

Abstract
Drug transporters govern the absorption, distribution, and elimination of pharmacologically active compounds. Members of the solute carrier and ATP binding-cassette drug transporter family mediate cellular drug uptake and efflux processes, thereby coordinating the vectorial movement of drugs across epithelial barriers. To exert their physiologic and pharmacological function in polarized epithelia, drug transporters must be targeted and stabilized to appropriate regions of the cell membrane (i.e., apical versus basolateral). Despite the critical importance of drug transporter membrane targeting, the mechanisms that underlie these processes are largely unknown. Several clinically significant drug transporters possess a recognition sequence that binds to PSD-95/Drosophila discs large/ZO-1 (PDZ) proteins. PDZ proteins, such as the Na+/H+ exchanger regulatory factor (NHERF) family, act to stabilize and organize membrane targeting of multiple transmembrane proteins, including many clinically relevant drug transporters. These PDZ proteins are normally abundant at apical membranes, where they tether membrane-delimited transporters. NHERF expression is particularly high at the apical membrane in polarized tissue such as intestinal, hepatic, and renal epithelia, tissues important to drug disposition. Several recent studies have highlighted NHERF proteins as determinants of drug transporter function secondary to their role in controlling membrane abundance and localization. Mounting evidence strongly suggests that NHERF proteins may have clinically significant roles in pharmacokinetics and pharmacodynamics of several pharmacologically active compounds and may affect drug action in cancer and chronic kidney disease. For these reasons, NHERF proteins represent a novel class of post-translational mediators of drug transport and novel targets for new drug development.
I. Introduction
Plasma membrane transport proteins play a central role in mediating the disposition or pharmacokinetics, i.e., absorption, distribution, and elimination, of a broad spectrum of endogenous and exogenous compounds including pharmacologically active compounds and clinically used medications (Nigam, 2015). Such transporters are now acknowledged also as clinically significant determinants of drug response, i.e., pharmacodynamics. Although broadly expressed, drug transporters exhibit highest abundance in intestine, liver, and kidney, tissues important for drug disposition. Furthermore, drug transporters expressed at the blood-brain barrier and the placenta serve an additional protective role to minimize exposure of the brain and fetus, respectively, to toxic compounds.
Solute carrier (SLC) and the ATP-binding cassette (ABC) transporters form the two major superfamilies of drug transporters. SLC transporters primarily mediate cellular uptake of drugs by facilitated diffusion or ion-coupled secondary active transport. ABC transporters function as primary energy-dependent active transporters and rely on the hydrolysis of ATP to energize cellular drug efflux. Functionally, SLC and ABC drug transporters work in concert to couple the vectorial movement of drugs and drug metabolites across epithelial barriers. The clinical impact of drug transporters in pharmacotherapy highlights the need to fully characterize their regulatory mechanisms.
Epithelial cells that form the intestinal, hepatic, and renal barriers display characteristic apical-basolateral membrane polarity. Drug transporters that are expressed in these cells must be targeted and stabilized at the appropriate membrane domain (i.e., apical or basolateral) to elicit their physiologic and pharmacologic function. A key role of drug transporters located at epithelial barriers is to limit systemic drug exposure. Oral drug absorption is accomplished by an array of drug transporters that are distributed throughout the intestinal epithelium. In the liver, transporters coordinate the hepatobiliary disposition of drugs and serve a principal role in biliary drug excretion. Together, intestinal and hepatic drug transporters play a major role in mediating drug bioavailability. Drug transporters expressed in renal proximal tubules move organic solutes across the proximal tubule epithelium. Drug transporters in the kidney are significant contributors to renal drug elimination and are involved in both secretion and reabsorption into and from the tubular lumen. At all of these epithelial barriers, drug transporters are localized to specific membrane domains, which is critical to their functional role in drug bioavailability, disposition, and response. Defining the mechanisms that determine drug transporter abundance and membrane-delimited localization is an emerging line of research.
PDZ proteins are named for the common structural domain shared with the postsynaptic density protein (PSD), Drosophila disc large tumor suppressor, and zonula occludens-1 protein (ZO-1). In addition to their involvement in the assembly of multiprotein signaling complexes, PDZ domain–containing proteins also regulate membrane abundance and the asymmetric sorting of proteins to specific membrane domains in polarized epithelia (Shenolikar and Weinman, 2001; Shenolikar et al., 2002; Padanyi et al., 2010). PDZ proteins recognize and bind to target proteins by specific internal and carboxy-terminal amino acid sequences, referred to as PDZ ligands (Songyang et al., 1997; Stricker et al., 1997). PDZ domains are classified based on the structural binding motif of the PDZ ligand with which they interact (Table 1). Class I PDZ proteins interact with ligands terminating in the sequence -X-[S/T]-X-ϕ, where X is promiscuous and ϕ is a hydrophobic residue, generally Leu, Ile, Val, or Met. Class II PDZ proteins prefer ligands terminating in -X-ϕ-X-ϕ and class III prefer a PDZ binding motif of the form -[D/E]-[K/R]-X-ϕ. However, PDZ proteins may also recognize internal peptide fragments from target proteins. Residues up to 18-amino-acids upstream from the carboxy terminus can participate in target recognition (Mahon and Segre, 2004). These noncanonical binding modes may also be important for establishing target specificities. Several clinically significant SLC and ABC drug transporters harbor a carboxy-terminal PDZ ligand and therefore may bind PDZ proteins. Mounting evidence highlights the impact of PDZ proteins in regulating the membrane abundance, localization, and function of a growing number of drug transporters (Karvar et al., 2014; Park et al., 2014; Zheng et al., 2014).
TABLE 1.
PDZ ligand recognition classification
| Class | C-Terminal Consensus Sequence of PDZ Liganda |
|---|---|
| I | -X-[S/T]-X-ϕ |
| II | -X- ϕ -X-ϕ |
| III | -[D/E]-[K/R]-X-ϕ |
X is permissive indicating any amino acid; S/T are serine and threonine, ϕ hydrophobic residue, typically L, I, V. D/E and K/R are aspartate/glutamate, and lysine/arginine, respectively.
The Na+/H+ exchanger regulatory factors (NHERFs) are a family of PDZ proteins that also act as mediators of drug transporter function secondary to their role in organizing drug transporter membrane abundance and localization. NHERF proteins alter drug transporter function in vitro, as well as drug absorption and disposition in vivo (Park et al., 2014). NHERF proteins are highly expressed at apical membranes of polarized epithelial cells of the kidney, intestine, and liver, where they interact with numerous drug transporters from both SLC and ABC superfamilies (Kato et al., 2004, 2006; Sugiura et al., 2011). The majority of reported interactions are mediated through canonical class I PDZ ligands, although interactions with drug transporters possessing class II and III PDZ ligands also have been described (Kato et al., 2004; Sugiura et al., 2008; Shimizu et al., 2011). Hence, NHERF proteins may have an indirect effect on drug response attributable to their influence on drug absorption and disposition in these important pharmacological barriers. Over the last decade, PDZ proteins have gained considerable attention as post-translational regulators of drug transporter function (Miyazaki et al., 2005; Kato et al., 2006; Noshiro et al., 2006; Sugiura et al., 2006, 2008, 2010, 2011; Choi et al., 2011; Park et al., 2014; Wang et al., 2014; Zheng et al., 2014). To highlight these new findings, we review the current knowledge of drug transporter regulation by PDZ proteins and address the association of PDZ proteins with drug disposition and response in select disease states.
II. Drug Transporter Structure, Function, and Tissue Location
A. Solute Carrier Drug Transporters
Solute carrier (SLC) drug transporters are classified into the SLC22A and SLCO gene families (uppercase denotes human genes or gene products and lowercase denotes rodent genes or gene products). The SLC22A genes include the organic anion transporters (OATs), organic cation transporters (OCTs), and organic carnitine transporters (OCTNs). The SLCO gene family consists of the organic anion-transporting polypeptides (OATPs). Members of both gene families are primarily involved in cellular drug uptake and are abundantly expressed at the epithelium in the liver, intestine, and kidney (Fig. 1) (Giacomini et al., 2010). Although most SLC superfamily drug transporters display overlapping substrate specificities, OATs, OCTs, and OCTNs transport anions, cations, and zwitterions, respectively, whereas OATPs transport larger hydrophobic anions.
Fig. 1.
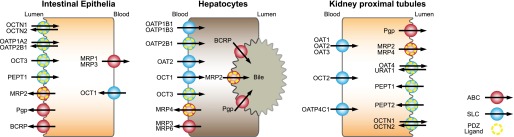
Selected drug transporters expressed at epithelial barriers of intestinal enterocytes, hepatocytes, and kidney proximal tubule cells. ABC transporters are colored red, SLC in blue. ABC or SLC transporters harboring a PDZ ligand are additionally depicted with an inner dotted yellow circle.
1. SLC22A Family.
Transporter proteins encoded by the SLC22A gene family include OCT1–3 (SLC22A1–3), OCTN1–2 (SLC22A4–5), OAT1–3 (SLC22A6–8), OAT4 (SLC22A11), and URAT1 (SLC22A12). The prototype membrane topology of the SLC22A family comprises 12 α-helical transmembrane domains (TMDs), an intracellular N terminus, a large extracellular loop between TMDs 1 and 2, which harbors several putative N-glycosylation sites, a large intracellular loop between TMDs 6 and 7 that contains additional glycosylation and phosphorylation sites and an intracellular C terminus (Fig. 2). Tissue expression and membrane localization of selected SLC22A drug transporters is summarized in Fig. 1. SLC22A drug transporters and selected substrates are summarized in Table 2.
Fig. 2.
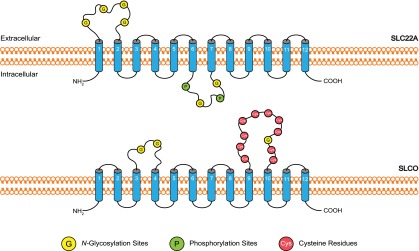
SLC drug transporter membrane topology. The prototype membrane topology of SLC22A family members (OCTs, OCTNs, OATs) is similar, as is SLCO for OATP family members. Blue cylinders represent predicted transmembrane helices. Putative N-glycosylation and phosphorylation sites and important cysteine residues are shown as yellow, green, or red circles, respectively.
TABLE 2.
Selected drug transporters
| Transporter | Superfamily | Family | Gene Name | PDZ Ligand | Selected Substrates |
|---|---|---|---|---|---|
| OCT1 | SLC | SLC22 | SLC22A1 | — | Metformin, oxaliplatin |
| OCT2 | SLC | SLC22 | SLC22A2 | — | Metformin, ranitidine, varinicline |
| OCT3 | SLC | SLC22 | SLC22A3 | R-S-H-L | Histamine, epinephrine |
| OCTN1 | SLC | SLC22 | SLC22A4 | L-T-A-F | Quinidine, verapamil, ipratropium, ergothioneine |
| OCTN2 | SLC | SLC22 | SLC22A5 | S-T-A-F | Carnitine, amisulpride, cephaloridine |
| OAT1 | SLC | SLC22 | SLC22A6 | — | Lamivudine, acyclovir, ciprofloxacin |
| OAT2 | SLC | SLC22 | SLC22A7 | — | Bumetanide, erythromycin, paclitaxel |
| OAT3 | SLC | SLC22 | SLC22A8 | — | Furosemide, bumetanide |
| OAT4 | SLC | SLC22 | SLC22A11 | S-T-S-L | Bumetanide, urate, methotrexate |
| URAT1 | SLC | SLC22 | SLC22A12 | S-T-Q-F | Urate |
| OATP1A2 | SLC | SLCO | SLCO1A2 | K-T-K-L | Statins, fexofenadine |
| OATP1B1 | SLC | SLCO | SLCO1B1 | — | Statins, repaglinide, olmesartan |
| OATP1B3 | SLC | SLCO | SLCO1B3 | — | Statins, telmisartan, fexofenadine |
| OATP1C1 | SLC | SLCO | SLCO1C1 | E-T-Q-L | Triiodothyronine, thyroxine |
| OATP2B1 | SLC | SLCO | SLCO2B1 | D-S-R-V | Statins, fexofenadine |
| OATP3A1 | SLC | SLCO | SLCO3A1 | E-S-V-L | Benzylpenicillin, vasopressin |
| OATP4A1 | SLC | SLCO | SLCO4A1 | Q-S-S-V | Estradiol-17β-glucuronide |
| OATP4C1 | SLC | SLCO | SLCO4C1 | — | Digoxin, methotrexate, sitagliptin |
| PEPT1 | SLC | SLC15 | SLC15A1 | Q-K-Q-M | Cephalexin, valacyclovir, enalapril |
| PEPT2 | SLC | SLC15 | SLC15A2 | K-T-K-L | Cephalexin, valacyclovir, enalapril |
| MDR1 (Pgp) | ABC | ABCB | ABCB1 | — | Digoxin, loperamide |
| BCRP | ABC | ABCG | ABCG2 | — | Statins, methotrexate |
| MRP1 | ABC | ABCC | ABCC1 | — | Citalopram, methotrexate, Leukotriene C4 |
| MRP2 | ABC | ABCC | ABCC2 | S-T-K-F | Olmesartan, methotrexate, etoposide |
| MRP3 | ABC | ABCC | ABCC3 | — | Estradiol-17β-glucuronide |
| MRP4 | ABC | ABCC | ABCC4 | E-T-A-L | Adefovir, topotecan, methotrexate |
| MRP7 | ABC | ABCC | ABCC10 | D-T-R-L | Docetaxel, paclitaxel, viscristine, vinblastine |
a. Organic cation transporters.
OCT1 (SLC22A1), OCT2 (SLC22A2), and OCT3 (SLC22A3) mediate transport of organic cationic drugs such as metformin, histamine H2 receptor antagonists, and platinum-based drugs. OCT1–3 share three common functional characteristics. First, substrate transport is bidirectional; that is, OCTs mediate both uptake and efflux processes. Second, transport occurs by passive facilitated diffusion that is driven by the negatively charged intracellular membrane voltage. The rate of substrate uptake and efflux therefore depends upon the electrochemical gradient and is sensitive to changes in the membrane potential. Third, OCT-mediated transport is independent of sodium and pH. OCTs are expressed in the kidney, intestine, and liver, where they function primarily as basolateral uptake transporters. However, OCT3 may be expressed at both basolateral and apical membranes (Muller et al., 2005; Nies et al., 2009). For example, OCT3 is localized to basolateral/sinusoidal membranes of hepatocytes and apical membranes of enterocytes. OCT3 is expressed in renal proximal and distal tubules, but additional studies are needed to define OCT3 membrane localization in the kidney (Wu et al., 2000b).
OCTs exhibit broad selectivity. Common substrates include hydrophilic, low molecular weight organic cations (Table 2). OCT1 and OCT2 are drug transporters of emerging clinical significance because of their potential influence on drug disposition and response. For example, OCT1 is an important factor in the pharmacotherapy of diabetes and chronic myeloid leukemia (Grinfeld et al., 2013; Koren-Michowitz et al., 2014; Mahrooz et al., 2015). OCT1 is thought to be liver specific; it is predominately expressed on basolateral/sinusoidal membranes of hepatocytes and mediates drug uptake (Gorboulev et al., 1997; Zhang et al., 1997). Although few endogenous OCT1 substrates have been identified, evidence suggests that hepatic OCT1 may mediate the uptake of biogenic amines such as serotonin (Boxberger et al., 2014). Moreover, commonly prescribed medications, such as diphenhydramine, fluoxetine, imatinib, and verapamil, possibly inhibit OCT1-dependent serotonin uptake, a finding highlighting the possibility that drugs may interfere with transport and elimination of endogenous substrates at the transporter level. OCT2 is found exclusively in the kidney at basolateral membranes of proximal tubule cells, where it facilitates secretion of drugs such as cisplatin and oxaliplatin (Gorboulev et al., 1997; Urakami et al., 2002; Iwata et al., 2012). OCT3, in contrast to OCT1 and OCT2, displays a much broader tissue expression pattern. In addition to expression in the intestine, liver, and kidney, OCT3 mRNA levels can be detected in the placenta, heart, brain, and skeletal muscle (Hayer-Zillgen et al., 2002). It is not known why OCT3 localizes to apical membranes in enterocytes but to basolateral membranes in hepatocytes. It is also not understood why OCT3 is capable of dual membrane localization that is tissue specific but OCT1 and OCT2 are restricted to basolateral membranes. Of the three OCT isoforms, only OCT3 contains a C-terminal PDZ-ligand (-RSHL). Therefore, the membrane localization of OCT3 may be determined by tissue- and domain-specific localization of PDZ proteins.
b. Organic cation/carnitine transporters.
The OCTN subfamily consists of OCTN1 (SLC22A4) and OCTN2 (SLC22A5). In addition to their physiologic role as uptake transporters for the zwitterion carnitine, members of the OCTNs are known also to interact with drugs such as levofloxacin, oxaliplatin, and cephaloridine (Hirano et al., 2008; Kano et al., 2009; Jong et al., 2011). Analogous to OCTs, OCTNs are able to perform bidirectional transport; however, there are subtle differences in the transport mechanisms between OCTN1 and OCTN2. For example, OCTN1 can function as an organic cation uniporter or an H+/organic cation antiporter, whereas OCTN2 functions as a sodium/carnitine cotransporter or may perform sodium-independent cation transport (Tamai et al., 1998, 2004; Wu et al., 1999; Ohashi et al., 2001; Grundemann et al., 2005). Although broadly expressed in multiple tissues, OCTN subfamily members display preferential tissue expression. For instance, OCTN1 is predominately expressed on apical membranes of renal proximal tubules but is found at much lower levels in other tissues such as the liver, small intestine, placenta, and brain (Tamai et al., 2000, 2004; Wu et al., 2000a). OCTN2 also is expressed on apical membranes of renal proximal tubule cells but its expression is not limited to polarized epithelia. It is found in several organs including the heart, brain, liver, testis, and skeletal muscle (Tamai et al., 1998, 2000). Because OCTN2 is believed to be the major OCTN regulating serum carnitine concentrations (Nezu et al., 1999), its wider expression may have evolved to ensure adequate carnitine absorption and distribution throughout the body. Carnitine is required for fatty acid transport in mitochondria for generation of metabolic energy. Underscoring its vital nature, primary carnitine deficiency, a disorder characterized by chronic muscle weakness, cardiomyopathy, hypoglycemia, and liver dysfunction, is caused by defects in OCTN2 activity (Tamai et al., 1998). The OCTN subfamily has been linked to other disease states as well. Several studies implicated OCTN1 and OCTN2 in the pathophysiology of inflammatory bowel disease, diabetes, and cancer (Peltekova et al., 2004; Okabe et al., 2008; Martini et al., 2012; Muoio et al., 2012; Pochini et al., 2013). By virtue of their association with several diseases, OCTNs are studied more in the context of pathophysiology and less for their potential pharmacological role as mediators of drug absorption and disposition. Nevertheless, OCTNs are involved in transport of organic cationic drugs and are potential sites for drug/carnitine interactions (Ganapathy et al., 2000). OCTN1 and OCTN2 mediate transport of drugs such as antipsychotics, antihypertensives, antibiotics, and drugs used in the treatment of neurologic disorders (Ganapathy et al., 2000; Grube et al., 2006; Urban et al., 2008; Nakamura et al., 2010; Jong et al., 2011). Both OCTN1 and OCTN2 contain C-terminal PDZ ligands (-LTAF and -STAF, respectively). The role of PDZ proteins and OCTN membrane abundance and function is summarized in section IV.
c. Organic anion transporters.
OATs mediate the systemic removal of organic anions including drugs, toxins, and endogenous metabolites. OAT1–3 (SLC22A6–8), OAT4 (SLC22A11), and URAT1 (SLC22A12) are deemed clinically significant human OAT isoforms due to their prominent role in the kidney as mediators of renal secretion and thus renal drug elimination (Giacomini et al., 2010; Burckhardt, 2012). Historically, OATs have been the targets of pharmacological strategies to extend limited supplies of drugs. The classic example of OAT-targeted interventions of this manner is the utilization of probenecid to conserve limited supplies of penicillin during World War II. Probenecid, an OAT inhibitor, increases serum concentrations of penicillin by significantly reducing OAT-dependent renal secretion, thus extending the half-life of penicillin and optimizing therapeutic efficacy by achieving concentrations above the minimum inhibitory concentration with a lower dose (Weiner et al., 1960). More recently, owing to its established inhibitory effects on OATs, probenecid has been suggested as an adjuvant to other medications that have been in short supply, such as the antiviral drug oseltamivir (Howton, 2006). Genetic variations encoding different OAT isoforms are also associated with altered transporter activity and impaired renal handling of OAT substrates (Bleasby et al., 2005; Xu et al., 2005; Erdman et al., 2006). For instance, genetic polymorphisms in OAT3 are associated with reduced renal clearance of cefotaxime, a cephalosporin antibiotic (Yee et al., 2013). Transcript variants of URAT1 may produce increases or decreases in renal tubular uric acid reabsorption, changes that result in hyperuricemia and hypouricemia, respectively (Wakida et al., 2005; Graessler et al., 2006).
