

The Cairns–Foster system appears to involve the stress-induced mutagenesis of nongrowing cells. However, the behavior of this system may not require mutagenesis but instead may rely on several aspects of selection.
Abstract
Selection detects mutants but does not cause mutations. Contrary to this dictum, Cairns and Foster plated a leaky lac mutant of Escherichia coli on lactose medium and saw revertant (Lac+) colonies accumulate with time above a nongrowing lawn. This result suggested that bacteria might mutagenize their own genome when growth is blocked. However, this conclusion is suspect in the light of recent evidence that revertant colonies are initiated by preexisting cells with multiple copies the conjugative F′lac plasmid, which carries the lac mutation. Some plated cells have multiple copies of the simple F′lac plasmid. This provides sufficient LacZ activity to support plasmid replication but not cell division. In nongrowing cells, repeated plasmid replication increases the likelihood of a reversion event. Reversion to lac+ triggers exponential cell growth leading to a stable Lac+ revertant colony. In 10% of these plated cells, the high-copy plasmid includes an internal tandem lac duplication, which provides even more LacZ activity—sufficient to support slow growth and formation of an unstable Lac+ colony. Cells with multiple copies of the F′lac plasmid have an increased mutation rate, because the plasmid encodes the error-prone (mutagenic) DNA polymerase, DinB. Without DinB, unstable and stable Lac+ revertant types form in equal numbers and both types arise with no mutagenesis. Amplification and selection are central to behavior of the Cairns–Foster system, whereas mutagenesis is a system-specific side effect or artifact caused by coamplification of dinB with lac. Study of this system has revealed several broadly applicable principles. In all populations, gene duplications are frequent stable genetic polymorphisms, common near-neutral mutant alleles can gain a positive phenotype when amplified under selection, and natural selection can operate without cell division when variability is generated by overreplication of local genome subregions.
AN INITIAL POINT TO CONSIDER
In considering the possibility of regulated mutagenesis, it is useful to consider a basic, but often forgotten truism. Most new mutations are near-neutral or deleterious types that are easily missed or ignored. Similarly, copy-number changes of any particular gene are extremely common (10−5/cell/division in Salmonella) but hard to detect. In contrast, rare beneficial mutations with big effects are obvious because selection allows them to increase in frequency. Genetic analysis has misdirected our attention toward these rare large-effect beneficial changes. It is easy to forget the “silent majority”—the near-neutral and deleterious changes—but it is important to remember that natural selection sees almost everything and is always watching.
THE MAJOR QUESTION UNDER CONSIDERATION
Bacterial genetics uses stringent selection conditions to detect rare mutants. Genetic analysis of bacteria was made possible by the demonstration that strong selection could reliably detect and enumerate preexisting mutants without contributing to their formation. Classical experiments by Luria and Delbrück (1943), Lederberg and Lederberg (1952), and Newcombe (1949) used lethal selections to show that mutants arise before selection and do not require growth limitation for their formation. These selections could only detect preexisting mutants and, therefore, could not show whether or not some adaptive mutations form at a higher rate in response to selection. However, the classic experiments were interpreted as evidence that selection does not affect mutation rates and led to the ultimate conclusion that all mutants arise as replication errors that are random with respect to functional or selective consequences.
Later experiments seemed to contradict this conclusion. In several systems, microbial growth was blocked by nonlethal conditions and mutant colonies accumulated over time above the lawn of plated cells (Shapiro 1984; Cairns et al. 1988; Hall 1988; Steele and Jinks-Robertson 1992; Maenhaut-Michel and Shapiro 1994; Sung and Yasbin 2002). The behavior of these systems was interpreted as evidence that nongrowing cells increased their own mutation rates to help them “get off the dime.” The cost of mutagenesis (deleterious mutations) was not evaluated and the possibility that cells grow under selection before mutation was not eliminated. If cells grow under selection, any observed mutants could result from unappreciated parent cell divisions or from proliferation of preexisting small-effect mutants rather than an increased mutation rate. When selection is imposed on a growing population, the effect of mutagenesis on mutant frequency is hard to quantify in the face of the enormous exponential effects of positive selection.
To avoid interference from selection, mutation rates are classically measured by focusing attention on mutant types that can grow well under some permissive growth conditions and can be detected later by stringent selection (reviewed by Rosche and Foster 2006). Genome-wide mutation rates have also been estimated by examining the whole genome sequence of populations that have been passed through repeated small bottlenecks to minimize accumulation of fitter mutants and loss of mutants that impair growth (Lee et al. 2012). These devices solve the rate measurement problem, but avoid the real issue—“do adaptive (beneficial) mutations occur at a higher rate under selection”?
The Cairns–Foster system (Cairns et al. 1988; Cairns and Foster 1991) was designed to solve these problems. The goal was to assess the formation rate of adaptive or beneficial mutations under selective conditions that prevent growth, but are not lethal. In interpreting the Cairns–Foster experiment, it is assumed that if cells are not dividing and chromosomes are not replicating then any increase in mutant frequency must be caused by mutagenesis and not selection. To eliminate growth, conditions were carefully adjusted (using scavenger cells, as described below). Selection was deliberately set at a stringency that is just enough to prevent growth (Cairns and Foster 1991). The parental cells do not grow, but are poised such that very minor improvements may allow growth to start. The system has several problems. There is no easy way to compare mutation rates in growing and nongrowing populations. During growth, one measures mutations/cell per generation. Without growth, one measures mutations/cell per unit time. It is hard to completely eliminate cryptic growth of a subset of the plated population or within slowly developing, partially revertant colonies. Selection never sleeps. We will suggest later that the problem is even worse—selection may even operate even when cell division is blocked.
PHENOMENOLOGY OF THE CAIRNS–FOSTER SYSTEM—HOW THE EXPERIMENT IS DONE
In the basic Escherichia coli tester strain, the lac operon is located on a low-copy F′lac plasmid (see Fig. 1) (Cairns and Foster 1991). The lacZ gene (β-galactosidase) is fused to the lacI (repressor) gene and transcribed from a constitutive (iQ) promoter. Expression of LacZ activity is prevented by a leaky +1 frameshift mutation within the lacI portion of the hybrid gene, which reduces the level of β-galactosidase (LacZ activity) to 2% of that seen in revertants (Foster 1994). Cells are pregrown in minimal glycerol medium, washed, and plated (108) on lactose medium, in which any residual growth is prevented by a 10-fold excess of scavenger cells with a lac deletion mutation. Scavengers cannot use lactose or revert, but can consume any residual nutrients in the agar or compounds excreted from revertant colonies (notably galactose). The number of scavenger cells is set to just barely prevent growth of the tester, which is poised on the brink of growth (Cairns and Foster 1991).
Figure 1.
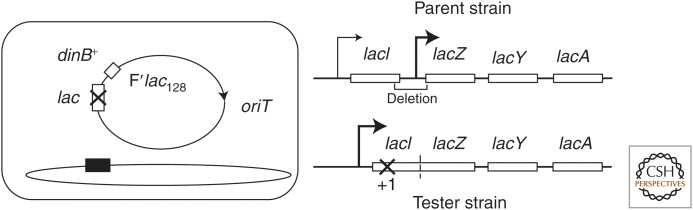
The strains used in the Cairns–Foster system. The Escherichia coli tester strain carries an F′lac plasmid with a mutant lac allele; the chromosomal lac region has been deleted. On the F′lac plasmid, the lacI and lacZ genes have been fused so they encode a single protein with β-galactosidase (LacZ) activity. This hybrid gene is transcribed from a constitutively expressed lacIQ promoter. The ability of the tester to grow on lactose is blocked by a +1 frameshift mutation within the lacI portion of the hybrid gene. This mutation is leaky and allows production of about 2% of the revertant enzyme activity (Cairns and Foster 1991). The F′lac plasmid also carries the dinB gene, which encodes an SOS-inducible error-prone (mutagenic) DNA repair polymerase. The transfer (tra) functions of plasmid F′lac128 that allow conjugation are constitutively expressed. Conjugal replication and transfer processes are initiated at a transfer origin (oriT) by the single-strand endonuclease TraI.
