

Abstract
Histamine is a developmentally highly conserved autacoid found in most vertebrate tissues. Its physiological functions are mediated by four 7-transmembrane G protein–coupled receptors (H1R, H2R, H3R, H4R) that are all targets of pharmacological intervention. The receptors display molecular heterogeneity and constitutive activity. H1R antagonists are long known antiallergic and sedating drugs, whereas the H2R was identified in the 1970s and led to the development of H2R-antagonists that revolutionized stomach ulcer treatment. The crystal structure of ligand-bound H1R has rendered it possible to design new ligands with novel properties. The H3R is an autoreceptor and heteroreceptor providing negative feedback on histaminergic and inhibition on other neurons. A block of these actions promotes waking. The H4R occurs on immuncompetent cells and the development of anti-inflammatory drugs is anticipated.
I. Introduction and Historical Perspective
Histamine pharmacology has experienced a renaissance over the last few decades, with the identification and cloning of the histamine H3 and H4 receptors, which doubles the members of the histamine receptor family. This has led to a massive increase in our understanding of the histamine systems in the whole body and recently resulted in the introduction of H3 receptor and H4 receptor drug leads into late-stage clinical development, with an ever expanding range of potential therapeutic applications. The molecular identification of the H3 receptor and H4 receptor, their attendant isoforms, and species variants have now clarified to some degree the pharmacological heterogeneity reported in the 1990s, reviewed in the previous Pharmacological Reviews article by Hill et al. (1997). This present review is dedicated to two of the foremost histamine receptor pharmacologists, Sir James Black and Walter Schunack, who sadly died at the beginning of 2010 and 2011, respectively. They provided the field with prototypical compounds and drugs, particularly in the H2 receptor and H3 receptor fields and contributed profoundly to our current understanding of histamine pharmacology.
Histamine (1) is an endogenous biogenic amine distributed ubiquitously in the body being present in high concentrations in the lungs, skin, and gastrointestinal tract (Fig. 1). Histamine is synthesized and stored at high concentrations within granules in so called "professional" cells, basophils and mast cells, where it is associated with heparin. Based on a sensitive high-performance liquid chromatography-mass spectrometry method, nonmast cell histamine occurs at high concentrations in enterchromaffin-like cells in the stomach, lymph nodes, and thymus, with modest levels in the liver, lung, and in varicosities of the histaminergic neurons in the brain (Zimmermann et al., 2011). Histamine acts as a neurotransmitter in the nervous system and as a local mediator in the gut, skin, and immune system. Histamine brings about complex physiologic changes, including neurotransmission, inflammation, smooth muscle contraction, dilatation of capillaries, chemotaxis, cytokine production, and gastric acid secretion. These biologic changes occur via four G protein–coupled receptor (GPCR) subtypes: H1 receptor, H2 receptor, H3 receptor, and H4 receptor. These seven-transmembrane domain GPCR proteins represent the largest family of membrane proteins in the human genome (Jacoby et al., 2006; Lagerstrom and Schioth, 2008) and have proven to be one of the most rewarding families of drug targets to date. All members, including the histamine receptors, share a common membrane topology, comprising an extracellular N terminus, an intracellular C terminus, and seven transmembrane (TM) helices interconnected by three intracellular loops and three extracellular loops. The relative concentrations of histamine required to activate respective histamine receptor subtypes are different. For example, H1 receptors and H2 receptors have relatively low affinity for histamine in comparison with H3 receptors and H4 receptors, thus the local concentrations of histamine and the presence of different receptor subtypes adds specificity to histamine responses.
Fig. 1.
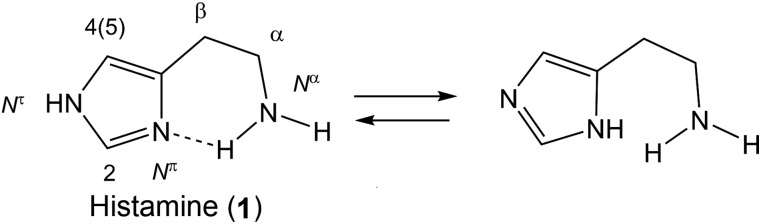
Histamine.
The classification of the histamine receptor family was historically based on pharmacological definitions but has subsequently relied upon the molecular biologic identification of new histamine receptor genes and the elucidation of four distinct histamine receptor polypeptide sequences. However, apparent molecular heterogeneity, through alternative splicing, has increased the number of potential receptor isoforms, particularly with the rat and human H3 receptor. This heterogeneity will be discussed in detail within this review. Moreover, with the availability of recombinant expression systems, new phenomena, including constitutive histamine receptor signaling and receptor oligomerization, have been shown for almost all of the histamine receptor subtypes (see next sections).
Constitutive GPCR activity is recognized for many GPCR family members and results in GPCR signaling without the need of an external agonist (Smit et al., 2007). This spontaneous GPCR signaling is thought to evolve from the conformational dynamics of GPCR proteins, resulting in equilibria between active and inactive receptor states. These equilibria can be altered by GPCR mutations, such as, e.g., in some inherited diseases (Smit et al., 2007), and by GPCR ligands. Agonists drive the equilibria toward active GPCR conformation(s), whereas so-called inverse agonists would favor the inactive conformations. Following this notion, many of the known GPCR antagonists (including the histamine receptor antagonists) have been reclassified as inverse agonists (Smit et al., 2007), whereas true (neutral) antagonists are difficult to identify for most GPCRs.
Oligomerization occurs in most if not all GPCRs, including several of the histamine receptor subtypes (see sections below). However, it is not clear whether this occurs in vivo in all cases and what might be the functional significance of this (Vischer et al., 2011). The majority of the studies have been performed with in vitro heterologous systems with recombinant receptors [e.g., H4 receptor (van Rijn et al., 2006)]. However, there is growing in vivo evidence for oligomerization for some classes of GPRCs, including class C GPRCs, for example, GABAB and metabotropic glutamate receptors. Structures comprising monomers, dimers, and higher-order oligomers of GPCRs have been shown to assemble into transient and/or stable homo-oligomeric and hetero-oligomeric macromolecular complexes. It is unclear whether the individual protomers within a homodimeric structure are always equivalent or play distinct roles in agonist occupancy and/or G protein activation, as appears to be the case in class C GPCRs. The current status of the field was recently nicely reviewed (Ferre et al., 2014). The increased molecular understanding of the histamine receptor proteins has been paralleled by developments in the field of histamine receptor pharmacology. The first histamine receptor antagonist drugs were developed in the 1930s and 1950s, over 20 years after their discovery in 1910 (Simons and Simons, 2011). The H2 receptor was discovered in the early 1970s (Powell and Brody, 1976) and the H3 receptor in the early 1980s (Arrang et al., 1983), and finally, the last member to join the group, the H4 receptor, was identified using molecular biologic techniques at the beginning of the millennium (Nakamura et al., 2000; Liu et al., 2001a; Morse et al., 2001; Nguyen et al., 2001; Zhu et al., 2001; O'Reilly et al., 2002).
The first two receptors have been exploited extremely successfully in the development of a range of “blockbuster” drugs. There is now growing anticipation that the two most recently discovered histamine receptor subtypes will yield further therapeutic success, because ligands for both recently entered clinical development, with the H3 receptor antagonist, pitolisant (Wakix) being filed for licensing by Bioprojet (Paris, France) in May 2014. The field waits to see if this filing is successful. In a similar fashion, although some positive results have been reported for H4 receptor ligands in early clinical trials, the first H4 receptor drug is yet to be filed and registered.
Gene-targeted histidine decarboxylase (HDC) knockout (KO) mice have provided a large amount of information regarding the role of histamine. These mice are viable, fertile, and display no gross abnormalities except for abnormal mast cells (Ohtsu et al., 2001). However, there are permanent changes in the cortical-electroencephalogram and sleep-wake cycle: At moments when high vigilance is required, mice lacking brain histamine are unable to remain awake, a prerequisite condition for responding to behavioral and cognitive challenges (Parmentier et al., 2002), changes in learning ability (Dere et al., 2003), and increased obesity in high-fat diet–fed mice (Haas et al., 2008).
The combinatorial roles of all four histamine receptors were studied in autoinflammatory disease of the nervous system in the search of an effective therapy for multiple sclerosis. H1 receptor, H2 receptor-KO mice developed less severe experimental autoimmune encephalitis (EAE) than H3 receptor-, H4 receptor-KO mice (Saligrama et al., 2012). Teuscher’s group (Saligrama et al., 2013) also compared the phenotypes of H1,2,3,4 receptor- and HDC-knockout mice; unexpectedly and interestingly they displayed opposite phenotypes.
This review will focus on the advances in our understanding of histamine pharmacology that have occurred since the last review published in 1997 (Hill et al., 1997). Histamine receptors can be accessed through the International Union of Basic and Clinical Pharmacology/British Pharmacological Society Guide to Pharmacology (Alexander et al., 2013; http://www.guidetopharmacology.org/GRAC/FamilyDisplayForward?familyId=33).
II. Histamine H1 Receptor
The first histamine receptor has been known since its pharmacological characterization using antagonists (Bovet and Staub, 1937) and agonists (Black et al., 1972). It was identified as a glycoprotein (Garbarg et al., 1985) and cloned in 1991 (Yamashita et al., 1991). It is very widely expressed throughout the body in epithelial, vascular, smooth vascular, neuronal, glial, and immune cells (Hill et al., 1997; Haas et al., 2008; Shimamura et al., 2011). This receptor is responsible for the early biologic method to determine histamine by contraction of guinea pig ileum preparations and most of the symptoms caused by histamine in allergy and asthma, except the inflammatory component.
A. Receptor Structure
As all four histamine receptors, the H1 receptor belongs to the GPCR family. The bovine H1 receptor cDNA was cloned in 1991 by expression cloning in the classic Xenopus oocyte system (Yamashita et al., 1991). This led rapidly to the cloning of the receptor cDNA in several species, including human in 1993 and 1994 (De Backer et al., 1993; Moguilevsky et al., 1994). The human gene was subsequently localized to chromosome 3p14-p21 (Le Coniat et al., 1994). A single H1 receptor polymorphism rs7651620 Glu270Gly has been identified that is relevant to African subjects only (Garcia-Martin et al., 2009; Micallef et al., 2013).
On the basis of immunoprecipitation, time-resolved fluorescence resonance transfer and mutagenesis approaches, there is strong evidence that the H1 receptor is expressed as both homomeric and oligomeric structures. Furthermore, evidence has been provided for the presence of robust domain-swapped H1 receptor dimers in which there is a reciprocal exchange of TM6 and 7 between the individual monomers in the dimer (Bakker, 2004).
Structural biology in GPCRs recently received tremendous input with the solved crystal structures of numerous GPCR family members, including many biogenic amine receptors (for a complete list, see, e.g., http://www.gpcr.org/7tm/ or http://zhanglab.ccmb.med.umich.edu/GPCRSD/). In 2011, the 3.1 Å resolution structure of a stabilized mutant human H1 receptor in complex with the first generation H1 receptor antagonist doxepin was reported (Shimamura et al., 2011). Crystallization was made possible by a replacement of the large third intracellular loop with T4-lysozyme fusion protein (H1 receptor-T4L) and numerous other technical advances. Doxepin stabilizes the H1 receptor in an inactive (R) state by preventing movements in the so-called “toggle switch” in TM6, an element proposed to be important in activation in many GPCRs. Obviously, on the basis of phylogenetic relationships, the H1 receptor shares more structural similarities with the other aminergic receptors than with the more distant GPCRs. Some common motifs in the TM region can be recognized [TM3: D(E)RY; TM6: CWxP; TM7: NPxxY], as well as a disulfide bond that connects the long extracellular loop 2 (ECL2) with the extracellular end of TM3 (Shimamura et al., 2011). Interestingly, the palmitoylation site at the end of TM7, which has often been recognized as a membrane anchor, is missing in H1 receptor (Fig. 2).
Fig. 2.

Overall 7TM X-ray structure of the H1 receptor (PDB ID 3RZE) bound with the tricyclic H1 receptor inverse agonist doxepin and a phosphate ion and a close-up on the binding site with residues surrounding doxepin and the phosphate ion. Residues are numbered as found in the human H1 receptor sequence and with the corresponding Ballesteros-Weinstein numbering.
The binding pocket of doxepin involves the highly conserved Asp1073.32 in TM3 and aromatic residues in TMs 5 and 6 (e.g., Phe4246.44, Trp4286.48, and Phe4326.52 (Shimamura et al., 2011), as previously had been assumed for related H1 receptor antagonists based on site-directed mutagenesis and homology modeling exercises (Ohta et al., 1994; Nonaka et al., 1998; Wieland et al., 1999). The strong hydrophobic interactions of the aromatic moieties of doxepin with Trp4286.48 may prevent movement of helix 6, which is generally seen as one of the hallmarks of GPCR activation. Doxepin binds relatively deep in the binding pocket defined by TM3, -5, and -6. Interestingly, in the X-ray structure an additional phosphate-anion binding site at the entrance of the ligand-binding pocket is observed as a novel feature. The phosphate anion is coordinated by Lys179ECL2, Lys1915.39, and His4507.35, and this binding pocket is suggested to play a role in the interaction with the second generation zwitterionic antihistamines (Shimamura et al., 2011). Evidence for a role of Lys1915.39 and extracellular domains of the H1 receptor in ligand–receptor interactions has earlier been obtained by mutagenesis and molecular dynamics studies (Wieland et al., 1999; Gillard et al., 2002a; Strasser and Wittmann, 2007; Wittmann et al., 2011b). Insights into the molecular features governing agonist-induced H1 receptor activation await the resolution of an active H1 receptor X-ray structure and currently still rely on molecular modeling and/or mutagenesis studies (Ohta et al., 1994; Jongejan et al., 2005; Strasser et al., 2008b; Sansuk et al., 2011). Histamine is thought to bind the H1 receptor with its protonated ethylamine side chain via Asp1073.32 (Ohta et al., 1994), whereas the imidazole ring is thought to interact with Asn1985.46 and Lys1915.39 (Leurs et al., 1994a, 1995a). The interaction of the protonated side chain with Asp1073.32 allows potentially for the release of Ser3.36, allowing it to act as a toggle switch and to interact with Asn7.45 in the active state of the receptor (Jongejan et al., 2005) (Fig. 3; Table 1).
Fig. 3.

Binding of histamine in the four different histamine receptors as based on mutagenesis data and docking studies in the H1 receptor X-ray structure and H2 receptor–H4 receptor homology models, based on the H1 receptor X-ray structure. Residues are numbered as found in the human sequences and with the corresponding Ballesteros-Weinstein numbering.
TABLE 1.
Overview human histamine receptor subtypes (hH1 receptor-hH4 receptor).
| hH1R | hH2R | hH3R | hH4R | |
|---|---|---|---|---|
| Chromosomal gene location | 3q25 | 5q35.2 | 20q13.33 | 18q11.2 |
| Amino acids | 487 | 359 | 445 | 390 |
| Isoforms | at least 20 (65, 66) | >3 | ||
| G protein coupling | Gαq/11 | Gαs | Gαi/o | Gαi/o |
| Constitutive activity | + | + | ++ | ++ |
| Signal transduction | PLC↑, Ca2+↑ | cAMP↑ | cAMP↓, Ca2+↑, MAPK↑ | cAMP↓, Ca2+↑, MAPK↑ |
| Tissue localizations | Ubiquitous (mainly lung, CNS, blood vessels) | Ubiquitous (mainly stomach, heart, CNS) | Neurons (CNS and PNS) | Bone marrow, hematopoietic cells |
| Physiologic relevance | Bronchoconstriction, vasodilation, food intake, sleep-wake regulation | Gastric acid secretion | Neurotransmitter release (→ sleep-wake regulation, attention/cognition, food intake) | Immune responses (→ chemotaxis, IL-, IFN-modulation) |
| Pathophysiological conditions | Allergic reactions, emesis, sleep-wake disorders | Gastric ulcers | Cognitive impairment, schizophrenia, sleep-wake disorders, epilepsy, pain, etc. | Inflammatory diseases (allergy, asthma, pruritus, arthritis), pain, etc. |
+ and ++, extent of constitutive activity; PL, phospholipase; PNS, peripheral nervous system.
B. Signal Mechanisms
The human H1 receptor produces its action mainly by coupling to Gq/11 proteins (Gutowski et al., 1991; Leopoldt et al., 1997; Selbach et al., 1997; Moniri et al., 2004), but also signals via Gi/o in some systems (Seifert et al., 1994; Wang and Kotlikoff, 2000), and the small G protein family, most likely through an indirect downstream effect (Mitchell and Mayeenuddin, 1998). Direct interaction of the H1 receptor with Gq/11 was recently visualized in real time in HELA cells endogenously expressing H1 receptor (Adjobo-Hermans et al., 2011).
The major signaling pathway for H1 receptor elicits activation of phospholipase C, which produces 1,2-diacylglycerol and inositol-1,4,5-trisphosphate, leading to activation of protein kinase C (PKC), catalyzing the Ser/Thr phosphorylation of multiple downstream mediators and release of calcium ions from intracellular stores, respectively (Hill et al., 1997). This leads to calcium ion entry through calcium channels, cation channels of the transient receptor potential channel type (Brown et al., 2002; Doreulee et al., 2003), and stimulation of a Na+/Ca2+ exchanger (Eriksson et al., 2001; Sergeeva et al., 2003). Nicotinic acid adenine dinucleotide phosphate (NAADP) has also been suggested as a potential second messenger in histamine-induced Ca2+ release from lysosome-like acidic compartments, functionally coupled to the endoplasmic reticulum via H1 receptor in endothelial cells. By using the human EA.hy926 endothelial cell line and primary human umbilical vein endothelial cells, selective H1 receptor activation increases intracellular NAADP levels, and Ca2+ release was shown to involve both acidic organelles and the endoplasmic reticulum. Furthermore, H1 receptor-induced von Willebrand factor secretion has been shown to require a specific Ca2+ signaling and NAADP mechanism (Esposito et al., 2011) (Fig. 4).
Fig. 4.

Schematic overview of the main signal transduction routes of the four different histamine receptors.
H1 receptors also mediate the production of arachidonic acid, nitric oxide, and cyclic GMP (Richelson, 1978; Snider et al., 1984; Leurs et al., 1994b; Prast and Philippu, 2001) through Gi/Go protein–mediated activation of phospholipase A2, [Ca2+]i-dependent nitric oxide (NO) synthases, and NO-dependent guanyl cyclases, respectively. NO is thought to be involved in the histamine-induced suppression of AMPA (α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid) receptor-mediated synaptic currents in supraoptic neurons through activation of H1 receptor. Histamine is believed to act initially at supraoptic cells (perhaps both neuronal and non-neuronal) to induce the production of NO, which then locally modulates synaptic transmission via a postsynaptic mechanism (Li and Hatton, 2000). Stimulation of the H1 receptor also increases endothelial NO synthase transcription in human vascular endothelial cells via a Ca2+/calmodulin-dependent protein kinase II (CaMKII) signaling pathway, which may be protective under normal conditions, but in contrast, may be deleterious under conditions of oxidative stress, when endothelial NO synthase can produce reactive oxygen species at the expense of NO (Li et al., 2003). Histamine, through H1 receptors, increases endothelial NO production as an endothelium-dependent vasodilator but acts as a vasoconstrictor in atherosclerotic coronary arteries.
In the mammalian brain, adrenal glands, and CHO cells, activation of the H1 receptor also stimulates adenylyl cyclase and consequently cAMP production, the canonical signaling pathway of the H2 receptor (see below). In rat brain, the cAMP production is dependent upon PKC activation and intra- and extracellular calcium ion levels (Marley et al., 1991). In the adrenal gland, H1 receptor activation of adenylyl cyclase is dependent on extracellular Ca2+ only (Marley et al., 1991). The stimulatory effects of histamine on cAMP production are most likely not mediated directly through Gs coupling but rather indirectly via an increase in [Ca2+]i and the release of activated Gβγ-subunits. For example, the neuronally expressed adenylyl cyclase 1 is a paradigm for a Ca2+/calmodulin-stimulated adenylyl cyclase (Sunahara and Taussig, 2002).
H1 receptor activation in transfected CHO cells increases forskolin-dependent cAMP production, which is independent of extracellular Ca2+ and PKC activation and is pertussis toxin–insensitive (Leurs et al., 1994b). Stimulation of H1 receptors activates adenylyl cyclase in CHO cells relies on the release of Gβγ-subunits from G proteins, thereby elevating intracellular cAMP concentrations (Maruko et al., 2005).
It is well established that histamine modulates cell proliferation through the activation of the H1 receptor. The H1 receptor can signal to the nucleus via PKCα subsequent to activation of phospholipase Cβ (Megson et al., 2001). Functional coupling of the H1 receptor to Gq-phospholipase C (PLC) leads to the activation of RhoA and Rac small GTPases through a direct interaction of activated Gα-subunits with p63RhoGEF and suggest distinct roles for Rho GTPases in the control of cell proliferation by histamine that has relevance in cancer biology (Valencia et al., 2001; Lutz et al., 2007; Notcovich et al., 2010). Moreover, also the canonical β-catenin pathway is activated downstream of the H1 receptor in a variety of cell types. Stimulation of the H1 receptor elicits transactivation of a T-cell factor/β-catenin–responsive construct in HeLa cells and in the SW-480 colon cell line. Furthermore, histamine treatment increases phosphorylation of glycogen synthase kinase 3-β in HeLa cells, murine macrophages, and DLD-1, HT-29, and SW-480 colon cell lines, whereas histamine also decreases the phosphorylated β-catenin content in HeLa cells and murine macrophages (Diks et al., 2003).
The H1 receptor displays constitutive receptor activation in the absence of agonists (Bakker et al., 2000; Jacoby et al., 2006), as measured by, e.g., inositol phosphate production and nuclear factor-κB reporter gene activation. This finding resulted in the reclassification of H1 receptor ligands; agonists activate the receptor (2–5), inverse agonists inhibit the constitutive activity and neutral antagonists inhibit the actions of both agonists and inverse agonists without modifying constitutive activity. All the “classical” antihistamines are indeed inverse agonists (Bakker et al., 2000) (6–16), while the experimental ligands, histabudifen and histapendifen are neutral antagonists (Govoni et al., 2003). These latter compounds may allow the study of in vivo relevance of H1 receptor constitutive antagonism, which remains to be elucidated. As with many GPCRs, the extent of H1 receptor constitutive activity depends on the specific parameter determined (Bakker et al., 2007; Appl et al., 2012). The high constitutive activity of the H1 receptor upon assessment of distal signaling parameters, i.e., reporter gene expression, may be explained by the fact that the concentration of effectors is limiting the cascade so that even a small number of constitutively active H1 receptor results in a high agonist-dependent signal. In fact, in native systems, there is little evidence to date for high constitutive H1 receptor activity.
A growing number of studies have also provided evidence for the concept of multiple signaling pathways activated by a single receptor (functional selectivity, biased signaling or ligand-specific receptor conformations) (Moniri and Booth, 2004; Galandrin et al., 2007; Kobilka and Deupi, 2007; Kenakin, 2011; Seifert et al., 2011). One of the first studies to support the concept of agonist-dependent functional selectivity at the H1 receptor used the two H1 receptor ligands, (±)-cis-5-phenyl-7-dimethylamino-5,6,7,8-tetrahydro-9H-benzocycloheptane (cis-PAB) and (−)-trans-1-phenyl-3-dimethylamino-1,2,3,4-tetrahydronaphthalene (trans-PAT) derived from the same chemical class (but with distinct stereochemistry) which were shown to display functional selectivity, the former stimulating cAMP production and the latter, PLC/inositol-1,4,5-trisphosphate metabolism (Moniri et al., 2004). However, as for constitutive H1 receptor signaling, currently there is no evidence for the importance of biased signaling in vivo.
The H1 receptor-mediated signaling cascade is known to desensitize quickly (Smit et al., 1992; McCreath et al., 1994) via both PKC-dependent and -independent mechanisms. GPCR kinase 2 is the principal GPCR kinase mediating agonist-induced H1 receptor desensitization in HEK293 cells (Iwata et al., 2005). The H1 receptor-mediated signaling, furthermore, can be inhibited by phorbol ester-induced PKC activation. Two amino acid residues (Ser396, Ser398) have been shown to be PKC phosphorylation sites by in vitro phosphorylation studies using a series of synthetic peptides. Moreover, treatment with phorbol ester decreased histamine-induced accumulation of inositol phosphates in CHO cells expressing the H1 receptor. Site-directed mutagenesis studies showed that Ser398 was the principle residue involved in PKC-mediated desensitization (Fujimoto et al., 1999) (Fig. 5).
Fig. 5.

Expression and receptor radioligand binding to cross sections of the rat brain. Data are shown for both mRNA distribution from in situ hybridization films and autoradiograms after labeling with radioactive ligands for each receptor. For identification of the brain areas, the following areas are marked in (A–D): 1, retrosplenial granular cortex; 2, primary somatosensory cortex; 3, entorhinal cortex; 4, CA1 area of the hippocampus; 5, dentate gyrus; 6, caudate putamen; 7, amygdala; 8, zona incerta; 9, lateral hypothalamic area; 10, arcuate nucleus; 11, dorsal lateral geniculate nucleus. Modified from Haas and Panula (2003).
C. Anatomic Framework
H1 receptors are found throughout the whole body and nervous system with considerable variations among species (Chang et al., 1979). H1 receptors are expressed in vascular and airway smooth muscle, chondrocytes, hepatocytes, endothelial cells, dendritic cells, monocytes, neutrophils, T and B cells (Jutel et al., 2001, 2009; Togias, 2003). The distribution in the brain is uneven, and because of the circuit organization of the central nervous system (CNS), the anatomy is particularly relevant. Some of the early studies on localization and physiologic/pharmacological implications of H1 receptors, discussed extensively in Hill et al. (1997), are only listed here: blood vessels (Barger and Dale, 1910; Dale and Laidlaw, 1910; Folkow et al., 1948; Black et al., 1972), other smooth muscle preparations (Marshall and Cherry, 1955; Ash and Schild, 1966; Black et al., 1972; Hill, 1990) and heart (Sakuma et al., 1988). The distribution of H1 receptors in different mammalian tissues was first studied using selective radioligands such as [3H]mepyramine (Hill et al., 1977), later using in situ hybridization (Fig. 5). [11C]Mepyramine (cf. 8) and [11C]doxepin have also proved useful for imaging H1 receptors in the living human brain (Villemagne et al., 1991; Yanai et al., 1992).
H1 receptor stimulation leads to endothelial cell contraction and thus increases vascular permeability, and the release of several bioactive substances including nitric oxide. H1 receptors also modulate the action of chromaffin cells in the adrenal medulla and lymphocytes (see Hill et al., 1997). An autoimmune disease locus, Bphs has been identified as H1 receptor (Ma et al., 2002). Endothelial H1 receptor signaling reduces blood-brain barrier permeability and susceptibility to autoimmune encephalomyelitis (Saligrama et al., 2012).
H1 receptors are widely distributed in mammalian brain (Hill, 1990; Schwartz et al., 1991). Particularly high densities are found in brain regions concerned with neuroendocrine, behavioral, and nutritional state control, including the periventricular, suprachiasmatic, and ventromedial nuclei of the hypothalamus, aminergic and cholinergic brainstem nuclei, thalamus, and cortex (Schwartz et al., 1991). In human brain, the highest [3H]mepyramine binding is found in the cerebral cortex and the infralimbic structures (Martinez-Mir et al., 1990), in keeping with the mapping of the H1 receptor using [125I]iodobolpyramine autoradiography in the guinea pig (Bouthenet et al., 1988). The distributions in rat (Palacios et al., 1981a) and guinea pig (Palacios et al., 1981b) are similar to each other and to humans with some exceptions: the guinea pig cerebellum shows high density (Chang et al., 1979; Hill and Young, 1980; Bouthenet et al., 1988; Ruat and Schwartz, 1989) in contrast to the low density in the rat, and the rat thalamus shows a clearly lower H1 receptor mRNA expression than the guinea pig (Lintunen et al., 1998). In most brain areas, there is overlap of H1 receptor binding sites and messenger ribonucleic acid levels except in hippocampus, where the pyramidal cell layer shows high mRNA expression and molecular layer high radioligand binding (Lintunen et al., 1998), and cerebellum in which the discrepancy is likely to reflect the presence of abundant H1 receptors in dendrites of pyramidal and Purkinje cells, respectively (Traiffort et al., 1994). In the hippocampus, both the H1 receptor mRNA expression and radioligand binding sites are in clear contrast with the low density of histamine-containing nerve fibers (Lintunen et al., 1998), which suggests that the turnover of histamine in hippocampus is rapid. Figure 5 shows the distribution of H1 receptor mRNA in the rat brain at midhypothalamic level in comparison with corresponding H1 receptor radioligand binding profiles. These patterns are also shown in comparison for H2 receptors and H3 receptors. Kainic acid–induced limbic seizures rapidly upregulate H1 receptor mRNA expression in the rat caudate, putamen, and granular layer of the dentate gyrus (Lintunen et al., 1998). This effect is in line with the findings on the protective role of histamine through H1 receptor versus kainic acid induced hippocampal damage (Kukko-Lukjanov et al., 2006). In immature mice, H1 receptor activation also plays a pivotal role in regulation of seizure intensity and duration, and seizure-induced neuronal damage (Kukko-Lukjanov et al., 2010). In human brain, higher densities of H1 receptor radioligand binding sites are found in neocortex, hippocampus, nucleus accumbens, thalamus, and posterior hypothalamus, whereas cerebellum and basal ganglia show lower densities (Chang et al., 1979; Kanba and Richelson, 1984; Martinez-Mir et al., 1990; Villemagne et al., 1991; Yanai et al., 1992). In human prefrontal cortex, the highest mRNA expression is found in deep layers IV, V and VI, with slightly higher levels in layer V than the other layers in most areas (Jin and Panula, 2005). The [3H]mepyramine binding levels is highest in layer III of the prefrontal cortex, suggesting that the apical dendrites of neurons in the deep laminae contain H1 receptors (Jin and Panula, 2005). In human thalamus, the anterior, medial, central and lateral nuclear region in dorsal and medial parts of the thalamus show higher H1 receptor mRNA expression than posterior and ventral parts (Jin et al., 2002). Although [3H]mepyramine is also detectable throughout thalamic nuclei, it is modest and many of the H1 receptor expressing neurons are likely to project to the cerebral cortex (Jin et al., 2002). With availability of appropriate positron emission tomography (PET) tracers ([11C]pyrilamine and [11C]doxepin) in the early 90s (Yanai et al., 1992), H1 receptor distribution and occupancy in humans have also been mapped using functional imaging techniques (Yanai and Tashiro, 2007) to study the sedative properties and blood-brain barrier permeability of H1 receptor antagonists (Tashiro and Yanai, 2007), aging (Yanai et al., 1992), and neuropsychiatric disorders, such as Alzheimer's disease, schizophrenia (Iwabuchi et al., 2005), and depression (Kano et al., 2004), in all of which H1 receptor binding was found to be lower than in age-matched healthy controls (Fig. 6).
Fig. 6.

Excitation of rat brainstem neurons by histamine through H1 receptor activation. (A) Inward current in a dorsal raphe neuron with increase in channel noise indicating (transient receptor potential channel-type) channel openings. (B) Increase in firing rate of 15 GABAergic (averaged) neurons in the substantia nigra (modified from Brown et al., 2002, and Korotkova et al., 2002).
D. Function
Calcium ion influx leads to depolarization and contraction of smooth muscle cells. This is a classic experimental pharmacological setting used in teaching and research. For example, in guinea pig ileum smooth muscle preparations, H1 receptor antagonists concentration-dependently inhibit histamine-induced contraction, which offers a simple and quantitative ex vivo testing system for H1 receptor ligands. Guinea pig trachea and rat aortic rings offer further smooth muscle preparations frequently used in pharmacological assay systems. Following the cloning of the H1 receptor, it became possible to express the receptor in heterologous systems and measure [Ca2+] levels or inositol phosphate hydrolysis directly in vitro. This has enabled further advances in understanding of H1 receptor signaling. Using transfected cells, it was shown that via both Gαq/11- and Gβγ-subunits human H1 receptors can activate nuclear factor-κB, a transcription factor prominent in inflammation (Bakker et al., 2001).
H1 receptor activation excites neurons in most brain regions, including brain stem (Lin et al., 1996; Barbara et al., 2002; Korotkova et al., 2005), hypothalamus (Tabarean, 2013), thalamus (McCormick and Williamson, 1991; Soria-Jasso et al., 1997; Zhou et al., 2006), amygdala, septum (Gorelova and Reiner, 1996; Xu et al., 2004), hippocampus (Selbach et al., 1997; Manahan-Vaughan et al., 1998), olfactory bulb (Jahn et al., 1995), and cortex (Reiner and Kamondi, 1994) through an increase of [Ca2+]i. Figure 6 illustrates examples of inward current and increased firing frequency. This can, however, also evoke hyperpolarization and inhibition by the opening of Ca2+-dependent K+ channels, for instance in the hippocampus. H1 receptor activation can thus evoke bidirectional and synergistic effects (Garbarg and Schwartz, 1988; Ruat et al., 1992; Bakker et al., 2004a; Dai et al., 2006), for example, reduction or amplification of H2 receptor actions depending on the timing and context of receptor activation (Baudry et al., 1975; Garbarg and Schwartz, 1988; McCormick and Williamson, 1989; Selbach et al., 1997).
Removal of the H1 receptor in KO mice (Hirai et al., 2004; Huang et al., 2006; Masaki and Yoshimatsu, 2006) produces immunologic (Jutel et al., 2001), metabolic, and behavioral state abnormalities similar to those observed in HDC-KO mice (Parmentier et al., 2002), indicating that the excitation mediated by H1 receptors is the major mechanism for the cortical activation during waking. This is also in keeping with the well known sedative action of the H1 receptor antagonists (Bovet, 1950), which function as inverse agonists, stabilizing the receptor in its inactive state (R) (Lin et al., 1996; Bakker et al., 2002, 2007; Jongejan et al., 2005). Many antidepressant and antipsychotic drugs also bind to the H1 receptor (Richelson, 1978; Kim et al., 2007). A recent analysis of rare copy variants in Tourette syndrome families indicated a strong association with histaminergic signaling, particularly within H1 receptor pathways (Fernandez et al., 2012).
E. H1-Selective Ligands
Despite the fact that histamine regulates numerous physiologic and pathophysiological effects via H1 receptors, the research area of the corresponding agonistic active compounds has been neglected for a long time. Modifications of the endogenous ligand led to 2-(thiazol-2-yl)ethanamine (2) and later to 2-(3-bromophenyl)histamine or 2-(3-(trifluoromethyl)phenyl)histamine (3) (Fig. 7). Although the well characterized 2-(thiazol-2-yl)ethanamine shows only a moderate efficacy of about 26% of that of histamine with a partial agonist behavior, it clearly demonstrated that the tautomeric shift on the imidazole nitrogen atoms is not an essential structural element. Also with other nonimidazole heterocyclic compounds such as the ergot derivative lisuride (partial) agonist properties intrinsic activity from 0.27 to 1.0 (intrinsic activity of histamine = 1.0) could be observed (Bakker et al., 2004b; Pertz et al., 2004). 2-Phenylhistamines were initially developed as H1 receptor ligands. However, a recent study revealed that the histamine receptor subtype is selectivity strongly affected by Nα-substitution. In contrast to the corresponding primary amines, the Nα-methylated phenylhistamines (KK62) have significantly higher affinity for the H4 receptor than the H1 receptor (Strasser et al., 2009; Wittmann et al., 2011a). Compounds with higher maximal efficacies than that of histamine could be obtained by histamine substitution in the 2-position by a diphenylpropyl moiety. This class of histaprodifens is supposed to simultaneously occupy an agonist as well as an antagonist binding site as shown by site-directed mutagenesis studies and molecular modeling techniques (Bruysters et al., 2004), showing slight improvement in efficacy from histaprodifen (4) over methylhistaprodifen to suprahistaprodifen (5) (Jongejan and Leurs, 2005). The distance of three methylene groups between the diphenyl structure and the imidazole ring seems to be crucial as shorter chains led to partial agonists and longer alkyl bridges to antagonists. Phenoprodifens, combining a phenylhistamine and histaprodifen partial structure represent a new class of H1 receptor partial agonists/neutral antagonists (Strasser et al., 2008a) (Fig. 7).
Fig. 7.

H1 receptor ligands.
The relatively low number of H1 receptor agonists is in contrast to the high number of different H1 receptor antagonists. Pharmacologically, they are divided into different generations because of their target profile and their unwanted side effects. Most of these compounds, if not all of them, are now pharmacologically more clearly defined as inverse agonists (Bakker et al., 2007), with the extent of inverse agonism being parameter dependent. The first generation “antihistamines” are structurally described as two aromatic elements linked through a mainly three membered bridge to a basic aliphatic tertiary amino functionality. Diphenhydramine (6), doxylamine (7), mepyramine (pyrilamine; 8), doxepine, dimetinden (9), and bamipine are a few examples based on this general construction pattern. These compounds have in common that they easily cross the blood-brain barrier causing sedation (Nicholson et al., 1991) and/or anticholinergic effects (Hill, 1990). At higher dosages, numerous examples of these compounds also display anesthetic effects that may have some therapeutic advantages on topical applications for urticaria or itch (Orhan et al., 2007; Murota and Katayama, 2011). However, it should also be noted that at high concentrations, first-generation H1 receptor antagonists, because of their cationic-amphiphilic properties, can also exhibit paradoxical proinflammatory effects that are mediated via a direct and receptor-independent activation of Gi proteins in cells of the immune system (Burde et al., 1996).
The class of (polycyclic) neuroleptics has been developed out of the lead structure of early H1 receptor antagonists. Therefore, it is not surprising that many of the neuroleptics still possess high H1 receptor antagonist affinities, causing sedation and weight gain. One may also assume that the antidepressants acting as selective inhibitors of neurotransmitter transporters have been detected by variation of the H1 receptor antagonist lead structure (e.g., diphenhydramine [6] versus fluoxetine), leading to a different pharmacological profile. It is clear that the members of family A of rhodopsin-like GPCRs share some sequence homology and therefore have comparable recognition areas for some structural motifs. These recurrently revealed fragments are termed privileged structures. The privileged structures can be detected in many compounds targeting the biogenic amine receptors in different combinations for receptor preference. With the H1 receptor, it is the diaryl element that may be incorporated in a tricyclic ring system, linked by the short chain to the amino group. By fulfilling all these requirements, the highly potent mepyramine (also called pyrilamine [8]) has often been used as reference antagonist and for radiolabeling studies (Tashiro and Yanai, 2007; Yanai and Tashiro, 2007) (see section II.C).
Ligand binding at the H1 receptor takes place in a stereoselective manner. The preference of one stereoisomer can be shown by different discriminative ratios for numerous compounds. Depending on the way of stereoselective orientation by stereocenter or stereoaxis and on the receptor interaction, the differences may be up to three orders of magnitude [e.g., E-configured (trans) triprolidine versus Z-configured isomer (cis)]. Point mutations in transmembrane helices IV and V show aromatic residues as potential stereoselective discriminators (Trp167, Phe433, Phe436) (Wieland et al., 1999). Although with cetirizine the difference between both enantiomers is only 2-fold, the “enantiomeric shift” has been exploited to make the more potent (R)-(−) enantiomer (levocetirizine; 11) (Gillard et al., 2002b; Chen, 2008). With the tricyclic compounds, including cyproheptadine or loratadine (14; Fig. 7), some conformational isomers have been observed. Normally these compounds undergo a rapid intraconversion, but in some cases the orientation of the piperidine moiety on the bend ring system remains stable and the remaining atropisomers (conformational enantiomers) can be separated (Remy et al., 1977; Randall et al., 1979). It is unclear if these effects can also be observed in vivo. Most investigations in this direction have been made on guinea pig systems and have been verified for human H1 receptors mostly for marketed compounds only.
In view of the sedative effects of the first generation antihistamines, their clinical usefulness has proven to be limited (see below), although some of the products are currently available as over-the-counter products as sleep aids. To target the important role of histamine in allergic conditions, the class of second-generation antihistamines has been developed. Second generation H1 receptor antagonists display substantially reduced central side effects, because they have a lower rate of penetration through the blood-brain barrier due to the presence of polar ionic structures or are high-affinity substrates for ATP-dependent P-glycoprotein or organic anion transport polypeptide efflux pumps (Devillier et al., 2008; Broccatelli et al., 2010). This approach has been realized by taking advantage of the active metabolites of antihistamines (e.g., hydroxyzine and cetirizine [cf. 11], terfenadine [12] and fexofenadine [13], ebastine and carebastine, and loratadine [14] and desloratadine [15] in Fig. 7) or by adding highly polar functional groups into the molecule, e.g., carboxylic acid (levocabastine, olopatidine, efletirizine [zwitterionic compounds] or acylguanidine (mizolastine [10]) (Fig. 7). Several of these compounds have successfully replaced the older first-generation H1 antagonists as therapeutic agents for allergic disorders (see next section).
In fact, it has been the detrimental interaction of several classic antihistamines with the cardiac hERG1 potassium channels that has triggered the development of these successful drugs. For terfenadine in combination with other CYP3A4 substrates or inhibitors (Bailey et al., 1998) or with liver dysfunction (Kamisako et al., 1995), the plasma levels are highly enhanced, leading to the inhibition of cardiac hERG1 potassium channels that then may lead to lethal arrhythmias as a result of QT prolongation (Salata et al., 1995; Lagrutta et al., 2008). Terfenadine (12) and astemizole were eventually withdrawn from the market (Estelle and Simons, 1999) in several, but not all, countries (you can still obtain terfenadine in Germany, for example) when they were associated with the risk of Torsades de Pointes and sudden death due to binding to hERG potassium channels. Although this has been questioned recently (Lu et al., 2012), these unfortunate developments led to fundamental changes in the drug discovery and development process. To predict potential adverse effects due to “antitarget” (“off-target”) binding, hERG screening is now undertaken at an early stage of the discovery process (e.g., Finlayson et al., 2004). After the terfenadine withdrawal it was found that the active zwitterionic metabolite of terfenadine (12), fexofenadine (13), although it has a slightly lower affinity for H1 receptors, is devoid of this QT-prolonging side effect and has an improved safety profile (Simpson and Jarvis, 2000). Fexofenadine is also less sedative, because it shows only a limited brain penetration, potentially because of its affinity for P-glycoprotein transporters (Molimard et al., 2004; Obradovic et al., 2007). Most of the second generation antihistamines (e.g., levo-cetirizine and olopatidine) share most of the properties of fexofenadine (13), including the zwitterionic nature. These compounds all show clearly improved safety profiles and have had reasonable therapeutic success.
F. Clinical Pharmacology
The prototypical “antihistamine” drugs chlorphenamine, clemastine, cyproheptadine, ketotifen, diphenhydramine (6), cyclizine, cinnarizine, buclizine, and promethazine (H1 receptor antagonists) have a long established place in the treatment of nausea and vomiting and allergy, specifically allergic rhinitis and conjunctivitis and anaphylactic shock, but not asthma (Bartho and Benko, 2013). Antihistamines have been formulated for local application to the nasal mucosa, eye, or skin (and together with mast cell stabilizers such as sodium cromoglicate can delay and diminish the symptoms precipitated by localized or systemic release of histamine) (Kari and Saari, 2012; Ridolo et al., 2014). Oral and topical antihistamines are used in the treatment of hay fever, insect bites/stings, and the combination of antipruritic and sedative effects is often beneficial in removing symptoms and in restoring sleep in seasonal allergy. The sedative side effects in the workplace, or while driving, are a significant problem with the first-generation of antihistamines and provided the biorational impetus for the development of congeners that were less brain permeant (the H1 receptor crystal structure explains well the differences between first- and second-generation H1 receptor antagonists, as discussed previously in section II.A). However, a recent study highlighted the need for caution in using such drugs in older hospitalized patients, with a suggested link to onset of delirium (Rothberg et al., 2013). It is disappointing that we still have no hypnotics that mimic or facilitate/extend physiologic sleep, bearing in mind the pivotal role biogenic amines underpin in sleep physiology, arousal, and circadian regulation (Lin et al., 2011a). Nevertheless, H1 receptor antagonists have been used rarely as CNS niche drugs. For example, H1 receptor antagonists have been used for their psychosomatic effects in dermatological conditions (Gupta and Gupta, 1996), cyproheptadine is used in pediatric migraine (Lewis et al., 2004a), and close structural congeners are used as neuroleptics and antidepressants (He et al., 2013; Sato et al., 2013).
Although the newer antihistamines generally display high H1 receptor affinity and higher selectivity versus nontargets than that of the older ones, side effects can still be observed (notably antimuscarinic effects and sedation when taken with alcohol or other depressant drugs (Simons and Simons, 1999; Yanai et al., 2011). With the patent expiry of many second-generation drugs and their widespread availability over the counter at low costs, the medicine agencies are looking for genuine innovation and novelty: one of the outcomes of this is the development of combination therapies, e.g., anti-inflammatory properties such as inhibition of neuropeptide or leukotriene signaling (Scannell et al., 2004; Beaton and Moree, 2010). One of the most recent compounds to enter this clinically and commercially important market is rupatadine (16) (Picado, 2006; Fantin et al., 2008), which blocks both peripheral H1 receptors and the platelet activation pathway, as a platelet-activating factor antagonist (Merlos et al., 1997). Efletirizine, however, was developed as a topical agent to fit the pharmacophores for H1 receptor antagonists and inhibitors of lipoxygenase (an enzyme involved in the production of leukotrienes) (Salmun, 2002; Lewis et al., 2004b) but was discontinued in the late phases of development. Regarding the H1 receptor antagonists already in the pharmacopoeia, few studies have been conducted on teratogenicity or embryotoxicity with high doses of antihistamines in animal testing, and it is recommended that antihistamines should not be used in pregnancy (Gilboa et al., 2014). The lack of any teratogenic effects reported thus far gives some confidence, but there should still be a note of caution because of the lack of controlled studies in children or pregnancy for the first-generation compounds (Church et al., 2010).
III. Histamine H2 Receptor
The H2 receptor was identified pharmacologically in 1972 based on the finding that several physiologic effects of histamine, including the stimulatory effect on gastric acid secretion, increase of heart rate, and inhibition of contraction of rat uterus were not antagonized by mepyramine (Black et al., 1972). Cloning of the H2 receptor allowed expression studies to be performed, which indicated strong expression in the stomach and brain (Gantz et al., 1991b). Clinically, H2 receptor antagonists transformed the treatment of dyspepsia, esophageal, and gastric ulcers, a trend that continued until these drugs were largely replaced by proton pump inhibitors. Apart from the brain and stomach, the H2 receptor is expressed in smooth muscle cells, chondrocytes, endothelial and epithelial cells, neutrophils, eosinophils, monocytes, macrophages, dendritic cells, T and B cells (Jutel et al., 2001, 2009).
A. Receptor Structure
Many years after the development of clinically useful H2 receptor antagonists, the H2 receptor was first cloned in 1991 by Gantz and colleagues from a canine genomic library, with subsequent rapid cloning of species orthologs from rat, mouse, guinea pig, and human over the subsequent four years (Gantz et al., 1991b; Ruat et al., 1991; Traiffort et al., 1995; Kobayashi et al., 1996). A single genomic clone was isolated that encoded a 1080-base pair (bp) open reading frame with a deduced 360-amino-acid polypeptide. The multiple transcription initiation sites of the human H2 receptor gene have been mapped and an 85-bp segment (−610 to −525 bp) immediately upstream of the initiation site was identified to exhibit a strong promoter activity in the gastric adenocarcinoma, MNK45, expressing the H2 receptor (Murakami et al., 1999). Several H2 receptor polymorphisms have been observed with changes, located in the gene promoter or in the coding region, including a nucleotide change leading to an Asn217Asp change (Orange et al., 1996a,b; Ito et al., 2000; Garcia-Martin et al., 2009; Micallef et al., 2013). Currently, no consensus has been obtained on a potential link of the Asn217Asp change to schizophrenia (Orange et al., 1996b; Ito et al., 2000)
Currently, there is no direct structural information available for the H2 receptor. Yet, the H2 receptor possesses many of the key prototypical amino acid residues common to the family A aminergic GPCRs, and homology modeling on the basis of the published H1 receptor X-ray structure and available mutagenesis data leads to reasonable models (Fig. 2) for the interaction of histamine with the receptor protein (Kooistra et al., 2013). Site-directed mutagenesis of residue Asp1173.32 in TM3 of the canine H2 receptor (= Asp983.32 in the human H2 receptor) led to complete loss of [3H]tiotidine (31) binding (Kelley et al., 2001). Mutation of either—alone or simultaneously—did not abolish histamine-induced cAMP but markedly reduced its efficacy. Asp1865.42, although it is essential for [3H]tiotidine antagonism, appears not to be crucial for cimetidine antagonism, indicating both shared and distinct sites (Gantz et al., 1992). On the basis of the mutagenesis data and molecular modeling using an H2 receptor model based on the H1 receptor X-ray structure, histamine is thought to bind with a different orientation in the binding pocket (Fig. 3).
B. Signal Transduction Mechanisms
The H2 receptor stimulates adenylyl cyclase–mediated cAMP formation in membrane preparations from various tissues as well as cAMP accumulation in various native cells (for review, see Hill, 1990, and Hill et al., 1997) and cells expressing recombinant H2 receptors (Alewijnse et al., 1998, 2000; Monczor et al., 2003). As for many adenylyl cyclase–coupled GPCRs, the H2 receptor stimulates cAMP production via the coupling to Gs proteins, as indicated by, for example, agonist-induced photoaffinity labeling. [α-32P]GTP azidoanilide showed that agonist-activation of H2 receptors results in labeling of Gsα-subunits with [α-32P]GTP azidoanilide in mammalian and insect cell expression systems (Kuhn et al., 1996; Leopoldt et al., 1997). Coupling to Gs proteins is furthermore indicated by studies with GPCR-Gα fusion proteins, which has permitted the analysis of H2 receptor/Gsα coupling with high sensitivity at the level of ternary complex formation (high-affinity complex between agonist-occupied receptor and nucleotide-free G protein), guanine nucleotide exchange, i.e., [35S]GTPγS binding and steady-state GTP hydrolysis, as well as adenylyl cyclase activation (Kelley et al., 2001; Wenzel-Seifert et al., 2001). H2 receptor-Gsα fusion proteins were also used to analyze ternary complex formation with guanidine-type agonists, which led to the suggestion that each agonist stabilizes a unique conformation in the H2 receptor (Kelley et al., 2001; Xie et al., 2006).
The H2 receptor does not only couple to Gs proteins but also to Gq/11 proteins, resulting in inositol phosphate formation and increases in cytosolic Ca2+ concentration in some, but not all, H2 receptor expressing cells (for previous reviews, see Hill, 1990, and Hill et al., 1997). Moreover, photoaffinity labeling studies with [γ-32P]GTP azidoanilide revealed that H2 receptor can activate both mammalian and insect cell Gq proteins in addition to Gs proteins (Kuhn et al., 1996; Leopoldt et al., 1997), whereas heterologous expression of H2 receptors in rat cardiomyocytes has also been reported to result in the interaction of recombinantly expressed H2 receptors with Gq/11, next to Gs proteins (Wellner-Kienitz et al., 2003). Interestingly, in this cell system also the Gs-cAMP-PKO pathway promotes increases in cytosolic Ca2+ concentration via the phosphorylation of L-type Ca2+ channels (Wellner-Kienitz et al., 2003).
The H2 receptor was the first histamine receptor subtype for which constitutive activation of adenylyl cyclase has been firmly established for both rat (Smit et al., 1996a) and human variants (Alewijnse et al., 1998, 2000; Monczor et al., 2003). Clinically used H2 receptor antagonists, including cimetidine (24) and ranitidine (25), behaved as inverse agonists, whereas up to now only burimamide has been described as neutral antagonist (Smit et al., 1996a). Interestingly, both rat and human H2 receptors are upregulated by prolonged receptor occupancy with inverse agonists (Smit et al., 1996a), most likely via both a reduction of cAMP-protein kinase A–mediated downregulation and a stabilization of an inherent structural instability of the H2 receptor (Alewijnse et al., 1998, 2000). So far the physiologic relevance and the structural basis of the constitutive activity of H2 receptors remain to be determined, but it is interesting to note that prolonged exposure of rabbit parietal cells to H2 receptor antagonists leads to an upregulation of H2 receptor protein, adenylyl cyclase, and ultimately to hypersecretion (Takeuchi et al., 1999). Moreover, the reported G649 allelic variant (Asn217Asn) of the human H2 receptor is reported to have reduced constitutive activity, lower histamine responses, and a diminished upregulation by ranitidine (Fukushima et al., 2001).
With respect to biased signaling, only limited information is currently available for the H2 receptor. A recent study reported that the inverse agonists ranitidine and tiotidine display positive efficacy toward extracellular signal-regulated kinase (ERK) 1/2 via a mechanism that involves Gβγ-subunits in both transfected HEK-293T cells and human gastric adenocarcinoma cells (Alonso et al., 2014). These findings suggest that H2 receptor biased signaling might be an important concept to consider.
As with many other GPCRs, the H2 receptor is desensitized and internalized upon agonist stimulation (Smit et al., 1996b; Fukushima et al., 1997). Truncations or replacements within the C-terminal tail of the receptor revealed that Thr315 was a crucial residue in agonist-dependent internalization but not desensitization (Smit et al., 1996b). Desensitization of the H2 receptor is suggested to involve both GPCR kinase 2 and 3 (Rodriguez-Pena et al., 2000; Fernandez et al., 2011). The C-terminal tail is required for membrane targeting through a putative palmitoylation-mediated mechanism (Fukushima et al., 1997), and the last 51 amino acids are reported to be crucial for internalization (Fernandez et al., 2008). Recently, the GTPase dynamin has been identified as a binding partner for the H2 receptor, both in vitro and in vivo (Xu et al., 2008). A role of dynamin, β-arrestin, and clathrin in H2 receptor internalization has indeed also been reported (Fernandez et al., 2008).
C. Anatomic Framework
In addition to stomach and brain, in which the expression level is high, the H2 receptor is expressed in smooth muscle cells, chondrocytes, endothelial and epithelial cells, neutrophils, eosinophils, monocytes, macrophages, dendritic cells, T and B cells (Jutel et al., 2009). The anatomically important uneven distribution of H2 receptors is described here to allow understanding of the role of histamine through H2 receptors in the regulation of gastric and neuronal functions.
Molecular cloning of the H2 receptor (Gantz et al., 1991a,b; Ruat et al., 1991) allowed detection and quantification of receptor mRNA in tissues. In the dog, which was the first species to be analyzed for the H2 receptor (Gantz et al., 1991b), high expression in the fundus of the stomach and moderate in the brain was reported. In the rat, Northern hybridization analysis has revealed high expression in various parts of the brain, including cerebral cortex, striatum, hippocampus, and hypothalamus. The highest mRNA expression in the rat brain was seen in brain stem (Ruat et al., 1991; Hirschfeld et al., 1992), where several other methods have suggested important physiologic actions. In another quantitative study, the expression in the stomach was reported to be 10-fold higher than that of several brain regions (Karlstedt et al., 2001). Expression in the frontal cortex, striatum, hippocampus, thalamus, hypothalamus, pons, and medulla was rather even, whereas the cerebellum expressed a significantly lower level of mRNA. In situ hybridization showed a much more even distribution than that detected for H1 receptors and H3 receptors; although almost all brain areas expressed H2 receptor mRNA, only a few areas including the piriform cortex, granule cell layer of the dentate gyrus, and hippocampal CA1, CA2, and CA3 showed high density of grains in the forebrain. The red nucleus expresses high levels of H2 receptor mRNA. In peripheral rat organs, the stomach expresses the highest amounts of H2 receptor mRNA (Traiffort et al., 1992b).
A detailed account on H2 receptor mRNA expression in the brain of the guinea pig (Vizuete et al., 1997) as revealed by in situ hybridization suggests widespread distribution in the cerebral cortex, particularly in layers III and V; hippocampal fields CA1, CA2, C3l; granular cell layer; and polymorph layer of the dentate gyrus, olfactory bulb, basal ganglia (particularly nucleus accumbens, caudate-putamen, islands of Calleja, olfactory tubercle and ventral pallidum). High expression is also characteristic of the amygdaloid complex, several thalamic, and hypothalamic nuclei. Distribution of H2 receptor mRNA and receptor radioligand binding in the rat brain are shown in Fig. 5 in comparison with H1 receptors and H3 receptors.
In human brain, H2 receptor radioligand binding is widely distributed with highest densities (measured using [125I]iodoaminopotentidine cf. 30) in the basal ganglia, hippocampus, amygdala, and cerebral cortex (Traiffort et al., 1992b). Lowest densities were detected in cerebellum and hypothalamus. A similar distribution was observed in guinea pig brain (Ruat et al., 1990). In human prefrontal cortex, cells in layer II show higher mRNA expression than the other layers, whereas the H2 receptor radioligand binding is highest in layers II and III (Jin and Panula, 2005).
In the gastric mucosa, where H2 receptor antagonists have long been known to exert inhibition of acid secretion, the H2 receptor localizes to parietal cells of the rat gastric mucosa, in more apical domains than the basal parts where HDC-expressing ECL cells, which are responsible for histaminergic regulation of parietal cell acid secretion, are localized (Panula and Wasowicz, 1993; Diaz et al., 1994). In the brain, H2 receptor expression and functions are not limited to neurons. There is a significant body of evidence suggesting the presence of H2 receptors in astrocytes (Kubo et al., 1991; Carman-Krzan and Lipnik-Stangelj, 2000) in vitro and in cultured brain endothelial cells (Karlstedt et al., 1999).
During embryonic development, H2 receptor mRNA is highly expressed in the rat brain from embryonic day 15 (Karlstedt et al., 2001). Particularly prominent expression is evident in the developing raphe area. From embryonic day 17, strong expression is found in superficial layers of the cortical plate and ventral hypothalamus. It is noteworthy that in rodents there is a distinct transient fetal histaminergic system in the raphe neurons (Auvinen and Panula, 1988; Vanhala et al., 1994), which implies that the fetal expression of H2 receptor mRNA may be functional during brain development.
Ligand binding methods for H2 receptors have also been useful to determine H2 receptor expression. Whereas early studies in guinea pig brain, lung, and transfected CHO cells have been successful using [3H]tiotidine (cf. 31) (Gajtkowski et al., 1983; Norris et al., 1984; Foreman et al., 1985; Gantz et al., 1991b), studies in rat brain and guinea pig mucosal cells were not successful (Maayani et al., 1982; Batzri and Harmon, 1986). The introduction of [125I]iodoaminopotentidine (cf. 30) was a major step forward, which provides reversible selective high-affinity binding with high sensitivity to membranes of, e.g., brain regions, and a suitable autoradiography radioligand (Hirschfeld et al., 1992). It has high affinity (KD = 0.3 nM) for the H2 receptor in brain membranes (Martinez-Mir et al., 1990; Ruat et al., 1990; Traiffort et al., 1992a) and transfected cells expressing rat or human H2 receptors (Traiffort et al., 1992b; Smit et al., 1996a). The compound has also been used for autoradiographic mapping of H2 receptors in the mammalian brain (Ruat et al., 1990; Traiffort et al., 1992a; Jin and Panula, 2005). High-affinity binding is detectable in widespread regions of rodent brain. In the guinea pig, high density of binding sites is characteristic of caudate putamen, nucleus accumbens, olfactory tubercle, superficial layers I–III of the cerebral cortex, superficial gray layer of the superior colliculus, and inferior olive. Moderate binding can be found in the deep layers of the cortex, the CA1–CA4 regions of the hippocampus, anterior and lateral nuclear groups of the thalamus, and some hypothalamic nuclei (Ruat et al., 1990). In a detailed mapping study of the ligand binding sites and comparison with mRNA expression in the guinea pig brain, the distribution of binding sites corresponded rather well with the distribution of histaminergic nerve fibers. In human prefrontal cortex H2 receptor binding is highest in layers II and III, although binding was detectable in all layers, and H2 receptor mRNA expression is highest in layer II (Jin and Panula, 2005). Discrepancies between RNA and ligand binding patterns corresponded generally to the expected difference in the translation site (cell bodies) and the site of action of the mature receptor protein (dendritic trees of neurons) (Vizuete et al., 1997) (Fig. 8).
Fig. 8.

