

Abstract
Lactoferrin is one of a number of multifunctional proteins that are present in or on all mucosal surfaces throughout the body. Levels of lactoferrin are consistently elevated in inflammatory diseases such as arthritis, inflammatory bowel diseases, corneal disease, and periodontitis. Single-nucleotide polymorphisms (SNPs) in lactoferrin have been shown to be present in individuals susceptible to Escherichia coli–induced travelers’ diarrhea and in tear fluid derived from virally associated corneal disease. Here, we review data showing a lactoferrin SNP in amino acid position 29 in the antimicrobial region of lactoferrin that acts against caries associated bacteria. This SNP was initially discovered in African American subjects with localized aggressive periodontitis (LAP) who had proximal bone loss but minimal proximal caries. Results were confirmed in a genetic association study of children from Brazil with this same SNP who showed a reduced level of caries. In vitro data indicate that lactoferrin from whole saliva derived from subjects with this SNP, recombinant human lactoferrin containing this SNP, or an 11-mer peptide designed for this SNP kills mutans streptococci associated with caries by >1 log. In contrast, the SNP has minimal effect on Gram-negative species associated with periodontitis. Moreover, periodontally healthy subjects homozygous for this lysine (K) SNP have lactoferrin in their saliva that kills mutans streptococci and have reduced proximal decay. The review summarizes data supporting the ecologic plaque hypothesis and suggests that a genetic variant in lactoferrin with K in position 29 when found in saliva and crevice fluid can influence community biofilm composition. We propose that, for caries, this SNP is ethnicity independent and protective by directly killing caries-provoking bacteria (reducing proximal decay). However, the clinical effect of this SNP in LAP is ethnicity dependent, destructive (increases LAP incidence), and complex with mechanisms still to be determined.
Keywords: dental caries, aggressive periodontitis, single nucleotide polymorphisms, streptococcus mutans, saliva, ecosystem
Introduction
Saliva continuously bathes oral tissues and can be considered the biofluid that sustains structural integrity in the oral cavity. On a daily basis, salivary glands produce approximately 1 to 1.5 L of saliva (Scannapieco 1994). Reduced salivary flow can have severe consequences (Mandel 1987). For example, patients with irradiated salivary glands have excessive caries and dry, leather-like cracking mucosa on the same side as the irradiated gland (Dreizen and Brown 1976). The importance of saliva has been highlighted in healthy subjects as well. Studies have shown that saliva modulates the interaction between the bacterial flora and oral tissues by means of its buffering capacity, ability to dilute and wash away substances that can antagonize oral tissues, and healing capabilities by supplying calcium to compromised enamel surfaces (Mandel 1987; Scannapieco 1994). In addition, saliva possesses a potent combination of antimicrobial factors that could affect bacteria, viruses, and fungi capable of attacking oral tissues (Sikorska et al. 2002). While several salivary components have been shown to possess attributes that can affect oral stability and modify dental disease, characterization of genetic variations in specific salivary proteins and the effect of these variants on oral disease outcomes have been slow to materialize (Helmerhorst and Oppenheim 2007). The focus of this review is on lactoferrin and its potential to affect both caries and periodontal disease. Special emphasis in this review is placed on genetic polymorphisms that exist in the antibacterial region of lactoferrin and the potential of these variants to influence the presentation of dental disease.
Lactoferrin
A Ubiquitous Biodiverse Molecule
Lactoferrin is typically found in saliva at concentrations of 1 to 7 µg/mL (Scannapieco 1994). It is also found in crevice fluid (Friedman et al. 1983). In addition to its “ubiquitous” presence, lactoferrin has been shown to have antimicrobial, antiviral, antifungal, anti-inflammatory, and anticancer properties (Farnaud and Evans 2003). Lactoferrin does not stand alone in its multifunctionality; however, its unique capability of binding iron has led to its characterization as a “metal iron chelator” (Gorr and Abdolhosseini 2011). Initially, lactoferrin was thought to have an indirect role in host defense because of its ability to sequester iron needed for bacterial survival. This bacteriostatic mechanism was thought to be lactoferrin’s primary function until it was demonstrated that iron-free lactoferrin had bactericidal activity that was independent of iron binding (Arnold et al. 1977; Arnold et al. 1981). Lactoferrin can also be viewed as a host defense protein that operates predominantly in the innate arm of the immune system, but it also affects adaptive immunity (Legrand et al. 2005, 2006).
Lactoferrin’s potential importance is illustrated by the fact that it is one of a handful of secreted multifunctional proteins that is present in or on all mucosal surfaces throughout the body. Specifically, lactoferrin is found in saliva, tears, nasal secretions, gastric secretions, milk, amniotic fluids, and so on (Farnaud and Evans 2003).