The course of an experiment is described in Figures 2 and 3. Tester cells (108) are grown nonselectively, washed, and plated at day 0. The Lac+ revertant colonies start appearing by day 2 and accumulate linearly for ∼5–6 days thereafter. During this time, the plated lawn shows little growth and this stasis is not a balance of death and growth (Foster 1994). The colonies appearing first—before day 2—are initiated by preexisting full lac+ revertants (with a compensating −1 frameshift mutation). Colonies that accumulate later, above a nongrowing lawn, are the critical revertants to explain. The yield of these late revertants depends on residual LacZ expression from the leaky mutant gene. This residual expression could, in principle, support very slow lawn growth or could simply supply energy needed for the reversion process, a point that will become critical. There are two types of late revertant colonies. Ninety percent of colonies appearing on day 5 contain cells with a stable lac+ mutation (a compensating −1 frameshift). The remaining colonies (10%) contain cells with a tandem amplification (10–100 copies) of the original mutant lac allele. Most of these are standard direct-order repeats with short (10 bp) junction sequences (or short junction [SJ] types). About 20% of unstables have tandem repeats arranged in alternating orientation (tandem inversion duplication [TID]). Stable types accumulate linearly, unstable types exponentially (Fig. 3).
Figure 2.

The Cairns–Foster reversion experiment. Cells of the strain described above are pregrown on glycerol, washed, and plated on lactose medium. The few revertant colonies that appear on lactose within 2 days are initiated by fully lac+ revertant cells that arose during prior nonselective pregrowth. More Lac+ colonies accumulate on the plate over the next 4 days. On day 5, 90% of new late colonies are made up of stable lac+ cells that have acquired a compensating frameshift mutation and, therefore, a functional lac+allele. The other 10% of revertant colonies are made up of cells with an amplified array of the original mutant lac allele. Amplification-bearing cells are unstable and frequently lose their Lac+ phenotype. When these revertants are streaked on rich, nonselective medium containing the chromogenic LacZ substrate X-gal, they form sectored (blue/white) colonies that reveal their frequent loss of ability to use lactose (see Fig. 3).
Figure 3.

The time course of accumulating stable and unstable revertants in the Cairns–Foster experiment. After the tester strain is plated on lactose medium (on day 0), revertant colonies accumulate over several days. Stable lac+ colonies accumulate linearly with time, whereas unstable Lac+ colonies accumulate exponentially with time. The genotype of the parent tester strain is diagrammed at the top right. Stable Lac+ colonies have acquired a compensating (−1) frameshift mutation and thereby a lac+ allele. Cells in unstable Lac+ revertant colonies have multiple copies (n) of the original leaky mutant lac allele, arranged either as tandem direct-order repeats (short junction [SJ]) or expanded tandem inversion duplications (TIDs) with copies in an alternating orientation. Cells with sufficient copies of the partially functional mutant lac allele can grow on lactose and form a sectored blue/white colony on rich X-gal medium, owing to loss of lac copies.
NUMEROLOGY OF THE CAIRNS–FOSTER SYSTEM—THE MUTATION PROBLEM TO BE SOLVED
The curious thing about the Cairns–Foster system is the abundance of revertant colonies and the paucity of evidence for any general mutagenesis that might cause their appearance. The lac → lac+ reversion rate is 10−8/cell/division during nonselective growth in liquid medium (Rosche and Foster 2006). A lawn of 108 plated cells produces 100 colonies over 5 to 6 days (about 100-fold more than expected) and does so with no apparent lawn growth and very little general mutagenesis. If every plated cell divided once, the number of revertants could be explained by a 100-fold increase in general mutation rate. However, the lawn of parent cells shows no increase in the frequency of unselected mutations (Torkelson et al. 1997; Rosche and Foster 1999; Slechta et al. 2002). The Lac+ revertant colonies show a 20-fold increase in the frequency of unselected associated mutations, which is unevenly distributed. Ninety percent of Lac+ revertants show no evidence of mutagenesis, whereas 10% have experienced about a 100-fold increase in genome-wide mutation frequency (Rosche and Foster 1999). It seems clear that some mutagenesis occurs but is insufficient to explain the revertants, and is distributed in interesting ways.
THREE SUGGESTIONS TO EXPLAIN THE UNEVEN DISTRIBUTION OF MUTAGENESIS
Several ideas have been proposed to explain how Lac+ revertant number can increase 100-fold with very little general genome-wide mutagenesis. The first two ideas below have been used, individually or together, as parts of models that explain the Lac+ mutants by stress-induced mutagenesis. The third idea (our favorite) suggests that stress does not induce mutagenesis at all. It attributes the modest observed increases in mutation rate to a nonessential artifactual side effect of growth under selection in this system.
Directed Mutagenesis—Stress Induces Predominantly Beneficial Mutations in All Cells of the Population
This model proposes a regulatory mechanism that senses the physiological problem and directs mutagenesis to sites that improve growth (Foster and Cairns 1992; Foster 1993). Direction to useful sites explains how the number of Lac+ revertants might increase more than the number of mutants at large. That is, the mechanism makes predominantly valuable changes. Although a mechanism to direct mutagenesis may seem difficult to imagine, clever ways of achieving this have been suggested (Stahl 1988). Those include an increase in transcription errors, or a failure in the postreplicative mismatch correction system. These possibilities were later rejected (Foster and Cairns 1992; Stahl 1992). One important early observation was that the recombinase RecA and some DNA synthesis are required for appearance of Lac+ revertants under selection (Cairns and Foster 1991; Foster and Cairns 1992). The selective-amplification model described below relies on RecA and has the effect of directing mutagenesis to the precise positions that limit growth (Roth et al. 1996).
Later experiments showed that mutagenesis is not directed in the strictest sense. Starvation of lac mutants on lactose stimulates selective reversion of mutations in other genes on the F′ plasmid (+1 frameshifts in tetA), not just in lac genes (Foster 1997). During starvation on lactose, tetracycline-resistant mutants (TetA+) accumulated at the same linear rate as did Lac+ revertants. This was evidence that limitation of growth on lactose does not direct mutations specifically to the lac operon, but also affects other sites close to lac on F′lac. The formation rate of both apparently directed mutation types can be enhanced by DinB if the dinB+ gene is also present on the plasmid that carries tetA and lac (see model below).
Hypermutable States—Stress Induces Genome-Wide Mutagenesis in a Subset (10−3) of the Plated Population
This model was suggested by Barry Hall (1990) and was supported by the discovery that Lac+ revertants have an increased probability of carrying associated unselected mutations (Torkelson et al. 1997; Rosche and Foster 1999; Slechta et al. 2002). The Lac+ revertant cells with associated mutations do not arise in cells with a heritable mutator mutation (Rosche and Foster 1999). The frequency of unselected mutants in the starved population is low, and the small fraction of those cells that were mutagenized has been estimated at about 1 cell in a 1000 (Rosche and Foster 1999; Bull et al. 2000). If the accumulated 100 Lac+ revertants were initiated by cells that arose in the mutagenized subset of cells, they would have to arise from 105 instead 108 plated cells. Generating 100 revertants from 105 cells (a rate of 10−3) would require a 105-fold increase in mutation rate over that measured in unselected cells. When inflicted on nongrowing cells, this intense mutagenesis is higher than can be achieved by any chemical mutagen and is expected to add about eight null mutations to each Lac+ genome (Roth et al. 2003). Thus, general mutagenesis alone cannot explain the revertants appearing in the Cairns–Foster system, but might contribute if combined with the directed mutagenesis described above.
Selective Improvement of Preexisting Small-Effect Mutants—Stress Is Not Mutagenic—Only an Agent of Selection
This model proposes that cells with multiple copies of the leaky mutant lac allele arise before selection and initiate the colonies that appear later on lactose plates. Because of the residual activity of the mutant lac allele, multiple copies of the mutant gene might provide sufficient energy to support growth or at least allow repeated plasmid replication under selection. Repeated replication of multiple lac target sites might enhance the likelihood of an improving mutation using only the basal mutation rate. This lac amplification may occur in slowly growing cells or in nondividing cells that continue to replicate their F′lac plasmid under selection.
The enhanced yield of Lac+ revertants under selection and the associated mutagenesis seen in 10% of Lac revertants is attributed in part to activity of the error-prone DinB polymerase. This polymerase is encoded by a gene located near lac on the F′lac plasmid, and, therefore, is subject to increased expression in strains with multiple F′lac copies or tandem duplications large enough to include both the lac and dinB genes.
