

Abstract
Introduction
The thyroid-stimulating hormone receptor (TSHR) is the essential molecule for thyroid growth and thyroid hormone production. Since it is also a key autoantigen in Graves’ disease and is involved in thyroid cancer pathophysiology, the targeting of the TSHR offers a logical model for disease control.
Areas covered
We review the structure and function of the TSHR and the progress in both small molecule ligands and TSHR antibodies for their therapeutic potential.
Expert opinion
Stabilization of a preferential conformation for the TSHR by allosteric ligands and TSHR antibodies with selective modulation of the signaling pathways is now possible. These tools may be the next generation of therapeutics for controlling the pathophysiological consequences mediated by the effects of the TSHR in the thyroid and other extrathyroidal tissues.
Keywords: autoantibodies, glycoprotein-hormone receptor, Graves’ disease, small molecule ligands, thyroid cancer, thyroid-stimulating hormone receptor
1. Introduction
G-protein-coupled receptors (GPCRs) transduce signals from the extracellular environment into the intracellular milieu leading to changes in cell function. The glycoprotein hormone receptor (GHR) family is one particular class of GPCRs, which includes the thyroid-stimulating hormone receptor (TSHR), the luteinizing hormone receptor (LHR) and the follicle-stimulating hormone receptor (FSHR) [1,2], which bind to their respective hormones to transduce the signal. The TSHR is a major human antigen in autoimmune thyroid disease [3,4] and has become a therapeutic target for the autoimmune hyperthyroidism known as Graves’ disease (GD) in which antigen-specific antibodies and T cells have been demonstrated. The TSHR is also an important participant in the development and dissemination of thyroid cancer and is a target in thyroid cancer diagnosis and treatment. In order to understand the different modalities of targeting this unique glycoprotein receptor, it is important to understand its structure and function.
2. Structure and function of the TSHR
The TSHR gene is located on chromosome 14q31 and contains 10 exons. The first 9 exons encode a large extracellular domain (ECD), while exon 10 codes for a transmembrane domain (TMD) (Figure 1A). The full-length receptor protein consists of 764 amino acids divided into a 21 amino-acid signal peptide at the N terminus, which is later cleaved from the mature protein, leaving the highly glycosylated ectodomain (ECD) of 394 amino acids incorporating 11 leucine-rich repeats (LRR) [5,6]. The ECD incorporating the LRRs has multiple TSH binding sites and also contains the major binding sites for many TSHR autoantibodies [5]. These antibodies may act as TSH agonists and are the cause of the hyperthyroid response in GD. Thyroid-stimulating autoantibodies bind to critical amino acids on the ECD and mimic the action of TSH leading to the hyperthyroidism [7]. Previous studies [8,9] have described a TSH ‘binding pocket’ on the ECD and a ‘handclasp’ binding mode of TSH into the concave surface of the ECD.
Figure 1.
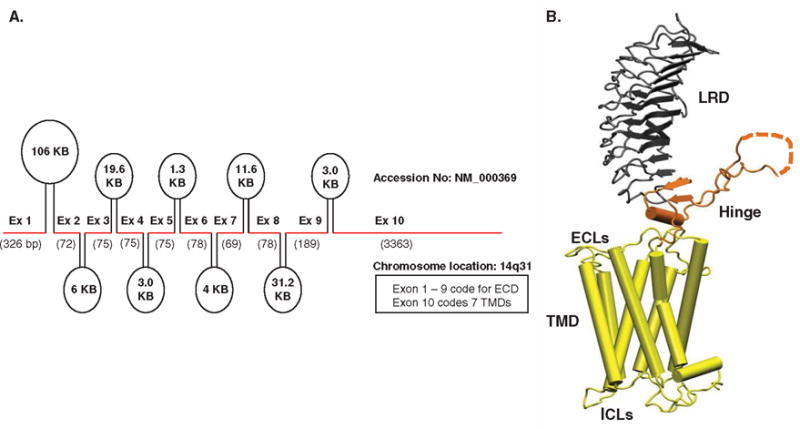
A. Genetic organization of the human TSHR receptor. The TSHR gene located on chromosome 14q31 consists of 10 exons as indicated here by the thick red line segments. The introns between these exons are indicated as ovals with their sizes marked within them. The first 9 exons of the TSHR gene code for the extracellular domain (ECD) and exon 10 codes for the transmembrane domain (TMD) of the receptor. B. Homology model of the TSH holoreceptor. The model highlights the tripartite structure of the TSHR. The ectodomain shown in gray is made up of 10 leucine-rich repeat domains (LRD with loops and β-pleated sheets obtained from the published crystal structure [9] (PDB: 3G04). The region connecting the LRD and TMD, known as the ‘hinge’ region has recently been crystallized for the FSH receptor and is shown as a looped structure (orange) with a helix conformation close to the carboxyl end of the LRD. The hinge in the TSHR has an additional sequence insert and is larger than in the FSH receptor. Therefore, amino acids 305 – 381 are missing in the illustrated model, and this insert is depicted as a closed dotted loop. The TMD (yellow), with its 7 helices, is depicted as cylindrical structures connected to each other by the specific TSHR intra- and extracellular loops (ICLs and ECLs). The TMD is the region that harbors the allosteric binding pockets for the SMLs.
A. Reproduced with permission from [95].
B. Reproduced with permission from [72].
ECL: Extracellular loops; ICL: Intracellular loops; SMLs: Small molecule ligands; TSHR: Thyroid-stimulating hormone receptor.
Initial insights into TSHR structure-function were first gained from human pathology. A variety of TSHR mutations were identified in thyroid tissues and thyroid tumors which revealed either constitutive activation or inactivation [10,11]. The partial crystal structure of the first 260 amino acids of the ECD bound to a stimulating TSHR antibody (S-TSHR-Ab) then extended this understanding of TSH binding and activation and added the detailed epitopes for TSHR autoantibodies [9]. These structures showed that the ECD is responsible for the specificity and high affinity of ligand binding and is considered the ‘orthosteric site’ of the receptor; meaning the primary ligand binding site. In addition, the distal half of the ECD is linked to the TMD by a region of 130 amino acids known as the ‘hinge region’. A partial crystal structure of the ‘hinge region’ of the FSHR [12] along with other mutational studies on the TSHR hinge has now clearly shown that this structural bridge undergoes conformational changes in response to ligand binding and is involved in the activation of the receptor. Hence, this region is now often referred to as the ‘signal-specific domain’ [6,13]. In addition, the membrane-embedded TMD, which consists of 349 amino acids, incorporates seven transmembrane helices (TMH) joined by extracellular (ECL) and intracellular loops and has a short intracytoplasmic tail which is crucial for the proper trafficking of the receptor to the plasma membrane (Figure 1B) [14,15].
The TSHR undergoes complex post-translational modifications, including intramolecular cleavage [16] and multimerization [17,18], in addition to generic palmitoylation, sulfation, glycosylation and phosphorylation [19]. Intramolecular cleavage of the ectodomain is likely to lead to increased receptor shedding and antigenic stimulation [16]. Although the predominant site(s) of dimer and multimer formation reside in the interfaces of the TMD [18,20,21], there is evidence that lower affinity interface sites also reside in the ectodomain as shown by biochemical and biophysical approaches [20] and for the FSHR by crystallization [12]. TSHR multimerization appears to be important in differential receptor reactivity and in the phenomenon of negative cooperativity [18]. Data show that TSHR multimers also regulate early events in receptor maturation and intracellular trafficking [20,22] and favor activation of Gq signaling [23]. We have previously shown that these monomeric and higher order complexes, which can bind autoantibodies [24], may be actually regulated by the binding of TSH and S-TSHR-Abs within specialized compartments of the plasma membrane known as ‘lipid rafts’ (Figure 2) [25]. Further, it has recently been shown that TSHR recycling involving clathrin-mediated endocytosis, via β-arrestins [26,27], leads to continued signaling from the endosomes [28] in addition to plasma membrane signaling [29,30]. These important structural and functional features of the TSHR may help explain some of the role of the TSHR in the pathophysiology of GD and as well as its role in a variety of extrathyroidal sites including the retro-orbit, skin and bone discussed below.
Figure 2. The TSHR resides in membrane rafts.
