

Abstract
G protein-coupled receptors have been well described to contribute to the regulation of glucose-stimulated insulin secretion (GSIS). The short-chain fatty acid-sensing G protein-coupled receptor, free fatty acid receptor 2 (FFAR2), is expressed in pancreatic β-cells, and in rodents, its expression is altered during insulin resistance. Thus, we explored the role of FFAR2 in regulating GSIS. First, assessing the phenotype of wild-type and Ffar2−/− mice in vivo, we observed no differences with regard to glucose homeostasis on normal or high-fat diet, with a marginally significant defect in insulin secretion in Ffar2−/− mice during hyperglycemic clamps. In ex vivo insulin secretion studies, we observed diminished GSIS from Ffar2−/− islets relative to wild-type islets under high-glucose conditions. Further, in the presence of acetate, the primary endogenous ligand for FFAR2, we observed FFAR2-dependent potentiation of GSIS, whereas FFAR2-specific agonists resulted in either potentiation or inhibition of GSIS, which we found to result from selective signaling through either Gαq/11 or Gαi/o, respectively. Lastly, in ex vivo insulin secretion studies of human islets, we observed that acetate and FFAR2 agonists elicited different signaling properties at human FFAR2 than at mouse FFAR2. Taken together, our studies reveal that FFAR2 signaling occurs by divergent G protein pathways that can selectively potentiate or inhibit GSIS in mouse islets. Further, we have identified important differences in the response of mouse and human FFAR2 to selective agonists, and we suggest that these differences warrant consideration in the continued investigation of FFAR2 as a novel type 2 diabetes target.
The short-chain fatty acid (SCFA), acetate, has been suggested to contribute to a wide range of metabolic effects (1). These effects were initially suggested by folk medicine based on the medicinal use of vinegar, of which acetate is the major component (2–5). As a result, multiple translational studies have explored the medicinal qualities of vinegar, or more directly, acetate. In the case of type 2 diabetes (T2D), vinegar has been observed to lead to an improvement in overall glucose control (2–4). Additional studies, directly focused on acetate, have reported that acetate contributes to metabolism by improving insulin sensitivity, suppressing body fat accumulation, and regulating glucose-stimulated insulin secretion (GSIS) (2, 5–8). However, the mechanism of acetate action in these studies has not been defined.
One potential mode of action for acetate is through the SCFA-specific G protein-coupled receptor (GPCR), free fatty acid receptor 2 (FFAR2) (GPR43) (9). Although this GPCR can be activated by multiple SCFAs, it demonstrates highest selectivity for acetate (9), which is also the predominant SCFA in peripheral blood (10). Thus far, studies of FFAR2 have suggested its role in mediating aspects of metabolism and energy balance (11–14), which is not surprising given its expression in multiple metabolically active tissues. Ffar2 is also expressed in pancreatic β-cells, as demonstrated through gene expression (15–17) and immunofluorescence studies (14).
Along with its expression in β-cells, gene expression studies by our group and others have observed increased expression of Ffar2 in mouse islets during the insulin resistant phase of pregnancy (15, 17). Further, data presented in a patent application have reported up-regulated expression of Ffar2 in islets from genetic mouse models of obesity and diabetes (ob/ob and db/db, respectively) (16). Finally, using a gene database that extensively profiled mouse gene expression in multiple tissues (islets, adipose, skeletal muscle, and hypothalamus) as a function of obesity, strain (diabetes susceptible and diabetes resistant), and age (18), increased expression of Ffar2 is observed, regardless of T2D susceptibility, in islets from obese mice, an effect that is largely absent in other tissues profiled (see Supplemental Figure 1). Because enhancement of GSIS is a primary adaptive mechanism by which β-cells respond to insulin resistance, these expression data suggest a role for FFAR2 in the β-cell response to insulin resistance through enhancement of GSIS.
Nutrients, such as long-chain fatty acids and amino acids, have well-recognized roles in regulating GSIS, commonly resulting in potentiation of GSIS through receptor-dependent as well as receptor-independent pathways (19). SCFAs, the endogenous ligands for FFAR2, are another class of nutrients that have been suggested to regulate GSIS (20). However, much less investigation has occurred into whether SCFAs contributes to GSIS; and most published reports are over 30 years old. Interestingly, no clear consensus has been reached in determining whether acetate or other SCFAs augment or inhibit GSIS. Some studies have reported that acetate augments GSIS (6, 7), whereas others have found inhibition of GSIS (21). Likewise, in studies of other SCFAs, no clear pattern has emerged; one study reported propionate-mediated inhibition of GSIS (22) and another found that butyrate enhances GSIS (23). The discovery of FFAR2 and another SCFA receptor (FFAR3) that are both expressed in β-cells suggests that at least some of the effects of SCFAs on GSIS could be mediated by these receptors. A recent study reported that SCFA signaling through FFAR2 and FFAR3 inhibits GSIS (14), and in a study by our group, we confirmed that FFAR3 signaling is a negative regulator of GSIS (24).
Considering the discrepancies in the literature concerning the effect of acetate on GSIS and the potential role of FFAR2 in contributing to how β-cells respond to insulin resistance, we have explored the role of FFAR2 signaling in GSIS in both mouse and human islets. Using multiple FFAR2 agonists, we found that FFAR2 signaling can either augment or inhibit GSIS in mouse islets. Interrogating the mechanism of signaling by FFAR2 agonists, we observed that these opposing effects were due to the ability of mouse FFAR2 to signal by divergent G protein signaling pathways. Finally, we identified important differences in the mechanism of acetate and FFAR2 agonist signaling between mouse and human FFAR2 (hFFAR2) orthologs, which resulted in significant interspecies differences in the effects of FFAR2 agonists on GSIS. Because GPCRs are an important therapeutic class of receptors for T2D (25–27), these data provide novel insight into how FFAR2 signaling can mediate insulin secretion, and suggest that more study is needed to inform the utility of FFAR2 as a translatable T2D target.
Materials and Methods
Animals
Heterozygous Ffar2 knockout mice (Ffar2+/−) on a C57BL/6J background (11, 28) were used to produce Ffar2 wild-type (WT) and Ffar2 knockout (Ffar2−/−) mice. Genotype was determined by PCR as previously described (11). Male mice were housed in a temperature-controlled facility with 12-hour light-dark cycle and ad libitum access to normal chow (NC) (LM-485; Harlan Laboratories) and water. For high-fat diet (HFD) experiments, mice were given ad libitum access to high-fat chow (TD.06414; Harlan Laboratories) beginning at 6 weeks of age. Energy density of NC diet is 3.1 kcal/g and 5.1 kcal/g for high-fat chow (with ∼60% calories from fat). The experiments described here were approved by the Institutional Animal Care and Use Committees at Northwestern University and the Centre de Recherche du Centre Hospitalier de l'Université de Montréal.