THE PHILOSOPHICAL CONUNDRUM POSED BY THE CAIRNS–FOSTER SYSTEM
Work on the Cairns–Foster system has generated a body of high-quality data. Parties to the debate accept most of these results. A few remaining conflicts will be discussed below. Despite their general acceptance, these results have been interpreted in diametrically opposite ways. One side concludes that cells have mechanisms to create mutations whenever growth is blocked (“adaptive mutation,” “stress-induced mutagenesis,” “directed mutation,” “stationary phase mutagenesis,” or “hypermutable states”). The other side (that is, us) interprets the same results as evidence that growth limitation does not change the mutation rate, but serves as an agent of selection that favors growth and improvement of preexisting partial revertants. The failure to resolve this issue may result from a tendency of both sides to test predictions that verify their point of view rather than doing experiments that decide between the two general possibilities. Experiments discussed below may prove decisive.
FACTORS THAT AFFECT THE YIELD OF STABLE AND UNSTABLE REVERTANTS—THE MAJOR FACTS
The accumulated body of information has been reviewed repeatedly, both from the viewpoint of stress-induced mutagenesis (Foster 1993, 1999, 2005, 2007; Rosenberg 2001; Hersh et al. 2004; Galhardo et al. 2007; Rosenberg et al. 2012) and from the position of selection alone (Roth and Andersson 2004; Roth et al. 2006; Andersson et al. 2011). Below, we describe the most striking findings. These results are agreed on but interpreted differently. Later, we will combine these results, resolve some data conflicts, and offer a comprehensive view of reversion in the Cairns–Foster system that has broader implications for evolutionary processes.
F′ Plasmid Transfer Replication
The yield of Lac+ revertants is essentially eliminated by mutations that inactivate the ability of the F′lac plasmid to transfer conjugatively to a recipient cell (Foster and Trimarchi 1995a; Galitski and Roth 1995; Radicella et al. 1995; Peters et al. 1996). This is most striking for strains lacking TraI—a plasmid protein with DNA nickase and helicase activities, both of which contribute to transfer replication and to reversion (Traxler and Minkley 1988; Foster and Trimarchi 1995a). During the reversion process, a Tn10 inserted on the F′ plasmid is subject to frequent loss, which is known be stimulated by single-strandedness (Syvanen et al. 1986). Because conjugal replication origin produces a single-strand product, frequent Tn10 loss suggested that reversion involves plasmid transfer or transfer-associated replication from the oriT transfer origin (Godoy and Fox 2000). However, transfer of the whole plasmid is seldom associated with reversion (Foster and Trimarchi 1995a,b; S Maisnier-Patin and JR Roth, unpubl.). These results do not eliminate the possibility that TraI initiates internal F′lac replication from the transfer origin (oriT) or contributes to transfer within a single cell or among daughter cells. The F-plasmid conjugation functions are needed for both stable and unstable Lac+ revertants (Ponder et al. 2005). Consistent with the above points, the effect of growth limitation on lac reversion depends on the leaky lac allele being located on the F′ plasmid (Slechta et al. 2003).
Recombination Proficiency
Lack of the RecA (DNA strand exchange) protein or the RecBC/RuvABC proteins (double-strand break repair) reduces revertant number severely (Cairns and Foster 1991; Harris et al. 1994). Other recombination defects cause a generally smaller reduction (He et al. 2006). The several schools of thought suggest that recombination either plays a role in initiating mutagenic DNA replication or contributes to the process of gene amplification. Ability to recombine, like ability to transfer, is required for both stable and unstable Lac+ revertant colonies.
The DinB Error-Prone DNA Repair Polymerase
This polymerase (also called Pol IV) belongs to the Y-family of translesion DNA polymerases and is part of the SOS response to DNA damage (Friedberg et al. 2005). DinB can efficiently replicate a template containing damaged bases that block normal replication, especially bulky lesions formed at the N2-position of deoxyguanosine (Jarosz et al. 2007; Walsh et al. 2011). DinB can also elongate misaligned primers and incorporate modified nucleotides (Nohmi 2006). Overexpression of DinB leads to spontaneous mutagenesis of undamaged DNA. In particular, a high level of DinB makes −1 mutations by template slippage in repetitive sequences (Kim et al. 2001; Nohmi 2006)—the type of sequence found at the site of the lac mutation in the Cairns system.
In the absence of DinB, the number of stable Lac+ revertants drops about 10-fold but the yield of unstable revertants is unaffected (McKenzie et al. 2001). Without DinB, stable and unstable revertants appear in equal numbers (a 1:1 instead of a 9:1 ratio) and total revertant yield is reduced about fivefold (see Fig. 4). Thus, DinB contributes substantially, but is not absolutely required for the accumulation of revertants under selection.
Figure 4.

Major contributors to revertant yield. A considerable body of data has been accumulated to test the effect of various chromosome- and plasmid-encoded functions on revertant yield. Top left shows dependence of revertant yield on the error-prone repair polymerase DinB (4- to 5-fold). Top center shows that, in a DinB+ strain, stable revertants are about 90% of total and unstable revertants are 10%. Top right shows that, without DinB, the number of stable revertants is reduced about 10-fold and becomes roughly equal to the number of unstable revertants (Foster 2000; McKenzie et al. 2001). That is, even without the mutagenic DinB polymerase, the number of stable revertants increases with time under selection. Two of the most important functions for reversion are TraI (bottom left), which nicks and unwinds plasmid DNA during conjugative transfer, and RecA, a strand-exchange protein that is central to homologous recombination. Mutants in recBC and ruvABC genes have similarly strong effects on revertant yield.
Duplication and Amplification of the lac Operon
Gene duplications form at a high rate and come to a high steady-state frequency during nonselective growth (Reams et al. 2010). This steady state is maintained by the balance between high formation rates on the one hand and high loss rates plus fitness cost on the other. Cells with higher amplification are expected to come to lower steady-state frequencies. Cells whose F′lac carries a large duplication (100 kb) of the plasmid lac region are carried at a high steady-state frequency of 1/500. These common lac duplications form on F′lac between flanking copies of IS3 (1.2 kb). Unstable Lac+ revertant colonies consist of cells with an amplification of a smaller lac region (20–30 kb). The duplication underlying these amplifications often form among repetitive extragenic palindromic (REP) elements (Bachellier et al. 1999; Kofoid et al. 2003; Slack et al. 2006; Kugelberg et al. 2010). The cells in unstable Lac+ revertant colonies grow under selection because they have 10–100 copies of a small region (lower fitness cost) that includes the leaky mutant lac allele.
The amplifications found in unstable Lac+ revertants are of two types. Some have tandem direct-order repeats with a short junction (SJ) sequence and appear to arise by an exchange among very short repeats (10 bp). Others have an array of tandem copies in alternating orientation (head-to-head and tail-to-tail) (Kugelberg et al. 2006, 2010; Slack et al. 2006). These are known as tandem inversion duplications (TIDs) (see Fig. 3). Both duplication types allow slow growth on lactose and expand into higher amplifications by unequal recombination among the directly repeated sequences (Kugelberg et al. 2006, 2010). Copy-number expansion is expected whenever the growth improvement provided by additional lac copies exceeds the fitness cost of the amplification.
TWO DIAMETRICALLY CONTRADICTORY MODELS—AND HOW THEY GREW
The two general models described below use the same basic facts outlined above, but explain reversion in the Cairns–Foster system in different ways. In the first, colonies are initiated on the selection plate when an evolved stress-induced cellular mechanism causes a discontinuous event in a nongrowing cell. This event either creates a new large-effect frameshift mutation in the lacI-lacZ gene or a sudden amplification of mutant lac copy number. In the second model, colonies are initiated by preexisting mutants with a lac duplication. These cells grow slowly at first and improve under selection. We will first describe one simple version of these two opposite models and then we will consider some data conflicts and present a unified model that we think accounts for all of the available information.
