00043 mutations/nucleotide, while the low fidelity variants presented roughly 3-fold more (0.00123 mutations/nucleotide each) (Fig. 3a). When all minority variants identified by ViVan were taken into account, regardless of their frequency (no cut-off threshold), the diversity of all three virus populations were significantly higher. Wild type virus, in particular, had higher than expected frequencies of 0.00107 mutations/nucleotide in the region previously sequenced by Gnädig et al., and 0.000996 mutations/nucleotide across the entire genome. These data suggest that the higher sensitivity of HTS identifies more extremely low-frequency variants than would otherwise be detected by other available methods. Indeed, when cut-off thresholds of 0.005 or 0.01 were set, ViVan generated more conservative mutation profiles that more closely resembled previously reported values for wild type and low fidelity variants obtained by molecular clone sequencing(Gnädig et al., 2012; Levi et al., 2010). Being able to identify subtle changes in population diversity is important for RNA viruses, because the naturally high mutation frequencies of these viruses confer susceptibility to conditions that further increase mutation rates above a maximal threshold. This increased mutation rate may result in extinction, also termed lethal mutagenesis (Bull et al., 2007; Vignuzzi et al., 2005).
Fig. 3.
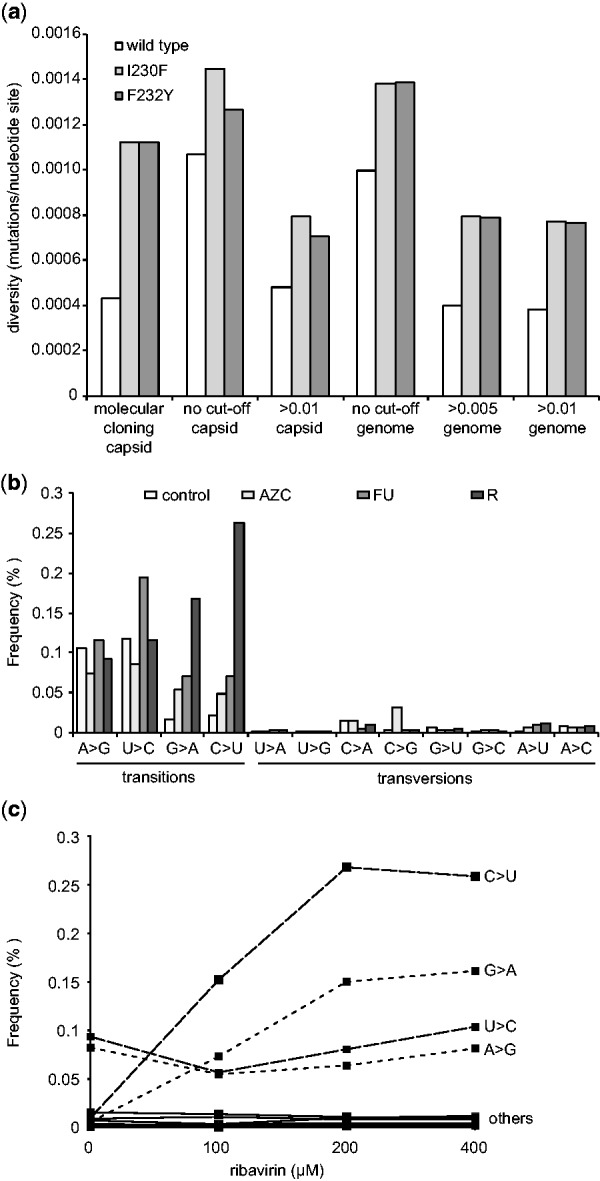
Monitoring population diversity and mutational profiles. (a) The diversity (mutations/nucleotide site) of wild type CBV3 and low fidelity variants I230F and F232Y as previously determined by molecular clone sequencing of a capsid coding region (Gnädig et al), were determined by deep sequencing across the same region (capsid) or across thewhole genome (genome), setting a minimal rate threshold (>0.005 or >0.01) or without a threshold (no cut-off) (b) Transition and transversion biases resulting from mutagen drug treatment. HeLa cells were infected with CVB3 and treated with the mutagens, 5-AZC, 5-FU or ribavirin (R) or left untreated (control). The frequency (% of total virus population) presenting specific transitions and transversions in deep sequence data were classified by ViVan. (c) Dose-dependent effects of mutagen treatment detected by deep sequencing. HeLa cells were infected with CVB3 with increasing concentrations of ribavirin (0, 100, 200 and 400 µM) and the dose dependent increase in the frequency (% of total virus population) of specific transition (C > U, G > A, U > C, A > G; dashed lines) and transversion (labeled ‘others’; solid lines) were determined by ViVan
Chemical agents inducing mutations, i.e. mutagens, are being explored as possible antiviral agents (Crotty and Andino, 2002; Graci and Cameron, 2008). An improved characterization of the effect of RNA mutagens and the genetic modulations they induce may facilitate their development as antiviral drugs. With this in mind, we used ViVan to compare virus progeny from cells infected with CVB3 and treated with known mutagens, with an untreated control infection. The drugs tested were ribavirin (R, 400 µM), 5-azacytidine (AZC, 300 µM) and 5-fluorouracil (FU, 150 µM). Each base analog is known to induce different types of mutations, with biases that can be considered genetic signatures of treatment: ribavirin promotes G to A and C to U transitions (Crotty et al., 2000), 5-AZC promotes G to A transitions and G to C, C to G and C to A transversions, and 5-FU promotes U to C and A to G transitions (Grande-Pérez et al., 2002) (Fig. 3b). Using our pipeline to quantify the mutational bias across the entire viral genome, the untreated sample revealed that transition mutations are more common than transversions, as expected, and that the most commonly occurring transition mutations were A to G and U to C, as previously reported (Levi et al., 2010). For treatment with AZC, a significant increase in G to A transitions and in the three transversions, particularly for C to G, was observed. The deep sequence profile for FU treatment revealed the most commonly induced mutations to be the expected U to C and A to G transitions. Finally, the ribavirin treated population also presents the expected G to A and C to U transitions with the highest frequencies. Another aspect of mutagenic drug treatment that can be readily demonstrated by ViVan is dosage-dependence, characterization of which would normally require classic sequencing of hundreds of molecular clones (Levi et al., 2010). We compared a CVB3 sample without treatment, with infected samples treated with three concentrations of ribavirin (100, 200 and 400 µM). The results confirmed a dose-dependent increase in the ribavirin-associated transition mutations (C to U and G to A) compared with the other transition (A to G and U to C) and transversion mutations, which showed no dose dependent effect (Fig. 3c).