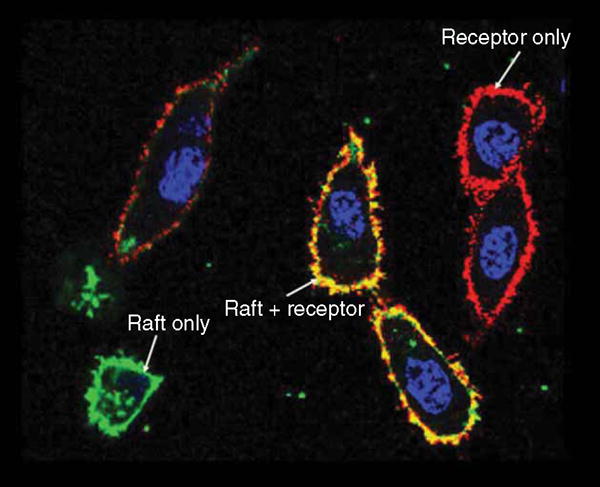
Shown here are lipid rafts stained with myristoylated-palmitoylated peptide from Lyn kinase fused to the N terminus of YFP (green) and TSHR labeled with antibody (RSR1) conjugated Alexa574 (red). Co-localization of receptors on the cell surface with the membrane rafts leads to a yellow color on these HEK 293 double transfected cells.
TSHR: Thyroid-stimulating hormone receptor.
3. TSHR physiology
The primary role of the TSHR is in thyroid cell growth and thyroid hormone synthesis and secretion. The TSHR is unique among the family of glycoprotein receptors because of its ‘constitutive’ signaling activity, although how this activity contributes to thyroid cell biology is unclear [31]. We do know that direct TSHR activation and coupling of G proteins leads to a cascade of complex intracellular events resulting in thyrocyte growth and hormone production when Gsα stimulates the production of adenyl cyclase and simultaneous Gq stimulation leads to PI3 kinase activation. The increase in cAMP generation and PI3 kinase, via protein kinase A (PKA) and phospholipase C (PLC), causes thyrocyte growth, differentiation and thyroid hormone synthesis [32]. Higher levels of TSHR stimulation activate the Gq protein leading to activation of a PLC-dependent pathway, the production of inositol triphosphate (IP3) and diacylglycerol (DAG) with subsequent production of hydrogen peroxide and iodination [33,34]. Protein kinase C, generated in response to DAG and PKA has a direct effect on gene transcription [35] via activation of the AKT signaling pathway, which leads to cell proliferation and survival. TSH or TSHR autoantibody binding to the receptor can also stimulate additional pathways which can lead to the activation of NF-κB [6,35,36].
4. Proteolytic cleavage
Another notable feature of the TSHR, which may be relevant to the initiation of the autoimmune process and the production of autoantibodies in GD, may come from the unique property of the receptor to undergo proteolytic cleavage [37]. The ECD of the receptor includes two distinct segments (residues 38 – 45 and 316 – 366) not seen in other GPCRs [5]. It is known that cysteine residue 41 contained in the 8 amino acids region of 38 – 45 is critical for TSH binding [38,39]. The second 50 amino acid unique region (316 – 366), which has defied crystallization, is lost from the TSHR by intramolecular cleavage [40] leading to the formation of two receptor subunits connected by disulfide bonds formed by cysteine residues [41]. Following cleavage of the cell surface TSHRs the disulfide bonds are broken down by protein disulfide isomerase with subsequent shedding of the α (or A) subunit of the receptor [42,43]. Studies have indicated that activation of the receptor by TSH can increase cell surface cleavage [44] and factors leading to up-regulation of metalloproteases can increase cell surface shedding of the receptor [45]. Therefore, this post-translational modification is an important source of intact antigen for immune stimulation in GD and targeting this phenomenon may have therapeutic implications.
5. Need for newer therapeutics
GD is one of the most common organ-specific autoimmune diseases and only affects humans. It has a prevalence in the female population of ~ 2% [46,47]. Although GD has been reasonably treated for more than a century using surgery and, later, radioiodine ablation [48], there has been very little progress in GD therapeutics for many years and anti-thyroid drugs with toxic side effects remain in widespread use. Furthermore, the treatment of Graves’ orbitopathy (GO) and pretibial myxedema (PTM), the two extrathyroidal complications of GD, remains extremely difficult and unsatisfactory.
The basic treatments of hyperthyroidism using the three available methods (antithyroid drugs, radioiodine and surgery) each have major drawbacks. Antithyroid drugs (propylthiouracil – PTU – and methimazole) have problems leading to bone marrow and liver failure [49] and congenital abnormalities [50,51] and both radioiodine ablation and surgery require the patient to take thyroid hormone replacement for the rest of their life. As the current therapeutic approaches in controlling thyroid disease are unsatisfactory, there is need for a concerted effort to develop newer approaches for controlling the activity of the receptor in GD.
It is also important to note that the TSHR is a target in the management of thyroid cancer [52]. At variance with other markers of thyroid differentiation, the TSHR is expressed in the majority of thyroid cancers and is an interesting therapeutic target. Currently, recombinant human TSH is used for detecting thyroglobulin release from metastatic thyroid cancer and for enhancing Radioiodine (RAI) uptake into thyroid glands [53–55]. In addition to the high cost of recombinant TSH, which is a large glycosylated complex protein, there has been difficulty in maintaining a steady supply of high-quality material and so a search for cheaper and more reliable TSHR agonists has been ongoing including the long search for more stably glycosylated superagonist TSH forms [56].
It is not difficult to define what an ideal therapeutic should be when targeting the TSHR (Table 1). Meeting some of these criteria are two classes of therapeutics – small molecules and designer TSHR antibodies.
Table 1.
The ideal GD drug.
In an ideal situation, any novel therapeutic designed to treat GD should meet the following requirements:
|
GD: Graves’ disease.
6. Development of small molecule TSH agonists
The TSHR is the major regulator of thyroid cell function and, therefore, stimulation and inhibition of this complex molecule has pathologic and therapeutic significance. Current modalities for influencing TSR function are limited to the availability of expensive recombinant human TSH (Thyrogen) used in the investigation and treatment of thyroid cancer and the potential of antibodies blocking the TSHR as a treatment for hyperthyroid GD. Hence, there has been a search for cheaper and perhaps more effective approaches to TSHR regulation.
The lead for developing small molecular ligands started with a range of bicyclic heteroaromatic compounds including thienopyrimidineses active as agonists against the LHR/Human chorionic gonadotropin receptor [57]. Following the discovery of thienopyrimidine – org41841 in 2002, the group of Marvin C Gershengorn, from the National Institutes of Health (NIH) predicted that org41841 might also activate the TSHR because of its homology to the LHR and subsequently found it to be a partial agonist for the TSHR [58]. They subsequently described the first specific TSHR agonist (NCG00161870) in 2009 (Table 2) [59] with relatively high affinity and potency. It showed selectivity because of its inability to activate the two closely related GHRs – the LHR and the FSHR. Molecular docking studies showed NCG00161870 binding to asparagine (Asn) 47 in helix 5 of the TMD and mutating Asp 47 obliterated its activity indicating that it acted allosterically. Our own extensive studies on the TSHR for several years lead us also to discover agonists to the TSHR by performing high-throughput screening (HTS) of large chemical libraries. Our efforts using a transcriptionally based luciferase assay based on TSHR transfected CHO cells [60] and screening 50,000 compounds revealed two novel and specific agonists against the TSHR which are also allosteric ligands with ‘drug-like’ characteristics (pharmacokinetics and pharmacodynamics -PKs/PDs) and showing in vivo potency in animal studies. These lead TSHR agonists (MS437 and MS438) (Table 2) have different binding sites than the agonist described earlier but are also allosteric ligands binding to the TMD (Figure 3) [60]. These data suggest that chemical modifications or ligands with novel scaffolds targeting signal-sensitive amino acids surrounding the allosteric binding sites within the TSHR may lead to agonists of even greater activity [61].
Table 2.