H2 receptor and cyclic AMP–mediated potentiation of excitation in rat hippocampal slices. (A) Block of the accommodation of firing in a CA1 pyramidal cell leading to a potentiation of the response to a depolarizing pulse. (B) Block of the IAHP current, which is responsible for a long-lasting Ca2+-dependent afterhyperpolarization and the accommodation of action potential firing. (C) Long-lasting potentiation of glutamatergic synaptic transmission (Schaffer collateral-CA1 pathway). The lower curve (open symbols) illustrates the pure H2 receptor mediated effect under mepyramine (8), the upper curve illustrates the boosting by H1 receptor signaling (which is sensitive to Li+). This long-lasting potentiation (several hours) has the characteristics of the protein kinase A–mediated LLTP (late phase of long-term potentiation) achieved here without high-frequency stimulation. H1 receptor activation alone causes no potentiation, rather an inhibitory effect presumably through activating a Ca2+-dependent K+-current (not shown here) (modified from Selbach et al., 1997).
D. Function
H2 receptor activation stimulates the production of cyclic AMP, protein kinase A and the transcription factor cAMP response element-binding protein, which are key regulators of neuronal physiology and plasticity (Haas and Konnerth, 1983; McCormick et al., 1991; Pedarzani and Storm, 1995; Selbach et al., 1997; Atzori et al., 2000). In the CNS, H2 receptor activation can inhibit nerve cells (Haas and Bucher, 1975), but the most intriguing action is a block of the long-lasting after-hyperpolarization and the accommodation of firing, an effect with a remarkably long duration leading to potentiation of excitation in rodent (Haas and Konnerth, 1983) and human brain (Haas and Panula, 2003) (Fig. 8). A slow excitation is also common (Greene and Haas, 1990), e.g., in oriens-alveus interneurons (Haas et al., 2008), where at the same time, the fast spiking frequency is cut by Kv3.2-containing potassium channel activation (Atzori et al., 2000). A long-lasting potentiation of synaptic transmission in the hippocampus is induced or markedly enhanced (Kostopoulos et al., 1988; Brown et al., 1995; Selbach et al., 1997; Haas and Panula, 2003). An increase of the hyperpolarization-activated inward current Ih (HCN2) opens the door to consciousness by depolarizing thalamic relay neurons (McCormick et al., 1991). A potassium current was blocked through H2 receptors in olfactory bulb interneurons (Jahn et al., 1995). Furthermore, H2 receptor activation can inhibit phospholipase A2 and release of arachidonic acid, which likely accounts for opposing physiologic responses elicited by H1 receptor and H2 receptor in many tissues (Traiffort et al., 1992b). As most H2 receptor ligands hardly pass the blood-brain barrier, the behavioral testing of H2 receptor functions in vivo is difficult, although zolantidine (29) may be useful (see section III.E). Mice deficient in H2 receptor function display cognitive deficits, an impairment in hippocampal long-term potentiation (Dai et al., 2007), and abnormalities in nociception (Mobarakeh et al., 2005, 2006), gastric, and immune functions (Jutel et al., 2001; Teuscher et al., 2004; Schneider et al., 2014a). The suppression of immune responses via the H2 receptor was not only shown in H2 receptor-deficient mice but also in human antigen presenting cells, where H2 receptor stimulation resulted in the downregulation of numerous cytokines (Gutzmer et al., 2005; Glatzer et al., 2013).
E. H2 Receptor-Selective Ligands
The first agonist with some selectivity for the H2 receptor was 4(5)-methylhistamine (17), which is actually now used as selective H4 receptor agonist (Durant et al., 1975; Lim et al., 2005) (Fig. 9). Other small molecule ligands used as H2 receptor agonists are the aminothiazole derivative amthamine (18) (Eriks et al., 1992) and the isothiourea compound dimaprit (19), which displays moderate affinity and selectivity (Garbarg et al., 1992). Increased affinity that is higher than that of histamine is found with the longer chained guanidine derivatives arpromidine (20) or impromidine (21), although both compounds simultaneously display some H3 receptor antagonist potencies. Whereas impromidine possesses a methylated imidazole ring as with 4-methylhistamine, arpromidine has the fluorinated analogs moiety of the diaryl binding motif of the H1 receptor antagonist pheniramine (Buschauer, 1989; Leurs et al., 1995a). The homohistamine elements with the guanidine structure on both compounds are mainly responsible for activation, whereas the other elements are affinity increasing moieties (Dove et al., 2004). In this respect, the chiral forms of sopromidine are sometimes used as helpful pharmacological tools, because the (R)-(−) enantiomer sopromidine (22) is a potent H2 receptor agonist, whereas the (S)-(+) isoform shows moderate antagonist properties in some assays.
Fig. 9.

H2 receptor ligands.
H2 receptor agonists are not used therapeutically, although arpromidine (20) and related imidazolylpropylguanidines were developed as positive inotropic vasodilators for the treatment of severe catecholamine-insensitive congestive heart failure. More recently, aiming at improved drug-like properties, the guanidine group was replaced by an acylguanidine moiety, resulting in substantially reduced basicity (by 4–5 orders of magnitude), but retaining H2 receptor agonistic activity (Ghorai et al., 2008). Unfortunately, as also found for arpromidine- and impromidine-like compounds, numerous NG-acylated imidazolylpropylguanidines (e.g., UR-AK24) proved to be poorly selective for the H2 receptor. Some N-acylguanidines show an overlapping (partial) agonist profile for H1 and H2 receptors (Xie et al., 2006). However, many compounds have high affinity for H3 receptor and H4 receptor and were, therefore, considered as lead structures in the design of new H3 receptor and H4 receptor ligands (see sections IV.D and V.D). Thus, the imidazolylpropylguanidine moiety can be considered a privileged structure for histamine receptor ligands. The selectivity problem was solved by bioisosteric replacement of the imidazol-4-yl with a 2-aminothiazol-5-yl residue, optionally bearing a 4-methyl substituent as in amthamine. 2-Aminothiazoles, such as UR-BIT24, are about equipotent to the corresponding imidazoles at the H2 receptor but devoid of noteworthy activities at H1 receptors, H3 receptors and H4 receptors (Kraus et al., 2009).
Very recently, the acylguanidine motif was incorporated into bivalent ligands for the H2 receptor, resulting in the most potent H2 receptor agonists reported so far (Birnkammer et al., 2012). Searching for potential pharmacological tools to study hypothetical H2 receptor dimers (Fukushima et al., 1997), two hetarylpropylguanidine moieties were connected by alkanedioyl residues varying in chain lengths (spacer lengths between 6 and 27 Å were covered). Interestingly, the highest potency resided in bivalent ligands with linkers that are, according to the model suggested by Porthoghese (2001) for opioid receptor dimers, too short to allow for simultaneous occupation of the orthosteric binding pockets of both protomers of an H2 receptor dimer. The optimal distance between the pharmacophoric groups rather suggest binding to the orthosteric and an accessory binding site of the same protomer. In accordance with the structure-activity relationships of monovalent acylguanidines, bivalent ligands comprising one or two imidazole moieties have also remarkably high activities at H3 receptors and H4 receptors, whereas homobivalent aminothiazoles are highly selective for the H2 receptor. The data from the guinea pig H2 receptor (GTPase assay, GTPγS binding assay) were largely consistent with the results from the isolated guinea pig right atrium. For instance, compounds with a decanedioyl spacer (UR-AK381, UR-AK480, UR-BIT82) were (nearly) full agonists and achieved up to 4000 times the potency of histamine at recombinant human and guinea pig H2 receptor (Sf9 cell membranes), respectively (Birnkammer et al., 2012) (Fig. 9; Table 1).
The strong relationship of ligand binding areas responsible for receptor (in)activation can also be seen with the first marketed H2 receptor antagonist cimetidine (24) introduced by Sir James Black (Black et al., 1972; Ganellin, 1981), because this compound with the highly polar cyanoguanidine element is comparable to the affinity increasing element in impromidine (Fig. 2). The reduced basicity of a highly polar core group can also be seen with ranitidine (nitroethendiamine) (25), famotidine (27) (sulfamoylamidine), roxatidine acetate (amide) (28), or the moderately polar compound zolantidine (aminobenzothiazole, 29), which have poor penetration into the brain with the exception of zolantidine (Baker, 2008). The imidazole moiety of cimetidine has been replaced by other bioisosteric heterocyclic moieties to reduce interaction with CYP3A4 enzymes and to overcome intellectual property issues. Although the H2 receptor antagonists have been replaced as first regimen therapeutics in peptic ulcer disease by the proton inhibitors, they are still commonly used as safe and reliable therapeutics. Ironically one of the first "selective" H2 receptor antagonists, the thiourea derivative burimamide (23) with low oral bioavailability used in the development of cimetidine, has also been used for the pharmacological characterization of H3 (antagonist) and H4 (agonist) receptors because of its affinities and properties at these receptor subtypes.
F. Clinical Pharmacology
The H2 receptor antagonists cimetidine (the first to be launched by Sir James Black’s team in 1977), ranitidine (25), nizatidine (26), and famotidine (27) are well established treatments for patients presenting with dyspepsia, gastric or duodenal ulcers, or gastroesophageal reflux disease (in the United States) (Hershcovici and Fass, 2011; Sigterman et al., 2013). Since the launch of antibiotic therapies for Helicobacter infections (based on the seminal work [Marshall and Warren, 1984; Marshall et al., 1988]) and the discovery of proton pump inhibitors (Olbe et al., 2003), the antisecretory H2 receptor antagonists are no longer alone at the frontline (with antacids) as symptomatic treatments. Alternating treatment with proton pump inhibitors (the preferred frontline treatment) and H2 receptor antagonists is used to treat both gastroesophageal reflux disease and more severe refractory reflux disease, e.g., esophageal ulceration or Barrett’s esophagus (Hershcovici and Fass, 2011). Treatment with H2 receptor drugs is generally considered safe and efficacious and is available to large numbers of patients (over the counter in pharmacies for some ligands and as prescription only for others). The symptom suppression elicited by H2 receptor drugs, however, may mask gastric cancer as has been reported for proton pump inhibitors (Wayman et al., 2000), and the drugs should be used with caution in patients with alarm symptoms. The safety profile, in part, reflects the polar nature of licensed H2 receptor antagonists, which makes them available in the periphery, but to a lesser extent in the CNS (qualified below). Although H2 receptor ligands (particularly agonists) have limited selectivity (particularly for H3/H4 receptors) H2 receptor-activation in the brain mediates important functions (Haas and Konnerth, 1983; Vizuete et al., 1997; Haas and Panula, 2003), and rare reports of CNS toxicity (including depression, confusion, hallucinations) indicate the drugs can enter the brain, particularly in the elderly (Moore and O'Keeffe, 1999), and in patients with renal impairment, although this is often clouded by polypharmacy and concomitant CNS disease. Therapeutic applications for CNS-permeant ligands like zolantidine (29) are so far rare, although there is a considerable body of preclinical research highlighting the involvement of H2 receptors in supraspinal nociception (Nalwalk and Hough, 1995; Nalwalk et al., 1995) and the potential of H2 receptor antagonists to enhance the effects of opiate analgesics (Mobarakeh et al., 2009) and to exert analgesic effects in models for cholestasis and pruritis (Hasanein, 2011). Further studies have indicated potential utility in psychotic disorders, and in a case report (Kaminsky et al., 1990), a positive therapeutic effect of oral famotidine was reported in a schizophrenic patient. Although one small open-label study (Whiteford et al., 1995) reported no significant effects, two larger studies (Rosse et al., 1990; Deutsch et al., 1993) reported significant therapeutic effects in a group of patients with schizophrenia. A recent randomized double-blind, placebo-controlled study with famotidine in treatment-resistant schizophrenia showed statistically significant improvement in the positive and negative syndrome scale (Meskanen et al., 2013). H2 receptor agonism blocks accommodation of firing and causes long-term potentiation in hippocampus and cortex (Selbach et al., 1997; Haas and Panula, 2003), which may lead to cognitive enhancement and be worthy of further study.
IV. Histamine H3 Receptor
Histamine and its receptors were generally known as mediators connected to allergic and inflammatory reactions before the detection of the histamine H3 receptor in 1983 proved its neglected neurotransmitter function as auto- as well as heteroreceptor at pre- and postsynaptic membranes and revealed its profound influence on different neurotransmitter balances. Although versatile pharmacological tools, localization of H3 receptor, and influence on brain functions have been described in the early years, the development line was curtailed for a while. Revitalization came from the cloning of the receptor and the detection of nonimidazole antagonists. Actually, the broad potential therapeutic indications due to the multiple neurotransmitter balance influenced by H3 receptors raise numerous and heterogeneous possibilities in pharmaceutical industry for innovative blockbusters as well as for pipe-dreams.
A. Receptor Structure
The H3 receptor was first described in 1983 on the basis of pharmacological evidence (Arrang et al., 1983). Because of the low overall homology of the H3 receptor with either of the previous cloned H1 receptors and H2 receptors, it was not until 1999 that the H3 receptor cDNA was finally cloned (Lovenberg et al., 1999). Lovenberg and colleagues used a partial cDNA clone (GPCR97) obtained from an Expressed Sequence Tags database that had significant general homology to biogenic amine receptors. The GPCR97 clone was used to probe a human thalamus cDNA library, which resulted in isolation of a full-length clone encoding a putative GPCR. Homology analysis showed the highest homology with M2 muscarinic acetylcholine receptors and overall low homology with all other biogenic amine receptors. The cDNA encoded an open reading frame of 445 amino acids, which upon heterologous expression was responsive to histamine with an H3 receptor pharmacological profile. The human H3 receptor gene has been mapped to chromosome 20q13.33 and is highly expressed in the CNS (Coge et al., 2001a; Tardivel-Lacombe et al., 2001). As with the other members of the histamine receptor family, H3 receptors are characterized by classic sequence motifs of the family A GPCRs binding biogenic amines. Currently no structural information is available, and all ligand-binding mode hypotheses are based on modeling efforts. Like for all biogenic amine receptors, the H3 receptor contains the conserved Asp residue in TM3 (Asp1143.32), which is suggested to be involved in the binding of the ethylamine side chain of histamine (Fig. 2) (Kooistra et al., 2013). The binding mode of histamine in a homology model for the H3 receptor furthermore suggests that the imidazole ring donates a hydrogen bond to E2065.46 in TM5, similar to the interaction with Asn1985.46 in H1 receptor. Because the E2065.46 can form stronger bonds with the Nτ-nitrogen of histamine than Asn1985.46, this might also explain the substantially higher affinity of histamine for H3 receptors (and also H4 receptors; see Fig. 3; Tables 1 and 2) (Kooistra et al., 2013).
TABLE 2.
Binding affinities for selected histamine receptor ligands
| Compound # | Ligand | hH1R pKia | hH2R pKib | hH3R pKic | hH4R pKid |
|---|---|---|---|---|---|
| 1 | Histamine | 4.6 | 5.1 | 8.2 | 8.1 |
| 2 | 2-(2-Thiazolyl)-ethylamine | 5.3 | <5 | ||
| 4 | Histaprodifen | 5.70 | <5 | ||
| 6 | Diphenhydramine | 7.89 | 5.80 | 4.60 | 4.37 |
| 8 | Mepyramine | 8.80 | 4.63 | <5 | <5 |
| 10 | Mizolastine | 9.10 | <5 | ||
| 11 | Levocetirizine | 8.15 | <6 | <6 | <4 |
| 12 | Terfenadine | 7.92 | <5 | <5 | |
| 13 | Fexofenadine | 7.54 | <5 | ||
| 14 | Loratadine | 7.21 | <5 | 4.74 | |
| 15 | Desloratadine | 8.40 | 6.45e | <5 | 5.10 |
| 17 | 4(5)-Methylhistamine | <5 | 5.10 | 4.72 | 7.73 |
| 18 | Amthamine | 6.82 | 5.30 | ||
| 19 | Dimaprit | <5 | 4.60 | 6.08 | 6.42 |
| 20 | Arpromidine | 6.5 | 6.3-8.0 | ||
| 21 | Impromidine | 5.2 | 7.64 | 6.99 | 7.76 |
| 23 | Burimamide | 4.3f | 5.41 | 7.36 | 6.97 |
| 24 | Cimetidine | 4.75 | 5.84 | 4.69 | 5.03 |
| 25 | Ranitidine | 4.47 | 6.67 | 4.89 | <5 |
| 27 | Famotidine | <5e | 7.56 | <5 | |
| 30 | Aminopotentidine | 4.60 | 7.40 | 7.10 | <5 |
| 31 | Tiotidine | <4 | 7.77 | <4f | <5 |
| 32 | Nα-Methylhistamine | 4.20 | 4.30 | 9.00 | 7.13 |
| 33 | (R)-α-Methylhistamine | <5 | <4 | 8.6 | 6.76 |
| 34 | Imetit | 4.30 | 5.10 | 9.29 | 8.48 |
| 35 | Immepip | 4.79 | <3.5 | 9.44 | 7.79 |
| 36 | Methimmepip | 9.00 | 5.70 | ||
| 38 | Proxyfan | 8.2 | 7.30 | ||
| 40 | Thioperamide | 3.88 | 4.27 | 7.6 | 7.04 |
| 41 | Clobenpropit | 5.45 | 5.47 | 9.0 | 7.98 |
| 42 | Ciproxifan | <5 | <5 | 7.1 | 5.73 |
| 45 | Pitolisant | 5.78 | 4.96 | 8.06 | <4 |
| 48 | Conessine | <5 | <5 | 8.3 | <5 |
| 55 | MK-0249 | <5 | <5 | 8.2 | <5 |
| 56 | Clozapine | 8.65 | 6.82 | <4 | 6.26 |
| 58 | JNJ-7777120 | <5 | <6 | 5.30 | 8.09 |
| 67 | ZPL-389378 | 6.73 | 8.31 | ||
| 72 | JNJ-39758979 | <6 | <6 | 6.0 | 7.9 |
| 74 | INCB-38579 | <5 | <5.3 | 6.39 | 8.3 |
| 75 | ST-1006 | <6 | <6 | 6.29 | 7.9 |
Data from Tran et al. (1978); Chang et al. (1979); Kanba and Richelson (1984); De Backer et al. (1993); Leurs et al. (1994a); Vollinga et al. (1994). Sharif et al. (1996); Merlos et al. (1997); Bakker et al. (2001); Gillard et al. (2002a); Esbenshade et al. (2003); Govoni et al. (2003); Thurmond et al. (2004, 2014a,b); Bielory et al. (2005); Xie et al. (2006); Ligneau et al.(2007); Nagase et al. (2008); Zhao et al. (2008); Sander et al. (2009); Rossbach et al. (2011); Appl et al. (2012); Shin et al. (2012); Savall et al. (2014); Where multiple values are reported, they have mostly been averaged.
Data from Harada et al. (1983); Gantz et al. (1991b); Leurs et al. (1994a, 1995b); Sharif et al. (1996); Kreutner et al. (2000); Saitoh et al. (2002); Esbenshade et al. (2003); Thurmond et al. (2004, 2014a,b); Bielory et al. (2005); Lim et al. (2005); Xie et al. (2006); Preuss et al. (2007); Nagase et al. (2008); Zhao et al. (2008); Sander et al. (2009); Rossbach et al. (2011); Shin et al. (2012); Savall et al. (2014); and Levocetirizine New Drug Application #22-064; Loratadine New Drug Application #21-165. Where multiple values are reported, they have mostly been averaged.
Data from West et al. (1990); Vollinga et al. (1994); Sharif et al. (1996); Kato et al. (1997); Lovenberg et al. (1999); Ligneau et al. (2000, 2007); Coge et al. (2001b); Liu et al. (2001a); Wieland et al. (2001); O'Reilly et al. (2002); Wellendorph et al. (2002); Wulff et al. (2002); Esbenshade et al. (2003); Thurmond et al. (2004, 2014a,b); Bielory et al. (2005); Lim et al. (2005); Gbahou et al. (2006); Nagase et al. (2008); Zhao et al. (2008);Sander et al. (2009); Mowbray et al. (2011); Rossbach et al. (2011); Appl et al. (2012); Shin et al. (2012); Andaloussi et al. (2013); Savall et al. (2014); and Levocetirizine New Drug Application #22-064. Where multiple values are reported, they have mostly been averaged.
Data from Ligneau et al., 2000; Liu et al., 2001b; Morse et al., 2001; Zhu et al., 2001; O'Reilly et al., 2002; Esbenshade et al., 2003; Thurmond et al., 2004, 2014a,b; Lim et al., 2005; Gbahou et al., 2006; Ligneau et al., 2007; Nagase et al., 2008; Zhao et al., 2008; Deml et al., 2009; Sander et al., 2009; Mowbray et al., 2011; Rossbach et al., 2011; Appl et al., 2012; Shin et al., 2012; Andaloussi et al., 2013; Savall et al., 2014. Where multiple values are reported, they have mostly been averaged.
Guinea pig.
Rat.
After the cloning of the human H3 receptor, the receptor was cloned in other species through sequence homology screening approaches (Lovenberg et al., 1999; Coge et al., 2001a,b). The rat full-length H3 receptor was cloned in 2000 (Lovenberg et al., 1999; Liu et al., 2000) followed by guinea pig (Tardivel-Lacombe et al., 2001), mouse (Chen et al., 2002), and monkey H3 receptor (Yao et al., 2003). The H3 receptor is approximately 92% conserved between the different species (Hancock et al., 2003). Despite this high conservation, substantial species differences can still sometimes be observed (e.g., Lovenberg et al., 2000).
Rare among GPCRs, the H3 receptor gene has been reported to consist of either three exons and two introns (Tardivel-Lacombe et al., 2001; Wiedemann et al., 2002) or four exons and three introns (Coge et al., 2001a). The diverse exon/intron junctions result in alternative splice sites producing multiple, distinct receptor isoforms (Drutel et al., 2001). The existence of multiple human H3 receptor isoform mRNAs has opened up possible explanations to account for the pharmacological heterogeneity reported in the 1980s and 1990s for the H3 receptor, within and across species. The reasons for this heterogeneity are complex and still not fully understood. H3 receptor isoforms have been identified in the rat (Drutel et al., 2001; Morisset et al., 2001), mouse (Rouleau et al., 2004; Krementsov et al., 2013), and guinea pig (Tardivel-Lacombe et al., 2000).
In all mammalian species tested thus far, the full-length H3 receptor encodes a polypeptide of 445 amino acids. The 445-amino-acid H3 receptor is an N-linked glycoprotein with a Mr 47,000 (monomeric) and Mr 90,000 (dimeric) based on recombinant H3 receptor 445 expressed in HEK293 cells and immunoblotting (Bakker et al., 2006). The number of H3 receptor isoforms possible is high because of simultaneous occurrence of multiple splicing events in the same H3 receptor mRNA. The mechanisms regulating alternative splicing of the H3 receptor gene remain to be investigated.
Approximately 20 shorter human isoforms have been identified to date with deletions in the N terminus, second TM domain, and first extracellular loop, intracellular loop or at the C terminus (regions A–D; see Fig. 10). (Leurs et al., 2005). Alternative splicing in regions A (red) and/or B (orange) results in partial deletion of the N-terminal domain and/or TM2 and EL1, respectively (not shown). Such splicing events will result in nonfunctional GPCR isoforms. In contrast, distinct splice events in region C (green) results in an IL3 (intracellular loop 3) of variable length and/or deletion of TM5, -6, and/or -7. Alternative splicing in region D (blue) adds eight additional residues to the C-terminal tail of H3 receptors. Finally, a frameshift after residue 171 results in a truncated receptor protein (magenta) (Leurs et al., 2005). Isoforms that have deletions in their third intracellular loop have attracted significant interest because of the coupling of the third intracellular loop with G proteins (Drutel et al., 2001). These isoforms show variation in their pharmacological profile (Hancock et al., 2003), such as agonist potencies, signaling properties and expression patterns (Drutel et al., 2001), and constitutive activity (Morisset et al., 2001; Bongers et al., 2007b).
Fig. 10.

Schematic overview of several 7TM and truncated H3 receptor isoforms. Alternative splicing in regions A (red) and/or B (orange) results in partial deletion of the N-terminal domain and/or TM2 and EL1, respectively (isoforms not shown). Distinct splice events in region C (green) results in an IL3 of variable length and/or deletion of TM5, -6, and/or -7. Alternative splicing in region D (blue) adds 8 additional residues to the C-terminal tail of H3R. Finally, a frameshift after residue 171 results in a truncated receptor protein (magenta). Truncated H3 receptor isoforms are indicated in gray.
Regional topographical differences for the isoform mRNAs has given rise to speculation that H3 receptor heterogeneity could underlie different activities and functions of the H3 receptor in specific brain regions (Coge et al., 2001a; Drutel et al., 2001). Furthermore, there is growing evidence that homo- and hetero-oligomerization of H3 receptor isoforms occur and yield a novel regulatory mechanism (Bakker, 2004). Not all of the isoforms are likely to be expressed at the surface as a receptor, e.g., rat isoforms H3D, H3E, and H3F. These isoforms have been shown to act as dominant negatives in vitro to either directly or indirectly control surface expression of the rat isoforms H3A, H3B, and H3C (Bakker et al., 2006). Differential and overlapping expression of rodent H3 receptor isoforms has been reported (Drutel et al., 2001). Immortalized rat brain vascular endothelial cells express both the full-length isoform H3A and H3C (Karlstedt et al., 2013) Previously, Coge et al. (2001a) had shown two human isoforms that varied from the full length in terms of binding and signal transduction. To determine the biologic role of the different isoforms, the pharmacological properties of the human H3 receptor431 and the human H3 receptor365 were compared with the full-length human H3 receptor445. The human H3 receptor431 was unable to bind [125I]iodoproxyfan because of a deletion in the second transmembrane. More recently, Bongers et al. (2007b) reported, based on mRNA levels, differential expression of the human H3 receptor365 and the human H3 receptor445, with the human H3 receptor365 displaying higher expression levels than the human H3 receptor445 in many brain structures. The human H3 receptor365 also displayed higher affinity and potency for H3 receptor agonists and conversely a lower potency and affinity for H3 receptor inverse agonists. The human H3 receptor365 also displayed higher constitutive signaling compared with the human H3 receptor445 in both [35S]GTPγS binding and cAMP functional assays. Lower expression patterns were observed for the human H3 receptor415, human H3 receptor413, and human H3 receptor329, which also bind H3 receptor ligands and exhibit subtle differences in coupling to signaling mechanisms. Protein expression of the various human isoforms in brain tissue awaits confirmation and encourages the development of isoform-selective antibodies.
Genetic polymorphisms have also been identified within the human H3 receptor gene, which may further contribute to the wide diversity of the H3 receptor pharmacology. Independent publications and GenBank submissions of the human H3 receptor sequences have either a glutamic acid or aspartic acid at position 19 (Hancock et al., 2003). Another polymorphism has been identified at position 280 (IL3), an alanine to valine substitution in a patient suffering with Shy-Drager syndrome, which is also known as neurologic orthostatic hypotension, a disease characterized by neuronal degeneration and autonomic failure. Abbott Laboratories (Abbott Park, IL) confirmed this finding as well as another polymorphism at position 197, a tyrosine to a cysteine substitution (Hancock et al., 2003). Several single amino acid polymorphisms have also been identified in the third intracellular loop in rat H3 receptors (Coge et al., 2001a). Similarly, polymorphic sites are thought to be present in the mouse H3 receptors because complete sequences do not match partial sequences (Goodearl et al., 1999). For most of the reported polymorphisms no functional data are available. Yet expression of the Shy-Drager syndrome linked Ala280Val H3 receptor mutant on CHO-K1 cells showed that this mutant receptor showed reduced signaling efficacy in both cAMP signaling and [35S]GTPγS assays (Flores-Clemente et al., 2013).
B. Signal Transduction Mechanisms
The H3 receptor mediates pertussis toxin-sensitive [35S]GTPγS binding in rat cerebral cortex membranes (Clark and Hill, 1996) and a variety of other brain areas and transfected cells (Rouleau et al., 2002; Bongers et al., 2007b). In addition, pertussis toxin-sensitive inhibition of N-type Ca2+ channels has been reported in human and guinea pig heart (for previous review, see Bongers et al., 2007a). These data point to the H3 receptor being a Gi/Go protein–coupled receptor. In agreement with this concept, the human and rat H3 receptors recombinantly expressed in, e.g., SK-N-MC neuroblastoma cells, are negatively coupled to cAMP accumulation (Lovenberg et al., 1999, 2000). The human H3 receptor and rat H3 receptor expressed in Sf9 insect cells couple equally well to Gi1, Gi2, Gi3, and Go1 as assessed by measurement of steady-state GTPase activity upon H3 receptor/G protein coexpression (Schnell et al., 2010).
Activation of Gi/o proteins by H3 receptors also results in the activation of mitogen-activated protein kinase (MAPK) pathways (Drutel et al., 2001; Giovannini et al., 2003). Using rat H3 receptor isoforms with different IL3 loops transiently expressed in COS-7 cells, Drutel et al. (2001) showed that H3 receptor agonists stimulate MAPK phosphorylation in a pertussis toxin–sensitive manner. Interestingly, the long (445-amino-acid) isoform H3A coupled considerably better to MAPK phosphorylation compared with the shorter H3B and H3C isoforms (Drutel et al., 2001). In contrast, the rat H3 receptor isoforms with deletions in the third intracellular loop were more effective to activate cAMP-responsive element-dependent transcription, compared with the full-length H3A receptor (Drutel et al., 2001). MAPK activation after H3 receptor activation is not restricted to recombinant systems but has also been observed in native tissue (Giovannini et al., 2003). In rat hippocampal CA3 cells MAPK activation via H3 receptor has been linked to memory improvement (Hill et al., 1997). Similarly, Gi/o activation via H3 receptors can activate PI3K, leading to the subsequent activation and phosphorylation of protein kinase B (also known as Akt). Activated protein kinase B subsequently phosphorylates and thereby inhibits the action of glycogen synthase kinase 3-β, a major tau kinase in the brain (Bongers et al., 2007c) linked to several diseases, e.g., Alzheimer’s disease.
Other pathways modulated by the H3 receptor include G16α-mediated increase in cytosolic calcium ion concentration (Krueger et al., 2005), inhibition of histamine synthesis and release (Torrent et al., 2005; Moreno-Delgado et al., 2006), decrease of the cytosolic Ca2+ concentration in sympathetic nerve endings (Silver et al., 2001, 2002), and inhibition of Na+/H+ exchange activity (Silver et al., 2001).
In addition to agonist-induced signaling, the H3 receptor also signals in an agonist-independent, constitutive manner, thereby being one of the few GPCRs for which constitutive signaling has been observed in vivo. Both the rat H3 receptor and human H3 receptor exhibit high constitutive activity, resulting in high basal GTPγS binding (Morisset et al., 2000; Rouleau et al., 2002). The high basal GTPγS binding is inhibited by inverse agonists such as thioperamide and ciproxifan. Moreover, constitutive activity of the H3 receptor results in inhibition of forskolin-stimulated cAMP accumulation in recombinant systems, agonists further decreasing cAMP accumulation, and inverse agonists enhancing cAMP accumulation (Wieland et al., 2001). Moreover, constitutive H3 receptor activation leads to increased arachidonic acid release and inhibition of neurotransmitter release in vivo (Morisset et al., 2000; Rouleau et al., 2002).
Like the other histamine receptors, H3 receptors are able to form homo- and heterodimers (Panula and Nuutinen, 2013). Whereas the biochemical evidence seems to indicate that the H3 receptor isoforms can form homo- and heterodimers, the direct relevance for signaling or functional responses has not been elucidated. Behavioral and signaling studies have provided more conclusive evidence that H3 receptor can form functional heterodimers with dopamine receptors (Ferrada et al., 2008, 2009). Activation of H3 receptors inhibits D1 receptor agonist SKF-81297–induced cAMP accumulation in slices of striatum (Sanchez-Lemus and Arias-Montano, 2004) and inhibits D1 receptor-dependent release of [3H]GABA from rat striatum (Arias-Montano et al., 2001). Furthermore, in transfected cells, H3 receptor or D1 receptor signaling can be blocked not only by the selective respective antagonist but also by a selective antagonist of the partner receptor (Ferrada et al., 2009), and H3 receptor or D1 receptor antagonists can block MAPK signaling induced by H3 receptor or D1 receptor agonists (Moreno et al., 2011) in the brain. In this context, it is interesting that histaminergic neurons, which project to the striatum, contain dopa-decarboxylase and can produce dopamine after uptake of dopa that can be released from nearby dopaminergic neurons or is available in patients treated with l-dopa for Parkinson’s disease (see Yanovsky et al., 2011).
C. Anatomic Framework
Cloning of the H3 receptor in 1999 allowed verification of the high expression of the receptor in the brain (Lovenberg et al., 1999). This first report also established that the H3 receptor is abundantly expressed in very important areas of the brain (Fig. 5), e.g., cerebral cortex, thalamus, caudate putamen, ventromedial nucleus of the hypothalamus, and several aminergic projection systems, including histaminergic tuberomamillary nucleus (TMN) neurons and the noradrenergic neurons of locus coeruleus. Subsequently, detailed analyses of H3 receptor mRNA expression in rat brain (Pillot et al., 2002b) and findings on differential expression of the H3 receptor isoforms (Drutel et al., 2001) have further contributed to the current understanding that the H3 receptor plays important roles in multiple functions in the CNS. In the cerebral cortex of the rat, intense labeling in in situ hybridization samples was characteristic of layers V in most cortical areas and in layer III (particularly in the secondary motor cortex), granular layer IV of the auditory cortex, and layer VI of the somatosensory and auditory cortices (Pillot et al., 2002a). Expression of the different splice forms in the rat brain shows differences that may be functionally significant: there are differences in expression patterns at least in the cerebral cortex, hippocampus, and cerebellum (Drutel et al., 2001). The distribution of H3 receptor mRNA and ligand binding in comparison with the H1 and H2 receptor are shown in Fig. 5.
In human brain, the expression of H3 receptor mRNA in the prefrontal cortex is higher in layer V than in any other layer in Brodmann’s areas 8–11,24, 32,44, 45, 46, and 47, i.e., in almost all areas investigated (Jin and Panula, 2005). In contrast, the [3H]Nα-methylhistamine binding in the same samples is highest in layers III and IV, suggesting that most of the receptors occur either on the dendrites of the pyramidal neurons or in the thalamocortical axons. The lowest binding density is seen in the superficial layer I and deep layer VI (Jin and Panula, 2005).
H3 receptor mRNA is expressed at a high intensity in the human dorsal thalamus, the only part of thalamus that projects to the cortex. High to moderate expression is found in most of the principle relay nuclei (anterior, lateral, and medial nuclei, lateral geniculate nucleus), midline, and internal medullary laminar nuclei, whereas the expression level is low in the medial geniculate nucleus, posterior thalamic region (pulvinar), and the ventral thalamus (reticular nucleus, zona incerta). The expression is particularly abundant in the associate relay areas that project to the prefrontal cortex, notably the mediodorsal thalamic nucleus (Jin et al., 2002). H3 receptor binding sites are detectable using [3H]Nα-methylhistamine also in human thalamus (Jin et al., 2002). The highest density of binding sites was seen in the midline nuclei (reuniens, paraventricular, subhabenula), mediodorsal nucleus, some of the internal medullary laminar nuclei (parafascicular, paratenial, fasciculosus), and parts of the pulvinar (medial, inferior, diffuse). The binding level is generally low in other parts of the thalamus. This distribution is in agreement with the concept that thalamic neurons, which express H3 receptors, project to the cortex, where radioligand binding is detected on presynaptic terminals.
In the basal ganglia, H3 receptor mRNA signal intensity is observed in decreasing order in the following structures: putamen > frontal cortex > globus pallidus externum > globus pallidus internum (Anichtchik et al., 2001). In the mesencephalon (at the level of the superior colliculus), in substantia nigra, H3 receptor mRNA in situ signal is very low or undetectable, whereas a specific signal is observed in the red nucleus. In the corpus striatum, the highest expression of H3 receptor mRNA is detected in the putamen, whereas in the paleostriatum the expression is low and barely higher than the nonspecific background (Anichtchik et al., 2001). In normal human brain, the H3 receptor radioligand binding level is found in decreasing order: substantia nigra > putamen > globus pallidus externus > globus pallidus internus > frontal cortex (Anichtchik et al., 2001). This distribution suggests that the striatonigral projection neurons express H3 receptors, which is likely to regulate the release of GABA within the substantia nigra. No evidence for expression of H3 receptors in human dopaminergic nigral neurons was found in one study (Anichtchik et al., 2001). In Parkinson’s disease, H3 receptor mRNA expression in the pallidum externum is elevated compared with the normal brains. In Parkinson’s disease substantia nigra, an increase of the receptor binding density has been detected (Anichtchik et al., 2001). This increase in receptor binding coincides with a clear increase in histamine content (Rinne et al., 2002) and histamine-immunoreactive nerve fibers (Anichtchik et al., 2000) in substantia nigra, suggesting that either the disease process itself, e.g., through an effect of decreased dopamine or associated medication, modifies the histaminergic system.
During embryonic development, the H3 receptor is expressed early (from embryonic days 14 and 15 onward) in the CNS of the rat (Heron et al., 2001; Karlstedt et al., 2003). The expression is prominent in the midbrain ventricular epithelium, in medulla oblongata, and spinal cord, which suggests developmental roles for H3 receptors, and in brown fat, which suggests a role in energy metabolism or development of these cells (Karlstedt et al., 2003). In another study, which used cRNA probes instead of an oligonucleotide probe, a more widespread distribution of H3 receptor mRNA in developing rat tissues was observed, with significant signals in spinal ganglia, salivary glands, respiratory epithelium, gastric and intestinal mucosa, skin, thymus, liver, heart, and kidney (Heron et al., 2001). This widespread distribution indicates that during development, H3 receptor functions may extend well beyond the nervous system.
Antibodies have been generated against synthetic peptides from the human and rat H3 receptors to localize the receptor protein in mammalian tissues. One of these antibodies detects immunoreactive species at 62 and 93 kDa in mouse brain (Chazot et al., 2001). Immunoreactivity was localized in pyramidal neurons of lamina V of the rodent cerebral cortex and in medium-sized neurons in the striatum, cerebellar Purkinje cells, and the pyramidal cell layer of the hippocampus (Chazot et al., 2001). The H3 receptor mRNA distribution in rat (Pillot et al., 2002a) and human (Jin and Panula, 2005) cortex is mainly in deep laminae, whereas the receptor radioligand binding is more even (Pillot et al., 2002a; Jin and Panula, 2005). The receptor immunoreactivity could be expected in the same areas as radioligand binding, which motivates further studies using parallel immunocytochemistry with in situ hybridization and/or radioligand binding analysis to verify the results. In adult rat stomach, the H3 receptor is found in ECL cells, which also express HDC (Grandi et al., 2008). In the sensory system of the rat and mouse, H3 receptor immunoreactivity resides in large Aδ fibers that terminate in Meissner’s corpuscles surrounded hair follicles (Cannon et al., 2007). In dorsal root ganglia, H3 receptor immunoreactivity was localized in medium-sized and large cells, which also expressed calcitonin gene-related peptide; H3 receptor KO mice did not show this expression, suggesting selectivity of labeling. The localization suggests that H3 receptor containing fibers are involved in high-threshold mechanical nociception. Epidermal Merkel cells and keratinocytes also express H3 receptor-like immunoreactivity. In the locus coeruleus, histamine activates, through H2 receptors, noradrenergic neurons that inhibit neuropathic pain. This descending inhibition may be facilitated through blocking the H3 receptor-mediated autoinhibition of histaminergic terminals in the locus coeruleus (Wei et al., 2014).
Although presence of the H3 receptor in rodent sensory ganglia on a subset of Aδ fibers and lack of reactivity in H3 receptor KO mice has been shown using immunocytochemistry (Cannon et al., 2007), H3 receptor expression in peripheral ganglia using in situ hybridization has not been systematically analyzed. Particularly many commercial antibodies against the H3 receptor are nonspecific: they detect multiple unrecognized bands in Western blot analysis and also react in tissues of H3 receptor KO mice. Thus the specific neuronal phenotypes in autonomous and sensory ganglia that express H3 receptors are still unknown in most cases.
The apparent high affinity of (R)-α-methylhistamine (33) for the H3 receptor has enabled the use of this compound as a radiolabeled probe (Arrang et al., 1987b). This compound has been successfully used to identify a single binding site in rat cerebral cortical membranes, which in phosphate buffer has the pharmacological characteristics of the H3 receptor (Arrang et al., 1987b, 1990). [3H](R)-α-Methylhistamine binds with high affinity (KD = 0.3 nM) to rat brain membranes, although the binding capacity is generally low (approximately 30 fmol/mg protein; Arrang et al., 1987b). It should be noted that this ligand has a moderate affinity for the H4 receptor (Liu et al., 2001b), and so the anatomic profile may be incomplete.
Radiolabeled H3 receptor antagonists have been available since the early 1990s. The first compound to be developed was [125I]iodophenpropit, which has been used to successfully label H3 receptors in rat brain membranes (Jansen et al., 1992). Inhibition curves for thioperamide and iodophenpropit were consistent with interaction with a single binding site, but H3 receptor agonists were able to discriminate high [4 nM for (R)-α-methylhistamine]- and low [0.2 mM for (R)-α-methylhistamine]-affinity binding sites (Jansen et al., 1992). More recently, [3H]GR16820 (Brown et al., 1994) and [125I]iodoproxyfan (Ligneau et al., 1994) have also proved useful as high-affinity radiolabeled H3 receptor antagonists. [125I]Iodoproxyfan (Stark et al., 1996) is the most potent and selective ligand available at the present time, with a KD of 65 pM (Ligneau et al., 1994). In rat striatum, in the presence of guanine nucleotides such as guanosine (3-thiotriphosphate) (GTPγS), 40% of the binding sites exhibited a 40-fold lower affinity for H3 receptor agonists, providing further evidence for a potential linkage of the H3 receptor to G proteins (Ligneau et al., 1994). [3H]Thioperamide and [3H]5-methylthioperamide have also been used to label H3 receptors in rat brain membranes (Yanai et al., 1994; Alves-Rodrigues et al., 1996). However, [3H]thioperamide (40) binds additionally to low-affinity, high-capacity, non-H3 receptor sites in this tissue (Alves-Rodrigues et al., 1996) and acts as a ligand for H4 receptors (Gbahou et al., 2006). Meanwhile different potent fluorescent H3 receptor antagonists have been developed that can be used for imaging purposes (Amon et al., 2006, 2007).
[35S]GTPγS binding assays can be used to detect receptor-dependent activation of G proteins (Wieland et al., 1994). This method can be used to locate anatomic patterns of G protein activation in tissues. In the rat brain, the histamine stimulated [35S]GTPγS binding corresponds to the H3 receptor radioligand binding pattern and responds to H3 receptor antagonists, suggesting that histamine-induced signals are due to activation of H3 receptors (Laitinen and Jokinen, 1998). By using this method, it has been shown that in hibernating ground squirrels the H3 receptors, which are upregulated in caudate putamen during hibernation bout, can be activated throughout the hibernation cycle (Sallmen et al., 2003a). This, together with the increased histamine turnover during hibernation (Sallmen et al., 1999), indicates that histamine may be a critical factor in maintaining the hibernation state.
In addition to data obtained from ligand binding and [35S]GTPγS binding studies, evidence for the localization of H3 receptors has also come from functional studies involving inhibition of neurotransmitter release and electrophysiology. The histaminergic neurons located in the tuberomamillary nucleus of the posterior hypothalamus project throughout the central nervous system. Histamine production and release is under feedback control through autoreceptors (H3 receptors); however, a large number of nonhistaminergic neurons, including their axon varicosities, carry H3 heteroreceptors. This makes the prediction of H3 receptor-mediated effects difficult but opens up an avenue to multiple interference with neural functions (Fig. 11).
Fig. 11.