3.3 Comparing between samples in evolutionary studies: identifying population-specific acquired mutations that correlate with phenotypic differences
For biologists interested in RNA virus evolution, a primary goal is to identify genotypes and mutations undergoing positive selection that would represent adaptive mutations to the experimental conditions, requiring comparison between numerous samples and controls. Precise comparison of viral population composition is difficult to carry out using common technologies as it requires either an immense amount of whole-genome sequencing of individuals from each population to be compared, or prior knowledge regarding the expected gene or allele targeted by selection. In order to demonstrate ViVan’s efficiency in pinpointing these functional mutational hotspots in viral populations, we analyzed deep sequence data of samples produced in a previously published experimental evolution study (Coffey and Vignuzzi, 2011). In this work, wild type chikungunya virus that normally cycles between mosquito and mammalian hosts, was adapted to either only mammalian host cells, or cycled between mammalian and mosquito cells (Coffey and Vignuzzi, 2011). We Applied ViVan on three sequenced samples: the initial virus population (stock virus); following seven passages in HeLa cells (p7 HeLa); and after alternating passages between HeLa cells and mosquito C6/36 cells (p7 alt). In the original study using molecular cloning data (Coffey and Vignuzzi, 2011), mammalian-host adapted virus presented the highest amount of genetic diversity compared with the initial starting population, while mammalian-mosquito passage had an intermediate diversity. Analysis with ViVan corroborated these observations. Setting a diversity threshold of >0.005 we found that the stock virus had a total of 305 minority variants. This diversity increased to 443 after serial passage in HeLa cells, and 419 when alternating between HeLa and C6 cells. In the original article, the presence of a more or less diverse set of neutralizing antibody-resistant variants was used as a surrogate for overall genetic diversity that could not be measured by classic sequencing. With our new approach, we were able to ascertain the variant rate across the entire E2 sequence containing these epitopes, detecting variants in extreme low rates. In correlation with the measured neutralizing antibody escape in the original studies, we noticed a significantly higher total variance in the p7 HeLa mammalian cell passaged E2 protein (1.6603) when compared with the mammalian-insect alternating passage (1.4262) (one tailed t-test, P = 0.00004) and the starting stock virus (1.3587) (one tailed t-test, P = 0.00018). Another powerful feature of the ViVan pipeline, is the ease with which it allows comparing and identifying minority variants in one group of samples that are common to that group and not found in a second group of samples. In analyzing this batch of samples, we assessed all the positions across the viral genome in which there was a significant variant in the alternating passage sample that had cycled through mosquito cells, which was not found in any of the other samples (that encountered only mammalian cells) above a rate of 0.001. We detected 396 such variants (Table 2 and Supplementary Data 1). Interestingly, only nine variants had frequencies above 1% of the total population, and the variant exhibiting the highest rate (62%), was an aspargine to lysine change, position 112 of the non-structural protein 4 (nsp4). It is tempting to speculate that this mutation in the nsp4 polymerase may represent a mechanism of adaptation to retain replicative capacity in disparate hosts.