Small molecule ligand agonists to the TSHR.
| Small molecule ligands | Chemical name | Action | EC50/IC50 | Binding site | Ref. |
|---|---|---|---|---|---|
| Org 41841 | N-tert-Butyl-5-amino-4-(3-methoxy-phenyl)-2-(methylthio)thieno[2,3-d] pyrimidine-6-carboxamide | Partial Agonist | 7.7 × 10−6 (7700 nM) | TM3,4,5,6 & 7 | [58] |
| NCG00161870 | N-4 (4-(5-(3-benzyl-5-hydrooxy-4-ox0-1,2,3,4-tetrahydroquinazolin-2-yl)-2-methoxybenzyloxy)acetamide | Agonist | 4 × 10−8 M (40 nM) | TM5 | [59] |
| MS437 | N,N-diisopropyl-2-phenyl-4 quinolinecarboxamide | Agonist | 1.3 × 10−7 M | TM3 | [60] |
| MS438 | Benzyl6methyl-2-oxo-4-[4-(trifluorome-thyl0phenyl]-1,2,3,4-tetrahydro-5-pyrimidinevarboxylate | Agonist | 5.3 × 10−8 M | TM3 | [60] |
TSHR: Thyroid-stimulating hormone receptor.
Figure 3. Docking of the TSH agonist molecules onto the homology model of the TSHR.
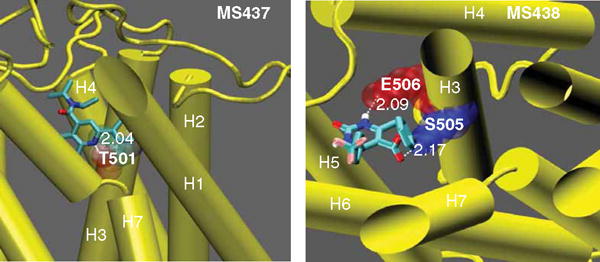
This figure shows intrahelical binding of MS437 and MS438 to TMH3. The molecule MS437 makes a single hydrogen bond with threonine 501 (T501) of TMH3 (left panel), whereas MS438 has two contacts points making hydrogen bonds with serine 505 (S505) and glutamic acid 506 (E506) of TMH3 in the receptor TMD (right panel).
Reproduced with permission from [60].
TMD: Transmembrane domain; TMH: Transmembrane helice; TSHR: Thyroid-stimulating hormone receptor.
7. Development of small molecule TSH antagonists
The ‘holy grail’ for treating GD remains a potent antagonist against the TSHR that would block stimulating TSHR auto-antibodies from activating the receptor without compromising TSH signaling. 1, 1, 1-trichloro-2,2-bis(p-chlorophenyl) ethane and Aroclor1254 (a complex mixture of polychlorinated biphenyls) were shown to have inverse agonist activity [62,63] but lacked specificity because of their ability to activate the cell by a post-receptor mechanism. Efforts towards a more selective molecule started in 2008 with the identification of a neutral antagonist (NIDDK-CEB-52) to the TSHR derived by chemical modification of the agonist NCG0016870 (Table 3) [64]. Although this first-generation antagonist was able to inhibit TSHR activation by TSH and TSHR-Abs in a model system and inhibited up-regulation of thyroid peroxidase in primary cultures, its potency was in the micromolar range. As the clinical need requires a more potent TSHR antagonist there have been efforts to develop small molecule ligands (SMLs) that have IC50 in the nano molar range (10−9 M). Van Koppen et al. [65] in 2012 had initially reported one such highly potent TSHR antagonist, but later, it was found to lack specificity and has not been developed further. A series of TSHR antagonists produced once again by chemical modification, this time of the scaffold of the NIDDK-CEB-52 analog [66], then identified a more potent molecule – ANTAG3 although still with an IC50 only in the 10−6 M range. Additional molecules were found that acted as inverse agonists inhibiting both constitutive and stimulatory functions of the TSHR and one appeared to be somewhat more potent (Table 3) and was capable of inhibiting cAMP production in Graves’ orbital fibroblasts [67]. Nevertheless, the in vivo thyroid inhibition that was obtained was only ~ 50% and is unlikely to be a clinically useful level of effect. Further potency improvement is still needed. PK/PD studies with these SMLs would likely help evaluate their potential. Finding additional novel antagonists to the TSHR would also be helped by the development of more specific and sensitive inhibition assays for HTS or more insight by the use of cheminformatics and chemical modifications.
Table 3.
Small molecule ligand antagonists to the TSHR.
| Small molecule ligands |
Chemical name | Action | EC50/IC*50 | Binding site | Ref. |
|---|---|---|---|---|---|
| NCGC242595 (NIDDK-CEB52) | 2-(3-(2,6-dimethylphenylthio)-4-methoxyphenyl)-3-(furan-2-ylmethy)-2,3-dihydroquinazolin-4(1H)-one | Neutral Antagonist | 4.2 × 10−6 M (4.2 μM) |
TM 3,5,6 and 7 | [64] |
| NCGC00242364 (ANTAG 3) | N-(4-(5-(3-(Furan-2-ylmethyl)-4-oxo-1,2,3,4-tetrahydroquina-olin-2-yl)-2-methoxybenzyloxy)-3,5-dimethylphenyl)acetamide | Antagonist | 2.1 × 10−6 M (2.1 μM) |
TMD | [93] |
| NCGC00161856 | 2-(3-((2,6-dimethylphenoxy) methyl)-4-methoxyphenyl)-3-(furan-2-ylmethyl)-2,3-dihydro-quinazolin-4(1H)-one | Inverse agonist | 3 × 10−6 M (3 μM) |
TMD | [93] |
| NCGC00229600 | 2-(3-((2,6-dimethylphenoxy) methyl)-4-methoxyphenyl)-3-(pyridin-3-ylmethyl)-2,3-dihy-droquinazolin-4(1H)-one | Inverse agonist | 30 × 10−6 M (30 μM) |
TMD | [66] |
| Org274179-0 | (S)-N-(1-acetyl-4-methyl-4-phenyl-1,2,3,4-tetrahydro-quinolin-6-yl)-3-(3-trifluoromethylphenyl)-propionamide | Inverse agonist | 9 10−9 M (9 nM) |
TMD | [94] |
All IC50 are based on inhibition of bovine TSH.
TMD: Transmembrane domain; TSHR: Thyroid-stimulating hormone receptor.
8. Mechanism of small molecule action
The large orthosteric site of the TSHR has multiple binding domains for autoantibodies [68] and TSH [69]. Clearly, stimulation or blockade of all such binding sites could not be achieved with one small molecule and so effective small molecules are more likely to activate or derail the signaling pathways rather than influencing the ligand–receptor interaction. This mechanism of action can be via allosteric modulation. ‘Allosteric’ means regulation from a distance and away from the orthosteric site. A small molecule binding to a receptor is capable of regulating its function through changes in receptor conformation as clearly seen in the β adrenergic receptor where the lengthening of TMH5 by two helical turns and a 14 Å outward movement of TMH6 is caused by the interaction of its endogenous ligand [70]. One possible mechanism by which the inhibition of signaling can be carried out by an SML antagonist is by stabilization of the ‘ionic lock’, a polar interaction between an arginine (R) located at the bottom of TMH3 which is part of a conserved, recurrent D/E-R-Y/W motif (aspartic acid/glutamate – arginine –tyrosine/tryptophan) and the partly conserved glutamate (E)/aspartic acid (D) at the bottom of TMH6 (Figure 4). On the other hand, molecules that are agonists to the TSHR may activate the receptor by destabilizing the ionic lock [70–72] and polar interaction between the helices. Therefore, such allosteric changes in the receptor may result in positive or negative modulation of the receptor and such molecules are commonly referred to as positive allosteric regulators or negative allosteric modulators. Such a generic mechanism of action may be true for TSH and TSHR-stimulating antibodies but the exact molecular evidence for conformational changes in the TMD as result of orthosteric binding is still awaited.
Figure 4. Mechanism of SML action.