Glucose tolerance test (GTT) and insulin tolerance test (ITT)
Oral GTT and ip GTT were performed on mice fasted overnight with glucose administered by ip injection or oral gavage (2 g/kg body weight or 1 g/kg for mice on HFD). ITTs were conducted on 6-hour fasted mice with insulin delivered by ip injection at 0.75 U/kg (Humalog; Eli Lilly). Blood was obtained from tail veins at multiple time points (0–120 min) for glucose determination (measured with a standard glucometer) and insulin levels by mouse anti-insulin enzyme-linked immunosorbent assay (ELISA) (ALPCO).
Metabolic assessment
Male mice aged 9–10 weeks were individually housed in automated metabolic cages (TSE Systems) for 5 days to monitor activity. After initial 48 hours of acclimatization, data were collected for 72 hours. Airflow through the cages was held at 0.25–0.35 L/min; 12-hour light, 12-hour dark cycles were maintained with ad libitum access to food and water. TSE LabMaster software recorded locomotor and metabolic activity in 30-minute intervals. Locomotor activity was monitored along x, y, and z axes. Metabolic activity was measured via indirect calorimetry recording maximal O2 consumption (VO2) and CO2 production (VCO2). VO2 and VCO2 values were normalized by the software to body weight in kilograms and are reported as mL/h·kg. Respiratory exchange ratio was calculated as VCO2/VO2. The data were analyzed for average light and dark activity.
Assessment of insulin secretion by hyperglycemic clamp
One-step hyperglycemic clamps were performed on conscious animals as previously described (29). A 20% dextrose solution was infused through the jugular vein to clamp plasma glucose at approximately 350 mg/dL for 60 minutes and was adjusted based on glucose measurements (Roche Accu-Check; Roche). At 60 minutes, an arginine bolus injection was performed (1 mmol/kg; Sandoz Canada) to assess the maximal insulin response. The glucose infusion rate (GIR) was calculated during the steady state (30–60 min into the clamp). Plasma samples were collected from the tail at several time points during the clamp for insulin measurements by ELISA (ALPCO).
Islet isolation and culture
Islets were isolated by collagenase digestion as previously described (30). Briefly, 3 mL of 0.8-mg/mL collagenase (Sigma Chemical Co) in Hanks' buffered salt solution (HBSS) was injected into the pancreas through the pancreatic duct. The inflated pancreas was excised, incubated for 17 minutes at 37°C, and passed through a 400-μm wire mesh. After digestion, pancreas was rinsed with ice-cold HBSS, and islets were separated by density gradient in Histopaque (Sigma). After several HBSS washes, islets were handpicked under a dissection microscope and left to recover overnight at 37°C in RPMI 1640 supplemented with 10% fetal bovine serum, 1% L-glutamine, and 1% penicillin/streptomycin.
Ex vivo insulin secretion
For each experiment, islets were isolated and pooled from 2 pancreata. For ex vivo GSIS assays, islets were first incubated in Krebs Ringer buffer (KRB) (130mM NaCl, 4.7mM KCl, 0.5mM NaH2PO4, 0.5mM MgSO4, 1.5mM CaCl2, 10mM HEPES, and 0.1% BSA; pH 7.4) for 30 minutes and then transferred to KRB medium supplemented with 2.8mM glucose for 60 minutes at 37°C. Groups of 5 islets were then batch incubated in 1 ml of KRB supplemented with either 16.7mM glucose or 16.7mM glucose with agonists for 60 minutes in a shaking water bath at 37°C. Agonists or GPCR pathway inhibitors included 100nM Exendin-4 (Sigma), 1mM sodium acetate (Sigma), 100μM propiolic acid (small carboxylic acid [SCA]14; Sigma), 100μM 2-butynoic acid (SCA15; Sigma), 100μM 4-chloro-α-(1-methylethyl)-N-2-thiazolylbenzeneacetamide (CMTB) (Tocris Bioscience), 100μM (S)-2-(4-chlorophenyl)-3,3-dimethyl-N-(5-phenylthiazol-2-yl)butanamide (CPTB) (Millipore), 300-ng/mL pertussis toxin (PTX) (Sigma), and/or 5μM U-73122 (Tocris Bioscience). At the end of the incubation period, an aliquot of the supernatant was sampled for measurement of insulin by an insulin ELISA (ALPCO).
Culture and treatment of human islets
Human islets were obtained through the Integrated Islet Distribution Program. After arrival, islets were immediately cultured in CMRL-1066 medium (Life Technologies) supplemented with 10% fetal bovine serum 1% penicillin/streptomycin, and 1% L-glutamine and used the next morning in experiments. All human islet preparations were handpicked before insulin secretion assays. The protocol for the insulin secretion studies were the same as described for mouse islets.
Islet insulin content
For measurement of total islet insulin content, 25 islets were collected from WT and Ffar2−/− mice (n = 4). Islets were sonicated in acid-ethanol solution (70% ethanol, 0.18M HCl) and incubated overnight at 4°C before an insulin ELISA was performed. Insulin content was normalized to total protein, as determined by bicinchoninic acid assay (Bio-Rad).
Assessment of Ca2+ mobilization in the pancreatic β-cell line, βTC3
Calcium mobilization was assessed in the βTC3 mouse β-cell line. Cells were seeded overnight and serum starved in a 96-well black wall/clear bottom plate. On the day of the experiment, cells were loaded with 5μM Fluo-8 (AAT Bioquest) and incubated at 37°C for 60 minutes. Compounds were added to achieve the final indicated concentrations and fluorescence was measured using a fluorometric imaging plate reader. In each experiment, each compound was tested in quadruplicate at multiple concentrations. The negative log of the maximal effect (pEmax) and pEC50 were obtained for each compound based on concentration-response curves generated using GraphPad Prism.
Semiquantitative RT-PCR analysis of FFAR2 and FFAR3 mRNA expression
Total RNA was isolated from cells followed by removal of genomic DNA by DNase I (Ambion; Applied Biosystems) and cDNA synthesis as previously described (15). Primers used for FFAR2, FFAR3, and β-action were as referenced in Hong et al (31).
Assessment of Ca2+ mobilization and cAMP accumulation by hFFAR2
For Ca2+ mobilization and cAMP accumulation, CHO-K1 cells were transfected with an expression vector containing full-length hFFAR2 cDNA (GenBank accession number NM_005306) with FLAG tag sequence at N terminus. The Ca2+ mobilization and cAMP assays were performed as previously described (32). Each of these assays was performed at Multispan, Inc.
Statistical analysis
Data were analyzed by Student's t test and expressed as mean ± SEM. Significance threshold was established as P ≤ .05.
Results
Ffar2−/− mice do not exhibit overt metabolic phenotype
First, we began by extensively profiling our Ffar2−/− model on NC and HFD. On NC, we observed no difference in weight gain, fasting glucose, or insulin levels between WT and Ffar2−/− males at 6 and 26 weeks (Table 1). Additionally, Ffar2−/− mice on NC showed no differences in oral or ip glucose tolerance (Supplemental Figure 2), nor did we observe any differences in metabolic parameters such as energy intake or expenditure, or VO2 and VCO2 during 48 hours in a mouse metabolic chamber (Supplemental Table 1). On HFD, which is known to induce obesity and insulin resistance, Ffar2−/− mice demonstrated no evidence of altered fasting glucose or insulin compared with WT (Table 1). Additionally, we observed no differences in ip and oral glucose tolerance, or insulin tolerance between WT and Ffar2−/− mice on HFD (Figure 1, A–C). However, Ffar2−/− mice gained less weight than WT controls on HFD, as previously reported by Bjursell et al (11) (Table 1).