Table 2.
Chikungunya alternating passages unique mutations
| Feature (gene) | Genome position | Reference allele | Read coverage | Variant allele | Amino acid position | Amino acid change | Variant rate |
|---|---|---|---|---|---|---|---|
| nsp4 | 6001 | C | 233 902 | A | 112 | N > K | 0.62075 |
| E1 | 10380 | A | 116 688 | C | 129 | synonymous | 0.18479 |
| nsp4 | 5950 | C | 112 016 | U | 95 | synonymous | 0.07434 |
| nsp4 | 6843 | A | 82 162 | G | 393 | D > G | 0.02060 |
| nsp3 | 4919 | A | 172 536 | C | 282 | synonymous | 0.01508 |
| E2 | 9701 | G | 294 074 | C | 387 | G > A | 0.01431 |
| nsp4 | 5910 | G | 95 074 | U | 82 | R > I | 0.01418 |
| nsp2 | 2234 | U | 77 920 | C | 185 | F > L | 0.01384 |
| 6K | 9967 | A | 179 958 | U | 53 | S > C | 0.01082 |
Using our pipeline, we were able to collect all the positions carrying any variant allele with a frequency >0.1% from every sequenced sample (the starting virus population (passage 1), eight cycles in Hela cells and alternating passages between Hela cells and mosquito C6 cells) and pinpoint unique positions across the viral genome in which variants were found only in the alternating passages positions. One of these variant positions, found in the viral polymerase protein (nsp4) demonstrated a high variant allele rate which may suggest functional relevance for survival in alternating host passages.
3.4 Comparing between samples in evolutionary studies: monitoring temporal changes and emerging variants during virus evolution
As RNA virus populations adapt to an environment, new mutations will amplify and eventually dominate the population to change the consensus sequence if the selective pressures remain constant over the long-term. However, with HTS technology the emergence of such mutations would expectedly be detectable long before dominating the viral population, a significant improvement to both experimental and clinical studies. As an example, the detection of low-frequency adaptive variants in an RNA viral population prior to and during antiretroviral drug treatment could result in more optimized HIV treatment protocols (Nájera et al., 1995). To validate ViVan in characterizing genome-wide temporal changes during virus evolution, we sequenced samples infected by three different coxsackie virus variants [WT, the A372V high fidelity, antimutator strain and the S299T low fidelity, mutator strain (Levi et al., 2010)] at three different time points (1, 60 and 120 replication cycles in HeLa cell culture), with each series performed as biologically independent triplicates. As with any RNA virus, we expected rapid generation of mutations throughout the viral population. We analyzed the variant rate for each sample and performed group comparisons based on time point and virus strain. The results demonstrated a significantly higher variation rates in the later passages, when comparing passage 1 against both passage 60 and 120, and comparing passage 60 against passage 120 (one tailed t-test, maximal P = 0.00016). This significant accumulation of mutations across viral genome was observed in all three coxsackie virus strains tested (Fig. 4) and is indicative of the temporal accumulation of variants in the sequenced viral population, regardless of the strain’s intrinsic replication fidelity. Further highlighting the sensitivity of this pipeline, the high fidelity A372V variant and the low fidelity S299T variant, respectively maintained lower and higher diversity throughout the 120 replication cycles and in all triplicate series. We then asked which variants specifically demonstrated a constant increase in rate, as passages progressed, that could be indicative of positive selection and could help elucidate functional determinants throughout a genome. For this purpose, we reviewed the accumulated changes in each coxsackie virus variant (WT, A372V, S299T) over the three time points. Using ViVan, we were able to collect all the positions across each variant’s genome which demonstrated a progressive increase in variant allele frequency. We then reviewed all the variants that either (i) accumulated in more than one population, or (ii) accumulated at the highest rate. Out of the four variants that accumulated in all three virus variants (Table 3), three occurred within viral capsid proteins VP3 and VP4, consistent with the higher evolutionary rates of viral structural proteins and the localization of many receptor binding and antigenic epitopes within the VP3 protein (Carson et al., 1997, 2011). The fourth variant occurred within the viral polymerase (3D; S452P).
Fig. 4.