Neuronal targets of histaminergic neurons. H1 receptor and H2 receptor are located on many neurons where they modulate the activity in several ways (see text). H3 receptors are located on histaminergic neuron somata, dendrites, and axons to control firing, histamine synthesis, and release acting as autoreceptors. As heteroreceptors they control transmitter release from a wide variety of nonhistaminergic axons and in some cases inhibit the activity of nonhistaminergic neurons. Modified from Haas and Panula (2003).
Candidate drugs as clinical PET tracers to both study the H3 receptor system and measure receptor occupancy of H3 receptor therapeutic compounds have been developed (Selivanova et al., 2010). [11C](1R,40R)-5-Methoxy-N-methyl-3-oxo-N-(2-piperidin-1-ylethyl)-3H-spiro[2-benzofuran-1,10-cyclohexane]-40-carboxamide and [18F](1R,40R)-5-(fluorodideuteromethoxy)-N-methyl-3-oxo-N-(2-piperidin-1-ylethyl)-3H-spiro[2-benzofuran-1,10-cyclohexane]-40-carboxamide bind specifically to sections of rhesus monkey and human brain, where substantia nigra, nucleus accumbens, and globus pallidus show strong signals. PET imaging on rhesus monkey brain shows strong signals in the striatum and frontal cortex and lower signals in the cerebellum (Hamill et al., 2009).
D. Function
The H3 receptor was first characterized as an autoreceptor regulating histamine synthesis and release from rat cerebral cortex, striatum, and hippocampus (Arrang et al., 1985a,b, 1987a, 1988a). H3 receptor-mediated inhibition of histamine release has also been observed in human cerebral cortex (Arrang et al., 1988b). Differences in the distribution of H3 receptor binding sites and the levels of histidine decarboxylase (an index of histaminergic nerve terminals) suggested at an early stage that H3 receptors were not confined to histamine-containing neurons within the mammalian CNS (Arrang et al., 1987a; van der Werf and Timmerman, 1989). This has been confirmed by the observations that H3 receptors can regulate serotonergic (Schlicker et al., 1988, 1989; Threlfell et al., 2004), noradrenergic (Schlicker et al., 1989), cholinergic (Clapham and Kilpatrick, 1992), and dopaminergic (Schlicker et al., 1992, 1993; Munari et al., 2013) neurotransmitter release in mammalian brain. H3 receptor activation inhibits the firing of the histamine neurons in the posterior hypothalamus through a mechanism distinct from autoreceptor functions found on other aminergic nuclei, namely a block of Ca2+-current, which is part of the pacemaker cycle in histaminergic neurons (Stevens et al., 2001) (Fig. 12). Electrophysiological evidence for reduction of excitatory transmitter release (glutamate) has been presented by Brown et al. (Brown and Reymann, 1996; Brown and Haas, 1999). Activation of H3 receptors inhibits neurotransmission also in the peripheral autonomous and sensory systems. Sympathetic neurotransmitter release is regulated by H3 receptor in guinea pig mesenteric artery (Ishikawa and Sperelakis, 1987), human saphenous vein (Molderings et al., 1992), guinea pig atria (Endou et al., 1994), human heart (Imamura et al., 1996), rat tail artery (Godlewski et al., 1997), and dog kidney (Yamasaki et al., 2001). Neuropeptide (tachykinins or calcitonin gene-related peptide) release from sensory C fibers from airways (Ichinose and Barnes, 1990), meninges (Matsubara et al., 1992), skin (Ohkubo and Shibata, 1995), heart (Imamura et al., 1996), and rat mesenteric arteries (Sun et al., 2011) is also controlled by H3 receptors. A modulation of acetylcholine, capsaicin, and substance P effects by H3 receptors in isolated perfused rabbit lungs has also been reported (Delaunois et al., 1995).
Fig. 12.

Electrophysiological actions of H3 receptor activation in brain slices. (A1) Typical regular firing of a histaminergic tuberomamillary neuron. (A2) Block of the H3-autoreceptor activation by thioperamide releases the inhibition and thus increases firing rate. (B) Ca2+-current in response to depolarization of histaminergic neuron is reduced by the H3 receptor-agonist R-α-methylhistamine (33). (C) Glutamatergic cortico-striatal synaptic transmission (co-str) is reduced by R-α-methylhistamine, an example of an H3-heteroreceptor action.
H3 receptor mRNA expression and G protein activation is increased in the basal ganglia of hibernating mammals during the hibernation period (Sallmen et al., 2003a) along with histamine levels and turnover (Sallmen et al., 1999), which suggests that histamine may be involved in regulation of hibernation or arousal from the bout. Indeed, infusion of histamine in the hippocampus of golden-mantled ground squirrel causes a delayed arousal (Sallmen et al., 2003b).
As autoreceptors on somata, dendrites, and axons of TMN neurons, constitutively active H3 receptors (Arrang et al., 2007) inhibit cell firing (Stevens et al., 2001), as well as histamine synthesis and release from varicosities (Arrang et al., 1983; Torrent et al., 2005; Moreno-Delgado et al., 2006). This autoinhibition by H3 receptor agonists [e.g., (R)-α-methylhistamine; 33] and its removal by H3 receptor antagonists/partial agonists can be demonstrated in recordings from TMN neurons. As presynaptic heteroreceptors, H3 receptors control the release of several other transmitters, including biogenic amines (Schlicker et al., 1994, 1999), acetylcholine (Blandina et al., 1996; Arrang, 2007), GABA (Jang et al., 2001), and glutamate; field potentials evoked by stimulation of glutamate fiber bundles, e.g., in hippocampus or striatum, show a marked decrease in the presence of an H3 receptor agonist (Brown and Haas, 1999; Doreulee et al., 2001) (Fig. 12). In this context, the H3 receptor has been shown as a potential therapeutic target for cerebral ischemia (Yan et al., 2014). Heterogeneity of the histaminergic neurons with respect to function and projections has been demonstrated by Giannoni et al. (2009); activation of histaminergic neurons by blocking the somatic autoreceptors causes differential release of histamine measured by microdialysis in different target regions of rats; cortex and nucleus basalis display strong increases of histamine release, but both the ventral and the dorsal striatum, regions receiving ample innervation too, do not show any such release (Blandina et al., 2012). H3 autoreceptors may thus have been absent, or not activated, in the histaminergic neurons projecting to these latter regions.
H3 receptor KO mice display changes in behavioral states, reduced locomotion (Toyota et al., 2002; Schneider et al., 2014b), a metabolic syndrome with hyperphagia, late-onset obesity, increased insulin and leptin levels (Toftegaard et al., 2003; Yoshimoto et al., 2006), and an increased severity of EAE, a model of multiple sclerosis (Teuscher et al., 2004, 2007). By comparing EAE-susceptible and -resistant mice strains as well as H3 receptor KO mice, a functional link between central H3 receptor signaling and peripheral immune responses was proposed (Krementsov et al., 2013).
With its unique pharmacological properties the H3 receptor is a major target for development of drugs against various disorders of the brain (Passani et al., 2007; Lin et al., 2011b). H3 receptor inverse agonists decrease alcohol intake in alcohol-preferring (Lintunen et al., 2001) and normal (Galici et al., 2011) rats and modulate alcohol-induced place preference and locomotory activation in mice (Nuutinen et al., 2010), but not amphetamine-induced place preference (Vanhanen et al., 2015), suggesting new possibilities to treat alcohol dependence.
Early studies on the role of H3 receptors on gastric acid secretion gave conflicting results as described by Barocelli and Ballabeni (2003). The well recognized role of histamine-producing enterochromaffin-like cells [ECL cells (Hakanson et al., 1986)] in gastric acid secretion and the role of H2 receptors prompted research using a number of different H3 receptor ligands and animal models. By using fistula cats and dogs, inhibition of acid secretion with (R)-α-methylhistamine (33) was found, but these results were not reproduced in some other models (Barocelli and Ballabeni 2003). One confounding factor was the imidazole nature of many studied ligands and their affinity to the H4 receptor. Recent results with more selective ligands immethridine and methimepip protected the gastric mucosa from HCl-induced damage, and the effect is antagonized by a selective H3 receptor antagonist A-331440 (Coruzzi et al., 2012). These results suggest that the H3 receptor-mediated effects are distinct from those mediated by H4 receptor (Coruzzi et al., 2012).
H3 receptors have been shown to relax the rabbit middle cerebral artery via an endothelium-dependent mechanism involving both nitric oxide and prostanoid release (Ea Kim et al., 1992). Finally, there is a report that H3 receptor activation can stimulate adrenocorticotropic hormone release from the pituitary cell line AtT-20 (Clark et al., 1992).
E. H3-Selective Ligands
A large number of potent ligands for the H3 receptor have been described, encompassing significant structural diversity. Many of these have been reported as useful pharmacological tools for in vitro or in vivo study and are described here. The number of ligands and structural genera is large, and a number of comprehensive reviews of the structure-activity relationship (SAR) and properties of H3 receptor ligands have been published (Celanire et al., 2005; Amon et al., 2007; Berlin and Boyce, 2007; Esbenshade et al., 2008; Sander et al., 2008; Gemkow et al., 2009; Tiligada et al., 2009; Lazewska and Kiec-Kononowicz, 2010; Berlin et al., 2011).
1. Agonists.
Histamine (1), 2-(1H-imidazol-4-yl) ethanamine, is of course the endogenous ligand for histamine receptors, and other imidazole analogs have proven to be useful tools for pharmacological studies (Fig. 13). The methylated analog [3H]Nα-methylhistamine (32) (Arrang et al., 1987a) is not selective for H3 receptors (versus H1 and H2 receptors) but because of its high affinity (rat H3 receptor Kd = 2 nM) (Arrang et al., 1983; Hill et al., 1997) and ready availability in tritiated form has found wide use in radioligand competition binding assays. (R)-α-Methylhistamine (33) is highly potent (Kd = 0.5 nM in rat brain) (Arrang et al., 1985b, 1987b; Hill et al., 1997) and is also used as an in vitro agonist tool. (R)-α-Methylhistamine does have significant H4 receptor activity (binding pKi of 6.85, human H4 receptor in transfected cells) (Liu et al., 2001a,b; Morse et al., 2001) and pKi = 6.1 at rat H4 receptor. This agonist also induces water drinking in rodents, a finding used as the basis of an in vivo pharmacological model (Clapham and Kilpatrick, 1993; Ligneau et al., 1998; Fox et al., 2002). Prodrugs of (R)-α-methylhistamine have been described with an improved pharmacokinetic profile (Krause et al., 2001; Stark et al., 2001). Imetit (34) is a readily available potent agonist (rat H3 receptor Ki = 0.1 nM) demonstrating the expected profile of an agonist in vivo, decreasing histamine release ex vivo and turnover (Eriks et al., 1992; Garbarg et al., 1992). More lipophilic ligands have also been described with improved properties. Immepip (35) has high affinity at H3 receptors but poor selectivity versus H4 receptors. However, methylation of methimmepip (36) gives an agent with excellent potency (human H3 receptor Ki = 1 nM), agonism (α of 0.9 versus histamine), and selectivity (>2000-fold selectivity over H1, H2 receptors and H4 receptors). Importantly, methimmepip profoundly reduced CNS microdialysate histamine levels at low doses (5 mg/kg i.p.) (Kitbunnadaj et al., 2005). Other lipophilic agonists have also been described in an extensive SAR study (Govoni et al., 2006), and an agonist (37) (human H3 receptor Ki = 0.17 nM, 77% efficacy) was active in vivo in mice in blocking aggression and stress (Ishikawa et al., 2010). Other H3 receptor agonists have been described with novel structural variations, including immethridine (human H3 receptor Ki = 0.9 nM, 90% agonist efficacy), an analog of immepip wherein the highly basic piperidine moiety is replaced by a less basic pyridinyl moiety (Kitbunnadaj et al., 2004). A particularly interesting ligand is the "protean" (Kenakin, 2002) agonist proxyfan (38) (Gbahou et al., 2003), a compound that does not fit neatly into a rigid categorization of agonist or antagonist, because its properties have been found to depend on the system assessing efficacy, with full agonism, neutral antagonism, and inverse agonism seen. Several other imidazole-containing H3 receptor ligands have been described, e.g., GT-2331 (39), with some having assay-dependent profiles (Tedford et al., 1999; Liu et al., 2004; Ito et al., 2006).
Fig. 13.

H3 receptor agonists.
2. Antagonists.
A number of named antagonist ligands have figured prominently in preclinical studies, with clear ability to release neurotransmitters and having efficacy in preclinical animal models. This has motivated continuing research on improved agents with potency, selectivity, and better drug likeness to enable clinical evaluation. Computer modeling strategies have been adopted widely in recent years for ligand docking and pharmacophore screening purposes (Schlegel et al., 2007; Levoin et al., 2008) (Fig. 14).
Fig. 14.

H3 receptor antagonists.
A number of antagonists have advanced to phase I testing and phase II efficacy trials for a number of conditions, including sleep disorders, cognitive disorders in schizophrenia, Alzheimer’s disease, attention deficit hyperactivity disorder, and allergic rhinitis.
The first H3 receptor antagonists were imidazole based, and before the identification of the H4 receptor, several were widely used “H3 receptor standards” for preclinical studies, also because of their availability from commercial sources, including thioperamide (40), clobenpropit (41), and ciproxifan (42). Thioperamide is highly potent at rat H3 receptors with Ki = 4.3 nM (Arrang et al., 1987a; Hill et al., 1997) that has been found broadly effective in vivo in a large number of behavioral models, including elevated plus maze learning, Morris water maze, Barnes maze (Miyazaki et al., 1995; Komater et al., 2005), and others. Clobenpropit (41) is likewise highly potent in vitro, with pA2 = 9.92 in the guinea pig ileum assay (Eriks et al., 1992) and activity in vivo, e.g., in models of working memory, seizure, and Alzheimer’s disease (Yokoyama et al., 1994; Zhang et al., 2003; Harada et al., 2004; Huang et al., 2004; Bardgett et al., 2011). Clobenpropit and thioperamide, however, also have potent activity at H4 receptors (Lim et al., 2009), and thioperamide also is active at 5HT3 receptors (Leurs et al., 1995b). Ciproxifan is a potent (rat H3 Ki = 0.5 nM) and selective (Ligneau et al., 1998; Stark et al., 2000) ligand with oral bioavailability, which is effective in a number of preclinical animal models and, thus, has found extensive use as an in vivo reference antagonist, for example, in attentional models, such as electroencephalogram-assessed waking and impulsivity (Komater et al., 2005; Hancock, 2006; Day et al., 2007). SCH-79687 (43) is a highly potent (rat H3 receptor Ki = 1.9 nM) antagonist that reduced congestion in animal models of allergic rhinitis when coadministered with an H1 receptor antagonist; evidence supports the hypothesis that the efficacy of H3 receptor antagonists is mediated through peripherally mediated release of norepinephrine from nasal mucosa, because this compound was virtually nonbrain penetrant with a brain/plasma ratio of 0.02 (Varty and Hey, 2002; McLeod et al., 2003).
Nonimidazoles have become the main focus of the H3 receptor antagonist design work in recent years, because these structures address the class-related issues noted with the early generation of imidazole-based agents, including cytochrome P450 inhibition, relatively poor CNS penetration, and incidence of off-target activity at H4 receptors or other receptors (Berlin et al., 2011). The first SAR study of potent nonimidazole antagonists disclosed UCL-1972 (44), with rat H3 receptor Ki of 39 nM. In this structure, the imidazole ring was replaced with a tertiary basic amine (Ganellin et al., 1995). Nonimidazoles as a class have proven much more supportive of combining cross species at rat and human receptors with good drug likeness (CNS penetration, minimal cytochrome P450 inhibition) (Ganellin et al., 1998; Meier et al., 2001). There has been rapid progress in the last decade in finding antagonists with excellent in vivo efficacy in diverse preclinical models, and several prominent compounds have been detailed in the literature. Of particular interest is pitolisant (45), also previously known as BF-2.649, BP-2649, or tiprolisant (cf. Meier et al., 2001). This potent (rat Ki = 17 nM, human Ki = 5 nM) antagonist entered clinical trials for a number of indications and has been well characterized preclinically as able to promote wakefulness in animals and humans (Ligneau et al., 2007; Lin et al., 2008; Schwartz, 2011). Antagonists with a second basic amine moiety have been widely described as highly potent, with JNJ-5207852 (46) having rat and human Ki of 1.2 and 0.6 nM, and the acetylene analog JNJ-10181457 (47) having a rat and human H3 receptor Ki of 7 and 1.2 nM. These two agents were reported to promote wakefulness in rat, mouse, and dog models (Barbier et al., 2004; Bonaventure et al., 2007), a property of H3 receptor antagonists as a class (Barbier and Bradbury, 2007). A number of synthetic diamine-containing H3 receptor antagonists have been described with high in vitro potency. One example of a potent agent is the natural product alkaloid conessine (48), with a human H3 receptor Ki of 5.4 nM, rat H3 receptor Ki of 25 nM. The core has been elaborated to analogs, whereas the potency and commercially availability of conessine itself suggest potential utility as a tool compound (Cowart et al., 2007; Santora et al., 2008; Zhao et al., 2008). The benzofuran ABT-239 (49) (human H3 receptor Ki = 0.45 nM, rat H3 receptor Ki = 1.35 nM) has been used as a reference antagonist with potent in vivo efficacy in a number of animal models of attention, cognition, schizophrenia, ethanol-associated learning deficit, and Alzheimer’s disease (Cowart et al., 2005; Fox et al., 2005; Varaschin et al., 2010; Bitner et al., 2011). Heterocyclic benzofuran A-688057 (50) was likewise highly brain penetrant, potent in vitro (human H3 receptor Ki = 0.5 nM, rat H3 receptor Ki = 3 nM) and effective in animal models in vivo at modest levels of H3 receptor occupancy (Cowart et al., 2007). GSK-189254 (51) (human H3 receptor Ki = 0.1 nM, rat H3 receptor Ki = 0.7 nM) has been described as broadly effective in a number of animal models, including attentional, memory, pain, and narcolepsy models (Guo et al., 2009; Medhurst et al., 2007; McGaraughty et al., 2012). GSK-189254 (51) has also been synthesized in an 11C-labeled form and used as a PET tracer to empirically measure H3 receptor occupancy of test compounds in intact animals and in the clinic (Ashworth et al., 2010). GSK-207040 (52) (human H3 Ki = 0.2 nM, rat H3 Ki = 0.8 nM) and GSK-334329 (53) (human H3 receptor Ki = 0.3 nM, rat H3 Ki = 0.8 nM) were both found effective in models of neuropathic pain and capsaicin-induced tactile allodynia and scopolamine-induced memory impairment (Medhurst et al., 2007). A separate report found efficacy in an osteoarthritis pain model for GSK-189254, GSK-334429, and ABT-239 (Hsieh et al., 2010b). Recently, the compound JNJ-39220675 (54) (human H3 receptor Ki = 1.4 nM) was found to reduce ethanol intake in ethanol-preferring rats (Galici et al., 2011) and was found to have decongestant efficacy in an early clinical trial in subjects with allergic rhinitis (Barchuk et al., 2013).
F. Clinical Pharmacology
After a decade of intensive studies (Ligneau et al., 2007; Lin et al., 2008; Schwartz, 2011), a range of H3 receptor inverse agonists/antagonists have entered advanced clinical development in the last 5 years, with the most advanced being pitolisant (BF2.649, 1-piperidine, hydrochloride, pitolisant, Wakix, 45) (Dauvilliers et al., 2013) having been officially filed for licensing by Bioprojet early in 2014 for use in refractory narcolepsy. This is the completion of an impressive research story spanning 40 years, initiated back in the mid-1970s, when histamine was first described as a neurotransmitter, and the pharmacological and molecular identification of the H3 receptor in the mid-1980s and late-1990s, respectively. Narcolepsy is characterized by excessive diurnal sleepiness and can occur alone or as part of other neurologic disorders (e.g., Parkinson’s disease). On the basis of the wake-inducing properties of H3 receptor blockade, pitolisant (45) was developed, together with an apparent ideal pharmacokinetic profile, where a once-a-day dose maintains attention over the day, but, importantly, allows individuals to sleep at night (Iannone et al., 2010; James et al., 2011; Schwartz, 2011). Preclinical studies have also suggested the lack of drug abuse liability for pitolisant in rodent and primate models, in contrast to modafinil (Uguen et al., 2013). In a double-blind, randomized, parallel-group controlled trial performed with 95 cases recruited across Europe in 32 sleep centers, pitolisant at doses up to 40 mg was efficacious on excessive daytime sleepiness compared with placebo. Over the 8-week treatment period, mean Epworth Sleepiness Scale score reductions were −3·4 in the placebo group, −5·8 in the pitolisant group, and −6·9 in the modafinil group. Pitolisant was well tolerated (with only one patient displaying a serious side effect, namely abdominal discomfort) compared with the previous gold standard, modafinil, where multiple issues arose (Dauvilliers et al., 2013). A recent retrospective study determined the benefits of pitolisant in patients with idiopathic hypersomnia and narcolepsy refractory to stimulants, including modafinil. Twenty-three to thirty-eight percent of the 78 patients receiving pitolisant (daily 5- to 50-mg dose) displayed a positive benefit/risk ratio (Leu-Semenescu et al., 2014).
A more modest study with an H3 receptor inverse agonist, MK-0249 (55) did not significantly affect maintenance of wakefulness test battery. However, the pattern of improvement on a range of subjective ratings and psychomotor performance end points suggested that MK-0249 (55) improved some aspects of cognition and performance not captured by the maintenance of wakefulness test (Herring et al., 2013). A number of other compounds developed by major players in this field have been investigated, with multiple trials listed in www.clinicaltrials.gov to date but yet to be reported. These include a series of phase I PET occupancy trials with PF-03654746, AZD5213, and GSK239512 distribution investigation after oral administration. H3 heteroreceptors modulate acetylcholine levels in cortical regions of the brain, a fact that suggests potential for H3 receptor blockade in reversing scopolamine-induced cognitive deficits. This was investigated in small acute clinical study, where 28 individuals completed a double-blind, placebo-controlled, five-period crossover study, and cognitive function was assessed using the Groton Maze Learning Task as the primary outcome measure. Interestingly, although these primary end points were not met at the extended time point (up to 12 hours), administration of either MK-3134 or donepezil improved performance at the 2-hour time point compared with scopolamine alone. Moreover, it appeared that the combination of MK-3134 and donepezil suppressed the scopolamine negative effect to a greater extent than either drug alone (Cho et al., 2011). Further clinical trials have been completed, focused on evaluation of the efficacy and safety of GSK239512 in Alzheimer's disease, using a randomized, double-blind, placebo-controlled study and cognitive impairment in schizophrenia. The initial report showed that GSK239512 displayed appropriate tolerability up to a maximum dose of 80 μg/day in patients with Alzheimer's disease, with some preliminary evidence for positive effects on attention and memory with modest effect sizes (Nathan et al., 2013). A larger follow study (over 190 mild/moderate Alzheimer's cases) reported GSK239512, up-titrated over 4 weeks (10–20–40–80 µg) followed by a 12-week maintenance phase, compared with sham controls, although it did not display improvement in working memory and other cognitive outcomes, however, it exhibited improved episodic memory, with a reasonable safety profile (Grove et al., 2014). Further large trials are required to investigate this differential responsiveness. A number of reports studying H3 receptor inverse agonists have indicated potential in vivo for anticonvulsive activity utilizing various models, including the maximal electroshock-induced and pentylenetetrazole-kindled seizure models in rodents (Sadek et al., 2014a,b).
In a randomized multicenter, double-blind, placebo- and active-controlled. parallel group study on adult attention-deficit hyperactivity syndrome on bavisant (JNJ-31001074), the drug did not differ from placebo, whereas atomoxetime hydrochloride and osmotic release oral system methylphenidate were effective (Weisler et al., 2012). A clinical trial with the same drug was also listed at clinicaltrials.gov but the trial has not been initiated. Thus, this indication for which there is experimental support from rodent studies and several drug candidates tested for safety and side effects in humans has not been tested. In a randomized crossover study the H3 receptor inverse agonist MK-0249 was not superior to placebo for the treatment of cognitive impairment in patients with schizophrenia (Egan et al., 2013). In conclusion, the expectations for extensive clinical use of H3 receptor inverse agonists have yet to be realized.
Peripheral indications have also been explored. In combination with fexofenadine, single doses of PF-03654746 produced a reduction in allergen-induced nasal symptoms in a small 20 patient trial using an acute nasal allergen challenge with a bolus of ragweed (Stokes et al., 2012).
V. Histamine H4 Receptor
The H4 receptor is the most recently discovered histamine receptor. The cloning of the H3 receptor provided a template for the search of other histamine receptors. This culminated with six independent groups reporting the cloning of the H4 receptor (Nakamura et al., 2000; Oda et al., 2000; Liu et al., 2001a; Morse et al., 2001; Nguyen et al., 2001; Zhu et al., 2001; O'Reilly et al., 2002). Since that time much research has focused on characterizing the receptor’s role in inflammatory process, because it originally appeared to be predominantly expressed in hematopoietic cells.
A. Receptor Structure
The H4 receptor displays similarities to the H3 receptor in terms of gene and protein secondary structure (Coge et al., 2001a,b). The human H4 receptor gene is present as a single copy per haploid genome on chromosome 18q11.2, spans more than 21 kbp, and contains three exons together with two large introns (7867 and >17,500 bp). The coding regions encode amino acids 1–65, 66–119, and 120–390, respectively. The transcript size of the gene is approximately 3.7 or 4.5 kb, which is explained by at least two polyadenylation sites 0.8 kb apart (Coge et al., 2001b). The open reading frame (1172 bp) encodes a 390-amino-acid polypeptide that forms the classic 7-transmembrane structure of the family A rhodopsin-like GPCR. A series of polymorphisms have been identified and confirmed by SNP analysis involving Caucasian, Chinese, Japanese, African-American, and Sub-Saharan African individuals, including V138A and R206H. The occurrence of A138 or H206 is 10-fold higher than the V138 or R206 version, respectively. Further SNPs have been found in intron 1, 33 in intron 2, and 13 in the 3′-untranslated region, as well as a frame-shift at the C terminus of the protein (http://www.ncbi.nlm.nih.gov/SNP).
It is possible that the H4 receptor can exist in dimeric or oligomeric forms. Based on immunoblotting techniques, various structural forms of the H4 receptor have been reported: 44 kDa (unglycosylated monomer), 46 kDa (glycosylated monomer), 85 kDa (glycosylated dimer), and >250 kDa (oligomeric structures) (Nguyen et al., 2001; van Rijn et al., 2006). Based on biochemical (immobilized metal affinity chromatography) and biophysical techniques (time-resolved fluorescence resonance transfer and BRET) in transfected cells, the human H4 receptor can exist as dimeric or oligomeric species (van Rijn et al., 2006). However given the uncertainties around the selectivity of H4 receptor antibodies (see section V.C) and lack of direct evidence in primary cells, it is still uncertain as to whether oligomerization occurs in native tissues and cells or whether it has physiologic consequences.
In addition to this, two human H4 receptor isoforms have been identified and designated H4 receptor 302 and H4 receptor 67, the former lacking residues 68–155, whereas the latter is prematurely terminated at residue 67. These isoforms are the result of alternative splicing, but do not bind histamine or inverse agonists. In heterologous expression systems, these isoforms have been shown to elicit dominant negative effects on the full-length isoform by decreasing the Bmax of [3H]histamine binding sites as well as retaining the full-length receptor intracellularly via oligomerization (van Rijn et al., 2008).
A study in human peripheral blood leukocytes showed an upregulation of the truncated isoform of the H4 receptor (H4R), but not the full-length H4 receptor during grass pollen season, suggesting a possible regulatory effect on H4 receptor signaling (Hodge et al., 2013). Whether these different isoforms are expressed in primary cells at the protein level and whether they are physiologically relevant is currently unknown. Moreover, whether there are also functional H4 receptor isoforms, as observed for the H3 receptor, is currently unknown. Yet the similar intron/exon structure might give rise to functional H4 receptor isoforms.
The identification of the human H4 receptor gene prompted the cloning of the rat, mouse, canine, guinea pig, and porcine H4 receptor orthologs (Oda et al., 2000; Liu et al., 2001b; Jiang et al., 2008). These are modestly homologous to the human ortholog, with sequence homology ranging from 67 and 72%. The monkey H4 receptor was subsequently cloned in 2005 and displays a high homology of 92% (Oda et al., 2005). Partial sequences of other species orthologs have been identified but not published. Species differences in pharmacology (ligand binding and signaling efficacy) are profound, also depending on readout systems (Nordemann et al., 2013), and several molecular features for the observed species differences have been elucidated (Liu et al., 2001b; Lim et al., 2008, 2010; Schnell et al., 2011).Whereas proximal readout systems such as [32P]GTPase or [35S]GTPS binding assays show pronounced differences between species orthologs, more distal reporter gene assays revealed less pronounced differences. Species splice isoforms have yet to be reported.
The H4 receptor possesses the protypical acidic residue (Asp943.32) in TM3, which interacts with the amine group of histamine (1; Fig. 3). In addition to the conserved aspartate, a glutamate residue is also present in TM5 (Glu 1825.46), consistent with the H3 receptor, and is another key histamine binding residue (Shin et al., 2002; Uveges et al., 2002). As discussed previously for the H3 receptor, the interaction of the imidazole ring with this glutamate residue is most likely responsible for the relatively high affinity of the H4 receptor for histamine. This amino acid is also important in binding smaller agonist ligands (e.g., VUF8430) (Lim et al., 2008). The H4 receptor also possesses cysteine residues that potentially form disulfide bridges spanning TM3 and ECL2. Interestingly, using a chimeric and site-directed mutagenesis approach it was shown that the ECL2 loop is also involved in the binding pocket. Phe169 in ECL2 was identified as the single amino acid responsible for the differences in histamine affinity between the human and rodent H4 receptor (Lim et al., 2008). Final confirmation of this suggestion awaits successful crystallization.
Interestingly, the H4 receptor does not contain the so-called “ionic lock” between the highly conserved R3.50 of the DRY motif at the end of TM3 and a negative charged D/E6.30 in TM6, which in many GPCRs is considered to hold the receptor in an inactive state (Smit et al., 2007). Yet, the H4 receptor contains an alanine residue at position 6.30, and mutation of this residue (e.g., into glutamate) does not really alter the constitutive signaling (Schneider et al., 2010a). A pseudoionic lock has been claimed for differentiating (partial) agonist from antagonist binding modes (Werner et al., 2010).
B. Signal Transduction Mechanisms
A detailed understanding of signal transduction mechanisms downstream of H4 receptor activation is emerging. Like the structurally related H3 receptor, the H4 receptor belongs to the class of Gi/Go-coupled GPCRs. The primary second messenger appears to be increased intracellular Ca2+ (Raible et al., 1994; Buckland et al., 2003; Hofstra et al., 2003; Dijkstra et al., 2007; Jemima et al., 2014), although activation of kinases has also be reported (Gutzmer et al., 2005, 2009; Desai and Thurmond, 2011; Ferreira et al., 2012; Mommert et al., 2012; Karlstedt et al., 2013). The best studied native cell type that expresses the H4 receptor is the human eosinophil. The H4 receptor-mediated activity in these cells such as cell shape change, Ca2+ mobilization, and actin polymerization is sensitive to inhibition by the Giα/Goα-ADP-ribosylating toxin pertussis toxin (Raible et al., 1994). Along the same lines, H4 receptor-mediated Ca2+ mobilization in mouse bone marrow–derived mast cells is also pertussis toxin sensitive (Hofstra et al., 2003). The pertussis toxin data suggest that the H4 receptor couples to Giα- or Goα-subunits; however, because myeloid cells express Giα-subunits (predominantly Giα2 and, to a lesser extent, Giα3, but not Giα1) and do not express Goα-subunits (Birnbaumer, 2007), it is very likely that Giα2- and Giα3 G protein α-subunits represent the cognate coupling partners of the H4 receptor in these native cells (Fig. 4).
Transfected cell systems also support the conclusion that the H4 receptor is a Gi-coupled GPCR. Expression of the H4 receptor in SK-N-MC neuroblastoma cells leads to ligand-dependent inhibition of forskolin-stimulated cAMP accumulation (Nakamura et al., 2000; Oda et al., 2000; Liu et al., 2001a; Lim et al., 2005). The inhibitory effect of the H4 receptor on cAMP accumulation is pertussis toxin sensitive, indicative for Gi protein coupling (Oda et al., 2000).
In further support of coupling of the H4 receptor to Giα proteins, to detect H4 receptor-mediated Ca2+ mobilization in a commonly used mammalian expression system such as HEK293 cells, the H4 receptor has to be cotransfected with the “universal” G protein G15α or G16α (Oda et al., 2000) or a chimeric Gqα protein in which the last five C-terminal amino acids are replaced by those of Giα/Goα-subunits (Coward et al., 1999; Liu et al., 2001a; Morse et al., 2001; Zhu et al., 2001; Jiang et al., 2008).
G protein coupling of the H4 receptor was further analyzed in the Sf9 insect cell expression system. One concern of this system is that the receptor expressed in Sf9 insect cells may differ from the H4 receptor expressed in mammalian cells. However, overall, it appears that the H4 receptor expressed in Sf9 insect cells and mammalian cells are similar (Schneider et al., 2009). The human H4 receptor couples only poorly, if at all, to insect cell G proteins as assessed by GTPγS binding and GTPase activity (Schneider et al., 2010b). Nonetheless, even in the absence of mammalian G proteins, the human H4 receptor exists in a state that exhibits high agonist affinity. Evidently, because G proteins are not required for this state, high-affinity [3H]histamine binding is GTPγS insensitive. These properties of the human H4 receptor are in marked contrast to several other Gi-coupled receptors, including the formyl peptide receptor that, like the H4 receptor, is expressed in myeloid cells (Ye et al., 2009). Moreover, GTPγS-insensitive [3H]histamine binding to the H4 receptor has also been observed for the H4 receptor expressed in mammalian cells (Lim et al., 2010). The lack of interaction with endogenous insect G proteins makes this an excellent system for exploring the interaction with mammalian G proteins. When reconstituted with various G protein α-subunits and the Gα1α2-complex, human H4 receptor exhibits clear preference for Giα2 (Schneider et al., 2009, 2010a), which is consistent with the fact that this is the most likely partner in eosinophils and mast cells. There is some coupling to Gα1 and Giα3, suggesting that these subunits may also be important in some cell types. However, the coupling to Goα, a G protein α-subunit predominantly expressed in neuronal and neuroendocrine cells (Birnbaumer, 2007), is poor (Schneider et al., 2009).
The human H4 receptor coupled to Giα2 exhibits high constitutive activity as is evident by high basal GTPγS binding and GTPase activity, relatively small agonist effects on guanine nucleotide exchange, and profound inhibitory effects of the inverse agonist thioperamide (Schneider et al., 2009). Amino acids F169 and S179 appear to play key roles for this high constitutive activity of the human H4 receptor (Wifling et al., 2015). Because the human H4 receptor exhibits high constitutive activity (Lim et al., 2005; Schneider et al., 2010b), H4 receptor agonists decrease cAMP accumulation, whereas H4 receptor inverse agonists increase cAMP accumulation (Lim et al., 2005; Kottke et al., 2011). In marked contrast to the situation with recombinant systems, there is little, if any, evidence as yet for constitutive activity of the H4 receptor in native systems.
In primary cells, it appears that the H4 receptor mediates signaling via increases in calcium (Raible et al., 1994; Buckland et al., 2003; Hofstra et al., 2003; Dijkstra et al., 2007; Jemima et al., 2014). In addition kinases such as ERK, phosphoinositide 3-kinase (PI3K), and p38 and the transcription factor activating protein-1 have been shown to be activated by the H4 receptor (Gutzmer et al., 2005, 2009; Desai and Thurmond, 2011; Ferreira et al., 2012; Mommert et al., 2012; Karlstedt et al., 2013).
The different H4 receptor species orthologs exhibit different pharmacological properties, perhaps related to a relatively low level of homology (Liu et al., 2001b; Jiang et al., 2008). For example, the affinity of histamine and various other ligands vary between the species. In addition, studies measuring inhibition of cAMP-dependent reporter gene expression and chimeric G protein–mediated increases in cytosolic calcium ion concentration or reporter gene expression indicated that not only the human H4 receptor, but also canine, murine, rat, and guinea pig H4 receptors couple to Gi proteins (Liu et al., 2001b; Jiang et al., 2008). In Sf9 cells, human, canine, rat, and murine H4 receptor all couple to Gi proteins as assessed by agonist-stimulated GTP hydrolysis (Schnell et al., 2011). These studies confirmed the previously observed pharmacological differences between H4 receptor species orthologs and also revealed substantial differences in constitutive activity of the four species orthologs. Specifically, only human H4 receptors, but not canine and murine H4 receptors, possess high constitutive activity as assessed by inhibitory effects of thioperamide on basal GTP hydrolysis (Schnell et al., 2011).
C. Anatomic Framework
Identification of the expression pattern of the H4 receptor has been hampered by several issues. mRNA levels can be used to determine if the H4 receptor message is expressed in a cell type or tissue; however, it does not provide evidence that the protein is expressed. Furthermore, it can be difficult to determine exactly which cell types express the RNA in tissues, and the purity level of primary cells can lead to false positive signals. Finally, the expression of RNA may depend on the environment, because it is known that message levels and activity change in response to inflammatory stimuli and to the state of the cell. For example, the H4 receptor was undetectable on human CD8+ T cells in one report (Ling et al., 2004), whereas another (Gantner et al., 2002) showed a function for the receptor in these cells by pharmacological means and mRNA expression; however, the level of expression varied from donor to donor. A major issue is the lack of a widely accepted and available antibody specific for the receptor. In particular the selectivity of the available antibodies has been questioned and care should be taken with interpreting expression data based on these antibodies. Specifically, one study (Beermann et al., 2012b), using stringent controls, showed that three commercially available antibodies directed against the human and mouse H4 receptor do not specifically recognize the H4 receptor in various receptor formats, including fluorescence-activated cell sorting and immunoblotting. This study included controls with epitope-tagged H4 receptors and H4 receptor knockout cells and is in line with a series of previous studies on various biogenic amine GPCRs indicating that many GPCR antibodies distributed commercially do not fulfill the criteria necessary for receptor expression studies (Michel et al., 2009). A general caveat for H4 receptor work, or any other field for that matter, is that one cannot rely solely on mRNA data, antibody data, or pharmacology data with a single ligand to prove the existence of the receptor. The best approach currently is to use a combination of techniques with the definitive answer being given by pharmacological activities defined by multiple ligands.
One of the best examples of using pharmacology to define the receptor function comes from the early work with eosinophils. In the late 1970s it was noted that histamine could induce eosinophil chemotaxis via non-H1 receptor, non-H2 receptor (Clark et al., 1975, 1977; Wadee et al., 1980). Later it was discovered that histamine-stimulated increases in intracellular Ca2+ in eosinophils were blocked by thioperamide, impromidine, and burimamide, but not by pyrilamine or cimetidine, suggesting at the time that the effects of histamine were H3 receptor mediated (Raible et al., 1994). However, other data did not match the known pharmacology of the H3 receptor, including the fact that (R)-α-methylhistamine was over an order of magnitude less potent than histamine. The cloning of the H4 receptor and its pharmacological characterization indicated that this pharmacology matched that of the new receptor and it was subsequently shown that indeed the H4 receptor mediates Ca2+ responses and chemotaxis of eosinophils (Buckland et al., 2003; Ling et al., 2004).
At the mRNA level, the H4 receptor appears to be most strongly expressed in bone marrow, but expression can also be detected in peripheral blood, spleen, thymus, as well as in lung, small intestine, colon, and heart (Nakamura et al., 2000; Oda et al., 2000; Liu et al., 2001a; Morse et al., 2001; Nguyen et al., 2001; Zhu et al., 2001). Some have also indicated H4 receptor expression in liver, brain, skeletal muscle, gastrointestinal tract, and epidermal tissue (Nakamura et al., 2000; Coge et al., 2001b; O'Reilly et al., 2002; Buckland et al., 2003; Sander et al., 2006; Rossbach et al., 2009, 2011; Yamaura et al., 2009). However, these organs also contain, to different extents, hematopoietic cells that may account for the mRNA signals. H4 receptor expression has been detected in synovial cells from rheumatoid arthritis and osteoarthritis patients and localized to both fibroblast-like and macrophage-like cells from rheumatoid arthritis synovial tissue, but this expression has not been confirmed by any functional data (Ikawa et al., 2005; Ohki et al., 2007; Grzybowska-Kowalczyk et al., 2008). The exact details of the expression of the receptor are still emerging and are complicated by the fact that the receptor levels and activity change in response to inflammatory stimuli and to the state of the cell. Furthermore, there is evidence that the receptor can be upregulated by inflammatory stimuli. For example, it was shown that the receptor expression on monocytes could be upregulated by interferon (IFN) γ treatment and that interleukin (IL)-4 upregulated the receptor expression on T cells (Dijkstra et al., 2007; Gutzmer et al., 2009). In addition it has been shown that the receptor is upregulated in all peripheral organs 24 hours after inducing sepsis in a mouse model and in the kidney of diabetic rats (Matsuda et al., 2010; Rosa et al., 2013).
Expression coupled with function defined by pharmacological methods has been shown on mast cells; eosinophils; neutrophils; dendritic cells; Langerhans cells; natural killer cells; monocytes; T cells, including γδT cells, T helper 1, 2, and 17 cells; keratinocytes; inflammatory dendritic epidermal cells; and fibroblasts (Gantner et al., 2002; Hofstra et al., 2003; Ling et al., 2004; Lippert et al., 2004; Gutzmer et al., 2005, 2009; Sander et al., 2006; Damaj et al., 2007; Dijkstra et al., 2007, 2008; Godot et al., 2007; Bäumer et al., 2008; Ikawa et al., 2008; Yamaura et al., 2009; Gschwandtner et al., 2010, 2011, 2012, 2013; Truta-Feles et al., 2010; Mommert et al., 2012; Glatzer et al., 2013; Mirzahosseini et al., 2013; Cowden et al., 2014; Czerner et al., 2014; Dib et al., 2014; Jemima et al., 2014). In addition, expression data, but as to date no functional data, have been reported in epithelial cells and basophils (Hofstra et al., 2003; Sander et al., 2006). However, care must be taken in interpreting all of these data even where expression appears to correlate with pharmacological activity. For example, some groups report mRNA in monocytes and CD8+ T cells whereas others do not, which may be attributed to intrasubject variability in expression or in different cell isolation and detection methods used (Gantner et al., 2002; Ling et al., 2004; Dijkstra et al., 2007; Werner et al., 2014). In addition, although most of the pharmacological data are compelling, some details require further scrutiny. For example, potency of thioperamide reported by Damaj et al. (2007) for natural killer cells and monocyte-derived dendritic cells is higher than that expected from binding data in recombinant systems (Liu et al., 2001a). A similar observation can be noted in the inhibition of CCL2 secretion in human monocytes, where the potency of clobenpropit is much higher than histamine in contrast to the affinity data in recombinant systems (Liu et al., 2001a; Dijkstra et al., 2007). These issues are clearly highlighted in the work of Gschwandtner et al. (2013) where six different H4 receptor agonists were shown to inhibit IL12p70 production in human monocytes, but in most cases this was only partially blocked by an H4 receptor antagonist. The situation is not as clear-cut as it first seems and warrants further investigation.
Recent studies provided evidence that genetic variations of the H4 receptor are associated with inflammatory diseases. Certain polymorphisms of the human H4 receptor gene that should result in an increased expression or signaling of the receptor were associated with atopic dermatitis (Yu et al., 2010b). Along this line, keratinocytes derived from patients with atopic dermatitis expressed higher levels of the H4 receptor mRNA compared with keratinocytes from patients without skin disease or psoriasis (Glatzer et al., 2013). In asthma, there appeared to be a link between three single nucleotide polymorphisms in the H4 receptor gene and infection-induced asthma (Simon et al., 2012). mRNA encoding the H4 receptor was significantly higher expressed and H4 receptor gene copy numbers were significantly amplified in patients with systemic lupus erythematosus compared with healthy controls. Moreover, H4 receptor gene copy numbers correlated to the incidence of arthritis, proteinuria, and antinuclear antibody abnormalities in systemic lupus erythematosus (Yu et al., 2010a).
There is some evidence that the H4 receptor may be expressed in the nervous system, but this remains controversial and needs further research. In general, results obtained with antibodies have not always been verified with parallel analyses of mRNA expression, and there is currently no consensus about expression of H4 receptor in various parts of the nervous system. In the human brain, expression of H4 receptor mRNA has been reported in the amygdala, cerebellum, corpus callosum, cortex frontal cortex, hippocampus, and thalamus in samples of a commercial mRNA panel (Strakhova et al., 2009b). Immunohistochemistry suggested that lamina I–VI of the murine and human cerebral cortex may express the H4 receptor with the strongest expression in layers IV and VI for the latter (Connelly et al., 2009). Expression has also been reported in the murine hippocampus, granule cell layer of the cerebellum, the thalamus, and in the spinal cord (Connelly et al., 2009; Strakhova et al., 2009b). The expression pattern in mouse and rat brains appeared to be similar to that in humans. However, quantitative reverse-transcription polymerase chain reaction and in situ hybridization data from many mammalian CNS areas are still not available to support the findings. In the rat, expression of the H4 receptor by immunohistochemistry was detected in approximately one-third of dorsal root ganglion neurons of all size classes. Immunoreactivity was also present in laminae I, II, and IV in spinal cord, in a pattern consistent with expression on primary afferent terminals (Strakhova et al., 2009b).
mRNA expression of both the full-length H4 receptor and one of the deletion forms has also been detected in a rat brain endothelial cell line using reverse-transcription polymerase chain reaction and sequencing (Karlstedt et al., 2013). The H4 receptor may also be involved in the vestibular system, because expression of the receptor has been reported in vestibular nucleus neurons and ganglia in rats (Desmadryl et al., 2012; Zhang et al., 2013). Overall, the presence of the H4 receptor in afferent pathways including dorsal root ganglion, spinal cord dorsal horn, and thalamus, as well as in other CNS areas, appears to be entirely consistent with a role in sensory signaling. The identity of the H4R-expressing sensory neurons in humans is yet to be established, but expression on skin innervating sensory neurons was shown in mice (Rossbach et al., 2011).
D. Function
One of the first functions ascribed to the H4 receptor was in mediating chemotaxis. Histamine acting via the H4 receptor has been shown to induce chemotaxis of a number of different cell types. In mast cells, histamine was shown to induce chemotaxis, and these effects were abrogated by treatment with H4 receptor antagonists but not antagonists to the other histamine receptors (Hofstra et al., 2003; Thurmond et al., 2004). Furthermore, neither response was seen in mast cells from H4R-deficient mice. Histamine also enhanced mast cell precursor chemotaxis to CXCL12 via a priming effect, but not to stem cell factor or leukotriene B4 (Godot et al., 2007). This enhanced chemotaxis was blocked by H4 receptor antagonists, but not antagonists for the H1 receptor or H2 receptor. In eosinophils, histamine or H4 receptor agonists induced chemotaxis, cell shape change, actin polymerization, and synergized with other chemotactic agents (O'Reilly et al., 2002; Buckland et al., 2003; Ling et al., 2004; Lim et al., 2005; Yu et al., 2010c). Furthermore, histamine activation of the H4 receptor induced adhesion molecule upregulation in eosinophils (Buckland et al., 2003; Ling et al., 2004). However, compared with eotaxin and formyl peptides, the stimulatory effects of histamine on eosinophil chemotaxis are small (Reher et al., 2012). Moreover, in contrast to the formyl peptide receptor, the H4 receptor does not mediate reactive oxygen species formation or eosinophil peroxidase release (Reher et al., 2012). Dendritic cells migration can also be modulated by the H4 receptor (Gutzmer et al., 2005; Damaj et al., 2007; Bäumer et al., 2008; Gschwandtner et al., 2010). Histamine-induced migration of dendritic cells in vitro or ex vivo and a H4R-dependent migration of dendritic cells was detected in vivo in mice during an allergic response (Cowden et al., 2010b). The migration of Langerhans cells has also been shown to be mediated by the H4 receptor (Gschwandtner et al., 2010). An ex vivo assay was used to show that the H4 receptor could mediate chemotaxis of Langerhans cells from epidermis from healthy donors or patients with bullous pemphigoid (Gschwandtner et al., 2010). In mice, injection of an H4 receptor agonist induced the migration of Langerhans cells in vivo (Gschwandtner et al., 2010). Chemotaxis of monocytes, γδ T cells, and fibroblasts has also been shown to be mediated via the H4 receptor (Damaj et al., 2007; Kohyama et al., 2010; Truta-Feles et al., 2010), although it was criticized (in particular for monocytes) that these data have only been published in single studies and have not been confirmed by other groups (Werner et al., 2014).
Migration of cells in response to H4 receptor stimulation has also been demonstrated in animal models or inflammation. The role of H1 receptor and H4 receptor in an ovalbumin specific murine dermatitis model was investigated with the adoptive transfer of ovalbumin-specific Th2-cells (Mahapatra et al., 2014). Treatment of recipient mice with either H1 receptor or H4 receptor antagonists resulted in a reduced migration of Th2 cells to sites of allergen challenge. Cytokine production in skin draining lymph nodes was only reduced by combined application of H1 receptor and H4 receptor antagonist, whereas mast cell counts were not altered by either H1 receptor, H4 receptor, or combined blockade. The paper does not state any results with regard to the effect of H1 receptor or H4 receptor blockade on skin inflammation.
Another study investigated carrageenan-induced pleurisy in mice (Ahmad et al., 2014). The administration of JNJ-7777120 (58) 1 hour before carrageenan application decreased both the number of T-cell subsets and GITR(+), GITR(+) IL-17A(+)–expressing T cells, and the production of Th1/Th17 cytokines, whereas the administration of 4-methylhistamine (17) increased both the numbers of CD4(+), CD25(+), CD4(+) CD25(+), GITR(+), GITR(+) IL-17A(+)–expressing T cells and the levels of T helper type 1 (Th1)/Th17 cytokines. JNJ-7777120 had anti-inflammatory effects, whereas 4-methyhistamine worsened inflammation in this model.
In addition to chemotaxis, the H4 receptor is also involved in cytokine release from a number of cell types. It has been shown that histamine can induce IL-16 production from CD8+ T cells (Gantner et al., 2002). This can be blocked by H2 receptor or H4 receptor antagonists but not by antagonists of the H1 receptor. The H4 receptor inhibited IL-12p70 release from human monocyte-derived dendritic cells and inflammatory dendritic epidermal cells (Gutzmer et al., 2005; Dijkstra et al., 2008). In mouse dendritic cells, Toll-like receptor (TLR)–stimulated production of IL-6, KC, MIP-1α, interferon-γ–induced protein 10 (IP-10), and tumor necrosis factor α (TNFα) was blocked by an H4 receptor antagonist and was attenuated in dendritic cells from H4R-deficient mice; however, in this case, IL-10 and IL-12p70 were unchanged (Dunford et al., 2006). Regulation of cytokine production from dendritic cells appears to impact their ability to polarize CD4+ T cells to a Th2 phenotype, and histamine acting via the H4 receptor may also mediate cross-presentation to CD8+ T cells (Dunford et al., 2006; Amaral et al., 2007). Histamine downregulated the production of CCL2 in human monocytes, Langerhans cells, inflammatory dendritic epidermal cells, and slan-dendritic cells (Dijkstra et al., 2007, 2008; Gschwandtner et al., 2011, 2012). Furthermore, other Th1-promoting cytokines and chemokines were downregulated in a variety of human antigen-presenting cells via H4 receptor stimulation, such as downregulation of TNFα, interferon α, and CXCL8 on plasmacytoid dendritic cells (Gschwandtner et al., 2011, 2012), and IP-10 on monocytes and myeloid dendritic cells (Glatzer et al., 2014). Stimulation of the H4 receptor induced IL-31 production in peripheral blood mononuclear cells (PBMC) or Th2 polarized T cells, and this production was significantly increased in PBMC from subjects with atopic dermatitis compared with healthy controls (Gutzmer et al., 2009). The receptor also mediated IL-4 and IFNγ production from murine natural killer T cells in vivo (Leite-de-Moraes et al., 2009). Cytokine and chemokine production in vivo has also been shown to be regulated by the H4 receptor in several disease models. Reduction in Th2 cytokines such as IL-4, IL-5, and IL-13 levels were observed after H4 receptor antagonist treatment in mouse models of asthma, allergic rhinitis, and allergic contact dermatitis (Takahashi et al., 2009; Cowden et al., 2010a; Seike et al., 2010). H4 receptor antagonist treatment reduced tissue TNFα levels in a rat colitis model, and several inflammatory cytokines and chemokines were reduced in a model of Th2-dependent dermal inflammation (Varga et al., 2005; Cowden et al., 2010a). IFNγ synthesis of murine T helper cells was also reduced by the H4 receptor antagonist (Vauth et al., 2012). The levels of lipid mediators are also affected by the H4 receptor, with antagonists showing reduction in leukotriene B4 and prostaglandin D2 in models of pleurisy and peritonitis, respectively (Takeshita et al., 2003; Strakhova et al., 2009a).
The H4 receptor also appears to be involved in IL-17 production from Th17 cells. Histamine and an H4R-selective agonist increased IL-17 message and protein secretion from human Th17 cells, and this effect could be blocked by an H4 receptor antagonist (Mommert et al., 2012). It has also been shown that H4 receptor antagonists can reduce IL-17 production from PBMC (peripheral blood mononuclear cells) after stimulation (Cowden et al., 2014). The same effect is seen in mice. Stimulation of whole blood with anti-CD3/CD28 and IL-23 led to an increase in IL-17 production in vitro, and this was decreased in blood taken from H4R-deficient mice or mice treated with an H4 receptor antagonist (Cowden et al., 2014). Furthermore, it was shown that Th17 cell development in vivo was inhibited in H4R-deficient mice or mice treated with an H4 receptor antagonist (Cowden et al., 2014). Finally, a reduction in IL-17 levels via H4 receptor blockade has been noted in mouse models of asthma, dermatitis, and arthritis (Dunford et al., 2006; Cowden et al., 2010b, 2014). Basophils have been implicated in enhancing Th17 responses in humans, and this effect appears to be mediated by H2 receptor and H4 receptor (Wakahara et al., 2012). In human neutrophils, engagement of the H4 receptor blocked the adhesion-dependent degranulation, indicating a possible anti-inflammatory effect of the H4 receptor on these cells (Dib et al., 2014).
In human and murine mast cells, the H4 receptor is not only involved in chemotaxis but also in other functions. H4 receptor stimulation in murine bone marrow–derived mast cells induced IL-6 production and increased lipopolysaccharide (LPS)-induced IL-6 production via ERK and PI3K activation (Desai and Thurmond, 2011). Stimulation of murine bone marrow–derived mast cells with an H4 receptor antagonist during the IgE sensitization phase inhibited the degranulation and IgE-induced upregulation of the high affinity IgE receptor FcεRI (Mirzahosseini et al., 2013). In human cord blood–derived mast cells, histamine and 4-methylhistamine (17) induced degranulation and the production of leukotrienes, and these effects were blocked by JNJ-7777120 (58) (Jemima et al., 2014). Furthermore, these cells responded to 4-methylhistamine with ERK and PI3K activation as well as the production of a variety of inflammatory mediators such as IL-6 (Jemima et al., 2014). In ex vivo animal models, human mastocytoma cells and bone marrow-derived murine mast cells, H4 receptor activation resulted in inhibition of renin release via activation of protein kinase C isotype-ε and aldehyde dehydrogenase type-2 (Aldi et al., 2014).
The in vitro effects on chemotaxis and mediator release from immune cells suggest that the H4 receptor should be involved in inflammatory responses. Indeed this has been borne out in several animal models. The first in vivo reports of H4 receptor activity involved neutrophilia models induced by the TLR ligand zymosan where H4 receptor antagonists were shown to be anti-inflammatory (Gantner et al., 2002; Thurmond et al., 2004). Effects on neutrophils can also be seen in a rat colitis model where treatment with H4 receptor antagonists reduced tissue damage, neutrophilia, and tissue TNFα levels (Fogel et al., 2005; Varga et al., 2005). Treatment with JNJ-7777120 was protective in a model of gastric ulceration (Adami et al., 2012). A link between TLR signaling and the H4 receptor may be a general effect as TNF production upon in vivo administration of LPS has been shown to be reduced in H4R-deficient mice or upon treatment with H4 receptor antagonists (Cowden et al., 2013). The role of the H4 receptor in both innate and adaptive immune responses may explain the protective effect of H4 receptor antagonists or genetic deficiency in the receptor in mouse models of arthritis (Nent et al., 2013; Cowden et al., 2014).
Eosinophilic inflammation appears also to be mediated by the H4 receptor. In mouse models of asthma, H4 receptor antagonists reduced eosinophilia and Th2 responses (Dunford et al., 2006; Deml et al., 2009; Cowden et al., 2010a; Somma et al., 2013). However, the story may not be as straightforward as it first appeared, because a locally administered H4R/H2 receptor agonist was protective in another mouse asthma study and these effects may have been mediated via enhanced recruitment of regulatory T cells via the H4 receptor (Morgan et al., 2007). In another study on EAE, H4 receptor knockout mice had a worse outcome compared with wild-type controls. This was attributed to altered migration and function of regulatory T cells in the H4 receptor knockouts (del Rio et al., 2012). Th2 responses can also be impacted in models of cutaneous inflammation. In most acute models driven by hapten application, the H4 receptor clearly mediated pruritic responses but did not have a role in the acute inflammation (Rossbach et al., 2009; Seike et al., 2010). However, with chronic application the situation appears to change and an H4 receptor antagonist was anti-inflammatory, whereas an H4 receptor agonist was proinflammatory (Seike et al., 2010). This may be a result of the switch to a more eosinophilic inflammation with the chronic treatment. Interestingly, the one acute hapten model that is sensitive to H4 receptor inhibition is the one where even the acute inflammation has a strong eosinophilic component. In this case, treatment with H4 receptor antagonists were anti-inflammatory and led to a reduction in the number of mast cells and eosinophils in the affected skin (Cowden et al., 2010b). In addition to effects on eosinophils, the H4 receptor also mediates keratinocyte proliferation, which may explain some of the effects seen in the model (Glatzer et al., 2013). Finally, the combination of H1 receptor and H4 receptor antagonists was more effective than either alone in two models of contact dermatitis (Matsushita et al., 2012; Ohsawa and Hirasawa, 2012).
Beyond the role in inflammatory response the H4 receptor has also been associated with neuronal responses, although this remains controversial (Schneider et al., 2014b). A study analyzed [35S]GTPγS binding and noradrenaline release in human, guinea pig, and mouse cortex (Feliszek et al., 2015). The H4 receptor agonist 4-methylhistamine (17) failed to affect these readouts, whereas the H3 receptor increased [35S]GTPγS binding and inhibited noradrenaline release. These readouts are limited in their coverage of potential neuronal effects of the H4 receptor, and so further studies are required. Nevertheless, the H4 receptor expression has been reported on neurons, suggesting that it can have an effect on sensory signaling. In whole cell clamp recordings of layer IV somatosensory cortex cells from mice, H4 receptor-induced hyperpolarization has been reported with a single H4 receptor agonist, antagonist, and respective dose (Connelly et al., 2009); confirmation is required with multiple ligands and a full concentration range. In rat vestibular neurons, treatment H4 receptor antagonist led to inhibitory effects on the action potential (Desmadryl et al., 2012). In addition it has been shown that the receptor also mediated action potential discharge in human submucosal neurons (Breunig et al., 2007). Single-cell Ca2+ imaging studies in mice recently showed that histamine induces a [Ca2+]i increase in a subset of (skin-specific) sensory neurons via activation of the H4 receptor. Dual activation of H1 receptor and H4 receptor on sensory neurons, which in turn results in the excitation of histamine-sensitive afferents, may underlie the sensation of itch (Rossbach et al., 2011). These effects may translate into the nociceptive and antipruritic activities seen in animal models (Bell et al., 2004; Coruzzi et al., 2007; Dunford et al., 2007; Altenbach et al., 2008; Cowart et al., 2008; Nakano et al., 2009; Rossbach et al., 2009; Yamaura et al., 2009; Cowden et al., 2010b; Hsieh et al., 2010a,b). In both cases the effects of H4 receptor antagonists appear to be mediated via modulation of neuronal activity. For example, the reduction in pruritic responses in H4R-deficient mice could not be reconstituted with bone marrow from wild-type animals (Dunford et al., 2007). Furthermore, compound 48/80 was shown to induce pruritic responses by directly activating neurons, and these effects were reduced by H4 receptor antagonists (Dunford et al., 2007).
Recently, the expression of all four histamine receptors has been demonstrated on microglia, which represents resident immune cells in the brain (Ferreira et al., 2012; Dong et al., 2014). Histamine induced microglia migration and inhibited LPS-induced IL-1β production via the H4 receptor in one study (Ferreira et al., 2012). In another study, histamine induced TNFα and IL-6 production, which could be blocked by H1 receptor and H4 receptor antagonists (Dong et al., 2014). Thus, neuronal functions could also be indirectly affected via histamine influence on microglia.
A number of other functions have been described for the H4 receptor. One study showed that the administration of histamine increased COX-2 expression and activity, cell proliferation, and vascular endothelial growth factor production in the COX-2-positive colon cancer cell lines HT29 and Caco-2, and treatment with the H2 receptor or H4 receptor antagonists prevented these effects (Cianchi et al., 2005). The H4 receptor has been shown to mediate cell cycle arrest in hematopoietic progenitor cells induced by growth factors (Petit-Bertron et al., 2009). In addition, activation of the receptor appears to inhibit proliferation of several human cancer cell lines (Massari et al., 2011, 2013). The release of adrenocorticotropic hormone from a mouse pituitary tumor cell line via H4 receptor activation has also been reported (Meng et al., 2008). Simultaneous activation of H4 receptor and H2 receptor has been reported to mediate sickle erythrocyte adhesion to endothelium (Wagner et al., 2006).
In a rat model of postinflammatory visceral hypersensitivity, colitis was induced by trinitrobenzenesulfonic acid, and after healing, visceral sensitivity was measured after administration of JNJ-7777120 (58) and/or levocetirizine (11) (Deiteren et al., 2014). Visceral sensitivity was reduced by JNJ-7777120 and levocetirizine, the combination of both had a synergistic effect.
E. H4 Receptor-Selective Ligands
Many known ligands of the H3 receptor, notably imidazole-containing compounds, also possess significant affinity for the H4 receptor. These include (R)-α-methylhistamine (33), imetit (34), immepip (35), and clobenpropit (41), which all act as agonists at the H4 receptor. In contrast, the H3 receptor neutral antagonist thioperamide (40) was shown to be an inverse agonist of the H4 receptor (Oda et al., 2000; Liu et al., 2001a,b; Lim et al., 2005).
1. Agonists.
The first described H4 receptor agonists were methylcyanoguanidine analogs of 2,5-disubstituted tetrahydrofuranylimidazoles. These compounds have moderate affinity for the H4 receptor and some selectivity over H3 receptor (Lim et al., 2005). The best example is OUP-16, with a binding Ki of 125 nM for the human H4 receptor and an 18-fold selectivity over H3 receptor (Hashimoto et al., 2003).
Although initially developed as a H2 receptor ligand, 4-methylhistamine (17) (Ki = 50 nM) is the most selective H4 receptor agonist currently reported, with more than 100-fold selectivity over the H1 receptor, H2 receptor and H3 receptor. Structural modification of dimaprit (a nonimidazole, mixed H2 receptor/H3 receptor/H4 receptor ligand; 19) led to the discovery of VUF-8430 (57), a full agonist at the H4 receptor with around 30-fold selectivity over the H3 receptor (Lim et al., 2006).
The antipsychotic drug clozapine (56) has affinity for a number of GPCRs but also possesses H4 receptor agonist activity. Minor structural modification of this tricyclic compound led to the identification of VUF-6884, a close analog with good affinity for the H4 receptor, shown to be more than 300-fold selective over the H3 receptor (Lim et al., 2006). The aminopyrimidine ligand ST-1006 (75) has been characterized as the most potent H4 receptor agonist reported to date, with a PEC50 of 8.95 (Gschwandtner et al., 2013) (Fig. 15).
Fig. 15.