In contrast to the OCTs, OATs transport anions into cells against the negatively charged electrochemical gradient and therefore are energy dependent. OATs transport substrates by exchanging an intracellular mono- or divalent anion, typically a dicarboxylate, for uptake of an extracellular anion (Burckhardt, 2012). The energy expenditure that drives this process comes indirectly from the Na+/K+-ATPase that maintains intracellular sodium concentration at low levels and creates a negative intracellular voltage. This favorable electrochemical gradient drives sodium-coupled uptake of dicarboxylate species by the sodium-dicarboxylate cotransporter (NaDC3). Together, the Na+/K+-ATPase, NaDC3, and OATs cooperate to facilitate the uptake and removal of organic anions. OATs are heavily expressed in the kidney, where they play a significant role in the uptake step of renal drug accumulation. Among the OAT family, OAT1 and OAT3 are the most extensively studied in the context of drug transport and have been shown to mediate the uptake of a wide variety of structurally diverse drug classes such as antihypertensive drugs, 3-hydroxy-3-methylglutaryl-co-enzyme A reductase inhibitors (statins), β-lactam antibiotics, H2 receptor antagonists, antineoplastic drugs, nonsteroidal anti-inflammatory drugs, and antiviral drugs (Burckhardt, 2012). However, OATs also transport multiple endogenous compounds and play complex physiologic roles (Mandal and Mount, 2015; Nigam et al., 2015). Recent evidence suggests that OAT3 not only handles transport of drugs and drug metabolites, but also may play a role in 1) pathways involved in the regulation of secondary metabolites, such as prostaglandins, steroids, and uric acid; 2) the metabolism and handling of gut microbiome products, as well as uremic toxins that accumulate in kidney disease; 3) handling of dietary flavonoids such as epicatechin; and 4) the tricarboxylic acid cycle and nucleotide and amino acid metabolism (Wu et al., 2013; Mandal and Mount, 2015; Nigam et al., 2015).
OAT1 and OAT3 are exclusively located at basolateral membranes in renal proximal tubule cells. In the kidney, all members of the OAT family localize to basolateral membranes with the exception of OAT4 and URAT1. OAT4 is unique in that it is expressed only in humans and is located at apical membranes of renal proximal tubule cells (Cha et al., 2000; Babu et al., 2002; Ekaratanawong et al., 2004). OAT4 is also expressed in the placenta but is restricted to basolateral membranes of syncytiotrophoblasts (Cha et al., 2000). This distinctive tissue-specific pattern of OAT4 membrane localization is similar to the dual-membrane expression patterns of OCT3. Like OCT3, OAT4 possesses a PDZ ligand (-STSL). Several studies suggest that multiple PDZ proteins may govern the membrane abundance and localization of OAT4 (Kato et al., 2004; Miyazaki et al., 2005; Zhou et al., 2008).
2. SLCO Family.
The SLCO gene family encodes the human OATPs. These proteins transport various organic amphiphilic compounds, the majority of which are large hydrophobic anions such as bile acids, steroid conjugates, and anionic peptides, as well as several drugs including statins, angiotensin II receptor antagonists, and angiotensin-converting enzyme inhibitors (Cha et al., 2000; Babu et al., 2002; Ekaratanawong et al., 2004; Zhou et al., 2008). Eleven human OATPs have been identified to date. Selected OATPs discussed in this manuscript are summarized in Table 2. There are six known human OATP families. Classification of OATPs is based on their amino acid similarities. OATPs within the same family share ≥40% amino acid sequence identity and are assigned root names followed by a numerical designation (OATP1–6). Subfamily members share ≥60% amino acid sequence identity and are designated with a letter following the numerical family designation. For instance, OATP family 2 (OATP2) has two subfamilies (OATP2A and OATP2B). A numerical designation after the subfamily heading is assigned to each member of a subfamily (OATP2A1 and OATP2B1). Size differences exist across members of the OATP family. OATPs have an average length of 710 amino acids but range from 643 amino acids (OATP2A1) to 848 amino acids (OATP5A1). However, all OATPs share a similar predicted membrane topology that comprises 12 α-helical transmembrane domains, intracellular N and C termini, and multiple glycosylation sites within the second and fifth extracellular loop. The fifth extracellular loop is larger and contains multiple conserved cysteine residues that participate in disulfide bonds (Fig. 2). In general, OATP-dependent transport involves bidirectional, sodium-independent exchange. OATPs couple the cellular uptake of anions with efflux of intracellular substances such as bicarbonate, reduced glutathione, or lactate. In the case of OATP2B1, transport is pH sensitive but only with selective solutes such as statins (Kobayashi et al., 2003; Nozawa et al., 2004; Varma et al., 2011). The precise mechanism of transport and how it relates to the physiologic and pharmacological role of individual OATPs remains unclear.
Tissue expression and membrane localization of selected SLCO drug transporters is summarized in Fig. 1. Some members of the OATP family display broad tissue expression. OATP1A2, OATP2A1, OATP2B1, OATP3A1, and OATP4A1 for instance are found in multiple tissues including the kidney, placenta, skeletal muscle, choroid plexus, and endothelial cells of the blood-brain barrier. Other OATPs exhibit a more restricted pattern. The major hepatic OATP isoforms include OATP1B1 and OATP1B3; OATP4C1 predominates in the kidney. Among the OATP family, OATP1B1, OATP1B3, OATP1A2, and OATP2B1 are considered clinically important determinants of pharmacokinetics due to their participation in transporting commonly used medications such as statins (Giacomini et al., 2010). In particular, OATP1B1 and OATP1B3 mediate the hepatic uptake of drugs, whereas OATP1A2 and OATP2B1 mediate intestinal drug absorption. The extensive expression of OATPs in the intestine and liver likely evolved as a protective mechanism to combat inadvertent ingestion of plants or foods containing toxins. An additional evolutionary advantage to OATP-dependent detoxification comes in the form of cooperativity with intestinal and hepatic metabolizing enzymes. As a means to provide protection against a wide spectrum of potentially harmful exogenous substances, intestinal and hepatic OATPs have adopted overlapping substrate specificity with intracellular cytochrome P450 (P450) enzymes. Together, OATPs cooperate with P450 enzymes in the intestine and liver to reduce the systemic exposure of xenobiotics by exposing substrates to intracellular P450s for subsequent metabolism. Evidence of this cooperative functionality is observed concerning the hepatic clearance of erythromycin, a commonly used probe to assess CYP3A function (Rivory et al., 2001). Erythromycin uptake into hepatocytes is mediated by OATPs (Sun et al., 2010; Lancaster et al., 2012). OATPs facilitate the uptake of erythromycin from the portal vein and into hepatocytes where metabolism by CYP3A occurs (Franke et al., 2011). Therefore, hepatic clearance of erythromycin is a function of OATP-dependent uptake and CYP3A function. Although these examples illustrate OATP-P450 cooperativity, they may not extend to other transporters and P450 proteins.
Although generally considered drug transporters, physiologic roles of OATPs also have been described. Members of the OATP family are involved in the transport of bilirubin (OATP1B1 and OATP1B3) and thyroid hormones (OATP1C1) (Keppler, 2014; Mayerl et al., 2014). In the liver, bilirubin metabolism and elimination is accomplished, in part, by the coordinated actions of OATPs and UDP glucuronosyltransferase-1 family, polypeptide-1. Similar to the proposed mechanism of OATP-P450 cooperative functionality and drug metabolism, hepatic OATP1B1 and OATP1B3 facilitate the uptake of unconjugated bilirubin, which is followed by glucuronic acid conjugation catalyzed by intracellular UDP glucuronosyltransferase-1 family, polypeptide-1 (Keppler, 2014). OATPs also may be involved in transporting endogenous substances that are otherwise impermeant and cannot traverse the cell membrane passively and, therefore, rely on transporters to enter cells. OATP1C1 has been implicated recently in the transport across the blood-brain barrier of thyroxine, the prohormone of triiodothyronine, a process thought to be important for proper brain development (Mayerl et al., 2014). The physiologic and pharmacological importance of renal OATPs, particularly the role of OATP4C1, is not fully appreciated.
Similar to other SLC transporter families, membrane localization of OATP isoforms is not uniform and in some cases is tissue dependent. Although most are located on basolateral membranes, OATP1A2 is exclusive to apical membranes, whereas OATP2B1 displays dual membrane localization. In the intestine, OATP1A2 and OATP2B1 are located on the apical brush-border membrane, where they contribute to drug absorption. In the liver, OATP1B1, OATP1B3, and OATP2B1 are located on basolateral/sinusoidal membranes, where they may be responsible for hepatic drug uptake (Kullak-Ublick et al., 2001; Kobayashi et al., 2003; Tamai, 2012). OATP1A2 is also found at apical membranes in endothelial cells of the blood-brain barrier, hepatocytes, and renal distal tubules (Lee et al., 2005). Analogous to the OCT and OAT families of drug transporters, some OATP isoforms harbor a C-terminal PDZ ligand and are binding partners with PDZ proteins (Kato et al., 2004; Wang et al., 2005; Zheng et al., 2014). Multiple PDZ proteins regulate the function of OATP1A2 (Zheng et al., 2014), which is discussed in section IV.
The OATP family is gaining considerable attention due to their central role in drug disposition and response (Link et al., 2008; Shitara et al., 2013). Undoubtedly, this recognition is underscored by the importance of OATPs on statin pharmacotherapy. Statins are orally administrated medications and considered mainstay therapy for the treatment of hypercholesterolemia. To elicit their lipid-lowering effects, statins must be taken up into hepatocytes. Thus, OATPs are not only critical determinants of statin pharmacokinetics but also for delivery to their site of action. Several OATPs are involved in the oral absorption and disposition of statins, particularly OATP1B1 (Hirano et al., 2006; Romaine et al., 2010; Prueksaritanont et al., 2014). All statins in clinical use are substrates for OATP1B1 (Niemi et al., 2011). Furthermore, OATP1B1 polymorphisms have been shown to alter the pharmacokinetics and pharmacodynamics of statins including pravastatin, atorvastatin, and simvastatin (Oh et al., 2013; Meyer Zu Schwabedissen et al., 2015). OATP1A2 has also been recognized as important for drug transport and disposition. Substrates for OATP1A2 include statins, human immonodeficiency virus–protease inhibitors, and fexofenadine (Kullak-Ublick et al., 1998). Selected OATP substrates are listed in Table 2. By virtue of their expanding role in pharmacokinetics and pharmacodynamics, characterizing OATP regulatory mechanisms in health and disease remains an area of focus.
3. Other Solute Carriers (SLC15A Family).
The SLC15A gene family encodes two human peptide transporters, PEPT1 (SLC15A1) and PEPT2 (SLC15A2). PEPT1 and PEPT2 function primarily as intestinal and renal peptide transporters, respectively. Both PEPT1 and PEPT2 are sodium-independent symporters that couple peptide transport with the movement of H+ down its electrochemical gradient. PEPT1 is a high-capacity, low-affinity transporter for di- and tripeptides but has negligible affinity for amino acids (Liang et al., 1995). PEPT1 is heavily expressed in the small intestine, where it localizes exclusively to the apical brush-border membranes of enterocytes (Liang et al., 1995). At the intestinal barrier, PEPT1 absorbs small peptides that are generated upon enzymatic breakdown of endogenous or ingested protein. PEPT1 also plays an important pharmacological role as a mediator of oral drug absorption and is a target for prodrug design (Hamman et al., 2005; Brandsch, 2013; Zhang et al., 2013). Hydrophilic drug compounds that are taken orally often have poor bioavailability secondary to limited intestinal membrane permeability. This barrier to pharmacotherapy and limitation to drug design has been circumvented by exploiting intestinal PEPT1. As a strategy to enhance the oral bioavailability of such compounds, prodrugs are formulated via amino acid esterification (Majumdar and Mitra, 2006). Chemical modifications of this type thus permit involvement of PEPT1 in drug uptake processes in the intestine. This approach has been used previously, most notably in the design of the nucleoside analog valacyclovir (Jacobson, 1993). Valacyclovir, a PEPT1 substrate and amino acid ester derivative of the active drug acyclovir, exhibits a 3- to 5-fold increase in bioavailability (Weller et al., 1993). Intestinal PEPT1 is also involved in the absorption of other peptidomimetic drug compounds and prodrugs, including β-lactam antibiotics and angiotensin converting-enzyme inhibitors (Daniel and Adibi, 1993; Ganapathy et al., 1995; Sugawara et al., 2000; Shu et al., 2001). PEPT2 is predominately expressed in renal proximal tubules and localizes to apical membranes, where it is involved in reabsorption of di- and tripeptides from the glomerular filtrate (Rubio-Aliaga et al., 2003). PEPT1 is also found at apical membranes in proximal tubules, where it may be involved in the reabsorption of peptides that are generated by the action of brush border peptidases on filtered proteins. Although both PEPT isoforms are expressed in kidney, PEPT1 is expressed at much lower levels, and therefore the relative contribution of PEPT1 to renal tubule reabsorption is likely less than that of PEPT2 (Liang et al., 1995). Furthermore, in contrast to PEPT1, PEPT2 is a high-affinity low-capacity transporter. This clear distinction in transport kinetics was described by revealing that PEPT1 and PEPT2 were responsible for the low-affinity (Km of 1.2 mM) and high-affinity (Km of 50 μM) transport of glycylsarcosine (Gly-Sar), respectively (Takahashi et al., 1998). It was later determined in studies comparing the contribution of PEPT1 and PEPT2 to the renal reabsorption of β-lactam antibiotics, that PEPT2 mediates high-affinity transport of amino β-lactam antibiotics such as amoxicillin, cephalexin, and cefadroxil, which contain an α-amino group, and that the involvement of PEPT1 is minimal in this regard (Takahashi et al., 1998; Inui et al., 2000). These findings strongly support that the pharmacological role of PEPT2 in the kidney might be to participate in reabsorption of amino β-lactam antibiotics, as well as other pharmacologically active compounds with similar chemical structures. Taken together, the expression pattern and transport kinetics of the PEPTs are not surprising if consideration is given to their physiologic roles. The abundant expression of PEPT1, a high-capacity transporter, at the apical membrane in enterocytes is strategically placed to ensure adequate dietary protein absorption. Conversely, the expression of PEPT2, a high-affinity transporter with specific substrate affinities, at apical membranes in renal proximal tubules cells facilitates tight control of compounds that undergo renal elimination. The pharmacological implications of PEPT1 and PEPT2 function may be to mediate the intestinal absorption and renal reabsorption of drugs, respectively.
Both PEPT isoforms diverge in their substrate affinity and transport capacity but share substrate specificity, transport mechanisms, and are exclusively located at apical membranes (Daniel and Adibi, 1993; Ganapathy et al., 1995; Liang et al., 1995; Leibach and Ganapathy, 1996; Saito et al., 1996; Lin et al., 1999). The mechanism for the exclusive apical localization of the PEPTs in the intestine and kidney is not clear. However, PEPT1 (-QKQM) contains a class III PDZ ligand, whereas PEPT2 (-KTKL) contains a class I PDZ-ligand and both PEPT1 and PEPT2 interact with PDZ proteins (Kato et al., 2004). Localization of PEPT1 at apical membranes in enterocytes from nherf3−/− mice is almost completely absent, indicating NHERF3 may play a role in PEPT1 regulation (Sugiura et al., 2008). Although multiple PDZ proteins have been shown to influence PEPT2 function, the role of PDZ proteins on PEPT1 function is less understood (Kato et al., 2004; Noshiro et al., 2006; Boehmer et al., 2008). These observations suggest that PDZ regulation of drug transporter membrane abundance may not solely depend on the presence of a class I PDZ-ligand. This may indicate that PDZ-drug transporter interactions are mediated through other binding motifs or facilitated by other cytoplasmic adapter proteins. A detailed analysis on the differences in PDZ regulation of PEPT1 and PEPT2 in the intestine and kidney, respectively, will address this disparity and may provide important clues lending to the complexity of PDZ function.
B. ATP Binding-Cassette Drug Transporters
The ATP binding cassette (ABC) superfamily represents the largest family of membrane transporters, comprising 49 members divided into 7 subfamilies (ABCA–ABCG) (Table 2). Members of subfamilies B, C, and G are the best studied in the context of drug transport and disposition. Specifically, the multidrug resistance protein (MDR1, ABCB1), multidrug resistance-associated proteins (MRPs, ABCCs), and breast cancer resistance protein (BCRP, ABCG2) are clinically significant mediators of multidrug resistance and response (Giacomini et al., 2010; Zamek-Gliszczynski et al., 2012; Hillgren et al., 2013). Drug resistance is a clinical problem in the treatment of many types of cancer. Resistance to several anticancer agents is caused, in part, by increased ABC drug transporter function (Kathawala et al., 2015). In tumors, ABC drug transporters actively extrude anticancer agents and prevent intracellular drug accumulation, resulting in poor drug response (Kim et al., 2015). In a similar manner, ABC drug transporters limit the systemic exposure of drugs and drug metabolites by preventing drug absorption and promoting drug elimination at anatomic barriers such as intestinal, hepatic, and renal epithelia, tissues where ABC drug transporter expression is high. The mechanism of transport is similar among all members in that they require ATP hydrolysis to energize cellular drug efflux. Most members share a membrane topology consisting of 12 hydrophobic TMDs that are divided into two distinct clusters, each containing six TMDs, and each with its own nucleotide-binding domain (NBD) (Fig. 3). MRP1, MRP2, MRP3, MRP6, and MRP7 harbor an additional cluster consisting of five TMDs near the N terminus (Fig. 3) (Choi and Yu, 2014). BCRP is unique among the ABC family in that it contains only six TMDs and one NBD (Fig. 3). For that reason BCRP is considered a “half-transporter.” ABC drug transporters expressed in the intestine, liver, and kidney mediate the cellular efflux of a vast array of structurally diverse drugs from several drug classes, including anticancer drugs, antibiotics, statins, and human immonodeficiency virus protease inhibitors, among many others (Cascorbi, 2011; Keppler, 2011; Mao and Unadkat, 2015). ABC drug transporters are also the molecular basis for clinically significant drug-drug interactions, which lead to changes in serum drug concentrations and consequently drug response (Konig et al., 2013). To limit drug exposure by promoting drug elimination in polarized epithelia, ABC drug transporters must be targeted to the membrane domain that complements this function, which in many cases is the apical membrane facing a luminal space. ABC drug transporters facilitate drug extrusion into bile (from hepatocytes), urine (from renal proximal tubule cells), and the intestinal lumen (from enterocytes). Tissue expression and membrane localization of selected ABC drug transporters is summarized in Fig. 1. ABC drug transporters and a list of selected substrates are summarized in Table 2.
Fig. 3.
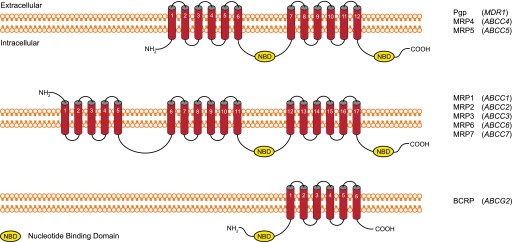
ABC drug transporter membrane topology. The membrane topology of ABC family drug transporters is not uniform but differs in the number of TMDs and NBDs. Red cylinders represent predicted transmembrane helices, and NBDs are depicted as yellow ovals for the ABC proteins indicated on the right, along with their corresponding human gene identification.
1. Multidrug Resistance Protein 1 (ABCB1).
The ABCB1 gene encodes MDR1, commonly referred to as P-glycoprotein (Pgp). Pgp was one of the first members of ABC drug transporters to be identified and has been well studied since it was first cloned and characterized nearly four decades ago (Juliano and Ling, 1976). Pgp is extensively distributed throughout the body with highest expression in epithelial cells with excretory roles such as intestinal, hepatic, and renal epithelia (Thiebaut et al., 1987). It is also expressed in epithelial cells lining the pancreatic ducts, as well as in endothelial cells of the blood-brain barrier (Thiebaut et al., 1987; Beaulieu et al., 1997). Pgp specifically localizes to apical membranes. The location of Pgp at the luminal surface supports its physiologic role to protect susceptible organs from a broad array of exogenous compounds and environmental toxins, including an equally diverse list of drugs and drug metabolites (Padowski and Pollack, 2010; Staud et al., 2010). Pgp actively extrudes intracellular substrates in an ATP-dependent manner and prevents tissue accumulation by promoting excretion. Pgp substrates include multiple drugs from structurally diverse classes such as immunosuppressants, antibiotics, statins, β-blockers, anticancer drugs, and the cardiac glycoside digoxin, among many others (Fromm, 2004; Cascorbi and Haenisch, 2010; Cascorbi, 2011).
Pgp is limited to apical membranes. The mechanism of its specific membrane domain localization is unknown. Pgp does not harbor a C-terminal PDZ ligand, and there is no evidence for interaction with PDZ proteins. Its homolog, the cholesterol efflux regulatory protein (ABCA1), contains a PDZ ligand (-ESYV) and is regulated by PDZ proteins (Okuhira et al., 2010). Although Pgp appears not to interact with PDZ proteins directly, radixin, a cytoskeletal adapter protein that interacts with PDZ proteins, plays an important role in regulating intestinal Pgp localization and function (Yano et al., 2013). This implies that PDZ proteins may be involved in regulating Pgp localization indirectly via interactions with radixin or other cytoskeletal adapter proteins such as ezrin or moesin. Regulation of Pgp function secondary to membrane abundance and localization is not well studied and may be another example of higher order complexity regarding PDZ function.