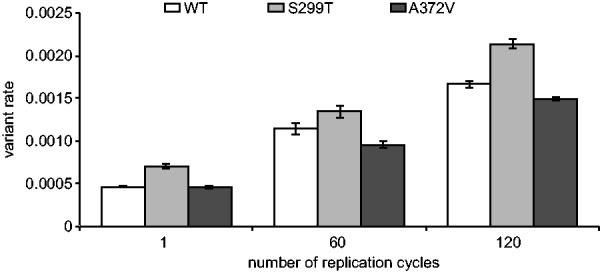
Temporal accumulation of variants found in viral populations. The total number of minority variants found in each CVB3 strain (wild type WT, high fidelity A372V and low fidelity S299T) was calculated for each sequenced passage (1, 60 and 120 replication cycles). Variation rates (the average proportion of variant alleles across covered bases in the genome sequence) ± SEM are shown, from three biological replicates, (P < 0.0001)
Table 3.
Allele rate change throughout replication passages
| Protein | AA position | AA change | Number of Population Samples | Virus variants |
|---|---|---|---|---|
| VP3 | 204 | A > V | 8 | WT 372 299 |
| VP4 | 17 | N > D | 6 | WT 372 299 |
| VP4 | 20 | G > S | 6 | WT 372 299 |
| 3D | 452 | S > P | 6 | WT 372 299 |
| 3B | 6 | V > L | 5 | WT 372 |
| VP2 | 138 | D > N | 4 | WT 299 |
| VP4 | 21 | N > D | 4 | WT 372 |
| VP4 | 15 | R > G | 4 | 372 299 |
| 2B | 11 | N > D | 4 | 372 299 |
| VP4 | 23 | I > T | 4 | 372 299 |
Implementing our method on one of the CVB wild-type samples, and recording the changes throughout the passages (1, 60 and 120), the positions demonstrating the highest increase in variant allele rate was detected. Out of the top 10 most changing positions, only four were non-structural, one synonymous in the viral protease (3C) one nonsynonymous in 2B and two non-synonymous in the viral polymerase (3D). The polymerase variants were then recognized as variants already known to modulate viral replication fidelity.
Interestingly, the only accumulated variant in the viral 3B protein was found in five different population sets (all of the A372V populations and two of the WT). Out of the variant positions, common to more than one population and having a rate >5% in passage 120 (n = 11; Table 4), seven were within viral capsid proteins, two in 3D, one in 3A and one in 3B. The 3D mutations are the known natural fidelity variants of the polymerase (Levi et al., 2010): S299T, known to decrease viral replication fidelity, and A372V, known to increase viral replication fidelity. By passage 120, the S299T variant, accumulated to 5 and 1% of the A372V and WT populations, respectively. A372V accumulated in all three WT populations with an average rate of 37% at passage 120.
Table 4.
Variant positions, common to more than one population and having a rate >5% in passage 120
| Protein | AA position | AA change | Number of population samples | Virus variants | Passage 1 average rate | Passage 60 average rate | Passage 120 average rate |
|---|---|---|---|---|---|---|---|
| VP3 | 234 | Q > E | 2 | WT | 0.0024 | 0.4682 | 0.6749 |
| 3D | 372 | A > V | 3 | WT | 0.0010 | 0.0256 | 0.3701 |
| VP2 | 137 | L > P | 2 | 372 | 0.0005 | 0.0959 | 0.3317 |
| VP3 | 204 | A > V | 8 | WT 372 299 | 0.0006 | 0.0462 | 0.2560 |
| 3A | 51 | T > A | 3 | 299 | 0.0013 | 0.0125 | 0.2477 |
| VP2 | 138 | D > N | 4 | WT 299 | 0.0003 | 0.0116 | 0.2206 |
| 3B | 6 | V > L | 5 | WT 372 | 0.0001 | 0.0113 | 0.1498 |
| VP4 | 21 | N > D | 4 | WT 372 | 0.0003 | 0.0310 | 0.1493 |
| VP4 | 17 | N > D | 6 | WT 372 299 | 0.0011 | 0.0088 | 0.1219 |
| VP4 | 15 | R > G | 4 | 372 299 | 0.0011 | 0.0036 | 0.0760 |
| 3D | 299 | S > T | 2 | WT 372 | 0.0004 | 0.0058 | 0.0626 |
Seven variants were within viral capsid proteins, two in 3D, one in 3A and one in 3B. The 3D mutations are the known natural fidelity variants of the polymerase: S299T, known to decrease viral replication fidelity, and A372V, known to increase viral replication fidelity. By passage 120, the S299T variant, accumulated to 5% and 1% of the A372V and WT populations respectively. A372V accumulated in all three WT populations with an average rate of 37% at passage 120. These observations suggest that viral populations may fine tune their mutation rates during their infection cycles by generating mixed populations of fidelity variants, so as to increase adaptability (fidelity decrease) while maintaining genetic integrity (fidelity increase).
These observations suggest that viral populations may fine tune their mutation rates during their infection cycles by generating mixed populations of fidelity variants, so as to increase adaptability (fidelity decrease) while maintaining genetic integrity (fidelity increase).