Table 1.
Phenotypic Assessment of Ffar2−/− Mice
| NC | High fat | ||||
|---|---|---|---|---|---|
| 6 weeks | 26 weeks | 6 weeks | 26 weeks | ||
| Weight (g) | WT | 22.0 ± 0.5 | 32.0 ± 0.7 | 21.0 ± 0.4 | 47.7 ± 0.7 |
| KO | 21.1 ± 0.3 | 32.7 ± 0.7 | 20.9 ± 0.4 | 43.3 ± 1.7a | |
| Fasting glucose (mg/dL) | WT | 82.9 ± 2.8 | 86.9 ± 2.4 | 84.9 ± 2.7 | 131.3 ± 6.0 |
| KO | 78.6 ± 2.3 | 88.2 ± 2.8 | 82.2 ± 3.8 | 122.0 ± 5.7 | |
| Fasting insulin (ng/mL) | WT | 0.5 ± 0.05 | 0.6 ± 0.03 | 0.6 ± 0.02 | 1.5 ± 0.2 |
| KO | 0.5 ± 0.03 | 0.8 ± 0.1 | 0.6 ± 0.03 | 1.13 ± 0.1 | |
Assessment of young (6 weeks) and aged (26 weeks) wild-type (WT) and Ffar2−/− (KO) mice on normal chow (WT, n = 12; ko, n = 12) and high fat (WT, n = 22; KO, n = 12) diets. Mice were weighed and fasting glucose and insulin measured at the indicated time points and conditions. P ≤ .05 between WT and KO mice at 26 weeks on high fat difet, mean ± SEM.
P = .01
Figure 1.

GTT and ITT with Ffar2−/− mice on HFD. A, ip and oral glucose (B) administered to overnight-fasted males on HFD at 26 weeks of age. Blood glucose was measured at 0, 15, 30, 60, and 120 minutes. C, Assessment of insulin tolerance in WT and Ffar2−/− mice on HFD at 26 weeks of age. For A–C, blood glucose was measured at 0, 15, 30, 60, and 120 minutes (n = 12–23), and circles represent WT, squares represent Ffar2−/−.
Given the absence of an overt metabolic phenotype in Ffar2−/− mice, we next sought to directly examine β-cell function in vivo using 1-hour hyperglycemic clamp in 13- to 16-week-old WT and Ffar2−/− mice on NC. Here, we observed decreased GIR (Figure 2A) and a trend, although not significant, toward decreased insulin secretion in response to glucose and arginine infusion in Ffar2−/− mice (Figure 2, B and C). Notably, the insulin sensitivity index was similar in Ffar2−/− and WT mice, suggesting that global Ffar2 deletion does not affect whole-body insulin sensitivity (Figure 2D). Thus, in our hands, our Ffar2−/− mouse model has no significant impairment in glucose homeostasis in vivo on NC or HFD but does exhibit a trend toward diminished insulin secretion during hyperglycemic clamp studies.
Figure 2.

Ffar2−/− mice exhibit trend toward decreased insulin secretion during hyperglycemic clamp. GIR (A) and insulin secretion (B) during 1-hour hyperglycemic clamp in WT and Ffar2−/− male mice maintained on NC diet (WT, n = 8; Ffar2−/−, n = 9). C, Insulin area under the curve (AUC) during hyperglycemic clamp studies. D, Insulin sensitivity index (M/I index) calculated by GIR (M) divided by the average 30- to 60-minute insulinemia (I). *, P < .05. For B, circles represent WT, squares represent Ffar2−/−. For A, C, and D, white bars represent WT, black bars represent Ffar2−/−.
FFAR2 contributes to ex vivo insulin secretion
Considering the above in vivo data and that islet-expressed GPCRs often contribute to GSIS (33), we examined insulin secretion during 1-hour static incubation assays using isolated islets from 10- to 14-week-old Ffar2−/− male mice and age-matched WT littermate controls. Examining GSIS at multiple glucose concentrations, we found that WT and Ffar2−/− islets both secreted insulin in a glucose-dependent manner. However, at high-glucose concentrations (16.7mM), Ffar2−/− islets secreted less insulin relative to WT islets, indicating diminished capacity for GSIS (Figure 3A). To determine whether this defect occurs in response to secretagogues other than glucose, we assessed GSIS in the presence of the glucagon-like peptide-1 (GLP-1) receptor agonist, Exendin-4, and observed no difference in GSIS between WT and Ffar2−/− islets, with an approximately 1.5-fold increase in insulin secretion with high glucose plus Exendin-4 compared with high glucose alone for both genotypes (1.5 ± 0.2, WT vs 1.5 ± 0.1, Ffar2−/−) (Figure 3A). Next, to test whether this diminished insulin secretion in Ffar2−/− islets was due to a difference in total islet insulin content between genotypes, we compared the insulin content of WT and Ffar2−/− islets and observed no differences (Figure 3B). Thus, although deletion of Ffar2 results in diminished GSIS at high-glucose concentrations compared with WT control, the overall ability of islets to secrete insulin in response to lower glucose concentrations and other secretagogues such as GLP-1 appears intact.
Figure 3.

FFAR2 signaling contributes to GSIS. A, Insulin secretion in response to increasing glucose concentrations, or 16.7mM glucose + 100nM Exendin-4 in isolated WT and Ffar2−/− islets during static insulin secretion assay (n ≥ 3). Insulin secretion is expressed as a percent of total insulin content. B, Total islet insulin content measured after acid ethanol extraction from 25 islets per replicate. Insulin content was normalized to total protein (n = 4). n.s., not significant. C, Insulin secretion from WT and Ffar2−/− islets in response to treatment with high glucose (16.7mM) alone or in combination with 1mM acetate (n ≥ 3). D, Insulin secretion from WT and Ffar2−/− islets in response to treatment with high glucose (16.7mM) alone or in combination with 100μM SCA14 or 100μM SCA15 (n ≥ 3). E, Insulin secretion from WT and Ffar2−/− islets in response to treatment with 16.7mM glucose or in combination with 100μM CMTB or 100μM CPTB (n ≥ 3). F, Insulin secretion from islets obtained from CD-1 mice in response to treatment with 16.7mM glucose or in combination with 1mM acetate, 100μM SCA14, 100μM SCA15, 100μM CMTB, or 100μM CPTB (n ≥ 3). For C–F, values are expressed as a fold change relative to WT islets treated with 16.7mM glucose alone. Asterisks represent significance between genotypes; daggers represent significance within a genotype between the indicated treatment conditions compared with 16.7mM glucose alone. White bars for WT, black bars for Ffar2−/−. *,†, P < .05; **,††, P < .01; ***,†††, P < .001; mean ± SEM.