H4 receptor ligands.
2. Antagonists.
The indole carboxamide compound JNJ-7777120 (1-[(5-chloro-1H-indol-2-yl) carbonyl]-4-methylpiperazine; 58) was the first published selective nonimidazole H4 antagonist. It was reported to bind to the human H4 receptor with high affinity (Ki = 4 nM) and has excellent selectivity over a panel of GPCRs, including the H1 receptor, H2 receptor and H3 receptor (Thurmond et al., 2004). JNJ-7777120 also possesses high affinity for the mouse and rat H4 receptor (Thurmond et al., 2004), and it has been used extensively as the reference compound to determine the role of H4 receptor antagonism in a variety of experimental animal models of disease (Stark, 2013).
Based on JNJ-7777120, a range of structurally diverse compound classes has evolved, as represented by the benzimidazole VUF-6002, also known as JNJ-10191584 (59) (Terzioglu et al., 2004) and thienopyrrole (60) analogs (Venable et al., 2005). Both compounds 59 and 60 demonstrated binding affinities close to that of JNJ-7777120 (Terzioglu et al., 2004; Venable et al., 2005). Similar compounds were published by Pfizer (New York, NY), as represented by compound 61, where the N-methylpiperazine moiety of 59 was replaced by an octahydropyrrolo[3,4-c]pyrrole group. This compound has low nanomolar affinity for the human H4 receptor (Lane et al., 2012). Further benzimidazole-containing compounds were described in a series of patents from Johnson & Johnson (New Brunswick, NJ) (Arienti et al., 2006; Lee-Dutra et al., 2006; Edwards et al., 2007a,b). The benzimidazole scaffold in these compounds is typically linked to a substituted phenyl or heterocycle. These are then connected by an alkyloxy or alkyl amine chain to a piperidine or (homo)piperazine moiety.
Quinoxaline derivatives such as compound 62 were found to possess excellent affinity for the human H4 receptor (Edwards and Venable, 2005). Further exploration of the SAR led to the identification of piperazin-1-yl quinoxalines, including VUF-10214 (63) and VUF-10148 (64), which retained good H4 receptor potency and demonstrated anti-inflammatory efficacy in a rat model of carrageenan-induced paw edema (Smits et al., 2008).
Aminopyrimidine analogs are the most widely published H4 receptor antagonists and it was Bayer (Leverkusen, North Rhine-Westphalia, Germany) who first disclosed two series of 2-aminopyrimidine compounds possessing significant H4 receptor functional activity, as exemplified by compound 65 (Sato et al., 2005). Subsequently, further 2-aminopyrimidine H4 receptor antagonists were published by Johnson & Johnson (66) (Cai et al., 2008), Pfizer (67) (Bell et al., 2007), Palau (Palau-solità i Plegamans, Barcelona, Spain) (68) (Carceller Gonzalez et al., 2007), UCB (Brussels, Belgium) (69) (Raphy et al., 2008), Abbott (70) (Altenbach et al., 2008; Cowart et al., 2008), Incyte (Wilmington, DE) (74), and academic institutions (Sander et al., 2009; Werner et al., 2010). ZPL-3893787 or formerly PF-3893787 (67) has completed phase I studies and is being developed by Ziarco Pharma (Sandwich, UK) as an oral treatment of atopic dermatitis (Liu, 2014). The other clinical stage compound JNJ-39758979 (72) significantly reduced histamine-induced itch in healthy human subjects (Kollmeier et al., 2014). INCB38579 (74) has demonstrated efficacy in preclinical models of inflammatory pain and pruritus (Shin et al., 2012).
Rotationally constrained analogs of the 2-aminopyrimidine class, including benzofuropyrimidines and benzothienopyrimidines, have also been shown to be potent H4 receptor antagonists. These compounds have been claimed by Argenta (Harlow, UK), Cellzome (Heidelberg, Germany) (Reid et al., 2007a,b,c), Johnson & Johnson (71) (Chavez et al., 2008), and Abbott (73) (Altenbach et al., 2008). Compound A-943931 (73) has shown efficacy in various animal models of pain, inflammation, and pruritus (Cowart et al., 2008).
JNJ-7777120 (58) has been broadly used as standard H4 receptor antagonist to delineate specific pathophysiological functions of the H4 receptor. In fact, JNJ-7777120 exhibits an excellent selectivity for the H4 receptor relative to the H1 receptor, H2 receptor and H3 receptor and numerous other GPCRs (Jablonowski et al., 2003). Most animal studies with JNJ-7777120 are compatible with the notion that the H4 receptor play a proinflammatory role in bronchial asthma, atopic dermatitis, and pruritus and that H4 receptor antagonism blocks those proinflammatory effects (Dunford et al., 2006; Deml et al., 2009; Rossbach et al., 2009).
However, the pharmacology of H4 receptor ligands is not clear cut and well understood and therefore care must be taken when interpreting the results from any single ligand. For example, Schnell et al. (2011) reconstituted H4 receptor species orthologs from human, rat, mouse, and canine with the G protein heterotrimer Giα2β1γ2 plus the regulator of G protein signaling RGS19 in Sf9 insect cell membranes and measured high-affinity steady-state GTPase activity as a parameter for receptor activation. At all four H4 receptor species orthologs, histamine acted as full agonist. At human H4 receptor, JNJ-7777120 exhibited partial inverse agonistic activity, whereas at the other H4 receptor species orthologs, JNJ-7777120 exhibited partial agonistic activity. Moreover, at canine H4 receptor, thioperamide displayed weak partial agonism, whereas at human H4 receptor, the compound was a strong inverse agonist, and at rat and mouse H4 receptor, thioperamide is a neutral antagonist or very weak inverse agonist. To make matters more complicated, Rosethorne and Charlton (2011) expressed the human H4 receptor in an osteosarcoma cell line and found that with respect to [35S]GTPγS binding, JNJ-7777120 behaved as a neutral antagonist or very weak inverse agonist. In marked contrast, with respect to translocation of beta-arrestin to the plasma membrane, JNJ-7777120 exhibited strong partial agonistic activity. Moreover, JNJ-7777120 induced a delayed activation of the mitogen-activated protein kinase pathway that is typical for β-arrestin–mediated signal transduction (Luttrell and Gesty-Palmer, 2010).
The reported biased H4 receptor signaling by JNJ-7777120 has in the meantime been extended to a broader range of benzimidazole analogs (Nijmeijer et al., 2013), but testing a variety of known H4 receptor ligands has resulted in the identification of biased ligands for both the G protein and β-arrestin pathways (Nijmeijer et al., 2012).
Although data such as these are currently restricted to transfected cell systems and have not been observed in primary cells, it points to the potential complex pharmacology of the H4 receptor and JNJ-7777120. As with any single ligand, researchers must use a combination of tools including agonist/antagonist pairs, ligands from different chemical classes, and/or knockout animals before making conclusions about the function of the receptor.
Discrepancies are also reported in vivo and in primary cells. In a murine model of allergic asthma, the effects of the H4 receptor antagonist JNJ-7777120, the H3 receptor antagonist JNJ-5207852, and the H3 receptor and H4 receptor antagonist thioperamide were compared when given during the sensitization phase in ovalbumin-induced allergic asthma (Neumann et al., 2013). After provocation, JNJ-7777120 resulted in reduced serum titers of ovalbumin-specific IgE, inflammatory infiltrations in the lungs, and eosinophils in the bronchoalveolar lavage as previously reported (Dunford et al., 2006). Thioperamide did not completely mimic the results seen with JNJ-7777120 because it only resulted in lower eosinophil counts in the bronchoalveolar lavage; JNJ-5207852 had no effect on the investigated parameters. The apparent disconnect between the results seen with JNJ-7777120 and thioperamide led the authors speculate that JNJ-7777120 could have effects other than blocking the H4 receptor. However, these differences can also be accounted for by the known nonspecific activity of thioperamide (such as dual H3 receptor/H4 receptor antagonism) or differences in pharmacokinetics. In monocytes as native human cells, a study compared the ability of different H4 receptor agonists (4-methylhistamine [14], UR-PI376, clobenpropit [41], VUF8430, and ST-1006 [75]) in their ability to suppress IFNγ + LPS-induced IL-12 production (Gschwandtner et al., 2013). All agonists had considerable lower potencies in this assay compared with published data from transfected cell lines, and the effect could be only partially blocked by JNJ-7777120 in the case of 4-methylhistamine, UR-PI376, clobenpropit, and VUF8430 but completely blocked the ST-1006 effect. Finally, a study compared the development of murine autoimmune encephalomyelitis in histidine-decarboxylase knockout mice lacking histamine and knockout mice lacking all four known histamine receptors (Saligrama et al., 2013). Whereas the encephalomyelitis was aggravated in histidine-decarboxylase knockout mice, symptoms were alleviated in H1 receptor–H4 receptor knockout mice. The authors speculated that there might be histamine signaling pathways independent of the known H1 receptor–H4 receptor.
F. Clinical Potential
Since its discovery at the turn of the millennium the therapeutic utility of targeting the H4 receptor has been a subject of intense research. It is remarkable that in such a short period of time compounds are already entering clinical development. This has culminated with the first clinical efficacy results being published in 2014.
Effects of H4 receptor ligand on pruritus have been among the first studied clinically. Histamine has long been known as a mediator that induces itch in normal human skin, and this pruritic response was shown to be further enhanced in patients with atopic dermatitis (Ikoma et al., 2003). In a mouse model of experimental pruritus, it was shown that both histamine and 4-methylhistamine (17) produced significantly less scratching in H4 receptor knockout mice when compared with wild-type animals (Dunford et al., 2007). In the same study, the histamine-induced pruritic response in wild-type mice was significantly reduced after oral treatment with the selective H4 receptor antagonist JNJ-7777120 (58). Administration of an H2 receptor antagonist, an H3 receptor antagonist, or the peripherally restricted H1 receptor antagonist fexofenadine (13) produced no reduction in the scratching response. Interestingly, the CNS-penetrant H1 receptor antagonist diphenhydramine (6) demonstrated some antipruritic activity, and the combination of diphenhydramine and JNJ-7777120 resulted in the total ablation of the histamine-induced pruritic response (Dunford et al., 2007).
The antipruritic property of H4 receptor antagonism in animal models appears to be broad based. Significant reduction in the pruritic response by JNJ-7777120 has been observed after challenges with allergen and compound 48/80 (Dunford et al., 2007), substance P (Yamaura et al., 2009), the haptens 2,4-dinitrochlorobenzene and toluene-2,4-diisocyanate (Rossbach et al., 2009), and fluorescein isothiocyanate (Cowden et al., 2010b). There is also increasing human evidence, including expression of the H4 receptor on skin mast cells (Lippert et al., 2004) and epidermal dendritic cells (Bäumer et al., 2008; Dijkstra et al., 2008). H4 receptor agonists have been shown to induce the production of IL-31 in PBMC and Th2 cells, a cytokine that is linked to the induction of pruritus in atopic dermatitis and the effect was blocked by JNJ-7777120 (Sonkoly et al., 2006; Grimstad et al., 2009; Gutzmer et al., 2009). The antipruritic effects of H4 receptor antagonist combined with anti-inflammatory effects in Th2-driven dermatitis models (Cowden et al., 2010b; Seike et al., 2010) indicate that compounds targeting the receptor should be beneficial in a number of pruritic conditions, such as atopic dermatitis and urticaria.
The preclinical findings have now been validated in the clinic using JNJ-39758979 (72), a potent and selective H4 receptor antagonist (Thurmond et al., 2014a). This compound showed excellent preclinical safety in rat and monkey 6-month toxicity studies, and an oral formulation was well tolerated, with the exception of nausea, in a phase I clinical study with healthy volunteers (Thurmond et al., 2014a). In a double-blind study the effect of a single dose of JNJ-39758979 (72), cetirizine (11), or placebo was tested on histamine-induced itch, wheal, and flare reactions. JNJ-39758979 significantly reduced itching but did not affect wheal and flare reactions, whereas cetirizine reduced both itch and wheal and flare (Kollmeier et al., 2014).
This clinical result points to therapeutic potential in pruritic conditions known to be mediated by histamine such as acute urticaria, allergic rhinitis, and allergic conjunctivitis. Furthermore, the combination of the human and preclinical data supports the potential in other pruritic conditions such as atopic dermatitis and psoriasis.
The clinical effects on histamine-induced pruritus along with preclinical data showing a role of for the H4 receptor in inflammation suggest that inflammatory skin disease may be attractive indications for ligands of the receptor. Preclinical data have shown that antagonism of the receptor is efficacious in several dermatitis models (Cowden et al., 2010b; Seike et al., 2010; Matsushita et al., 2012; Ohsawa and Hirasawa, 2012; Thurmond et al., 2014a). In addition, the receptor has been shown to mediate inflammatory activity of many cell types implicated in atopic dermatitis and psoriasis.
Allergic airway diseases, such as asthma and allergic rhinitis, have also been suggested to be potential indications for H4 receptor ligands. Histamine has long been documented to be released in the human lung during an asthmatic response (Casale et al., 1987). However, H1 receptor antagonists are only weakly efficacious in treating this inflammatory airway disease (Simons, 1999; Thurmond et al., 2008), and there is little evidence supporting a clinical benefit with H2 receptor or H3 receptor antagonists. Although the lack of effect may imply that histamine is not important in asthma, this seems unlikely given its bronchoconstrictive and proinflammatory actions. Instead the lack of efficacy of H1 receptor antagonists may indicate the involvement of another histamine receptor. Certainly the expression of the H4 receptor on immune cell types known to be involved in asthma (such as eosinophils, mast cells, T lymphocytes, and dendritic cells), coupled with its involvement in inflammatory processes including chemotaxis and cytokine production, support the concept that targeting this receptor may provide therapeutic benefits.
In addition to data derived from cellular expression and in vitro functional studies, results obtained from animal models have provided further evidence to support a role for the H4 receptor in modulating airway inflammation. After allergen challenge of sensitized H4 receptor KO mice, there was a significant decrease in infiltrating lung eosinophils and lymphocytes compared with sensitized wild-type animals (Dunford et al., 2006). Ex vivo restimulation of T lymphocytes from the H4R-deficient animals showed significant decreases in a range of proinflammatory cytokines and indicated that the receptor was mediating Th2 responses in this model (Dunford et al., 2006). This dampening of airway inflammation has also been observed after the administration of H4 receptor antagonists in a number of animal models (Dunford et al., 2006; Deml et al., 2009; Cowden et al., 2010a; Thurmond et al., 2014a). In addition to the anti-inflammatory effects in the lung, JNJ-7777120 was also shown to improve lung function in these mouse models (Cowden et al., 2010a). In contrast, intratracheal administration of the H4 receptor agonist 4-methylhistamine was found to be protective in a mouse model of allergic asthma, potentially through recruitment of regulatory T lymphocytes (Morgan et al., 2007). However, the 4-methylhistamine data are difficult to interpret, because in addition to its agonist effects on the H4 receptor, 4-methylhistamine (17) also exhibits agonist effects on the H2 receptor (Black et al., 1972). Although H1 receptor antagonists per se do not play a role in bronchial asthma, it is possible that there are effects when combined with H4 receptor antagonists. Specifically, when applied alone in the sensitization phase, the H1 receptor antagonist mepyramine exhibited no beneficial effect on eosinophilia in the bronchoalveolar lavage fluid in a mouse asthma model (Deml et al., 2009). However, mepyramine (8) potentiated the therapeutic effects of JNJ-7777120 (58) in this disease model. Based on these data, the development of dual H1 receptor/H4 receptor antagonists may be desirable. But again and by analogy to the data with 4-methylhistamine (Morgan et al., 2007), the situation may be more complex, particularly when applied during the provocation phase, because mepyramine (8) antagonized the effects of JNJ-7777120 (Beermann et al., 2012a).
The H4 receptor may also play a role in mediating upper airway inflammation. In patients with chronic rhinosinusitis, there is a significant increase in the level of H4 receptor expression in nasal polyp tissue when compared with normal nasal mucosal tissue (Jokuti et al., 2007). In a mouse model of allergic rhinitis, JNJ-7777120 (58) was effective in significantly reducing various upper airway inflammatory symptoms, including sneezing and nasal rubbing, when the compound when given either orally or intranasally (Takahashi et al., 2009). Clinical studies in asthma and allergic rhinitis have been reported for the H4 receptor antagonists JNJ-39758979 (72), UR-63325, and PF-03893787 (now known as ZPL-3893787 [67]), so the question on whether H4 receptor antagonism would provide therapeutic benefits in inflammatory airway diseases may soon be answered once the data from these studies are published.
In addition to inflammatory diseases, a role for the H4 receptor in nociception was recently suggested. Both JNJ-7777120 (58) and its benzimidazole analog VUF6002 (59) were effective in increasing paw withdrawal latency in a rat model of carrageenan-induced thermal hyperalgesia (Coruzzi et al., 2007). Additional work has shown that this compound was as efficacious as the nonsteroidal anti-inflammatory drug diclofenac in this model (Hsieh et al., 2010a). Similar effects have also been shown with other H4 receptor antagonists in this model where once again the maximum effect was similar to diclofenac (Altenbach et al., 2008; Cowart et al., 2008; Liu et al., 2008). The amino-pyrimidine H4 receptor antagonist A-943931 (73) was also effective in the carrageenan model, as well as in a spinal nerve ligation model of neuropathic pain (Altenbach et al., 2008). Effects have also been seen in more chronic models. JNJ-7777120 (58) was efficacious in a subchronic inflammatory pain model induced by complete Freunds adjuvant (Hsieh et al., 2010a). Effects were also seen in a skin incision model of postoperative pain using mechanical allodynia as a readout (Hsieh et al., 2010a). In this case, the highest dose of JNJ-7777120 yielded an effect approaching that of morphine. The compound was also tested in model of osteoarthritis joint pain (Hsieh et al., 2010a). This model uses intra-articular injection of sodium monoiodoacetate, and pain was assessed by the hindlimb grip force. JNJ-7777120 was delivered 20 days after sodium monoiodoacetate injection 30 minutes before the assessment. The compound was efficacious and improved the grip force by 47% compared with a 62% improvement with celecoxib. H4 receptor antagonists have also shown efficacy in models of neuropathic pain. In pain models in rats induced by chronic constriction of the sciatic or spinal nerve, JNJ-7777120 showed a reversal that was better than that observed with gabapentin (Hsieh et al., 2010a). Furthermore, the effect was maintained upon dosing for 8 days, indicating that no tolerance development (Hsieh et al., 2010a). Other H4 receptor antagonists have also shown activity in this model (Cowart et al., 2008). Although the mechanism of action of these antinociceptive effects are not clear, the recent observation of H4 receptor expression in the spinal cord, dorsal root ganglion, and certain regions of the brain may offer an explanation (Strakhova et al., 2009b).
In conclusion, the H4 receptor has emerged as a novel and attractive drug target for a range of disease indications, in particular in disorders associated with inflammation. However, there are still many open questions regarding the function of the receptor. Key among these are the exact expression profile of the receptor and the function, if any, in the nervous system. The recent advancement of H4 receptor compounds into clinical development will undoubtedly aid our understanding of therapeutic utilities in targeting this newest member of the histamine receptor family.
VI. Other Responses to Histamine
A. Histamine-Gated Channels
Ionotropic histamine receptors are common in mollusks and arthropods but have not been unequivocally identified in vertebrates (McCaman and Weinreich, 1985; Gisselmann et al., 2002). The ionotropic histamine receptor in invertebrates is a chloride channel related to the glycine and the GABAA receptor (Fleck et al., 2012). In vertebrates it may turn out to be a GABAA-type receptor with a particular subunit composition. Among the many sites for allosteric modulators of the GABAA receptor there may also be a histamine-sensitive example. Saras et al. (2008) reported that histamine can directly open homomultimeric channels composed of GABAA receptor β-subunits in which GABA is only a weak partial agonist in Xenopus oocytes (Fig. 16A). In heteromultimeric channels composed of α1β2- or α1β2γ2-subunits histamine is not an agonist but potentiates the GABA response. These effects have yet to be shown in native neurons.
Fig. 16.

Histamine gating chloride-channels. (A) Responses of artificially expressed GABAA receptor β3-homomeric channels in xenopus oocytes to GABA, tele-methylhistamine, histidine, and histamine all at 3 mM. (B) Thalamic GABAergic neuron is inhibited and hyperpolarized by histamine in a slice from ferret brain. An increased Cl−-conductance (reversal potential −73 mV) displays H2 receptor pharmacology, see text. Modified from Lee et al. (2004) and Saras et al. (2008) with permission.
The axon varicosities of aminergic, including histaminergic, neurons rarely form classic synapses in vertebrates and, apart from nicotinic ACh and 5HT3 receptors, the biogenic amines act on metabotropic receptors. Nevertheless there are indications for histamine-gated chloride channels in hypothalamus (Hatton and Yang, 2001) and thalamus (Lee et al., 2004) on oxytocin neurons in the supraoptic nucleus, such a conductance is blocked by the chloride channel blocker picrotoxin (not the GABAA antagonist bicuculline) and H2 receptor antagonists, not mediated by a G protein. Stimulation of histamine neurons evokes fast inhibitory postsynaptic potentials (IPSPs) that reverse at the chloride equilibrium (Fig. 16B). Hatton and Yang (2001) suggested an ionotropic action and ruled out GABA release from histaminergic axons. Because histamine neurons express GABA, GABA release could have been responsible for these IPSPs.
GABAergic interneurons in the thalamus are surrounded by histaminergic fibers and histamine evokes an inhibitory chloride conductance mediated by a H2-related receptor but not cyclic AMP (Lee et al., 2004). GABAergic activity of imidazole compounds (Haas et al., 1973), in particular imidazole-derived H2R-antagonists (Lakoski et al., 1983), has been described. Unequivocal evidence for histaminergic IPSPs mediated by chloride currents has been obtained in paired recordings from the histaminergic C2 neuron and a follower neuron in Aplysia (McCaman and Weinreich, 1985) (Fig. 17).
Fig. 17.