4 Discussion
It is becoming increasingly clear that studying only the consensus sequence of an RNA virus insufficiently summarizes the viral population. Often, increases in fitness and changes in adaptability are observed without changes in the consensus sequence (Coffey et al., 2008; Sanz-Ramos et al., 2008). This hints that minority variants within the viral population are responsible for this effect. Virus diversity represents a pool of randomly generated minority variants, available for adaptation and gradually amplified in frequency through selection. Only with the advent of HTS has studying such minorities become feasible but we are missing standard validated tools to mine, analyze and compare this information from multiple samples. Currently available tools require a high degree of computational savoir-faire, and for more detailed RNA virus population analysis, the user must write additional scripts.
Our computational pipeline has several advantages over other methods. We use a robust algorithm, based on each variant allele's initial rate and read qualities, to differentiate between sequencing-introduced errors and actual population variants, facilitating accurate variant assessment even at extremely low rates. Another advantage of the pipeline is the set of different outputs we provide for analysis (Fig. 1, Supplementary Data 2). First, we provide a table of the synonymous and non-synonymous changes for each significantly variable position, organized by gene and position for the whole viral genome. Second, we provide a battery of metrics including nucleotide substitution matrix, transition/transversion frequencies and variant allele rates. We also include the consensus changes found to be different from the original reference, enabling reconstruction of the genuine consensus for a given sample. Third, we provide three files with pairwise comparisons supplying the mutations found to be different or common among samples. These outputs are rapid and powerful, enabling comprehensive analysis of large data sets. Importantly, we have made ViVan available and accessible for users without computational proficiency in the virology community through an easy-to-use web server, enabling a complete analysis given raw deep sequencing data.
Using low fidelity CVB3 variants that generate subtle, yet biologically confirmed, differences in mutant composition, we showed that detected variation frequencies correlated with the mutation frequencies obtained by molecular cloning in the original paper (Gnädig et al., 2012). We also showed that sequence analysis of mutagen treated virus populations can identify the specific transition/transversion footprints for three different mutagens, as well as reveal dose-dependent effects. The increased ease and sensitivity of this approach could help identify new compounds with antiviral mutagenic activity and distinguish statistically significant changes that would otherwise be overlooked by classic methods. This may help answer the debate as to whether mutagens such as ribavirin have mutagenic activity at physiological concentrations, as in the treatment of chronic HCV infection (Dusheiko et al., 1996).
In addition to comparing samples from different environmental conditions, we determined the sensitivity of the pipeline in detecting temporal changes in minority variant composition within the same virus population. We serially passaged wild type coxsackie, as well as naturally occurring high (A372V) and low (S299T) fidelity strains in a cell line to which the virus is already well adapted, to favor a general expansion of more neutral or high fitness variants in this highly permissive environment. Gene by gene analysis of variance corroborated with previous data on picornaviruses, where variability and mutation is seemingly most tolerated in the structural proteins (P1 region) and the P3 non-structural region; while the non-structural P2 region is less variable (Kistler et al., 2007). A very interesting and unexpected observation was the emergence of fidelity altering mutations in late passages of the wild type strain that is generally considered to be genetically stable. Unlike other fidelity altering mutations isolated in our lab (Gnädig et al., 2012), these two alleles exist in some CVB3 isolates (Levi et al., 2010), but the natural emergence of fidelity variants has not been previously observed. That they emerge within the wild type population during longer-term experimental evolution raises the intriguing possibility that fidelity modulation may occur in natural settings, according to host environment. Such modulation of fidelity would be reminiscent of environment-dependent changes in bacterial mutation rates (Sniegowski et al., 1997). Our method is intended for high coverage short read data, it does not incorporate information regarding variants detected on the same read and therefore does not support haplotype reconstruction (Eriksson et al., 2008; Zagordi et al., 2011). Our method is also limited to per-position analysis and therefore it cannot directly account for compensatory mutations. If one wishes to identify such associated variants, we suggest an initial analysis using ViVan in order to identify significant variants followed by a deeper assessment of the data in regards to adjacent variants found within a read length of each other. The examples presented in this work were performed using virus references matching the molecular clone (plasmid) that is later used to produce the viruses themselves. These references are extremely well characterized and serve as a good template for variant analysis. In cases where such appropriate references are not available, we recommend either (i) an initial ViVan run on the data and then a re-run using the modified consensus sequence produced, or (ii) an initial assembly of the viral genome using appropriate assembly tools (Bankevich et al., 2012; Simpson et al., 2009; Zerbino and Birney, 2008) and using the assembled genome as input for ViVan.