Pgp is widely acknowledged as a clinically significant mediator of pharmacokinetics and pharmacodynamics because of its high expression in tissues important for drug disposition and well established list of substrates and inhibitors (Giacomini et al., 2010). Pgp is implicated in several clinically significant drug-drug interactions. The quindine-digoxin interaction is a classic example of a Pgp mediated drug-drug interaction (Bigger and Leahey, 1982). The antiarrhythmic drug quinidine is an established Pgp inhibitor. When coadministered with the cardiac glycoside digoxin, a Pgp substrate, patients are at risk for digoxin-induced toxicity, including the development of cardiac arrhythmias (Gessman et al., 1983). This adverse drug event is partially attributed to an increase in the systemic exposure of digoxin secondary to impaired Pgp-dependent renal elimination (De Lannoy et al., 1992). Other clinically significant Pgp interactions have been documented (Aszalos, 2007). For these reasons, regulatory agencies such as the Food and Drug Administration and European Medicines Agency suggest that all new drug candidates be screened for in vitro Pgp substrate and inhibition liability.
2. Multidrug Resistance Proteins (ABCCs).
The MRP family of drug transporters is encoded by the ABCC gene subfamily. The MRP family consists of 9 proteins (MRP1–9). Analogous to other members of the ABC superfamily of drug transporters, MRPs mediate cellular drug efflux and play a central role in protecting tissues from accumulation of toxic compounds. MRPs are also responsible for transporting several endogenous physiologic molecules, illustrating that their function is not purely pharmacological. For example, MRP1 mediates the transport of reduced and oxidized glutathione and the proinflammatory mediator cysteinyl leukotriene C4 (Cole, 2014). The physiologic role of MRP4 is to regulate intracellular concentrations of cyclic nucleotides, such as cAMP and cGMP, by functioning as a nucleotide efflux transporter. MRP4 regulation of cAMP was recently implicated in the mechanism of drug-induced diarrhea and in fibroblast cell migration (Sinha et al., 2013; Moon et al., 2015). In addition to their pharmacological role as mediators of drug disposition, dysfunctional MRPs are responsible for the development of pathophysiological conditions. For example, sequence variations that result in loss of functional MRP2 and MRP6 are the molecular basis for Dubin-Johnson syndrome (DJS) and pseudoxanthoma elasticum (PXE), respectively (Kartenbeck et al., 1996; Toh et al., 1999; Bergen et al., 2007). DJS is characterized by dysfunctional hepatic secretion of conjugated bilirubin into the bile. The loss of functional MRP2 on the apical/canalicular membrane of hepatocytes impairs biliary excretion of conjugated bilirubin, which results in elevated serum concentrations of bilirubin and the development of DJS (Devgun et al., 2012). PXE is characterized by mineralization and eventual degradation of elastic fibers in connective tissue, which can lead to loss of vascular tone and premature arteriosclerosis (Li et al., 2009). Although the molecular pathologies of PXE are still unclear, mutations in MRP6 cause PXE (Bergen et al., 2007). The physiologic MRP6 substrates that are involved in the development of PXE remain unknown. In accordance with their functional role, MRPs are prominently expressed at anatomic barriers, with high expression in the intestine, liver, kidney, and blood-brain barrier.
The MRPs display overlapping substrate specificity for many endogenous and exogenous lipophilic organic anions. MRP drug substrates include statins, angiotensin II receptor antagonists, antiviral drugs, and chemotherapeutic agents (Keppler, 2011). Within the MRP family, MRP2 and MRP4 are deemed the most clinically significant to drug development because of their established influence on drug disposition and expanding list of drug substrates (Giacomini et al., 2010; Zamek-Gliszczynski et al., 2012; Hillgren et al., 2013). Several commonly used medications are substrates for MRP2 dependent transport including methotrexate, olmesartan, and etoposide (Gerk and Vore, 2002). In the kidney, MRP4 plays a principal role in the renal luminal efflux of acyclic nucleotide reverse transcriptase inhibitors such as adefovir and tenofovir (Imaoka et al., 2007). An important clinical limitation to the usage of adefovir and tenofovir is the development of nephrotoxicity, which has been proposed to be partially attributable to a reduction in MRP4-dependent renal secretion (Izzedine et al., 2005). MRP2 and MRP4 seem to be responsible for drug transport, whereas other members of the MRP family are involved in transport of endogenous substrates. Although both MRP2 and MRP4 are expressed in the liver and kidney, MRP2 is considered the major hepatic isoform, whereas MRP4 predominates in the kidney. Interestingly, both hepatic and renal MRP2 localize to apical membranes, whereas kidney MRP4 is found at apical membranes but at basolateral surfaces in hepatocytes. The mechanism responsible for the difference in membrane localization of MRP4 in the liver versus the kidney is not known. Both MRP2 (-STKF) and MPR4 (-ETAL) contain C-terminal PDZ ligands. Multiple PDZ proteins regulate MRP2 and MPR4 function secondary to preservation of membrane abundance and organization of membrane localization (Hoque et al., 2009; Karvar et al., 2014; Park et al., 2014). In addition, members of the ezrin/radixin/moesin/merlin (ERM) family have been shown to regulate MRP2 function. Radixin selectively modulates the functional expression of hepatic MRP2 but not other MRP isoforms (Kikuchi et al., 2002; He et al., 2012). Other studies show that both radixin and ezrin are required for MRP2 apical membrane localization (Yang et al., 2007). The role of radixin and ezrin in targeting MRP2 to apical membranes is believed to be secondary to preventing internalization (Yang et al., 2007; Kojima et al., 2008; Rost et al., 2008; Saeki et al., 2011). Considering PDZ proteins are known to interact with the ERM family, these observations may indicate that MRPs are subject to tissue-specific PDZ regulation dictated by distinct patterns of tissue expression and colocalization of PDZ proteins with selected ERM proteins. Therefore, tissue-specific ERM proteins may influence the nonredundant physiologic and pharmacological roles of PDZ proteins in different tissues. Determining how PDZ proteins cooperate with cytoskeletal adapter proteins to regulate membrane abundance, localization, and function of target proteins in different tissues, such as drug transporters, may address these issues.
3. Breast Cancer Resistance Protein (ABCG2).
Breast cancer resistance protein (BCRP) is the second member of the G subfamily of ABC drug transporters and is encoded by ABCG2. BCRP was originally identified in several breast cancer cell lines, where it confers resistance to multiple anticancer drugs (Dietel et al., 1990; Taylor et al., 1991; Nakagawa et al., 1992; Futscher et al., 1994; Kellner et al., 1997). It was later cloned, characterized, and assigned to the G subfamily of ABC drug transporters (Doyle et al., 1998). BCRP contains only six TMDs and one NBD and thus is considered a half transporter because it diverges from the prototypical membrane topology of other ABC drug transporters. Recent evidence shows that BCRP forms homodimers or homooligomers in intact cells (Ni et al., 2010). Covalent linkages mediated by cysteine residues in the third extracellular loop are believed to be responsible for BCRP dimerization (Shigeta et al., 2010). However, dimerization is not a requirement for BCRP function (Kage et al., 2005; Shigeta et al., 2010). The expression pattern of BCRP complements its role as an efflux drug transporter. BCRP is highly expressed at the luminal surface of intestinal and hepatic epithelia where it attenuates intestinal drug absorption and promotes biliary drug excretion (Hillgren et al., 2013). The highest expression of BCRP is detected in placental tissue, specifically on apical syncytiotrophoblast membranes. Other tissues with high expression of BCRP include the blood-brain barrier, testis, and lactating mammary tissue (Jani et al., 2014). In the kidney, BCRP localizes to apical membranes of proximal tubule epithelial cells; however, its level of expression is less compared with the intestine and liver (Mao and Unadkat, 2015). BCRP transports a broad array of both endogenous and exogenous compounds, including several marketed drugs such as anticancer agents, nucleoside analogs, and statins. A list of selected BCRP substrates is summarized in Table 2.
Recent clinical pharmacogenetic studies have emphasized the importance of BCRP to drug disposition and response, which has prompted the Food and Drug Administration and European Medicines Agency to recommend that all investigational new drugs be tested for BCRP substrate and inhibitor liability, except for substrates that fall into biopharmaceutical classification system 1, drugs with high solubility and high permeability. Loss of function BCRP polymorphisms that result in decreased drug clearance and increased drug exposure and toxicity have been reported. For example, in carriers of BCRP polymorphisms, the systemic exposure of several drugs such as atorvastatin, rosuvastatin, sunitinib, and sulfasalazine is significantly increased (Mizuno et al., 2012; Giacomini et al., 2013; Lee et al., 2015). In the case of sunitinib, carriers of BCRP polymorphisms also experience a higher incidence of sunitinib-induced toxicity (Mizuno et al., 2012). In addition to altering pharmacokinetics and pharmacodynamics, BCRP polymorphisms are associated with disease states such as Alzheimer’s disease and gout (Matsuo et al., 2011b; Feher et al., 2013; Takada et al., 2014). The association between BCRP and these disease states is hypothesized to be secondary to BCRP dependent handling of amyloid beta in Alzheimer’s disease and uric acid in patients with gout (Matsuo et al., 2011a,b; Abuznait and Kaddoumi, 2012).
BCRP membrane location in polarized cells is restricted to apical membranes. The mechanism that underlies the restricted apical membrane localization of BCRP has yet to be defined. Despite the fact that BCRP (-KKYS) does not harbor a PDZ ligand, NHERF3 regulates intestinal BCRP function and plays an important role in localizing BCRP to apical membranes in enterocytes (Shimizu et al., 2011). NHERF3 also regulates intestinal PEPT1, which harbors a class III PDZ ligand (-QKQM) (Sugiura et al., 2008; Shimizu et al., 2011). These observations suggest that the interaction between NHERF3 and BCRP differs from those of other drug transporters, which interact with NHERFs via a canonical PDZ ligand. The mechanism of NHERF3 regulation of BCRP may involve the presence of a noncanonical PDZ-binding motif. The NHERF3-interacting binding sequence in BCRP is unclear. Because of its extensive tissue distribution, expanding list of drug substrates, and association with altered pharmacokinetics and pathophysiology, BCRP has been recognized as a clinically relevant drug transporter of emerging importance.
III. Tissue and Cellular Localization of PSD-95/Drosophila Discs Large/ZO-1 Proteins
PDZ domains are the most abundant protein-protein interaction modules in humans (Ponting et al., 1997; Feng and Zhang, 2009). Canonical PDZ domains consist of 80- to 90-amino-acid residues that form six β-stands (βA to βF) and two α-helices (αA and αB) that are arranged in a three-dimensional globular structure (Karthikeyan et al., 2001). Binding of a protein ligand to the PDZ domain occurs within a hydrophobic binding pocket that is created by the βB strand, the αA helix, and the loop connecting the αA and βB strands. This carboxylate-binding loop contains a GLGF core motif or a related sequence such as GYGF, as is the case with the NHERF family of PDZ proteins (Ponting et al., 1997). PDZ domains interact with target proteins via specific internal or carboxy-terminal amino acid residues, commonly referred to as PDZ ligands. Carboxy- terminal PDZ ligands are a short stretch of amino acids approximately three to four residues in length, although upstream sequences affect affinity and specificity of binding. The amino acid sequence that comprises the PDZ ligand is conventionally numbered starting from the last amino acid at the extreme carboxy terminus and is assigned position zero (P0).
PDZ recognition motifs are grouped into three classes (class I–III) according to the consensus sequence of the PDZ ligand for which they bind. Table 1 summarizes the classification of PDZ ligands. Binding specificity among the three classes is determined, in part, by the interaction between the first amino acid residue in the βB-helix of the PDZ domain and the amino acid at position two (P2) of the PDZ ligand in the target protein. There are over 150 PDZ domain-containing proteins and more than 250 distinct PDZ domains in the human proteome. Individual PDZ proteins may contain multiple PDZ domains as well as other interaction modules such as ezrin-binding domains (EBDs). PDZ proteins lack intrinsic catalytic activity and therefore function primarily as scaffold proteins, where they execute a range of biologic functions that include establishing and maintaining cellular polarity, assembling multiprotein signaling complexes, and anchoring transmembrane proteins, including transporters, to the actin cytoskeleton via interactions with the ERM family of adapters (Ponting et al., 1997; Wang et al., 2007, 2010, 2012; Georgescu et al., 2014; Zheng et al., 2014).
NHERF proteins are the most well studied PDZ proteins that are expressed in polarized epithelia. The NHERF family consists of four structurally related proteins (NHERF1–4) (Fig. 4). NHERF1 and NHERF2 contain two tandem nonidentical PDZ domains and an EBD, whereas NHERF3 and NHERF4 have four PDZ domains but lack an EBD (Seidler et al., 2009). NHERF proteins play an important role in organizing signaling complexes, controlling apical membrane trafficking, and coupling apical membrane transporters and receptors with other PDZ targets (Wang et al., 2007; Klenk et al., 2010; Georgescu et al., 2014). Although NHERF isoforms share these common functional characteristics, they perform distinct physiologic functions. For example, both NHERF1 and NHERF3 are important for phosphate homeostasis, yet in nherf1−/− and nherf3−/− mice, only nherf1−/− mice exhibit prominent phosphate wasting, suggesting that NHERF1 and NHERF3 play different physiologic roles (Shenolikar et al., 2002; Kocher et al., 2003; Giral et al., 2011). NHERF proteins also exhibit differential tissue expression. NHERF1 and NHERF2, for instance, are abundantly expressed and widely distributed, with NHERF2 displaying a more restricted tissue distribution compared with NHERF1. NHERF1 is highly expressed on the apical aspect of polarized epithelia in the kidney, liver, and placenta (Reczek et al., 1997). NHERF1 also is found in regions of the gastrointestinal tract (small intestine and colon), gastric parietal cells, and brain (Weinman et al., 1995; Reczek et al., 1997). Although NHERF2 is coexpressed with NHERF1 in the kidney, its distribution within the kidney differs. NHERF1 is found in proximal tubules, whereas NHERF2 is expressed in the glomeruli, collecting duct, and throughout the renal vasculature and is absent from the proximal tubule. The highest expression of NHERF2 is in lung, including pulmonary alveoli. NHERF3 is expressed at the brush-border membrane of proximal tubule cells, the intestinal epithelium, and is also found in the liver (Custer et al., 1997; Kocher et al., 1998; Gisler et al., 2001). NHERF1 and NHERF3 predominantly localize to the apical aspect of polarized epithelia, whereas the localization of NHERF2 is variable, including both apical membranes and a diffuse subcellular cytosolic distribution among different tissues. NHERF4 represents the isoform with the most restricted tissue distribution. Significant levels of NHERF4 are found in the kidney and gastrointestinal tract. Within these tissues, NHERF4 is localized to the apical surface and subapical regions throughout the cytoplasm (Gisler et al., 2001; Donowitz et al., 2005).
Fig. 4.
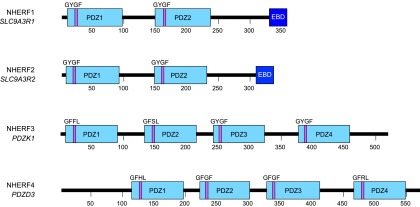
Schematic representation of human NHERF isoforms. PDZ domains are indicated by the light blue rectangular boxes and EBDs by dark blue boxes. The amino acid residues that constitute the core binding-motif are noted and shown as a purple bar indicating their location within the respective PDZ domain.
NHERF proteins can form homo- and heterodimers or undergo oligomerization. Dimerization is thought to be mediated either by intermolecular interactions between PDZ domains or by an intramolecular head-to-tail interaction between the carboxy terminus and a PDZ domain within the same protein, as is the case with NHERF1 (Lau and Hall, 2001; Shenolikar et al., 2001; Morales et al., 2007). Head-to-tail dimerization of NHERF1 is mediated by the interaction between the carboxy terminus, which itself serves as a PDZ-ligand (-FSNL), with PDZ2 (Morales et al., 2007; Wang et al., 2012). Although the biologic significance of NHERF dimerization is not fully appreciated, a recent study showed that heterodimerization of NHERF2 and NHERF3 is required for the inhibition of Na+/H+ exchanger-3 by carbachol (Yang et al., 2014). The formation of a large scaffolding complex comprising multiple NHERF isoforms because of dimerization may give rise to an elaborate network of cytoplasmic scaffold proteins. Formation of such a complex may dictate unique tissue-specific NHERF function. Elucidating the physiologic importance of NHERF dimerization as it relates to function may aid in our understanding of the in vivo roles of the NHERF family.
Despite the structural similarities in PDZ domains among the NHERF family, target proteins that harbor PDZ ligands, including drug transporters, display different affinities for NHERF isoforms (Hoque and Cole, 2008; Wang et al., 2010; Park et al., 2014). Such differences in affinity may partially explain the nonredundant physiologic functions of the NHERF family, as well as the fact that several tissues express multiple NHERF isoforms (Voltz et al., 2001). Additionally, because of the fact that NHERF proteins can form homo- and heterodimers, some NHERF isoforms may operate independently, whereas other physiologic processes may require NHERF cooperativity. In the latter scenario, NHERF proteins would bind to target proteins directly via one PDZ domain, reserving the remaining PDZ domain(s) to interact with other NHERF isoforms that in turn are responsible for anchoring the entire multiprotein complex to the actin cytoskeleton through interactions with the ERM family. Further studies are needed to determine the physiologic and potential pharmacological relevance of NHERF dimerization, cooperativity, and regulation of drug transport.
Given that NHERF expression parallels that of drug transporters and that several drug transporters have been identified as NHERF binding partners, these proteins constitute a novel class of post-translational mediators of drug transport. For this reason, among all PDZ proteins, the NHERF proteins are the most extensively studied in the context of drug transporter regulation.
IV. Role of PSD-95/Drosophila Discs Large/ZO-1 Proteins in Regulation of Cell Membrane Abundance, Localization, and Function of Drug Transporters
The widely acknowledged clinical significance of drug transporters has led to a surge in studying their regulatory mechanisms. Recently, the mechanisms that underlie drug transporter membrane organization and abundance have gained considerable interest. To coordinate the vectorial movement of drugs across polarized epithelia, drug transporters must be sorted and stabilized to the appropriate membrane domain. By virtue of the fact that drug transporters display unique tissue-specific apical or basolateral membrane localization, domain organization is not random. If the mechanisms that dictate membrane organization and stability are faulty, drug transporters may localize to the wrong membrane domain or display decreased membrane abundance. The consequences of faulty membrane targeting could result in an overall reduction in transporter function, which may then alter the pharmacokinetics and pharmacodynamics of drugs that use transporter pathways for elimination. The mechanism of NHERF1-dependent renal phosphate excretion, secondary to its role in stabilizing the sodium phosphate cotransporter-2a (NaPi-2a) to the apical membrane in renal proximal tubule cells, best illustrates this. In the absence of NHERF1, the membrane abundance of NaPi-2a decreases, preventing phosphate reabsorption from the glomerular filtrate and thus promoting renal phosphate excretion (Shenolikar et al., 2002). Exploring NHERF in the setting of drug transport may address disparities surrounding drug transporter function and drug response. Emerging evidence has shown that PDZ proteins, namely the NHERF family, play a critical role in the localization, stabilization, and functional regulation of select SLC and ABC drug transporters (Table 3) (Sugiura et al., 2011).
TABLE 3.
NHERF-dependent regulation of drug transporter function
Uppercase denotes human genes or gene products and lowercase denotes rodent genes or gene products.
| Transporter | NHERF Isoform | Experimental Model | Substrate | Transporter Function | Reference |
|---|---|---|---|---|---|
| OCTN2 | NHERF3 NHERF4 | HEK293 | Carnitine | Increase | Kato et al., 2005 |
| Watanabe et al., 2006 | |||||
| Octn2 | Nherf3 | Nherf3−/− mice | Carnitine | Decrease | Sugiura et al., 2008 |
| OAT4 | NHERF1 NHERF3 | HEK293 | Estrone-3-sulfate | Increase | Miyazaki et al., 2005 |
| LLC-PK1 | Zhou et al., 2008 | ||||
| URAT1 | NHERF3 | HEK293 | Urate | Increase | Anzai et al., 2004 |
| OATP1A2 | NHERF1 NHERF3 | HEK293 | Estrone-3-sulfate | Increase | Zheng et al., 2014 |
| Oatp1a1 | Nherf3 | Nherf3−/− mice | Estrone-3-sulfate | Increase | Sugiura et al., 2010 |
| PEPT2 | NHERF2 | Xenopus oocytes | Gly-Gly | Increase | Boehmer et al., 2008 |
| PEPT2 | NHERF3 | HEK293 | Gly-Sar | Increase | Noshiro et al., 2006 |
| Sugiura et al., 2006 | |||||
| Pept1 | Nherf3 | Nherf3−/− mice | Cephalexin | Decrease | Sugiura et al., 2008 |
| MRP2 | NHERF1 | WIF-B | CMFDA | Increase | Karvar et al., 2014 |
| MRP4 | NHERF1 | HeLa | 6-Mercaptopurine, 9-[2-(phosphonylmethoxy) ethyl]-adenine | Decrease | Hoque and Cole, 2008 |
| MRP4 | NHERF3 | HEK293 | Adefovir | Increase | Park et al., 2014 |
| Mrp4 | Nherf3 | Nherf3−/− mice | Adefovir | Decrease | Park et al., 2014 |
| Bcrp | Nherf3 | Nherf3−/− | Cimetidine | Decrease | Shimizu et al., 2011 |
CMFDA, 5-chloromethylfluorescein diacetate.