Direct Effect on Microbial Activities
On a structural level, lactoferrin is composed of 2 homologous lobes—an N-lobe (amino acid residues 1 to 333) and a C-lobe (amino acid residues 334 to 692)—each containing a central pit area that accepts and retains iron (Fig. 1). The tail end of the N-terminal region of the glycoprotein can be thought of as a stem of lactoferrin’s bilobed flower-like structure (Baker and Baker 2009). This region consists of 47 amino acids that demonstrate a direct antimicrobial function. Residues 1 to 11 and 22 to 47 are the prominent actors against bacteria and yeast (Arnold et al. 1981). Many believe that residues 1 to 11 (termed LF-1) are genetically linked to residues 22 to 47 (termed lactoferricin), although most attribute the greatest potency to an 11-mer region—amino acids 21 (F) to 31 (R), FQWQRN MRKVR—within the lactoferricin moiety (Odell et al. 1996). There is universal agreement that lactoferrin’s antimicrobial activity occurs in its apo- or iron-limited condition (Arnold et al. 1977).
Figure 1.
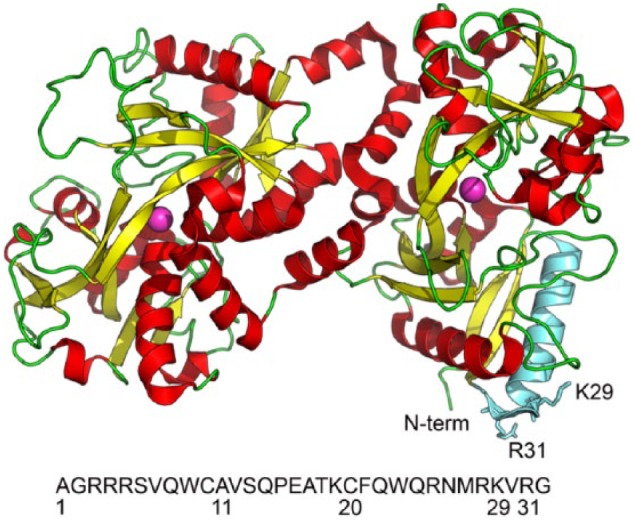
Structure of lactoferrin in 3 dimensions. Region in green represents the N-terminus where the antimicrobial activity is seen (residues 1 to 47). Two lobes show the iron-binding region (identified by the purple ball) in pit area. N-terminus is labeled (N-term), and the amino acid of interest is the lysine region (K) in position 29. Single-nucleotide polymorphism described in text occurs as shift from K to R in position 29 (not shown in this illustration). Illustration also depicts arginine (R) in position 31. Amino acids 1 to 32 are presented in letter form to allow for orientation of N-terminus. LF-1 region is seen as amino acids 1 to 11, and the lactoferricin region occupies amino acids 22 to 47; the region of interest extends from 21 to 31.
In the case of Gram-negative bacteria, lactoferrin’s antimicrobial action is thought to occur through the binding of lactoferricin directly or indirectly to the lipid A portion of lipopolysaccharide (LPS; Appelmelk et al. 1994). It appears that amino acids 28 to 34 (RKVRGPP) in the loop region of the N-terminus are critical for binding to lipid A (Fig. 2; Elass-Rochard et al. 1995). Evidence indicates that positively charged lysine (K) and/or arginine (R) residues from lactoferricin interact with negatively charged regions of lipid A or lipoteichoic acid (in the case of Gram positives), which causes disorientation of the bacterial outer membrane structure. Further, tryptophan (W) or valine (V) residues in lactoferrin interact with other hydrophobic membrane residues to disturb microbial membrane stability (Naidu et al. 1993). Lactoferrin’s N-terminus is cationic and hydrophobic. It appears as if electrostatic and hydrophobic interactions are key elements that determine lactoferrin’s antimicrobial activity (Nibbering et al. 2001). Other cationic peptides show similar effects on cell membrane integrity so that lactoferrin’s effects are not unique (Gorr and Abdolhosseini 2011).
Figure 2.