In this article, we have presented a new bioinformatic pipeline for the study of HTS data. We hope this tool will help standardize and facilitate the analysis of data this technology provides. As we have illustrated with our experimental work, it can be used for studies ranging from the more practical, detection of resistant mutations and effects of antiviral treatments, to the more theoretical temporal characterization of the population in evolutionary studies. Our analysis pipeline provides an extremely low allele rate cut-off threshold to determine statistically significant minority variants, as well as several metrics and statistics on population diversity, transitions and transversions bias, synonymous and non-synonymous mutation distributions, gene-by-gene and whole-genome analysis. Furthermore, it readily performs group sample comparisons, a feature not currently available to the scientific community interested in experimental evolution or analysis of clinical and field samples. This set of outputs, coupled with an online web server, sets ViVan as an extensive analysis tool which can be readily used by the virology community.
Supplementary Material
Acknowledgements
We thank C. Barbezange, L. Coffey, L. Levi, N. Gnädig, S. Beaucourt for previous work that was characterized in this study. This work was performed in partial fulfillment of the requirements for a PhD degree of O. Isakov at the Sackler Faculty of Medicine, Tel Aviv University.
Funding
This study was supported in part by a fellowship from the Edmond J. Safra Center for Bioinformatics at Tel-Aviv University, by a grant from the France-Israel High Council for Research Scientific and Technological Cooperation and from the European Union Seventh Framework Programme [FP7/2007-2013] under Grant Agreement n°278433-PREDEMICS. The Shomron laboratory is supported by the Israel Cancer Research Fund (ICRF), Research Career Development Award (RCDA); Wolfson Family Charitable Fund; Earlier.org—Friends for an Earlier Breast Cancer Test; Claire and Amedee Maratier Institute for the Study of Blindness and Visual Disorders; I-CORE Program of the Planning and Budgeting Committee, The Israel Science Foundation [41/11]; The Israeli Ministry of Defense, Office of Assistant Minister of Defense for Chemical, Biological, Radiological and Nuclear (CBRN) Defense; Foundation Fighting Blindness; Saban Family Foundation, Melanoma Research Alliance; Binational Science Foundation (BSF); ICRF Acceleration Grant; Israel Cancer Association (ICA); Donation from the Kateznik K. Association Holocaust; Margot Stoltz Foundation through the Faculty of Medicine grants of Tel-Aviv University; The Varda and Boaz Dotan Research Center in Hemato-Oncology, Idea Grant. M. Vignuzzi and H. Blanc are supported in part by the ERC Starting [242719]. A.B. was supported by the ANR-09-JCJC-0118. O. Isakov and D. Golan are supported in part by the Colton Family Foundation.
Conflict of Interest: none declared.
References
- Acevedo A., et al. (2013) Mutational and fitness landscapes of an RNA virus revealed through population sequencing. Nature. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ahlquist P. (2002) RNA-dependent RNA polymerases, viruses, and RNA silencing. Science, 296, 1270–1273. [DOI] [PubMed] [Google Scholar]
- Archer J., et al. (2012a) Analysis of high-depth sequence data for studying viral diversity: a comparison of next generation sequencing platforms using Segminator II. BMC Bioinformatics, 13, 47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Archer J., et al. (2012b) Use of four next-generation sequencing platforms to determine HIV-1 coreceptor tropism. PLoS One, 7, e49602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bankevich A., et al. (2012) SPAdes: a new genome assembly algorithm and its applications to single-cell sequencing. J. Comput. Biol., 19, 455–477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barzon L., et al. (2011) Applications of next-generation sequencing technologies to diagnostic virology. Int. J. Mol. Sci., 12, 7861–7884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beerenwinkel N., Zagordi O. (2011) Ultra-deep sequencing for the analysis of viral populations. Curr. Opin. Virol., 1, 413–418. [DOI] [PubMed] [Google Scholar]
- Bull J.J., et al. (2007) Theory of lethal mutagenesis for viruses. J. Virol., 81, 2930–2939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bull R.A., et al. (2011) Sequential bottlenecks drive viral evolution in early acute hepatitis C virus infection. PLoS Pathog., 7, e1002243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Capobianchi M.R., et al. (2013) Next-generation sequencing technology in clinical virology. Clin. Microbiol. Infect., 19, 15–22. [DOI] [PubMed] [Google Scholar]
- Carson S.D., et al. (2011) Variations of coxsackievirus B3 capsid primary structure, ligands, and stability are selected for in a coxsackievirus and adenovirus receptor-limited environment. J. Virol., 85, 3306–3314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carson S.D., et al. (1997) Purification of the putative coxsackievirus B receptor from HeLa cells. Biochem. Biophys. Res. Commun., 233, 325–328. [DOI] [PubMed] [Google Scholar]