A. Solute Carrier Drug Transporters
1. Organic Carnitine Transporters.
Although the molecular mechanisms of membrane targeting of the OCT and OCTN family are not well characterized, NHERF2, NHERF3, and NHERF4 have been shown to interact with and/or directly regulate OCTN2 function via modulating membrane abundance (Kato et al., 2005; Sugiura et al., 2006, 2008; Watanabe et al., 2006). A pull-down study using recombinant C-terminal proteins identified an interaction between apically localized OCTN1 and OCTN2 with both NHERF2 and NHERF3 but not with OCT1 and OCT2, which in contrast, do not harbor PDZ ligands and exclusively localize to basolateral membranes (Kato et al., 2005). Additional studies revealed that the interaction required the presence of the last four amino acids of OCTN1 and OCTN2 (-LTAF and -STAF, respectively), both of which are class I PDZ ligands. The interaction of NHERF3 with the C terminus of OCTN2 was confirmed in a pull-down study using kidney brush-border membrane vesicles. Subsequent immunohistochemical analysis revealed that NHERF3 and OCTN2 colocalize in mouse kidney brush border membranes. Furthermore, NHERF3 increased OCTN2 mediated uptake of carnitine, a well established OCTN2 probe substrate. This result could be explained by a 6-fold increase in carnitine transport capacity in vitro. Modulation of OCTN2 function by NHERF3 was not observed upon deletion of the last four amino acids of OCTN2 (-STAF), supporting that these two proteins interact directly via the PDZ ligand of OCTN2 (Kato et al., 2005). An in vivo study performed in nherf3−/− mice found that absorption of carnitine was decreased compared with wild-type mice (Sugiura et al., 2008). Immunohistochemical analysis revealed that the apical localization of intestinal Octn2 was reduced in nherf3−/− mice compared with wild-type mice (Sugiura et al., 2008). Collectively, these studies suggest a role for NHERF3 in regulating OCTN2 membrane abundance and function in the kidney and intestine. NHERF2 may have a similar role.
Coexpression of OCTN2 and NHERF4 in HEK293 cells also increased the uptake of carnitine (Watanabe et al., 2006). Subsequent kinetic analysis revealed a 2-fold increase in transport capacity but a minimal effect on carnitine specificity for OCTN2, suggesting that the increase in OCTN2 function was not due to changes in carnitine affinity but rather a greater number of OCTNs involved in transport. Furthermore, upon deletion of the last four amino acids in OCTN2 (-STAF), the effect of NHERF4 on OCTN2 was abolished (Watanabe et al., 2006), signifying that the mechanism is dependent on the presence of the PDZ ligand. However, these results were not observed when NHERF4 was coexpressed with OCT3 or OCTN1, despite the fact that both OCT3 and OCTN1 harbor class I PDZ ligands (-RSHL and -LTAF, respectively). These data indicate that NHERF4 might operate by a mechanism that is reliant on structural characteristics other than the existence of a PDZ ligand alone.
Taken together, NHERF proteins may differentially modulate OCTN function secondary to preservation of membrane abundance. These data also point to an uncharacterized NHERF mechanism that operates independent of a PDZ ligand. This hints to involvement of other cytoplasmic adaptor/scaffold proteins or noncanonical binding motifs affecting NHERF binding affinities and thus function. Given that OCTNs have described roles in pharmacokinetics, pharmacodynamics, and pathophysiology, characterizing NHERF-mediated OCTN function may lead to a more detailed mechanistic explanation of the pharmacological and pathophysiological role of OCTNs.
2. Organic Anion Transporters.
The OATs play clinically important roles in the disposition of several pharmacologically active compounds as well as many endogenous substances. OAT4 and URAT1 are highly expressed in the kidney and are exclusively found at apical membranes, whereas other renal OAT isoforms (OAT1–3) are found at the basolateral membrane (Burckhardt, 2012). Among the OAT family, OAT4 and URAT1 contain class I PDZ ligands (-STSL and -STQF, respectively) and have been shown to be binding partners with NHERF1 and NHERF3. Evidence suggests that NHERF1 and NHERF3 play a role in regulating OAT4 and URAT1 function (Anzai et al., 2004; Miyazaki et al., 2005; Zhou et al., 2008; Zhang et al., 2010a). Yeast two-hybrid studies and surface plasmon resonance confirmed OAT4 binds both NHERF1 and NHERF3 and that the interaction is mediated by the C-terminal region of OAT4 and the PDZ1 domain of NHERF1 and both PDZ1 and PDZ4 domains in NHERF3 (Miyazaki et al., 2005). The association of OAT4 with NHERF1 and NHERF3 depends on the presence of the C-terminal PDZ ligand of OAT4 (-STSL). Furthermore, association of OAT4 with NHERF1 or NHERF3 enhanced transport of estrone-3-sulfate, an established OAT4 probe substrate, in HEK293 cells. Upon deletion of the OAT4 PDZ ligand, this functional effect was abolished (Miyazaki et al., 2005). These data indicate that NHERF1 and NHERF3 may affect OAT4 function through interactions mediated by specific PDZ domains.
The effect of NHERF proteins on OAT4 function may be tissue specific (Zhou et al., 2008). In a comparison of NHERF-mediated OAT4 function in LLC-PK1 versus BeWo cells, a kidney and placental cell line, respectively, NHERF1 and NHERF3 only enhanced OAT4-dependent estrone-3-sulfate uptake in LLC-PK1 cells but not BeWo cells. This suggests that the effect of NHERF1 and NHERF3 may be cell or tissue specific. These results further support the hypothesis that NHERF proteins display tissue-specific function (Zhou et al., 2008). To evaluate the mechanism of NHERF1-mediated OAT4 regulation, Zhang et al. (2010a) showed that protein kinase C (PKC), which has previously been shown to inhibit NHERF1 function via phosphorylation, downregulates OAT4 activity. Further studies in COS-7 cells revealed that NHERF1 attenuates OAT4 internalization (Zhang et al., 2010a). These studies demonstrate that the mechanism of NHERF1-mediated OAT4 regulation may be secondary to preventing OAT4 internalization.
URAT1 has also been studied in the context of NHERF-mediated regulation. Similar to OAT4, the C-terminal PDZ ligand of URAT1 (-STQF) is required for interaction with NHERF3 and for NHERF3-dependent functional regulation (Anzai et al., 2004). In HEK293 cells, urate transport was increased by NHERF3, an effect that was abolished upon deletion of the URAT1 C-terminal PDZ ligand. Kinetic analysis revealed that this effect was due to an increase in the capacity of urate transport rather than a change in urate specificity for URAT1. Subsequent coimmunoprecipitation studies showed that URAT1 colocalizes with NHERF3 at the apical brush border membrane in renal proximal tubule cells and the presence of the URAT1 PDZ ligand is required for this interaction (Anzai et al., 2004).
Together, these studies highlight an important role for NHERF1 and NHERF3 in regulating OAT4 and URAT1 function. Although both transporters have been proposed to play important pharmacological roles, they also have been shown to mediate the renal secretion and reabsorption of uric acid (Graessler et al., 2006; Sakiyama et al., 2014). This implies that in addition to potential effects on renal drug secretion, NHERF proteins may also influence uric acid homeostasis and may have an underlying role in the pathophysiology of gout.
3. Organic Anion-Transporting Polypeptides.
Five members of the OATP family harbor a class I PDZ ligand at their C terminus (Table 2). Evolutionarily, the OATPs are poorly conserved and orthologs in rodents may not exist in humans, making findings from studies performed in animal models difficult to translate clinically. Although multiple studies indicate that NHERF proteins play a role in regulating rodent Oatps (Wang et al., 2005, 2014; Sugiura et al., 2010; Choi et al., 2011), there is little information regarding how NHERF proteins affect human OATP isoforms.
OATP1A2 was the first identified human OATP isoform and has since been well studied and found to play an important role in the cellular uptake of a variety of drugs and endogenous substances. Recently, NHERF1 and NHERF3 were found to regulate OATP1A2 function by modulating protein internalization and enhancing membrane stability. The influence of NHERF1 and NHERF3 on OATP1A2 function was assessed using HEK293 cells that coexpressed NHERF1 or NHERF3 (Zheng et al., 2014). Similar to studies in OATs, estrone-3-sulfate uptake was used as a surrogate for OATP1A2 function. Both NHERF1 and NHERF3 were found to significantly enhance E1S uptake. Overexpression of NHERF1 or NHERF3 leads to an increase in OATP1A2 membrane abundance and was proposed to be the underlying basis for the observed increase in OATP1A2 function. Subsequent studies revealed that the mechanism for the apparent increase in OATP1A2 membrane abundance was due to a decrease in its internalization via a clathrin-dependent, but caveolin-independent, manner (Zheng et al., 2014).
NHERF proteins may have similar effects on the other human OATPs that contain C-terminal PDZ ligands (Table 2). Particular attention should be given to OATP2B1 because of its established functional role in the intestine as an uptake transporter for several drugs including fexofenadine and statins. OATP2B1 also uniquely displays dual-membrane specificity, apical in enterocytes and basolateral in hepatocytes (Fig. 1). This may indicate that if NHERF proteins are involved in regulating OATP2B1, they may do so differently in the intestine than in the liver. Investigating the mechanism that determines intestinal versus hepatic OATP2B1 membrane targeting may provide clues on how NHERF proteins, or other PDZ proteins, function in different tissues.
4. Peptide Transporters.
PEPT1 and PEPT2 are restricted to apical membranes in the intestine and kidney. Recent evidence shows that NHERF2 and NHERF3 interact with PEPT2 and enhance transport activity by preserving PEPT2 membrane abundance (Kato et al., 2004; Noshiro et al., 2006; Sugiura et al., 2006; Boehmer et al., 2008). A preliminary screening of several drug transporters and PDZ proteins identified a potent interaction between PEPT2 and NHERF3 (Kato et al., 2004). Studies using yeast two-hybrid assays and surface plasmon resonance confirmed the interaction between PEPT2 and NHERF3 is mediated by the PDZ2 and PDZ3 domains (Noshiro et al., 2006). The influence of NHERF3 on PEPT2 transport was subsequently assessed in HEK293 cells using Gly-Sar uptake as a surrogate of PEPT2 function (Noshiro et al., 2006; Sugiura et al., 2006). Coexpression of NHERF3 in HEK293 cells augmented PEPT2 function and was associated with an increase in transport capacity. Coexpression with NHERF3 increased PEPT2 membrane abundance and was proposed to account for the increase in function. Sugiura et al. (2006) later evaluated PEPT2 by assessing Gly-Sar uptake in HEK293 cells stably expressing NHERF3 constructs with a mutation in the PDZ2 domain. Although HEK293 cells overexpressing the NHERF3 mutant displayed a decrease in Gly-Sar uptake compared with those overexpressed with wild-type NHERF3, the results were not significant. Identifying the effects of genetic polymorphisms in NHERF proteins on drug transport may help clarify which drug transporters are subject to NHERF regulation. Revealing which PDZ domains are important for drug transporter regulation also may provide detail on the structural requirements of NHERF function.
Despite similarities in structure and domain organization between NHERF1 and NHERF2, expression studies performed in Xenopus oocytes have shown that NHERF2, but not NHERF1, enhance PEPT2 membrane abundance and function. This is another example of NHERF target-proteins displaying differential binding affinities for NHERF isoforms. The effect of NHERF2 on PEPT2 was not observed upon deletion of the C-terminal PDZ ligand of PEPT2 (-KTKL), suggesting that the PEPT2 PDZ ligand is a requirement for interaction with NHERF2 (Boehmer et al., 2008). Why NHERF1 does not produce similar effects, despite its similarity with NHERF2, is uncertain.
Collectively, these studies indicate that NHERF proteins modulate PEPT2 function and therefore may play important physiologic and pharmacological roles in oligopeptide and peptide-like drug transport in the kidney. However, several questions remain regarding the regulation of PEPT membrane targeting. For instance, it is unclear why PEPT2 preferentially binds NHERF2 and NHERF3, but not NHERF1, especially considering NHERF1 and NHERF3 are highly expressed in the kidney, where PEPT2 is also prominently expressed. A closer inspection of PEPT2 regulation by NHERF proteins in the kidney may reveal cooperative or opposing roles for NHERF1 and NHERF3. This uncertainty is an opportunity to flesh out details regarding regulation mediated by multiple NHERF isoforms. Alternatively, NHERFs might regulate PEPT transport indirectly. NHERF3 may regulate PEPT1 as described (Kato et al., 2006). PDZ proteins, such as NHERFs, may influence drug transport indirectly by regulating proteins that are responsible for producing electrochemical gradients necessary for dissipative transport. For example, PEPT1 utilizes an inwardly directed H+ gradient that is produced by the Na+/H+ exchanger (NHE) to provide the driving force for substrate uptake. NHERFs may indirectly regulate PEPT1 by directly regulating NHE or recruiting NHE in close proximity to PEPT1 to ensure an adequate H+ gradient is achieved. Discovering the nature of the functional coupling of PEPT1 and NHE by NHERFs, as well as differences in NHERF-dependent PEPT membrane targeting, may broaden our understanding of NHERF action and drug transport. These studies will aid in understanding how PEPTs are organized at cell membranes and lead to new discoveries regarding how PDZ proteins, including NHERF proteins, interact with each other to perform nonredundant physiologic functions in different tissues.
B. ATP Binding-Cassette Drug Transporters
1. Multidrug Resistance Proteins.
NHERF1 and NHERF3 have been reported to regulate the membrane abundance, localization, and function of MRP2 (-STKF) and MRP4 (-ETAL), both of which contain a class I C-terminal PDZ ligand. The initial discovery that NHERF proteins interact with MRPs resulted from studies that focused on elucidating the cellular mechanisms responsible for multidrug resistance in cancer after it was found that NHERF3 is upregulated in selected tumors of epithelial origin (Kocher et al., 1998, 1999). This prompted an investigation of potential NHERF3 binding partners. Expression studies and yeast two-hybrid assays performed in human carcinomas confirmed an increase in NHERF3 mRNA and revealed that NHERF3 interacts with the C-terminal portion of MRP2 (Kocher et al., 1999). Later, it was revealed that MRP2 interacts with all four NHERF isoforms (NHERF1–4) and that phosphorylation of Ser1542 in the MRP2 PDZ binding motif significantly enhanced binding to NHERF1 and NHERF4; the functional consequence was not assessed (Hegedus et al., 2003). Because of these findings, others have investigated the role of intracellular kinases, such as PKC, and membrane localization of MRP2. For example, specific PKC isoforms, such as conventional PKCα (cPKCα), novel PKCδ (nPKCδ), nPKCε, and atypical PKCζ (aPKCζ), appear to be involved in regulating hepatic MRP2 membrane recycling at the apical/canalicular membrane (Beuers et al., 2003; Ito et al., 2005; Schonhoff et al., 2008; Stross et al., 2009; Park et al., 2012). It is still unclear if these PKC-mediated phosphorylation events result from direct phosphorylation of MRP2, NHERF proteins, or members of the ERM family, such as radixin, known to interact with and influence MRP2 function (Suda et al., 2011, 2015). Investigating phosphorylation events important for drug transporter regulation represents an emerging line of research that intersects with the physiologic and pharmacological role of PDZ proteins.
To describe further the role of NHERF1-dependent MRP2 regulation in the liver, nherf1−/− mice have been employed. In livers obtained from nherf1−/− mice, Mrp2 mRNA was unchanged but protein levels were significantly decreased compared with wild-type mice, suggesting post-translational changes in Mrp2 (Li et al., 2010). Immunofluorescent staining of Mrp2 in hepatocytes from nherf1−/− mice showed that although a pool of Mrp2 remained at the apical/canalicular membrane, the signal was noticeably weaker. Bile flow in nherf1−/− mice was reduced by 70% compared with wild-type mice, with a 50% and 60% reduction in glutathione and glutathione-methylfluorescein biliary excretion in isolated hepatocytes. Bile acid and bilirubin excretion, surrogates of hepatic MRP2 function, were unchanged (Li et al., 2010). Studies performed in nherf1−/− provide strong evidence of the importance of NHERF1 in targeting and stabilizing hepatic MRP2 to the apical/canalicular membrane. In a more recent study, fluorescent-tagged mutant constructs were used in WIF-B cells, a polarized hepatocyte cell line, to study localization and function of NHERF1 (Karvar et al., 2014). Fluorescence microscopy was used to visualize the cellular distribution of NHERF1, MRP2, and radixin. A point mutation in the radixin binding site of NHERF1 (F335R) and deletion mutations (PDZ1 and PDZ2) were used to assess NHERF1 membrane association, and a 5-chloromethylfluorescein diacetate assay was used to characterize MRP2 function. It was later confirmed that only overexpression with the radixin binding site mutant (F335R) and the PDZ1 deletion mutant resulted in decreased NHERF1 membrane association and MRP2 function (Karvar et al., 2014). These results support that the functional importance of NHERF1 depends on its interaction with MRP2 and the PDZ1 domain and demonstrate that NHERF1 is important for the distributional dynamics of MRP2 in the liver. Although NHERF3 was the initial NHERF isoform identified to interact with MRP2 (Kocher et al., 1999), subsequent studies performed in kidney proximal tubule cells of nherf3−/− mice revealed no change in expression or cellular distribution of Mrp2 (Kocher et al., 2003). This indicates that NHERF3 is not involved in MRP2 membrane targeting. Alternatively, functional compensation by other PDZ proteins or cytoplasmic adapters may preserve MRP2 membrane abundance in the absence of NHERF3 and thus be confounding these results.
The antiviral drug adefovir is an established MRP4 probe substrate and is often used as a surrogate of MRP4 function. As noted earlier, MRP4 is critical for the renal elimination of adefovir (Imaoka et al., 2007). NHERF1 and NHERF3 also regulate MRP4 membrane localization and function; however, they appear to have opposing roles, because downregulation of NHERF1 increases MRP4 function, whereas NHERF3 stabilizes MRP4 at apical membranes and increases function (Hoque and Cole, 2008; Hoque et al., 2009; Park et al., 2014). Knock down of NHERF1 by small interfering RNA in HeLa cells increases MRP4 membrane abundance and reduces cellular accumulation of the MRP4 probe substrates, 6-mercaptopurine, and 9-[2-(phosphonylmethoxy) ethyl]-adenine (Hoque and Cole, 2008). The authors attributed these results to a decrease in MRP4 internalization mediated by NHERF1. It was later demonstrated that MRP4 localizes to either apical or basolateral membranes depending on the cell type and the degree of NHERF1 expression (Hoque et al., 2009). This study found that MRP4 localizes to basolateral membranes in MDCKI cells, where NHERF1 expression is low, whereas MRP4 localizes to apical membranes in LLC-PK1 cells, which express a higher amount of NHERF1 (Hoque et al., 2009). Although these studies provide proof-of-principle, in vitro cell systems are limited and may not represent the endogenous role(s) of NHERF proteins and drug transporter regulation. Furthermore, PDZ regulation of drug transporters is not likely to depend on a single PDZ isoform but rather multiple isoforms, as well as on members of the ERM family. For these reasons, extrapolating these data to in vivo systems will advance our understanding of PDZ regulatory processes.
In contrast to NHERF1, NHERF3 is critical for MRP4-dependent efflux in the kidney. The role of NHERF3 on MRP4 membrane targeting and function was assessed in an elegantly conducted study using nherf3−/− mice and HEK293 cells (Park et al., 2014). MRP4 protein expression, membrane abundance, and function decreased in nherf3−/− mice compared with wild-type mice. A pharmacokinetic study evaluated the role of NHERF3 on MRP4-dependent renal drug elimination (Park et al., 2014). Mice were administered an intravenous injection of [3H]adefovir, an established prototype MRP4 probe substrate that undergoes negligible metabolism and is predominately eliminated via the kidney. Plasma clearance of [3H]adefovir was significantly decreased in nherf3−/− mice compared with wild-type mice. This result was accompanied by a significant increase in the [3H]adefovir concentration area under the curve (AUC0-inf), which is reflective of the systemic exposure of [3H]adefovir. Furthermore, the concentration of [3H]adefovir in kidneys was significantly higher in nherf3−/− mice compared with wild-type mice, suggesting impaired MRP4-mediated efflux from renal proximal tubules (Park et al., 2014). Collectively, these pharmacokinetic changes suggest that NHERF3 plays a crucial role in MRP4-dependent renal drug elimination as a regulator of MRP4 apical membrane insertion in proximal tubule epithelial cells and thus is an indirect contributor to drug clearance via the kidney. Immunoprecipitation assays using transfected HEK293 cells revealed that the interaction between NHERF3 and MRP4 is mediated by the C-terminal portion of MRP4 (-ETAL) and the PDZ1 domain of NHERF3 (Park et al., 2014). When coexpressed in HEK293 cells, surface biotinylation and internalization assays revealed that NHERF3 increases MRP4 membrane stability and reduces MRP4 internalization (Park et al., 2014). Pharmacokinetic studies that use nherf1−/− and nherf3−/− mice represent the next stage in translating the pharmacological significance of NHERF proteins and drug transport.