Stress-Induced Mutagenesis
In this model, none of the plated tester cells grow under selection but all remain alive and respond to starvation by inducing a set of genes that lead to DinB-dependent mutagenesis. Increased expression of DinB is mediated by LexA, repressor of the SOS DNA repair system, and by RpoS, activator of genes during slow growth (Layton and Foster 2003; Lombardo et al. 2004; Galhardo et al. 2009). Although the tester cells are not dividing, the leakiness of the lac allele is thought to give enough energy for occasional firing of the vegetative plasmid origin on F′ and DNA synthesis. If a replication fork encounters a nick produced at the conjugational replication origin oriT by the endonuclease TraI, the fork collapses creating a double-strand break. Normally, such ends are processed in an error-free manner by RecA/BC-dependent recombination. However, during growth limitation, the model proposes that repair becomes mutagenic because RpoS and DinB proteins are up-regulated. Both of these proteins are crucial to the formation of the point mutations (−1 frameshifts) that produce a lac+ allele and a stable revertant colony. In other tester cells, starvation causes a sudden lac amplification, and the multiple mutant lac alleles allow growth and formation of an unstable revertant colony. In this model, both stable and unstable revertant types are independently initiated by discrete events that are induced under selection by a stress-induced mechanism. These events all occur in nongrowing cells.
Amplification Under Selection
In the initial form of this model, the reversion process starts with a duplication-bearing cell formed during nonselective pregrowth before plating. Such cells are extremely common (1/500) and grow very slowly under selection. Their growth rate increases progressively by expansion of the tandem array of lac alleles. Ultimately, the mutant lac allele is replicated a sufficient number of times that one copy acquires a reversion event to lac+. This can occur at the standard unenhanced mutation rate. As soon as a frameshift mutation generates a revertant lac+ allele, growth accelerates and a stable revertant colony forms. If a deletion removes a junction of the initial duplication, the repeat size is reduced. This lowers the fitness cost of amplification, allowing higher amplification and formation of an unstable revertant colony.
This model requires no mutagenesis, but does benefit from multiple copies of the dinB+ gene. Mutation rates do increase in colonies whose lac amplification includes the nearby dinB gene, which happens to lie only 16 kb away from lac on the F′lac plasmid. This model was developed based on experiments with the Salmonella version of this system in which the lac mutation supports higher lawn growth. The model seems to explain the behavior of Salmonella, but does not account for behavior of the E. coli system in which much less growth occurs under selection.
FOUR DATA CONFLICTS THAT HAVE DELAYED A FINAL DECISION
Below are four questions whose answers should help decide between “stress-induced mutagenesis” and “amplification under selection.” We think that these answers are near.
Are Revertants Initiated before or after Selection?
A decisive question regarding these models is whether revertant colonies are initiated by cells that arise on the selection plate, possibly induced by stress. Alternatively, colonies could be initiated by weak Lac+ cells that form before plating as proposed by the selection models. One of the strongest arguments in favor of “stress-induced mutagenesis” is the outcome of a Luria–Delbrück fluctuation test applied to reversion in the Cairns–Foster system (Cairns and Foster 1991). Multiple parallel cultures were plated independently on selective medium, and day 5 revertant yields were found to show a Poisson distribution. The absence of “jackpots” characteristic of a Luria–Delbrück distribution was interpreted as evidence that Lac+ colonies cannot be initiated before selection but must arise on the selection plate. This result is opposite to that of classical experiments showing that mutants detected by laboratory selections arise before selection (Luria and Delbrück 1943; Newcombe 1949; Lederberg and Lederberg 1952). The results were taken as evidence that the Lac+ colonies arising in the Cairns–Foster system were initiated on the plate by mutations formed in response to selection or “stress.”
Since the time those fluctuation tests were done, it has become clear that duplication mutations are immune to Luria–Delbrück fluctuation tests (Reams et al. 2010). The frequency of duplications and higher amplifications in unselected cultures comes to a steady-state frequency that is maintained by a balance between the high rate of gene duplication formation (10−5/cell/generation) on the one hand, and the even higher loss rate (typically 10−2/cell/generation) plus the fitness cost of the duplication on the other hand. The forces that drive the frequency of cells with a gene duplication (or amplification) toward steady state act on any aberrant frequency caused by timing of formation events. This minimizes the occurrence of jackpots. Because these steady states obscure fluctuation, a different test was required to determine whether cells with preexisting duplications initiate revertant colonies that appear under selection.
Recent evidence suggests that revertant colonies are initiated by cells that form before selection and thus cannot be stress induced (Sano et al. 2014). This was determined by placing the tetA gene near lac on the F′lac plasmid. Multiple copies of tetA are toxic when induced by the analogue anhydrotetracycline (AnTc). If cells with multiple copies of lac initiate revertants, many of those cells should also have multiple copies of the nearby tetA gene and be sensitive to inhibition by AnTc. Reducing the frequency of high-copy variant cells (lowering their steady-state frequency) should reduce the number of revertant colonies seen later under selection. When cultures of the tester strain with tetA on the F′lac plasmid were pregrown in the presence of AnTc, the number of Lac+ revertants was reduced sharply. The revertant number is restored by a few additional generations of growth in the absence of AnTc. Unexpectedly, the reduction in revertant yield was seen regardless of the position of tetA on the F′lac plasmid. This suggested that the critical cells have multiple copies of the whole F′lac plasmid, rather than an amplification of a small lac region. This finding is parallel with the “amplification under selection” model, except that amplification occurs at the level of plasmid copy number instead of by tandem repeats of only the lac region. Cells with multiple copies of the F′lac plasmid initiate both the stable and unstable Lac+ revertants. If all revertants are initiated before selection, we conclude they cannot be “stress-induced.”
Must dinB Be Located on the F′lac Plasmid?
In the original “amplification under selection” model, expression of DinB is increased by selective coamplification of the dinB and lac genes. This requires that the two genes be located close together, as they happen to be in the original tester strain. In contrast, “mutagenesis” models” propose that stress increases the level of DinB protein by transcriptional gene regulatory mechanisms (e.g., RpoS, LexA) and gene position should be irrelevant. We found a clear effect of dinB gene position on revertant yield—the Lac+ revertant number was severely decreased by removing the dinB+ allele from its position cis to lac on the F′lac plasmid (Slechta et al. 2003). Functionality of the chromosomal dinB allele was not required. The conflict arose when the same experiment (performed in another laboratory) showed no effect of position (McKenzie et al. 2001). We have reconstructed all of the critical strains and still get our original result—a full yield of stable revertants requires a functional dinB+ allele located on the same plasmid as lac. Resolving this “he said, she said” stand-off will require more testing, but we are convinced that dinB gene position is critical and observed mutagenesis requires coamplification of dinB with lac.
Is There Growth before Mutation Under Selection?
Mutagenesis models depend on mutants arising in nongrowing cells. In contrast, our original amplification–selection model relies on growth under selection. Most other systems used to support stress-induced mutagenesis have not eliminated growth under selection, making it hard to decide what is happening (Mittelman 2013). In the Cairns–Foster system, supporters of mutagenesis have taken pains to show that cells are not growing (see below). Supporters of selective amplification argue that these tests have missed important growth within developing colonies, which is systematically missed by assays of the plate population. The question of growth could be decisive.
A considerable body of evidence supports the belief that mutations arise in nongrowing cells. This evidence must be accounted for in any final model. Notice that the preponderance of evidence below argues against growth:
The total number of parental cells in the lawn (outside of colonies) does not increase for 4 days. The constant lawn population is not a balance between growth and death (Foster 1994). However, a large population (108) is being tested so one doubling would be hard to detect and yet would provide 10−8 opportunities for mutation. In addition, these tests do not assess growth within the developing colony.
If growth occurs within colonies, as suggested by the original amplification model, then the number of visible colonies is expected to increase exponentially because an exponentially increasing number of precursor cells is present in each colony. The graphs describing revertant accumulation show an upward flex in some experiments but, in general, this accumulation is linear with time, consistent with very little general population growth over 4 days. This argues against growth within the developing colony before the reversion event.
Newcomb spreading experiments did not show evidence that lac+ revertants arise from cells growing under selection before mutation (Newcombe 1949). This classic experiment is a contemporary of the Luria–Delbrück fluctuation test and the Lederbergs’ replica-printing experiment, which originally showed that mutations are not caused by the selection. Newcombe plated bacteria cells on nonselective medium and allowed time for a few divisions. He then spread the population of the plate before imposing a selection (phage resistance). If resistant mutants were present in the plated population and grew before selection (which they did), the act of spreading multiple cells within a developing colony should seed multiple colonies that could be detected after selection was imposed. Spreading should not increase selected mutant number if mutations are induced by exposure to selection.