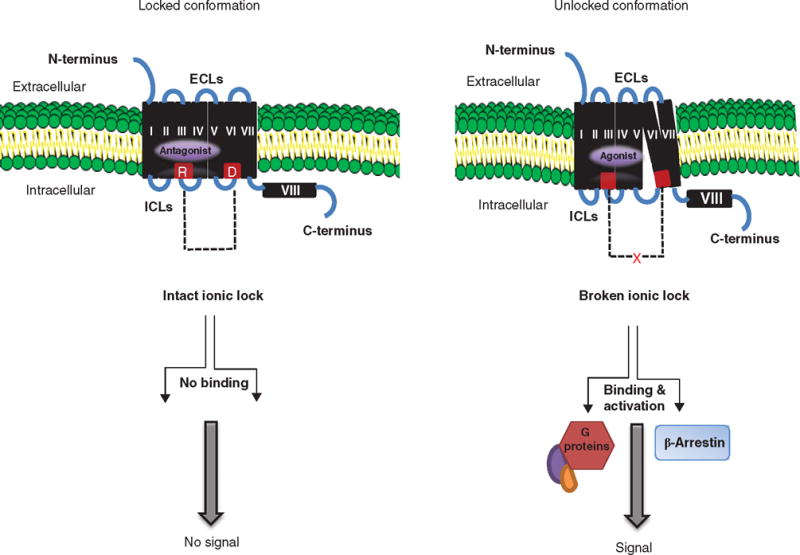
The basal state of TSHR activity is hypothesized to be constrained by polar interactions between different TMHs forming an ‘ionic lock’ between an arginine (R) at the base of transmembrane TMH3 and partly conserved residues such as glutamate (E) or aspartic acid (D) at the base of TM6 as shown in red (left panel). When an SML antagonist binds to the allosteric pocket (purple) on the TMD, the ionic lock remains intact even if TSH ligand binds to the ectodomain because there can be no outward movement of TMH6. Hence, the basal conformation of the receptor is locked and stabilized. In contrast, when a SML agonist binds to the allosteric pocket (right panel), there is an outward movement of TMH6 as a result of destabilization of the ‘ionic lock’ and loss of constrained polar interactions. This movement causes conformational changes in the ICL, which, in turn, allow binding of G proteins and β-arrestin activation leading to signal transduction.
Reproduced with permission from [72].
ECL: Extracellular loops; ICL: Intracellular loops; SML: Small molecule ligands; TMD: Transmembrane domain; TMH: Transmembrane helice; TSHR: Thyroid-stimulating hormone receptor.
Crystallography and molecular modeling approaches have provided insight into critical aspects of the binding and activation process of both endogenous ligands and SMLs [71] which have provided structural explanations for the role of specific sequences and residues within the TMDs of these receptors. The TSHR TMD is rich with known activating and inactivating mutations in the different helices as well as the intracellular and ECL. Model-driven site-directed mutagenesis studies [61,73] have indicated that these signaling-sensitive amino acids often correspond with SML binding sites for agonist and antagonist SMLs, distributed mostly in TMH 1 – 7 and ECL2 forming two main structural clusters [73]. The greater diversity of these allosteric binding sites, in contrast to orthosteric sites, has been of great advantage in not only redesigning molecules by chemical modifications in order to improve their potency and specificity, but also provided more discriminative pharmacophores [73]. Therefore, the ‘hot spots’, both activating and inactivating mutations, within the TSHR TMD offer the possibility of finding newer more powerful agonists and antagonists using molecular modeling and HTS.
9. TSHR antibodies
The classical biochemical features of hyperthyroid GD (elevated thyroid hormone levels and low to undetectable TSH) arise from the action of stimulating TSHR autoantibodies, which are oligoclonal and usually belong to the IgG1 subclass. Their action is to stimulate the synthesis of iodinized thyroglobulin with subsequent release of thyroid hormones. In spite of actions similar to TSH, S-TSHR-Abs harbor some distinct features including the different pharmacodynamics of IgG molecules resulting in a prolonged action when compared to TSH itself, and hence their original name of long-acting thyroid stimulators. These antibodies are easily detectable in patients but not in normal healthy controls [74], and we now know that they may come in three different flavors: stimulating, blocking and neutral antibodies depending on how they interact with the TSHR ectodomain and all three may be seen in the same patient (Figure 5).
Figure 5. Three types of TSHR-Abs.
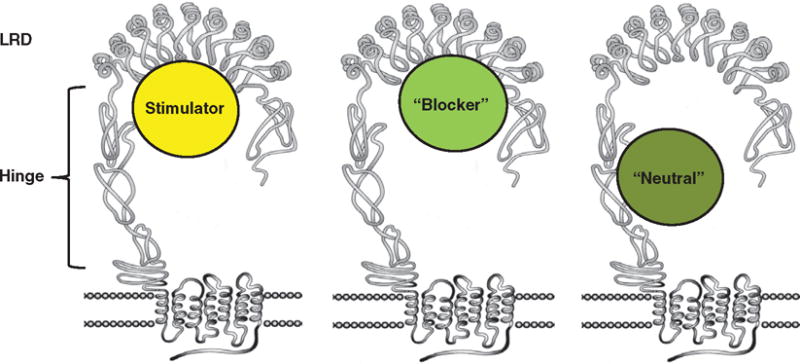
The receptor stimulating antibodies and blocking antibodies as described bind to the leucine-rich domain in the ectodomain. Stimulating antibody lead to activation of Gs and cAMP, whereas the blocking antibodies inhibit TSH from binding. The neutrals bind to the ‘hinge region’ of the receptor and are capable of MAPK signaling. We have made three different types of hamster mAbs to the TSHR. Stimulating antibody binds with the TSHR ectodomain, whereas the neutral variety recognizes hinge region. We are interested to study their vesicular trafficking and signaling.
TSHR: Thyroid-stimulating hormone receptor.
TSHR antibody binding can lead to selective signaling via activation of different G proteins because the TSHR, unlike other GPCRs, is promiscuous in the recruitment and activation of all four subfamilies of G proteins (Gs, Gi/o, Gq/11 and G12/13). S-TSHR-Abs induce thyroid epithelial cell proliferation and function via both Gs- and Gq/11-coupled signaling pathways, while ‘blocking’ antibodies (B-TSHR-Abs) inhibit the action of TSH but may also act as weak agonists. In contrast, the antibody type poorly named ‘neutral’ TSHR-Abs (N-TSHR-Abs) are unable to activate cAMP via Gsα but are capable of initiating a cascade of signaling imprints resulting in programmed cell death [75]. The conformational binding site for S-TSHR-Abs and some B-TSHR-Abs mainly involves the LRR region of the TSHR ECD [9,75,76]. In contrast, the linear epitopes recognized by N-TSHR-Abs are often confined to the hinge region of the ectodomain (especially residues 316 – 366) [75] suggesting a possible role for proteolytic cleavage in the generation of antibodies with linear epitopes. The frequency of N-TSHR-Abs in GD has been reported as ranging from 30 to 90% [75,77–82] but their pathophysiological significance remains poorly characterized and their presence is not routinely measured in the clinical situation.
10. Monoclonal TSHR antibodies
mAbs are a promising and rapidly growing category of targeted therapeutics entering clinical study. Although ~ 150 mAbs in various categories have entered clinical study the need for antibodies as therapeutics in combating GD remains lacking even after the discovery of the first thyroid-stimulating antibody 50 years ago. The generation of murine and human mAbs with the characteristics of TSHR-Abs [83,84] followed by the crystallization of human-stimulating and blocking mAbs bound to a partial ectodomain of the TSHR [9,76] has shown the precise binding sites of these antibodies and advanced our understanding of their possible mode of action. On binding to the receptor, the stimulating antibody induces a conformational change to the holoreceptor leading to activation of Gsα, which in-turn leads to generation of cAMP within the cells. Conversely, blocking antibodies keep the receptor from becoming activated, whereas neutral antibodies bind to the hinge region of the receptor and are incapable of generating cAMP. Recent studies from our own laboratory have shown these neutral antibodies to be MAPK activators [75].
To date, two human mAbs with TSHR agonist activity (M22 and K1-18) and one human mAb (K1-70) with antagonist activity have been characterized in the laboratory of Bernard Rees Smith. Although M22 and K1-70 bind to the concave surface of TSHR 1–260, as shown in the crystal structure, it was also clear that the K1-70 Fab binding sites were positioned more N terminally than the M22 Fab sites simply indicating that all antibodies have their own unique epitopes. Importantly, our studies using the approach of antibody protection and mass spectrometry on the entire ectodomain (TSHR 1–412) have indicated that there are additional binding sites outside the TSHR 1–260 region and implicating the role of the hinge region in activation of the receptor [13]. The lack of an active or inactive crystal structure of the full holoreceptor leaves gaps in our complete understanding of the conformational changes in the TSHR following binding of these autoantibodies.