Several questions remain regarding how NHERF1 and NHERF3 affect MRP4 function and why they seemingly exhibit opposite actions. It is unclear why MRP4 localizes to basolateral membranes in hepatocytes rather than the apical membrane as it does in renal proximal tubules. Although NHERF proteins are likely involved in MRP4 membrane organization and function, members of the ERM family may also direct MRP4 targeting as they do with MRP2. Investigating the interaction between NHERF proteins, MRP2, MRP4, and members of the ERM family may provide additional details regarding tissue-specific mechanisms.
V. PSD-95/Drosophila Discs Large/ZO-1 Proteins and Drug Action in Disease States
A. Cancer
The ABC superfamily of drug transporters is well known in oncology for their role in conferring multidrug resistance (MDR) (Gottesman et al., 2002). ABC transporters actively efflux a broad array of chemotherapeutic agents such as taxanes, camptothecins, and platinum agents, thus contributing to decreased intracellular drug accumulation in tumor cells and ultimately failure of chemotherapy (Gottesman et al., 2002). Pgp (MDR1), MRP1 (ABCC1), and BCRP (ABCG2), the ABC superfamily, play important roles in MDR (Doyle et al., 1998; Kunická and Souček, 2014; Wu et al., 2014). Overexpression of these transporters is well documented in many types of cancer and considered to be the underlying mechanism for chemoresistance attributable to ABC drug efflux in tumors (Szakacs et al., 2006; Teodori et al., 2006; Wu et al., 2014). Substrate-induced induction is one mechanism involved in the upregulation of ABC drug transporters, where chemotherapeutic drugs enhance chemoresistance through nuclear receptors involved in the transcriptional expression of efflux drug transporters (Chen, 2010; Herraez et al., 2012; Manceau et al., 2012; Oda et al., 2013). For example, the pregnane X receptor (PXR) is a nuclear receptor that binds promiscuously with many structurally diverse ligands, including anticancer drugs such as paclitaxel (Harmsen et al., 2009, 2010). The expression of PXR has been characterized in many cancers where it is thought to be involved in resistance to chemotherapy. Upon ligand activation, PXR forms a heterodimer with other nuclear receptors, such as the retinoid X receptor, and induces the expression of target genes involved in drug resistance. It has been clearly demonstrated that PXR directly regulates Pgp expression (Harmsen et al., 2010). To address the problem of MDR in cancer, novel host- and tumor-mediated pathways continue to be explored in an effort to identify potential new therapeutic targets.
The clinical contribution to MDR from other drug transporters remains uncertain. However, in an evaluation of ABC drug transporters in a panel of tumor cell lines from the US National Cancer Institute, over half of the members that comprise the ABC drug transporter family have been implicated as playing a role in conferring MDR, suggesting multiple drug transporters are likely involved (Szakacs et al., 2004). As a means to combat MDR in cancer patients, targeting ABC drug transporters through pharmacological inhibition has been attempted for several years but with poor results (Turk and Szakacs, 2009; Karthikeyan and Hoti, 2015). Because of their clinical contribution to MDR, studies aimed at understanding the regulatory mechanisms of transporters focus heavily on changes in gene and protein expression. Studies that investigate post-translational modification of drug transporters in cancer remain an area of research that is underrepresented. Future studies aimed at expanding our knowledge of drug transporter regulation in cancer outside the realm of transcriptional regulation are warranted.
The NHERF proteins, particularly NHERF1, have been implicated in different types of cancers. Although its role as a tumor suppressor and/or oncogenic protein remains controversial, overexpression of NHERF1 in several cancers, such as breast cancer, schwannoma, and hepatocellular carcinoma has been described (Stemmer-Rachamimov et al., 2001; Fraenzer et al., 2003; Shibata et al., 2003; Cardone et al., 2007). In addition, tumor stage, metastatic progression, poor prognosis, and estrogen receptor status are significantly associated with NHERF1 overexpression (Cardone et al., 2007; Song et al., 2007). Furthermore, NHERF1 upregulation in conditions of serum deprivation and hypoxia suggest that NHERF1 function may increase in a tumor microenvironment (Cardone et al., 2007). Although NHERF1 expression increases in human cancer, its cellular distribution appears to shift from the plasma membrane to nuclear and cytoplasmic regions (Stemmer-Rachamimov et al., 2001; Fraenzer et al., 2003; Cardone et al., 2007). The functional consequences of the observed shift in the cellular distribution of NHERF1 in cancer continue to be investigated.
The mechanisms of MDR in cancer are likely multifactorial. In addition to alterations in transcriptional expression, post-translational modification of drug transporters may also be affected. The reported changes in NHERF1 expression in cancer combined with the newly described role of NHERF1 in drug transporter function underscore NHERF1 as an attractive and novel MDR target that is worthy of investigation. A critical evaluation of the role of NHERF1, as well as other NHERF isoforms, and drug transporter function in cancer may offer mechanistic insight into chemoresistance. Overexpression of select drug transporters, such as those believed to play major roles in cancer (Pgp, MRP1, and BCRP), may represent only one of several mechanisms that contribute to MDR. Although not considered to confer significantly to MDR, ABC drug transporters that contain class I PDZ ligands, such as MRP2 and MRP4, are implicated in chemoresistance (Kool et al., 1997; Zhang et al., 2010b). Therefore, the observed reduction in drug action may be partially attributable to altered membrane targeting and/or abundance of drug transporters involved in efflux of chemotherapeutic agents. Faulty membrane targeting may not result in observable changes in mRNA or protein expression of drug transporters in cancer, and therefore those transporters subject to NHERF regulation may be overlooked despite their potential involvement in conferring resistance. Turning attention to NHERF1 as a mediator of drug transporter regulation in cancer may lead to a more complete understanding of MDR while also highlighting NHERF1 as a potential new target.
B. Chronic Kidney Disease
A well known consequence of chronic kidney disease (CKD) is reduced renal drug clearance. Dosing guidelines have been developed for drugs that undergo renal elimination, where doses are adjusted according to some measure of kidney function (i.e., creatinine clearance or glomerular filtration rate) based on the premise that renal clearance declines in proportion to kidney function (Nielsen et al., 2014). Despite the development of renal dosing guidelines, patients with CKD experience high rates of adverse drug events (Zaidenstein et al., 2002). However, both renal and extra-renal drug clearance mediated by key drug transporters is altered in patients with CKD (Nolin et al., 2009). This undoubtedly contributes to the high variability in drug response and the increased frequency of adverse drug events observed in CKD patients (Matzke and Frye, 1997; Bates et al., 1999; Zaidenstein et al., 2002; Nolin and Unruh, 2010). Changes in drug transporter gene and protein expression in both renal and extrarenal tissues do not consistently correlate with the expected changes in transporter function in the setting of CKD. For instance, intestinal Pgp function is reduced ex vivo in the intestine of 5/6th nephrectomized rats, an experimental model of CKD (Veau et al., 2001). Interestingly, no change in Pgp mRNA or protein expression was observed, indicating that changes in transcriptional expression could not explain the reduction in function. Later, others confirmed and extended these results to other transporters. In 5/6th nephrectomized rats, a significant decrease in Pgp, Mrp2, and Mrp3 protein expression was observed with no change in mRNA (Naud et al., 2007). These discrepancies are not limited to the intestine. Additional studies utilizing the same experimental animal model of CKD show that several hepatic and renal drug transporters, such as members from the Oatp, Oat, Oct, Pgp, and Mrp families, also display changes in expression that do not correlate with function (Laouari et al., 2001; Ji et al., 2002; Naud et al., 2008; Tsujimoto et al., 2008). However, data across studies yield highly variable and inconsistent results, which makes interpretation difficult. Furthermore, despite these important findings, there is currently no clear mechanism as to the cause. These discrepancies and lack of a definitive mechanism for changes in transporter function in CKD limit the extrapolation of these data into the clinical setting.
To date, studies aimed at evaluating changes in gene and protein expression alone in CKD have not reconciled this disparity. Many studies attribute uremic toxins, metabolic byproducts that accumulate in uremia, as a major cause of reduced drug transporter function in CKD (Barreto et al., 2014; Masereeuw et al., 2014). Uremic toxins are believed to competitively inhibit drug transport and to downregulate transporter expression (Reyes and Benet, 2011; Sato et al., 2014). However, uremic toxins do not begin to accumulate until end-stage renal disease and therefore may only explain changes in drug transporter function in patients with end-state renal disease. Moreover, hundreds, if not thousands of uremic toxins may exist (Vanholder et al., 2003; Masereeuw et al., 2014; Massy, 2014). Determining which toxin(s) have the greatest impact on transporter function has also proved difficult. The mechanisms responsible for reduced drug transporter function may be more complex and begin in earlier stages of CKD, well before reduced kidney function limits uremic toxin elimination. Novel mechanistic hypotheses that address altered drug transporter function in CKD are needed to advance our understanding of this field and ultimately develop dosing strategies to prevent adverse events in this patient population.
CKD represents a disease state for which exploration of NHERF-dependent drug transporter regulation may provide novel insights into altered transporter function (Fig. 5). How drug transporter function changes in CKD from the standpoint of NHERF-dependent transport has yet to be investigated. This is likely because only recently has research in drug transporter membrane targeting gained interest. The NHERF family is abundantly expressed in the intestine, liver, and kidney, tissues that also highly express transporters. To date, there are no data on changes in NHERF function in CKD. Consideration should be given to understanding how NHERF function may change over the course of CKD and the implications on drug transport.
Fig. 5.

Model of NHERF regulation of MRP4-mediated renal proximal tubule adefovir secretion. Under physiologic conditions (A), NHERF1 or NHERF3 (NHERF1/3) tether MRP4 to apical membranes. Adefovir is taken up across basolateral membranes by organic acid transporters OAT1 and OAT3 and exits the cell across apical membranes into the urine, The coordinated actions of basolateral OAT1 and OAT3 and apical MRP4 drug transporters assure little cytoplasmic drug accumulation. In chronic kidney disease (B), NHERF1/3 are downregulated. This reduces the abundance of MRP4 at apical membranes, with attendant cellular drug accumulation potentially leading to toxic concentrations. The illustration is based on recent findings (Crean et al., 2014) and unpublished observations.
VI. Conclusions and Future Directions
The functional regulation of drug transporters by the NHERF family continues to capture the attention of those who investigate the underlying mechanisms that govern pharmacokinetics and pharmacodynamics. This relatively novel understanding of NHERF is still in its infancy, because several questions remain surrounding the NHERF family and regulation of drug transport. Future studies in this area should focus on addressing 1) the NHERF isoforms affecting drug transport and which transporters are the most susceptible to regulation by NHERF; 2) the different mechanisms of NHERF-directed membrane targeting depending on tissue type; 3) the functional importance of NHERF dimerization and cooperativity as it relates to drug transport; 4) the role of the ERM family in NHERF/drug transporter interactions; and 5) the role of NHERF proteins in MDR in cancer and altered drug transport function in CKD. Additional pharmacokinetic studies using specific phenotypic probe substrates of drug transport performed in nherf1−/− and nherf3−/− mice would provide an excellent model to begin addressing these gaps in our understanding. However, data performed in mouse models eventually must be translated to humans. The inability to assess NHERF noninvasively in humans represents an important limitation to this work. However, mutations in the human NHERF1 (SLC9A3R1) gene do exist (Karim et al., 2008). Clinical pharmacokinetic studies performed in patients who carry polymorphisms in the NHERF1 (SLC9A3R1) gene may provide a means to address this limitation.
Acknowledgments
The authors thank Lisa M. Christopher for contributing to figure preparation.
Abbreviations
- ABC
ATP binding-cassette
- BCRP
breast cancer resistance protein
- CKD
chronic kidney disease
- DJS
Dubin-Johnson syndrome
- EBD
ezrin binding domain
- ERM
ezrin/radixin/moesin/merlin
- Gly-Sar
glycylsarcosine
- MDR
multidrug resistance
- MDR1
multidrug resistance protein 1
- MRP
multidrug resistance-associated protein
- NBD
nucleotide binding domain
- NHE
Na+/H+ exchanger
- NHERF
Na+/H+ exchange regulatory factor
- OAT
organic anion transporter
- OATP
organic anion-transporting polypeptide
- OCT
organic cation transporter
- OCTN
organic carnitine transporters
- P450
cytochrome P450
- PDZ
PSD-95/Drosophila discs large/ZO-1
- PEPT
peptide transporter
- Pgp
P-glycoprotein
- PKC
protein kinase C
- PSD
postsynaptic density protein
- PXE
pseudoxanthoma elasticum
- PXR
pregnane X receptor
- SLC
solute carrier
- TMD
transmembrane domain
Authorship Contributions
Wrote or contributed to the writing of the manuscript: Walsh, Friedman, Nolin.
Footnotes
Original research was supported the National Institutes of Health National Institute of Diabetes and Digestive and Kidney Diseases [Grants R01-DK054171 and R01-DK069998 (to P.A.F.)]; and the National Institutes of Health National Institute of General Medical Sciences [Grant R01-GM107122 (to T.D.N.)]. D.R.W. received support from the Department of Pharmacy and Therapeutics, University of Pittsburgh School of Pharmacy.
References
- Abuznait AH, Kaddoumi A. (2012) Role of ABC transporters in the pathogenesis of Alzheimer’s disease. ACS Chem Neurosci 3:820–831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anzai N, Miyazaki H, Noshiro R, Khamdang S, Chairoungdua A, Shin HJ, Enomoto A, Sakamoto S, Hirata T, Tomita K, et al. (2004) The multivalent PDZ domain-containing protein PDZK1 regulates transport activity of renal urate-anion exchanger URAT1 via its C terminus. J Biol Chem 279:45942–45950. [DOI] [PubMed] [Google Scholar]
- Aszalos A. (2007) Drug-drug interactions affected by the transporter protein, P-glycoprotein (ABCB1, MDR1) II. Clinical aspects. Drug Discov Today 12:838–843. [DOI] [PubMed] [Google Scholar]
- Babu E, Takeda M, Narikawa S, Kobayashi Y, Enomoto A, Tojo A, Cha SH, Sekine T, Sakthisekaran D, Endou H. (2002) Role of human organic anion transporter 4 in the transport of ochratoxin A. Biochim Biophys Acta 1590:64–75. [DOI] [PubMed] [Google Scholar]
- Barreto FC, Stinghen AE, de Oliveira RB, Franco AT, Moreno AN, Barreto DV, Pecoits-Filho R, Drüeke TB, Massy ZA. (2014) The quest for a better understanding of chronic kidney disease complications: an update on uremic toxins. J Bras Neurol 36:221–235. [DOI] [PubMed] [Google Scholar]
- Bates DW, Miller EB, Cullen DJ, Burdick L, Williams L, Laird N, Petersen LA, Small SD, Sweitzer BJ, Vander Vliet M, et al. ADE Prevention Study Group (1999) Patient risk factors for adverse drug events in hospitalized patients. Arch Intern Med 159:2553–2560. [DOI] [PubMed] [Google Scholar]
- Beaulieu E, Demeule M, Ghitescu L, Béliveau R. (1997) P-glycoprotein is strongly expressed in the luminal membranes of the endothelium of blood vessels in the brain. Biochem J 326:539–544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bergen AA, Plomp AS, Hu X, de Jong PT, Gorgels TG. (2007) ABCC6 and pseudoxanthoma elasticum. Pflugers Arch 453:685–691. [DOI] [PubMed] [Google Scholar]
- Beuers U, Denk GU, Soroka CJ, Wimmer R, Rust C, Paumgartner G, Boyer JL. (2003) Taurolithocholic acid exerts cholestatic effects via phosphatidylinositol 3-kinase-dependent mechanisms in perfused rat livers and rat hepatocyte couplets. J Biol Chem 278:17810–17818. [DOI] [PubMed] [Google Scholar]
- Bigger JT, Jr, Leahey EB., Jr (1982) Quinidine and digoxin. An important interaction. Drugs 24:229–239. [DOI] [PubMed] [Google Scholar]
- Bleasby K, Hall LA, Perry JL, Mohrenweiser HW, Pritchard JB. (2005) Functional consequences of single nucleotide polymorphisms in the human organic anion transporter hOAT1 (SLC22A6). J Pharmacol Exp Ther 314:923–931. [DOI] [PubMed] [Google Scholar]
- Boehmer C, Palmada M, Klaus F, Jeyaraj S, Lindner R, Laufer J, Daniel H, Lang F. (2008) The peptide transporter PEPT2 is targeted by the protein kinase SGK1 and the scaffold protein NHERF2. Cell Physiol Biochem 22:705–714. [DOI] [PubMed] [Google Scholar]
- Boxberger KH, Hagenbuch B, Lampe JN. (2014) Common drugs inhibit human organic cation transporter 1 (OCT1)-mediated neurotransmitter uptake. Drug Metab Dispos 42:990–995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brandsch M. (2013) Drug transport via the intestinal peptide transporter PepT1. Curr Opin Pharmacol 13:881–887. [DOI] [PubMed] [Google Scholar]
- Burckhardt G. (2012) Drug transport by organic anion transporters (OATs). Pharmacol Ther 136:106–130. [DOI] [PubMed] [Google Scholar]
- Cardone RA, Bellizzi A, Busco G, Weinman EJ, Dell’Aquila ME, Casavola V, Azzariti A, Mangia A, Paradiso A, Reshkin SJ. (2007) The NHERF1 PDZ2 domain regulates PKA-RhoA-p38-mediated NHE1 activation and invasion in breast tumor cells. Mol Biol Cell 18:1768–1780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cascorbi I. (2011) P-glycoprotein: tissue distribution, substrates, and functional consequences of genetic variations. Handb Exp Pharmacol 201:261–283. [DOI] [PubMed] [Google Scholar]