If colonies are initiated by rare preexisting cells that grow under selection before reversion, then respreading the lawn shortly after plating should increase the number of revertants detected after several days under selection. This happened in Newcomb’s experiment, indicating that the mutant clones arose and grew before selection. In the Cairns–Foster experiment, this experiment would test whether preexisting slow-growing cells grow before acquiring a full lac+ reversion mutation, as suggested by the amplification model. This experiment is difficult because spreading must be performed after some growth has occurred but before any developing clone has acquired a full lac+ allele. It is also crucial to avoid preexisting Lac+ cells, which will certainly grow and produce many colonies after spreading. The experiment was tried by Foster (1994) and by us (S Quiñones-Soto and Y Toofan, unpubl.). Both attempts failed to provide evidence of preexisting cells that grow before reversion.
Are Amplifications Remodeled Under Selection?
The original “amplification–selection” model proposed that revertants were initiated by cells with a duplication or amplification of the large 100-kb lac region among IS3 copies. These cells were suggested to grow and amplify a bit under selection and then acquire either a point mutation to lac+ (aided by their multiple copies of DinB) or a join point deletion. The join point deletion reduces the size of the repeated unit and thereby the fitness cost so as to allow high amplification under selection. Without growth, this remodeling process seems unlikely.
Since then, we have found evidence that the common large IS3-mediated duplications make no contribution to reversion and the shorter duplications (see Fig. 3) that underlie the amplifications in unstable revertants actually arise during nonselective pregrowth (Kugelberg et al. 2010). These short duplications are sufficiently frequent in the plated population to explain all of the unstable Lac+ revertants (Reams et al. 2012). If SJ and TID duplications arise by remodeling of a preexisting duplication, this must all occur during growth before selection. Because unstable revertants are initiated by cells that arise before plating, they cannot be “stress-induced” and are essentially already remodeled before selection. We will propose below that the precursors of unstable revertants grow immediately after plating and improve under selection by further expansion of their amplified array. As expected for exponentially growing clones that become visible with time, the number of unstable revertants increases exponentially over a series of days (Fig. 4). It seems clear that growth is central to development of these colonies and they are initiated by cells that arise before selection. The open question of growth involves primarily the early history of stable Lac+ revertants.
THE DEEPER MEANING OF GROWTH AND MUTAGENESIS
The question of growth under selection is critical because growth and mutagenesis are essentially trade-offs in trying to account for the 100 revertants that arise in this system. If growth is truly eliminated by selection, as seems likely, mutagenesis of some kind seems essential to explain the observed mutations. If growth occurs, then the exponential expansion may provide sufficient acts of replication to produce the revertants using the standard unenhanced basal mutation rate. We have resisted the idea of mutagenesis because the idea of an evolved mechanism to increase the genome-wide mutation rate temporarily seemed numerically unsanitary. Mathematical modeling of the “hypermutable state” model suggested that temporary up-regulation of genome-wide mutagation rate in a subset of the population is unlikely to be useful long term because of the high cost of deleterious mutations and the persistence of deleterious mutations after any episode of hypermutability (Roth et al. 2003). This modeling does not eliminate the mechanistically problematic possibility of “directed mutation.”
Our persistent confidence that growth must occur but somehow escape the tests listed above was based on the finding that stable Lac+ revertant colonies include rare cells that are unstable Lac+ (Hendrickson et al. 2002). The growth and amplification model predicts that stable revertant colonies should contain a few unstable precursor cells with an amplified mutant lac allele. Such ancestral cells were identified by their formation of sectored (blue/white) colonies when streaked on rich X-gal medium and were taken as evidence for amplification and growth within the developing colony before reversion. These unstable Lac+ cells represented about one cell in 1000 stable Lac+ cells (Hendrickson et al. 2002). These sectored colonies are visually indistinguishable from the extensively characterized cells in unstable Lac+ colonies, which carry an extensive lac amplification. However, Hastings et al. (2004) reported that the rare unstable cells within otherwise stable Lac+ colonies showed an unstable Lac+ phenotype that is not heritable. We have confirmed their observation and found that the rare unstable cells do not have lac amplifications (which are heritable) but seem to have multiple copies of the F′lac plasmid, some lac+ and some lac. This is an extremely unstable (poorly heritable) situation attributed to the mechanisms that control plasmid copy number and distribution. This result suggested that lac+ revertant alleles might not arise in cells growing with amplification, but rather in cells with multiple copies of the whole F′lac plasmid. This is consistent with the evidence above that revertants are initiated by cells with multiple plasmids copies. These results reopened the question of growth, because cells with multiple plasmid copies seem too unstable to form extensive clones under selection but might persist when division is blocked.
Without growth it is difficult to explain the revertant number without some mutagenesis.
A NEW MODEL RESOLVES THE CONFLICTS
This model is consistent with the entire body of information accumulated by both sides of the Cairns debate and outlined above. In this model, reversion starts before selection (with a plasmid copy-number increase) and the critical frameshift mutations occur under selection without prior growth and with no programmed increase in mutation rate:
The major feature of this model is that both stable and unstable Lac+ revertants are initiated by cells with an increased F′lac plasmid copy number. These cells arise before selection so the revertant colonies they initiate cannot be stress induced.
Although the initiator cells with multiple copies of F′lac arise before selection, their precise structure is unclear. These cells seem to have evaded standard plasmid copy-number controls. These plasmids may have transfer replication forks that have switched to rolling circle replication and produce double-stranded linear products that may be degraded as they are made. Alternatively, actual transfer might be occurring as suggested in earlier studies (Galitski and Roth 1995; Radicella et al. 1995; Peters et al. 1996; Godoy and Fox 2000). Transfer could inject a single-strand end that becomes double stranded, recombines, and initiates a rolling circle replication fork using the recipient plasmid as template. Additional replication of lac in these nongrowing cells may result from nicks introduced at oriT, which become double-strand breaks when hit by a replication fork, as suggested previously (Foster et al. 1996; Harris et al. 1996). Regardless of the nature of these cells, they replicate lac repeatedly over the course of several days using energy provided by the leaky lac allele. Each act of lac replication provides an opportunity for reversion. The DNA single strands exposed during this process induce an SOS response. The absence of cell growth induces the RpoS regulon. These two features have been suggested previously (Foster 2007; Galhardo et al. 2007).
After plating, these initiator cells do not grow exponentially, because they lack sufficient energy to fire their chromosomal replication forks. They use the energy provided by multiple lac copies to repeatedly replicate and repair their plasmids. The plasmid replication (from an unregulated origin) exploits available energy and allows continued copying of the plasmid in nondividing cells. The failure of cells to divide leaves them unable to easily reduce their plasmid copy number until growth resumes.
Formation of stable revertants (−1 frameshifts) is enhanced about 10-fold by the presence of a dinB+ gene anywhere on the F′lac plasmid. An increased level of DinB protein results from multiple dinB genes on the several plasmid copies. Mutations to lac+ form during overreplication of lac on the multiple plasmid copies. This replication alone is sufficient for some reversion, but the revertant yield is enhanced about 10-fold because DinB is amplified as part of the plasmid. Because cells are not dividing, the main chromosome is not replicating and is not subject to DinB mutagenesis.
The events described above do not reflect a process that is stress induced in a standard sense. Selective conditions prevent cell division but allow repeated local overreplication of the F′lac plasmid. This overreplication is excessive only because cell division and chromosome replication are blocked. Extra plasmid copies do not segregate.
When any one of the many copies of the mutant lac allele acquires a lac+ reversion event (−1 frameshift), energy is immediately supplied, allowing resumption of cell division and chromosome replication. All plasmids with nonrevertant lac copies are lost by segregation as the cell divides and copy-number controls come into play. The revertant F′lac+ copy is selectively maintained in cells with a normalized copy number. This stable lac+ colony contains no cells with a tandem lac+ amplification, but may include a few cells that have not yet diluted out all of their unreverted F′lac copies. These explain the rare cells in a stable Lac+ colony that show nonheritable unstable Lac+ phenotypes.