11. TSHR antibodies as therapeutics
There is great promise in using antibodies as therapeutics targeting the TSHR to counteract the effects of GD by blocking stimulating antibodies from binding to the receptor and if possible not curtailing the action of endogenous TSH. To date, this approach has been considered too costly to develop when simple treatments such as antithyroid drugs are available and when the complications from such treatment remain unknown. While this logic may also apply to SMLs, the production of mAbs is far more expensive, but should still be pursued because of their predictable success in controlling thyroid function and their potential in GO. The use of clever design and small size nanobodies that contain the unique structural and functional properties of naturally occurring heavy-chain antibodies does offer the prospect of a simpler approach. The nanobody was originally developed following the discovery that camels and llamas possess fully functional antibodies that lack light chains. These heavy-chain antibodies contain a single variable domain (VHH) and two constant domains (CH2 and CH3). Importantly, the cloned and isolated VHH domain is a perfectly stable polypeptide harboring the full antigen-binding capacity of the original heavy-chain antibody. These newly discovered VHH domains with their unique structural and functional properties may form the basis of a new generation of therapeutic antibodies for GD.
12. Extrathyroidal TSHRs
Expression of the TSHR is not restricted to the thyroid but is expressed in a wide variety of tissues such as bone, fibroblasts, adipocytes, immune cells and brain [74]. We need to consider the consequences of such expression when considering the TSHR as a therapeutic target, although only limited data are available.
A well-established role of the receptor is in bone cells where it acts as a negative regulator of osteclast activity and a positive regulator of osteoblasts leading to an osteoprotective influence and where long-term TSHR blockade may induce bone loss [85]. In adipocytes, TSH is known to induce adipogenesis as seen in the retro-orbit [86] and other model systems [87].
The TSHR has attracted a lot of attention in the retro-orbit [88]. This is because the Grave’s disease Triad consists of thyroid disease with a dermopathy referred to as PTM and a retro-orbital inflammatory condition referred to as GO. There is considerable evidence that retro-orbital expression of the TSHR in the fibroblasts and adipocytes behind the eye may contribute to this difficult to treat orbitopathy and serum TSHR-Ab levels tend to correlate with eye disease. It has been hypothesized that blockade of the receptor may be a useful mode of therapy [89]. However, it is not clear that just inhibiting TSHR signaling, rather than removing the TSHR antigen from immune presentation, would help such a disease. One way such blockade could help is by reducing TSHR-Ab-induced cytokine release from retro-orbital fibroblasts. Such cytokines contribute to glycosaminoglycan generation and disrupt the osmotic pressure behind the eyes causing muscle fiber damage and swelling [90,91].
13. Expert opinion
The TSHR located on the basolateral surface of thyrocytes plays a key role in the growth and function of the thyroid gland. It is the primary antigenic target of T cells and unique stimulating autoantibodies in the autoimmune hyperthyroidism known as GD and undergoes a variety of genomic and somatic activating and inactivating mutations. In addition, the role of the TSHR in regulating the expression and activation of the sodium-iodide symporter in the uptake of radioiodine is of paramount importance in the detection and ablation of thyroid cancer. Furthermore, added to this conventional role of the TSHR are the recent discoveries of the widespread distribution of the receptor in extrathyroidal tissues such as adipocytes, thymocytes, fibroblasts, bone cells and more. The involvement of extrathyroidal manifestations, especially in GO and PTMa, also implicates these sites as important therapeutic targets.
The pioneering technical advances in drug screening methods using automated HTS and in silico development combined with the structural and functional insight of GPCRs facilitated by crystallization have led to a surge in drug discovery associated with GPCRs such as the TSHR. We believe that stabilization of a preferential conformation of the TSHR by allosteric ligands and mAbs with selective modulation of the signaling pathways may be the next generation of therapeutics for controlling the pathophysiological consequences in the thyroid and other extrathyroidal tissues. The first small molecule agonists have paved the way for structural modifications to either activate it or inhibit its signaling with even greater potency. The ease with which such small molecules can be produced for therapeutic use and the potential for chemical modification makes such small molecules highly attractive compared to mAbs. However, parallel technological developments in the cloning of CDR regions from specific antibodies, the use of humanized mice, and the use of nanobodies which have the potential to permeate cell membranes due to their small size may also enhance the potential of mAbs as another therapeutic option in targeting the receptor. The recent development of TSH containing nanoliposomes for thyroid cell targeting may also prove to be an important avenue for further development [92]. However, unlike other GHRs (such as the LH and FSH receptors) the TSHR poses a more challenging therapeutic target due to its ability to exist in different forms on the cell surface secondary to post translational cleavage and shedding. Additionally, the ability of the receptor to exist as higher order forms (multimers) and the selective signaling that may result due to manipulation of this heterogenous receptor repertoire are challenges that have to be incorporated and addressed when targeting the receptor in specific tissues. Therefore, the structural uniqueness and the complexity of the TSHR cannot be ignored in the development of therapeutics targeting the receptor in order to modulate its antigenicity and signaling capacity.
In summary, there remain exciting opportunities in the arena of thyroid diseases, especially GD, and these selective pharmacological tools will almost certainly contribute to the investigation of the physiologic and pathologic roles of the TSHR. Clearly, we have much to learn about the pharmacology and physiology of TSHR active small molecules and monocloanal antibodies and the journey is only just starting. As Winston Churchill famously stated, it is only ‘the beginning of the beginning’.
Article highlights.
Structure-function of the TSH receptor.
Small molecule therapeutics targeting the TSH receptor.
TSH receptor antibodies as therapeutic tools.
This box summarizes key points contained in the article.
Footnotes
Declaration of interest
The authors state no conflict of interest and have received no payment in preparation of this manuscript.
Bibliography
Papers of special note have been highlighted as either of interest (•) or of considerable interest (••) to readers.
- 1.Caltabiano G, Campillo M, De Leener A, et al. The specificity of binding of glycoprotein hormones to their receptors. Cell Mol Life Sci. 2008;65(16):2484–92. doi: 10.1007/s00018-008-8002-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Vassart G, Costagliola S. G protein-coupled receptors: mutations and endocrine diseases. Nat Rev Endocrinol. 2011;7(6):362–72. doi: 10.1038/nrendo.2011.20. [DOI] [PubMed] [Google Scholar]