Signaling via FFAR2 mediates insulin secretion
The ligands for FFAR2, SCFAs, have previously been suggested to regulate GSIS, although the mechanism is unclear (23). The predominant SCFA in circulation (34) and the major endogenous ligand for FFAR2 is acetate. Therefore, we investigated the role of acetate in regulating GSIS at concentrations similar to its physiologic concentrations in mice and humans (1) and observed that addition of acetate to high glucose resulted in a greater than 2-fold potentiation of GSIS in WT but not Ffar2−/− islets (Figure 3C), suggesting that acetate contributes to GSIS, at least in part, by signaling through FFAR2.
Because acetate could affect GSIS through receptor-independent mechanisms, we wanted to verify and more precisely interrogate how FFAR2 signaling affects GSIS. To do this, we used several previously described FFAR2-selective agonists including 2 SCA derivatives referred to here as: SCA14 and SCA15 (35) and 2 phenylacetamide derivatives: CMTB (36) and CPTB (37), in ex vivo insulin secretion assays. Surprisingly, addition of these agonists to high-glucose media resulted in either potentiation (by SCA14 and SCA15) or inhibition (by CMTB) of GSIS in WT islets (Figure 3, D and E). For compounds SCA14 and SCA15, we observed receptor-dependent potentiation of GSIS, because these compounds did not increase insulin secretion with Ffar2−/− islets (Figure 3D). Conversely, treatment with CMTB strongly inhibited GSIS, whereas treatment with CPTB resulted in slight, although not significant inhibition of GSIS, as compared with treatment with high glucose only (Figure 3E). Interestingly, CMTB and CPTB maintained their inhibitory effect in Ffar2−/− islets, suggesting that their influence on GSIS is not entirely FFAR2-dependent. Finally, because our results conflict with recently published data (14), we tested whether our observations were dependent on the particular genetic background of the mouse. Therefore, we again tested the effect of acetate and FFAR2 agonists on GSIS in the commonly used CD-1 mouse model. Similar to what is observed in Figure 3, C–E, acetate and SCA14 significantly enhanced GSIS; SCA15 marginally increased GSIS, and CMTB inhibited GSIS in islets isolated from CD-1 mice relative to high glucose (Figure 3F).
FFAR2 differentially regulates GSIS via Gαq/11 and Gαi/o
FFAR2 has been described to couple to Gαq/11 and Gαi/o pathways (20). In the β-cell, these pathways exert opposing effects on GSIS, with signaling via Gαq/11 leading to Ca2+ mobilization and enhanced insulin secretion and Gαi/o signaling leading to cAMP inhibition and diminished GSIS (Figure 4A) (33). The GSIS-potentiating effect of acetate, SCA14, and SCA15 suggests that these compounds signal at FFAR2 through Gαq/11. To test this, we developed a Ca2+ mobilization assay in the mouse β-cell line, βTC3, which we found expresses Ffar2 but not the related receptor, Ffar3 (Figure 4B). Because FFAR2 and FFAR3 overlap in ligand preference, this cell line offers the benefit of allowing us to test specific effects of FFAR2 agonists, without the confounding expression of Ffar3. As seen in Figure 4C, acetate induced a robust Ca2+ mobilization response in this assay. Further, we observed a dose-dependent Ca2+ mobilization by acetate, SCA14, and SCA15 but not CPTB or CMTB (Table 2). No Ca2+ mobilization was observed with CPTB and CMTB. This observation is consistent with the potentiating effects of acetate, SCA14, and SCA15 being mediated by FFAR2 signaling through Gαq/11.
Figure 4.
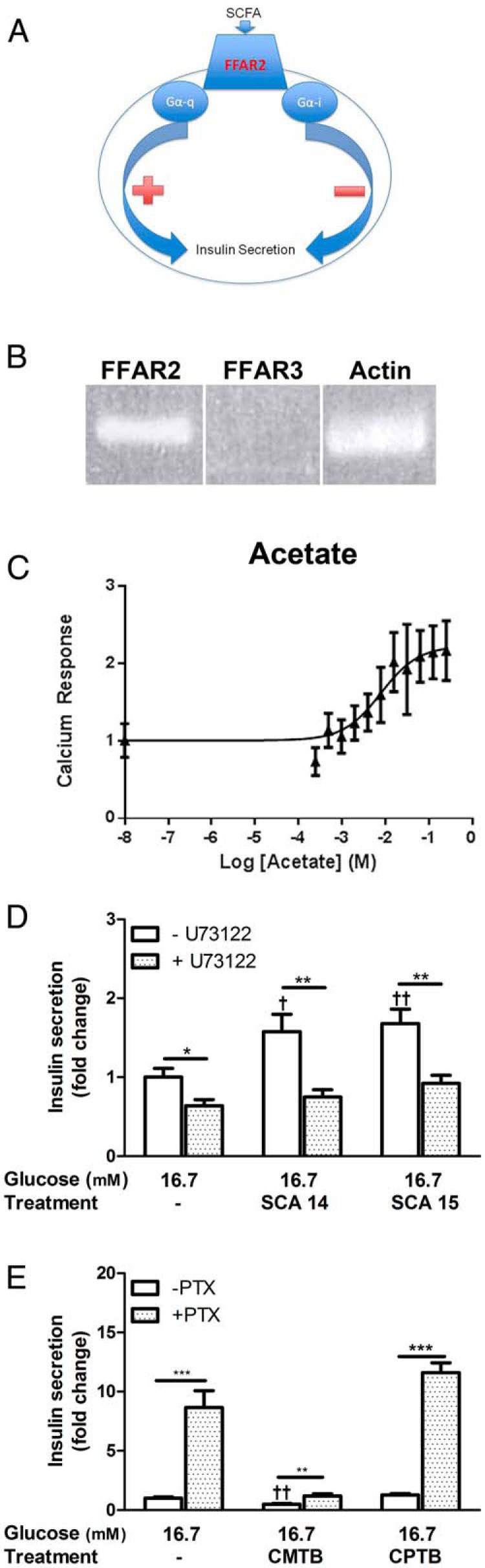
FFAR2 differentially regulates GSIS via Gαq/11 and Gαi/o. A, Schematic depicting signaling pathways mediated by FFAR2. B, RT-PCR expression of FFAR2 and FFAR3 in βTC3 cells, β-actin shown as a reference. C, Dose-response curve measuring calcium mobilization in the βTC3 cell line after treatment with acetate at the indicated concentrations. D, Insulin secretion in response to SCA14 and SCA15 after pretreatment with 5μM U73122. E, Insulin secretion in response to CMTB and CPTB after pretreatment with 300-ng/mL PTX. For D and E, insulin secretion is expressed as a fold change relative to WT islets treated with 16.7mM glucose only. Asterisks represent significance between inhibitor treated and untreated islets; daggers represent significance within a condition as compared with 16.7mM glucose alone. For D and E, white bars for untreated, hash bars for inhibitor treated. *,†, P < .05; **,††, P < .01; ***,†††, P < .001; mean ± SEM.