Ionic mechanism of histaminergic inhibitory postsynaptic potentials (ipsps) in aplysia. Paired recording in the histaminergic C2 neuron from aplysia and a follower neuron. Five action potentials are elicited (A) and the response is registered at different voltage levels (B, from −50 to −90 mV). Histamine is also puffed on the follower neuron at these different voltage levels (C). A fast component reverses at the equilibrium potential for Cl−, between −60 and −70 mV, a slow component near −90 mV, close to the K+-equilibrium potential. Modified from McCaman and Weinreich (1985).
B. Allosteric Modulation of N-Methyl-d-Aspartate Receptor
A second-messenger-mediated modulation of ionotropic receptors is common for several transmitters: facilitation of N-methyl-d-aspartate (NMDA) receptors through PKC and a reduction of the Mg2+-block have been described as a result of H1 receptor activation (Payne and Neuman, 1997). However, histamine also directly facilitates NMDA receptors and enhances excitatory transmission (Bekkers, 1993), likely through their polyamine modulatory site (Vorobjev et al., 1993). This action is occluded by spermidine and is pH sensitive (Saybasili et al., 1995; Yanovsky et al., 1995). In a slightly acidified environment (pH, 7.0), but not at pH 7.4, the late NMDA component of extra- and intracellularly registered excitatory postsynaptic potentials in hippocampal slices is enhanced by histamine (Fig. 18). Such shifts in pH occur during intense nervous discharges, e.g., during seizures or after tetanic stimulation, and in hypoxic conditions. The effect is not mediated by any of the known histamine receptors but can be mimicked by the histamine metabolite 1-methyl-histamine and is selective for the GluN2B subunit of the NMDA receptor (Williams, 1994), which plays a central role in synaptic plasticity. The H3 receptor antagonists clobenpropit and iodophenpropit have been identified as NMDA-antagonists selective for the GluN2B subunit (Hansen et al., 2010). Histamine also potentiates NMDA-induced [3H]noradrenaline release from hippocampal synaptosomes through an allosteric site distinct from the polyamine site (Faucard et al., 2006). An action of the diamine histamine on the polyamine site of the NMDA receptor may have been predicted from the cross-reaction histamine-spermidine, jeopardizing the early attempts to label histaminergic neurons through fluorescence histology that was available for the other biogenic amines (Green, 1964).
Fig. 18.
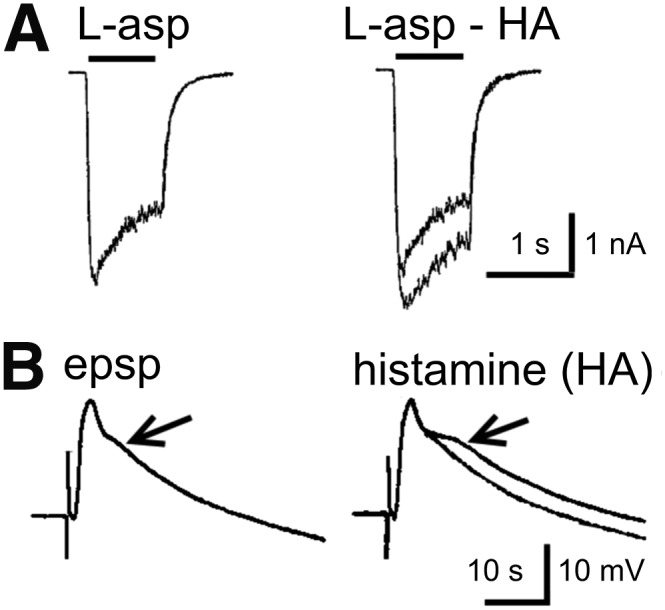
Histamine potentiates NMDA current. (A) Potentiation of NMDA-current in whole cell clamp with 1-second application of l-aspartate. (B) NMDA component (arrows) of excitatory postsynaptic potential (epsp) in hippocampal pyramidal cell. Modified from Vorobjev et al. (1993) and Yanovsky et al. (1995).
VII. Future Perspectives
Histamine pharmacology is as exciting now as it was back in the early 1970s when there was only one histamine receptor. Recent advances in our knowledge and understanding of histamine pharmacology, with the identification of the H3 and H4 receptors and their isoforms over the last decade and the solving of the H1 receptor structure in 2011, bode well for potential new improved “antihistamines” and future clinical exploitation of these newer histamine receptors. The spectrum of physiologic and pathophysiological roles of histamine has been expanded profoundly over the last decade, with growing evidence in feeding behaviors, sleep physiology, cognitive function, fear memory, pain physiology for the H3 receptor, and chronic inflammation, autoimmune function, gastrointestinal endocrine and salivary exocrine function, pain and itch biology, diabetic nephropathy, and cancer biology for the H4 receptor.
There is still much to understand. The full therapeutic potential of H3 receptor and H4 receptor ligands is just starting to emerge as clinical trial data are published. Furthermore, being the newest histamine receptor, there are many outstanding questions with regard to the characterization of the H4 receptor. In particular it is still not completely clear as to the full complement of cells that express the receptor and whether the receptor has functions beyond inflammation such as in the central or peripheral nervous systems. In addition there is much left to learn about all four receptors with regard to the physiologic and pharmacological relevance of functional selectivity, constitutive activity, splice variants, and oligomerization of the histamine receptor family. Because numerous receptor (hetero)oligomerizations and receptor crosstalks have been reported, one may speculate on the functional properties of numerous compounds with a combined or multitargeted profile with in vivo application and its adoptive processes.
Acknowledgments
The authors thank Drs. Chris van de Graaf and Albert Kooistra for the illustrations in Fig. 2 of the H1 receptor overall structure and the binding modes of histamine in the X-ray structure (H1 receptor) or homology models (H2 receptor–H4 receptor) of the four different receptor subtypes (Fig. 3).
Abbreviations
- bp
base pair
- CNS
central nervous system
- EAE
experimental autoimmune encephalitis
- ECL
extracellular loop
- ERK
extracellular signal-regulated kinase
- GPCR
G protein–coupled receptor
- HDC
histidine decarboxylase
- IFN
interferon
- IL
interleukin
- IPSP
inhibitory postsynaptic potential
- KO
knockout
- LPS
lipopolysaccharide
- MAPK
mitogen-activated protein kinase
- NAADP
nicotinic acid adenine dinucleotide phosphate
- NMDA
N-methyl-d-aspartate
- NO
nitric oxide
- PBMC
peripheral blood mononuclear cells
- PET
positron emission tomography
- PKA
protein kinase A
- PKC
protein kinase C
- PLC
phospholipase C
- SAR
structure-activity relationship
- TLR
Toll-like receptor
- TM
transmembrane
- TNFα
tumor necrosis factor α
Authorship Contributions
Wrote or contributed to the writing of the manuscript: Panula, Chazot, Cowart, Gutzmer, Leurs, Liu, Stark, Thurmond, Haas.
Footnotes
P.P. received financial support from The Academy of Finland, The Sigrid Juselius Foundation, The Finnish Foundation for Alcohol Studies and (previously) a lecture compensation from Abbot Laboratories. P.C. received financial support from Gedeon Richter and GSK, compensation from Abbott Laboratories for a lecture and consultancy support from Ziarco Pharma Ltd. M.C. is an employee of AbbVie. R.G. received support from DFG [German Research Foundation GU434/6-1] and project support by Johnson & Johnson. R.L. received financial support from the Netherlands Organisation for Scientific Research (NWO), Abbott Laboratories, Pfizer, and UCB Pharma, and is a founder of Griffin Discoveries. W.L.S.L. is a founder of Ziarco Pharma Limited. R.L.T. is an employee of Janssen Research & Development. H.S. is one of the inventors of pitolisant. NC-IUPHAR is supported in part by Wellcome Trust Grant 099156/Z/12/Z. Original work by P.P., P.C., R.G., R.L., and H.S. was supported by European Cooperation in Science and Technology Action BM0806.
This review is dedicated to the memory of Professor Sir James Black, FRS, Nobel laureate, who died on 22 March 2010, and Professor Walter Schunack, who died on 6 April 2011.
References
- Adami M, Pozzoli C, Menozzi A, Bertini S, Passeri B, Cantoni AM, Smits R, de Esch I, Leurs R, Coruzzi G. (2012) Effects of histamine H4 receptor ligands in a mouse model of gastric ulceration. Pharmacology 89:287–294. [DOI] [PubMed] [Google Scholar]
- Adjobo-Hermans MJ, Goedhart J, van Weeren L, Nijmeijer S, Manders EM, Offermanns S, Gadella TW., Jr (2011) Real-time visualization of heterotrimeric G protein Gq activation in living cells. BMC Biol 9:32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ahmad SF, Zoheir KM, Abdel-Hamied HE, Alrashidi I, Attia SM, Bakheet SA, Ashour AE, Abd-Allah AR. (2014) Role of a histamine 4 receptor as an anti-inflammatory target in carrageenan-induced pleurisy in mice. Immunology 142:374–383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aldi S, Takano K, Tomita K, Koda K, Chan NY-K, Marino A, Salazar-Rodriguez M, Thurmond RL, Levi R. (2014) Histamine H4-receptors inhibit mast cell renin release in ischemia/reperfusion via protein kinase C ε-dependent aldehyde dehydrogenase type-2 activation. J Pharmacol Exp Ther 349:508–517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alewijnse AE, Smit MJ, Hoffmann M, Verzijl D, Timmerman H, Leurs R. (1998) Constitutive activity and structural instability of the wild-type human H2 receptor. J Neurochem 71:799–807. [DOI] [PubMed] [Google Scholar]
- Alewijnse AE, Timmerman H, Jacobs EH, Smit MJ, Roovers E, Cotecchia S, Leurs R. (2000) The effect of mutations in the DRY motif on the constitutive activity and structural instability of the histamine H(2) receptor. Mol Pharmacol 57:890–898. [PubMed] [Google Scholar]
- Alexander SP, Benson HE, Faccenda E, Pawson AJ, Sharman JL, Spedding M, Peters JA, Harmar AJ, CGTP Collaborators (2013) The Concise Guide to PHARMACOLOGY 2013/14: G protein-coupled receptors. Br J Pharmacol 170:1459–1581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alonso N, Monczor F, Echeverría E, Davio C, Shayo C, Fernández N. (2014) Signal transduction mechanism of biased ligands at histamine H2 receptors. Biochem J 459:117–126. [DOI] [PubMed] [Google Scholar]
- Altenbach RJ, Adair RM, Bettencourt BM, Black LA, Fix-Stenzel SR, Gopalakrishnan SM, Hsieh GC, Liu H, Marsh KC, McPherson MJ, et al. (2008) Structure-activity studies on a series of a 2-aminopyrimidine-containing histamine H4 receptor ligands. J Med Chem 51:6571–6580. [DOI] [PubMed] [Google Scholar]
- Alves-Rodrigues A, Leurs R, Wu TS, Prell GD, Foged C, Timmerman H. (1996) [3H]-thioperamide as a radioligand for the histamine H3 receptor in rat cerebral cortex. Br J Pharmacol 118:2045–2052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amaral MM, Davio C, Ceballos A, Salamone G, Cañones C, Geffner J, Vermeulen M. (2007) Histamine improves antigen uptake and cross-presentation by dendritic cells. J Immunol 179:3425–3433. [DOI] [PubMed] [Google Scholar]
- Amon M, Ligneau X, Camelin JC, Berrebi-Bertrand I, Schwartz JC, Stark H. (2007) Highly potent fluorescence-tagged nonimidazole histamine H3 receptor ligands. ChemMedChem 2:708–716. [DOI] [PubMed] [Google Scholar]
- Amon M, Ligneau X, Schwartz JC, Stark H. (2006) Fluorescent non-imidazole histamine H3 receptor ligands with nanomolar affinities. Bioorg Med Chem Lett 16:1938–1940. [DOI] [PubMed] [Google Scholar]
- Andaloussi M, Lim HD, van der Meer T, Sijm M, Poulie CB, de Esch IJ, Leurs R, Smits RA. (2013) A novel series of histamine H4 receptor antagonists based on the pyrido[3,2-d]pyrimidine scaffold: comparison of hERG binding and target residence time with PF-3893787. Bioorg Med Chem Lett 23:2663–2670. [DOI] [PubMed] [Google Scholar]
- Anichtchik OV, Peitsaro N, Rinne JO, Kalimo H, Panula P. (2001) Distribution and modulation of histamine H(3) receptors in basal ganglia and frontal cortex of healthy controls and patients with Parkinson’s disease. Neurobiol Dis 8:707–716. [DOI] [PubMed] [Google Scholar]
- Anichtchik OV, Rinne JO, Kalimo H, Panula P. (2000) An altered histaminergic innervation of the substantia nigra in Parkinson’s disease. Exp Neurol 163:20–30. [DOI] [PubMed] [Google Scholar]
- Appl H, Holzammer T, Dove S, Haen E, Strasser A, Seifert R. (2012) Interactions of recombinant human histamine H₁R, H₂R, H₃R, and H₄R receptors with 34 antidepressants and antipsychotics. Naunyn Schmiedebergs Arch Pharmacol 385:145–170. [DOI] [PubMed] [Google Scholar]
- Arias-Montaño JA, Floran B, Garcia M, Aceves J, Young JM. (2001) Histamine H(3) receptor-mediated inhibition of depolarization-induced, dopamine D(1) receptor-dependent release of [(3)H]-gamma-aminobutryic acid from rat striatal slices. Br J Pharmacol 133:165–171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arienti KL, Breitenbucher JG, Buzard DJ, Edwards JP, Hack MD, Khatuya H, Kindrachuk DE, Lee A, and Venable JD (2006) inventors, Janssen Pharmaceutica N.V., assignee. Benzoimidazole compounds. Patent UA84576 (C2). 2004 Sept 29.
- Arrang JM. (2007) [The histamine H3 receptor: a new target for the treatment of arousal and cognitive disorders]. Ann Pharm Fr 65:275–284. [DOI] [PubMed] [Google Scholar]
- Arrang JM, Devaux B, Chodkiewicz JP, Schwartz JC. (1988a) H3-receptors control histamine release in human brain. J Neurochem 51:105–108. [DOI] [PubMed] [Google Scholar]
- Arrang JM, Garbarg M, Lancelot JC, Lecomte JM, Pollard H, Robba M, Schunack W, Schwartz JC. (1987a) Highly potent and selective ligands for histamine H3-receptors. Nature 327:117–123. [DOI] [PubMed] [Google Scholar]
- Arrang JM, Garbarg M, Lancelot JC, Lecomte JM, Pollard H, Robba M, Schunack W, Schwartz JC. (1988b) Highly potent and selective ligands for a new class H3 of histamine receptor. Invest Radiol 23 (Suppl 1):S130–S132. [DOI] [PubMed] [Google Scholar]
- Arrang JM, Garbarg M, Quach TT, Dam Trung Tuong M, Yeramian E, Schwartz JC. (1985a) Actions of betahistine at histamine receptors in the brain. Eur J Pharmacol 111:73–84. [DOI] [PubMed] [Google Scholar]
- Arrang JM, Garbarg M, Schwartz JC. (1983) Auto-inhibition of brain histamine release mediated by a novel class (H3) of histamine receptor. Nature 302:832–837. [DOI] [PubMed] [Google Scholar]
- Arrang JM, Garbarg M, Schwartz JC. (1985b) Autoregulation of histamine release in brain by presynaptic H3-receptors. Neuroscience 15:553–562. [DOI] [PubMed] [Google Scholar]
- Arrang JM, Garbarg M, Schwartz JC. (1987b) Autoinhibition of histamine synthesis mediated by presynaptic H3-receptors. Neuroscience 23:149–157. [DOI] [PubMed] [Google Scholar]
- Arrang JM, Morisset S, Gbahou F. (2007) Constitutive activity of the histamine H3 receptor. Trends Pharmacol Sci 28:350–357. [DOI] [PubMed] [Google Scholar]
- Arrang JM, Roy J, Morgat JL, Schunack W, Schwartz JC. (1990) Histamine H3 receptor binding sites in rat brain membranes: modulations by guanine nucleotides and divalent cations. Eur J Pharmacol 188:219–227. [DOI] [PubMed] [Google Scholar]
- Ash AS, Schild HO. (1966) Receptors mediating some actions of histamine. Br Pharmacol Chemother 27:427–439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ashworth S, Rabiner EA, Gunn RN, Plisson C, Wilson AA, Comley RA, Lai RYK, Gee AD, Laruelle M, Cunningham VJ. (2010) Evaluation of C-11-GSK189254 as a novel radioligand for the H-3 receptor in humans using PET. J Nucl Med 51:1021–1029. [DOI] [PubMed] [Google Scholar]
- Atzori M, Lau D, Tansey EP, Chow A, Ozaita A, Rudy B, McBain CJ. (2000) H2 histamine receptor-phosphorylation of Kv3.2 modulates interneuron fast spiking. Nat Neurosci 3:791–798. [DOI] [PubMed] [Google Scholar]
- Auvinen S, Panula P. (1988) Development of histamine-immunoreactive neurons in the rat brain. J Comp Neurol 276:289–303. [DOI] [PubMed] [Google Scholar]
- Bailey DG, Malcolm J, Arnold O, Spence JD. (1998) Grapefruit juice-drug interactions. Br J Clin Pharmacol 46:101–110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baker JG. (2008) A study of antagonist affinities for the human histamine H2 receptor. Br J Pharmacol 153:1011–1021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bakker RA. (2004) Histamine H3-receptor isoforms. Inflamm Res 53:509–516. [DOI] [PubMed] [Google Scholar]
- Bakker RA, Dees G, Carrillo JJ, Booth RG, López-Gimenez JF, Milligan G, Strange PG, Leurs R. (2004a) Domain swapping in the human histamine H1 receptor. J Pharmacol Exp Ther 311:131–138. [DOI] [PubMed] [Google Scholar]
- Bakker RA, Lozada AF, van Marle A, Shenton FC, Drutel G, Karlstedt K, Hoffmann M, Lintunen M, Yamamoto Y, van Rijn RM, et al. (2006) Discovery of naturally occurring splice variants of the rat histamine H3 receptor that act as dominant-negative isoforms. Mol Pharmacol 69:1194–1206. [DOI] [PubMed] [Google Scholar]
- Bakker RA, Nicholas MW, Smith TT, Burstein ES, Hacksell U, Timmerman H, Leurs R, Brann MR, Weiner DM. (2007) In vitro pharmacology of clinically used central nervous system-active drugs as inverse H(1) receptor agonists. J Pharmacol Exp Ther 322:172–179. [DOI] [PubMed] [Google Scholar]
- Bakker RA, Schoonus SB, Smit MJ, Timmerman H, Leurs R. (2001) Histamine H(1)-receptor activation of nuclear factor-kappa B: roles for G beta gamma- and G alpha(q/11)-subunits in constitutive and agonist-mediated signaling. Mol Pharmacol 60:1133–1142. [DOI] [PubMed] [Google Scholar]
- Bakker RA, Timmerman H, Leurs R. (2002) Histamine receptors: specific ligands, receptor biochemistry, and signal transduction. Clin Allergy Immunol 17:27–64. [PubMed] [Google Scholar]
- Bakker RA, Weiner DM, ter Laak T, Beuming T, Zuiderveld OP, Edelbroek M, Hacksell U, Timmerman H, Brann MR, Leurs R. (2004b) 8R-lisuride is a potent stereospecific histamine H1-receptor partial agonist. Mol Pharmacol 65:538–549. [DOI] [PubMed] [Google Scholar]
- Bakker RA, Wieland K, Timmerman H, Leurs R. (2000) Constitutive activity of the histamine H(1) receptor reveals inverse agonism of histamine H(1) receptor antagonists. Eur J Pharmacol 387:R5–R7. [DOI] [PubMed] [Google Scholar]
- Bárbara A, Aceves J, Arias-Montaño JA. (2002) Histamine H1 receptors in rat dorsal raphe nucleus: pharmacological characterisation and linking to increased neuronal activity. Brain Res 954:247–255. [DOI] [PubMed] [Google Scholar]
- Barbier AJ, Berridge C, Dugovic C, Laposky AD, Wilson SJ, Boggs J, Aluisio L, Lord B, Mazur C, Pudiak CM, et al. (2004) Acute wake-promoting actions of JNJ-5207852, a novel, diamine-based H3 antagonist. Br J Pharmacol 143:649–661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barbier AJ, Bradbury MJ. (2007) Histaminergic control of sleep-wake cycles: recent therapeutic advances for sleep and wake disorders. CNS Neurol Disord Drug Targets 6:31–43. [DOI] [PubMed] [Google Scholar]
- Barchuk WT, Salapatek AM, Ge T, D’Angelo P, Liu X. (2013) A proof-of-concept study of the effect of a novel H3-receptor antagonist in allergen-induced nasal congestion. J Allergy Clin Immunol 132:838–846, e1–e6. [DOI] [PubMed] [Google Scholar]
- Bardgett ME, Davis NN, Schultheis PJ, Griffith MS. (2011) Ciproxifan, an H3 receptor antagonist, alleviates hyperactivity and cognitive deficits in the APP Tg2576 mouse model of Alzheimer’s disease. Neurobiol Learn Mem 95:64–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barger G, Dale HH. (1910) Chemical structure and sympathomimetic action of amines. J Physiol 41:19–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barocelli E, Ballabeni V. (2003) Histamine in the control of gastric acid secretion: a topic review. Pharmacol Res 47:299–304. [DOI] [PubMed] [Google Scholar]
- Bartho L, Benko R. (2013) Should antihistamines be re-considered as antiasthmatic drugs as adjuvants to anti-leukotrienes? Eur J Pharmacol 701:181–184. [DOI] [PubMed] [Google Scholar]
- Batzri S, Harmon JW. (1986) Is [3H]-tiotidine a specific ligand for the H2-receptor? Pharmacology 32:241–247. [DOI] [PubMed] [Google Scholar]
- Baudry M, Martres MP, Schwartz JC. (1975) H1 and H2 receptors in the histamine-induced accumulation of cyclic AMP in guinea pig brain slices. Nature 253:362–364. [DOI] [PubMed] [Google Scholar]
- Bäumer W, Wendorff S, Gutzmer R, Werfel T, Dijkstra D, Chazot P, Stark H, Kietzmann M. (2008) Histamine H4 receptors modulate dendritic cell migration through skin—immunomodulatory role of histamine. Allergy 63:1387–1394. [DOI] [PubMed] [Google Scholar]
- Beaton G, Moree WJ. (2010) The expanding role of H1 antihistamines: a patent survey of selective and dual activity compounds 2005-2010. Expert Opin Ther Pat 20:1197–1218. [DOI] [PubMed] [Google Scholar]
- Beermann S, Glage S, Jonigk D, Seifert R, Neumann D. (2012a) Opposite effects of mepyramine on JNJ 7777120-induced amelioration of experimentally induced asthma in mice in sensitization and provocation. PLoS ONE 7:e30285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beermann S, Seifert R, Neumann D. (2012b) Commercially available antibodies against human and murine histamine H₄-receptor lack specificity. Naunyn Schmiedebergs Arch Pharmacol 385:125–135. [DOI] [PubMed] [Google Scholar]
- Bekkers JM. (1993) Enhancement by histamine of NMDA-mediated synaptic transmission in the hippocampus. Science 261:104–106. [DOI] [PubMed] [Google Scholar]
- Bell AS, Lane CAL, Mowbray CE, Selby MD, Swain NA, Williams DH. (2007) Preparation of Substituted 2,4-Diaminopyrimidines as Histamine H4 Receptor Ligands, Pfizer Limited, UK. [Google Scholar]
- Bell JK, McQueen DS, Rees JL. (2004) Involvement of histamine H4 and H1 receptors in scratching induced by histamine receptor agonists in Balb C mice. Br J Pharmacol 142:374–380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berlin M, Boyce CW. (2007) Recent advances in the development of histamine H3 antagonists. Expert Opin Ther Pat 17:675–687. [DOI] [PubMed] [Google Scholar]
- Berlin M, Boyce CW, Ruiz MdeL. (2011) Histamine H3 receptor as a drug discovery target. J Med Chem 54:26–53. [DOI] [PubMed] [Google Scholar]
- Bielory L, Lien KW, Bigelsen S. (2005) Efficacy and tolerability of newer antihistamines in the treatment of allergic conjunctivitis. Drugs 65:215–228. [DOI] [PubMed] [Google Scholar]
- Birnbaumer L. (2007) The discovery of signal transduction by G proteins: a personal account and an overview of the initial findings and contributions that led to our present understanding. Biochim Biophys Acta 1768:756–771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Birnkammer T, Spickenreither A, Brunskole I, Lopuch M, Kagermeier N, Bernhardt G, Dove S, Seifert R, Elz S, Buschauer A. (2012) The bivalent ligand approach leads to highly potent and selective acylguanidine-type histamine H₂ receptor agonists. J Med Chem 55:1147–1160. [DOI] [PubMed] [Google Scholar]
- Bitner RS, Markosyan S, Nikkel AL, Brioni JD. (2011) In-vivo histamine H3 receptor antagonism activates cellular signaling suggestive of symptomatic and disease modifying efficacy in Alzheimer’s disease. Neuropharmacology 60:460–466. [DOI] [PubMed] [Google Scholar]
- Black JW, Duncan WA, Durant CJ, Ganellin CR, Parsons EM. (1972) Definition and antagonism of histamine H 2 -receptors. Nature 236:385–390. [DOI] [PubMed] [Google Scholar]
- Blandina P, Giorgetti M, Bartolini L, Cecchi M, Timmerman H, Leurs R, Pepeu G, Giovannini MG. (1996) Inhibition of cortical acetylcholine release and cognitive performance by histamine H3 receptor activation in rats. Br J Pharmacol 119:1656–1664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blandina P, Munari L, Provensi G, Passani MB. (2012) Histamine neurons in the tuberomamillary nucleus: a whole center or distinct subpopulations? Front Syst Neurosci 6:33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonaventure P, Letavic M, Dugovic C, Wilson S, Aluisio L, Pudiak C, Lord B, Mazur C, Kamme F, Nishino S, et al. (2007) Histamine H3 receptor antagonists: from target identification to drug leads. Biochem Pharmacol 73:1084–1096. [DOI] [PubMed] [Google Scholar]
- Bongers G, Bakker RA, Leurs R. (2007a) Molecular aspects of the histamine H3 receptor. Biochem Pharmacol 73:1195–1204. [DOI] [PubMed] [Google Scholar]
- Bongers G, Krueger KM, Miller TR, Baranowski JL, Estvander BR, Witte DG, Strakhova MI, van Meer P, Bakker RA, Cowart MD, et al. (2007b) An 80-amino acid deletion in the third intracellular loop of a naturally occurring human histamine H3 isoform confers pharmacological differences and constitutive activity. J Pharmacol Exp Ther 323:888–898. [DOI] [PubMed] [Google Scholar]
- Bongers G, Sallmen T, Passani MB, Mariottini C, Wendelin D, Lozada A, Marle Av, Navis M, Blandina P, Bakker RA, et al. (2007c) The Akt/GSK-3beta axis as a new signaling pathway of the histamine H(3) receptor. J Neurochem 103:248–258. [DOI] [PubMed] [Google Scholar]
- Bouthenet ML, Ruat M, Sales N, Garbarg M, Schwartz JC. (1988) A detailed mapping of histamine H1-receptors in guinea-pig central nervous system established by autoradiography with [125I]iodobolpyramine. Neuroscience 26:553–600. [DOI] [PubMed] [Google Scholar]
- Bovet D. (1950) Introduction to antihistamine agents and antergan derivative. Ann N Y Acad Sci 50:1089–1126. [DOI] [PubMed] [Google Scholar]
- Bovet D, Staub AM. (1937) Action protection des ethers phenoliques au cours de l’intoxication histaminique. CR Seances Acad Sci 124:547–549. [Google Scholar]
- Breunig E, Michel K, Zeller F, Seidl S, Weyhern CW, Schemann M. (2007) Histamine excites neurones in the human submucous plexus through activation of H1, H2, H3 and H4 receptors. J Physiol 583:731–742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Broccatelli F, Carosati E, Cruciani G, Oprea TI. (2010) Transporter-mediated efflux influences CNS side effects: ABCB1, from antitarget to target. Mol Inform 29:16–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown RE, Fedorov NB, Haas HL, Reymann KG. (1995) Histaminergic modulation of synaptic plasticity in area CA1 of rat hippocampal slices. Neuropharmacology 34:181–190. [DOI] [PubMed] [Google Scholar]
- Brown RE, Haas HL. (1999) On the mechanism of histaminergic inhibition of glutamate release in the rat dentate gyrus. J Physiol 515:777–786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown RE, Reymann KG. (1996) Histamine H3 receptor-mediated depression of synaptic transmission in the dentate gyrus of the rat in vitro. J Physiol 496:175–184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown RE, Sergeeva OA, Eriksson KS, Haas HL. (2002) Convergent excitation of dorsal raphe serotonin neurons by multiple arousal systems (orexin/hypocretin, histamine and noradrenaline). J Neurosci 22:8850–8859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown RH, Zerhouni EA, Hirshman CA. (1994) Reversal of bronchoconstriction by inhaled nitric oxide. Histamine versus methacholine. Am J Respir Crit Care Med 150:233–237. [DOI] [PubMed] [Google Scholar]
- Bruysters M, Pertz HH, Teunissen A, Bakker RA, Gillard M, Chatelain P, Schunack W, Timmerman H, Leurs R. (2004) Mutational analysis of the histamine H1-receptor binding pocket of histaprodifens. Eur J Pharmacol 487:55–63. [DOI] [PubMed] [Google Scholar]
- Buckland KF, Williams TJ, Conroy DM. (2003) Histamine induces cytoskeletal changes in human eosinophils via the H4 receptor. Br J Pharmacol 140:1117–1127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burde R, Dippel E, Seifert R. (1996) Receptor-independent G protein activation may account for the stimulatory effects of first-generation H1-receptor antagonists in HL-60 cells, basophils, and mast cells. Biochem Pharmacol 51:125–131. [DOI] [PubMed] [Google Scholar]
- Buschauer A. (1989) Synthesis and in vitro pharmacology of arpromidine and related phenyl(pyridylalkyl)guanidines, a potential new class of positive inotropic drugs. J Med Chem 32:1963–1970. [DOI] [PubMed] [Google Scholar]
- Cai H, Chavez F, Edwards JP, Fitzgerald AE, Liu J, Mani NS, Neff DK, Rizzolio MC, Savall BM, Smith DM, et al. (2008) inventors, Janssen Pharmaceutica N.V., assignee. Preparation of 2-aminopyrimidine modulators of the histamine H4 receptor, Belg.Patent BRPI0807815 (A2). 2008 Feb 14.
- Cannon KE, Chazot PL, Hann V, Shenton F, Hough LB, Rice FL. (2007) Immunohistochemical localization of histamine H3 receptors in rodent skin, dorsal root ganglia, superior cervical ganglia, and spinal cord: potential antinociceptive targets. Pain 129:76–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carceller Gonzalez E, Salas Solana J, Soliva Soliva R, Medina Fuentes EM, and Marti Via J (2007) inventors, Palau Pharma, assignee. Preparation of diaminopyrimidines as modulators of histamine H4 receptor activity, Patent UA 101963 (C2). 2007 Dec 21.
- Carman-Krzan M, Lipnik-Stangelj M. (2000) Molecular properties of central and peripheral histamine H1 and H2 receptors. Pflugers Arch 439:R131–132. [PubMed] [Google Scholar]
- Casale TB, Wood D, Richerson HB, Trapp S, Metzger WJ, Zavala D, Hunninghake GW. (1987) Elevated bronchoalveolar lavage fluid histamine levels in allergic asthmatics are associated with methacholine bronchial hyperresponsiveness. J Clin Invest 79:1197–1203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Celanire S, Wijtmans M, Talaga P, Leurs R, de Esch IJ. (2005) Keynote review: histamine H3 receptor antagonists reach out for the clinic. Drug Discov Today 10:1613–1627. [DOI] [PubMed] [Google Scholar]
- Chang RS, Tran VT, Snyder SH. (1979) Heterogeneity of histamine H1-receptors: species variations in [3H]mepyramine binding of brain membranes. J Neurochem 32:1653–1663. [DOI] [PubMed] [Google Scholar]
- Chavez F, Curtis MP, Edwards JP, Gomez L, Grice CA, Kearney AM, Savall BM, Fitzgerald AE, Liu J, and Mani NS (2008), inventors, Janssen Pharmaceutica, N.V., assignee. Preparation of benzofuro- and benzothienopyrimidine modulators of the histamine H4 receptor, Belg. Patent UA101963 WO2008008359 (C2). 2007 Jul 10.
- Chazot PL, Hann V, Wilson C, Lees G, Thompson CL. (2001) Immunological identification of the mammalian H3 histamine receptor in the mouse brain. Neuroreport 12:259–262. [DOI] [PubMed] [Google Scholar]
- Chen C. (2008) Physicochemical, pharmacological and pharmacokinetic properties of the zwitterionic antihistamines cetirizine and levocetirizine. Curr Med Chem 15:2173–2191. [DOI] [PubMed] [Google Scholar]
- Chen D, Zhao CM, Al-Haider W, Håkanson R, Rehfeld JF, Kopin AS. (2002) Differentiation of gastric ECL cells is altered in CCK2 receptor-deficient mice. Gastroenterology 123:577–585. [DOI] [PubMed] [Google Scholar]
- Cho W, Maruff P, Connell J, Gargano C, Calder N, Doran S, Fox-Bosetti S, Hassan A, Renger J, Herman G, et al. (2011) Additive effects of a cholinesterase inhibitor and a histamine inverse agonist on scopolamine deficits in humans. Psychopharmacology (Berl) 218:513–524. [DOI] [PubMed] [Google Scholar]
- Church MK, Maurer M, Simons FE, Bindslev-Jensen C, van Cauwenberge P, Bousquet J, Holgate ST, Zuberbier T, Global Allergy and Asthma European Network (2010) Risk of first-generation H1-antihistamines: a GA2LEN position paper. Allergy 65:459–466. [DOI] [PubMed] [Google Scholar]
- Cianchi F, Cortesini C, Schiavone N, Perna F, Magnelli L, Fanti E, Bani D, Messerini L, Fabbroni V, Perigli G, et al. (2005) The role of cyclooxygenase-2 in mediating the effects of histamine on cell proliferation and vascular endothelial growth factor production in colorectal cancer. Clin Cancer Res 11:6807–6815. [DOI] [PubMed] [Google Scholar]
- Clapham J, Kilpatrick GJ. (1992) Histamine H3 receptors modulate the release of [3H]-acetylcholine from slices of rat entorhinal cortex: evidence for the possible existence of H3 receptor subtypes. Br J Pharmacol 107:919–923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clapham J, Kilpatrick GJ. (1993) Histamine H3 receptor-mediated modulation of water consumption in the rat. Eur J Pharmacol 232:99–103. [DOI] [PubMed] [Google Scholar]
- Clark EA, Hill SJ. (1996) Sensitivity of histamine H3 receptor agonist-stimulated [35S]GTP gamma[S] binding to pertussis toxin. Eur J Pharmacol 296:223–225. [DOI] [PubMed] [Google Scholar]
- Clark MA, Korte A, Myers J, Egan RW. (1992) High affinity histamine H3 receptors regulate ACTH release by AtT-20 cells. Eur J Pharmacol 210:31–35. [DOI] [PubMed] [Google Scholar]
- Clark RAF, Gallin JI, Kaplan AP. (1975) The selective eosinophil chemotactic activity of histamine. J Exp Med 142:1462–1476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clark RAF, Sandler JA, Gallin JI, Kaplan AP. (1977) Histamine modulation of eosinophil migration. J Immunol 118:137–145. [PubMed] [Google Scholar]
- Cogé F, Guénin SP, Audinot V, Renouard-Try A, Beauverger P, Macia C, Ouvry C, Nagel N, Rique H, Boutin JA, et al. (2001a) Genomic organization and characterization of splice variants of the human histamine H3 receptor. Biochem J 355:279–288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cogé F, Guénin SP, Rique H, Boutin JA, Galizzi JP. (2001b) Structure and expression of the human histamine H4-receptor gene. Biochem Biophys Res Commun 284:301–309. [DOI] [PubMed] [Google Scholar]
- Connelly WM, Shenton FC, Lethbridge N, Leurs R, Waldvogel HJ, Faull RL, Lees G, Chazot PL. (2009) The histamine H4 receptor is functionally expressed on neurons in the mammalian CNS. Br J Pharmacol 157:55–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coruzzi G, Adami M, Guaita E, de Esch IJP, Leurs R. (2007) Antiinflammatory and antinociceptive effects of the selective histamine H4-receptor antagonists JNJ7777120 and VUF6002 in a rat model of carrageenan-induced acute inflammation. Eur J Pharmacol 563:240–244. [DOI] [PubMed] [Google Scholar]
- Coruzzi G, Adami M, Pozzoli C. (2012) Role of histamine H4 receptors in the gastrointestinal tract. Front Biosci (Schol Ed) 4:226–239 Schol Ed. [DOI] [PubMed] [Google Scholar]
- Coward P, Chan SDH, Wada HG, Humphries GM, Conklin BR. (1999) Chimeric G proteins allow a high-throughput signaling assay of Gi-coupled receptors. Anal Biochem 270:242–248. [DOI] [PubMed] [Google Scholar]
- Cowart M, Faghih R, Curtis MP, Gfesser GA, Bennani YL, Black LA, Pan L, Marsh KC, Sullivan JP, Esbenshade TA, et al. (2005) 4-(2-[2-(2(R)-methylpyrrolidin-1-yl)ethyl]benzofuran-5-yl)benzonitrile and related 2-aminoethylbenzofuran H3 receptor antagonists potently enhance cognition and attention. J Med Chem 48:38–55. [DOI] [PubMed] [Google Scholar]
- Cowart M, Gfesser GA, Browman KE, Faghih R, Miller TR, Milicic I, Baranowski JL, Krueger KM, Witte DG, Molesky AL, et al. (2007) Novel heterocyclic-substituted benzofuran histamine H3 receptor antagonists: in vitro properties, drug-likeness, and behavioral activity. Biochem Pharmacol 73:1243–1255. [DOI] [PubMed] [Google Scholar]
- Cowart MD, Altenbach RJ, Liu H, Hsieh GC, Drizin I, Milicic I, Miller TR, Witte DG, Wishart N, Fix-Stenzel SR, et al. (2008) Rotationally constrained 2,4-diamino-5,6-disubstituted pyrimidines: a new class of histamine H4 receptor antagonists with improved druglikeness and in vivo efficacy in pain and inflammation models. J Med Chem 51:6547–6557. [DOI] [PubMed] [Google Scholar]
- Cowden JM, Riley JP, Ma JY, Thurmond RL, Dunford PJ. (2010a) Histamine H4 receptor antagonism diminishes existing airway inflammation and dysfunction via modulation of Th2 cytokines. Respir Res 11:86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cowden JM, Yu F, Banie H, Farahani M, Ling P, Nguyen S, Riley JP, Zhang M, Zhu J, Dunford PJ, et al. (2014) The histamine H4 receptor mediates inflammation and Th17 responses in preclinical models of arthritis. Ann Rheum Dis 73:600–608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cowden JM, Yu F, Challapalli M, Huang JF, Kim S, Fung-Leung WP, Ma JY, Riley JP, Zhang M, Dunford PJ, et al. (2013) Antagonism of the histamine H4 receptor reduces LPS-induced TNF production in vivo. Inflamm Res 62:599–607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cowden JM, Zhang M, Dunford PJ, Thurmond RL. (2010b) The histamine H4 receptor mediates inflammation and pruritus in Th2-dependent dermal inflammation. J Invest Dermatol 130:1023–1033. [DOI] [PubMed] [Google Scholar]
- Czerner CP, Klos A, Seifert R, Neumann D. (2014) Histamine induces chemotaxis and phagocytosis in murine bone marrow-derived macrophages and RAW 264.7 macrophage-like cells via histamine H4-receptor. Inflamm Res 63:239–247. [DOI] [PubMed] [Google Scholar]
- Dai H, Kaneko K, Kato H, Fujii S, Jing Y, Xu A, Sakurai E, Kato M, Okamura N, Kuramasu A, et al. (2007) Selective cognitive dysfunction in mice lacking histamine H1 and H2 receptors. Neurosci Res 57:306–313. [DOI] [PubMed] [Google Scholar]
- Dai H, Zhang Z, Zhu Y, Shen Y, Hu W, Huang Y, Luo J, Timmerman H, Leurs R, Chen Z. (2006) Histamine protects against NMDA-induced necrosis in cultured cortical neurons through H receptor/cyclic AMP/protein kinase A and H receptor/GABA release pathways. J Neurochem 96:1390–1400. [DOI] [PubMed] [Google Scholar]
- Dale HH, Laidlaw PP. (1910) The physiological action of beta-iminazolylethylamine. J Physiol 41:318–344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Damaj BB, Becerra CB, Esber HJ, Wen Y, Maghazachi AA. (2007) Functional expression of H4 histamine receptor in human natural killer cells, monocytes, and dendritic cells. J Immunol 179:7907–7915. [DOI] [PubMed] [Google Scholar]
- Dauvilliers Y, Bassetti C, Lammers GJ, Arnulf I, Mayer G, Rodenbeck A, Lehert P, Ding CL, Lecomte JM, Schwartz JC, HARMONY I study group (2013) Pitolisant versus placebo or modafinil in patients with narcolepsy: a double-blind, randomised trial. Lancet Neurol 12:1068–1075. [DOI] [PubMed] [Google Scholar]
- Day M, Pan JB, Buckley MJ, Cronin E, Hollingsworth PR, Hirst WD, Navarra R, Sullivan JP, Decker MW, Fox GB. (2007) Differential effects of ciproxifan and nicotine on impulsivity and attention measures in the 5-choice serial reaction time test. BiochemPharmacol 73:1123–1134. [DOI] [PubMed] [Google Scholar]
- De Backer MD, Gommeren W, Moereels H, Nobels G, Van Gompel P, Leysen JE, Luyten WH. (1993) Genomic cloning, heterologous expression and pharmacological characterization of a human histamine H1 receptor. Biochem Biophys Res Commun 197:1601–1608. [DOI] [PubMed] [Google Scholar]
- Deiteren A, De Man JG, Ruyssers NE, Moreels TG, Pelckmans PA, De Winter BY. (2014) Histamine H4 and H1 receptors contribute to postinflammatory visceral hypersensitivity. Gut 63:1873–1882. [DOI] [PubMed] [Google Scholar]
- Delaunois A, Gustin P, Garbarg M, Ansay M. (1995) Modulation of acetylcholine, capsaicin and substance P effects by histamine H3 receptors in isolated perfused rabbit lungs. Eur J Pharmacol 277:243–250. [DOI] [PubMed] [Google Scholar]
- del Rio R, Noubade R, Saligrama N, Wall EH, Krementsov DN, Poynter ME, Zachary JF, Thurmond RL, Teuscher C. (2012) Histamine H4 receptor optimizes T regulatory cell frequency and facilitates anti-inflammatory responses within the central nervous system. J Immunol 188:541–547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deml KF, Beermann S, Neumann D, Strasser A, Seifert R. (2009) Interactions of histamine H1-receptor agonists and antagonists with the human histamine H4-receptor. Mol Pharmacol 76:1019–1030. [DOI] [PubMed] [Google Scholar]
- Dere E, De Souza-Silva MA, Topic B, Spieler RE, Haas HL, Huston JP. (2003) Histidine-decarboxylase knockout mice show deficient nonreinforced episodic object memory, improved negatively reinforced water-maze performance, and increased neo- and ventro-striatal dopamine turnover. Learn Mem 10:510–519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Desai P, Thurmond RL. (2011) Histamine H₄ receptor activation enhances LPS-induced IL-6 production in mast cells via ERK and PI3K activation. Eur J Immunol 41:1764–1773. [DOI] [PubMed] [Google Scholar]
- Desmadryl G, Gaboyard-Niay S, Brugeaud A, Travo C, Broussy A, Saleur A, Dyhrfjeld-Johnsen J, Wersinger E, Chabbert C. (2012) Histamine H4 receptor antagonists as potent modulators of mammalian vestibular primary neuron excitability. Br J Pharmacol 167:905–916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deutsch SI, Rosse RB, Kendrick KA, Fay-McCarthy M, Collins JP, Jr, Wyatt RJ. (1993) Famotidine adjunctive pharmacotherapy for schizophrenia: preliminary data. Clin Neuropharmacol 16:518–524. [PubMed] [Google Scholar]
- Devillier P, Roche N, Faisy C. (2008) Clinical pharmacokinetics and pharmacodynamics of desloratadine, fexofenadine and levocetirizine : a comparative review. Clin Pharmacokinet 47:217–230. [DOI] [PubMed] [Google Scholar]
- Diaz J, Vizuete ML, Traiffort E, Arrang JM, Ruat M, Schwartz JC. (1994) Localization of the histamine H2 receptor and gene transcripts in rat stomach: back to parietal cells. Biochem Biophys Res Commun 198:1195–1202. [DOI] [PubMed] [Google Scholar]
- Dib K, Perecko T, Jenei V, McFarlane C, Comer D, Brown V, Katebe M, Scheithauer T, Thurmond RL, Chazot PL, et al. (2014) The histamine H4 receptor is a potent inhibitor of adhesion-dependent degranulation in human neutrophils. J Leukoc Biol 96:411–418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dijkstra D, Leurs R, Chazot P, Shenton FC, Stark H, Werfel T, Gutzmer R. (2007) Histamine downregulates monocyte CCL2 production through the histamine H4 receptor. J Allergy Clin Immunol 120:300–307. [DOI] [PubMed] [Google Scholar]
- Dijkstra D, Stark H, Chazot PL, Shenton FC, Leurs R, Werfel T, Gutzmer R. (2008) Human inflammatory dendritic epidermal cells express a functional histamine H4 receptor. J Invest Dermatol 128:1696–1703. [DOI] [PubMed] [Google Scholar]
- Diks SH, Hardwick JC, Diab RM, van Santen MM, Versteeg HH, van Deventer SJ, Richel DJ, Peppelenbosch MP. (2003) Activation of the canonical beta-catenin pathway by histamine. J Biol Chem 278:52491–52496. [DOI] [PubMed] [Google Scholar]
- Dong H, Zhang W, Zeng X, Hu G, Zhang H, He S, Zhang S. (2014) Histamine induces upregulated expression of histamine receptors and increases release of inflammatory mediators from microglia. Mol Neurobiol 49:1487–1500. [DOI] [PubMed] [Google Scholar]
- Doreulee N, Sergeeva OA, Yanovsky Y, Chepkova AN, Selbach O, Gödecke A, Schrader J, Haas HL. (2003) Cortico-striatal synaptic plasticity in endothelial nitric oxide synthase deficient mice. Brain Res 964:159–163. [DOI] [PubMed] [Google Scholar]
- Doreulee N, Yanovsky Y, Flagmeyer I, Stevens DR, Haas HL, Brown RE. (2001) Histamine H3 receptors depress synaptic transmission in the corticostriatal pathway. Neuropharmacology 40:106–113. [DOI] [PubMed] [Google Scholar]
- Dove S, Elz S, Seifert R, Buschauer A. (2004) Structure-activity relationships of histamine H2 receptor ligands. Mini Rev Med Chem 4:941–954. [DOI] [PubMed] [Google Scholar]
- Drutel G, Peitsaro N, Karlstedt K, Wieland K, Smit MJ, Timmerman H, Panula P, Leurs R. (2001) Identification of rat H3 receptor isoforms with different brain expression and signaling properties. Mol Pharmacol 59:1–8. [PubMed] [Google Scholar]
- Dunford PJ, O’Donnell N, Riley JP, Williams KN, Karlsson L, Thurmond RL. (2006) The histamine H4 receptor mediates allergic airway inflammation by regulating the activation of CD4+ T cells. J Immunol 176:7062–7070. [DOI] [PubMed] [Google Scholar]
- Dunford PJ, Williams KN, Desai PJ, Karlsson L, McQueen D, Thurmond RL. (2007) Histamine H4 receptor antagonists are superior to traditional antihistamines in the attenuation of experimental pruritus. J Allergy Clin Immunol 119:176–183. [DOI] [PubMed] [Google Scholar]