- 3.Adams DD, Purves HD. The role of thyrotrophin in hyperthyroidism and exophthalmos. Metabolism. 1957;6(1):26–35. [PubMed] [Google Scholar]
- 4•.Smith BR, Hall R. Thyroid-stimulating immunoglobulins in Graves’ disease. Lancet. 1974;2(7878):427–31. doi: 10.1016/s0140-6736(74)91815-7. Extensive review describing the TSH receptor in detail and its interaction with autoantibodies. [DOI] [PubMed] [Google Scholar]
- 5.Rapoport B, Chazenbalk GD, Jaume JC, et al. The thyrotropin (TSH) receptor: interaction with TSH and autoantibodies. Endocr Rev. 1998;19(6):673–716. doi: 10.1210/edrv.19.6.0352. [DOI] [PubMed] [Google Scholar]
- 6••.Kleinau G, Neumann S, Gruters A, et al. Novel insights on thyroid-stimulating hormone receptor signal transduction. Endocr Rev. 2013;34(5):691–724. doi: 10.1210/er.2012-1072. A comprehensive review on TSHR signaling with current understanding of its structure. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kriss JP, Pleshakov V, Chien JR. Isolation and identification of the long-acting thyroid stimulator and its relation to hyperthyroidism and circumscribed pretibial myxedema. J Clin Endocrinol Metab. 1964;24:1005–28. doi: 10.1210/jcem-24-10-1005. [DOI] [PubMed] [Google Scholar]
- 8.Jeffreys J, Depraetere H, Sanders J, et al. Characterization of the thyrotropin binding pocket. Thyroid. 2002;12(12):1051–61. doi: 10.1089/105072502321085144. [DOI] [PubMed] [Google Scholar]
- 9••.Sanders J, Chirgadze DY, Sanders P, et al. Crystal structure of the TSH receptor in complex with a thyroid-stimulating autoantibody. Thyroid. 2007;17(5):395–410. doi: 10.1089/thy.2007.0034. Important study describing the crystal of the TSHR ectodomain. [DOI] [PubMed] [Google Scholar]
- 10.Biebermann H, Winkler F, Kleinau G. Genetic defects, thyroid growth and malfunctions of the TSHR in pediatric patients. Front Biosci. 2010;15:913–33. doi: 10.2741/3654. [DOI] [PubMed] [Google Scholar]
- 11.Wonerow P, Neumann S, Gudermann T, et al. Thyrotropin receptor mutations as a tool to understand thyrotropin receptor action. J Mol Med. 2001;79(12):707–21. doi: 10.1007/s001090100279. [DOI] [PubMed] [Google Scholar]
- 12.Jiang X, Fischer D, Chen X, et al. Evidence for Follicle-stimulating Hormone Receptor as a Functional Trimer. J Biol Chem. 2014;289(20):14273–82. doi: 10.1074/jbc.M114.549592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Mizutori Y, Chen CR, McLachlan SM, et al. The thyrotropin receptor hinge region is not simply a scaffold for the leucine-rich domain but contributes to ligand binding and signal transduction. Mol Endocrinol. 2008;22(5):1171–82. doi: 10.1210/me.2007-0407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kosugi S, Mori T. Cysteine-699, a possible palmitoylation site of the thyrotropin receptor, is not crucial for cAMP or phosphoinositide signaling but is necessary for full surface expression. Biochem Biophys Res Commun. 1996;221(3):636–40. doi: 10.1006/bbrc.1996.0648. [DOI] [PubMed] [Google Scholar]
- 15.Tanaka K, Nagayama Y, Nishihara E, et al. Palmitoylation of human thyrotropin receptor: slower intracellular trafficking of the palmitoylation-defective mutant. Endocrinology. 1998;139(2):803–6. doi: 10.1210/endo.139.2.5911. [DOI] [PubMed] [Google Scholar]
- 16.Tanaka K, Chazenbalk GD, McLachlan SM, et al. The shed thyrotropin receptor is primarily a carboxyl terminal truncated form of the A subunit, not the entire A subunit. Mol Cell Endocrinol. 1999;150(1–2):113–19. doi: 10.1016/s0303-7207(99)00018-0. [DOI] [PubMed] [Google Scholar]
- 17•.Latif R, Graves P, Davies TF. Oligomerization of the human thyrotropin receptor: fluorescent protein-tagged hTSHR reveals post-translational complexes. J Biol Chem. 2001;276(48):45217–24. doi: 10.1074/jbc.M103727200. The original paper that reports oligomerization of the TSH receptor and its function. [DOI] [PubMed] [Google Scholar]
- 18•.Urizar E, Montanelli L, Loy T, et al. Glycoprotein hormone receptors: link between receptor homodimerization and negative cooperativity. EMBO J. 2005;24(11):1954–64. doi: 10.1038/sj.emboj.7600686. The original paper that reports oligomerization of the TSH receptor and its function. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kursawe R, Paschke R. Modulation of TSHR signaling by posttranslational modifications. Trends Endocrinol Metab. 2007;18(5):199–207. doi: 10.1016/j.tem.2007.05.002. [DOI] [PubMed] [Google Scholar]
- 20.Latif R, Michalek K, Davies TF. Subunit interactions influence TSHR multimerization. Mol Endocrinol. 2010;24(10):2009–18. doi: 10.1210/me.2010-0001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Latif R, Ali MR, Mezei M, Davies TF. Transmembrane domains of attraction on the TSH receptor. Endocrinology. 2015;156(2):488–98. doi: 10.1210/en.2014-1509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Calebiro D, de Filippis T, Lucchi S, et al. Intracellular entrapment of wild-type TSH receptor by oligomerization with mutants linked to dominant TSH resistance. Hum Mol Genet. 2005;14(20):2991–3002. doi: 10.1093/hmg/ddi329. [DOI] [PubMed] [Google Scholar]
- 23.Allen MD, Neumann S, Gershengorn MC. Occupancy of both sites on the thyrotropin (TSH) receptor dimer is necessary for phosphoinositide signaling. FASEB J. 2011;25(10):3687–94. doi: 10.1096/fj.11-188961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Graves PN, Vlase H, Davies TF. Folding of the recombinant human thyrotropin (TSH) receptor extracellular domain: identification of folded monomeric and tetrameric complexes that bind TSH receptor autoantibodies. Endocrinology. 1995;136(2):521–7. doi: 10.1210/endo.136.2.7530646. [DOI] [PubMed] [Google Scholar]
- 25.Latif R, Ando T, Davies TF. Lipid rafts are triage centers for multimeric and monomeric thyrotropin receptor regulation. Endocrinology. 2007;148(7):3164–75. doi: 10.1210/en.2006-1580. [DOI] [PubMed] [Google Scholar]
- 26.Frenzel R, Voigt C, Paschke R. The human thyrotropin receptor is predominantly internalized by β-arrestin 2. Endocrinology. 2006;147(6):3114–22. doi: 10.1210/en.2005-0687. [DOI] [PubMed] [Google Scholar]
- 27.Werthmann RC, Volpe S, Lohse MJ, et al. Persistent cAMP signaling by internalized TSH receptors occurs in thyroid but not in HEK293 cells. FASEB J. 2012;26(5):2043–8. doi: 10.1096/fj.11-195248. [DOI] [PubMed] [Google Scholar]
- 28.Calebiro D, Nikolaev VO, Gagliani MC, et al. Persistent cAMP-signals triggered by internalized G-protein-coupled receptors. PLoS Biol. 2009;7(8):e1000172. doi: 10.1371/journal.pbio.1000172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Chazenbalk GD, Nagayama Y, Kaufman KD, et al. The functional expression of recombinant human thyrotropin receptors in nonthyroidal eukaryotic cells provides evidence that homologous desensitization to thyrotropin stimulation requires a cell-specific factor. Endocrinology. 1990;127(3):1240–4. doi: 10.1210/endo-127-3-1240. [DOI] [PubMed] [Google Scholar]
- 30.Nagayama Y, Chazenbalk GD, Takeshita A, et al. Studies on homologous desensitization of the thyrotropin receptor in 293 human embryonal kidney cells. Endocrinology. 1994;135(3):1060–5. doi: 10.1210/endo.135.3.8070347. [DOI] [PubMed] [Google Scholar]
- 31.Zhang M, Tong KP, Fremont V, et al. The extracellular domain suppresses constitutive activity of the transmembrane domain of the human TSH receptor: implications for hormone-receptor interaction and antagonist design. Endocrinology. 2000;141(9):3514–17. doi: 10.1210/endo.141.9.7790. [DOI] [PubMed] [Google Scholar]
- 32.Zaballos MA, Garcia B, Santisteban P. Gbetagamma dimers released in response to thyrotropin activate phosphoinositide 3-kinase and regulate gene expression in thyroid cells. Mol Endocrinol. 2008;22(5):1183–99. doi: 10.1210/me.2007-0093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Allgeier A, Offermanns S, Van Sande J, et al. The human thyrotropin receptor activates G-proteins Gs and Gq/11. J Biol Chem. 1994;269(19):13733–5. [PubMed] [Google Scholar]