Table 2.
Potency and Efficacy of FFAR2 Agonists at Mouse FFAR2
| mFFAR2 | ||
|---|---|---|
| Calcium | ||
| Acetate | pEC50 | 5.00 ± 0.01 (n = 13) |
| pEmax | 51.17 ± 13.06 | |
| SCA14 | pEC50 | 6.03 ± 0.52 (n = 5) |
| pEmax | 59.42 ± 26.50 | |
| SCA15 | pEC50 | 3.28 ± 0.432 (n = 3) |
| pEmax | 72.09 ± 27.91 | |
| CMTB | pEC50 | 6.53 ± 0.11 (n = 4) |
| pEmax | 34.07 ± 11.40 | |
| CPTB | pEC50 | 6.19 ± 0.24 (n = 4) |
| pEmax | 44.76 ± 12.58 |
pEC50 and pEmax values for each FFAR2 agonist in the calcium mobilization assays conducted in the βTC3 mouse β cell line is shown. NR refers to no response, where this indicates the absence of a calcium flux response to the agonsit during a multiple concentration titration. Shown are mean ± SEM.
To further dissect the signaling pathways of these agonists, we conducted ex vivo GSIS assays with WT islets that were pretreated with the phospholipase C-β (PLCβ) inhibitor, U73122. Inhibition of this key downstream effector of Gαq/11 abolished the GSIS-potentiating effect of SCA14 and SCA15 in WT islets, further supporting the hypothesis that SCA14 and SCA15 signal by FFAR2 through Gαq/11 to enhance GSIS (Figure 4D). Next, to examine whether the inhibitory effect on GSIS from CMTB and CPTB is mediated by biased signaling through Gαi/o, GSIS assays were conducted after pretreatment of WT islets with PTX, which prevents receptor coupling to Gαi/o. Pretreatment with PTX significantly alleviated the inhibition of GSIS by CMTB and CPTB in WT islets, demonstrating that inhibition by these compounds is, in part, mediated by Gαi/o signaling (Figure 4E). Thus, these data illustrate that FFAR2 agonists can signal via different G protein pathways, which translates to disparate effects on GSIS.
FFAR2-mediated GSIS is not significantly altered by HFD
As described before, our group and others have reported increased islet expression of Ffar2 in insulin resistant states (15–18). Therefore, we explored whether Ffar2 up-regulation in islets leads to altered GSIS in response to FFAR2 agonists. After 6–8 weeks on HFD, we verified that Ffar2 expression was increased in WT islets (Figure 5A). Next, using islets isolated from these HFD-fed Ffar2−/− and WT mice, we conducted ex vivo insulin secretion experiments. Here, we observed that HFD-WT islets responded to acetate and FFAR2 agonists in a similar receptor-dependent manner to islets from WT mice on NC, with the exception of CPTB (Figure 5B), which enhanced, rather than inhibited, GSIS with WT islets. Thus, HFD lead to increased islet expression of Ffar2 in WT islets, with the insulin secretion response to acetate and FFAR2 agonists largely unaltered, with the exception of CPTB.
Figure 5.

Effects of FFAR2 signaling in islets from HFD-fed mice and human islets. A, Ffar2 expression in islets isolated from NC and HFD-fed mice. Expression was measured after 6–8 weeks of HFD and is expressed relative to GAPDH (*, P < .05). B, Insulin secretion from HFD-fed WT and Ffar2−/− islets in response to treatment with high glucose (16.7mM) alone or in combination with 1mM acetate or FFAR2-specific agonists (100μM SCA14, 100μM SCA15, 100μM CMTB, or 100μM CPTB). For these insulin secretion experiments, values are expressed as a fold change relative to the high glucose alone condition within each genotype. White bars represent WT, black bars represent Ffar2−/−. Asterisks represent significance between genotypes; daggers represent significance within a genotype between the indicated treatment conditions compared with 16.7mM glucose alone. C, Insulin secretion from human islets in response to treatment with high glucose (16.7mM) alone or in combination with 1mM acetate or FFAR2-specific agonists (100μM SCA14, 100μM SCA15, 100μM CMTB, or 100μM CPTB) (n = 7). D, Insulin secretion from human islets in response to treatment with high glucose (16.7mM) alone or in combination with 1mM acetate or FFAR2-specific agonists (100μM SCA14, 100μM SCA15, 100μM CMTB, or 100μM CPTB) after pretreatment with 300-ng/mL PTX (n = 5). *,†, P < .05; **,††, P < .01; ***,†††, P < .001; mean ± SEM.
FFAR2 exhibits interspecies variation in signaling properties in response to agonists
To explore the effect of FFAR2 signaling in human islets, we conducted GSIS experiments with human islets obtained from the Integrated Islet Distribution Program (see donor characteristics) (Supplemental Table 2). Here, we observed that acetate, SCA14, SCA15, and CPTB did not alter GSIS (Figure 5C), whereas CMTB significantly inhibited GSIS (Figure 5C). Interspecies differences in ligand preference and constitutive activity of FFAR2 have been well described (38), and may offer an explanation for the differences in agonist response between mouse and human. Thus, we next set out to explore whether these agonists signal by hFFAR2 through the Gαq/11 and Gαi/o pathways as noted with mouse islets, using in vitro Ca2+ mobilization assays to assess Gαq/11 signaling and cAMP accumulation assays to assess Gαi/o signaling (Supplemental Figure 3, A and B). Interestingly, in these assays, we found that each agonist activated both Gαq/11 and Gαi/o via hFFAR2 (Table 3). Thus, signaling via hFFAR2 by these agonists can simultaneously activate 2 pathways known to have opposing effects on GSIS.
Table 3.
Potency and Efficacy of FFAR2 Agonists at Human FFAR2
| hFFAR2 | |||
|---|---|---|---|
| Calcium | cAMP | ||
| Acetate | pEC50 | 5.00 ± 0.01 (n = 7) | 4.44 ± 0.21 (n = 6) |
| pEmax | 51.17 ± 13.06 | 2.78 ± 0.32 | |
| SCA14 | pEC50 | 6.03 ± 0.52 (n = 5) | 5.85 ± 0.17 (n = 4) |
| pEmax | 59.42 ± 26.50 | 3.42 ± 0.40 | |
| SCA15 | pEC50 | 3.28 ± 0.43 (n = 3) | 3.44 ± 0.08 (n = 4) |
| pEmax | 72.09 ± 27.91 | 3.72 ± 0.34 | |
| CMTB | pEC50 | 6.53 ± 0.11 (n = 4) | 6.27 ± 0.32 (n = 5) |
| pEmax | 34.07 ± 11.40 | 2.39 ± 0.23 | |
| CPTB | pEC50 | 6.19 ± 0.24 (n = 6) | 6.43 ± 0.27 (n = 5) |
| pEmax | 44.76 ± 12.58 | 1.96 ± 0.22 | |
pEC50 and pEmax for each agonsit in hFFAR2 calcium mobilization and cAMP accumation assays is shown.