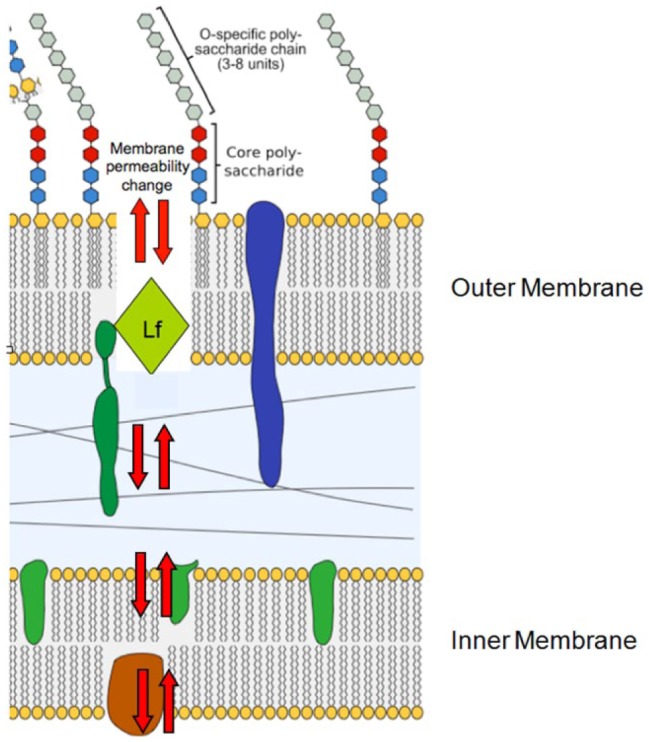
Illustration of proposed direct effect of lactoferricin on Gram-negative cell wall. Gram-negative cell wall is composed of 3 units: an outer O-specific polysaccharide that determines antigenic specificity, a core polysaccharide, and a lipid A inner structure (also known as endotoxin). Lactoferrin initially depolarizes the membrane by attacking the region where fatty acid tail joins polar head of lipid A. Lipid bilayer is composed of upper region head and tail and a lower region that consists of a tail-to-head configuration. Head is represented as circle or hexagon; fatty acid/hydrophobic tail is represented as squiggly lines beneath head. Lactoferricin insertion between leaflets of the bilayer creates holes in membrane and increased membrane permeability resulting in disturbance of membrane function. Lactoferricin participates in creating physical holes (red arrows) and induces production of hydrolases that degrade the cell wall. This activity causes destabilization of the membrane and leads to redistribution of membrane lipids between leaflets of the bilayer. Lf, lactoferrin.
Indirect Effects: Host Modulation
Lactoferrin’s role in immune responsiveness is complex. Lactoferrin can effect Toll-like receptors (TLRs) at 2 levels: (1) by associating with LPS directly prior to its interaction with LPS binding protein (LBP) and (2) by interfacing with membrane-bound CD14 (see Fig. 3). Lactoferrin and other cationic peptides are capable of neutralizing LPS’s interaction with host defense cells (Scott et al. 2000; Rosenfeld et al. 2006). LPS interacts with TLRs prominent on cell surfaces that are known to initiate innate immune responsiveness (Kawai and Akira 2005). LPS interaction is thought to be mediated by the association of lipid A with serum-circulating LBP, which brings the LPS–LBP complex in contact with membrane-bound CD14. These CD14 receptors are found on the surface of monocytes, polymorphonuclear neutrophils, dendritic cells, and so on (Miyake 2004). LPS is then transferred from CD14 to membrane-bound myeloid-differentiating protein (MD-2; Visintin et al. 2006). Neither CD14 or MD-2 alone or in complex can transduce signals from the cell membrane; however, transfer of the LPS–MD-2 complex to TLR-4 causes dimerization of 2 adjacent TLR-4 membrane receptors (Ren et al. 2005). This dimerized TLR-4 complex destabilizes the cell membrane (Fig. 3). Membrane perturbation causes a chain reaction leading to early- and late-phase NF-kappa B translocation from the cytoplasm into the nucleus, resulting in signaling and release of inflammatory cytokines. Membrane perturbation reduction by lactoferrin results in reduced cytokine production (Abreu et al. 2001).
Figure 3.

Indirect effect of lactoferrin by means of host modulation. (A) Lactoferrin can interfere with the ability of lipopolysaccharide-binding protein (LBP) found in serum and emanating from the liver to bind to soluble lipopolysaccharide (LPS). Lactoferrin can also interfere with the transfer of LPS to soluble CD14. (B) Lactoferrin can also interfere with LPS transfer from LBP to membrane bound mCD14. CD14 receptors are found on the surface of monocytes, polymorphonuclear neutrophils, dendritic cells. (C) Transfer of LBP bound LPS from CD14 to myeloid differentiating protein 2 (MD-2) occurs. LPS transfer to MD-2 allows for association of LPS with Toll-like 4 receptors for dimerization. (D) This dimerization of Toll-like receptors causes membrane perturbation and cell signaling resulting in cytokine production. Lf, lactoferrin; TLR4, Toll-like receptor 4.