- Durant GJ, Ganellin CR, Parsons ME. (1975) Chemical differentiation of histamine H1- and H2-receptor agonists. J Med Chem 18:905–909. [DOI] [PubMed] [Google Scholar]
- Ea Kim L, Javellaud J, Oudart N. (1992) Endothelium-dependent relaxation of rabbit middle cerebral artery to a histamine H3-agonist is reduced by inhibitors of nitric oxide and prostacyclin synthesis. Br J Pharmacol 105:103–106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edwards JP, Kindrachuk DE, and Venable JD (2007a) inventors, Janssen Pharmaceutica, N.V., assignee. Preparation of benzoimidazol-2-yl pyridines as modulators of the histamine H4 receptor. Patent US20060788190P 20060331.
- Edwards JP, Savall BM, and Shah CR (2007b) inventors, Janssen Pharmaceutica, N.V., assignee. Preparation of indoles and benzimidazoles as histamine H4 receptor modulators. Patent US20060790421P 20060407.
- Edwards JP and Venable JD (2005) inventors, Janssen Pharmaceutica, N.V., assignee. Quinoxaline compounds. Patent US20030507176P. 2005 Mar 31.
- Egan MF, Zhao X, Gottwald R, Harper-Mozley L, Zhang Y, Snavely D, Lines C, Michelson D. (2013) Randomized crossover study of the histamine H3 inverse agonist MK-0249 for the treatment of cognitive impairment in patients with schizophrenia. Schizophr Res 146:224–230. [DOI] [PubMed] [Google Scholar]
- Endou M, Poli E, Levi R. (1994) Histamine H3-receptor signaling in the heart: possible involvement of Gi/Go proteins and N-type Ca2+ channels. J Pharmacol Exp Ther 269:221–229. [PubMed] [Google Scholar]
- Eriks JC, van der Goot H, Sterk GJ, Timmerman H. (1992) Histamine H2-receptor agonists. Synthesis, in vitro pharmacology, and qualitative structure-activity relationships of substituted 4- and 5-(2-aminoethyl)thiazoles. J Med Chem 35:3239–3246. [DOI] [PubMed] [Google Scholar]
- Eriksson KS, Sergeeva O, Brown RE, Haas HL. (2001) Orexin/hypocretin excites the histaminergic neurons of the tuberomammillary nucleus. J Neurosci 21:9273–9279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Esbenshade TA, Browman KE, Bitner RS, Strakhova M, Cowart MD, Brioni JD. (2008) The histamine H3 receptor: an attractive target for the treatment of cognitive disorders. Br J Pharmacol 154:1166–1181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Esbenshade TA, Krueger KM, Miller TR, Kang CH, Denny LI, Witte DG, Yao BB, Fox GB, Faghih R, Bennani YL, et al. (2003) Two novel and selective nonimidazole histamine H3 receptor antagonists A-304121 and A-317920: I. In vitro pharmacological effects. J Pharmacol Exp Ther 305:887–896. [DOI] [PubMed] [Google Scholar]
- Esposito B, Gambara G, Lewis AM, Palombi F, D’Alessio A, Taylor LX, Genazzani AA, Ziparo E, Galione A, Churchill GC, et al. (2011) NAADP links histamine H1 receptors to secretion of von Willebrand factor in human endothelial cells. Blood 117:4968–4977. [DOI] [PubMed] [Google Scholar]
- Estelle F, Simons R. (1999) H1-receptor antagonists: safety issues. Ann Allergy Asthma Immunol 83:481–488. [DOI] [PubMed] [Google Scholar]
- Fantin S, Maspero J, Bisbal C, Agache I, Donado E, Borja J, Mola O, Izquierdo I, international Rupatadine study group (2008) A 12-week placebo-controlled study of rupatadine 10 mg once daily compared with cetirizine 10 mg once daily, in the treatment of persistent allergic rhinitis. Allergy 63:924–931. [DOI] [PubMed] [Google Scholar]
- Faucard R, Armand V, Héron A, Cochois V, Schwartz JC, Arrang JM. (2006) N-methyl-D-aspartate receptor antagonists enhance histamine neuron activity in rodent brain. J Neurochem 98:1487–1496. [DOI] [PubMed] [Google Scholar]
- Feliszek M, Speckmann V, Schacht D, von Lehe M, Stark H, Schlicker E. (2015) A search for functional histamine H4 receptors in the human, guinea pig and mouse brain. Naunyn Schmiedebergs Arch Pharmacol 388:11–17. [DOI] [PubMed] [Google Scholar]
- Fernandez N, Gottardo FL, Alonso MN, Monczor F, Shayo C, Davio C. (2011) Roles of phosphorylation-dependent and -independent mechanisms in the regulation of histamine H2 receptor by G protein-coupled receptor kinase 2. J Biol Chem 286:28697–28706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fernandez N, Monczor F, Baldi A, Davio C, Shayo C. (2008) Histamine H2 receptor trafficking: role of arrestin, dynamin, and clathrin in histamine H2 receptor internalization. Mol Pharmacol 74:1109–1118. [DOI] [PubMed] [Google Scholar]
- Fernandez TV, Sanders SJ, Yurkiewicz IR, Ercan-Sencicek AG, Kim YS, Fishman DO, Raubeson MJ, Song Y, Yasuno K, Ho WS, et al. (2012) Rare copy number variants in tourette syndrome disrupt genes in histaminergic pathways and overlap with autism. Biol Psychiatry 71:392–402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferrada C, Ferré S, Casadó V, Cortés A, Justinova Z, Barnes C, Canela EI, Goldberg SR, Leurs R, Lluis C, et al. (2008) Interactions between histamine H3 and dopamine D2 receptors and the implications for striatal function. Neuropharmacology 55:190–197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferrada C, Moreno E, Casadó V, Bongers G, Cortés A, Mallol J, Canela EI, Leurs R, Ferré S, Lluís C, et al. (2009) Marked changes in signal transduction upon heteromerization of dopamine D1 and histamine H3 receptors. Br J Pharmacol 157:64–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferré S, Casadó V, Devi LA, Filizola M, Jockers R, Lohse MJ, Milligan G, Pin JP, Guitart X. (2014) G protein-coupled receptor oligomerization revisited: functional and pharmacological perspectives. Pharmacol Rev 66:413–434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferreira R, Santos T, Gonçalves J, Baltazar G, Ferreira L, Agasse F, Bernardino L. (2012) Histamine modulates microglia function. J Neuroinflammation 9:90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Finlayson K, Witchel HJ, McCulloch J, Sharkey J. (2004) Acquired QT interval prolongation and HERG: implications for drug discovery and development. Eur J Pharmacol 500:129–142. [DOI] [PubMed] [Google Scholar]
- Fleck MW, Thomson JL, Hough LB. (2012) Histamine-gated ion channels in mammals? Biochem Pharmacol 83:1127–1135. [DOI] [PubMed] [Google Scholar]
- Flores-Clemente C, Osorio-Espinoza A, Escamilla-Sánchez J, Leurs R, Arias JM, Arias-Montaño JA. (2013) A single-point mutation (Ala280Val) in the third intracellular loop alters the signalling properties of the human histamine H₃ receptor stably expressed in CHO-K1 cells. Br J Pharmacol 170:127–135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fogel WA, Wagner W, Sasiak K, Stasiak A. (2005) The role of histamine in experimental ulcerative colitis in rats. Inflamm Res 54 (Suppl 1):S68–S69. [DOI] [PubMed] [Google Scholar]
- Folkow B, Frost J, Uvnas B. (1948) Action of adrenaline, nor-adrenaline and some other sympathomimetic drugs on the muscular, cutaneous and splanchnic vessels of the cat. Acta Physiol Scand 15:412–420. [DOI] [PubMed] [Google Scholar]
- Foreman JC, Rising TJ, Webber SE. (1985) A study of the histamine H2-receptor mediating relaxation of the parenchymal lung strip preparation of the guinea-pig. Br J Pharmacol 86:465–473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fox GB, Esbenshade TA, Pan JB, Browman KE, Zhang M, Ballard ME, Radek RJ, Miner H, Bitner RS, Krueger KM, et al. (2005) Selective H3 receptor (H3R) blockade: broad efficacy in cognition and schizophrenia. Inflamm Res 54 (Suppl 1):S23–S24. [DOI] [PubMed] [Google Scholar]
- Fox GB, Pan JB, Esbenshade TA, Bitner RS, Nikkel AL, Miller T, Kang CH, Bennani YL, Black LA, Faghih R, et al. (2002) Differential in vivo effects of H3 receptor ligands in a new mouse dipsogenia model. Pharmacol Biochem Behav 72:741–750. [DOI] [PubMed] [Google Scholar]
- Fujimoto K, Ohta K, Kangawa K, Kikkawa U, Ogino S, Fukui H. (1999) Identification of protein kinase C phosphorylation sites involved in phorbol ester-induced desensitization of the histamine H1 receptor. Mol Pharmacol 55:735–742. [PubMed] [Google Scholar]
- Fukushima Y, Asano T, Saitoh T, Anai M, Funaki M, Ogihara T, Katagiri H, Matsuhashi N, Yazaki Y, Sugano K. (1997) Oligomer formation of histamine H2 receptors expressed in Sf9 and COS7 cells. FEBS Lett 409:283–286. [DOI] [PubMed] [Google Scholar]
- Fukushima Y, Saitoh T, Anai M, Tsukuda K, Onishi Y, Sakoda H, Inukai K, Ogihara T, Funaki M, Ono H, et al. (2001) G649, an allelic variant of the human H2 receptor with low basal activity, is resistant to upregulation upon antagonist exposure. Pharmacogenomics J 1:78–83. [DOI] [PubMed] [Google Scholar]
- Gajtkowski GA, Norris DB, Rising TJ, Wood TP. (1983) Specific binding of 3H-tiotidine to histamine H2 receptors in guinea pig cerebral cortex. Nature 304:65–67. [DOI] [PubMed] [Google Scholar]
- Galandrin S, Oligny-Longpré G, Bouvier M. (2007) The evasive nature of drug efficacy: implications for drug discovery. Trends Pharmacol Sci 28:423–430. [DOI] [PubMed] [Google Scholar]
- Galici R, Rezvani AH, Aluisio L, Lord B, Levin ED, Fraser I, Boggs J, Welty N, Shoblock JR, Motley ST, et al. (2011) JNJ-39220675, a novel selective histamine H3 receptor antagonist, reduces the abuse-related effects of alcohol in rats. Psychopharmacology (Berl) 214:829–841. [DOI] [PubMed] [Google Scholar]
- Ganellin CR, Fkyerat A, Hosseini SK, Khalaf YS, Piripitsi A, Tertiuk W, Arrang JM, Garbarg M, Ligneau X, Schwartz JC. (1995) Structure-activity studies with histamine H3-receptor ligands. J Pharm Belg 50:179–187. [PubMed] [Google Scholar]
- Ganellin CR, Leurquin F, Piripitsi A, Arrang JM, Garbarg M, Ligneau X, Schunack W, Schwartz JC. (1998) Synthesis of potent non-imidazole histamine H3-receptor antagonists. Arch Pharm (Weinheim) 331:395–404. [DOI] [PubMed] [Google Scholar]
- Ganellin R. (1981) 1980 Award in Medicinal Chemistry: Medicinal chemistry and dynamic structure-activity analysis in the discovery of drugs acting at histamine H2 receptors. J Med Chem 24:913–920. [DOI] [PubMed] [Google Scholar]
- Gantner F, Sakai K, Tusche MW, Cruikshank WW, Center DM, Bacon KB. (2002) Histamine h4 and h2 receptors control histamine-induced interleukin-16 release from human CD8+ T cells. J Pharmacol Exp Ther 303:300–307. [DOI] [PubMed] [Google Scholar]
- Gantz I, DelValle J, Wang LD, Tashiro T, Munzert G, Guo YJ, Konda Y, Yamada T. (1992) Molecular basis for the interaction of histamine with the histamine H2 receptor. J Biol Chem 267:20840–20843. [PubMed] [Google Scholar]
- Gantz I, Munzert G, Tashiro T, Schäffer M, Wang L, DelValle J, Yamada T. (1991a) Molecular cloning of the human histamine H2 receptor. Biochem Biophys Res Commun 178:1386–1392. [DOI] [PubMed] [Google Scholar]
- Gantz I, Schäffer M, DelValle J, Logsdon C, Campbell V, Uhler M, Yamada T. (1991b) Molecular cloning of a gene encoding the histamine H2 receptor. Proc Natl Acad Sci USA 88:429–433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garbarg M, Arrang JM, Rouleau A, Ligneau X, Tuong MD, Schwartz JC, Ganellin CR. (1992) S-[2-(4-imidazolyl)ethyl]isothiourea, a highly specific and potent histamine H3 receptor agonist. J Pharmacol Exp Ther 263:304–310. [PubMed] [Google Scholar]
- Garbarg M, Schwartz JC. (1988) Synergism between histamine H1- and H2-receptors in the cAMP response in guinea pig brain slices: effects of phorbol esters and calcium. Mol Pharmacol 33:38–43. [PubMed] [Google Scholar]
- Garbarg M, Yeramian E, Korner M, Schwartz J-C. (1985) Biochemical studies of cerebral H1-receptors, in Frontiers in Histamine Research, Vol. 51 (Ganellin CR, Schwartz JC, eds) pp 19–25, Advances in the Biosciences, Elsevier, UK. [Google Scholar]
- García-Martín E, Ayuso P, Martínez C, Blanca M, Agúndez JA. (2009) Histamine pharmacogenomics. Pharmacogenomics 10:867–883. [DOI] [PubMed] [Google Scholar]
- Gbahou F, Rouleau A, Morisset S, Parmentier R, Crochet S, Lin JS, Ligneau X, Tardivel-Lacombe J, Stark H, Schunack W, et al. (2003) Protean agonism at histamine H3 receptors in vitro and in vivo. Proc Natl Acad Sci USA 100:11086–11091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gbahou F, Vincent L, Humbert-Claude M, Tardivel-Lacombe J, Chabret C, Arrang JM. (2006) Compared pharmacology of human histamine H3 and H4 receptors: structure-activity relationships of histamine derivatives. Br J Pharmacol 147:744–754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gemkow MJ, Davenport AJ, Harich S, Ellenbroek BA, Cesura A, Hallett D. (2009) The histamine H3 receptor as a therapeutic drug target for CNS disorders. Drug Discov Today 14:509–515. [DOI] [PubMed] [Google Scholar]
- Ghorai P, Kraus A, Keller M, Götte C, Igel P, Schneider E, Schnell D, Bernhardt G, Dove S, Zabel M, et al. (2008) Acylguanidines as bioisosteres of guanidines: NG-acylated imidazolylpropylguanidines, a new class of histamine H2 receptor agonists. J Med Chem 51:7193–7204. [DOI] [PubMed] [Google Scholar]
- Giannoni P, Passani MB, Nosi D, Chazot PL, Shenton FC, Medhurst AD, Munari L, Blandina P. (2009) Heterogeneity of histaminergic neurons in the tuberomammillary nucleus of the rat. Eur J Neurosci 29:2363–2374. [DOI] [PubMed] [Google Scholar]
- Gilboa SM, Ailes EC, Rai RP, Anderson JA, Honein MA. (2014) Antihistamines and birth defects: a systematic review of the literature. Expert Opin Drug Saf 13:1667–1698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gillard M, Van Der Perren C, Moguilevsky N, Massingham R, Chatelain P. (2002a) Binding characteristics of cetirizine and levocetirizine to human H1 histamine receptors: contribution of Lys191 and Thr194. Mol Pharmacol 61:391–399. [DOI] [PubMed] [Google Scholar]
- Gillard M, Van der Perren C, Moguilevsky N, Massingham R, Chatelain P. (2002b) Major role for the carboxylic function of cetirizine and levocetirizine in their binding characteristics to human H1-histamine-receptors. Inflamm Res 51 (Suppl 1):S79–S80. [DOI] [PubMed] [Google Scholar]
- Giovannini MG, Efoudebe M, Passani MB, Baldi E, Bucherelli C, Giachi F, Corradetti R, Blandina P. (2003) Improvement in fear memory by histamine-elicited ERK2 activation in hippocampal CA3 cells. J Neurosci 23:9016–9023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gisselmann G, Pusch H, Hovemann BT, Hatt H. (2002) Two cDNAs coding for histamine-gated ion channels in D. melanogaster. Nat Neurosci 5:11–12. [DOI] [PubMed] [Google Scholar]
- Glatzer F, Gschwandtner M, Ehling S, Rossbach K, Janik K, Klos A, Bäumer W, Kietzmann M, Werfel T, Gutzmer R. (2013) Histamine induces proliferation in keratinocytes from patients with atopic dermatitis through the histamine 4 receptor. J Allergy Clin Immunol 132:1358–1367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glatzer F, Mommert S, Köther B, Gschwandtner M, Stark H, Werfel T, Gutzmer R. (2014) Histamine downregulates the Th1-associated chemokine IP-10 in monocytes and myeloid dendritic cells. Int Arch Allergy Immunol 163:11–19. [DOI] [PubMed] [Google Scholar]
- Godlewski G, Malinowska B, Schlicker E, Bucher B. (1997) Identification of histamine H3 receptors in the tail artery from normotensive and spontaneously hypertensive rats. J Cardiovasc Pharmacol 29:801–807. [DOI] [PubMed] [Google Scholar]
- Godot V, Arock M, Garcia G, Capel F, Flys C, Dy M, Emilie D, Humbert M. (2007) H4 histamine receptor mediates optimal migration of mast cell precursors to CXCL12. J Allergy Clin Immunol 120:827–834. [DOI] [PubMed] [Google Scholar]
- Goodearl ADJ, Glucksmann MA, Xie M, and Distefano P (1999) inventors, Millenium Pharm Inc, assignee. G-protein coupled receptors and uses therefor. Patent WO1999028470 A1. 1999 Jun 10.
- Gorelova N, Reiner PB. (1996) Histamine depolarizes cholinergic septal neurons. J Neurophysiol 75:707–714. [DOI] [PubMed] [Google Scholar]
- Govoni M, Bakker RA, van de Wetering I, Smit MJ, Menge WM, Timmerman H, Elz S, Schunack W, Leurs R. (2003) Synthesis and pharmacological identification of neutral histamine H1-receptor antagonists. J Med Chem 46:5812–5824. [DOI] [PubMed] [Google Scholar]
- Govoni M, Lim HD, El-Atmioui D, Menge WM, Timmerman H, Bakker RA, Leurs R, De Esch IJ. (2006) A chemical switch for the modulation of the functional activity of higher homologues of histamine on the human histamine H3 receptor: effect of various substitutions at the primary amino function. J Med Chem 49:2549–2557. [DOI] [PubMed] [Google Scholar]
- Grandi D, Shenton FC, Chazot PL, Morini G. (2008) Immunolocalization of histamine H3 receptors on endocrine cells in the rat gastrointestinal tract. Histol Histopathol 23:789–798. [DOI] [PubMed] [Google Scholar]
- Green JP. (1964) Histamine and the nervous system. Fed Proc 23:1095–1102. [PubMed] [Google Scholar]
- Greene RW, Haas HL. (1990) Effects of histamine on dentate granule cells in vitro. Neuroscience 34:299–303. [DOI] [PubMed] [Google Scholar]
- Grimstad O, Sawanobori Y, Vestergaard C, Bilsborough J, Olsen UB, Grønhøj-Larsen C, Matsushima K. (2009) Anti-interleukin-31-antibodies ameliorate scratching behaviour in NC/Nga mice: a model of atopic dermatitis. Exp Dermatol 18:35–43. [DOI] [PubMed] [Google Scholar]
- Grove RA, Harrington CM, Mahler A, Beresford I, Maruff P, Lowy MT, Nicholls AP, Boardley RL, Berges AC, Nathan PJ, et al. (2014) A randomized, double-blind, placebo-controlled, 16-week study of the H3 receptor antagonist, GSK239512 as a monotherapy in subjects with mild-to-moderate Alzheimer’s disease. Curr Alzheimer Res 11:47–58. [DOI] [PubMed] [Google Scholar]
- Grzybowska-Kowalczyk A, Maslinska D, Wojciechowska M, Szukiewicz D, Wojtecka-Lukasik E, Paradowska A, Maldyk P, Maslinski S. (2008) Expression of histamine H4 receptor in human osteoarthritic synovial tissue. Inflamm Res 57 (Suppl 1):S63–S64. [DOI] [PubMed] [Google Scholar]
- Gschwandtner M, Bunk H, Köther B, Thurmond RL, Kietzmann M, Werfel T, Bäumer W, Gutzmer R. (2012) Histamine down-regulates IL-27 production in antigen-presenting cells. J Leukoc Biol 92:21–29. [DOI] [PubMed] [Google Scholar]
- Gschwandtner M, Koether B, Werfel T, Stark H, Gutzmer R. (2013) Profiling of histamine H4 receptor agonists in native human monocytes. Br J Pharmacol 170:136–143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gschwandtner M, Rossbach K, Dijkstra D, Bäumer W, Kietzmann M, Stark H, Werfel T, Gutzmer R. (2010) Murine and human Langerhans cells express a functional histamine H4 receptor: modulation of cell migration and function. Allergy 65:840–849. [DOI] [PubMed] [Google Scholar]
- Gschwandtner M, Schäkel K, Werfel T, Gutzmer R. (2011) Histamine H4 receptor activation on human slan-dendritic cells down-regulates their pro-inflammatory capacity. Immunology 132:49–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo RX, Anaclet C, Roberts JC, Parmentier R, Zhang M, Guidon G, Buda C, Sastre JP, Feng JQ, Franco P, et al. (2009) Differential effects of acute and repeat dosing with the H3 antagonist GSK189254 on the sleep-wake cycle and narcoleptic episodes in Ox−/− mice. Br J Pharmacol 157:104–117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gupta MA, Gupta AK. (1996) Psychodermatology: an update. J Am Acad Dermatol 34:1030–1046. [DOI] [PubMed] [Google Scholar]
- Gutowski S, Smrcka A, Nowak L, Wu DG, Simon M, Sternweis PC. (1991) Antibodies to the alpha q subfamily of guanine nucleotide-binding regulatory protein alpha subunits attenuate activation of phosphatidylinositol 4,5-bisphosphate hydrolysis by hormones. J Biol Chem 266:20519–20524. [PubMed] [Google Scholar]
- Gutzmer R, Diestel C, Mommert S, Köther B, Stark H, Wittmann M, Werfel T. (2005) Histamine H4 receptor stimulation suppresses IL-12p70 production and mediates chemotaxis in human monocyte-derived dendritic cells. J Immunol 174:5224–5232. [DOI] [PubMed] [Google Scholar]
- Gutzmer R, Mommert S, Gschwandtner M, Zwingmann K, Stark H, Werfel T. (2009) The histamine H4 receptor is functionally expressed on T(H)2 cells. J Allergy Clin Immunol 123:619–625. [DOI] [PubMed] [Google Scholar]
- Haas H, Panula P. (2003) The role of histamine and the tuberomamillary nucleus in the nervous system. Nat Rev Neurosci 4:121–130. [DOI] [PubMed] [Google Scholar]
- Haas HL, Anderson EG, Hösli L. (1973) Histamine and metabolites: their effects and interactions with convulsants on brain stem neurones. Brain Res 51:269–278. [DOI] [PubMed] [Google Scholar]
- Haas HL, Bucher UM. (1975) Histamine H2-receptors on single central neurones. Nature 255:634–635. [DOI] [PubMed] [Google Scholar]
- Haas HL, Konnerth A. (1983) Histamine and noradrenaline decrease calcium-activated potassium conductance in hippocampal pyramidal cells. Nature 302:432–434. [DOI] [PubMed] [Google Scholar]
- Haas HL, Sergeeva OA, Selbach O. (2008) Histamine in the nervous system. Physiol Rev 88:1183–1241. [DOI] [PubMed] [Google Scholar]
- Håkanson R, Böttcher G, Ekblad E, Panula P, Simonsson M, Dohlsten M, Hallberg T, Sundler F. (1986) Histamine in endocrine cells in the stomach. A survey of several species using a panel of histamine antibodies. Histochemistry 86:5–17. [DOI] [PubMed] [Google Scholar]
- Hamill TG, Lin LS, Hagmann W, Liu P, Jewell J, Sanabria S, Eng W, Ryan C, Fong TM, Connolly B, et al. (2009) PET imaging studies in rhesus monkey with the cannabinoid-1 (CB1) receptor ligand [11C]CB-119. Mol Imaging Biol 11:246–252. [DOI] [PubMed] [Google Scholar]
- Hancock AA. (2006) The challenge of drug discovery of a GPCR target: analysis of preclinical pharmacology of histamine H3 antagonists/inverse agonists. Biochem Pharmacol 71:1103–1113. [DOI] [PubMed] [Google Scholar]
- Hancock AA, Esbenshade TA, Krueger KM, Yao BB. (2003) Genetic and pharmacological aspects of histamine H3 receptor heterogeneity. Life Sci 73:3043–3072. [DOI] [PubMed] [Google Scholar]
- Hansen KB, Mullasseril P, Dawit S, Kurtkaya NL, Yuan H, Vance KM, Orr AG, Kvist T, Ogden KK, Le P, et al. (2010) Implementation of a fluorescence-based screening assay identifies histamine H3 receptor antagonists clobenpropit and iodophenpropit as subunit-selective N-methyl-D-aspartate receptor antagonists. J Pharmacol Exp Ther 333:650–662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harada C, Hirai T, Fujii Y, Harusawa S, Kurihara T, Kamei C. (2004) Intracerebroventricular administration of histamine H3 receptor antagonists decreases seizures in rat models of epilepsia. Methods Find Exp Clin Pharmacol 26:263–270. [PubMed] [Google Scholar]
- Harada M, Terai M, Maeno H. (1983) Effect of a new potent H2-receptor antagonist 3[[[2-[(diaminomethylene)amino]-4-thiazolyl]methyl]thio]-N2- sulfamoylpropionamidine (YM-11170) on gastric mucosal histamine-sensitive adenylate cyclase from guinea pig. Biochem Pharmacol 32:1635–1640. [DOI] [PubMed] [Google Scholar]
- Hasanein P. (2011) Two histamine H2 receptor antagonists, zolantidine and cimetidine, modulate nociception in cholestatic rats. J Psychopharmacol 25:281–288. [DOI] [PubMed] [Google Scholar]
- Hashimoto T, Harusawa S, Araki L, Zuiderveld OP, Smit MJ, Imazu T, Takashima S, Yamamoto Y, Sakamoto Y, Kurihara T, et al. (2003) A selective human H4-receptor agonist: (−)-2-cyano-1-methyl-3-[(2R,5R)-5- [1H-imidazol-4(5)-yl]tetrahydrofuran-2-y] methylguanidine. J Med Chem 46:3162–3165. [DOI] [PubMed] [Google Scholar]
- Hatton GI, Yang QZ. (2001) Ionotropic histamine receptors and H2 receptors modulate supraoptic oxytocin neuronal excitability and dye coupling. J Neurosci 21:2974–2982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He M, Deng C, Huang XF. (2013) The role of hypothalamic H1 receptor antagonism in antipsychotic-induced weight gain. CNS Drugs 27:423–434. [DOI] [PubMed] [Google Scholar]
- Héron A, Rouleau A, Cochois V, Pillot C, Schwartz JC, Arrang JM. (2001) Expression analysis of the histamine H3 receptor in developing rat tissues. Mech Dev 105:167–173. [DOI] [PubMed] [Google Scholar]
- Herring WJ, Liu K, Hutzelmann J, Snavely D, Snyder E, Ceesay P, Lines C, Michelson D, Roth T. (2013) Alertness and psychomotor performance effects of the histamine-3 inverse agonist MK-0249 in obstructive sleep apnea patients on continuous positive airway pressure therapy with excessive daytime sleepiness: a randomized adaptive crossover study. Sleep Med 14:955–963. [DOI] [PubMed] [Google Scholar]
- Hershcovici T, Fass R. (2011) Gastro-oesophageal reflux disease: beyond proton pump inhibitor therapy. Drugs 71:2381–2389. [DOI] [PubMed] [Google Scholar]
- Hill SJ. (1990) Distribution, properties, and functional characteristics of three classes of histamine receptor. Pharmacol Rev 42:45–83. [PubMed] [Google Scholar]
- Hill SJ, Ganellin CR, Timmerman H, Schwartz JC, Shankley NP, Young JM, Schunack W, Levi R, Haas HL. (1997) International Union of Pharmacology. XIII. Classification of histamine receptors. Pharmacol Rev 49:253–278. [PubMed] [Google Scholar]
- Hill SJ, Young JM. (1980) Histamine H1-receptors in the brain of the guinea-pig and the rat: differences in ligand binding properties and regional distribution. Br J Pharmacol 68:687–696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hill SJ, Young JM, Marrian DH. (1977) Specific binding of 3H-mepyramine to histamine H1 receptors in intestinal smooth muscle. Nature 270:361–363. [DOI] [PubMed] [Google Scholar]
- Hirai T, Okuma C, Harada C, Mio M, Ohtsu H, Watanabe T, Kamei C. (2004) Development of amygdaloid kindling in histidine decarboxylase-deficient and histamine H1 receptor-deficient mice. Epilepsia 45:309–313. [DOI] [PubMed] [Google Scholar]
- Hirschfeld J, Buschauer A, Elz S, Schunack W, Ruat M, Traiffort E, Schwartz JC. (1992) Iodoaminopotentidine and related compounds: a new class of ligands with high affinity and selectivity for the histamine H2 receptor. J Med Chem 35:2231–2238. [DOI] [PubMed] [Google Scholar]
- Hodge E, Chang WYC, Selby KA, Hall IP, Sayers I. (2013) Effects of atopy and grass pollen season on histamine H4 receptor expression in human leukocytes. Ann Allergy Asthma Immunol 111:38–44. [DOI] [PubMed] [Google Scholar]
- Hofstra CL, Desai PJ, Thurmond RL, Fung-Leung W-P. (2003) Histamine H4 receptor mediates chemotaxis and calcium mobilization of mast cells. J Pharmacol Exp Ther 305:1212–1221. [DOI] [PubMed] [Google Scholar]
- Hsieh GC, Chandran P, Salyers AK, Pai M, Zhu CZ, Wensink EJ, Witte DG, Miller TR, Mikusa JP, Baker SJ, et al. (2010a) H4 receptor antagonism exhibits anti-nociceptive effects in inflammatory and neuropathic pain models in rats. Pharmacol Biochem Behav 95:41–50. [DOI] [PubMed] [Google Scholar]
- Hsieh GC, Honore P, Pai M, Wensink EJ, Chandran P, Salyers AK, Wetter JM, Zhao C, Liu H, Decker MW, et al. (2010b) Antinociceptive effects of histamine H3 receptor antagonist in the preclinical models of pain in rats and the involvement of central noradrenergic systems. Brain Res 1354:74–84. [DOI] [PubMed] [Google Scholar]
- Huang YW, Hu WW, Chen Z, Zhang LS, Shen HQ, Timmerman H, Leurs R, Yanai K. (2004) Effect of the histamine H3-antagonist clobenpropit on spatial memory deficits induced by MK-801 as evaluated by radial maze in Sprague-Dawley rats. Behav Brain Res 151:287–293. [DOI] [PubMed] [Google Scholar]
- Huang ZL, Mochizuki T, Qu WM, Hong ZY, Watanabe T, Urade Y, Hayaishi O. (2006) Altered sleep-wake characteristics and lack of arousal response to H3 receptor antagonist in histamine H1 receptor knockout mice. Proc Natl Acad Sci USA 103:4687–4692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iannone R, Palcza J, Renger JJ, Calder N, Cerchio K, Gottesdiener K, Hargreaves R, Dijk DJ, Boyle J, Murphy MG. (2010) Acute alertness-promoting effects of a novel histamine subtype-3 receptor inverse agonist in healthy sleep-deprived male volunteers. Clin Pharmacol Ther 88:831–839. [DOI] [PubMed] [Google Scholar]
- Ichinose M, Barnes PJ. (1990) Histamine H3 receptors modulate antigen-induced bronchoconstriction in guinea pigs. J Allergy Clin Immunol 86:491–495. [DOI] [PubMed] [Google Scholar]
- Ikawa Y, Shiba K, Ohki E, Mutoh N, Suzuki M, Sato H, Ueno K. (2008) Comparative study of histamine H4 receptor expression in human dermal fibroblasts. J Toxicol Sci 33:503–508. [DOI] [PubMed] [Google Scholar]
- Ikawa Y, Suzuki M, Shiono S, Ohki E, Moriya H, Negishi E, Ueno K. (2005) Histamine H4 receptor expression in human synovial cells obtained from patients suffering from rheumatoid arthritis. Biol Pharm Bull 28:2016–2018. [DOI] [PubMed] [Google Scholar]
- Ikoma A, Rukwied R, Ständer S, Steinhoff M, Miyachi Y, Schmelz M. (2003) Neuronal sensitization for histamine-induced itch in lesional skin of patients with atopic dermatitis. Arch Dermatol 139:1455–1458. [DOI] [PubMed] [Google Scholar]
- Imamura M, Smith NC, Garbarg M, Levi R. (1996) Histamine H3-receptor-mediated inhibition of calcitonin gene-related peptide release from cardiac C fibers. A regulatory negative-feedback loop. Circ Res 78:863–869. [DOI] [PubMed] [Google Scholar]
- Ishikawa M, Shinei R, Yokoyama F, Yamauchi M, Oyama M, Okuma K, Nagayama T, Kato K, Kakui N, Sato Y. (2010) Role of hydrophobic substituents on the terminal nitrogen of histamine in receptor binding and agonist activity: development of an orally active histamine type 3 receptor agonist and evaluation of its antistress activity in mice. J Med Chem 53:3840–3844. [DOI] [PubMed] [Google Scholar]
- Ishikawa S, Sperelakis N. (1987) A novel class (H3) of histamine receptors on perivascular nerve terminals. Nature 327:158–160. [DOI] [PubMed] [Google Scholar]
- Ito C, Morisset S, Krebs MO, Olié JP, Lôo H, Poirier MF, Lannfelt L, Schwartz JC, Arrang JM. (2000) Histamine H2 receptor gene variants: lack of association with schizophrenia. Mol Psychiatry 5:159–164. [DOI] [PubMed] [Google Scholar]
- Ito S, Yoshimoto R, Miyamoto Y, Mitobe Y, Nakamura T, Ishihara A, MacNeil DJ, Kanatani A, Tokita S. (2006) Detailed pharmacological characterization of GT-2331 for the rat histamine H3 receptor. Eur J Pharmacol 529:40–46. [DOI] [PubMed] [Google Scholar]
- Iwabuchi K, Ito C, Tashiro M, Kato M, Kano M, Itoh M, Iwata R, Matsuoka H, Sato M, Yanai K. (2005) Histamine H1 receptors in schizophrenic patients measured by positron emission tomography. Eur Neuropsychopharmacol 15:185–191. [DOI] [PubMed] [Google Scholar]
- Iwata K, Luo J, Penn RB, Benovic JL. (2005) Bimodal regulation of the human H1 histamine receptor by G protein-coupled receptor kinase 2. J Biol Chem 280:2197–2204. [DOI] [PubMed] [Google Scholar]
- Jablonowski JA, Grice CA, Chai W, Dvorak CA, Venable JD, Kwok AK, Ly KS, Wei J, Baker SM, Desai PJ, et al. (2003) The first potent and selective non-imidazole human histamine H4 receptor antagonists. J Med Chem 46:3957–3960. [DOI] [PubMed] [Google Scholar]
- Jacoby E, Bouhelal R, Gerspacher M, Seuwen K. (2006) The 7 TM G-protein-coupled receptor target family. ChemMedChem 1:761–782. [DOI] [PubMed] [Google Scholar]
- Jahn K, Haas HL, Hatt H. (1995) Patch clamp study of histamine activated potassium currents on rabbit olfactory bulb neurons. Naunyn Schmiedebergs Arch Pharmacol 352:386–393. [DOI] [PubMed] [Google Scholar]
- James LM, Iannone R, Palcza J, Renger JJ, Calder N, Cerchio K, Gottesdiener K, Hargreaves R, Murphy MG, Boyle J, et al. (2011) Effect of a novel histamine subtype-3 receptor inverse agonist and modafinil on EEG power spectra during sleep deprivation and recovery sleep in male volunteers. Psychopharmacology (Berl) 215:643–653. [DOI] [PubMed] [Google Scholar]
- Jang IS, Rhee JS, Watanabe T, Akaike N, Akaike N. (2001) Histaminergic modulation of GABAergic transmission in rat ventromedial hypothalamic neurones. J Physiol 534:791–803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jansen FP, Rademaker B, Bast A, Timmerman H. (1992) The first radiolabeled histamine H3 receptor antagonist, [125I]iodophenpropit: saturable and reversible binding to rat cortex membranes. Eur J Pharmacol 217:203–205. [DOI] [PubMed] [Google Scholar]
- Jemima EA, Prema A, Thangam EB. (2014) Functional characterization of histamine H4 receptor on human mast cells. Mol Immunol 62:19–28. [DOI] [PubMed] [Google Scholar]
- Jiang W, Lim HD, Zhang M, Desai P, Dai H, Colling PM, Leurs R, Thurmond RL. (2008) Cloning and pharmacological characterization of the dog histamine H4 receptor. Eur J Pharmacol 592:26–32. [DOI] [PubMed] [Google Scholar]
- Jin CY, Kalimo H, Panula P. (2002) The histaminergic system in human thalamus: correlation of innervation to receptor expression. Eur J Neurosci 15:1125–1138. [DOI] [PubMed] [Google Scholar]
- Jin CY, Panula P. (2005) The laminar histamine receptor system in human prefrontal cortex suggests multiple levels of histaminergic regulation. Neuroscience 132:137–149. [DOI] [PubMed] [Google Scholar]
- Jókúti A, Hellinger E, Hellinger A, Darvas Z, Falus A, Thurmond RL, Hirschberg A. (2007) Histamine H4 receptor expression is elevated in human nasal polyp tissue. Cell Biol Int 31:1367–1370. [DOI] [PubMed] [Google Scholar]
- Jongejan A, Bruysters M, Ballesteros JA, Haaksma E, Bakker RA, Pardo L, Leurs R. (2005) Linking agonist binding to histamine H1 receptor activation. Nat Chem Biol 1:98–103. [DOI] [PubMed] [Google Scholar]
- Jongejan A, Leurs R. (2005) Delineation of receptor-ligand interactions at the human histamine H1 receptor by a combined approach of site-directed mutagenesis and computational techniques - or - how to bind the H1 receptor. Arch Pharm (Weinheim) 338:248–259. [DOI] [PubMed] [Google Scholar]
- Jutel M, Akdis M, Akdis CA. (2009) Histamine, histamine receptors and their role in immune pathology. Clin Exp Allergy 39:1786–1800. [DOI] [PubMed] [Google Scholar]
- Jutel M, Watanabe T, Klunker S, Akdis M, Thomet OA, Malolepszy J, Zak-Nejmark T, Koga R, Kobayashi T, Blaser K, et al. (2001) Histamine regulates T-cell and antibody responses by differential expression of H1 and H2 receptors. Nature 413:420–425. [DOI] [PubMed] [Google Scholar]
- Kaminsky R, Moriarty TM, Bodine J, Wolf DE, Davidson M. (1990) Effect of famotidine on deficit symptoms of schizophrenia. Lancet 335:1351–1352. [DOI] [PubMed] [Google Scholar]
- Kamisako T, Adachi Y, Nakagawa H, Yamamoto T. (1995) Torsades de pointes associated with terfenadine in a case of liver cirrhosis and hepatocellular carcinoma. Intern Med 34:92–95. [DOI] [PubMed] [Google Scholar]
- Kanba S, Richelson E. (1984) Histamine H1 receptors in human brain labelled with [3H]doxepin. Brain Res 304:1–7. [DOI] [PubMed] [Google Scholar]
- Kano M, Fukudo S, Tashiro A, Utsumi A, Tamura D, Itoh M, Iwata R, Tashiro M, Mochizuki H, Funaki Y, et al. (2004) Decreased histamine H1 receptor binding in the brain of depressed patients. Eur J Neurosci 20:803–810. [DOI] [PubMed] [Google Scholar]
- Kari O, Saari KM. (2012) Diagnostics and new developments in the treatment of ocular allergies. Curr Allergy Asthma Rep 12:232–239. [DOI] [PubMed] [Google Scholar]
- Karlstedt K, Ahman MJ, Anichtchik OV, Soinila S, Panula P. (2003) Expression of the H3 receptor in the developing CNS and brown fat suggests novel roles for histamine. Mol Cell Neurosci 24:614–622. [DOI] [PubMed] [Google Scholar]
- Karlstedt K, Jin C, Panula P. (2013) Expression of histamine receptor genes Hrh3 and Hrh4 in rat brain endothelial cells. Br J Pharmacol 170:58–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karlstedt K, Sallmén T, Eriksson KS, Lintunen M, Couraud PO, Joó F, Panula P. (1999) Lack of histamine synthesis and down-regulation of H1 and H2 receptor mRNA levels by dexamethasone in cerebral endothelial cells. J Cereb Blood Flow Metab 19:321–330. [DOI] [PubMed] [Google Scholar]
- Karlstedt K, Senkas A, Ahman M, Panula P. (2001) Regional expression of the histamine H2 receptor in adult and developing rat brain. Neuroscience 102:201–208. [DOI] [PubMed] [Google Scholar]
- Kato M, Nishida A, Aga Y, Kita J, Kudo Y, Narita H, Endo T. (1997) Pharmacokinetic and pharmacodynamic evaluation of central effect of the novel antiallergic agent betotastine besilate. Arzneimittelforschung 47:1116–1124. [PubMed] [Google Scholar]
- Kelley MT, Bürckstümmer T, Wenzel-Seifert K, Dove S, Buschauer A, Seifert R. (2001) Distinct interaction of human and guinea pig histamine H2-receptor with guanidine-type agonists. Mol Pharmacol 60:1210–1225. [DOI] [PubMed] [Google Scholar]
- Kenakin T. (2002) Drug efficacy at G protein-coupled receptors. Annu Rev Pharmacol Toxicol 42:349–379. [DOI] [PubMed] [Google Scholar]
- Kenakin T. (2011) Functional selectivity and biased receptor signaling. J Pharmacol Exp Ther 336:296–302. [DOI] [PubMed] [Google Scholar]
- Kim SF, Huang AS, Snowman AM, Teuscher C, Snyder SH. (2007) From the Cover: Antipsychotic drug-induced weight gain mediated by histamine H1 receptor-linked activation of hypothalamic AMP-kinase. Proc Natl Acad Sci USA 104:3456–3459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kitbunnadaj R, Hashimoto T, Poli E, Zuiderveld OP, Menozzi A, Hidaka R, de Esch IJ, Bakker RA, Menge WM, Yamatodani A, et al. (2005) N-substituted piperidinyl alkyl imidazoles: discovery of methimepip as a potent and selective histamine H3 receptor agonist. J Med Chem 48:2100–2107. [DOI] [PubMed] [Google Scholar]
- Kitbunnadaj R, Zuiderveld OP, Christophe B, Hulscher S, Menge WM, Gelens E, Snip E, Bakker RA, Celanire S, Gillard M, et al. (2004) Identification of 4-(1H-imidazol-4(5)-ylmethyl)pyridine (immethridine) as a novel, potent, and highly selective histamine H3 receptor agonist. J Med Chem 47:2414–2417. [DOI] [PubMed] [Google Scholar]
- Kobayashi T, Inoue I, Jenkins NA, Gilbert DJ, Copeland NG, Watanabe T. (1996) Cloning, RNA expression, and chromosomal location of a mouse histamine H2 receptor gene. Genomics 37:390–394. [DOI] [PubMed] [Google Scholar]
- Kobilka BK, Deupi X. (2007) Conformational complexity of G-protein-coupled receptors. Trends Pharmacol Sci 28:397–406. [DOI] [PubMed] [Google Scholar]
- Kohyama T, Yamauchi Y, Takizawa H, Kamitani S, Kawasaki S, Nagase T. (2010) Histamine stimulates human lung fibroblast migration. Mol Cell Biochem 337:77–81. [DOI] [PubMed] [Google Scholar]
- Kollmeier A, Francke K, Chen B, Dunford PJ, Greenspan AJ, Xia Y, Xu XL, Zhou B, Thurmond RL. (2014) The histamine H₄ receptor antagonist, JNJ 39758979, is effective in reducing histamine-induced pruritus in a randomized clinical study in healthy subjects. J Pharmacol Exp Ther 350:181–187. [DOI] [PubMed] [Google Scholar]
- Komater VA, Buckley MJ, Browman KE, Pan JB, Hancock AA, Decker MW, Fox GB. (2005) Effects of histamine H3 receptor antagonists in two models of spatial learning. Behav Brain Res 159:295–300. [DOI] [PubMed] [Google Scholar]
- Kooistra AJ, Kuhne S, de Esch IJ, Leurs R, de Graaf C. (2013) A structural chemogenomics analysis of aminergic GPCRs: lessons for histamine receptor ligand design. Br J Pharmacol 170:101–126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Korotkova TM, Haas HL, Brown RE. (2002) Histamine excites GABAergic cells in the rat substantia nigra and ventral tegmental area in vitro. Neurosci Lett 320:133–136. [DOI] [PubMed] [Google Scholar]
- Korotkova TM, Sergeeva OA, Ponomarenko AA, Haas HL. (2005) Histamine excites noradrenergic neurons in locus coeruleus in rats. Neuropharmacology 49:129–134. [DOI] [PubMed] [Google Scholar]
- Kostopoulos G, Psarropoulou C, Haas HL. (1988) Membrane properties, response to amines and to tetanic stimulation of hippocampal neurons in the genetically epileptic mutant mouse tottering. Exp Brain Res 72:45–50. [DOI] [PubMed] [Google Scholar]
- Kottke T, Sander K, Weizel L, Schneider EH, Seifert R, Stark H. (2011) Receptor-specific functional efficacies of alkyl imidazoles as dual histamine H3/H4 receptor ligands. Eur J Pharmacol 654:200–208. [DOI] [PubMed] [Google Scholar]
- Kraus A, Ghorai P, Birnkammer T, Schnell D, Elz S, Seifert R, Dove S, Bernhardt G, Buschauer A. (2009) NG-acylated aminothiazolylpropylguanidines as potent and selective histamine H2 receptor agonists. ChemMedChem 4:232–240. [DOI] [PubMed] [Google Scholar]
- Krause M, Stark H, Schunack W. (2001) Azomethine prodrugs of (R)-alpha-methylhistamine, a highly potent and selective histamine H3-receptor agonist. Curr Med Chem 8:1329–1340. [DOI] [PubMed] [Google Scholar]
- Krementsov DN, Wall EH, Martin RA, Subramanian M, Noubade R, Del Rio R, Mawe GM, Bond JP, Poynter ME, Blankenhorn EP, et al. (2013) Histamine H3 receptor integrates peripheral inflammatory signals in the neurogenic control of immune responses and autoimmune disease susceptibility. PLoS ONE 8:e62743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kreutner W, Hey JA, Anthes J, Barnett A, Young S, Tozzi S. (2000) Preclinical pharmacology of desloratadine, a selective and nonsedating histamine H1 receptor antagonist. 1st communication: receptor selectivity, antihistaminic activity, and antiallergenic effects. Arzneimittelforschung 50:345–352. [DOI] [PubMed] [Google Scholar]
- Krueger KM, Witte DG, Ireland-Denny L, Miller TR, Baranowski JL, Buckner S, Milicic I, Esbenshade TA, Hancock AA. (2005) G protein-dependent pharmacology of histamine H3 receptor ligands: evidence for heterogeneous active state receptor conformations. J Pharmacol Exp Ther 314:271–281. [DOI] [PubMed] [Google Scholar]
- Kubo A, Fukui H, Inagaki N, Kanamura A, Wada H. (1991) Histamine-induced cyclic AMP accumulation in type-1 and type-2 astrocytes in primary culture. Eur J Pharmacol 208:249–253. [DOI] [PubMed] [Google Scholar]
- Kühn B, Schmid A, Harteneck C, Gudermann T, Schultz G. (1996) G proteins of the Gq family couple the H2 histamine receptor to phospholipase C. Mol Endocrinol 10:1697–1707. [DOI] [PubMed] [Google Scholar]
- Kukko-Lukjanov TK, Lintunen M, Jalava N, Laurén HB, Lopez-Picon FR, Michelsen KA, Panula P, Holopainen IE. (2010) Involvement of histamine 1 receptor in seizure susceptibility and neuroprotection in immature mice. Epilepsy Res 90:8–15. [DOI] [PubMed] [Google Scholar]
- Kukko-Lukjanov TK, Soini S, Taira T, Michelsen KA, Panula P, Holopainen IE. (2006) Histaminergic neurons protect the developing hippocampus from kainic acid-induced neuronal damage in an organotypic coculture system. J Neurosci 26:1088–1097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lagerström MC, Schiöth HB. (2008) Structural diversity of G protein-coupled receptors and significance for drug discovery. Nat Rev Drug Discov 7:339–357. [DOI] [PubMed] [Google Scholar]
- Lagrutta AA, Trepakova ES, Salata JJ. (2008) The hERG channel and risk of drug-acquired cardiac arrhythmia: an overview. Curr Top Med Chem 8:1102–1112. [DOI] [PubMed] [Google Scholar]
- Laitinen JT, Jokinen M. (1998) Guanosine 5′-(gamma-[35S]thio)triphosphate autoradiography allows selective detection of histamine H3 receptor-dependent G protein activation in rat brain tissue sections. J Neurochem 71:808–816. [DOI] [PubMed] [Google Scholar]