Considering the simultaneous activation of Gαq/11 and Gαi/o pathways via hFFAR2, we set out to examine the effect of FFAR2 agonists after PTX-mediated uncoupling of the receptor from Gαi/o. Of note, PTX treatment did not alleviate the CMTB inhibitory effect on GSIS (Figure 5D). However, it did result in enhanced GSIS with SCA15. These data indicate that acetate and FFAR2 agonists can signal via hFFAR2. However, the signaling biases and effect on GSIS of FFAR2 agonists are not maintained across species, likely due to interspecies variation in receptor pharmacology.
Discussion
Many GPCRs are expressed in β-cells and contribute to the regulation of insulin secretion (27). The discovery of an acetate-specific GPCR that is expressed in β-cells raises the possibility that acetate can regulate insulin secretion via FFAR2. Tang et al (14) recently reported that acetate signaling through FFAR2 and FFAR3 is a negative regulator of GSIS, acting through a PTX-sensitive Gαi/o pathway. A recent publication from our group confirms the finding of FFAR3 as a negative regulator of GSIS by PTX-sensitive Gαi/o (24). However, here, our data indicate that in mouse islets, signaling via FFAR2 can either potentiate or inhibit GSIS, depending on the G protein pathways activated (Gαq/11 or Gαi/o, respectively). When explored in human islets, acetate and FFAR2 agonists, with the exception of CMTB, did not affect GSIS. Using hFFAR2 signaling in vitro assays, we confirmed that acetate and each FFAR2 agonist elicited robust activation of hFFAR2. As reflected in the lack of enhanced or inhibited GSIS with the exception of CMTB, acetate and FFAR2 agonists did not have a bias for Gαq/11 or Gαi/o signaling. Of particular interest, CMTB maintained its ability to inhibit GSIS in human islets in the presence of PTX. These data would suggest 2 possibilities: first, that CMTB does not act at FFAR2 or any GPCR coupled to the PTX-sensitive Gαi/o pathway; or second, that FFAR2 can couple to Gαz, a PTX-insensitive member of the Gαi/o pathway that is also known to inhibit GSIS (39). Regardless, these data illustrate the ability of FFAR2 to regulate GSIS by signaling through multiple G protein pathways.
As apparent from our data with mouse and human islets, the likely reason we and others have observed that acetate augments GSIS, while others, including Tang et al (14), observe inhibitory effects of acetate on GSIS is that FFAR2 signals via multiple G protein pathways. Moreover, it is likely that G protein switching between Gαq/11 and Gαi/o can occur with FFAR2 under different conditions, a phenomenon well described with other GPCRs (40) such as the β-adrenergic receptor in heart tissue (41). Other possibilities that account for the varying effects of acetate on GSIS include differences in experimental conditions (which may affect G protein coupling) and also that acetate may act through anaplerotic pathways, in addition to receptor mediated pathways, confounding the interpretation of results. Taken together, our findings highlight the need for future studies to address what factors control FFAR2 signaling in β-cells, in order to gain greater insight into its role in insulin secretion.
Another key difference between our study and that of Tang et al (14) is that they reported no difference in GSIS between WT and Ffar2−/− islets, whereas in our hands, genetic deletion of Ffar2 results in diminished GSIS at higher glucose concentrations. Additionally, in a separate study, we recently reported that genetic deletion of Ffar3 results in enhanced GSIS at higher glucose concentrations (24). As suggested in that report, reasons for the discrepancy between our findings and those of Tang et al (14) may stem from the use of different strategies to create each mouse model, with each knockout approach leading to different downstream genetic effects that may influence insulin secretion. Additionally, Tang et al (14) did not examine insulin secretion at multiple glucose concentrations as we did here and also in our recent report (24).
Considering the complexity of FFAR2 signaling, it is not surprising that we found that mouse and human islets responded differently to acetate and FFAR2 agonists in ex vivo GSIS assays. This finding warrants close attention in future studies, especially if FFAR2 is to be considered as a therapeutic T2D target. The pharmacology underlying these interspecies differences has been well described, with hFFAR2 demonstrating constitutive activity, whereas mouse FFAR2 does not (38). This property of constitutive activity at hFFAR2 may minimize the effect of receptor agonists in β-cells, because the receptor may be almost fully active, resulting in little overall change in activity with addition of agonist. Moreover, the potency of various SCFAs at FFAR2 and FFAR3 differs between mouse and human orthologs. Although the potency of acetate and propionate at mouse FFAR2 is approximately equal, hFFAR2 demonstrates the highest affinity for acetate as compared with other SCFAs (38). These differences, along with the unexplored possibility of species differences in specific G protein signaling bias, may account for the interspecies variation in FFAR2 agonist response. A separate variable is that human islets were obtained from donors with wide variation in body mass index (see Supplemental Table 2). Given the association between Ffar2 expression and obesity and/or insulin resistance, it is possible that variations in the expression of Ffar2 with changing levels of obesity may have meaningful effects on GSIS and the insulin secretion response to acetate.
Next, the in vivo phenotyping studies that have been published thus far have observed multiple opposing outcomes. For example, our in vivo findings align with previously published findings of Bjursell et al (11), but our data are inconsistent with the report of Tolhurst et al (12), who reported impaired oral glucose tolerance with Ffar2−/− mice due to impaired GLP-1 secretion. In addition to the dual coupling properties of FFAR2, additional factors could be contributing to the inconsistencies in mouse model studies. First, 2 Ffar2−/− models on different genetic backgrounds have been made, raising the possibility of phenotypic variation due to genetic background, or due to the approach used to knockout Ffar2. Additionally, the closely related receptor FFAR3, which has a similar expression pattern and is activated by similar ligands as FFAR2, is located in close genetic proximity to Ffar2 gene and differences in the methodology used to generate each Ffar2−/− model may alter the expression and/or activity of Ffar3 (42). Further, SCFAs are produced by microbial fermentation of dietary fiber, and it has been reported that differences in gut microbiome composition (between animal facilities) may manifest in differing phenotypes (13, 28, 43). These factors underscore the need for well-controlled studies, which incorporate tissue-specific knockouts and well-characterized FFAR2-specific agonists in order to more clearly define the role of FFAR2 in metabolism.
Given these numerous conflicting findings, the role of FFAR2 as a novel diabetes target remains unclear. Although our group and others have shown that Ffar2 expression is increased in insulin resistant states (15–17), the implications of these changes to islet function are not clear. Here, Ffar2 expression increased after HFD but did not confer any significant change in the islet response to glucose, acetate, or most FFAR2 agonists. However, because multiple independent groups have observed the regulation of Ffar2 expression in response to insulin resistance, the implications, if any, should be further explored. Additionally, future studies are needed to assess whether Ffar2 expression is altered by obesity or T2D in human islets and whether these changes lead to meaningful effects on insulin secretion in response to glucose, acetate, or FFAR2 agonists. These later studies will be important in determining the translational utility of FFAR2 as a viable T2D treatment option. Along with these studies, an understanding of what leads to changes in G protein coupling with FFAR2 will also help guide our understanding of the therapeutic value of FFAR2.