The unstable Lac+ revertants are also initiated by preexisting cells with multiple copies of an F′lac plasmid, but the plasmid in these cells carry an internal lac duplication (SJ or TID). Cells with multiple copies of a duplication-bearing F′lac plasmid have enough energy to fire their replication origins and divide. Their plasmid copy number can be maintained in part by continued replication (requiring Tra functions), but drops progressively by unequal plasmid partitioning during growth. As plasmid copy number returns toward normal, the lac amplification expands and progressively improves growth. These cells ultimately produce a colony of unstable Lac+ colonies.
In principle, this model is identical to the initial selected tandem amplification model described above except that: (1) Plasmid copy number increases before selection and is maintained without growth. Stable elevated plasmids copy numbers in nongrowing cells take the place of tandem copy amplification during growth under selection. (2) Local overreplication of the whole plasmid in nongrowing cells replaces cell growth to provide opportunities for reversion.
Preexisting cells with multiple copies of an F′lac plasmid may be equivalent to the rare “hypermutable” cells in mutagenesis models. Under selection, cells in this state replicate their F plasmid and lac region in the presence of high levels of DinB, but do not replicate their chromosome, which is not generally mutagenized.
Mutagenesis in this model is “directed” in two senses. Only the F′lac replicates in the presence of elevated DinB levels. Following a rare act of reversion, the population of unreverted lac alleles that allowed this event is lost and only the rare lac+ allele is retained selectively. This has the effect of directing mutagenesis to the precise base pairs that limit ability to grow on lactose (Roth et al. 1996). This point will be discussed more below.
RELEVANCE OF THE CAIRNS–FOSTER SYSTEM TO EVOLUTION UNDER SELECTION
Since the advent of the Cairns–Foster system, it has become clear that copy-number variations are the most common genetic changes in natural populations and in populations of metazoan somatic cells (reviewed by Reams and Roth 2014). Selective gene amplification underlies progression of many malignancies (Albertson 2006). Our original selection–amplification model for the Cairns system involved growth and improvement under selection. This first model seems to explain the Salmonella version of the Cairns–Foster system and has been experimentally verified for a variety of other biological situations, including evolution of bacterial antibiotic resistance (Nilsson et al. 2006; Sandegren and Andersson 2009; Sun et al. 2009; Paulander et al. 2010; Pranting and Andersson 2011) and adaptation of poxvirus to host defenses (Elde et al. 2012). Most generally, the selective amplification model offers an explanation for evolution of novel genes under selection (Bergthorsson et al. 2007) and has been experimentally shown to produce a new gene within 3000 generations of growth under selection (Näsvall et al. 2012). Thus, the Cairns–Foster system has suggested a broadly relevant biological process.
Despite these successes, it now appears that our original model does not quite explain the E. coli version of the Cairns–Foster system in which very little growth occurs. Resolution of several conflicts led to the new model described above, which does explain the E. coli system. This new model may also provide answers to questions with broad evolutionary relevance.
How Does Selected Amplification Mimic Mutagenesis?
Despite a considerable fitness cost, duplications and amplifications are held as stable polymorphisms in unselected populations (Reams et al. 2010). These steady-state frequencies are high because duplications and amplifications form at a high rate. Steady-state frequencies increase when selection favors cells with multiple copies of some near-neutral allele. Amplification of such an allele can provide a selective value that exceeds the cost of the duplication itself. This allows lineages with amplifications to expand and add copies of a growth-limiting allele to the genome. These cells grow faster and replicate more copies of the rate-limiting allele at each division. Each act of replication provides an opportunity for further mutations that improve the functionality of the limiting allele. In the Cairns–Foster system, the mutant lac allele has a very low ability to support growth on lactose and is, in a sense, a near-neutral allele that can provide a beneficial phenotype when amplified. Thus, selection gives the appearance of mutagenesis that actually results from replicating more copies of the target allele (see Fig. 5).
Figure 5.

How lac amplification enhances revertant yield without mutagenesis. Each act of lac replication provides an opportunity for a reversion event (frameshift). A cell with only one lac copy has one opportunity to revert for each cell division or plasmid replication (see left column). As the lac allele amplifies, each cell gains additional chances for a reversion event with no increase in mutation rate (right column). If the amplified array improves growth on lactose, then the lineage on the right expands faster than that on the left (without an amplification) and this growth also adds to the likelihood of a reversion event. In effect, amplification directs mutations to the exact base pairs that limit growth, because once a revertant allele forms, selection holds only that allele, whereas the nonrevertants alleles are no longer selectively retained. The likelihood of an unselected mutation near lac is the same with and without selection, because only the revertant allele is kept in the genome.
How Can Selection Appear to Direct Mutation to Critical Sites?
Direction of mutation to selectively valuable sites is a side effect of amplification under selection. As soon as an improving mutation occurs in any one of the multiple lac copies, selection holds the improved allele and allows loss of other copies of the same gene. Deleterious alleles of the gene in question are selectively lost from the lineage. The apparent directedness is caused by the dynamic control of gene copy number—loss and reamplification occurs continuously and affects genes carried in either tandem arrays or on multicopy plasmids. In essence, the multiple gene copies in an amplified array (or multiple plasmid copies in a cell) constitute a population that is under selection to improve. The probability that a final revertant plasmid has an associated unselected mutation is the same as the probability of that associated mutation arising in any single copy of the gene with no selection or amplification. Thus, amplification enhances the likelihood that some lac+ mutation will occur, but does not enhance the likelihood of an associated unselected mutation, because selection fixes only the revertant allele. Mutations are, in effect, directed to the exact base pairs that provide the selected phenotype and are not made more likely at other sites.
Natural Selection without Reproduction
It is generally assumed that natural selection requires cell division because improvement (or purification) is attributed to differential reproductive success of cells with different genotypes. In the Cairns–Foster system, the plated cell population is subjected to selection that prevents growth (just barely). The growth-limiting gene (on the F′lac plasmid) is repeatedly copied until an improving mutation occurs. A revertant allele triggers exponential growth and is selectively held, whereas nonrevertant alleles are lost by segregation as soon as cell division starts and the population expands.
The increase in lac copy number occurs before selection, by some loss of plasmid copy-number control. On lactose medium, cells cannot divide and have no way to reduce their copy number. They can, however, replicate their plasmids and use DNA repair mechanisms to heal plasmid breaks introduced by the TraI protein and/or recombine DNA fragments with recipient plasmids during redundant conjugation. Thus, the plasmid is a subset of the genome that is overreplicated in nondividing cells. Cells are trapped in this state until a reversion event occurs.
Mutagenesis by overreplication may be relevant to progression of some malignancies (Albertson 2006). Somatic cells that grow very little may repeatedly repair regions of their genome surrounding fragile sites, which are subject to frequent breakage. In this situation, nongrowing cells are under strong selection to grow and may repeatedly replicate specific regions of their genome during break repair. This may be mimicked in the Cairns–Foster system in which breaks induced at oriT stimulate mutagenesis of the F′lac plasmid. We submit that the phenomenon of break-induced mutagenesis (Shee et al. 2011; Rosenberg et al. 2012) may reflect repeated repair and local overreplication in nongrowing cells and involve no increase in mutation rate.
CONCLUDING REMARKS
At first glance, the Cairns–Foster system appears simple and seems to show stress-induced mutagenesis of nongrowing cells. This conflicts with the classical conclusion that selection and mutation are independent. Deeper inspection reveals that the behavior of this system does not require mutagenesis but relies on several aspects of selection. The apparent direction of mutations to valuable sites results from selective amplification of the growth-limiting gene and selective retention of the improved allele (a form of selection). The modest increase in general mutation rate caused by DinB is an artifact owing to the chance proximity of the dinB and lac genes, which enables selective coamplification. Cells in a hypermutable state are those with multiple copies of the F′lac plasmid that includes lac and dinB. These cells arise before selection and are not stress induced.
The response of populations to selection can be rapid when it exploits two of the most common mutation types known. Duplications and amplifications form frequently and are held in populations as stable genetic polymorphisms. Near-neutral beneficial alleles are extremely common and gain significant value when amplified. Selection thus gives the appearance of mutagenesis by driving rapid progressive genetic adaptation with no programmed change in mutation rate.