- Cascorbi I, Haenisch S. (2010) Pharmacogenetics of ATP-binding cassette transporters and clinical implications. Methods Mol Biol 596:95–121. [DOI] [PubMed] [Google Scholar]
- Cha SH, Sekine T, Kusuhara H, Yu E, Kim JY, Kim DK, Sugiyama Y, Kanai Y, Endou H. (2000) Molecular cloning and characterization of multispecific organic anion transporter 4 expressed in the placenta. J Biol Chem 275:4507–4512. [DOI] [PubMed] [Google Scholar]
- Chen T. (2010) Overcoming drug resistance by regulating nuclear receptors. Adv Drug Deliv Rev 62:1257–1264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi JH, Murray JW, Wolkoff AW. (2011) PDZK1 binding and serine phosphorylation regulate subcellular trafficking of organic anion transport protein 1a1. Am J Physiol Gastrointest Liver Physiol 300:G384–G393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi YH, Yu AM. (2014) ABC transporters in multidrug resistance and pharmacokinetics, and strategies for drug development. Curr Pharm Des 20:793–807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cole SP. (2014) Multidrug resistance protein 1 (MRP1, ABCC1), a “multitasking” ATP-binding cassette (ABC) transporter. J Biol Chem 289:30880–30888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crean D, Bellwon P, Aschauer L, Limonciel A, Moenks K, Hewitt P, Schmidt T, Herrgen K, Dekant W, Lukas A, et al. (2014) Development of an in vitro renal epithelial disease state model for xenobiotic toxicity testing. Toxicol In Vitro DOI: [published ahead of print]. [DOI] [PubMed] [Google Scholar]
- Custer M, Spindler B, Verrey F, Murer H, Biber J. (1997) Identification of a new gene product (diphor-1) regulated by dietary phosphate. Am J Physiol 273:F801–F806. [DOI] [PubMed] [Google Scholar]
- Daniel H, Adibi SA. (1993) Transport of beta-lactam antibiotics in kidney brush border membrane. Determinants of their affinity for the oligopeptide/H+ symporter. J Clin Invest 92:2215–2223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Lannoy IA, Koren G, Klein J, Charuk J, Silverman M. (1992) Cyclosporin and quinidine inhibition of renal digoxin excretion: evidence for luminal secretion of digoxin. Am J Physiol 263:F613–F622. [DOI] [PubMed] [Google Scholar]
- Devgun MS, El-Nujumi AM, O’Dowd GJ, Barbu V, Poupon R. (2012) Novel mutations in the Dubin-Johnson syndrome gene ABCC2/MRP2 and associated biochemical changes. Ann Clin Biochem 49:609–612. [DOI] [PubMed] [Google Scholar]
- Dietel M, Arps H, Lage H, Niendorf A. (1990) Membrane vesicle formation due to acquired mitoxantrone resistance in human gastric carcinoma cell line EPG85-257. Cancer Res 50:6100–6106. [PubMed] [Google Scholar]
- Donowitz M, Cha B, Zachos NC, Brett CL, Sharma A, Tse CM, Li X. (2005) NHERF family and NHE3 regulation. J Physiol 567:3–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doyle LA, Yang W, Abruzzo LV, Krogmann T, Gao Y, Rishi AK, Ross DD. (1998) A multidrug resistance transporter from human MCF-7 breast cancer cells. Proc Natl Acad Sci USA 95:15665–15670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ekaratanawong S, Anzai N, Jutabha P, Miyazaki H, Noshiro R, Takeda M, Kanai Y, Sophasan S, Endou H. (2004) Human organic anion transporter 4 is a renal apical organic anion/dicarboxylate exchanger in the proximal tubules. J Pharmacol Sci 94:297–304. [DOI] [PubMed] [Google Scholar]
- Erdman AR, Mangravite LM, Urban TJ, Lagpacan LL, Castro RA, de la Cruz M, Chan W, Huang CC, Johns SJ, Kawamoto M, et al. (2006) The human organic anion transporter 3 (OAT3; SLC22A8): genetic variation and functional genomics. Am J Physiol Renal Physiol 290:F905–F912. [DOI] [PubMed] [Google Scholar]
- Fehér Á, Juhász A, László A, Pákáski M, Kálmán J, Janka Z. (2013) Association between the ABCG2 C421A polymorphism and Alzheimer’s disease. Neurosci Lett 550:51–54. [DOI] [PubMed] [Google Scholar]
- Feng W, Zhang M. (2009) Organization and dynamics of PDZ-domain-related supramodules in the postsynaptic density. Nat Rev Neurosci 10:87–99. [DOI] [PubMed] [Google Scholar]
- Fraenzer JT, Pan H, Minimo L, Jr, Smith GM, Knauer D, Hung G. (2003) Overexpression of the NF2 gene inhibits schwannoma cell proliferation through promoting PDGFR degradation. Int J Oncol 23:1493–1500. [PubMed] [Google Scholar]
- Franke RM, Lancaster CS, Peer CJ, Gibson AA, Kosloske AM, Orwick SJ, Mathijssen RH, Figg WD, Baker SD, Sparreboom A. (2011) Effect of ABCC2 (MRP2) transport function on erythromycin metabolism. Clin Pharmacol Ther 89:693–701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fromm MF. (2004) Importance of P-glycoprotein at blood-tissue barriers. Trends Pharmacol Sci 25:423–429. [DOI] [PubMed] [Google Scholar]
- Futscher BW, Abbaszadegan MR, Domann F, Dalton WS. (1994) Analysis of MRP mRNA in mitoxantrone-selected, multidrug-resistant human tumor cells. Biochem Pharmacol 47:1601–1606. [DOI] [PubMed] [Google Scholar]
- Ganapathy ME, Brandsch M, Prasad PD, Ganapathy V, Leibach FH. (1995) Differential recognition of beta -lactam antibiotics by intestinal and renal peptide transporters, PEPT 1 and PEPT 2. J Biol Chem 270:25672–25677. [DOI] [PubMed] [Google Scholar]
- Ganapathy ME, Huang W, Rajan DP, Carter AL, Sugawara M, Iseki K, Leibach FH, Ganapathy V. (2000) beta-lactam antibiotics as substrates for OCTN2, an organic cation/carnitine transporter. J Biol Chem 275:1699–1707. [DOI] [PubMed] [Google Scholar]
- Georgescu MM, Cote G, Agarwal NK, White CL., 3rd (2014) NHERF1/EBP50 controls morphogenesis of 3D colonic glands by stabilizing PTEN and ezrin-radixin-moesin proteins at the apical membrane. Neoplasia 16:365–374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerk PM, Vore M. (2002) Regulation of expression of the multidrug resistance-associated protein 2 (MRP2) and its role in drug disposition. J Pharmacol Exp Ther 302:407–415. [DOI] [PubMed] [Google Scholar]
- Gessman L, Danilo P, Jr, Rosen MR. (1983) An electrophysiologic study of the digoxin—quinidine interaction. J Clin Pharmacol 23:16–23. [DOI] [PubMed] [Google Scholar]
- Giacomini KM, Balimane PV, Cho SK, Eadon M, Edeki T, Hillgren KM, Huang SM, Sugiyama Y, Weitz D, Wen Y, et al. International Transporter Consortium (2013) International Transporter Consortium commentary on clinically important transporter polymorphisms. Clin Pharmacol Ther 94:23–26. [DOI] [PubMed] [Google Scholar]
- Giacomini KM, Huang SM, Tweedie DJ, Benet LZ, Brouwer KL, Chu X, Dahlin A, Evers R, Fischer V, Hillgren KM, et al. International Transporter Consortium (2010) Membrane transporters in drug development. Nat Rev Drug Discov 9:215–236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giral H, Lanzano L, Caldas Y, Blaine J, Verlander JW, Lei T, Gratton E, Levi M. (2011) Role of PDZK1 protein in apical membrane expression of renal sodium-coupled phosphate transporters. J Biol Chem 286:15032–15042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gisler SM, Stagljar I, Traebert M, Bacic D, Biber J, Murer H. (2001) Interaction of the type IIa Na/Pi cotransporter with PDZ proteins. J Biol Chem 276:9206–9213. [DOI] [PubMed] [Google Scholar]
- Gorboulev V, Ulzheimer JC, Akhoundova A, Ulzheimer-Teuber I, Karbach U, Quester S, Baumann C, Lang F, Busch AE, Koepsell H. (1997) Cloning and characterization of two human polyspecific organic cation transporters. DNA Cell Biol 16:871–881. [DOI] [PubMed] [Google Scholar]
- Gottesman MM, Fojo T, Bates SE. (2002) Multidrug resistance in cancer: role of ATP-dependent transporters. Nat Rev Cancer 2:48–58. [DOI] [PubMed] [Google Scholar]
- Graessler J, Graessler A, Unger S, Kopprasch S, Tausche AK, Kuhlisch E, Schroeder HE. (2006) Association of the human urate transporter 1 with reduced renal uric acid excretion and hyperuricemia in a German Caucasian population. Arthritis Rheum 54:292–300. [DOI] [PubMed] [Google Scholar]
- Grinfeld J, Gerrard G, Alikian M, Alonso-Dominguez J, Ale S, Valgañon M, Nteliopoulos G, White D, Marin D, Hedgley C, et al. (2013) A common novel splice variant of SLC22A1 (OCT1) is associated with impaired responses to imatinib in patients with chronic myeloid leukaemia. Br J Haematol 163:631–639. [DOI] [PubMed] [Google Scholar]
- Link E, Parish S, Armitage J, Bowman L, Heath S, Matsuda F, Gut I, Lathrop M, Collins R, SEARCH Collaborative Group (2008) SLCO1B1 variants and statin-induced myopathy—a genomewide study. N Engl J Med 359:789–799. [DOI] [PubMed] [Google Scholar]
- Grube M, Meyer zu Schwabedissen HE, Präger D, Haney J, Möritz KU, Meissner K, Rosskopf D, Eckel L, Böhm M, Jedlitschky G, et al. (2006) Uptake of cardiovascular drugs into the human heart: expression, regulation, and function of the carnitine transporter OCTN2 (SLC22A5). Circulation 113:1114–1122. [DOI] [PubMed] [Google Scholar]
- Gründemann D, Harlfinger S, Golz S, Geerts A, Lazar A, Berkels R, Jung N, Rubbert A, Schömig E. (2005) Discovery of the ergothioneine transporter. Proc Natl Acad Sci USA 102:5256–5261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamman JH, Enslin GM, Kotzé AF. (2005) Oral delivery of peptide drugs: barriers and developments. BioDrugs 19:165–177. [DOI] [PubMed] [Google Scholar]
- Harmsen S, Meijerman I, Beijnen JH, Schellens JH. (2009) Nuclear receptor mediated induction of cytochrome P450 3A4 by anticancer drugs: a key role for the pregnane X receptor. Cancer Chemother Pharmacol 64:35–43. [DOI] [PubMed] [Google Scholar]
- Harmsen S, Meijerman I, Febus CL, Maas-Bakker RF, Beijnen JH, Schellens JH. (2010) PXR-mediated induction of P-glycoprotein by anticancer drugs in a human colon adenocarcinoma-derived cell line. Cancer Chemother Pharmacol 66:765–771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hayer-Zillgen M, Brüss M, Bönisch H. (2002) Expression and pharmacological profile of the human organic cation transporters hOCT1, hOCT2 and hOCT3. Br J Pharmacol 136:829–836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He XJ, Wang WR, Zhang Y, Yang Q. (2012) The effect of radixin knockdown on the expression and efflux function of MRP2 in SGC-7901 cells. Eur J Pharm Sci 46:426–434. [DOI] [PubMed] [Google Scholar]
- Hegedüs T, Sessler T, Scott R, Thelin W, Bakos E, Váradi A, Szabó K, Homolya L, Milgram SL, Sarkadi B. (2003) C-terminal phosphorylation of MRP2 modulates its interaction with PDZ proteins. Biochem Biophys Res Commun 302:454–461. [DOI] [PubMed] [Google Scholar]
- Herraez E, Gonzalez-Sanchez E, Vaquero J, Romero MR, Serrano MA, Marin JJ, Briz O. (2012) Cisplatin-induced chemoresistance in colon cancer cells involves FXR-dependent and FXR-independent up-regulation of ABC proteins. Mol Pharm 9:2565–2576. [DOI] [PubMed] [Google Scholar]
- Hillgren KM, Keppler D, Zur AA, Giacomini KM, Stieger B, Cass CE, Zhang L, International Transporter Consortium (2013) Emerging transporters of clinical importance: an update from the International Transporter Consortium. Clin Pharmacol Ther 94:52–63. [DOI] [PubMed] [Google Scholar]
- Hirano M, Maeda K, Shitara Y, Sugiyama Y. (2006) Drug-drug interaction between pitavastatin and various drugs via OATP1B1. Drug Metab Dispos 34:1229–1236. [DOI] [PubMed] [Google Scholar]
- Hirano T, Yasuda S, Osaka Y, Asari M, Kobayashi M, Itagaki S, Iseki K. (2008) The inhibitory effects of fluoroquinolones on L-carnitine transport in placental cell line BeWo. Int J Pharm 351:113–118. [DOI] [PubMed] [Google Scholar]
- Hoque MT, Cole SP. (2008) Down-regulation of Na+/H+ exchanger regulatory factor 1 increases expression and function of multidrug resistance protein 4. Cancer Res 68:4802–4809. [DOI] [PubMed] [Google Scholar]
- Hoque MT, Conseil G, Cole SP. (2009) Involvement of NHERF1 in apical membrane localization of MRP4 in polarized kidney cells. Biochem Biophys Res Commun 379:60–64. [DOI] [PubMed] [Google Scholar]
- Howton JC. (2006) Probenecid with oseltamivir for human influenza A (H5N1) virus infection? N Engl J Med 354:879–880. [DOI] [PubMed] [Google Scholar]
- Imaoka T, Kusuhara H, Adachi M, Schuetz JD, Takeuchi K, Sugiyama Y. (2007) Functional involvement of multidrug resistance-associated protein 4 (MRP4/ABCC4) in the renal elimination of the antiviral drugs adefovir and tenofovir. Mol Pharmacol 71:619–627. [DOI] [PubMed] [Google Scholar]
- Inui K, Terada T, Masuda S, Saito H. (2000) Physiological and pharmacological implications of peptide transporters, PEPT1 and PEPT2. Nephrol Dial Transplant 15 (Suppl 6):11–13. [DOI] [PubMed] [Google Scholar]
- Ito K, Wakabayashi T, Horie T. (2005) Mrp2/Abcc2 transport activity is stimulated by protein kinase Calpha in a baculo virus co-expression system. Life Sci 77:539–550. [DOI] [PubMed] [Google Scholar]
- Iwata K, Aizawa K, Kamitsu S, Jingami S, Fukunaga E, Yoshida M, Yoshimura M, Hamada A, Saito H. (2012) Effects of genetic variants in SLC22A2 organic cation transporter 2 and SLC47A1 multidrug and toxin extrusion 1 transporter on cisplatin-induced adverse events. Clin Exp Nephrol 16:843–851. [DOI] [PubMed] [Google Scholar]
- Izzedine H, Launay-Vacher V, Deray G. (2005) Antiviral drug-induced nephrotoxicity. Am J Kidney Dis 45:804–817. [DOI] [PubMed] [Google Scholar]
- Jacobson MA. (1993) Valaciclovir (BW256U87): the L-valyl ester of acyclovir. J Med Virol (Suppl 1):150–153. [DOI] [PubMed] [Google Scholar]
- Jani M, Ambrus C, Magnan R, Jakab KT, Beéry E, Zolnerciks JK, Krajcsi P. (2014) Structure and function of BCRP, a broad specificity transporter of xenobiotics and endobiotics. Arch Toxicol 88:1205–1248. [DOI] [PubMed] [Google Scholar]
- Ji L, Masuda S, Saito H, Inui K. (2002) Down-regulation of rat organic cation transporter rOCT2 by 5/6 nephrectomy. Kidney Int 62:514–524. [DOI] [PubMed] [Google Scholar]
- Jong NN, Nakanishi T, Liu JJ, Tamai I, McKeage MJ. (2011) Oxaliplatin transport mediated by organic cation/carnitine transporters OCTN1 and OCTN2 in overexpressing human embryonic kidney 293 cells and rat dorsal root ganglion neurons. J Pharmacol Exp Ther 338:537–547. [DOI] [PubMed] [Google Scholar]
- Juliano RL, Ling V. (1976) A surface glycoprotein modulating drug permeability in Chinese hamster ovary cell mutants. Biochim Biophys Acta 455:152–162. [DOI] [PubMed] [Google Scholar]
- Kage K, Fujita T, Sugimoto Y. (2005) Role of Cys-603 in dimer/oligomer formation of the breast cancer resistance protein BCRP/ABCG2. Cancer Sci 96:866–872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kano T, Kato Y, Ito K, Ogihara T, Kubo Y, Tsuji A. (2009) Carnitine/organic cation transporter OCTN2 (Slc22a5) is responsible for renal secretion of cephaloridine in mice. Drug Metab Dispos 37:1009–1016. [DOI] [PubMed] [Google Scholar]
- Karim Z, Gérard B, Bakouh N, Alili R, Leroy C, Beck L, Silve C, Planelles G, Urena-Torres P, Grandchamp B, et al. (2008) NHERF1 mutations and responsiveness of renal parathyroid hormone. N Engl J Med 359:1128–1135. [DOI] [PubMed] [Google Scholar]
- Kartenbeck J, Leuschner U, Mayer R, Keppler D. (1996) Absence of the canalicular isoform of the MRP gene-encoded conjugate export pump from the hepatocytes in Dubin-Johnson syndrome. Hepatology 23:1061–1066. [DOI] [PubMed] [Google Scholar]
- Karthikeyan S, Hoti SL. (2015) Development of fourth generation ABC inhibitors from natural products: a novel approach to overcome cancer multidrug resistance. Anticancer Agents Med Chem 15:605–615. [DOI] [PubMed] [Google Scholar]
- Karthikeyan S, Leung T, Birrane G, Webster G, Ladias JA. (2001) Crystal structure of the PDZ1 domain of human Na(+)/H(+) exchanger regulatory factor provides insights into the mechanism of carboxyl-terminal leucine recognition by class I PDZ domains. J Mol Biol 308:963–973. [DOI] [PubMed] [Google Scholar]
- Karvar S, Suda J, Zhu L, Rockey DC. (2014) Distribution dynamics and functional importance of NHERF1 in regulation of Mrp-2 trafficking in hepatocytes. Am J Physiol Cell Physiol 307:C727–C737. [DOI] [PubMed] [Google Scholar]
- Kathawala RJ, Gupta P, Ashby CR, Jr, Chen ZS. (2015) The modulation of ABC transporter-mediated multidrug resistance in cancer: a review of the past decade. Drug Resist Updat 18:1–17. [DOI] [PubMed] [Google Scholar]
- Kato Y, Sai Y, Yoshida K, Watanabe C, Hirata T, Tsuji A. (2005) PDZK1 directly regulates the function of organic cation/carnitine transporter OCTN2. Mol Pharmacol 67:734–743. [DOI] [PubMed] [Google Scholar]
- Kato Y, Watanabe C, Tsuji A. (2006) Regulation of drug transporters by PDZ adaptor proteins and nuclear receptors. Eur J Pharm Sci 27:487–500. [DOI] [PubMed] [Google Scholar]
- Kato Y, Yoshida K, Watanabe C, Sai Y, Tsuji A. (2004) Screening of the interaction between xenobiotic transporters and PDZ proteins. Pharm Res 21:1886–1894. [DOI] [PubMed] [Google Scholar]
- Kellner U, Hutchinson L, Seidel A, Lage H, Danks MK, Dietel M, Kaufmann SH. (1997) Decreased drug accumulation in a mitoxantrone-resistant gastric carcinoma cell line in the absence of P-glycoprotein. Int J Cancer 71:817–824. [DOI] [PubMed] [Google Scholar]
- Keppler D. (2011) Multidrug resistance proteins (MRPs, ABCCs): importance for pathophysiology and drug therapy. Handb Exp Pharmacol 201:299–323. [DOI] [PubMed] [Google Scholar]
- Keppler D. (2014) The roles of MRP2, MRP3, OATP1B1, and OATP1B3 in conjugated hyperbilirubinemia. Drug Metab Dispos 42:561–565. [DOI] [PubMed] [Google Scholar]
- Kikuchi S, Hata M, Fukumoto K, Yamane Y, Matsui T, Tamura A, Yonemura S, Yamagishi H, Keppler D, Tsukita S, et al. (2002) Radixin deficiency causes conjugated hyperbilirubinemia with loss of Mrp2 from bile canalicular membranes. Nat Genet 31:320–325. [DOI] [PubMed] [Google Scholar]
- Kim JK, Jeon HY, Kim H. (2015) The molecular mechanisms underlying the therapeutic resistance of cancer stem cells. Arch Pharm Res 38:389–401. [DOI] [PubMed] [Google Scholar]
- Klenk C, Vetter T, Zürn A, Vilardaga JP, Friedman PA, Wang B, Lohse MJ. (2010) Formation of a ternary complex among NHERF1, beta-arrestin, and parathyroid hormone receptor. J Biol Chem 285:30355–30362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kobayashi D, Nozawa T, Imai K, Nezu J, Tsuji A, Tamai I. (2003) Involvement of human organic anion transporting polypeptide OATP-B (SLC21A9) in pH-dependent transport across intestinal apical membrane. J Pharmacol Exp Ther 306:703–708. [DOI] [PubMed] [Google Scholar]
- Kocher O, Comella N, Gilchrist A, Pal R, Tognazzi K, Brown LF, Knoll JH. (1999) PDZK1, a novel PDZ domain-containing protein up-regulated in carcinomas and mapped to chromosome 1q21, interacts with cMOAT (MRP2), the multidrug resistance-associated protein. Lab Invest 79:1161–1170. [PubMed] [Google Scholar]
- Kocher O, Comella N, Tognazzi K, Brown LF. (1998) Identification and partial characterization of PDZK1: a novel protein containing PDZ interaction domains. Lab Invest 78:117–125. [PubMed] [Google Scholar]