- 34.Laurent E, Mockel J, Van Sande J, et al. Dual activation by thyrotropin of the phospholipase C and cyclic AMP cascades in human thyroid. Mol Cell Endocrinol. 1987;52(3):273–8. doi: 10.1016/0303-7207(87)90055-4. [DOI] [PubMed] [Google Scholar]
- 35••.Morshed SA, Latif R, Davies TF. Characterization of thyrotropin receptor antibody-induced signaling cascades. Endocrinology. 2009;150(1):519–29. doi: 10.1210/en.2008-0878. Review detailing the role of TSH receptor signaling in cancer. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Garcia-Jimenez C, Santisteban P. TSH signalling and cancer. Arq Bras Endocrinol Metabol. 2007;51(5):654–71. doi: 10.1590/s0004-27302007000500003. [DOI] [PubMed] [Google Scholar]
- 37.Russo D, Nagayama Y, Chazenbalk GD, et al. Role of amino acids 261–418 in proteolytic cleavage of the extracellular region of the human thyrotropin receptor. Endocrinology. 1992;130(4):2135–8. doi: 10.1210/endo.130.4.1547731. [DOI] [PubMed] [Google Scholar]
- 38.Wadsworth HL, Russo D, Nagayama Y, et al. Studies on the role of amino acids 38–45 in the expression of a functional thyrotropin receptor. Mol Endocrinol. 1992;6(3):394–8. doi: 10.1210/mend.6.3.1584215. [DOI] [PubMed] [Google Scholar]
- 39.Wadsworth HL, Chazenbalk GD, Nagayama Y, et al. An insertion in the human thyrotropin receptor critical for high affinity hormone binding. Science. 1990;249(4975):1423–5. doi: 10.1126/science.2169649. [DOI] [PubMed] [Google Scholar]
- 40.Chazenbalk GD, Jaume JC, McLachlan SM, et al. Engineering the human thyrotropin receptor ectodomain from a non-secreted form to a secreted, highly immunoreactive glycoprotein that neutralizes autoantibodies in Graves’ patients’ sera. J Biol Chem. 1997;272(30):18959–65. doi: 10.1074/jbc.272.30.18959. [DOI] [PubMed] [Google Scholar]
- 41.Loosfelt H, Pichon C, Jolivet A, et al. Two-subunit structure of the human thyrotropin receptor. Proc Natl Acad Sci USA. 1992;89(9):3765–9. doi: 10.1073/pnas.89.9.3765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Couet J, de Bernard S, Loosfelt H, et al. Cell surface protein disulfide-isomerase is involved in the shedding of human thyrotropin receptor ectodomain. Biochemistry. 1996;35(47):14800–5. doi: 10.1021/bi961359w. [DOI] [PubMed] [Google Scholar]
- 43.Couet J, Sar S, Jolivet A, et al. Shedding of human thyrotropin receptor ectodomain. Involvement of a matrix metalloprotease. J Biol Chem. 1996;271(8):4545–52. doi: 10.1074/jbc.271.8.4545. [DOI] [PubMed] [Google Scholar]
- 44.Latif R, Ando T, Davies TF. Monomerization as a prerequisite for intramolecular cleavage and shedding of the thyrotropin receptor. Endocrinology. 2004;145(12):5580–8. doi: 10.1210/en.2004-0797. [DOI] [PubMed] [Google Scholar]
- 45.Misrahi M, Couet J, Milgrom E. Mechanisms of shedding of a soluble form of the TSH receptor. Ann Endocrinol (Paris) 1997;58(5):365–9. [PubMed] [Google Scholar]
- 46.Boelaert K. Thyroid gland: revised guidelines for the management of thyroid cancer. Nat Rev Endocrinol. 2010;6(4):185–6. doi: 10.1038/nrendo.2010.17. [DOI] [PubMed] [Google Scholar]
- 47.Boelaert K. Treatment of Graves’ disease with antithyroid drugs: current perspectives. Thyroid. 2010;20(9):943–6. doi: 10.1089/thy.2010.1654. [DOI] [PubMed] [Google Scholar]
- 48.Galofre JC, Duntas LH, Premawardhana LD, et al. Advances in graves’ disease. J Thyroid Res. 2012;2012:809231. doi: 10.1155/2012/809231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Bahn RS, Burch HS, Cooper DS, et al. The Role of Propylthiouracil in the Management of Graves’ Disease in Adults: report of a meeting jointly sponsored by the American Thyroid Association and the Food and Drug Administration. Thyroid. 2009;19(7):673–4. doi: 10.1089/thy.2009.0169. [DOI] [PubMed] [Google Scholar]
- 50.Karlsson FA, Axelsson O, Melhus H. Severe embryopathy and exposure to methimazole in early pregnancy. J Clin Endocrinol Metab. 2002;87(2):947–9. doi: 10.1210/jcem.87.2.8120. [DOI] [PubMed] [Google Scholar]
- 51.Bahn RS, Burch HB, Cooper DS, et al. Hyperthyroidism and other causes of thyrotoxicosis: management guidelines of the American Thyroid Association and American Association of Clinical Endocrinologists. Endocr Pract. 2011;17(3):456–520. doi: 10.4158/ep.17.3.456. [DOI] [PubMed] [Google Scholar]
- 52.Haugen BR, Pacini F, Reiners C, et al. A comparison of recombinant human thyrotropin and thyroid hormone withdrawal for the detection of thyroid remnant or cancer. J Clin Endocrinol Metab. 1999;84(11):3877–85. doi: 10.1210/jcem.84.11.6094. [DOI] [PubMed] [Google Scholar]
- 53.Huber GK, Fong P, Concepcion ES, et al. Recombinant human thyroid-stimulating hormone: initial bioactivity assessment using human fetal thyroid cells. J Clin Endocrinol Metab. 1991;72(6):1328–31. doi: 10.1210/jcem-72-6-1328. [DOI] [PubMed] [Google Scholar]
- 54.Ladenson PW, Braverman LE, Mazzaferri EL, et al. Comparison of administration of recombinant human thyrotropin with withdrawal of thyroid hormone for radioactive iodine scanning in patients with thyroid carcinoma [see comments] N Engl J Med. 1997;337(13):888–96. doi: 10.1056/NEJM199709253371304. [DOI] [PubMed] [Google Scholar]
- 55.Mazzaferri EL, Kloos RT. Is diagnostic iodine-131 scanning with recombinant human TSH useful in the follow-up of differentiated thyroid cancer after thyroid ablation? J Clin Endocrinol Metab. 2002;87(4):1490–8. doi: 10.1210/jcem.87.4.8338. [DOI] [PubMed] [Google Scholar]
- 56.Mueller S, Kleinau G, Szkudlinski MW, et al. The superagonistic activity of bovine thyroid-stimulating hormone (TSH) and the human TR1401 TSH analog is determined by specific amino acids in the hinge region of the human TSH receptor. J Biol Chem. 2009;284(24):16317–24. doi: 10.1074/jbc.M109.005710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Guo T. Small molecule agonists and antagonists for the LH and FSH receptors. Expert Opinion Ther Targets. 2005;15(11):1555–63. [Google Scholar]
- 58.Jaschke H, Neumann S, Moore S, et al. A low molecular weight agonist signals by binding to the transmembrane domain of thyroid-stimulating hormone receptor (TSHR) and luteinizing hormone/chorionic gonadotropin receptor (LHCGR) J Biol Chem. 2006;281(15):9841–4. doi: 10.1074/jbc.C600014200. [DOI] [PubMed] [Google Scholar]
- 59•.Neumann S, Huang W, Titus S, et al. Small-molecule agonists for the thyrotropin receptor stimulate thyroid function in human thyrocytes and mice. Proc Natl Acad Sci USA. 2009;106(30):12471–6. doi: 10.1073/pnas.0904506106. Study characterizing two novel small molecule TSHR agonists. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Latif R, Ali MR, Ma R, et al. New small molecule agonists to the thyrotropin receptor. Thyroid. 2015;25(1):51–62. doi: 10.1089/thy.2014.0119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Kleinau G, Haas AK, Neumann S, et al. Signaling-sensitive amino acids surround the allosteric ligand binding site of the thyrotropin receptor. FASEB J. 2010;24(7):2347–54. doi: 10.1096/fj.09-149146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Rossi M, Dimida A, Dell’anno MT, et al. The thyroid disruptor 1,1,1-trichloro-2,2-bis(p-chlorophenyl)-ethane appears to be an uncompetitive inverse agonist for the thyrotropin receptor. J Pharmacol Exp Ther. 2007;320(1):465–74. doi: 10.1124/jpet.106.113613. [DOI] [PubMed] [Google Scholar]
- 63.Rossi M, Dimida A, Ferrarini E, et al. Presence of a putative steroidal allosteric site on glycoprotein hormone receptors. Eur J Pharmacol. 2009;623(1–3):155–9. doi: 10.1016/j.ejphar.2009.09.029. [DOI] [PubMed] [Google Scholar]