ACKNOWLEDGMENTS
We thank our laboratory mates Emiko Sano, John Paul Aboubechara, Semarhy Quiñones-Soto, Andrew Reams, Eric Kofoid, and Douglas Huseby for ideas and critique. We thank Ivy Roush for allowing us to cite her unpublished work. We thank colleagues Wolf Heyer, Neil Hunter, and Stephen Kowalczykowski for suggestions. We thank collaborators Dan Andersson and Diarmaid Hughes of Uppsala University for constant encouragement. Most of all, we thank John Cairns and our loyal opposition—Susan Rosenberg and Pat Foster—for their persistent and incisive defense of stress-induced mutagenesis. Their models have provided constant stimulation even though we have remained dubious that they are correct. The model described here for the Cairns system involves no programmed or induced mutagenesis. Selection alone mimics all of the previously suggested mechanisms—directed mutagenesis, hypermutable states, and amplification during growth under selection.
REFERENCES
- Albertson DG. 2006. Gene amplification in cancer. Trends Genet 22: 447–455. [DOI] [PubMed] [Google Scholar]
- Andersson DI, Hughes D, Roth JR. 2011. The origin of mutants under selection: Interactions of mutation, growth, and selection. In EcoSal-Escherichia coli and Salmonella: Cellular and molecular biology (ed. Slauch JM, et al. ). ASM, Washington, DC. [DOI] [PubMed] [Google Scholar]
- Bachellier S, Clement JM, Hofnung M. 1999. Short palindromic repetitive DNA elements in enterobacteria: A survey. Res Microbiol 150: 627–639. [DOI] [PubMed] [Google Scholar]
- Bergthorsson U, Andersson DI, Roth JR. 2007. Ohno’s dilemma: Evolution of new genes under continuous selection. Proc Natl Acad Sci 104: 17004–17009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bull HJ, McKenzie GJ, Hastings PJ, Rosenberg SM. 2000. Evidence that stationary-phase hypermutation in the Escherichia coli chromosome is promoted by recombination. Genetics 154: 1427–1437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cairns J, Foster PL. 1991. Adaptive reversion of a frameshift mutation in Escherichia coli. Genetics 128: 695–701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cairns J, Overbaugh J, Miller S. 1988. The origin of mutants. Nature 335: 142–145. [DOI] [PubMed] [Google Scholar]
- Elde NC, Child SJ, Eickbush MT, Kitzman JO, Rogers KS, Shendure J, Geballe AP, Malik HS. 2012. Poxviruses deploy genomic accordions to adapt rapidly against host antiviral defenses. Cell 150: 831–841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Foster PL. 1993. Adaptive mutation: The uses of adversity. Annu Rev Microbiol 47: 467–504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Foster PL. 1994. Population dynamics of a Lac− strain of Escherichia coli during selection for lactose utilization. Genetics 138: 253–261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Foster PL. 1997. Nonadaptive mutations occur on the F′ episome during adaptive mutation conditions in Escherichia coli. J Bacteriol 179: 1550–1554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Foster PL. 1999. Mechanisms of stationary phase mutation: A decade of adaptive mutation. Annu Rev Genet 33: 57–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Foster PL. 2000. Adaptive mutation in Escherichia coli. Cold Spring Harb Symp Quant Biol 65: 21–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Foster PL. 2005. Stress responses and genetic variation in bacteria. Mutat Res 569: 3–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Foster PL. 2007. Stress-induced mutagenesis in bacteria. Crit Rev Biochem Mol Biol 42: 373–397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Foster PL, Cairns J. 1992. Mechanisms of directed mutation. Genetics 131: 783–789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Foster PL, Trimarchi JM. 1995a. Adaptive reversion of an episomal frameshift mutation in Escherichia coli requires conjugal functions but not actual conjugation. Proc Natl Acad Sci 92: 5487–5490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Foster PL, Trimarchi JM. 1995b. Conjugation is not required for adaptive reversion of an episomal frameshift mutation in Escherichia coli. J Bacteriol 177: 6670–6671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Foster PL, Trimarchi JM, Maurer RA. 1996. Two enzymes, both of which process recombination intermediates, have opposite effects on adaptive mutation in Escherichia coli. Genetics 142: 25–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Friedberg EC, Walker GC, Siede W, Wood RD, Schultz A. 2005. DNA repair and mutagenesis. ASM, Washington, DC. [Google Scholar]
- Galhardo RS, Hastings PJ, Rosenberg SM. 2007. Mutation as a stress response and the regulation of evolvability. Crit Rev Biochem Mol Biol 42: 399–435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galhardo RS, Do R, Yamada M, Friedberg EC, Hastings PJ, Nohmi T, Rosenberg SM. 2009. DinB upregulation is the sole role of the SOS response in stress-induced mutagenesis in Escherichia coli. Genetics 182: 55–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galitski T, Roth JR. 1995. Evidence that F plasmid transfer replication underlies apparent adaptive mutation. Science 268: 421–423. [DOI] [PubMed] [Google Scholar]
- Godoy VG, Fox MS. 2000. Transposon stability and a role for conjugational transfer in adaptive mutability. Proc Natl Acad Sci 97: 7393–7398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hall BG. 1988. Adaptive evolution that requires multiple spontaneous mutations. I: Mutations involving an insertion sequence. Genetics 120: 887–897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hall BG. 1990. Spontaneous point mutations that occur more often when advantageous than when neutral. Genetics 126: 5–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harris RS, Longerich S, Rosenberg SM. 1994. Recombination in adaptive mutation. Science 264: 258–260. [DOI] [PubMed] [Google Scholar]
- Harris RS, Ross KJ, Rosenberg SM. 1996. Opposing roles of the Holliday junction processing systems of Escherichia coli in recombination-dependent adaptive mutation. Genetics 142: 681–691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hastings PJ, Slack A, Petrosino JF, Rosenberg SM. 2004. Adaptive amplification and point mutation are independent mechanisms: Evidence for various stress-inducible mutation mechanisms. PLoS Biol 2: e399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He AS, Rohatgi PR, Hersh MN, Rosenberg SM. 2006. Roles of E. coli double-strand-break-repair proteins in stress-induced mutation. DNA Repair (Amst) 5: 258–273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hendrickson H, Slechta ES, Bergthorsson U, Andersson DI, Roth JR. 2002. Amplification-mutagenesis: Evidence that “directed” adaptive mutation and general hypermutability result from growth with a selected gene amplification. Proc Natl Acad Sci 99: 2164–2169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hersh MN, Ponder RG, Hastings PJ, Rosenberg SM. 2004. Adaptive mutation and amplification in Escherichia coli: Two pathways of genome adaptation under stress. Res Microbiol 155: 352–359. [DOI] [PubMed] [Google Scholar]
- Jarosz DF, Beuning PJ, Cohen SE, Walker GC. 2007. Y-family DNA polymerases in Escherichia coli. Trends Microbiol 15: 70–77. [DOI] [PubMed] [Google Scholar]
- Kim SR, Matsui K, Yamada M, Gruz P, Nohmi T. 2001. Roles of chromosomal and episomal dinB genes encoding DNA pol IV in targeted and untargeted mutagenesis in Escherichia coli. Mol Genet Genomics 266: 207–215. [DOI] [PubMed] [Google Scholar]