Many therapeutic options for the treatment of T2D have adverse side effects, and not all patients are responsive to the currently available options (27, 44). Thus, there remains a need for the development of new T2D drugs. Significant precedent for targeting GPCRs as a therapeutic target for T2D exists (27). The data presented here demonstrate that the GPCR, FFAR2, can mediate insulin secretion, and thus, FFAR2 warrants consideration as a potentially novel therapeutic T2D target. If validated as a viable therapeutic target, the ability of FFAR2 to signal by divergent pathways may be exploited in the construction of biased agonists. However, effects of FFAR2 agonists on mouse and human islets were markedly different, likely owing to species differences in the pharmacologic properties of FFAR2 (38). Thus, comprehensive, in-depth studies on the pharmacology and function of mouse and hFFAR2 in the β-cell will be critical to developing FFAR2 into a promising therapeutic target.
Acknowledgments
We thank the Rodent Metabolic Phenotyping core facility of the Centre de Recherche du Centre Hospitalier de l'Université de Montréal for help with hyperglycemic clamps. V.P. holds the Canada Research Chair in Diabetes and Pancreatic β-Cell Function.
This work was supported by the Department of Veterans Affairs, Veterans Health Administration, Office of Research and Development, Career Development Grant 1IK2BX001587-01 (to B.T.L.). M.P. is supported by an American Heart Association Postdoctoral Fellowship 15POST22410016. S.R.V. is supported by the Northwestern University Program in Endocrinology, Diabetes and Hormone Action Grant NIH T32 DK007169 and by the Northwestern University Cellular and Molecular Basis of Disease Training Grant NIH T32 GM08061. R.G.M. is supported by National Institutes of Health Grants R01 DK060581 and UC4 DK104166. T.A. was supported by a salary award from Fonds de Recherche Québec-Santé. B.W. is supported by National Institutes of Health Grant R01 DK085129.
Disclosure Summary: The authors have nothing to disclose.
Footnotes
- CMTB
- 4-chloro-α-(1-methylethyl)-N-2-thiazolylbenzeneacetamide
- CPTB
- (S)-2-(4-chlorophenyl)-3,3-dimethyl-N-(5-phenylthiazol-2-yl)butanamide
- FFAR2
- free fatty acid receptor 2
- GIR
- glucose infusion rate
- GLP-1
- glucagon-like peptide-1
- GPCR
- G protein-coupled receptor
- GSIS
- glucose-stimulated insulin secretion
- GTT
- glucose tolerance test
- HBSS
- Hanks' buffered salt solution
- HFD
- high-fat diet
- hFFAR2
- human FFAR2
- ITT
- insulin tolerance test
- KRB
- Krebs Ringer buffer
- NC
- normal chow
- pEmax
- negative log of the maximal effect
- SCA
- small carboxylic acid
- SCFA
- short-chain fatty acid
- PTX
- pertussis toxin
- T2D
- type 2 diabetes
- VCO2
- CO2 production
- VO2
- O2 consumption
- WT
- wild-type.
References
- 1. Layden BT, Angueira AR, Brodsky M, Durai V, Lowe WL., Jr Short chain fatty acids and their receptors: new metabolic targets. Transl Res. 2013;161(3):131–140. [DOI] [PubMed] [Google Scholar]
- 2. Johnston CS, Kim CM, Buller AJ. Vinegar improves insulin sensitivity to a high-carbohydrate meal in subjects with insulin resistance or type 2 diabetes. Diabetes Care. 2004;27(1):281–282. [DOI] [PubMed] [Google Scholar]
- 3. Johnston CS, Steplewska I, Long CA, Harris LN, Ryals RH. Examination of the antiglycemic properties of vinegar in healthy adults. Ann Nutr Metab. 2010;56(1):74–79. [DOI] [PubMed] [Google Scholar]
- 4. White AM, Johnston CS. Vinegar ingestion at bedtime moderates waking glucose concentrations in adults with well-controlled type 2 diabetes. Diabetes Care. 2007;30(11):2814–2815. [DOI] [PubMed] [Google Scholar]
- 5. Kondo T, Kishi M, Fushimi T, Ugajin S, Kaga T. Vinegar intake reduces body weight, body fat mass, and serum triglyceride levels in obese Japanese subjects. Biosci Biotechnol Biochem. 2009;73(8):1837–1843. [DOI] [PubMed] [Google Scholar]
- 6. Shah JH, Wongsurawat N, Aran PP. Effect of ethanol on stimulus-induced insulin secretion and glucose tolerance. A study of mechanisms. Diabetes. 1977;26(4):271–277. [DOI] [PubMed] [Google Scholar]
- 7. Patel DG, Singh SP. Effect of ethanol and its metabolites on glucose mediated insulin release from isolated islets of rats. Metabolism. 1979;28(1):85–89. [DOI] [PubMed] [Google Scholar]
- 8. Yamashita H, Fujisawa K, Ito E, et al. Improvement of obesity and glucose tolerance by acetate in type 2 diabetic Otsuka Long-Evans Tokushima fatty (OLETF) rats. Biosci Biotechnol Biochem. 2007;71(5):1236–1243. [DOI] [PubMed] [Google Scholar]
- 9. Brown AJ, Goldsworthy SM, Barnes AA, et al. The orphan G protein-coupled receptors GPR41 and GPR43 are activated by propionate and other short chain carboxylic acids. J Biol Chem. 2003;278(13):11312–11319. [DOI] [PubMed] [Google Scholar]
- 10. Priyadarshini M, Thomas A, Reisetter AC, et al. Maternal short-chain fatty acids are associated with metabolic parameters in mothers and newborns. Transl Res. 2014;164(2):153–157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Bjursell M, Admyre T, Göransson M, et al. Improved glucose control and reduced body fat mass in free fatty acid receptor 2-deficient mice fed a high-fat diet. Am J Physiol Endocrinol Metab. 2011;300(1):E211–E220. [DOI] [PubMed] [Google Scholar]
- 12. Tolhurst G, Heffron H, Lam YS, et al. Short-chain fatty acids stimulate glucagon-like peptide-1 secretion via the G-protein-coupled receptor FFAR2. Diabetes. 2012;61(2):364–371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Kimura I, Ozawa K, Inoue D, et al. The gut microbiota suppresses insulin-mediated fat accumulation via the short-chain fatty acid receptor GPR43. Nat Commun. 2013;4:1829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Tang C, Ahmed K, Gille A, et al. Loss of FFA2 and FFA3 increases insulin secretion and improves glucose tolerance in type 2 diabetes. Nat Med. 2015;21(2):173–177. [DOI] [PubMed] [Google Scholar]
- 15. Layden BT, Durai V, Newman MV, et al. Regulation of pancreatic islet gene expression in mouse islets by pregnancy. J Endocrinol. 2010;207(3):265–279. [DOI] [PubMed] [Google Scholar]
- 16. Leonard J, Chu ZL, Bruce MA, Boatman PD. Pat PCT/US2005/039551. WO 2006/052566. A2 2006. [Google Scholar]