- Coffey L.L., et al. (2011) Arbovirus high fidelity variant loses fitness in mosquitoes and mice. Proc. Natl Acad. Sci. USA, 108, 16038–16043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coffey L.L., et al. (2008) Arbovirus evolution in vivo is constrained by host alternation. Proc. Natl Acad. Sci. USA, 105, 6970–6975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coffey L.L., Vignuzzi M. (2011) Host alternation of chikungunya virus increases fitness while restricting population diversity and adaptability to novel selective pressures. J. Virol., 85, 1025–1035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crotty S., Andino R. (2002) Implications of high RNA virus mutation rates: lethal mutagenesis and the antiviral drug ribavirin. Microbes Infect., 4, 1301–1307. [DOI] [PubMed] [Google Scholar]
- Crotty S., et al. (2001) RNA virus error catastrophe: direct molecular test by using ribavirin. Proc. Natl Acad. Sci. USA, 98, 6895–6900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crotty S., et al. (2000) The broad-spectrum antiviral ribonucleoside ribavirin is an RNA virus mutagen. Nat. Med., 6, 1375–1379. [DOI] [PubMed] [Google Scholar]
- Delwart E. (2013) A roadmap to the human virome. PLoS Pathog., 9, e1003146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Didelot X., et al. (2012) Transforming clinical microbiology with bacterial genome sequencing. Nat. Rev. Genet., 13, 601–612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dusheiko G., et al. (1996) Ribavirin treatment for patients with chronic hepatitis C: results of a placebo-controlled study. J. Hepatol., 25, 591–598. [DOI] [PubMed] [Google Scholar]
- Eriksson N., et al. (2008) Viral population estimation using pyrosequencing. PLoS Comput. Biol., 4, e1000074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Escobar-Gutiérrez A., et al. (2012) Identification of hepatitis C virus transmission using a next-generation sequencing approach. J. Clin. Microbiol., 50, 1461–1463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flaherty P., et al. (2012) Ultrasensitive detection of rare mutations using next-generation targeted resequencing. Nucleic Acids Res., 40, e2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Foulongne V., et al. (2012) Human skin microbiota: high diversity of DNA viruses identified on the human skin by high throughput sequencing. PLoS One, 7, e38499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghedin E., et al. (2012) Presence of oseltamivir-resistant pandemic A/H1N1 minor variants before drug therapy with subsequent selection and transmission. J. Infect. Dis., 206, 1504–1511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gnädig N.F., et al. (2012) Coxsackievirus B3 mutator strains are attenuated in vivo. Proc. Natl Acad. Sci. USA, 109, E2294–E2303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Graci J.D., Cameron C.E. (2008) Therapeutically targeting RNA viruses via lethal mutagenesis. Future Virol., 3, 553–566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grad Y.H., et al. (2014) Within-host whole-genome deep sequencing and diversity analysis of human respiratory syncytial virus infection reveals dynamics of genomic diversity in the absence and presence of immune pressure. J. Virol., 88, 7286–7293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Graham R.L., et al. (2012) A live, impaired-fidelity coronavirus vaccine protects in an aged, immunocompromised mouse model of lethal disease. Nat. Med., 18, 1820–1826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grande-Pérez A., et al. (2002) Molecular indetermination in the transition to error catastrophe: systematic elimination of lymphocytic choriomeningitis virus through mutagenesis does not correlate linearly with large increases in mutant spectrum complexity. Proc. Natl Acad. Sci. USA, 99, 12938–12943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo Y., et al. (2012) The effect of strand bias in Illumina short-read sequencing data. BMC Genomics, 13, 666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hurwitz B.L., Sullivan M.B. (2013) The pacific ocean virome (POV): a marine viral metagenomic dataset and associated protein clusters for quantitative viral ecology. PLoS One, 8, e57355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jabara C.B., et al. (2011) Accurate sampling and deep sequencing of the HIV-1 protease gene using a Primer ID. Proc. Natl Acad. Sci. USA, 108, 20166–20171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kinde I., et al. (2011) Detection and quantification of rare mutations with massively parallel sequencing. Proc. Natl Acad. Sci. USA, 108, 9530–9535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kistler A.L., et al. (2007) Genome-wide diversity and selective pressure in the human rhinovirus. Virol. J., 4, 40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lauring A.S., Andino R. (2010) Quasispecies Theory and the Behavior of RNA Viruses. PLoS Pathog., 6, e1001005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levi L.I., et al. (2010) Fidelity variants of RNA dependent rna polymerases uncover an indirect, mutagenic activity of amiloride compounds. PLoS Pathog., 6, e1001163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H., Durbin R. (2009) Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics, 25, 1754–1760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H., et al. (2009) The sequence alignment/map format and SAMtools. Bioinformatics, 25, 2078–2079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Love T.M.T., et al. (2010) Mathematical modeling of ultradeep sequencing data reveals that acute CD8+ T-lymphocyte responses exert strong selective pressure in simian immunodeficiency virus-infected macaques but still fail to clear founder epitope sequences. J. Virol. 