- Lakoski JM, Aghajanian GK, Gallager DW. (1983) Interaction of histamine H2-receptor antagonists with GABA and benzodiazepine binding sites in the CNS. Eur J Pharmacol 88:241–245. [DOI] [PubMed] [Google Scholar]
- Lane CA, Hay D, Mowbray CE, Paradowski M, Selby MD, Swain NA, Williams DH. (2012) Synthesis of novel histamine H4 receptor antagonists. Bioorg Med Chem Lett 22:1156–1159. [DOI] [PubMed] [Google Scholar]
- Lazewska D, Kiec-Kononowicz K. (2010) Recent advances in histamine H3 receptor antagonists/inverse agonists. Expert Opin Ther Pat 20:1147–1169. [DOI] [PubMed] [Google Scholar]
- Le Coniat M, Traiffort E, Ruat M, Arrang JM, Berger R. (1994) Chromosomal localization of the human histamine H1-receptor gene. Hum Genet 94:186–188. [DOI] [PubMed] [Google Scholar]
- Lee-Dutra A, Arienti KL, Buzard DJ, Hack MD, Khatuya H, Desai PJ, Nguyen S, Thurmond RL, Karlsson L, Edwards JP, et al. (2006) Identification of 2-arylbenzimidazoles as potent human histamine H4 receptor ligands. Bioorg Med Chem Lett 16:6043–6048. [DOI] [PubMed] [Google Scholar]
- Lee KH, Broberger C, Kim U, McCormick DA. (2004) Histamine modulates thalamocortical activity by activating a chloride conductance in ferret perigeniculate neurons. Proc Natl Acad Sci USA 101:6716–6721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leite-de-Moraes MC, Diem S, Michel ML, Ohtsu H, Thurmond RL, Schneider E, Dy M. (2009) Cutting edge: histamine receptor H4 activation positively regulates in vivo IL-4 and IFN-gamma production by invariant NKT cells. J Immunol 182:1233–1236. [DOI] [PubMed] [Google Scholar]
- Leopoldt D, Harteneck C, Nürnberg B. (1997) G proteins endogenously expressed in Sf 9 cells: interactions with mammalian histamine receptors. Naunyn Schmiedebergs Arch Pharmacol 356:216–224. [DOI] [PubMed] [Google Scholar]
- Leu-Semenescu S, Nittur N, Golmard JL, Arnulf I. (2014) Effects of pitolisant, a histamine H3 inverse agonist, in drug-resistant idiopathic and symptomatic hypersomnia: a chart review. Sleep Med 15:681–687. [DOI] [PubMed] [Google Scholar]
- Leurs R, Bakker RA, Timmerman H, de Esch IJ. (2005) The histamine H3 receptor: from gene cloning to H3 receptor drugs. Nat Rev Drug Discov 4:107–120. [DOI] [PubMed] [Google Scholar]
- Leurs R, Smit MJ, Meeder R, Ter Laak AM, Timmerman H. (1995a) Lysine200 located in the fifth transmembrane domain of the histamine H1 receptor interacts with histamine but not with all H1 agonists. Biochem Biophys Res Commun 214:110–117. [DOI] [PubMed] [Google Scholar]
- Leurs R, Smit MJ, Tensen CP, Ter Laak AM, Timmerman H. (1994a) Site-directed mutagenesis of the histamine H1-receptor reveals a selective interaction of asparagine207 with subclasses of H1-receptor agonists. Biochem Biophys Res Commun 201:295–301. [DOI] [PubMed] [Google Scholar]
- Leurs R, Traiffort E, Arrang JM, Tardivel-Lacombe J, Ruat M, Schwartz JC. (1994b) Guinea pig histamine H1 receptor. II. Stable expression in Chinese hamster ovary cells reveals the interaction with three major signal transduction pathways. J Neurochem 62:519–527. [DOI] [PubMed] [Google Scholar]
- Leurs R, Tulp MT, Menge WM, Adolfs MJ, Zuiderveld OP, Timmerman H. (1995b) Evaluation of the receptor selectivity of the H3 receptor antagonists, iodophenpropit and thioperamide: an interaction with the 5-HT3 receptor revealed. Br J Pharmacol 116:2315–2321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levoin N, Calmels T, Poupardin-Olivier O, Labeeuw O, Danvy D, Robert P, Berrebi-Bertrand I, Ganellin CR, Schunack W, Stark H, et al. (2008) Refined docking as a valuable tool for lead optimization: application to histamine H3 receptor antagonists. Arch Pharm (Weinheim) 341:610–623. [DOI] [PubMed] [Google Scholar]
- Lewis DW, Diamond S, Scott D, Jones V. (2004a) Prophylactic treatment of pediatric migraine. Headache 44:230–237. [DOI] [PubMed] [Google Scholar]
- Lewis TA, Bayless L, Eckman JB, Ellis JL, Grewal G, Libertine L, Marie Nicolas J, Scannell RT, Wels BF, Wenberg K, et al. (2004b) 5-lipoxygenase inhibitors with histamine H1 receptor antagonist activity. Bioorg Med Chem Lett 14:2265–2268. [DOI] [PubMed] [Google Scholar]
- Li M, Luo X, Chen L, Zhang J, Hu J, Lu B. (2003) Co-localization of histamine and dopamine-beta-hydroxylase in sympathetic ganglion and release of histamine from cardiac sympathetic terminals of guinea-pig. Auton Autacoid Pharmacol 23:327–333. [DOI] [PubMed] [Google Scholar]
- Li Z, Hatton GI. (2000) Histamine suppresses non-NMDA excitatory synaptic currents in rat supraoptic nucleus neurons. J Neurophysiol 83:2616–2625. [DOI] [PubMed] [Google Scholar]
- Ligneau X, Garbarg M, Vizuete ML, Diaz J, Purand K, Stark H, Schunack W, Schwartz JC. (1994) [125I]iodoproxyfan, a new antagonist to label and visualize cerebral histamine H3 receptors. J Pharmacol Exp Ther 271:452–459. [PubMed] [Google Scholar]
- Ligneau X, Lin J, Vanni-Mercier G, Jouvet M, Muir JL, Ganellin CR, Stark H, Elz S, Schunack W, Schwartz J. (1998) Neurochemical and behavioral effects of ciproxifan, a potent histamine H3-receptor antagonist. J Pharmacol Exp Ther 287:658–666. [PubMed] [Google Scholar]
- Ligneau X, Morisset S, Tardivel-Lacombe J, Gbahou F, Ganellin CR, Stark H, Schunack W, Schwartz JC, Arrang JM. (2000) Distinct pharmacology of rat and human histamine H3 receptors: role of two amino acids in the third transmembrane domain. Br J Pharmacol 131:1247–1250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ligneau X, Perrin D, Landais L, Camelin JC, Calmels TP, Berrebi-Bertrand I, Lecomte JM, Parmentier R, Anaclet C, Lin JS, et al. (2007) BF2.649 [1-3-[3-(4-Chlorophenyl)propoxy]propylpiperidine, hydrochloride], a nonimidazole inverse agonist/antagonist at the human histamine H3 receptor: Preclinical pharmacology. J Pharmacol Exp Ther 320:365–375. [DOI] [PubMed] [Google Scholar]
- Lim HD, de Graaf C, Jiang W, Sadek P, McGovern PM, Istyastono EP, Bakker RA, de Esch IJP, Thurmond RL, Leurs R. (2010) Molecular determinants of ligand binding to H4R species variants. Mol Pharmacol 77:734–743. [DOI] [PubMed] [Google Scholar]
- Lim HD, Istyastono EP, van de Stolpe A, Romeo G, Gobbi S, Schepers M, Lahaye R, Menge WM, Zuiderveld OP, Jongejan A, et al. (2009) Clobenpropit analogs as dual activity ligands for the histamine H3 and H4 receptors: synthesis, pharmacological evaluation, and cross-target QSAR studies. Bioorg Med Chem 17:3987–3994. [DOI] [PubMed] [Google Scholar]
- Lim HD, Jongejan A, Bakker RA, Haaksma E, de Esch IJ, Leurs R. (2008) Phenylalanine 169 in the second extracellular loop of the human histamine H4 receptor is responsible for the difference in agonist binding between human and mouse H4 receptors. J Pharmacol Exp Ther 327:88–96. [DOI] [PubMed] [Google Scholar]
- Lim HD, Smits RA, Bakker RA, van Dam CM, de Esch IJ, Leurs R. (2006) Discovery of S-(2-guanidylethyl)-isothiourea (VUF 8430) as a potent nonimidazole histamine H4 receptor agonist. J Med Chem 49:6650–6651. [DOI] [PubMed] [Google Scholar]
- Lim HD, van Rijn RM, Ling P, Bakker RA, Thurmond RL, Leurs R. (2005) Evaluation of histamine H1-, H2-, and H3-receptor ligands at the human histamine H4 receptor: identification of 4-methylhistamine as the first potent and selective H4 receptor agonist. J Pharmacol Exp Ther 314:1310–1321. [DOI] [PubMed] [Google Scholar]
- Lin JS, Anaclet C, Sergeeva OA, Haas HL. (2011a) The waking brain: an update. Cell Mol Life Sci 68:2499–2512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin JS, Dauvilliers Y, Arnulf I, Bastuji H, Anaclet C, Parmentier R, Kocher L, Yanagisawa M, Lehert P, Ligneau X, et al. (2008) An inverse agonist of the histamine H3 receptor improves wakefulness in narcolepsy: studies in orexin−/− mice and patients. Neurobiol Dis 30:74–83. [DOI] [PubMed] [Google Scholar]
- Lin JS, Hou Y, Sakai K, Jouvet M. (1996) Histaminergic descending inputs to the mesopontine tegmentum and their role in the control of cortical activation and wakefulness in the cat. J Neurosci 16:1523–1537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin JS, Sergeeva OA, Haas HL. (2011b) Histamine H3 receptors and sleep-wake regulation. J Pharmacol Exp Ther 336:17–23. [DOI] [PubMed] [Google Scholar]
- Ling P, Ngo K, Nguyen S, Thurmond RL, Edwards JP, Karlsson L, Fung-Leng WP. (2004) Histamine H4 receptor mediates eosinophil chemotaxis with cell shape change and adhesion molecule upregulation. Br J Pharmacol 142:161–172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lintunen M, Hyytiä P, Sallmen T, Karlstedt K, Tuomisto L, Leurs R, Kiianmaa K, Korpi ER, Panula P. (2001) Increased brain histamine in an alcohol-preferring rat line and modulation of ethanol consumption by H3 receptor mechanisms. FASEB J 15:1074–1076. [DOI] [PubMed] [Google Scholar]
- Lintunen M, Sallmen T, Karlstedt K, Fukui H, Eriksson KS, Panula P. (1998) Postnatal expression of H1-receptor mRNA in the rat brain: correlation to L-histidine decarboxylase expression and local upregulation in limbic seizures. Eur J Neurosci 10:2287–2301. [DOI] [PubMed] [Google Scholar]
- Lippert U, Artuc M, Grützkau A, Babina M, Guhl S, Haase I, Blaschke V, Zachmann K, Knosalla M, Middel P, et al. (2004) Human skin mast cells express H2 and H4, but not H3 receptors. J Invest Dermatol 123:116–123. [DOI] [PubMed] [Google Scholar]
- Liu C, Ma X, Jiang X, Wilson SJ, Hofstra CL, Blevitt J, Pyati J, Li X, Chai W, Carruthers N, et al. (2001a) Cloning and pharmacological characterization of a fourth histamine receptor (H4) expressed in bone marrow. Mol Pharmacol 59:420–426. [DOI] [PubMed] [Google Scholar]
- Liu C, Ma XJ, Lovenberg TW. (2000) Alternative splicing of the histamine H3 receptor mRNA at the third cytoplasmic loop is not detectable in humans. Brain Res Mol Brain Res 83:145–150. [DOI] [PubMed] [Google Scholar]
- Liu C, Wilson SJ, Kuei C, Lovenberg TW. (2001b) Comparison of human, mouse, rat, and guinea pig histamine H4 receptors reveals substantial pharmacological species variation. J Pharmacol Exp Ther 299:121–130. [PubMed] [Google Scholar]
- Liu H, Altenbach RJ, Carr TL, Chandran P, Hsieh GC, Lewis LG, Manelli AM, Milicic I, Marsh KC, Miller TR, et al. (2008) Cis-4-(piperazin-1-yl)-5,6,7a,8,9,10,11,11a-octahydrobenzofuro[2,3-h]quina zolin-2-amine (A-987306), a new histamine H4R antagonist that blocks pain responses against carrageenan-induced hyperalgesia. J Med Chem 22:7094–7098. [DOI] [PubMed] [Google Scholar]
- Liu H, Kerdesky FA, Black LA, Fitzgerald M, Henry R, Esbenshade TA, Hancock AA, Bennani YL. (2004) An efficient multigram synthesis of the potent histamine H3 antagonist GT-2331 and the reassessment of the absolute configuration. J Org Chem 69:192–194. [DOI] [PubMed] [Google Scholar]
- Liu WL. (2014) Histamine H4 receptor antagonists for the treatment of inflammatory disorders. Drug Discov Today 19:1222–1225. [DOI] [PubMed] [Google Scholar]
- Lovenberg TW, Pyati J, Chang H, Wilson SJ, Erlander MG. (2000) Cloning of rat histamine H3 receptor reveals distinct species pharmacological profiles. J Pharmacol Exp Ther 293:771–778. [PubMed] [Google Scholar]
- Lovenberg TW, Roland BL, Wilson SJ, Jiang X, Pyati J, Huvar A, Jackson MR, Erlander MG. (1999) Cloning and functional expression of the human histamine H3 receptor. Mol Pharmacol 55:1101–1107. [PubMed] [Google Scholar]
- Lu HR, Hermans AN, Gallacher DJ. (2012) Does terfenadine-induced ventricular tachycardia/fibrillation directly relate to its QT prolongation and Torsades de Pointes? Br J Pharmacol 166:1490–1502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luttrell LM, Gesty-Palmer D. (2010) Beyond desensitization: physiological relevance of arrestin-dependent signaling. Pharmacol Rev 62:305–330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lutz S, Shankaranarayanan A, Coco C, Ridilla M, Nance MR, Vettel C, Baltus D, Evelyn CR, Neubig RR, Wieland T, et al. (2007) Structure of Galphaq-p63RhoGEF-RhoA complex reveals a pathway for the activation of RhoA by GPCRs. Science 318:1923–1927. [DOI] [PubMed] [Google Scholar]
- Ma RZ, Gao J, Meeker ND, Fillmore PD, Tung KS, Watanabe T, Zachary JF, Offner H, Blankenhorn EP, Teuscher C. (2002) Identification of Bphs, an autoimmune disease locus, as histamine receptor H1. Science 297:620–623. [DOI] [PubMed] [Google Scholar]
- Maayani S, Hough LB, Weinstein H, Green JP. (1982) Response of the histamine h2-receptor in brain to antidepressant drugs. Adv Biochem Psychopharmacol 31:133–147. [PubMed] [Google Scholar]
- Mahapatra S, Albrecht M, Behrens B, Jirmo A, Behrens G, Hartwig C, Neumann D, Raap U, Bähre H, Herrick C, et al. (2014) Delineating the role of histamine-1- and -4-receptors in a mouse model of Th2-dependent antigen-specific skin inflammation. PLoS ONE 9:e87296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manahan-Vaughan D, Reymann KG, Brown RE. (1998) In vivo electrophysiological investigations into the role of histamine in the dentate gyrus of the rat. Neuroscience 84:783–790. [DOI] [PubMed] [Google Scholar]
- Marley PD, Thomson KA, Jachno K, Johnston MJ. (1991) Histamine-induced increases in cyclic AMP levels in bovine adrenal medullary cells. Br J Pharmacol 104:839–846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marshall BJ, Goodwin CS, Warren JR, Murray R, Blincow ED, Blackbourn SJ, Phillips M, Waters TE, Sanderson CR. (1988) Prospective double-blind trial of duodenal ulcer relapse after eradication of Campylobacter pylori. Lancet 2:1437–1442. [DOI] [PubMed] [Google Scholar]
- Marshall BJ, Warren JR. (1984) Unidentified curved bacilli in the stomach of patients with gastritis and peptic ulceration. Lancet 1:1311–1315. [DOI] [PubMed] [Google Scholar]
- Marshall SF and Cherry JW (1955) Smooth muscle tumors of the alimentary canal. Surg Clin North Am Lahey Clinic No.:719–33. [DOI] [PubMed]
- Martinez-Mir MI, Pollard H, Moreau J, Arrang JM, Ruat M, Traiffort E, Schwartz JC, Palacios JM. (1990) Three histamine receptors (H1, H2 and H3) visualized in the brain of human and non-human primates. Brain Res 526:322–327. [DOI] [PubMed] [Google Scholar]
- Maruko T, Nakahara T, Sakamoto K, Saito M, Sugimoto N, Takuwa Y, Ishii K. (2005) Involvement of the βγ subunits of G proteins in the cAMP response induced by stimulation of the histamine H1 receptor. Naunyn Schmiedebergs Arch Pharmacol 372:153–159. [DOI] [PubMed] [Google Scholar]
- Masaki T, Yoshimatsu H. (2006) The hypothalamic H1 receptor: a novel therapeutic target for disrupting diurnal feeding rhythm and obesity. Trends Pharmacol Sci 27:279–284. [DOI] [PubMed] [Google Scholar]
- Massari NA, Medina VA, Cricco GP, Martinel Lamas DJ, Sambuco L, Pagotto R, Ventura C, Ciraolo PJ, Pignataro O, Bergoc RM, et al. (2013) Antitumor activity of histamine and clozapine in a mouse experimental model of human melanoma. J Dermatol Sci 72:252–262. [DOI] [PubMed] [Google Scholar]
- Massari NA, Medina VA, Martinel Lamas DJ, Cricco GP, Croci M, Sambuco L, Bergoc RM, Rivera ES. (2011) Role of H4 receptor in histamine-mediated responses in human melanoma. Melanoma Res 21:395–404. [DOI] [PubMed] [Google Scholar]
- Matsubara T, Moskowitz MA, Huang Z. (1992) UK-14,304, R(−)-alpha-methyl-histamine and SMS 201-995 block plasma protein leakage within dura mater by prejunctional mechanisms. Eur J Pharmacol 224:145–150. [DOI] [PubMed] [Google Scholar]
- Matsuda N, Teramae H, Futatsugi M, Takano K, Yamamoto S, Tomita K, Suzuki T, Yokoo H, Koike K, Hattori Y. (2010) Up-regulation of histamine H4 receptors contributes to splenic apoptosis in septic mice: counteraction of the antiapoptotic action of nuclear factor-kappaB. J Pharmacol Exp Ther 332:730–737. [DOI] [PubMed] [Google Scholar]
- Matsushita A, Seike M, Okawa H, Kadawaki Y, Ohtsu H. (2012) Advantages of histamine H4 receptor antagonist usage with H1 receptor antagonist for the treatment of murine allergic contact dermatitis. Exp Dermatol 21:714–715. [DOI] [PubMed] [Google Scholar]
- McCaman RE, Weinreich D. (1985) Histaminergic synaptic transmission in the cerebral ganglion of Aplysia. J Neurophysiol 53:1016–1037. [DOI] [PubMed] [Google Scholar]
- McCormick DA, Pape HC, Williamson A. (1991) Actions of norepinephrine in the cerebral cortex and thalamus: implications for function of the central noradrenergic system. Prog Brain Res 88:293–305. [DOI] [PubMed] [Google Scholar]
- McCormick DA, Williamson A. (1989) Convergence and divergence of neurotransmitter action in human cerebral cortex. Proc Natl Acad Sci USA 86:8098–8102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCormick DA, Williamson A. (1991) Modulation of neuronal firing mode in cat and guinea pig LGNd by histamine: possible cellular mechanisms of histaminergic control of arousal. J Neurosci 11:3188–3199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCreath G, Hall IP, Hill SJ. (1994) Agonist-induced desensitization of histamine H1 receptor-mediated inositol phospholipid hydrolysis in human umbilical vein endothelial cells. Br J Pharmacol 113:823–830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGaraughty S, Chu KL, Cowart MD, Brioni JD. (2012) Antagonism of supraspinal histamine H3 receptors modulates spinal neuronal activity in neuropathic rats. J Pharmacol Exp Ther 343:13–20. [DOI] [PubMed] [Google Scholar]
- McLeod RL, Rizzo CA, West RE, Jr, Aslanian R, McCormick K, Bryant M, Hsieh Y, Korfmacher W, Mingo GG, Varty L, et al. (2003) Pharmacological characterization of the novel histamine H3-receptor antagonist N-(3,5-dichlorophenyl)-N′-[[4-(1H-imidazol-4-ylmethyl)phenyl]-methyl]-urea (SCH 79687). J Pharmacol Exp Ther 305:1037–1044. [DOI] [PubMed] [Google Scholar]
- Medhurst AD, Atkins AR, Beresford IJ, Brackenborough K, Briggs MA, Calver AR, Cilia J, Cluderay JE, Crook B, Davis JB, et al. (2007) GSK189254, a novel H3 receptor antagonist that binds to histamine H3 receptors in Alzheimer’s disease brain and improves cognitive performance in preclinical models. J Pharmacol Exp Ther 321:1032–1045. [DOI] [PubMed] [Google Scholar]
- Megson AC, Walker EM, Hill SJ. (2001) Role of protein kinase Calpha in signaling from the histamine H1 receptor to the nucleus. Mol Pharmacol 59:1012–1021. [DOI] [PubMed] [Google Scholar]
- Meier G, Apelt J, Reichert U, Grassmann S, Ligneau X, Elz S, Leurquin F, Ganellin CR, Schwartz JC, Schunack W, et al. (2001) Influence of imidazole replacement in different structural classes of histamine H3-receptor antagonists. Eur J Pharm Sci 13:249–259. [DOI] [PubMed] [Google Scholar]
- Meng J, Ma X, Li M, Jia M, Luo X. (2008) Histamine H4 receptors regulate ACTH release in AtT-20 cells. Eur J Pharmacol 587:336–338. [DOI] [PubMed] [Google Scholar]
- Merlos M, Giral M, Balsa D, Ferrando R, Queralt M, Puigdemont A, García-Rafanell J, Forn J. (1997) Rupatadine, a new potent, orally active dual antagonist of histamine and platelet-activating factor (PAF). J Pharmacol Exp Ther 280:114–121. [PubMed] [Google Scholar]
- Meskanen K, Ekelund H, Laitinen J, Neuvonen PJ, Haukka J, Panula P, Ekelund J. (2013) A randomized clinical trial of histamine 2 receptor antagonism in treatment-resistant schizophrenia. J Clin Psychopharmacol 33:472–478. [DOI] [PubMed] [Google Scholar]
- Micallef S, Stark H, Sasse A. (2013) Polymorphisms and genetic linkage of histamine receptors. Life Sci 93:487–494. [DOI] [PubMed] [Google Scholar]
- Michel MC, Wieland T, Tsujimoto G. (2009) How reliable are G-protein-coupled receptor antibodies? Naunyn Schmiedebergs Arch Pharmacol 379:385–388. [DOI] [PubMed] [Google Scholar]
- Mirzahosseini A, Dalmadi B, Csutora P. (2013) Histamine receptor H4 regulates mast cell degranulation and IgE induced FcεRI upregulation in murine bone marrow-derived mast cells. Cell Immunol 283:38–44. [DOI] [PubMed] [Google Scholar]
- Mitchell J, Mayeenuddin LH. (1998) Purification, G protein activation, and partial amino acid sequence of a novel phospholipase C from squid photoreceptors. Biochemistry 37:9064–9072. [DOI] [PubMed] [Google Scholar]
- Miyazaki S, Imaizumi M, Onodera K. (1995) Effects of thioperamide, a histamine H3-receptor antagonist, on a scopolamine-induced learning deficit using an elevated plus-maze test in mice. Life Sci 57:2137–2144. [DOI] [PubMed] [Google Scholar]
- Mobarakeh JI, Takahashi K, Sakurada S, Kuramasu A, Yanai K. (2006) Enhanced antinociceptive effects of morphine in histamine H2 receptor gene knockout mice. Neuropharmacology 51:612–622. [DOI] [PubMed] [Google Scholar]
- Mobarakeh JI, Takahashi K, Sakurada S, Nishino S, Watanabe H, Kato M, Naghdi N, Yanai K. (2005) Enhanced antinociception by intracerebroventricularly administered orexin A in histamine H1 or H2 receptor gene knockout mice. Pain 118:254–262. [DOI] [PubMed] [Google Scholar]
- Mobarakeh JI, Takahashi K, Yanai K. (2009) Enhanced morphine-induced antinociception in histamine H3 receptor gene knockout mice. Neuropharmacology 57:409–414. [DOI] [PubMed] [Google Scholar]
- Moguilevsky N, Varsalona F, Noyer M, Gillard M, Guillaume JP, Garcia L, Szpirer C, Szpirer J, Bollen A. (1994) Stable expression of human H1-histamine-receptor cDNA in Chinese hamster ovary cells. Pharmacological characterisation of the protein, tissue distribution of messenger RNA and chromosomal localisation of the gene. Eur J Biochem 224:489–495. [DOI] [PubMed] [Google Scholar]
- Molderings GJ, Weissenborn G, Schlicker E, Likungu J, Göthert M. (1992) Inhibition of noradrenaline release from the sympathetic nerves of the human saphenous vein by presynaptic histamine H3 receptors. Naunyn Schmiedebergs Arch Pharmacol 346:46–50. [DOI] [PubMed] [Google Scholar]
- Molimard M, Diquet B, Benedetti MS. (2004) Comparison of pharmacokinetics and metabolism of desloratadine, fexofenadine, levocetirizine and mizolastine in humans. Fundam Clin Pharmacol 18:399–411. [DOI] [PubMed] [Google Scholar]
- Mommert S, Gschwandtner M, Koether B, Gutzmer R, Werfel T. (2012) Human memory Th17 cells express a functional histamine H4 receptor. Am J Pathol 180:177–185. [DOI] [PubMed] [Google Scholar]
- Monczor F, Fernandez N, Legnazzi BL, Riveiro ME, Baldi A, Shayo C, Davio C. (2003) Tiotidine, a histamine H2 receptor inverse agonist that binds with high affinity to an inactive G-protein-coupled form of the receptor. Experimental support for the cubic ternary complex model. Mol Pharmacol 64:512–520. [DOI] [PubMed] [Google Scholar]
- Moniri NH, Booth RG. (2004) Functional heterogeneity of histamine H1 receptors. Inflamm Res 53 (Suppl 1):S71–S72. [DOI] [PubMed] [Google Scholar]
- Moniri NH, Covington-Strachan D, Booth RG. (2004) Ligand-directed functional heterogeneity of histamine H1 receptors: novel dual-function ligands selectively activate and block H1-mediated phospholipase C and adenylyl cyclase signaling. J Pharmacol Exp Ther 311:274–281. [DOI] [PubMed] [Google Scholar]
- Moore AR, O’Keeffe ST. (1999) Drug-induced cognitive impairment in the elderly. Drugs Aging 15:15–28. [DOI] [PubMed] [Google Scholar]
- Moreno-Delgado D, Torrent A, Gómez-Ramírez J, de Esch I, Blanco I, Ortiz J. (2006) Constitutive activity of H3 autoreceptors modulates histamine synthesis in rat brain through the cAMP/PKA pathway. Neuropharmacology 51:517–523. [DOI] [PubMed] [Google Scholar]
- Moreno E, Hoffmann H, Gonzalez-Sepúlveda M, Navarro G, Casadó V, Cortés A, Mallol J, Vignes M, McCormick PJ, Canela EI, et al. (2011) Dopamine D1-histamine H3 receptor heteromers provide a selective link to MAPK signaling in GABAergic neurons of the direct striatal pathway. J Biol Chem 286:5846–5854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morgan RK, McAllister B, Cross L, Green DS, Kornfeld H, Center DM, Cruikshank WW. (2007) Histamine 4 receptor activation induces recruitment of FoxP3+ T cells and inhibits allergic asthma in a murine model. J Immunol 178:8081–8089. [DOI] [PubMed] [Google Scholar]
- Morisset S, Sasse A, Gbahou F, Héron A, Ligneau X, Tardivel-Lacombe J, Schwartz JC, Arrang JM. (2001) The rat H3 receptor: gene organization and multiple isoforms. Biochem Biophys Res Commun 280:75–80. [DOI] [PubMed] [Google Scholar]
- Morisset S, Traiffort E, Arrang JM, Schwartz JC. (2000) Changes in histamine H3 receptor responsiveness in mouse brain. J Neurochem 74:339–346. [DOI] [PubMed] [Google Scholar]
- Morse KL, Behan J, Laz TM, West RE, Jr, Greenfeder SA, Anthes JC, Umland S, Wan Y, Hipkin RW, Gonsiorek W, et al. (2001) Cloning and characterization of a novel human histamine receptor. J Pharmacol Exp Ther 296:1058–1066. [PubMed] [Google Scholar]
- Mowbray CE, Bell AS, Clarke NP, Collins M, Jones RM, Lane CA, Liu WL, Newman SD, Paradowski M, Schenck EJ, et al. (2011) Challenges of drug discovery in novel target space. The discovery and evaluation of PF-3893787: a novel histamine H4 receptor antagonist. Bioorg Med Chem Lett 21:6596–6602. [DOI] [PubMed] [Google Scholar]
- Munari L, Provensi G, Passani MB, Blandina P. (2013) Selective brain region activation by histamine H₃ receptor antagonist/inverse agonist ABT-239 enhances acetylcholine and histamine release and increases c-Fos expression. Neuropharmacology 70:131–140. [DOI] [PubMed] [Google Scholar]
- Murakami H, Sun-Wada GH, Matsumoto M, Nishi T, Wada Y, Futai M. (1999) Human histamine H2 receptor gene: multiple transcription initiation and tissue-specific expression. FEBS Lett 451:327–331. [DOI] [PubMed] [Google Scholar]
- Murota H, Katayama I. (2011) Assessment of antihistamines in the treatment of skin allergies. Curr Opin Allergy Clin Immunol 11:428–437. [DOI] [PubMed] [Google Scholar]
- Nagase T, Mizutani T, Ishikawa S, Sekino E, Sasaki T, Fujimura T, Ito S, Mitobe Y, Miyamoto Y, Yoshimoto R, et al. (2008) Synthesis, structure-activity relationships, and biological profiles of a quinazolinone class of histamine H3 receptor inverse agonists. J Med Chem 51:4780–4789. [DOI] [PubMed] [Google Scholar]
- Nakamura T, Itadani H, Hidaka Y, Ohta M, Tanaka K. (2000) Molecular cloning and characterization of a new human histamine receptor, HH4R. Biochem Biophys Res Commun 279:615–620. [DOI] [PubMed] [Google Scholar]
- Nakano Y, Takahashi Y, Ono R, Kurata Y, Kagawa Y, Kamei C. (2009) Role of histamine H4 receptor in allergic conjunctivitis in mice. Eur J Pharmacol 608:71–75. [DOI] [PubMed] [Google Scholar]
- Nalwalk JW, Hough LB. (1995) Importance of histamine H2 receptors in restraint-morphine interactions. Life Sci 57:PL153–PL158. [DOI] [PubMed] [Google Scholar]
- Nalwalk JW, Koch JE, Barke KE, Bodnar RJ, Hough LB. (1995) Modulation of morphine antinociception by the brain-penetrating H2 antagonist zolantidine: detailed characterization in five nociceptive test systems. Pharmacol Biochem Behav 50:421–429. [DOI] [PubMed] [Google Scholar]
- Nathan PJ, Boardley R, Scott N, Berges A, Maruff P, Sivananthan T, Upton N, Lowy MT, Nestor PJ, Lai R. (2013) The safety, tolerability, pharmacokinetics and cognitive effects of GSK239512, a selective histamine H₃ receptor antagonist in patients with mild to moderate Alzheimer’s disease: a preliminary investigation. Curr Alzheimer Res 10:240–251. [DOI] [PubMed] [Google Scholar]
- Nent E, Frommholz D, Gajda M, Bräuer R, Illges H. (2013) Histamine 4 receptor plays an important role in auto-antibody-induced arthritis. Int Immunol 25:437–443. [DOI] [PubMed] [Google Scholar]
- Neumann D, Beermann S, Burhenne H, Glage S, Hartwig C, Seifert R. (2013) The dual H3/4R antagonist thioperamide does not fully mimic the effects of the ‘standard’ H4R antagonist JNJ 7777120 in experimental murine asthma. Naunyn Schmiedebergs Arch Pharmacol 386:983–990. [DOI] [PubMed] [Google Scholar]
- Nguyen T, Shapiro DA, George SR, Setola V, Lee DK, Cheng R, Rauser L, Lee SP, Lynch KR, Roth BL, et al. (2001) Discovery of a novel member of the histamine receptor family. Mol Pharmacol 59:427–433. [DOI] [PubMed] [Google Scholar]
- Nicholson AN, Pascoe PA, Turner C, Ganellin CR, Greengrass PM, Casy AF, Mercer AD. (1991) Sedation and histamine H1-receptor antagonism: studies in man with the enantiomers of chlorpheniramine and dimethindene. Br J Pharmacol 104:270–276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nijmeijer S, Vischer HF, Rosethorne EM, Charlton SJ, Leurs R. (2012) Analysis of multiple histamine H₄ receptor compound classes uncovers Gαi protein- and β-arrestin2-biased ligands. Mol Pharmacol 82:1174–1182. [DOI] [PubMed] [Google Scholar]
- Nijmeijer S, Vischer HF, Sirci F, Schultes S, Engelhardt H, de Graaf C, Rosethorne EM, Charlton SJ, Leurs R. (2013) Detailed analysis of biased histamine H₄ receptor signalling by JNJ 7777120 analogues. Br J Pharmacol 170:78–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nonaka H, Otaki S, Ohshima E, Kono M, Kase H, Ohta K, Fukui H, Ichimura M. (1998) Unique binding pocket for KW-4679 in the histamine H1 receptor. Eur J Pharmacol 345:111–117. [DOI] [PubMed] [Google Scholar]
- Nordemann U, Wifling D, Schnell D, Bernhardt G, Stark H, Seifert R, Buschauer A. (2013) Luciferase reporter gene assay on human, murine and rat histamine H4 receptor orthologs: correlations and discrepancies between distal and proximal readouts. PLoS ONE 8:e73961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Norris DB, Gajtkowski GA, Rising TJ. (1984) Histamine H2-binding studies in the guinea-pig brain. Agents Actions 14:543–545. [DOI] [PubMed] [Google Scholar]
- Notcovich C, Diez F, Tubio MR, Baldi A, Kazanietz MG, Davio C, Shayo C. (2010) Histamine acting on H1 receptor promotes inhibition of proliferation via PLC, RAC, and JNK-dependent pathways. Exp Cell Res 316:401–411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nuutinen S, Karlstedt K, Aitta-Aho T, Korpi ER, Panula P. (2010) Histamine and H3 receptor-dependent mechanisms regulate ethanol stimulation and conditioned place preference in mice. Psychopharmacology (Berl) 208:75–86. [DOI] [PubMed] [Google Scholar]
- O’Reilly M, Alpert R, Jenkinson S, Gladue RP, Foo S, Trim S, Peter B, Trevethick M, Fidock M. (2002) Identification of a histamine H4 receptor on human eosinophils—role in eosinophil chemotaxis. J Recept Signal Transduct Res 22:431–448. [DOI] [PubMed] [Google Scholar]
- Obradovic T, Dobson GG, Shingaki T, Kungu T, Hidalgo IJ. (2007) Assessment of the first and second generation antihistamines brain penetration and role of P-glycoprotein. Pharm Res 24:318–327. [DOI] [PubMed] [Google Scholar]
- Oda T, Matsumoto S, Matsumoto M, Takasaki J, Kamohara M, Soga T, Hiyama H, Kobori M, Katoh M. (2005) Molecular cloning of monkey histamine H4 receptor. J Pharmacol Sci 98:319–322. [DOI] [PubMed] [Google Scholar]
- Oda T, Morikawa N, Saito Y, Masuho Y, Matsumoto S. (2000) Molecular cloning and characterization of a novel type of histamine receptor preferentially expressed in leukocytes. J Biol Chem 275:36781–36786. [DOI] [PubMed] [Google Scholar]
- Ohki E, Suzuki M, Aoe T, Ikawa Y, Negishi E, Ueno K. (2007) Expression of histamine H4 receptor in synovial cells from rheumatoid arthritic patients. Biol Pharm Bull 30:2217–2220. [DOI] [PubMed] [Google Scholar]
- Ohkubo T, Shibata M. (1995) ATP-sensitive K+ channels mediate regulation of substance P release via the prejunctional histamine H3 receptor. Eur J Pharmacol 277:45–49. [DOI] [PubMed] [Google Scholar]
- Ohsawa Y, Hirasawa N. (2012) The antagonism of histamine H1 and H4 receptors ameliorates chronic allergic dermatitis via anti-pruritic and anti-inflammatory effects in NC/Nga mice. Allergy 67:1014–1022. [DOI] [PubMed] [Google Scholar]
- Ohta K, Hayashi H, Mizuguchi H, Kagamiyama H, Fujimoto K, Fukui H. (1994) Site-directed mutagenesis of the histamine H1 receptor: roles of aspartic acid107, asparagine198 and threonine194. Biochem Biophys Res Commun 203:1096–1101. [DOI] [PubMed] [Google Scholar]
- Ohtsu H, Tanaka S, Terui T, Hori Y, Makabe-Kobayashi Y, Pejler G, Tchougounova E, Hellman L, Gertsenstein M, Hirasawa N, et al. (2001) Mice lacking histidine decarboxylase exhibit abnormal mast cells. FEBS Lett 502:53–56. [DOI] [PubMed] [Google Scholar]
- Olbe L, Carlsson E, Lindberg P. (2003) A proton-pump inhibitor expedition: the case histories of omeprazole and esomeprazole. Nat Rev Drug Discov 2:132–139. [DOI] [PubMed] [Google Scholar]
- Orange PR, Heath PR, Wright SR, Pearson RC. (1996a) Allelic variations of the human histamine H2 receptor gene. Neuroreport 7:1293–1296. [DOI] [PubMed] [Google Scholar]
- Orange PR, Heath PR, Wright SR, Ramchand CN, Kolkeiwicz L, Pearson RC. (1996b) Individuals with schizophrenia have an increased incidence of the H2R649G allele for the histamine H2 receptor gene. Mol Psychiatry 1:466–469. [PubMed] [Google Scholar]
- Orhan ME, Yüksel U, Bilgin F, Doğrul A. (2007) Comparison of the local anesthetic effects of chlorpheniramine, midazolam, lidocaine, and normal saline after intradermal injection. Med Sci Monit 13:PI7–PI11. [PubMed] [Google Scholar]
- Palacios JM, Wamsley JK, Kuhar MJ. (1981a) The distribution of histamine H1-receptors in the rat brain: an autoradiographic study. Neuroscience 6:15–37. [DOI] [PubMed] [Google Scholar]
- Palacios JM, Wamsley JK, Kuhar MJ. (1981b) GABA benzodiazepine and histamine-H1 receptors in the guinea pig cerebellum: effects of kainic acid injections studied by autoradiographic methods. Brain Res 214:155–162. [DOI] [PubMed] [Google Scholar]
- Panula P, Nuutinen S. (2013) The histaminergic network in the brain: basic organization and role in disease. Nat Rev Neurosci 14:472–487. [DOI] [PubMed] [Google Scholar]
- Panula P, Wasowicz K. (1993) Action of antiulcer drugs. Science 262:1454–1455, author reply 1454–1455. [DOI] [PubMed] [Google Scholar]
- Parmentier R, Ohtsu H, Djebbara-Hannas Z, Valatx JL, Watanabe T, Lin JS. (2002) Anatomical, physiological, and pharmacological characteristics of histidine decarboxylase knock-out mice: evidence for the role of brain histamine in behavioral and sleep-wake control. J Neurosci 22:7695–7711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Passani MB, Giannoni P, Bucherelli C, Baldi E, Blandina P. (2007) Histamine in the brain: beyond sleep and memory. Biochem Pharmacol 73:1113–1122. [DOI] [PubMed] [Google Scholar]
- Payne GW, Neuman RS. (1997) Effects of hypomagnesia on histamine H1 receptor-mediated facilitation of NMDA responses. Br J Pharmacol 121:199–204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pedarzani P, Storm JF. (1995) Protein kinase A-independent modulation of ion channels in the brain by cyclic AMP. Proc Natl Acad Sci USA 92:11716–11720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pertz HH, Elz S, Schunack W. (2004) Structure-activity relationships of histamine H1-receptor agonists. Mini Rev Med Chem 4:935–940. [DOI] [PubMed] [Google Scholar]
- Petit-Bertron AF, Machavoine F, Defresne MP, Gillard M, Chatelain P, Mistry P, Schneider E, Dy M. (2009) H4 histamine receptors mediate cell cycle arrest in growth factor-induced murine and human hematopoietic progenitor cells. PLoS ONE 4:e6504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei H, Jin CY, Viisanen H, You HJ, Pertovaara A. (2014) Histamine in the locus coeruleus promotes descending noradrenergic inhibition of neuropathic hypersensitivity. Pharmacol Res 90:58–66. [DOI] [PubMed] [Google Scholar]
- Picado C. (2006) Rupatadine: pharmacological profile and its use in the treatment of allergic disorders. Expert Opin Pharmacother 7:1989–2001. [DOI] [PubMed] [Google Scholar]
- Pillot C, Heron A, Cochois V, Tardivel-Lacombe J, Ligneau X, Schwartz JC, Arrang JM. (2002a) A detailed mapping of the histamine H3 receptor and its gene transcripts in rat brain. Neuroscience 114:173–193. [DOI] [PubMed] [Google Scholar]
- Pillot C, Ortiz J, Héron A, Ridray S, Schwartz JC, Arrang JM. (2002b) Ciproxifan, a histamine H3-receptor antagonist/inverse agonist, potentiates neurochemical and behavioral effects of haloperidol in the rat. J Neurosci 22:7272–7280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Portoghese PS. (2001) From models to molecules: opioid receptor dimers, bivalent ligands, and selective opioid receptor probes. J Med Chem 44:2259–2269. [DOI] [PubMed] [Google Scholar]
- Powell JR, Brody MJ. (1976) Participation of H1 and H2 histamine receptors in physiological vasodilator responses. Am J Physiol 231:1002–1009. [DOI] [PubMed] [Google Scholar]
- Prast H, Philippu A. (2001) Nitric oxide as modulator of neuronal function. Prog Neurobiol 64:51–68. [DOI] [PubMed] [Google Scholar]
- Preuss H, Ghorai P, Kraus A, Dove S, Buschauer A, Seifert R. (2007) Mutations of Cys-17 and Ala-271 in the human histamine H2 receptor determine the species selectivity of guanidine-type agonists and increase constitutive activity. J Pharmacol Exp Ther 321:975–982. [DOI] [PubMed] [Google Scholar]
- Raible DG, Lenahan T, Fayvilevich Y, Kosinski R, Schulman ES. (1994) Pharmacologic characterization of a novel histamine receptor on human eosinophils. Am J Respir Crit Care Med 149:1506–1511. [DOI] [PubMed] [Google Scholar]
- Randall WC, Anderson PS, Cresson EL, Hunt CA, Lyon TF, Rittle KE, Remy DC, Springer JP, Hirshfield JM, Hoogsteen K, et al. (1979) Synthesis, assignment of absolute configuration, and receptor binding studies relevant to the neuroleptic activities of a series of chiral 3-substituted cyproheptadine atropisomers. J Med Chem 22:1222–1230. [DOI] [PubMed] [Google Scholar]
- Raphy G, Watson RJ, Hannah D, Pegurier C, Ortmans I, Lock CJ, Knight RL, and Owen DA (2008) inventors, UCB Pharma, S.A., assignee. Preparation of novel 2-aminopyrimidine derivatives as histamine receptor H4 antagonists. Patent EP20060019518 20060919.
- Reher TM, Neumann D, Buschauer A, Seifert R. (2012) Incomplete activation of human eosinophils via the histamine H4-receptor: evidence for ligand-specific receptor conformations. Biochem Pharmacol 84:192–203. [DOI] [PubMed] [Google Scholar]
- Reid A, Wilson F, Dyke H, Price S, Cramp S. (2007a) Preparation of Aminopyrimidine Eenantiomers for Treatment of Inflammation, Cellzome UK Ltd., Stevenage, UK. [Google Scholar]
- Reid A, Wilson F, Dyke H, Price S, and Cramp S (2007b) inventors, Cellzome UK Ltd., assignee. Preparation of azetidine amino benzo[4,5]furo[3,2-d]pyrimidine derivatives as modulators of histamine H4 receptor for treatment of inflammatory disorders, EP1860109 (A1)
- Reid A, Wilson F, Dyke H, Price S, Cramp S.(2007c) Preparation of Benzo[4,5]furo[3,2-d]Pyrimidine Derivatives as Modulators of Histamine H4 Receptor for Treatment of Inflammatory Ddisorders, Cellzome UK Ltd., Stevenage, UK. [Google Scholar]
- Reiner PB, Kamondi A. (1994) Mechanisms of antihistamine-induced sedation in the human brain: H1 receptor activation reduces a background leakage potassium current. Neuroscience 59:579–588. [DOI] [PubMed] [Google Scholar]
- Remy DC, Rittle KE, Hunt CA, Anderson PS, Engelhardt EL, Clineschmidt BV, Scriabine A. (1977) (+)- and (−)-3-Methoxycyproheptadine. A comparative evaluation of the antiserotonin, antihistaminic, anticholinergic, and orexigenic properties with cyproheptadine. J Med Chem 20:1681–1684. [DOI] [PubMed] [Google Scholar]
- Richelson E. (1978) Tricyclic antidepressants block histamine H1 receptors of mouse neuroblastoma cells. Nature 274:176–177. [DOI] [PubMed] [Google Scholar]
- Ridolo E, Montagni M, Melli V, Braido F, Incorvaia C, Canonica GW. (2014) Pharmacotherapy of allergic rhinitis: current options and future perspectives. Expert Opin Pharmacother 15:73–83. [DOI] [PubMed] [Google Scholar]
- Rinne JO, Anichtchik OV, Eriksson KS, Kaslin J, Tuomisto L, Kalimo H, Röyttä M, Panula P. (2002) Increased brain histamine levels in Parkinson’s disease but not in multiple system atrophy. J Neurochem 81:954–960. [DOI] [PubMed] [Google Scholar]
- Rodriguez-Pena MS, Timmerman H, Leurs R. (2000) Modulation of histamine H2 receptor signalling by G-protein-coupled receptor kinase 2 and 3. Br J Pharmacol 131:1707–1715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosa AC, Grange C, Pini A, Katebe MA, Benetti E, Collino M, Miglio G, Bani D, Camussi G, Chazot PL, et al. (2013) Overexpression of histamine H4 receptors in the kidney of diabetic rat. Inflamm Res 62:357–365. [DOI] [PubMed] [Google Scholar]
- Rosethorne EM, Charlton SJ. (2011) Agonist-biased signaling at the histamine H4 receptor: JNJ7777120 recruits β-arrestin without activating G proteins. Mol Pharmacol 79:749–757. [DOI] [PubMed] [Google Scholar]
- Rossbach K, Nassenstein C, Gschwandtner M, Schnell D, Sander K, Seifert R, Stark H, Kietzmann M, Bäumer W. (2011) Histamine H1, H3 and H4 receptors are involved in pruritus. Neuroscience 190:89–102. [DOI] [PubMed] [Google Scholar]
- Rossbach K, Wendorff S, Sander K, Stark H, Gutzmer R, Werfel T, Kietzmann M, Bäumer W. (2009) Histamine H4 receptor antagonism reduces hapten-induced scratching behaviour but not inflammation. Exp Dermatol 18:57–63. [DOI] [PubMed] [Google Scholar]
- Rosse RB, Schwartz BL, Leighton MP, Davis RE, Deutsch SI. (1990) An open-label trial of milacemide in schizophrenia: an NMDA intervention strategy. Clin Neuropharmacol 13:348–354. [DOI] [PubMed] [Google Scholar]
- Rothberg MB, Herzig SJ, Pekow PS, Avrunin J, Lagu T, Lindenauer PK. (2013) Association between sedating medications and delirium in older inpatients. J Am Geriatr Soc 61:923–930. [DOI] [PubMed] [Google Scholar]
- Rouleau A, Héron A, Cochois V, Pillot C, Schwartz JC, Arrang JM. (2004) Cloning and expression of the mouse histamine H3 receptor: evidence for multiple isoforms. J Neurochem 90:1331–1338. [DOI] [PubMed] [Google Scholar]
- Rouleau A, Ligneau X, Tardivel-Lacombe J, Morisset S, Gbahou F, Schwartz JC, Arrang JM. (2002) Histamine H3-receptor-mediated [35S]GTP gamma[S] binding: evidence for constitutive activity of the recombinant and native rat and human H3 receptors. Br J Pharmacol 135:383–392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruat M, Schwartz JC. (1989) Photoaffinity labeling and electrophoretic identification of the H1-receptor: comparison of several brain regions and animal species. J Neurochem 53:335–339. [DOI] [PubMed] [Google Scholar]
- Ruat M, Traiffort E, Arrang JM, Leurs R, Schwartz JC. (1991) Cloning and tissue expression of a rat histamine H2-receptor gene. Biochem Biophys Res Commun 179:1470–1478. [DOI] [PubMed] [Google Scholar]
- Ruat M, Traiffort E, Bouthenet ML, Schwartz JC, Hirschfeld J, Buschauer A, Schunack W. (1990) Reversible and irreversible labeling and autoradiographic localization of the cerebral histamine H2 receptor using [125I]iodinated probes. Proc Natl Acad Sci USA 87:1658–1662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruat M, Traiffort E, Garbarg M, Schwartz JC, Demonchaux P, Ife RJ, Tertiuk W, Ganellin CR. (1992) Design of an affinity matrix for purification of the histamine H1 receptor from guinea pig cerebellum. J Neurochem 58:350–356. [DOI] [PubMed] [Google Scholar]