- Kocher O, Pal R, Roberts M, Cirovic C, Gilchrist A. (2003) Targeted disruption of the PDZK1 gene by homologous recombination. Mol Cell Biol 23:1175–1180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kojima H, Sakurai S, Yoshiji H, Uemura M, Yoshikawa M, Fukui H. (2008) The role of radixin in altered localization of canalicular conjugate export pump Mrp2 in cholestatic rat liver. Hepatol Res 38:202–210. [DOI] [PubMed] [Google Scholar]
- König J, Müller F, Fromm MF. (2013) Transporters and drug-drug interactions: important determinants of drug disposition and effects. Pharmacol Rev 65:944–966. [DOI] [PubMed] [Google Scholar]
- Kool M, de Haas M, Scheffer GL, Scheper RJ, van Eijk MJ, Juijn JA, Baas F, Borst P. (1997) Analysis of expression of cMOAT (MRP2), MRP3, MRP4, and MRP5, homologues of the multidrug resistance-associated protein gene (MRP1), in human cancer cell lines. Cancer Res 57:3537–3547. [PubMed] [Google Scholar]
- Koren-Michowitz M, Buzaglo Z, Ribakovsky E, Schwarz M, Pessach I, Shimoni A, Beider K, Amariglio N, le Coutre P, Nagler A. (2014) OCT1 genetic variants are associated with long term outcomes in imatinib treated chronic myeloid leukemia patients. Eur J Haematol 92:283–288. [DOI] [PubMed] [Google Scholar]
- Kullak-Ublick GA, Fisch T, Oswald M, Hagenbuch B, Meier PJ, Beuers U, Paumgartner G. (1998) Dehydroepiandrosterone sulfate (DHEAS): identification of a carrier protein in human liver and brain. FEBS Lett 424:173–176. [DOI] [PubMed] [Google Scholar]
- Kullak-Ublick GA, Ismair MG, Stieger B, Landmann L, Huber R, Pizzagalli F, Fattinger K, Meier PJ, Hagenbuch B. (2001) Organic anion-transporting polypeptide B (OATP-B) and its functional comparison with three other OATPs of human liver. Gastroenterology 120:525–533. [DOI] [PubMed] [Google Scholar]
- Kunická T, Souček P. (2014) Importance of ABCC1 for cancer therapy and prognosis. Drug Metab Rev 46:325–342. [DOI] [PubMed] [Google Scholar]
- Lancaster CS, Bruun GH, Peer CJ, Mikkelsen TS, Corydon TJ, Gibson AA, Hu S, Orwick SJ, Mathijssen RH, Figg WD, et al. (2012) OATP1B1 polymorphism as a determinant of erythromycin disposition. Clin Pharmacol Ther 92:642–650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laouari D, Yang R, Veau C, Blanke I, Friedlander G. (2001) Two apical multidrug transporters, P-gp and MRP2, are differently altered in chronic renal failure. Am J Physiol Renal Physiol 280:F636–F645. [DOI] [PubMed] [Google Scholar]
- Lau AG, Hall RA. (2001) Oligomerization of NHERF-1 and NHERF-2 PDZ domains: differential regulation by association with receptor carboxyl-termini and by phosphorylation. Biochemistry 40:8572–8580. [DOI] [PubMed] [Google Scholar]
- Lee CA, O'Connor MA, Ritchie TK, Galetin A, Cook JA, Ragueneau-Majlessi I, Ellens H, Feng B, Taub ME, Paine MF, et al. (2015) Breast cancer resistance protein (ABCG2) in clinical pharmacokinetics and drug interactions: practical recommendations for clinical victim and perpetrator drug-drug interaction study design. Drug Metab Dispos 43:490–509. [DOI] [PubMed] [Google Scholar]
- Lee W, Glaeser H, Smith LH, Roberts RL, Moeckel GW, Gervasini G, Leake BF, Kim RB. (2005) Polymorphisms in human organic anion-transporting polypeptide 1A2 (OATP1A2): implications for altered drug disposition and central nervous system drug entry. J Biol Chem 280:9610–9617. [DOI] [PubMed] [Google Scholar]
- Leibach FH, Ganapathy V. (1996) Peptide transporters in the intestine and the kidney. Annu Rev Nutr 16:99–119. [DOI] [PubMed] [Google Scholar]
- Li M, Wang W, Soroka CJ, Mennone A, Harry K, Weinman EJ, Boyer JL. (2010) NHERF-1 binds to Mrp2 and regulates hepatic Mrp2 expression and function. J Biol Chem 285:19299–19307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Q, Jiang Q, Pfendner E, Váradi A, Uitto J. (2009) Pseudoxanthoma elasticum: clinical phenotypes, molecular genetics and putative pathomechanisms. Exp Dermatol 18:1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liang R, Fei YJ, Prasad PD, Ramamoorthy S, Han H, Yang-Feng TL, Hediger MA, Ganapathy V, Leibach FH. (1995) Human intestinal H+/peptide cotransporter. Cloning, functional expression, and chromosomal localization. J Biol Chem 270:6456–6463. [DOI] [PubMed] [Google Scholar]
- Lin CJ, Akarawut W, Smith DE. (1999) Competitive inhibition of glycylsarcosine transport by enalapril in rabbit renal brush border membrane vesicles: interaction of ACE inhibitors with high-affinity H+/peptide symporter. Pharm Res 16:609–615. [DOI] [PubMed] [Google Scholar]
- Mahon MJ, Segre GV. (2004) Stimulation by parathyroid hormone of a NHERF-1-assembled complex consisting of the parathyroid hormone I receptor, phospholipase Cbeta, and actin increases intracellular calcium in opossum kidney cells. J Biol Chem 279:23550–23558. [DOI] [PubMed] [Google Scholar]
- Mahrooz A, Parsanasab H, Hashemi-Soteh MB, Kashi Z, Bahar A, Alizadeh A, Mozayeni M. (2015) The role of clinical response to metformin in patients newly diagnosed with type 2 diabetes: a monotherapy study. Clin Exp Med 15:159–165. [DOI] [PubMed] [Google Scholar]
- Majumdar S, Mitra AK. (2006) Chemical modification and formulation approaches to elevated drug transport across cell membranes. Expert Opin Drug Deliv 3:511–527. [DOI] [PubMed] [Google Scholar]
- Manceau S, Giraud C, Declèves X, Batteux F, Chéreau C, Chouzenoux S, Scherrmann JM, Weill B, Perrot JY, Tréluyer JM. (2012) Expression and induction by dexamethasone of ABC transporters and nuclear receptors in a human T-lymphocyte cell line. J Chemother 24:48–55. [DOI] [PubMed] [Google Scholar]
- Mandal AK, Mount DB. (2015) The molecular physiology of uric acid homeostasis. Annu Rev Physiol 77:323–345. [DOI] [PubMed] [Google Scholar]
- Mao Q, Unadkat JD. (2015) Role of the breast cancer resistance protein (BCRP/ABCG2) in drug transport—an update. AAPS J 17:65–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martini M, Ferrara AM, Giachelia M, Panieri E, Siminovitch K, Galeotti T, Larocca LM, Pani G. (2012) Association of the OCTN1/1672T variant with increased risk for colorectal cancer in young individuals and ulcerative colitis patients. Inflamm Bowel Dis 18:439–448. [DOI] [PubMed] [Google Scholar]
- Masereeuw R, Mutsaers HA, Toyohara T, Abe T, Jhawar S, Sweet DH, Lowenstein J. (2014) The kidney and uremic toxin removal: glomerulus or tubule? Semin Nephrol 34:191–208. [DOI] [PubMed] [Google Scholar]
- Massy ZA. (2014) The role of lipids and uremic toxins in cardiovascular disease in CKD. Clin Exp Nephrol 18:255–256. [DOI] [PubMed] [Google Scholar]
- Matsuo H, Takada T, Ichida K, Nakamura T, Nakayama A, Suzuki H, Hosoya T, Shinomiya N. (2011a) ABCG2/BCRP dysfunction as a major cause of gout. Nucleosides Nucleotides Nucleic Acids 30:1117–1128. [DOI] [PubMed] [Google Scholar]
- Matsuo H, Takada T, Ichida K, Nakamura T, Nakayama A, Takada Y, Okada C, Sakurai Y, Hosoya T, Kanai Y, et al. (2011b) Identification of ABCG2 dysfunction as a major factor contributing to gout. Nucleosides Nucleotides Nucleic Acids 30:1098–1104. [DOI] [PubMed] [Google Scholar]
- Matzke GR, Frye RF. (1997) Drug administration in patients with renal insufficiency. Minimising renal and extrarenal toxicity. Drug Saf 16:205–231. [DOI] [PubMed] [Google Scholar]
- Mayerl S, Müller J, Bauer R, Richert S, Kassmann CM, Darras VM, Buder K, Boelen A, Visser TJ, Heuer H. (2014) Transporters MCT8 and OATP1C1 maintain murine brain thyroid hormone homeostasis. J Clin Invest 124:1987–1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meyer zu Schwabedissen HE, Albers M, Baumeister SE, Rimmbach C, Nauck M, Wallaschofski H, Siegmund W, Völzke H, Kroemer HK. (2015) Function-impairing polymorphisms of the hepatic uptake transporter SLCO1B1 modify the therapeutic efficacy of statins in a population-based cohort. Pharmacogenet Genomics 25:8–18. [DOI] [PubMed] [Google Scholar]
- Miyazaki H, Anzai N, Ekaratanawong S, Sakata T, Shin HJ, Jutabha P, Hirata T, He X, Nonoguchi H, Tomita K, et al. (2005) Modulation of renal apical organic anion transporter 4 function by two PDZ domain-containing proteins. J Am Soc Nephrol 16:3498–3506. [DOI] [PubMed] [Google Scholar]
- Mizuno T, Fukudo M, Terada T, Kamba T, Nakamura E, Ogawa O, Inui K, Katsura T. (2012) Impact of genetic variation in breast cancer resistance protein (BCRP/ABCG2) on sunitinib pharmacokinetics. Drug Metab Pharmacokinet 27:631–639. [DOI] [PubMed] [Google Scholar]
- Moon C, Zhang W, Ren A, Arora K, Sinha C, Yarlagadda S, Woodrooffe K, Schuetz JD, Valasani KR, de Jonge HR, et al. (2015) Compartmentalized Accumulation of cAMP near Complexes of Multidrug Resistance Protein 4 (MRP4) and Cystic Fibrosis Transmembrane Conductance Regulator (CFTR) Contributes to Drug-induced Diarrhea. J Biol Chem 290:11246–11257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morales FC, Takahashi Y, Momin S, Adams H, Chen X, Georgescu MM. (2007) NHERF1/EBP50 head-to-tail intramolecular interaction masks association with PDZ domain ligands. Mol Cell Biol 27:2527–2537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Müller J, Lips KS, Metzner L, Neubert RH, Koepsell H, Brandsch M. (2005) Drug specificity and intestinal membrane localization of human organic cation transporters (OCT). Biochem Pharmacol 70:1851–1860. [DOI] [PubMed] [Google Scholar]
- Muoio DM, Noland RC, Kovalik JP, Seiler SE, Davies MN, DeBalsi KL, Ilkayeva OR, Stevens RD, Kheterpal I, Zhang J, et al. (2012) Muscle-specific deletion of carnitine acetyltransferase compromises glucose tolerance and metabolic flexibility. Cell Metab 15:764–777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakagawa M, Schneider E, Dixon KH, Horton J, Kelley K, Morrow C, Cowan KH. (1992) Reduced intracellular drug accumulation in the absence of P-glycoprotein (mdr1) overexpression in mitoxantrone-resistant human MCF-7 breast cancer cells. Cancer Res 52:6175–6181. [PubMed] [Google Scholar]
- Nakamura T, Nakanishi T, Haruta T, Shirasaka Y, Keogh JP, Tamai I. (2010) Transport of ipratropium, an anti-chronic obstructive pulmonary disease drug, is mediated by organic cation/carnitine transporters in human bronchial epithelial cells: implications for carrier-mediated pulmonary absorption. Mol Pharm 7:187–195. [DOI] [PubMed] [Google Scholar]
- Naud J, Michaud J, Boisvert C, Desbiens K, Leblond FA, Mitchell A, Jones C, Bonnardeaux A, Pichette V. (2007) Down-regulation of intestinal drug transporters in chronic renal failure in rats. J Pharmacol Exp Ther 320:978–985. [DOI] [PubMed] [Google Scholar]
- Naud J, Michaud J, Leblond FA, Lefrancois S, Bonnardeaux A, Pichette V. (2008) Effects of chronic renal failure on liver drug transporters. Drug Metab Dispos 36:124–128. [DOI] [PubMed] [Google Scholar]
- Nezu J, Tamai I, Oku A, Ohashi R, Yabuuchi H, Hashimoto N, Nikaido H, Sai Y, Koizumi A, Shoji Y, et al. (1999) Primary systemic carnitine deficiency is caused by mutations in a gene encoding sodium ion-dependent carnitine transporter. Nat Genet 21:91–94. [DOI] [PubMed] [Google Scholar]
- Ni Z, Mark ME, Cai X, Mao Q. (2010) Fluorescence resonance energy transfer (FRET) analysis demonstrates dimer/oligomer formation of the human breast cancer resistance protein (BCRP/ABCG2) in intact cells. Int J Biochem Mol Biol 1:1–11. [PMC free article] [PubMed] [Google Scholar]
- Nielsen AL, Henriksen DP, Marinakis C, Hellebek A, Birn H, Nybo M, Søndergaard J, Nymark A, Pedersen C. (2014) Drug dosing in patients with renal insufficiency in a hospital setting using electronic prescribing and automated reporting of estimated glomerular filtration rate. Basic Clin Pharmacol Toxicol 114:407–413. [DOI] [PubMed] [Google Scholar]
- Niemi M, Pasanen MK, Neuvonen PJ. (2011) Organic anion transporting polypeptide 1B1: a genetically polymorphic transporter of major importance for hepatic drug uptake. Pharmacol Rev 63:157–181. [DOI] [PubMed] [Google Scholar]
- Nies AT, Koepsell H, Winter S, Burk O, Klein K, Kerb R, Zanger UM, Keppler D, Schwab M, Schaeffeler E. (2009) Expression of organic cation transporters OCT1 (SLC22A1) and OCT3 (SLC22A3) is affected by genetic factors and cholestasis in human liver. Hepatology 50:1227–1240. [DOI] [PubMed] [Google Scholar]
- Nigam SK. (2015) What do drug transporters really do? Nat Rev Drug Discov 14:29–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nigam SK, Bush KT, Martovetsky G, Ahn SY, Liu HC, Richard E, Bhatnagar V, Wu W. (2015) The organic anion transporter (OAT) family: a systems biology perspective. Physiol Rev 95:83–123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nolin TD, Frye RF, Le P, Sadr H, Naud J, Leblond FA, Pichette V, Himmelfarb J. (2009) ESRD impairs nonrenal clearance of fexofenadine but not midazolam. J Am Soc Nephrol 20:2269–2276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nolin TD, Unruh ML. (2010) Clinical relevance of impaired nonrenal drug clearance in ESRD. Semin Dial 23:482–485. [DOI] [PubMed] [Google Scholar]
- Noshiro R, Anzai N, Sakata T, Miyazaki H, Terada T, Shin HJ, He X, Miura D, Inui K, Kanai Y, et al. (2006) The PDZ domain protein PDZK1 interacts with human peptide transporter PEPT2 and enhances its transport activity. Kidney Int 70:275–282. [DOI] [PubMed] [Google Scholar]
- Nozawa T, Imai K, Nezu J, Tsuji A, Tamai I. (2004) Functional characterization of pH-sensitive organic anion transporting polypeptide OATP-B in human. J Pharmacol Exp Ther 308:438–445. [DOI] [PubMed] [Google Scholar]
- Oda K, Nishimura T, Higuchi K, Ishido N, Ochi K, Iizasa H, Sai Y, Tomi M, Nakashima E. (2013) Estrogen receptor α induction by mitoxantrone increases Abcg2 expression in placental trophoblast cells. J Pharm Sci 102:3364–3372. [DOI] [PubMed] [Google Scholar]
- Oh ES, Kim CO, Cho SK, Park MS, Chung JY. (2013) Impact of ABCC2, ABCG2 and SLCO1B1 polymorphisms on the pharmacokinetics of pitavastatin in humans. Drug Metab Pharmacokinet 28:196–202. [DOI] [PubMed] [Google Scholar]
- Ohashi R, Tamai I, Nezu Ji J, Nikaido H, Hashimoto N, Oku A, Sai Y, Shimane M, Tsuji A. (2001) Molecular and physiological evidence for multifunctionality of carnitine/organic cation transporter OCTN2. Mol Pharmacol 59:358–366. [DOI] [PubMed] [Google Scholar]
- Okabe M, Szakács G, Reimers MA, Suzuki T, Hall MD, Abe T, Weinstein JN, Gottesman MM. (2008) Profiling SLCO and SLC22 genes in the NCI-60 cancer cell lines to identify drug uptake transporters. Mol Cancer Ther 7:3081–3091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okuhira K, Fitzgerald ML, Tamehiro N, Ohoka N, Suzuki K, Sawada J, Naito M, Nishimaki-Mogami T. (2010) Binding of PDZ-RhoGEF to ATP-binding cassette transporter A1 (ABCA1) induces cholesterol efflux through RhoA activation and prevention of transporter degradation. J Biol Chem 285:16369–16377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Padányi R, Xiong Y, Antalffy G, Lór K, Pászty K, Strehler EE, Enyedi A. (2010) Apical scaffolding protein NHERF2 modulates the localization of alternatively spliced plasma membrane Ca2+ pump 2B variants in polarized epithelial cells. J Biol Chem 285:31704–31712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Padowski JM, Pollack GM. (2010) Pharmacokinetic and pharmacodynamic implications of P-glycoprotein modulation. Methods Mol Biol 596:359–384. [DOI] [PubMed] [Google Scholar]
- Park J, Kwak JO, Riederer B, Seidler U, Cole SP, Lee HJ, Lee MG. (2014) Na⁺/H⁺ exchanger regulatory factor 3 is critical for multidrug resistance protein 4-mediated drug efflux in the kidney. J Am Soc Nephrol 25:726–736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park SW, Schonhoff CM, Webster CR, Anwer MS. (2012) Protein kinase Cδ differentially regulates cAMP-dependent translocation of NTCP and MRP2 to the plasma membrane. Am J Physiol Gastrointest Liver Physiol 303:G657–G665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peltekova VD, Wintle RF, Rubin LA, Amos CI, Huang Q, Gu X, Newman B, Van Oene M, Cescon D, Greenberg G, et al. (2004) Functional variants of OCTN cation transporter genes are associated with Crohn disease. Nat Genet 36:471–475. [DOI] [PubMed] [Google Scholar]
- Pochini L, Scalise M, Galluccio M, Indiveri C. (2013) OCTN cation transporters in health and disease: role as drug targets and assay development. J Biomol Screen 18:851–867. [DOI] [PubMed] [Google Scholar]
- Ponting CP, Phillips C, Davies KE, Blake DJ. (1997) PDZ domains: targeting signalling molecules to sub-membranous sites. BioEssays 19:469–479. [DOI] [PubMed] [Google Scholar]
- Prueksaritanont T, Chu X, Evers R, Klopfer SO, Caro L, Kothare PA, Dempsey C, Rasmussen S, Houle R, Chan G, et al. (2014) Pitavastatin is a more sensitive and selective organic anion-transporting polypeptide 1B clinical probe than rosuvastatin. Br J Clin Pharmacol 78:587–598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reczek D, Berryman M, Bretscher A. (1997) Identification of EBP50: A PDZ-containing phosphoprotein that associates with members of the ezrin-radixin-moesin family. J Cell Biol 139:169–179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reyes M, Benet LZ. (2011) Effects of uremic toxins on transport and metabolism of different biopharmaceutics drug disposition classification system xenobiotics. J Pharm Sci 100:3831–3842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rivory LP, Slaviero KA, Hoskins JM, Clarke SJ. (2001) The erythromycin breath test for the prediction of drug clearance. Clin Pharmacokinet 40:151–158. [DOI] [PubMed] [Google Scholar]
- Romaine SP, Bailey KM, Hall AS, Balmforth AJ. (2010) The influence of SLCO1B1 (OATP1B1) gene polymorphisms on response to statin therapy. Pharmacogenomics J 10:1–11. [DOI] [PubMed] [Google Scholar]
- Rost D, Kloeters-Plachky P, Stiehl A. (2008) Retrieval of the rat canalicular conjugate export pump Mrp2 is associated with a rearrangement of actin filaments and radixin in bile salt-induced cholestasis. Eur J Med Res 13:314–318. [PubMed] [Google Scholar]
- Rubio-Aliaga I, Frey I, Boll M, Groneberg DA, Eichinger HM, Balling R, Daniel H. (2003) Targeted disruption of the peptide transporter Pept2 gene in mice defines its physiological role in the kidney. Mol Cell Biol 23:3247–3252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saeki J, Sekine S, Horie T. (2011) LPS-induced dissociation of multidrug resistance-associated protein 2 (Mrp2) and radixin is associated with Mrp2 selective internalization in rats. Biochem Pharmacol 81:178–184. [DOI] [PubMed] [Google Scholar]
- Saito H, Terada T, Okuda M, Sasaki S, Inui K. (1996) Molecular cloning and tissue distribution of rat peptide transporter PEPT2. Biochim Biophys Acta 1280:173–177. [DOI] [PubMed] [Google Scholar]
- Sakiyama M, Matsuo H, Shimizu S, Nakashima H, Nakayama A, Chiba T, Naito M, Takada T, Suzuki H, Hamajima N, et al. (2014) A common variant of organic anion transporter 4 (OAT4/SLC22A11) gene is associated with renal underexcretion type gout. Drug Metab Pharmacokinet 29:208–210. [DOI] [PubMed] [Google Scholar]