84, 5802–5814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Macalalad A.R., et al. (2012) Highly sensitive and specific detection of rare variants in mixed viral populations from massively parallel sequence data. PLoS Comput. Biol., 8, e1002417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mangul S., et al. (2014) Accurate viral population assembly from ultra-deep sequencing data. Bioinformatics, 30, i329–i337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martínez F., et al. (2012) Ultradeep sequencing analysis of population dynamics of virus escape mutants in RNAi-mediated resistant plants. Mol. Biol. Evol., 29, 3297–3307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McElroy K., et al. (2013) Accurate single nucleotide variant detection in viral populations by combining probabilistic clustering with a statistical test of strand bias. BMC Genomics, 14, 501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nájera I., et al. (1995) Pol gene quasispecies of human immunodeficiency virus: mutations associated with drug resistance in virus from patients undergoing no drug therapy. J. Virol., 69, 23–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Radford A.D., et al. (2012) Application of next-generation sequencing technologies in virology. J. Gen. Virol., 93, 1853–1868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roux S., et al. (2012) Assessing the diversity and specificity of two freshwater viral communities through metagenomics. PLoS One, 7, e33641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanjuán R., et al. (2010) Viral mutation rates. J. Virol., 84, 9733–9748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanz-Ramos M., et al. (2008) Hidden virulence determinants in a viral quasispecies in vivo. J. Virol., 82, 10465–10476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmitt M.W., et al. (2012) Detection of ultra-rare mutations by next-generation sequencing. Proc. Natl Acad. Sci. USA, 109, 14508–14513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Selleri M., et al. (2012) Detection of haemagglutinin D222 polymorphisms in influenza A(H1N1)pdm09-infected patients by ultra-deep pyrosequencing. Clin. Microbiol. Infect., [DOI] [PubMed] [Google Scholar]
- Shomron N. (2013) Genetics research: jumping into the deep end of the pool. Genet. Res. (Camb), 95, 1–3. [DOI] [PubMed] [Google Scholar]
- Simpson J.T., et al. (2009) ABySS: a parallel assembler for short read sequence data. Genome Res., 19, 1117–1123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sniegowski P.D., et al. (1997) Evolution of high mutation rates in experimental populations of E. coli.. Nature, 387, 703–705. [DOI] [PubMed] [Google Scholar]
- Steinhauer D.A., Holland J.J. (1987) Rapid Evolution of RNA Viruses. Annu. Rev. Microbiol., 41, 409–431. [DOI] [PubMed] [Google Scholar]
- Töpfer A., et al. (2013) Sequencing approach to analyze the role of quasispecies for classical swine fever. Virology, 438, 14–19. [DOI] [PubMed] [Google Scholar]
- Vignuzzi M., et al. (2005) Quasispecies diversity determines pathogenesis through cooperative interactions in a viral population. Nature, 439, 344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watson S.J., et al. (2013) Viral population analysis and minority-variant detection using short read next-generation sequencing. Philos. Trans. R. Soc. Lond., B, Biol. Sci., 368, 20120205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Willerth S.M., et al. (2010) Development of a low bias method for characterizing viral populations using next generation sequencing technology. PLoS One, 5, e13564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilm A., et al. (2012) LoFreq: a sequence-quality aware, ultra-sensitive variant caller for uncovering cell-population heterogeneity from high-throughput sequencing datasets. Nucleic Acids Res., 40, 11189–11201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woo H.-J., Reifman J. (2012) A quantitative quasispecies theory-based model of virus escape mutation under immune selection. Proc. Natl Acad. Sci. USA, 109, 12980–12985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wright C.F., et al. (2011) Beyond the consensus: dissecting within-host viral population diversity of foot-and-mouth disease virus by using next-generation genome sequencing. J. Virol., 85, 2266–2275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu N.C., et al. (2014) High-throughput profiling of influenza A virus hemagglutinin gene at single-nucleotide resolution. Sci. Rep., 4, 4942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang X., et al. (2013) V-Phaser 2: variant inference for viral populations. BMC Genomics, 14, 674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yin L., et al. (2012) High-resolution deep sequencing reveals biodiversity, population structure, and persistence of HIV-1 quasispecies within host ecosystems. Retrovirology, 9, 108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zagordi O., et al. (2011) ShoRAH: estimating the genetic diversity of a mixed sample from next-generation sequencing data. BMC Bioinformatics, 12, 119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zerbino D.R., Birney E. (2008) Velvet: algorithms for de novo short read assembly using de Bruijn graphs. Genome Res., 18, 821–829. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.