- 64.Neumann S, Kleinau G, Costanzi S, et al. A low-molecular-weight antagonist for the human thyrotropin receptor with therapeutic potential for hyperthyroidism. Endocrinology. 2008;149(12):5945–50. doi: 10.1210/en.2008-0836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.van Koppen CJ, Zaman GJ, Timmers CM, et al. A signaling-selective, nanomolar potent allosteric low molecular weight agonist for the human luteinizing hormone receptor. Naunyn Schmiedebergs Arch Pharmacol. 2008;378(5):503–14. doi: 10.1007/s00210-008-0318-3. [DOI] [PubMed] [Google Scholar]
- 66.Neumann S, Eliseeva E, McCoy JG, et al. A new small-molecule antagonist inhibits Graves’ disease antibody activation of the TSH receptor. J Clin Endocrinol Metab. 2011;96(2):548–54. doi: 10.1210/jc.2010-1935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Neumann S, Pope A, Geras-Raaka E, et al. A drug-like antagonist inhibits thyrotropin receptor-mediated stimulation of cAMP production in Graves’ orbital fibroblasts. Thyroid. 2012;22(8):839–43. doi: 10.1089/thy.2011.0520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Rees Smith B, Sanders J, Evans M. TSH receptor - autoantibody interactions. Horm Metab Res. 2009;41(6):448–55. doi: 10.1055/s-0029-1220913. [DOI] [PubMed] [Google Scholar]
- 69.Nunez Miguel R, Sanders J, Chirgadze DY, et al. Thyroid stimulating autoantibody M22 mimics TSH binding to the TSH receptor leucine rich domain: a comparative structural study of protein-protein interactions. J Mol Endocrinol. 2009;42(5):381–95. doi: 10.1677/JME-08-0152. [DOI] [PubMed] [Google Scholar]
- 70.Benovic JL. G-protein-coupled receptors signal victory. Cell. 2012;151(6):1148–50. doi: 10.1016/j.cell.2012.11.015. [DOI] [PubMed] [Google Scholar]
- 71.Audet M, Bouvier M. Restructuring G-protein- coupled receptor activation. Cell. 2012;151(1):14–23. doi: 10.1016/j.cell.2012.09.003. [DOI] [PubMed] [Google Scholar]
- 72.Davies TF, Ali MR, Latif R. Allosteric modulators hit the TSH receptor. Endocrinology. 2014;155(1):1–5. doi: 10.1210/en.2013-2079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Haas AK, Kleinau G, Hoyer I, et al. Mutations that silence constitutive signaling activity in the allosteric ligand-binding site of the thyrotropin receptor. Cell Mol Life Sci. 2011;68(1):159–67. doi: 10.1007/s00018-010-0451-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Davies T, Marians R, Latif R. The TSH receptor reveals itself. J Clin Invest. 2002;110(2):161–4. doi: 10.1172/JCI16234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Morshed SA, Ando T, Latif R, Davies TF. Neutral antibodies to the TSH receptor are present in Graves’ disease and regulate selective signaling cascades. Endocrinology. 2010;151(11):5537–49. doi: 10.1210/en.2010-0424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76•.Sanders P, Young S, Sanders J, et al. Crystal structure of the TSH receptor (TSHR) bound to a blocking-type TSHR autoantibody. J Mol Endocrinol. 2011;46(2):81–99. doi: 10.1530/JME-10-0127. Recent review outlining the autoimmune mechanism in Graves’ disease. [DOI] [PubMed] [Google Scholar]
- 77.Morshed SA, Latif R, Davies TF. Delineating the autoimmune mechanisms in Graves’ disease. Immunol Res. 2012;54(1–3):191–203. doi: 10.1007/s12026-012-8312-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Vlase H, Graves PN, Magnusson RP, et al. Human autoantibodies to the thyrotropin receptor: recognition of linear, folded, and glycosylated recombinant extracellular domain. J Clin Endocrinol Metab. 1995;80(1):46–53. doi: 10.1210/jcem.80.1.7829638. [DOI] [PubMed] [Google Scholar]
- 79.Mori T, Sugawa H, Piraphatdist T, et al. A synthetic oligopeptide derived from human thyrotropin receptor sequence binds to Graves’ immunoglobulin and inhibits thyroid stimulating antibody activity but lacks interactions with TSH. Biochem Biophys Res Commun. 1991;178(1):165–72. doi: 10.1016/0006-291x(91)91794-d. [DOI] [PubMed] [Google Scholar]
- 80.Endo T, Ohmori M, Ikeda M, et al. Heterogeneous responses of recombinant human thyrotropin receptor to immunoglobulins from patients with Graves’ disease. Biochem Biophys Res Commun. 1992;186(3):1391–6. doi: 10.1016/s0006-291x(05)81560-8. [DOI] [PubMed] [Google Scholar]
- 81.Nagy EV, Burch HB, Mahoney K, et al. Graves’ IgG recognizes linear epitopes in the human thyrotropin receptor. Biochem Biophys Res Commun. 1992;188(1):28–33. doi: 10.1016/0006-291x(92)92345-x. [DOI] [PubMed] [Google Scholar]
- 82.Takai O, Desai RK, Seetharamaiah GS, et al. Prokaryotic expression of the thyrotropin receptor and identification of an immunogenic region of the protein using synthetic peptides. Biochem Biophys Res Commun. 1991;179(1):319–26. doi: 10.1016/0006-291x(91)91372-j. [DOI] [PubMed] [Google Scholar]
- 83.Ando T, Latif R, Pritsker A, et al. A monoclonal thyroid-stimulating antibody. J Clin Invest. 2002;110(11):1667–74. doi: 10.1172/JCI16991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Sanders J, Evans M, Premawardhana LD, et al. Human monoclonal thyroid stimulating autoantibody. Lancet. 2003;362(9378):126–8. doi: 10.1016/s0140-6736(03)13866-4. [DOI] [PubMed] [Google Scholar]
- 85.Abe E, Marians RC, Yu W, et al. TSH is a negative regulator of skeletal remodeling. Cell. 2003;115(2):151–62. doi: 10.1016/s0092-8674(03)00771-2. [DOI] [PubMed] [Google Scholar]
- 86.Kumar S, Nadeem S, Stan MN, et al. A stimulatory TSH receptor antibody enhances adipogenesis via phosphoinositide 3-kinase activation in orbital preadipocytes from patients with Graves’ ophthalmopathy. J Mol Endocrinol. 2011;46(3):155–63. doi: 10.1530/JME-11-0006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Lu M, Lin RY. TSH stimulates adipogenesis in mouse embryonic stem cells. J Endocrinol. 2008;196(1):159–69. doi: 10.1677/JOE-07-0452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Bahn RS. TSH receptor expression in orbital tissue and its role in the pathogenesis of Graves’ ophthalmopathy. J Endocrinol Invest. 2004;27(3):216–20. doi: 10.1007/BF03345269. [DOI] [PubMed] [Google Scholar]
- 89.Rees Smith B, Nordmeyer P, Sanders J, et al. Eye signs of graves’ disease and blocking-type thyrotropin receptor autoantibodies. Thyroid. 2006;16(1):97–8. doi: 10.1089/thy.2006.16.97. [DOI] [PubMed] [Google Scholar]
- 90.Hansen C, Rouhi R, Forster G, et al. Increased sulfatation of orbital glycosaminoglycans in Graves’ ophthalmopathy. J Clin Endocrinol Metab. 1999;84(4):1409–13. doi: 10.1210/jcem.84.4.5609. [DOI] [PubMed] [Google Scholar]
- 91.Bahn RS. Graves’ ophthalmopathy. N Engl J Med. 2010;362(8):726–38. doi: 10.1056/NEJMra0905750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92•.Paolino D, Cosco D, Gaspari M, et al. Targeting the thyroid gland with thyroid-stimulating hormone (TSH)-nanoliposomes. Biomaterials. 2014;35(25):7101–9. doi: 10.1016/j.biomaterials.2014.04.088. Study characterizing a potent TSH receptor antagonist. [DOI] [PubMed] [Google Scholar]
- 93.Neumann S, Nir EA, Eliseeva E, et al. A selective TSH receptor antagonist inhibits stimulation of thyroid function in female mice. Endocrinology. 2014;155(1):310–14. doi: 10.1210/en.2013-1835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.van Koppen CJ, de Gooyer ME, Karstens WJ, et al. Mechanism of action of a nanomolar potent, allosteric antagonist of the thyroid-stimulating hormone receptor. Br J Pharmacol. 2012;165(7):2314–24. doi: 10.1111/j.1476-5381.2011.01709.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Davies TF, Yin X, Latif R. The genetics of the thyroid stimulating hormone receptor: history and relevance. Thyroid. 2010;20(7):727–36. doi: 10.1089/thy.2010.1638. [DOI] [PMC free article] [PubMed] [Google Scholar]