- Sato T, Yamaguchi H, Kogawa T, Abe T, Mano N. (2014) Organic anion transporting polypeptides 1B1 and 1B3 play an important role in uremic toxin handling and drug-uremic toxin interactions in the liver. J Pharm Pharm Sci 17:475–484. [DOI] [PubMed] [Google Scholar]
- Schonhoff CM, Gillin H, Webster CR, Anwer MS. (2008) Protein kinase Cdelta mediates cyclic adenosine monophosphate-stimulated translocation of sodium taurocholate cotransporting polypeptide and multidrug resistant associated protein 2 in rat hepatocytes. Hepatology 47:1309–1316. [DOI] [PubMed] [Google Scholar]
- Seidler U, Singh AK, Cinar A, Chen M, Hillesheim J, Hogema B, Riederer B. (2009) The role of the NHERF family of PDZ scaffolding proteins in the regulation of salt and water transport. Ann N Y Acad Sci 1165:249–260. [DOI] [PubMed] [Google Scholar]
- Shenolikar S, Minkoff CM, Steplock DA, Evangelista C, Liu M, Weinman EJ. (2001) N-terminal PDZ domain is required for NHERF dimerization. FEBS Lett 489:233–236. [DOI] [PubMed] [Google Scholar]
- Shenolikar S, Voltz JW, Minkoff CM, Wade JB, Weinman EJ. (2002) Targeted disruption of the mouse NHERF-1 gene promotes internalization of proximal tubule sodium-phosphate cotransporter type IIa and renal phosphate wasting. Proc Natl Acad Sci USA 99:11470–11475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shenolikar S, Weinman EJ. (2001) NHERF: targeting and trafficking membrane proteins. Am J Physiol Renal Physiol 280:F389–F395. [DOI] [PubMed] [Google Scholar]
- Shibata T, Chuma M, Kokubu A, Sakamoto M, Hirohashi S. (2003) EBP50, a beta-catenin-associating protein, enhances Wnt signaling and is over-expressed in hepatocellular carcinoma. Hepatology 38:178–186. [DOI] [PubMed] [Google Scholar]
- Shigeta J, Katayama K, Mitsuhashi J, Noguchi K, Sugimoto Y. (2010) BCRP/ABCG2 confers anticancer drug resistance without covalent dimerization. Cancer Sci 101:1813–1821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shimizu T, Sugiura T, Wakayama T, Kijima A, Nakamichi N, Iseki S, Silver DL, Kato Y. (2011) PDZK1 regulates breast cancer resistance protein in small intestine. Drug Metab Dispos 39:2148–2154. [DOI] [PubMed] [Google Scholar]
- Shitara Y, Maeda K, Ikejiri K, Yoshida K, Horie T, Sugiyama Y. (2013) Clinical significance of organic anion transporting polypeptides (OATPs) in drug disposition: their roles in hepatic clearance and intestinal absorption. Biopharm Drug Dispos 34:45–78. [DOI] [PubMed] [Google Scholar]
- Shu C, Shen H, Hopfer U, Smith DE. (2001) Mechanism of intestinal absorption and renal reabsorption of an orally active ace inhibitor: uptake and transport of fosinopril in cell cultures. Drug Metab Dispos 29:1307–1315. [PubMed] [Google Scholar]
- Sinha C, Ren A, Arora K, Moon CS, Yarlagadda S, Zhang W, Cheepala SB, Schuetz JD, Naren AP. (2013) Multi-drug resistance protein 4 (MRP4)-mediated regulation of fibroblast cell migration reflects a dichotomous role of intracellular cyclic nucleotides. J Biol Chem 288:3786–3794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song J, Bai J, Yang W, Gabrielson EW, Chan DW, Zhang Z. (2007) Expression and clinicopathological significance of oestrogen-responsive ezrin-radixin-moesin-binding phosphoprotein 50 in breast cancer. Histopathology 51:40–53. [DOI] [PubMed] [Google Scholar]
- Songyang Z, Fanning AS, Fu C, Xu J, Marfatia SM, Chishti AH, Crompton A, Chan AC, Anderson JM, Cantley LC. (1997) Recognition of unique carboxyl-terminal motifs by distinct PDZ domains. Science 275:73–77. [DOI] [PubMed] [Google Scholar]
- Staud F, Ceckova M, Micuda S, Pavek P. (2010) Expression and function of p-glycoprotein in normal tissues: effect on pharmacokinetics. Methods Mol Biol 596:199–222. [DOI] [PubMed] [Google Scholar]
- Stemmer-Rachamimov AO, Wiederhold T, Nielsen GP, James M, Pinney-Michalowski D, Roy JE, Cohen WA, Ramesh V, Louis DN. (2001) NHE-RF, a merlin-interacting protein, is primarily expressed in luminal epithelia, proliferative endometrium, and estrogen receptor-positive breast carcinomas. Am J Pathol 158:57–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stricker NL, Christopherson KS, Yi BA, Schatz PJ, Raab RW, Dawes G, Bassett DE, Jr, Bredt DS, Li M. (1997) PDZ domain of neuronal nitric oxide synthase recognizes novel C-terminal peptide sequences. Nat Biotechnol 15:336–342. [DOI] [PubMed] [Google Scholar]
- Stross C, Keitel V, Winands E, Häussinger D, Kubitz R. (2009) Expression and localization of atypical PKC isoforms in liver parenchymal cells. Biol Chem 390:235–244. [DOI] [PubMed] [Google Scholar]
- Suda J, Rockey DC, Karvar S. (2015) Phosphorylation dynamics of radixin in hypoxia-induced hepatocyte injury. Am J Physiol Gastrointest Liver Physiol 308:G313–G324. [DOI] [PubMed] [Google Scholar]
- Suda J, Zhu L, Karvar S. (2011) Phosphorylation of radixin regulates cell polarity and Mrp-2 distribution in hepatocytes. Am J Physiol Cell Physiol 300:C416–C424. [DOI] [PubMed] [Google Scholar]
- Sugawara M, Huang W, Fei YJ, Leibach FH, Ganapathy V, Ganapathy ME. (2000) Transport of valganciclovir, a ganciclovir prodrug, via peptide transporters PEPT1 and PEPT2. J Pharm Sci 89:781–789. [DOI] [PubMed] [Google Scholar]
- Sugiura T, Kato Y, Kubo Y, Tsuji A. (2006) Mutation in an adaptor protein PDZK1 affects transport activity of organic cation transporter OCTNs and oligopeptide transporter PEPT2. Drug Metab Pharmacokinet 21:375–383. [DOI] [PubMed] [Google Scholar]
- Sugiura T, Kato Y, Wakayama T, Silver DL, Kubo Y, Iseki S, Tsuji A. (2008) PDZK1 regulates two intestinal solute carriers (Slc15a1 and Slc22a5) in mice. Drug Metab Dispos 36:1181–1188. [DOI] [PubMed] [Google Scholar]
- Sugiura T, Otake T, Shimizu T, Wakayama T, Silver DL, Utsumi R, Nishimura T, Iseki S, Nakamichi N, Kubo Y, et al. (2010) PDZK1 regulates organic anion transporting polypeptide Oatp1a in mouse small intestine. Drug Metab Pharmacokinet 25:588–598. [DOI] [PubMed] [Google Scholar]
- Sugiura T, Shimizu T, Kijima A, Minakata S, Kato Y. (2011) PDZ adaptors: their regulation of epithelial transporters and involvement in human diseases. J Pharm Sci 100:3620–3635. [DOI] [PubMed] [Google Scholar]
- Sun H, Frassetto LA, Huang Y, Benet LZ. (2010) Hepatic clearance, but not gut availability, of erythromycin is altered in patients with end-stage renal disease. Clin Pharmacol Ther 87:465–472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szakács G, Annereau JP, Lababidi S, Shankavaram U, Arciello A, Bussey KJ, Reinhold W, Guo Y, Kruh GD, Reimers M, et al. (2004) Predicting drug sensitivity and resistance: profiling ABC transporter genes in cancer cells. Cancer Cell 6:129–137. [DOI] [PubMed] [Google Scholar]
- Szakács G, Paterson JK, Ludwig JA, Booth-Genthe C, Gottesman MM. (2006) Targeting multidrug resistance in cancer. Nat Rev Drug Discov 5:219–234. [DOI] [PubMed] [Google Scholar]
- Takada T, Ichida K, Matsuo H, Nakayama A, Murakami K, Yamanashi Y, Kasuga H, Shinomiya N, Suzuki H. (2014) ABCG2 dysfunction increases serum uric acid by decreased intestinal urate excretion. Nucleosides Nucleotides Nucleic Acids 33:275–281. [DOI] [PubMed] [Google Scholar]
- Takahashi K, Nakamura N, Terada T, Okano T, Futami T, Saito H, Inui KI. (1998) Interaction of beta-lactam antibiotics with H+/peptide cotransporters in rat renal brush-border membranes. J Pharmacol Exp Ther 286:1037–1042. [PubMed] [Google Scholar]
- Tamai I. (2012) Oral drug delivery utilizing intestinal OATP transporters. Adv Drug Deliv Rev 64:508–514. [DOI] [PubMed] [Google Scholar]
- Tamai I, Nakanishi T, Kobayashi D, China K, Kosugi Y, Nezu J, Sai Y, Tsuji A. (2004) Involvement of OCTN1 (SLC22A4) in pH-dependent transport of organic cations. Mol Pharm 1:57–66. [DOI] [PubMed] [Google Scholar]
- Tamai I, Ohashi R, Nezu J, Yabuuchi H, Oku A, Shimane M, Sai Y, Tsuji A. (1998) Molecular and functional identification of sodium ion-dependent, high affinity human carnitine transporter OCTN2. J Biol Chem 273:20378–20382. [DOI] [PubMed] [Google Scholar]
- Tamai I, Ohashi R, Nezu JI, Sai Y, Kobayashi D, Oku A, Shimane M, Tsuji A. (2000) Molecular and functional characterization of organic cation/carnitine transporter family in mice. J Biol Chem 275:40064–40072. [DOI] [PubMed] [Google Scholar]
- Taylor CW, Dalton WS, Parrish PR, Gleason MC, Bellamy WT, Thompson FH, Roe DJ, Trent JM. (1991) Different mechanisms of decreased drug accumulation in doxorubicin and mitoxantrone resistant variants of the MCF7 human breast cancer cell line. Br J Cancer 63:923–929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Teodori E, Dei S, Martelli C, Scapecchi S, Gualtieri F. (2006) The functions and structure of ABC transporters: implications for the design of new inhibitors of Pgp and MRP1 to control multidrug resistance (MDR). Curr Drug Targets 7:893–909. [DOI] [PubMed] [Google Scholar]
- Thiebaut F, Tsuruo T, Hamada H, Gottesman MM, Pastan I, Willingham MC. (1987) Cellular localization of the multidrug-resistance gene product P-glycoprotein in normal human tissues. Proc Natl Acad Sci USA 84:7735–7738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toh S, Wada M, Uchiumi T, Inokuchi A, Makino Y, Horie Y, Adachi Y, Sakisaka S, Kuwano M. (1999) Genomic structure of the canalicular multispecific organic anion-transporter gene (MRP2/cMOAT) and mutations in the ATP-binding-cassette region in Dubin-Johnson syndrome. Am J Hum Genet 64:739–746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsujimoto M, Kinoshita Y, Hirata S, Otagiri M, Ohtani H, Sawada Y. (2008) Effects of uremic serum and uremic toxins on hepatic uptake of digoxin. Ther Drug Monit 30:576–582. [DOI] [PubMed] [Google Scholar]
- Türk D, Szakács G. (2009) Relevance of multidrug resistance in the age of targeted therapy. Curr Opin Drug Discov Devel 12:246–252. [PubMed] [Google Scholar]
- Urakami Y, Akazawa M, Saito H, Okuda M, Inui K. (2002) cDNA cloning, functional characterization, and tissue distribution of an alternatively spliced variant of organic cation transporter hOCT2 predominantly expressed in the human kidney. J Am Soc Nephrol 13:1703–1710. [DOI] [PubMed] [Google Scholar]
- Urban TJ, Brown C, Castro RA, Shah N, Mercer R, Huang Y, Brett CM, Burchard EG, Giacomini KM. (2008) Effects of genetic variation in the novel organic cation transporter, OCTN1, on the renal clearance of gabapentin. Clin Pharmacol Ther 83:416–421. [DOI] [PubMed] [Google Scholar]
- Vanholder R, De Smet R, Glorieux G, Argilés A, Baurmeister U, Brunet P, Clark W, Cohen G, De Deyn PP, Deppisch R, et al. European Uremic Toxin Work Group (EUTox) (2003) Review on uremic toxins: classification, concentration, and interindividual variability. Kidney Int 63:1934–1943. [DOI] [PubMed] [Google Scholar]
- Varma MV, Rotter CJ, Chupka J, Whalen KM, Duignan DB, Feng B, Litchfield J, Goosen TC, El-Kattan AF. (2011) pH-sensitive interaction of HMG-CoA reductase inhibitors (statins) with organic anion transporting polypeptide 2B1. Mol Pharm 8:1303–1313. [DOI] [PubMed] [Google Scholar]
- Veau C, Leroy C, Banide H, Auchère D, Tardivel S, Farinotti R, Lacour B. (2001) Effect of chronic renal failure on the expression and function of rat intestinal P-glycoprotein in drug excretion. Nephrol Dial Transplant 16:1607–1614. [DOI] [PubMed] [Google Scholar]
- Voltz JW, Weinman EJ, Shenolikar S. (2001) Expanding the role of NHERF, a PDZ-domain containing protein adapter, to growth regulation. Oncogene 20:6309–6314. [DOI] [PubMed] [Google Scholar]
- Wakida N, Tuyen DG, Adachi M, Miyoshi T, Nonoguchi H, Oka T, Ueda O, Tazawa M, Kurihara S, Yoneta Y, et al. (2005) Mutations in human urate transporter 1 gene in presecretory reabsorption defect type of familial renal hypouricemia. J Clin Endocrinol Metab 90:2169–2174. [DOI] [PubMed] [Google Scholar]
- Wang B, Ardura JA, Romero G, Yang Y, Hall RA, Friedman PA. (2010) Na/H exchanger regulatory factors control parathyroid hormone receptor signaling by facilitating differential activation of G(alpha) protein subunits. J Biol Chem 285:26976–26986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang B, Bisello A, Yang Y, Romero GG, Friedman PA. (2007) NHERF1 regulates parathyroid hormone receptor membrane retention without affecting recycling. J Biol Chem 282:36214–36222. [DOI] [PubMed] [Google Scholar]
- Wang B, Means CK, Yang Y, Mamonova T, Bisello A, Altschuler DL, Scott JD, Friedman PA. (2012) Ezrin-anchored protein kinase A coordinates phosphorylation-dependent disassembly of a NHERF1 ternary complex to regulate hormone-sensitive phosphate transport. J Biol Chem 287:24148–24163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang P, Wang JJ, Xiao Y, Murray JW, Novikoff PM, Angeletti RH, Orr GA, Lan D, Silver DL, Wolkoff AW. (2005) Interaction with PDZK1 is required for expression of organic anion transporting protein 1A1 on the hepatocyte surface. J Biol Chem 280:30143–30149. [DOI] [PubMed] [Google Scholar]
- Wang WJ, Murray JW, Wolkoff AW. (2014) Oatp1a1 requires PDZK1 to traffic to the plasma membrane by selective recruitment of microtubule-based motor proteins. Drug Metab Dispos 42:62–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watanabe C, Kato Y, Sugiura T, Kubo Y, Wakayama T, Iseki S, Tsuji A. (2006) PDZ adaptor protein PDZK2 stimulates transport activity of organic cation/carnitine transporter OCTN2 by modulating cell surface expression. Drug Metab Dispos 34:1927–1934. [DOI] [PubMed] [Google Scholar]
- Weiner IM, Washington JA, 2nd, Mudge GH. (1960) On the mechanism of action of probenecid on renal tubular secretion. Bull Johns Hopkins Hosp 106:333–346. [PubMed] [Google Scholar]
- Weinman EJ, Steplock D, Wang Y, Shenolikar S. (1995) Characterization of a protein cofactor that mediates protein kinase A regulation of the renal brush border membrane Na(+)-H+ exchanger. J Clin Invest 95:2143–2149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weller S, Blum MR, Doucette M, Burnette T, Cederberg DM, de Miranda P, Smiley ML. (1993) Pharmacokinetics of the acyclovir pro-drug valaciclovir after escalating single- and multiple-dose administration to normal volunteers. Clin Pharmacol Ther 54:595–605. [DOI] [PubMed] [Google Scholar]
- Wu CP, Hsiao SH, Luo SY, Tuo WC, Su CY, Li YQ, Huang YH, Hsieh CH. (2014) Overexpression of human ABCB1 in cancer cells leads to reduced activity of GSK461364, a specific inhibitor of polo-like kinase 1. Mol Pharm 11:3727–3736. [DOI] [PubMed] [Google Scholar]
- Wu W, Jamshidi N, Eraly SA, Liu HC, Bush KT, Palsson BO, Nigam SK. (2013) Multispecific drug transporter Slc22a8 (Oat3) regulates multiple metabolic and signaling pathways. Drug Metab Dispos 41:1825–1834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu X, George RL, Huang W, Wang H, Conway SJ, Leibach FH, Ganapathy V. (2000a) Structural and functional characteristics and tissue distribution pattern of rat OCTN1, an organic cation transporter, cloned from placenta. Biochim Biophys Acta 1466:315–327. [DOI] [PubMed] [Google Scholar]
- Wu X, Huang W, Ganapathy ME, Wang H, Kekuda R, Conway SJ, Leibach FH, Ganapathy V. (2000b) Structure, function, and regional distribution of the organic cation transporter OCT3 in the kidney. Am J Physiol Renal Physiol 279:F449–F458. [DOI] [PubMed] [Google Scholar]
- Wu X, Huang W, Prasad PD, Seth P, Rajan DP, Leibach FH, Chen J, Conway SJ, Ganapathy V. (1999) Functional characteristics and tissue distribution pattern of organic cation transporter 2 (OCTN2), an organic cation/carnitine transporter. J Pharmacol Exp Ther 290:1482–1492. [PubMed] [Google Scholar]
- Xu G, Bhatnagar V, Wen G, Hamilton BA, Eraly SA, Nigam SK. (2005) Analyses of coding region polymorphisms in apical and basolateral human organic anion transporter (OAT) genes [OAT1 (NKT), OAT2, OAT3, OAT4, URAT (RST)]. Kidney Int 68:1491–1499. [DOI] [PubMed] [Google Scholar]
- Yang J, Singh V, Chen TE, Sarker R, Xiong L, Cha B, Jin S, Li X, Tse CM, Zachos NC, et al. (2014) NHERF2/NHERF3 protein heterodimerization and macrocomplex formation are required for the inhibition of NHE3 activity by carbachol. J Biol Chem 289:20039–20053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Q, Onuki R, Nakai C, Sugiyama Y. (2007) Ezrin and radixin both regulate the apical membrane localization of ABCC2 (MRP2) in human intestinal epithelial Caco-2 cells. Exp Cell Res 313:3517–3525. [DOI] [PubMed] [Google Scholar]
- Yano K, Tomono T, Sakai R, Kano T, Morimoto K, Kato Y, Ogihara T. (2013) Contribution of radixin to P-glycoprotein expression and transport activity in mouse small intestine in vivo. J Pharm Sci 102:2875–2881. [DOI] [PubMed] [Google Scholar]
- Yee SW, Nguyen AN, Brown C, Savic RM, Zhang Y, Castro RA, Cropp CD, Choi JH, Singh D, Tahara H, et al. (2013) Reduced renal clearance of cefotaxime in asians with a low-frequency polymorphism of OAT3 (SLC22A8). J Pharm Sci 102:3451–3457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zaidenstein R, Eyal S, Efrati S, Akivison L, Michowitz MK, Nagornov V, Golik A. (2002) Adverse drug events in hospitalized patients treated with cardiovascular drugs and anticoagulants. Pharmacoepidemiol Drug Saf 11:235–238. [DOI] [PubMed] [Google Scholar]
- Zamek-Gliszczynski MJ, Hoffmaster KA, Tweedie DJ, Giacomini KM, Hillgren KM. (2012) Highlights from the International Transporter Consortium second workshop. Clin Pharmacol Ther 92:553–556. [DOI] [PubMed] [Google Scholar]
- Zhang L, Dresser MJ, Gray AT, Yost SC, Terashita S, Giacomini KM. (1997) Cloning and functional expression of a human liver organic cation transporter. Mol Pharmacol 51:913–921. [DOI] [PubMed] [Google Scholar]
- Zhang Q, Pan Z, You G. (2010a) Regulation of human organic anion transporter 4 by protein kinase C and NHERF-1: altering the endocytosis of the transporter. Pharm Res 27:589–596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Sun J, Sun Y, Wang Y, He Z. (2013) Prodrug design targeting intestinal PepT1 for improved oral absorption: design and performance. Curr Drug Metab 14:675–687. [DOI] [PubMed] [Google Scholar]
- Zhang YH, Wu Q, Xiao XY, Li DW, Wang XP. (2010b) Silencing MRP4 by small interfering RNA reverses acquired DDP resistance of gastric cancer cell. Cancer Lett 291:76–82. [DOI] [PubMed] [Google Scholar]
- Zheng J, Chan T, Cheung FS, Zhu L, Murray M, Zhou F. (2014) PDZK1 and NHERF1 regulate the function of human organic anion transporting polypeptide 1A2 (OATP1A2) by modulating its subcellular trafficking and stability. PLoS ONE 9:e94712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou F, Xu W, Tanaka K, You G. (2008) Comparison of the interaction of human organic anion transporter hOAT4 with PDZ proteins between kidney cells and placental cells. Pharm Res 25:475–480. [DOI] [PubMed] [Google Scholar]