- 17. Rieck S, White P, Schug J, et al. The transcriptional response of the islet to pregnancy in mice. Mol Endocrinol. 2009;23:1702–1712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Keller MP, Choi Y, Wang P, et al. A gene expression network model of type 2 diabetes links cell cycle regulation in islets with diabetes susceptibility. Genome Res. 2008;18(5):706–716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Prentki M, Matschinsky FM, Madiraju SR. Metabolic signaling in fuel-induced insulin secretion. Cell Metab. 2013;18(2):162–185. [DOI] [PubMed] [Google Scholar]
- 20. Le Poul E, Loison C, Struyf S, et al. Functional characterization of human receptors for short chain fatty acids and their role in polymorphonuclear cell activation. J Biol Chem. 2003;278(28):25481–25489. [DOI] [PubMed] [Google Scholar]
- 21. Tiengo A, Valerio A, Molinari M, Meneghel A, Lapolla A. Effect of ethanol, acetaldehyde, and acetate on insulin and glucagon secretion in the perfused rat pancreas. Diabetes. 1981;30(9):705–709. [DOI] [PubMed] [Google Scholar]
- 22. Ximenes HM, Hirata AE, Rocha MS, Curi R, Carpinelli AR. Propionate inhibits glucose-induced insulin secretion in isolated rat pancreatic islets. Cell Biochem Funct. 2007;25(2):173–178. [DOI] [PubMed] [Google Scholar]
- 23. Montague W, Taylor KW. Regulation of insulin secretion by short chain fatty acids. Nature. 1968;217(5131):853. [DOI] [PubMed] [Google Scholar]
- 24. Priyadarshini M, Layden BT. FFAR3 modulates insulin secretion and global gene expression in mouse islets. Islets. 2015. DOI: 10.1080/19382014.2015.1045182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Burant CF, Viswanathan P, Marcinak J, et al. TAK-875 versus placebo or glimepiride in type 2 diabetes mellitus: a phase 2, randomised, double-blind, placebo-controlled trial. Lancet. 2012;379(9824):1403–1411. [DOI] [PubMed] [Google Scholar]
- 26. Araki T, Hirayama M, Hiroi S, Kaku K. GPR40-induced insulin secretion by the novel agonist TAK-875: first clinical findings in patients with type 2 diabetes. Diabetes Obes Metab. 2012;14(3):271–278. [DOI] [PubMed] [Google Scholar]
- 27. Ahrén B. Islet G protein-coupled receptors as potential targets for treatment of type 2 diabetes. Nat Rev Drug Discov. 2009;8(5):369–385. [DOI] [PubMed] [Google Scholar]
- 28. Maslowski KM, Vieira AT, Ng A, et al. Regulation of inflammatory responses by gut microbiota and chemoattractant receptor GPR43. Nature. 2009;461(7268):1282–1286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Fergusson G, Ethier M, Guévremont M, et al. Defective insulin secretory response to intravenous glucose in C57Bl/6J compared to C57Bl/6N mice. Mol Metab. 2014;3(9):848–854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Chen X, Zhang X, Larson CS, Baker MS, Kaufman DB. In vivo bioluminescence imaging of transplanted islets and early detection of graft rejection. Transplantation. 2006;81(10):1421–1427. [DOI] [PubMed] [Google Scholar]
- 31. Hong YH, Nishimura Y, Hishikawa D, et al. Acetate and propionate short chain fatty acids stimulate adipogenesis via GPCR43. Endocrinology. 2005;146(12):5092–5099. [DOI] [PubMed] [Google Scholar]
- 32. Whittle BJ, Silverstein AM, Mottola DM, Clapp LH. Binding and activity of the prostacyclin receptor (IP) agonists, treprostinil and iloprost, at human prostanoid receptors: treprostinil is a potent DP1 and EP2 agonist. Biochem Pharmacol. 2012;84(1):68–75. [DOI] [PubMed] [Google Scholar]
- 33. Winzell MS, Ahrén B. G-protein-coupled receptors and islet function-implications for treatment of type 2 diabetes. Pharmacol Ther. 2007;116(3):437–448. [DOI] [PubMed] [Google Scholar]
- 34. Pomare EW, Branch WJ, Cummings JH. Carbohydrate fermentation in the human colon and its relation to acetate concentrations in venous blood. J Clin Invest. 1985;75(5):1448–1454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Schmidt J, Smith NJ, Christiansen E, et al. Selective orthosteric free fatty acid receptor 2 (FFA2) agonists: identification of the structural and chemical requirements for selective activation of FFA2 versus FFA3. J Biol Chem. 2011;286(12):10628–10640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Lee T, Schwandner R, Swaminath G, et al. Identification and functional characterization of allosteric agonists for the G protein-coupled receptor FFA2. Mol Pharmacol. 2008;74(6):1599–1609. [DOI] [PubMed] [Google Scholar]
- 37. Wang Y, Jiao X, Kayser F, et al. The first synthetic agonists of FFA2: discovery and SAR of phenylacetamides as allosteric modulators. Bioorg Med Chem Lett. 2010;20(2):493–498. [DOI] [PubMed] [Google Scholar]
- 38. Hudson BD, Tikhonova IG, Pandey SK, Ulven T, Milligan G. Extracellular ionic locks determine variation in constitutive activity and ligand potency between species orthologs of the free fatty acid receptors FFA2 and FFA3. J Biol Chem. 2012;287(49):41195–41209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Kimple ME, Joseph JW, Bailey CL, et al. Gαz negatively regulates insulin secretion and glucose clearance. J Biol Chem. 2008;283(8):4560–4567. [DOI] [PubMed] [Google Scholar]
- 40. Daaka Y, Luttrell LM, Lefkowitz RJ. Switching of the coupling of the β2-adrenergic receptor to different G proteins by protein kinase A. Nature. 1997;390(6655):88–91. [DOI] [PubMed] [Google Scholar]
- 41. Xiao RP. β-Adrenergic signaling in the heart: dual coupling of the β2-adrenergic receptor to G(s) and G(i) proteins. Sci STKE. 2001;2001(104):re15. [DOI] [PubMed] [Google Scholar]
- 42. Sawzdargo M, George SR, Nguyen T, Xu S, Kolakowski LF, O'Dowd BF. A cluster of four novel human G protein-coupled receptor genes occurring in close proximity to CD22 gene on chromosome 19q13.1. Biochem Biophys Res Commun. 1997;239(2):543–547. [DOI] [PubMed] [Google Scholar]
- 43. Dewulf EM, Cani PD, Neyrinck AM, et al. Inulin-type fructans with prebiotic properties counteract GPR43 overexpression and PPARγ-related adipogenesis in the white adipose tissue of high-fat diet-fed mice. J Nutr Biochem. 2011;22(8):712–722. [DOI] [PubMed] [Google Scholar]
- 44. Defronzo RA. Banting Lecture. From the triumvirate to the ominous octet: a new paradigm for the treatment of type 2 diabetes mellitus. Diabetes. 2009;58(4):773–795. [DOI] [PMC free article] [PubMed] [Google Scholar]