- Kofoid E, Bergthorsson U, Slechta ES, Roth JR. 2003. Formation of an F′ plasmid by recombination between imperfectly repeated chromosomal Rep sequences: A closer look at an old friend (F′128 pro lac). J Bacteriol 185: 660–663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kugelberg E, Kofoid E, Reams AB, Andersson DI, Roth JR. 2006. Multiple pathways of selected gene amplification during adaptive mutation. Proc Natl Acad Sci 103: 17319–17324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kugelberg E, Kofoid E, Andersson DI, Lu Y, Mellor J, Roth FP, Roth JR. 2010. The tandem inversion duplication in Salmonella enterica: Selection drives unstable precursors to final mutation types. Genetics 185: 65–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Layton JC, Foster PL. 2003. Error-prone DNA polymerase IV is controlled by the stress-response sigma factor, RpoS, in Escherichia coli. Mol Microbiol 50: 549–561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lederberg J, Lederberg EM. 1952. Replica plating and indirect selection of bacterial mutants. J Bacteriol 63: 399–406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee H, Popodi E, Tang H, Foster PL. 2012. Rate and molecular spectrum of spontaneous mutations in the bacterium Escherichia coli as determined by whole-genome sequencing. Proc Natl Acad Sci 109: E2774–E2783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lombardo MJ, Aponyi I, Rosenberg SM. 2004. General stress response regulator RpoS in adaptive mutation and amplification in Escherichia coli. Genetics 166: 669–680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luria SE, Delbrück M. 1943. Mutations of bacteria from virus sensitivity to virus resistance. Genetics 28: 491–511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maenhaut-Michel G, Shapiro JA. 1994. The roles of starvation and selective substrates in the emergence of araB-lacZ fusion clones. EMBO J 13: 5229–5239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKenzie G, Lee P, Lombardo M-J, Hastings P, Rosenberg S. 2001. SOS mutator DNA polymerase IV functions in adaptive mutation and not adaptive amplification. Molecular Cell 7: 571–579. [DOI] [PubMed] [Google Scholar]
- Mittelman D. 2013. Stress-induced mutagenesis. Springer, New York. [Google Scholar]
- Näsvall J, Sun L, Roth JR, Andersson DI. 2012. Real-time evolution of new genes by innovation, amplification, and divergence. Science 338: 384–387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Newcombe HB. 1949. Origin of bacterial variants. Nature 164: 150. [DOI] [PubMed] [Google Scholar]
- Nilsson AI, Zorzet A, Kanth A, Dahlstrom S, Berg OG, Andersson DI. 2006. Reducing the fitness cost of antibiotic resistance by amplification of initiator tRNA genes. Proc Natl Acad Sci 103: 6976–6981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nohmi T. 2006. Environmental stress and lesion-bypass DNA polymerases. Annu Rev Microbiol 60: 231–253. [DOI] [PubMed] [Google Scholar]
- Paulander W, Andersson DI, Maisnier-Patin S. 2010. Amplification of the gene for isoleucyl-tRNA synthetase facilitates adaptation to the fitness cost of mupirocin resistance in Salmonella enterica. Genetics 185: 305–312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peters JE, Bartoszyk IM, Dheer S, Benson SA. 1996. Redundant homosexual F transfer facilitates selection-induced reversion of plasmid mutations. J Bacteriol 178: 3037–3043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ponder RG, Fonville NC, Rosenberg SM. 2005. A switch from high-fidelity to error-prone DNA double-strand break repair underlies stress-induced mutation. Mol Cell 19: 791–804. [DOI] [PubMed] [Google Scholar]
- Pranting M, Andersson DI. 2011. Escape from growth restriction in small colony variants of Salmonella typhimurium by gene amplification and mutation. Mol Microbiol 79: 305–315. [DOI] [PubMed] [Google Scholar]
- Radicella JP, Park PU, Fox MS. 1995. Adaptive mutation in Escherichia coli: A role for conjugation. Science 268: 418–420. [DOI] [PubMed] [Google Scholar]
- Reams AB, Roth JR. 2014. Mechanisms of gene duplication and amplification. In Recombination mechanisms (ed. Kowalczykowski S, Hunter N, Heyer W-D). Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reams AB, Kofoid E, Savageau M, Roth JR. 2010. Duplication frequency in a population of Salmonella enterica rapidly approaches steady state with or without recombination. Genetics 184: 1077–1094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reams AB, Kofoid E, Kugelberg E, Roth JR. 2012. Multiple pathways of duplication formation with and without recombination (RecA) in Salmonella enterica. Genetics 192: 397–415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosche WA, Foster PL. 1999. The role of transient hypermutators in adaptive mutation in Escherichia coli. Proc Natl Acad Sci 96: 6862–6867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosche WA, Foster PA. 2006. Methods for determining spontaneous mutation rates. Methods Enzymol 409: 195–213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosenberg S. 2001. Evolving responsively: Adaptive mutation. Nat Rev Genet 2: 504–514. [DOI] [PubMed] [Google Scholar]
- Rosenberg SM, Shee C, Frisch RL, Hastings PJ. 2012. Stress-induced mutation via DNA breaks in Escherichia coli: A molecular mechanism with implications for evolution and medicine. Bioessays 34: 885–892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roth JR, Andersson DI. 2004. Amplification-mutagenesis—How growth under selection contributes to the origin of genetic diversity and explains the phenomenon of adaptive mutation. Res Microbiol 155: 342–351. [DOI] [PubMed] [Google Scholar]
- Roth JR, Benson N, Galitski T, Haack K, Lawrence JG, Miesel L. 1996. Rearrangement of the bacterial chromosome: Formation and applications. In Escherichia coli and Salmonella (ed. Neidhardt FC), pp. 2256–2276. ASM, Washington, DC. [Google Scholar]
- Roth JR, Kofoid E, Roth FP, Berg OG, Seger J, Andersson DI. 2003. Regulating general mutation rates: Examination of the hypermutable state model for Cairnisian adaptive mutation. Genetics 163: 1483–1496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roth JR, Kugelberg E, Reams AB, Kofoid E, Andersson DI. 2006. Origin of mutations under selection: The adaptive mutation controversy. Annu Rev Microbiol 60: 477–501. [DOI] [PubMed] [Google Scholar]
- Sandegren L, Andersson DI. 2009. Bacterial gene amplification: Implications for the evolution of antibiotic resistance. Nat Rev Microbiol 7: 578–588. [DOI] [PubMed] [Google Scholar]
- Sano E, Maisnier-Patin S, Aboubechara J, Quiñones-Soto S, Roth JR. 2014. Plasmid copy number underlies adaptive mutability in bacteria. Genetics 198: 919–933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shapiro JA. 1984. Observations on the formation of clones containing araB-lacZ cistron fusions. Mol Gen Genet 194: 79–90. [DOI] [PubMed] [Google Scholar]
- Shee C, Gibson JL, Darrow MC, Gonzalez C, Rosenberg SM. 2011. Impact of a stress-inducible switch to mutagenic repair of DNA breaks on mutation in Escherichia coli. Proc Natl Acad Sci 108: 13659–13664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Slack A, Thornton PC, Magner DB, Rosenberg SM, Hastings PJ. 2006. On the mechanism of gene amplification induced under stress in Escherichia coli. PLoS Genet 2: e48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Slechta ES, Liu J, Andersson DI, Roth JR. 2002. Evidence that selected amplification of a bacterial lac frameshift allele stimulates Lac+ reversion (adaptive mutation) with or without general hypermutability. Genetics 161: 945–956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Slechta ES, Bunny KL, Kugelberg E, Kofoid E, Andersson DI, Roth JR. 2003. Adaptive mutation: General mutagenesis is not a programmed response to stress but results from rare coamplification of dinB with lac. Proc Natl Acad Sci 100: 12847–12852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stahl FW. 1988. News and views: A unicorn in the garden. Nature 355: 112–113. [DOI] [PubMed] [Google Scholar]
- Stahl F. 1992. Unicorns revisited. Genetics 132: 865–867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steele DF, Jinks-Robertson S. 1992. An examination of adaptive reversion in Saccharomyces cerevisiae. Genetics 132: 9–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun S, Berg OG, Roth JR, Andersson DI. 2009. Contribution of gene amplification to evolution of increased antibiotic resistance in Salmonella typhimurium. Genetics 182: 1183–1195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sung HM, Yasbin RE. 2002. Adaptive, or stationary-phase, mutagenesis, a component of bacterial differentiation in Bacillus subtilis. J Bacteriol 184: 5641–5653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Syvanen M, Hopkins JD, Griffin TJt, Liang TY, Ippen-Ihler K, Kolodner R. 1986. Stimulation of precise excision and recombination by conjugal proficient F′ plasmids. Mol Gen Genet 203: 1–7. [DOI] [PubMed] [Google Scholar]
- Torkelson J, Harris RS, Lombardo M-J, Nagendran J, Thulin C, Rosenberg SM. 1997. Genome-wide hypermutation in a subpopulation of stationary-phase cells underlies recombination-dependent adaptive mutation. EMBO J 16: 3303–3311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Traxler BA, Minkley EG Jr. 1988. Evidence that DNA helicase I and oriT site-specific nicking are both functions of the F TraI protein. J Mol Biol 204: 205–209. [DOI] [PubMed] [Google Scholar]
- Walsh JM, Hawver LA, Beuning PJ. 2011. Escherichia coli Y family DNA polymerases. Front Biosci (Landmark Ed) 16: 3164–3182. [DOI] [PubMed] [Google Scholar]