- Sadek B, Kuder K, Subramanian D, Shafiullah M, Stark H, Lażewska D, Adem A, Kieć-Kononowicz K. (2014a) Anticonvulsive effect of nonimidazole histamine H3 receptor antagonists. Behav Pharmacol 25:245–252. [DOI] [PubMed] [Google Scholar]
- Sadek B, Schwed JS, Subramanian D, Weizel L, Walter M, Adem A, Stark H. (2014b) Non-imidazole histamine H3 receptor ligands incorporating antiepileptic moieties. Eur J Med Chem 77:269–79. [DOI] [PubMed] [Google Scholar]
- Saitoh T, Fukushima Y, Otsuka H, Ishikawa M, Tamai M, Takahashi H, Mori H, Asano T, Anai M, Ishikawa T, et al. (2002) Effects of N-alpha-methyl-histamine on human H2 receptors expressed in CHO cells. Gut 50:786–789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sakuma I, Gross SS, Levi R. (1988) Positive inotropic effect of histamine on guinea pig left atrium: H1-receptor-induced stimulation of phosphoinositide turnover. J Pharmacol Exp Ther 247:466–472. [PubMed] [Google Scholar]
- Salata JJ, Jurkiewicz NK, Wallace AA, Stupienski RF, 3rd, Guinosso PJ, Jr, Lynch JJ., Jr (1995) Cardiac electrophysiological actions of the histamine H1-receptor antagonists astemizole and terfenadine compared with chlorpheniramine and pyrilamine. Circ Res 76:110–119. [DOI] [PubMed] [Google Scholar]
- Saligrama N, Case LK, del Rio R, Noubade R, Teuscher C. (2013) Systemic lack of canonical histamine receptor signaling results in increased resistance to autoimmune encephalomyelitis. J Immunol 191:614–622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saligrama N, Noubade R, Case LK, del Rio R, Teuscher C. (2012) Combinatorial roles for histamine H1-H2 and H3-H4 receptors in autoimmune inflammatory disease of the central nervous system. Eur J Immunol 42:1536–1546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sallmen T, Beckman AL, Stanton TL, Eriksson KS, Tarhanen J, Tuomisto L, Panula P. (1999) Major changes in the brain histamine system of the ground squirrel Citellus lateralis during hibernation. J Neurosci 19:1824–1835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sallmen T, Lozada AF, Anichtchik OV, Beckman AL, Panula P. (2003a) Increased brain histamine H3 receptor expression during hibernation in golden-mantled ground squirrels. BMC Neurosci 4:24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sallmen T, Lozada AF, Beckman AL, Panula P. (2003b) Intrahippocampal histamine delays arousal from hibernation. Brain Res 966:317–320. [DOI] [PubMed] [Google Scholar]
- Salmun LM. (2002) Antihistamines in late-phase clinical development for allergic disease. Expert Opin Investig Drugs 11:259–273. [DOI] [PubMed] [Google Scholar]
- Sánchez-Lemus E, Arias-Montaño JA. (2004) Histamine H3 receptor activation inhibits dopamine D1 receptor-induced cAMP accumulation in rat striatal slices. Neurosci Lett 364:179–184. [DOI] [PubMed] [Google Scholar]
- Sander K, Kottke T, Stark H. (2008) Histamine H3 receptor antagonists go to clinics. Biol Pharm Bull 31:2163–2181. [DOI] [PubMed] [Google Scholar]
- Sander K, Kottke T, Tanrikulu Y, Proschak E, Weizel L, Schneider EH, Seifert R, Schneider G, Stark H. (2009) 2,4-Diaminopyrimidines as histamine H4 receptor ligands—Scaffold optimization and pharmacological characterization. Bioorg Med Chem 17:7186–7196. [DOI] [PubMed] [Google Scholar]
- Sander LE, Lorentz A, Sellge G, Coëffier M, Neipp M, Veres T, Frieling T, Meier PN, Manns MP, Bischoff SC. (2006) Selective expression of histamine receptors H1R, H2R, and H4R, but not H3R, in the human intestinal tract. Gut 55:498–504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sansuk K, Deupi X, Torrecillas IR, Jongejan A, Nijmeijer S, Bakker RA, Pardo L, Leurs R. (2011) A structural insight into the reorientation of transmembrane domains 3 and 5 during family A G protein-coupled receptor activation. Mol Pharmacol 79:262–269. [DOI] [PubMed] [Google Scholar]
- Santora VJ, Covel JA, Hayashi R, Hofilena BJ, Ibarra JB, Pulley MD, Weinhouse MI, Sengupta D, Duffield JJ, Semple G, et al. (2008) A new family of H3 receptor antagonists based on the natural product Conessine. Bioorg Med Chem Lett 18:1490–1494. [DOI] [PubMed] [Google Scholar]
- Saras A, Gisselmann G, Vogt-Eisele AK, Erlkamp KS, Kletke O, Pusch H, Hatt H. (2008) Histamine action on vertebrate GABAA receptors: direct channel gating and potentiation of GABA responses. J Biol Chem 283:10470–10475. [DOI] [PubMed] [Google Scholar]
- Sato H, Ito C, Tashiro M, Hiraoka K, Shibuya K, Funaki Y, Iwata R, Matsuoka H, Yanai K. (2013) Histamine H₁ receptor occupancy by the new-generation antidepressants fluvoxamine and mirtazapine: a positron emission tomography study in healthy volunteers. Psychopharmacology (Berl) 230:227–234. [DOI] [PubMed] [Google Scholar]
- Sato H, Tanaka K, Shimazaki M, Urbahns K, Sakai K, Gantner F, and Bacon K (2005) inventors, Bayer Healthcare A.G., assignee. Preparation of 2-aminopyrimidine derivatives as histamine H4 antagonists for the treatment of asthma, WO2004EP13322 20041124.
- Savall BM, Chavez F, Tays K, Dunford PJ, Cowden JM, Hack MD, Wolin RL, Thurmond RL, Edwards JP. (2014) Discovery and SAR of 6-alkyl-2,4-diaminopyrimidines as histamine H₄ receptor antagonists. J Med Chem 57:2429–2439. [DOI] [PubMed] [Google Scholar]
- Saybasili H, Stevens DR, Haas HL. (1995) pH-dependent modulation of N-methyl-D-aspartate receptor-mediated synaptic currents by histamine in rat hippocampus in vitro. Neurosci Lett 199:225–227. [DOI] [PubMed] [Google Scholar]
- Scannell RT, Differding E, Talaga P. (2004) Dual acting antihistaminergic agents. Mini Rev Med Chem 4:923–933. [DOI] [PubMed] [Google Scholar]
- Schlegel B, Laggner C, Meier R, Langer T, Schnell D, Seifert R, Stark H, Höltje HD, Sippl W. (2007) Generation of a homology model of the human histamine H3 receptor for ligand docking and pharmacophore-based screening. J Comput Aided Mol Des 21:437–453. [DOI] [PubMed] [Google Scholar]
- Schlicker E, Behling A, Lümmen G, Göthert M. (1992) Histamine H3A receptor-mediated inhibition of noradrenaline release in the mouse brain cortex. Naunyn Schmiedebergs Arch Pharmacol 345:489–493. [DOI] [PubMed] [Google Scholar]
- Schlicker E, Betz R, Göthert M. (1988) Histamine H3 receptor-mediated inhibition of serotonin release in the rat brain cortex. Naunyn Schmiedebergs Arch Pharmacol 337:588–590. [DOI] [PubMed] [Google Scholar]
- Schlicker E, Fink K, Detzner M, Göthert M. (1993) Histamine inhibits dopamine release in the mouse striatum via presynaptic H3 receptors. J Neural Transm 93:1–10. [DOI] [PubMed] [Google Scholar]
- Schlicker E, Fink K, Hinterthaner M, Göthert M. (1989) Inhibition of noradrenaline release in the rat brain cortex via presynaptic H3 receptors. Naunyn Schmiedebergs Arch Pharmacol 340:633–638. [DOI] [PubMed] [Google Scholar]
- Schlicker E, Kathmann M, Detzner M, Exner HJ, Göthert M. (1994) H3 receptor-mediated inhibition of noradrenaline release: an investigation into the involvement of Ca2+ and K+ ions, G protein and adenylate cyclase. Naunyn Schmiedebergs Arch Pharmacol 350:34–41. [DOI] [PubMed] [Google Scholar]
- Schlicker E, Werthwein S, Zentner J. (1999) Histamine H3 receptor-mediated inhibition of noradrenaline release in the human brain. Fundam Clin Pharmacol 13:120–122. [DOI] [PubMed] [Google Scholar]
- Schneider EH, Schnell D, Papa D, Seifert R. (2009) High constitutive activity and a G-protein-independent high-affinity state of the human histamine H4-receptor. Biochemistry 48:1424–1438. [DOI] [PubMed] [Google Scholar]
- Schneider EH, Schnell D, Strasser A, Dove S, Seifert R. (2010a) Impact of the DRY motif and the missing “ionic lock” on constitutive activity and G-protein coupling of the human histamine H4 receptor. J Pharmacol Exp Ther 333:382–392. [DOI] [PubMed] [Google Scholar]
- Schneider EH, Strasser A, Thurmond RL, Seifert R. (2010b) Structural requirements for inverse agonism and neutral antagonism of indole-, benzimidazole-, and thienopyrrole-derived histamine H4 receptor ligands. J Pharmacol Exp Ther 334:513–521. [DOI] [PubMed] [Google Scholar]
- Schneider EH, Neumann D, Seifert R. (2014a) Modulation of behavior by the histaminergic system: lessons from H1R-and H2R-deficient mice. Neurosci Biobehav Rev 42:252–266. [DOI] [PubMed] [Google Scholar]
- Schneider EH, Neumann D, Seifert R. (2014b) Modulation of behavior by the histaminergic system: lessons from HDC-, H3R- and H4R-deficient mice. Neurosci Biobehav Rev 47:101–121. [DOI] [PubMed] [Google Scholar]
- Schnell D, Brunskole I, Ladova K, Schneider EH, Igel P, Dove S, Buschauer A, Seifert R. (2011) Expression and functional properties of canine, rat, and murine histamine H₄ receptors in Sf9 insect cells. Naunyn Schmiedebergs Arch Pharmacol 383:457–470. [DOI] [PubMed] [Google Scholar]
- Schnell D, Strasser A, Seifert R. (2010) Comparison of the pharmacological properties of human and rat histamine H3-receptors. Biochem Pharmacol 80:1437–1449. [DOI] [PubMed] [Google Scholar]
- Schwartz JC. (2011) The histamine H3 receptor: from discovery to clinical trials with pitolisant. Br J Pharmacol 163:713–721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwartz JC, Arrang JM, Garbarg M, Pollard H, Ruat M. (1991) Histaminergic transmission in the mammalian brain. Physiol Rev 71:1–51. [DOI] [PubMed] [Google Scholar]
- Seifert R, Hagelüken A, Höer A, Höer D, Grünbaum L, Offermanns S, Schwaner I, Zingel V, Schunack W, Schultz G. (1994) The H1 receptor agonist 2-(3-chlorophenyl)histamine activates Gi proteins in HL-60 cells through a mechanism that is independent of known histamine receptor subtypes. Mol Pharmacol 45:578–586. [PubMed] [Google Scholar]
- Seifert R, Schneider EH, Dove S, Brunskole I, Neumann D, Strasser A, Buschauer A. (2011) Paradoxical stimulatory effects of the “standard” histamine H4-receptor antagonist JNJ7777120: the H4 receptor joins the club of 7 transmembrane domain receptors exhibiting functional selectivity. Mol Pharmacol 79:631–638. [DOI] [PubMed] [Google Scholar]
- Seike M, Furuya K, Omura M, Hamada-Watanabe K, Matsushita A, Ohtsu H. (2010) Histamine H4 receptor antagonist ameliorates chronic allergic contact dermatitis induced by repeated challenge. Allergy 65:319–326. [DOI] [PubMed] [Google Scholar]
- Selbach O, Brown RE, Haas HL. (1997) Long-term increase of hippocampal excitability by histamine and cyclic AMP. Neuropharmacology 36:1539–1548. [DOI] [PubMed] [Google Scholar]
- Selivanova SV, Honer M, Combe F, Isensee K, Stark H, Krämer SD, Schubiger PA, Ametamey SM. (2012) Radiofluorinated histamine H3 receptor antagonist as a potential probe for in vivo PET imaging: radiosynthesis and pharmacological evaluation. Bioorg Med Chem 20:2889–2896. [DOI] [PubMed] [Google Scholar]
- Sergeeva OA, Amberger BT, Eriksson KS, Scherer A, Haas HL. (2003) Co-ordinated expression of 5-HT2C receptors with the NCX1 Na+/Ca2+ exchanger in histaminergic neurones. J Neurochem 87:657–664. [DOI] [PubMed] [Google Scholar]
- Sharif NA, Xu SX, Yanni JM. (1996) Olopatadine (AL-4943A): ligand binding and functional studies on a novel, long acting H1-selective histamine antagonist and anti-allergic agent for use in allergic conjunctivitis. J Ocul Pharmacol Ther 12:401–407. [DOI] [PubMed] [Google Scholar]
- Shimamura T, Shiroishi M, Weyand S, Tsujimoto H, Winter G, Katritch V, Abagyan R, Cherezov V, Liu W, Han GW, et al. (2011) Structure of the human histamine H1 receptor complex with doxepin. Nature 475:65–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shin N, Coates E, Murgolo NJ, Morse KL, Bayne M, Strader CD, Monsma FJ., Jr (2002) Molecular modeling and site-specific mutagenesis of the histamine-binding site of the histamine H4 receptor. Mol Pharmacol 62:38–47. [DOI] [PubMed] [Google Scholar]
- Shin N, Covington M, Bian D, Zhuo J, Bowman K, Li Y, Soloviev M, Qian DQ, Feldman P, Leffet L, et al. (2012) INCB38579, a novel and potent histamine H₄ receptor small molecule antagonist with anti-inflammatory pain and anti-pruritic functions. Eur J Pharmacol 675:47–56. [DOI] [PubMed] [Google Scholar]
- Sigterman KE, van Pinxteren B, Bonis PA, Lau J, Numans ME. (2013) Short-term treatment with proton pump inhibitors, H2-receptor antagonists and prokinetics for gastro-oesophageal reflux disease-like symptoms and endoscopy negative reflux disease. Cochrane Database Syst Rev 5:CD002095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silver RB, Mackins CJ, Smith NC, Koritchneva IL, Lefkowitz K, Lovenberg TW, Levi R. (2001) Coupling of histamine H3 receptors to neuronal Na+/H+ exchange: a novel protective mechanism in myocardial ischemia. Proc Natl Acad Sci USA 98:2855–2859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silver RB, Poonwasi KS, Seyedi N, Wilson SJ, Lovenberg TW, Levi R. (2002) Decreased intracellular calcium mediates the histamine H3-receptor-induced attenuation of norepinephrine exocytosis from cardiac sympathetic nerve endings. Proc Natl Acad Sci USA 99:501–506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simon T, Semsei AF, Ungvári I, Hadadi E, Virág V, Nagy A, Vangor MS, László V, Szalai C, Falus A. (2012) Asthma endophenotypes and polymorphisms in the histamine receptor HRH4 gene. Int Arch Allergy Immunol 159:109–120. [DOI] [PubMed] [Google Scholar]
- Simons FE. (1999) Is antihistamine (H1-receptor antagonist) therapy useful in clinical asthma? Clin Exp Allergy 29 (Suppl 3):98–104. [DOI] [PubMed] [Google Scholar]
- Simons FE, Simons KJ. (1999) Clinical pharmacology of new histamine H1 receptor antagonists. Clin Pharmacokinet 36:329–352. [DOI] [PubMed] [Google Scholar]
- Simons FE, Simons KJ. (2011) Histamine and H1-antihistamines: celebrating a century of progress. J Allergy Clin Immunol 128:1139–1150. [DOI] [PubMed] [Google Scholar]
- Simpson K, Jarvis B. (2000) Fexofenadine: a review of its use in the management of seasonal allergic rhinitis and chronic idiopathic urticaria. Drugs 59:301–321. [DOI] [PubMed] [Google Scholar]
- Smit MJ, Bloemers SM, Leurs R, Tertoolen LG, Bast A, de Laat SW, Timmerman H. (1992) Short-term desensitization of the histamine H1 receptor in human HeLa cells: involvement of protein kinase C dependent and independent pathways. Br J Pharmacol 107:448–455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smit MJ, Leurs R, Alewijnse AE, Blauw J, Van Nieuw Amerongen GP, Van De Vrede Y, Roovers E, Timmerman H. (1996a) Inverse agonism of histamine H2 antagonist accounts for upregulation of spontaneously active histamine H2 receptors. Proc Natl Acad Sci USA 93:6802–6807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smit MJ, Timmerman H, Hijzelendoorn JC, Fukui H, Leurs R. (1996b) Regulation of the human histamine H1 receptor stably expressed in Chinese hamster ovary cells. Br J Pharmacol 117:1071–1080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smit MJ, Vischer HF, Bakker RA, Jongejan A, Timmerman H, Pardo L, Leurs R. (2007) Pharmacogenomic and structural analysis of constitutive G protein-coupled receptor activity. Annu Rev Pharmacol Toxicol 47:53–87. [DOI] [PubMed] [Google Scholar]
- Smits RA, Lim HD, Hanzer A, Zuiderveld OP, Guaita E, Adami M, Coruzzi G, Leurs R, de Esch IJ. (2008) Fragment based design of new H4 receptor-ligands with anti-inflammatory properties in vivo. J Med Chem 51:2457–2467. [DOI] [PubMed] [Google Scholar]
- Snider RM, McKinney M, Forray C, Richelson E. (1984) Neurotransmitter receptors mediate cyclic GMP formation by involvement of arachidonic acid and lipoxygenase. Proc Natl Acad Sci USA 81:3905–3909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Somma T, Cinci L, Formicola G, Pini A, Thurmond R, Ennis M, Bani D, Masini E. (2013) A selective antagonist of histamine H₄ receptors prevents antigen-induced airway inflammation and bronchoconstriction in guinea pigs: involvement of lipocortin-1. Br J Pharmacol 170:200–213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sonkoly E, Muller A, Lauerma AI, Pivarcsi A, Soto H, Kemeny L, Alenius H, Dieu-Nosjean MC, Meller S, Rieker J, et al. (2006) IL-31: a new link between T cells and pruritus in atopic skin inflammation. J Allergy Clin Immunol 117:411–417. [DOI] [PubMed] [Google Scholar]
- Soria-Jasso LE, Bahena-Trujillo R, Arias-Montaño JA. (1997) Histamine H1 receptors and inositol phosphate formation in rat thalamus. Neurosci Lett 225:117–120. [DOI] [PubMed] [Google Scholar]
- Stark H. (2013) Histamine H4 receptor. A novel drug target for immunoregulation and inflammation, Versita-deGruyter, London. [Google Scholar]
- Stark H, Ligneau X, Sadek B, Ganellin CR, Arrang JM, Schwartz JC, Schunack W. (2000) Analogues and derivatives of ciproxifan, a novel prototype for generating potent histamine H3-receptor antagonists. Bioorg Med Chem Lett 10:2379–2382. [DOI] [PubMed] [Google Scholar]
- Stark H, Purand K, Hüls A, Ligneau X, Garbarg M, Schwartz JC, Schunack W. (1996) [125I]iodoproxyfan and related compounds: a reversible radioligand and novel classes of antagonists with high affinity and selectivity for the histamine H3 receptor. J Med Chem 39:1220–1226. [DOI] [PubMed] [Google Scholar]
- Stark H, Sippl W, Ligneau X, Arrang JM, Ganellin CR, Schwartz JC, Schunack W. (2001) Different antagonist binding properties of human and rat histamine H3 receptors. Bioorg Med Chem Lett 11:951–954. [DOI] [PubMed] [Google Scholar]
- Stevens DR, Eriksson KS, Brown RE, Haas HL. (2001) The mechanism of spontaneous firing in histamine neurons. Behav Brain Res 124:105–112. [DOI] [PubMed] [Google Scholar]
- Stokes JR, Romero FA, Jr, Allan RJ, Phillips PG, Hackman F, Misfeldt J, Casale TB. (2012) The effects of an H3 receptor antagonist (PF-03654746) with fexofenadine on reducing allergic rhinitis symptoms. J Allergy Clin Immunol 129:409–412. [DOI] [PubMed] [Google Scholar]
- Strakhova MI, Cuff CA, Manelli AM, Carr TL, Witte DG, Baranowski JL, Vortherms TA, Miller TR, Rundell L, McPherson MJ, et al. (2009a) In vitro and in vivo characterization of A-940894: a potent histamine H4 receptor antagonist with anti-inflammatory properties. Br J Pharmacol 157:44–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strakhova MI, Nikkel AL, Manelli AM, Hsieh GC, Esbenshade TA, Brioni JD, Bitner RS. (2009b) Localization of histamine H4 receptors in the central nervous system of human and rat. Brain Res 1250:41–48. [DOI] [PubMed] [Google Scholar]
- Strasser A, Striegl B, Wittmann HJ, Seifert R. (2008a) Pharmacological profile of histaprodifens at four recombinant histamine H1 receptor species isoforms. J Pharmacol Exp Ther 324:60–71. [DOI] [PubMed] [Google Scholar]
- Strasser A, Wittmann HJ. (2007) Analysis of the activation mechanism of the guinea-pig Histamine H1-receptor. J Comput Aided Mol Des 21:499–509. [DOI] [PubMed] [Google Scholar]
- Strasser A, Wittmann HJ, Kunze M, Elz S, Seifert R. (2009) Molecular basis for the selective interaction of synthetic agonists with the human histamine H1-receptor compared with the guinea pig H1-receptor. Mol Pharmacol 75:454–465. [DOI] [PubMed] [Google Scholar]
- Strasser A, Wittmann HJ, Seifert R. (2008b) Ligand-specific contribution of the N terminus and E2-loop to pharmacological properties of the histamine H1-receptor. J Pharmacol Exp Ther 326:783–791. [DOI] [PubMed] [Google Scholar]
- Sun P, Takatori S, Jin X, Koyama T, Tangsucharit P, Li S, Zamami Y, Kitamura Y, Kawasaki H. (2011) Histamine H3 receptor-mediated modulation of perivascular nerve transmission in rat mesenteric arteries. Eur J Pharmacol 655:67–73. [DOI] [PubMed] [Google Scholar]
- Sunahara RK, Taussig R. (2002) Isoforms of mammalian adenylyl cyclase: multiplicities of signaling. Mol Interv 2:168–184. [DOI] [PubMed] [Google Scholar]
- Tabarean IV. (2013) Functional pharmacology of H1 histamine receptors expressed in mouse preoptic/anterior hypothalamic neurons. Br J Pharmacol 170:415–425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takahashi Y, Kagawa Y, Izawa K, Ono R, Akagi M, Kamei C. (2009) Effect of histamine H4 receptor antagonist on allergic rhinitis in mice. Int Immunopharmacol 9:734–738. [DOI] [PubMed] [Google Scholar]
- Takeshita K, Sakai K, Bacon KB, Gantner F. (2003) Critical role of histamine H4 receptor in leukotriene B4 production and mast cell-dependent neutrophil recruitment induced by zymosan in vivo. J Pharmacol Exp Ther 307:1072–1078. [DOI] [PubMed] [Google Scholar]
- Takeuchi K, Kajimura M, Kodaira M, Lin S, Hanai H, Kaneko E. (1999) Up-regulation of H2 receptor and adenylate cyclase in rabbit parietal cells during prolonged treatment with H2-receptor antagonists. Dig Dis Sci 44:1703–1709. [DOI] [PubMed] [Google Scholar]
- Tardivel-Lacombe J, Morisset S, Gbahou F, Schwartz JC, Arrang JM. (2001) Chromosomal mapping and organization of the human histamine H3 receptor gene. Neuroreport 12:321–324. [DOI] [PubMed] [Google Scholar]
- Tardivel-Lacombe J, Rouleau A, Héron A, Morisset S, Pillot C, Cochois V, Schwartz JC, Arrang JM. (2000) Cloning and cerebral expression of the guinea pig histamine H3 receptor: evidence for two isoforms. Neuroreport 11:755–759. [DOI] [PubMed] [Google Scholar]
- Tashiro M, Yanai K. (2007) Molecular imaging of histamine receptors in the human brain. Brain Nerve 59:221–231. [PubMed] [Google Scholar]
- Tedford CE, Phillips JG, Gregory R, Pawlowski GP, Fadnis L, Khan MA, Ali SM, Handley MK, Yates SL. (1999) Development of trans-2-[1H-imidazol-4-yl] cyclopropane derivatives as new high-affinity histamine H3 receptor ligands. J Pharmacol Exp Ther 289:1160–1168. [PubMed] [Google Scholar]
- Terzioglu N, van Rijn RM, Bakker RA, De Esch IJ, Leurs R. (2004) Synthesis and structure-activity relationships of indole and benzimidazole piperazines as histamine H4 receptor antagonists. Bioorg Med Chem Lett 14:5251–5256. [DOI] [PubMed] [Google Scholar]
- Teuscher C, Poynter ME, Offner H, Zamora A, Watanabe T, Fillmore PD, Zachary JF, Blankenhorn EP. (2004) Attenuation of Th1 effector cell responses and susceptibility to experimental allergic encephalomyelitis in histamine H2 receptor knockout mice is due to dysregulation of cytokine production by antigen-presenting cells. Am J Pathol 164:883–892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Teuscher C, Subramanian M, Noubade R, Gao JF, Offner H, Zachary JF, Blankenhorn EP. (2007) Central histamine H3 receptor signaling negatively regulates susceptibility to autoimmune inflammatory disease of the CNS. Proc Natl Acad Sci USA 104:10146–10151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Threlfell S, Cragg SJ, Kalló I, Turi GF, Coen CW, Greenfield SA. (2004) Histamine H3 receptors inhibit serotonin release in substantia nigra pars reticulata. J Neurosci 24:8704–8710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thurmond RL, Chen B, Dunford PJ, Greenspan AJ, Karlsson L, La D, Ward P, Xu XL. (2014a) Clinical and preclinical characterization of the histamine H4 receptor antagonist JNJ-39758979. J Pharmacol Exp Ther 349:176–184. [DOI] [PubMed] [Google Scholar]
- Thurmond RL, Desai PJ, Dunford PJ, Fung-Leung WP, Hofstra CL, Jiang W, Nguyen S, Riley JP, Sun S, Williams KN, et al. (2004) A potent and selective histamine H4 receptor antagonist with anti-inflammatory properties. J Pharmacol Exp Ther 309:404–413. [DOI] [PubMed] [Google Scholar]
- Thurmond RL, Gelfand EW, Dunford PJ. (2008) The role of histamine H1 and H4 receptors in allergic inflammation: the search for new antihistamines. Nat Rev Drug Discov 7:41–53. [DOI] [PubMed] [Google Scholar]
- Thurmond RL, Kazerouni K, Chaplan SR, Greenspan AJ. (2014b) Peripheral Neuronal Mechanism of Itch: Histamine and Itch, in Itch: Mechanisms and Treatment (Carstens E, Akiyama T, eds), CRC Press, Boca Raton, FL. [PubMed] [Google Scholar]
- Tiligada E, Zampeli E, Sander K, Stark H. (2009) Histamine H3 and H4 receptors as novel drug targets. Expert Opin Investig Drugs 18:1519–1531. [DOI] [PubMed] [Google Scholar]
- Toftegaard CL, Knigge U, Kjaer A, Warberg J. (2003) The role of hypothalamic histamine in leptin-induced suppression of short-term food intake in fasted rats. Regul Pept 111:83–90. [DOI] [PubMed] [Google Scholar]
- Togias A. (2003) H1-receptors: localization and role in airway physiology and in immune functions. J Allergy Clin Immunol 112(4, Suppl)S60–S68. [DOI] [PubMed] [Google Scholar]
- Torrent A, Moreno-Delgado D, Gómez-Ramírez J, Rodríguez-Agudo D, Rodríguez-Caso C, Sánchez-Jiménez F, Blanco I, Ortiz J. (2005) H3 autoreceptors modulate histamine synthesis through calcium/calmodulin- and cAMP-dependent protein kinase pathways. Mol Pharmacol 67:195–203. [DOI] [PubMed] [Google Scholar]
- Toyota H, Dugovic C, Koehl M, Laposky AD, Weber C, Ngo K, Wu Y, Lee DH, Yanai K, Sakurai E, et al. (2002) Behavioral characterization of mice lacking histamine H3 receptors. Mol Pharmacol 62:389–397. [DOI] [PubMed] [Google Scholar]
- Traiffort E, Leurs R, Arrang JM, Tardivel-Lacombe J, Diaz J, Schwartz JC, Ruat M. (1994) Guinea pig histamine H1 receptor. I. Gene cloning, characterization, and tissue expression revealed by in situ hybridization. J Neurochem 62:507–518. [DOI] [PubMed] [Google Scholar]
- Traiffort E, Pollard H, Moreau J, Ruat M, Schwartz JC, Martinez-Mir MI, Palacios JM. (1992a) Pharmacological characterization and autoradiographic localization of histamine H2 receptors in human brain identified with [125I]iodoaminopotentidine. J Neurochem 59:290–299. [DOI] [PubMed] [Google Scholar]
- Traiffort E, Ruat M, Arrang JM, Leurs R, Piomelli D, Schwartz JC. (1992b) Expression of a cloned rat histamine H2 receptor mediating inhibition of arachidonate release and activation of cAMP accumulation. Proc Natl Acad Sci USA 89:2649–2653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Traiffort E, Vizuete ML, Tardivel-Lacombe J, Souil E, Schwartz JC, Ruat M. (1995) The guinea pig histamine H2 receptor: gene cloning, tissue expression and chromosomal localization of its human counterpart. Biochem Biophys Res Commun 211:570–577. [DOI] [PubMed] [Google Scholar]
- Tran VT, Chang RS, Snyder SH. (1978) Histamine H1 receptors identified in mammalian brain membranes with [3H]mepyramine. Proc Natl Acad Sci USA 75:6290–6294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Truta-Feles K, Lagadari M, Lehmann K, Berod L, Cubillos S, Piehler S, Herouy Y, Barz D, Kamradt T, Maghazachi A, et al. (2010) Histamine modulates γδ-T lymphocyte migration and cytotoxicity, via Gi and Gs protein-coupled signalling pathways. Br J Pharmacol 161:1291–1300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uguen M, Perrin D, Belliard S, Ligneau X, Beardsley PM, Lecomte JM, Schwartz JC. (2013) Preclinical evaluation of the abuse potential of Pitolisant, a histamine H₃ receptor inverse agonist/antagonist compared with Modafinil. Br J Pharmacol 169:632–644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uveges AJ, Kowal D, Zhang Y, Spangler TB, Dunlop J, Semus S, Jones PG. (2002) The role of transmembrane helix 5 in agonist binding to the human H3 receptor. J Pharmacol Exp Ther 301:451–458. [DOI] [PubMed] [Google Scholar]
- Valencia S, Hernández-Angeles A, Soria-Jasso LE, Arias-Montaño JA. (2001) Histamine H1 receptor activation inhibits the proliferation of human prostatic adenocarcinoma DU-145 cells. Prostate 48:179–187. [DOI] [PubMed] [Google Scholar]
- van der Werf JF, Timmerman H. (1989) The histamine H3 receptor: a general presynaptic histaminergic regulatory system? Trends Pharmacol Sci 10:159–162. [DOI] [PubMed] [Google Scholar]
- van Rijn RM, Chazot PL, Shenton FC, Sansuk K, Bakker RA, Leurs R. (2006) Oligomerization of recombinant and endogenously expressed human histamine H4 receptors. Mol Pharmacol 70:604–615. [DOI] [PubMed] [Google Scholar]
- van Rijn RM, van Marle A, Chazot PL, Langemeijer E, Qin Y, Shenton FC, Lim HD, Zuiderveld OP, Sansuk K, Dy M, et al. (2008) Cloning and characterization of dominant negative splice variants of the human histamine H4 receptor. Biochem J 414:121–131. [DOI] [PubMed] [Google Scholar]
- Vanhala A, Yamatodani A, Panula P. (1994) Distribution of histamine-, 5-hydroxytryptamine-, and tyrosine hydroxylase-immunoreactive neurons and nerve fibers in developing rat brain. J Comp Neurol 347:101–114. [DOI] [PubMed] [Google Scholar]
- Vanhanen J, Kinnunen M, Nuutinen S, Panula P. (2015) Histamine H3 receptor antagonist JNJ-39220675 modulates locomotor responses but not place conditioning by dopaminergic drugs. Psychopharmacology (Berl) 232:1143–1153. [DOI] [PubMed] [Google Scholar]
- Varaschin RK, Akers KG, Rosenberg MJ, Hamilton DA, Savage DD. (2010) Effects of the cognition-enhancing agent ABT-239 on fetal ethanol-induced deficits in dentate gyrus synaptic plasticity. J Pharmacol Exp Ther 334:191–198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Varga C, Horvath K, Berko A, Thurmond RL, Dunford PJ, Whittle BJR. (2005) Inhibitory effects of histamine H4 receptor antagonists on experimental colitis in the rat. Eur J Pharmacol 522:130–138. [DOI] [PubMed] [Google Scholar]
- Varty LM, Hey JA. (2002) Histamine H3 receptor activation inhibits neurogenic sympathetic vasoconstriction in porcine nasal mucosa. Eur J Pharmacol 452:339–345. [DOI] [PubMed] [Google Scholar]
- Vauth M, Möhner D, Beermann S, Seifert R, Neumann D. (2012) Histamine via the histamine H₂-receptor reduces α-CD3-induced interferon-γ synthesis in murine CD4+ T cells in an indirect manner. J Interferon Cytokine Res 32:185–190. [DOI] [PubMed] [Google Scholar]
- Venable JD, Cai H, Chai W, Dvorak CA, Grice CA, Jablonowski JA, Shah CR, Kwok AK, Ly KS, Pio B, et al. (2005) Preparation and biological evaluation of indole, benzimidazole, and thienopyrrole piperazine carboxamides: potent human histamine h4 antagonists. J Med Chem 48:8289–8298. [DOI] [PubMed] [Google Scholar]
- Villemagne VL, Dannals RF, Sánchez-Roa PM, Ravert HT, Vazquez S, Wilson AA, Natarajan TK, Wong DF, Yanai K, Wagner HN., Jr (1991) Imaging histamine H1 receptors in the living human brain with carbon-11-pyrilamine. J Nucl Med 32:308–311. [PubMed] [Google Scholar]
- Vischer HF, Watts AO, Nijmeijer S, Leurs R. (2011) G protein-coupled receptors: walking hand-in-hand, talking hand-in-hand? Br J Pharmacol 163:246–260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vizuete ML, Traiffort E, Bouthenet ML, Ruat M, Souil E, Tardivel-Lacombe J, Schwartz JC. (1997) Detailed mapping of the histamine H2 receptor and its gene transcripts in guinea-pig brain. Neuroscience 80:321–343. [DOI] [PubMed] [Google Scholar]
- Vollinga RC, de Koning JP, Jansen FP, Leurs R, Menge WM, Timmerman H. (1994) A new potent and selective histamine H3 receptor agonist, 4-(1H-imidazol-4-ylmethyl)piperidine. J Med Chem 37:332–333. [DOI] [PubMed] [Google Scholar]
- Vorobjev VS, Sharonova IN, Walsh IB, Haas HL. (1993) Histamine potentiates N-methyl-D-aspartate responses in acutely isolated hippocampal neurons. Neuron 11:837–844. [DOI] [PubMed] [Google Scholar]
- Wadee AA, Anderson R, Sher R. (1980) In vitro effects of histamine on eosinophil migration. Int Arch Allergy Appl Immunol 63:322–329. [DOI] [PubMed] [Google Scholar]
- Wagner MC, Eckman JR, Wick TM. (2006) Histamine increases sickle erythrocyte adherence to endothelium. Br J Haematol 132:512–522. [DOI] [PubMed] [Google Scholar]
- Wakahara K, Baba N, Van VQ, Bégin P, Rubio M, Ferraro P, Panzini B, Wassef R, Lahaie R, Caussignac Y, et al. (2012) Human basophils interact with memory T cells to augment Th17 responses. Blood 120:4761–4771. [DOI] [PubMed] [Google Scholar]
- Wang YX, Kotlikoff MI. (2000) Signalling pathway for histamine activation of non-selective cation channels in equine tracheal myocytes. J Physiol 523:131–138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wayman J, Hayes N, Raimes SA, Griffin SM. (2000) Prescription of proton pump inhibitors before endoscopy. A potential cause of missed diagnosis of early gastric cancers. Arch Fam Med 9:385–388. [DOI] [PubMed] [Google Scholar]
- Weisler RH, Pandina GJ, Daly EJ, Cooper K, Gassmann-Mayer C, 31001074-ATT2001 Study Investigators (2012) Randomized clinical study of a histamine H3 receptor antagonist for the treatment of adults with attention-deficit hyperactivity disorder. CNS Drugs 26:421–434. [DOI] [PubMed] [Google Scholar]
- Wellendorph P, Goodman MW, Burstein ES, Nash NR, Brann MR, Weiner DM. (2002) Molecular cloning and pharmacology of functionally distinct isoforms of the human histamine H3 receptor. Neuropharmacology 42:929–940. [DOI] [PubMed] [Google Scholar]
- Wellner-Kienitz MC, Bender K, Meyer T, Pott L. (2003) Coupling to Gs and Gq/11 of histamine H2 receptors heterologously expressed in adult rat atrial myocytes. Biochim Biophys Acta 1642:67–77. [DOI] [PubMed] [Google Scholar]
- Wenzel-Seifert K, Kelley MT, Buschauer A, Seifert R. (2001) Similar apparent constitutive activity of human histamine H2-receptor fused to long and short splice variants of Gsalpha. J Pharmacol Exp Ther 299:1013–1020. [PubMed] [Google Scholar]
- Werner K, Neumann D, Buschauer A, Seifert R. (2014) No evidence for histamine H4-receptor in human monocytes. J Pharmacol Exp Ther 351:519–526. [DOI] [PubMed] [Google Scholar]
- Werner T, Sander K, Tanrikulu Y, Kottke T, Proschak E, Stark H, Schneider G. (2010) In silico characterization of ligand binding modes in the human histamine H4 receptor and their impact on receptor activation. ChemBioChem 11:1850–1855. [DOI] [PubMed] [Google Scholar]
- West RE, Jr, Zweig A, Granzow RT, Siegel MI, Egan RW. (1990) Biexponential kinetics of (R)-alpha-[3H]methylhistamine binding to the rat brain H3 histamine receptor. J Neurochem 55:1612–1616. [DOI] [PubMed] [Google Scholar]
- Whiteford HA, Stedman TJ, McGrath JJ, Welham J, Pond S. (1995) An open-label study of famotidine as a treatment for schizophrenia. J Psychiatry Neurosci 20:239–240. [PMC free article] [PubMed] [Google Scholar]
- Wiedemann P, Bönisch H, Oerters F, Brüss M. (2002) Structure of the human histamine H3 receptor gene (HRH3) and identification of naturally occurring variations. J Neural Transm 109:443–453. [DOI] [PubMed] [Google Scholar]
- Wieland C, Jakobs KH, Wieland T. (1994) Altered guanine nucleoside triphosphate binding to transducin by cholera toxin-catalysed ADP-ribosylation. Cell Signal 6:487–492. [DOI] [PubMed] [Google Scholar]
- Wieland K, Bongers G, Yamamoto Y, Hashimoto T, Yamatodani A, Menge WM, Timmerman H, Lovenberg TW, Leurs R. (2001) Constitutive activity of histamine h3 receptors stably expressed in SK-N-MC cells: display of agonism and inverse agonism by H3 antagonists. J Pharmacol Exp Ther 299:908–914. [PubMed] [Google Scholar]
- Wieland K, Laak AM, Smit MJ, Kühne R, Timmerman H, Leurs R. (1999) Mutational analysis of the antagonist-binding site of the histamine H1 receptor. J Biol Chem 274:29994–30000. [DOI] [PubMed] [Google Scholar]
- Wifling D, Löffel K, Nordemann U, Strasser A, Bernhardt G, Dove S, Seifert R, Buschauer A. (2015) Molecular determinants for the high constitutive activity of the human histamine H4 receptor: functional studies on orthologues and mutants. Br J Pharmacol 172:785–798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams K. (1994) Subunit-specific potentiation of recombinant N-methyl-D-aspartate receptors by histamine. Mol Pharmacol 46:531–541. [PubMed] [Google Scholar]
- Wittmann HJ, Elz S, Seifert R, Straber A. (2011a) N (α)-Methylated phenylhistamines exhibit affinity to the hH4R-a pharmacological and molecular modelling study. Naunyn Schmiedebergs Arch Pharmacol 384:287–299. [DOI] [PubMed] [Google Scholar]
- Wittmann HJ, Seifert R, Strasser A. (2011b) Influence of the N-terminus and the E2-loop onto the binding kinetics of the antagonist mepyramine and the partial agonist phenoprodifen to H1R. Biochem Pharmacol 82:1910–1918. [DOI] [PubMed] [Google Scholar]
- Wulff BS, Hastrup S, Rimvall K. (2002) Characteristics of recombinantly expressed rat and human histamine H3 receptors. Eur J Pharmacol 453:33–41. [DOI] [PubMed] [Google Scholar]
- Xie SX, Ghorai P, Ye QZ, Buschauer A, Seifert R. (2006) Probing ligand-specific histamine H1- and H2-receptor conformations with NG-acylated Imidazolylpropylguanidines. J Pharmacol Exp Ther 317:139–146. [DOI] [PubMed] [Google Scholar]
- Xu AJ, Kuramasu A, Maeda K, Kinoshita K, Takayanagi S, Fukushima Y, Watanabe T, Yanagisawa T, Sukegawa J, Yanai K. (2008) Agonist-induced internalization of histamine H2 receptor and activation of extracellular signal-regulated kinases are dynamin-dependent. J Neurochem 107:208–217. [DOI] [PubMed] [Google Scholar]
- Xu C, Michelsen KA, Wu M, Morozova E, Panula P, Alreja M. (2004) Histamine innervation and activation of septohippocampal GABAergic neurones: involvement of local ACh release. J Physiol 561:657–670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamasaki T, Tamai I, Matsumura Y. (2001) Activation of histamine H3 receptors inhibits renal noradrenergic neurotransmission in anesthetized dogs. Am J Physiol Regul Integr Comp Physiol 280:R1450–R1456. [DOI] [PubMed] [Google Scholar]
- Yamashita M, Fukui H, Sugama K, Horio Y, Ito S, Mizuguchi H, Wada H. (1991) Expression cloning of a cDNA encoding the bovine histamine H1 receptor. Proc Natl Acad Sci USA 88:11515–11519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamaura K, Oda M, Suwa E, Suzuki M, Sato H, Ueno K. (2009) Expression of histamine H4 receptor in human epidermal tissues and attenuation of experimental pruritus using H4 receptor antagonist. J Toxicol Sci 34:427–431. [DOI] [PubMed] [Google Scholar]
- Yan H, Zhang X, Hu W, Ma J, Hou W, Zhang X, Wang X, Gao J, Shen Y, Lv J, et al. (2014) Histamine H3 receptors aggravate cerebral ischaemic injury by histamine-independent mechanisms. Nat Commun 5:3334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yanai K, Ryu JH, Sakai N, Takahashi T, Iwata R, Ido T, Murakami K, Watanabe T. (1994) Binding characteristics of a histamine H3-receptor antagonist, [3H]S-methylthioperamide: comparison with [3H](R)alpha-methylhistamine binding to rat tissues. Jpn J Pharmacol 65:107–112. [DOI] [PubMed] [Google Scholar]
- Yanai K, Tashiro M. (2007) The physiological and pathophysiological roles of neuronal histamine: an insight from human positron emission tomography studies. Pharmacol Ther 113:1–15. [DOI] [PubMed] [Google Scholar]
- Yanai K, Watanabe T, Yokoyama H, Meguro K, Hatazawa J, Itoh M, Iwata R, Ishiwata K, Takahashi T, Ido T. (1992) Histamine H1 receptors in human brain visualized in vivo by [11C]doxepin and positron emission tomography. Neurosci Lett 137:145–148. [DOI] [PubMed] [Google Scholar]
- Yanai K, Zhang D, Tashiro M, Yoshikawa T, Naganuma F, Harada R, Nakamura T, Shibuya K, Okamura N. (2011) Positron emission tomography evaluation of sedative properties of antihistamines. Expert Opin Drug Saf 10:613–622. [DOI] [PubMed] [Google Scholar]
- Yanovsky Y, Li S, Klyuch BP, Yao Q, Blandina P, Passani MB, Lin JS, Haas HL, Sergeeva OA. (2011) L-Dopa activates histaminergic neurons. J Physiol 589:1349–1366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yanovsky Y, Reymann K, Haas HL. (1995) pH-dependent facilitation of synaptic transmission by histamine in the CA1 region of mouse hippocampus. Eur J Neurosci 7:2017–2020. [DOI] [PubMed] [Google Scholar]
- Yao BB, Sharma R, Cassar S, Esbenshade TA, Hancock AA. (2003) Cloning and pharmacological characterization of the monkey histamine H3 receptor. Eur J Pharmacol 482:49–60. [DOI] [PubMed] [Google Scholar]
- Ye RD, Boulay F, Wang JM, Dahlgren C, Gerard C, Parmentier M, Serhan CN, Murphy PM. (2009) International Union of Basic and Clinical Pharmacology. LXXIII. Nomenclature for the formyl peptide receptor (FPR) family. Pharmacol Rev 61:119–161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yokoyama H, Onodera K, Maeyama K, Sakurai E, Iinuma K, Leurs R, Timmerman H, Watanabe T. (1994) Clobenpropit (VUF-9153), a new histamine H3 receptor antagonist, inhibits electrically induced convulsions in mice. Eur J Pharmacol 260:23–28. [DOI] [PubMed] [Google Scholar]
- Yoshimoto R, Miyamoto Y, Shimamura K, Ishihara A, Takahashi K, Kotani H, Chen AS, Chen HY, Macneil DJ, Kanatani A, et al. (2006) Therapeutic potential of histamine H3 receptor agonist for the treatment of obesity and diabetes mellitus. Proc Natl Acad Sci USA 103:13866–13871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu B, Shao Y, Li P, Zhang J, Zhong Q, Yang H, Hu X, Chen B, Peng X, Wu Q, et al. (2010a) Copy number variations of the human histamine H4 receptor gene are associated with systemic lupus erythematosus. Br J Dermatol 163:935–940. [DOI] [PubMed] [Google Scholar]
- Yu B, Shao Y, Zhang J, Dong XL, Liu WL, Yang H, Liu L, Li MH, Yue CF, Fang ZY, et al. (2010b) Polymorphisms in human histamine receptor H4 gene are associated with atopic dermatitis. Br J Dermatol 162:1038–1043. [DOI] [PubMed] [Google Scholar]
- Yu F, Wolin RL, Wei J, Desai PJ, McGovern PM, Dunford PJ, Karlsson L, Thurmond RL. (2010c) Pharmacological characterization of oxime agonists of the histamine H4 receptor. J Receptor Ligand Channel Res 3:37–49. [Google Scholar]
- Zhang L, Chen Z, Ren K, Leurs R, Chen J, Zhang W, Ye B, Wei E, Timmerman H. (2003) Effects of clobenpropit on pentylenetetrazole-kindled seizures in rats. Eur J Pharmacol 482:169–175. [DOI] [PubMed] [Google Scholar]
- Zhang XY, Yu L, Zhuang QX, Peng SY, Zhu JN, Wang JJ. (2013) Postsynaptic mechanisms underlying the excitatory action of histamine on medial vestibular nucleus neurons in rats. Br J Pharmacol 170:156–169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao C, Sun M, Bennani YL, Gopalakrishnan SM, Witte DG, Miller TR, Krueger KM, Browman KE, Thiffault C, Wetter J, et al. (2008) The alkaloid conessine and analogues as potent histamine H3 receptor antagonists. J Med Chem 51:5423–5430. [DOI] [PubMed] [Google Scholar]
- Zhou FW, Xu JJ, Zhao Y, LeDoux MS, Zhou FM. (2006) Opposite functions of histamine H1 and H2 receptors and H3 receptor in substantia nigra pars reticulata. J Neurophysiol 96:1581–1591. [DOI] [PubMed] [Google Scholar]
- Zhu Y, Michalovich D, Wu H, Tan KB, Dytko GM, Mannan IJ, Boyce R, Alston J, Tierney LA, Li X, et al. (2001) Cloning, expression, and pharmacological characterization of a novel human histamine receptor. Mol Pharmacol 59:434–441. [DOI] [PubMed] [Google Scholar]
- Zimmermann AS, Burhenne H, Kaever V, Seifert R, Neumann D. (2011) Systematic analysis of histamine and N-methylhistamine concentrations in organs from two common laboratory mouse strains: C57Bl/6 and Balb/c. Inflamm Res 60:1153–1159. [DOI] [PubMed] [Google Scholar]