Lactoferrin can also interact with soluble CD14 (Wurfel et al. 1995). When bound to soluble CD14, lactoferrin competes with LPS and reduces the chance of the LPS–CD14 complex to induce a downstream inflammatory response, which can include endothelial cell expression of E-selectin, ICAM-1, and IL-8 (Baveye et al. 2000). In this manner, lactoferrin can neutralize or reduce production of cell-modulated inflammatory cytokines. Once again, these activities do not exclusively belong to lactoferrin. For example, LL-37 has many similar properties (Gorr and Abdolhosseini 2011). Single-nucleotide polymorphisms (SNPs) in lactoferrin have been shown to alter its ability to modify inflammatory diseases (see below; Mohamed et al. 2007). In addition, lactoferrin modulates adaptive immune responsiveness by assisting in recruitment of “sentinel” cells, promoting maturation of T-cell precursors to competent T-helper cells, and promoting differentiation of immature B cells into efficient antigen-presenting cells (Actor et al. 2009).
Association with Inflammatory Diseases
Lactoferrin marker levels, mostly attributed to polymorphonuclear neutrophils, are consistently elevated in inflammatory diseases such as arthritis, inflammatory bowel disease, ulcerative colitis, and periodontitis (Adonogianaki et al. 1993; Legrand et al. 2005; Glimvall et al. 2012). Animal experiments illustrate the potency of lactoferrin as a protective molecule. Lactoferrin knockout mice challenged with oral microbial pathogens succumb to inflammatory disease that can be reversed by adding lactoferrin back to the knockout mice (Velusamy et al. 2013). Furthermore, lactoferrin can reduce Staphylococcus aureus–induced joint inflammation by both killing S. aureus and modulating its inflammatory effects (Guillen et al. 2000). Lactoferrin-related SNPs appear to alter susceptibility to infection. Thus, SNPs in lactoferrin (in amino acid position 632) and/or in the lactoferrin receptor protein CD14 (in position −159 in the CD14 promoter region) have been shown to be present in subjects susceptible to Escherichia coli–induced travelers’ diarrhea (Mohamed et al. 2007; Mohamed et al. 2011). These effects are ethnicity dependent. A different SNP in lactoferrin (region 561) was found in tear fluid in subjects with virally based corneal disease (Keijser et al. 2008). Although more work needs to be done, parallels can be seen in studies of inflammatory bowel disease. In inflammatory bowel disease, lactoferrin SNPs in different alleles affect different ethnicities and different genders in their susceptibility to disease (Sidhu et al. 2010). In effect, less lactoferrin will lead to more LPS TLR-stimulated cytokine activity (Elass-Rochard et al. 1995).
Association with Periodontitis and Caries
Dental plaque is a multispecies biofilm composed of a mixture of Gram-positive and Gram-negative bacteria that develops sequentially in a metabolically interactive manner (Marsh 2003). Typically, dental caries–related biofilms are composed of elevated levels of Gram-positive, acid-producing species, while periodontitis-related biofilms are generally more mixed in composition but comprise higher levels of Gram-negative species (Griffen et al. 2012; Gross et al. 2012). With more sophisticated DNA approaches at hand, it has become obvious that biofilms related to both caries and periodontitis are more complex than previously proposed. While during the caries process the microbiota becomes less diverse and more focused on organisms that can survive in an acid environment, the periodontitis flora becomes more diverse as disease progresses (Griffen et al. 2012). Concerning caries, mutans streptococci still predominate but other acid-producing microbes require consideration (Gross et al. 2012).
With respect to lactoferrin, a series of seminal experiments demonstrated that suboptimal levels of lactoferrin effect biofilm development (Singh et al. 2002). The lactoferrin effect was not attributed to interference with cell growth but to lactoferrin’s ability to sequester or chelate iron required for biofilm development (Singh et al. 2002). While my colleagues and I showed, in a limited study population (Fine et al. 2002), that African American adolescents with localized aggressive periodontitis (LAP) have lactoferrin containing minimal iron, the effects of apo-lactoferrin on caries- or periodontitis-related biofilms have not be studied in any depth to date.
As for caries, it has been proposed that lactoferrin’s effect can be independent or in concert with lysozyme, lactoperoxidase, and IgA (Rudney et al. 1999). In vitro evidence shows that apo-lactoferrin kills Streptococcus mutans (Arnold et al. 1981). Ex vivo studies showed that saliva from subjects with LAP can kill S. mutans and other acid-producing, caries-associated bacteria (Fine, Furgang, et al. 2013).
As for periodontitis, subjects with LAP were shown to have high levels of salivary lactoferrin with low levels of iron (Fine et al. 2002). Iron-saturated lactoferrin has been shown to block binding of Porphyromonas gingivalis and Aggregatibacter actinomycetemcomitans to epithelial cells (Alugupalli and Kalfas 1995). While more recent studies have shown that apo-lactoferrin has little effect on A. actinomycetemcomitans growth or binding of well-maintained recent clinical isolates, the effect of apo-lactoferrin on A. actinomycetemcomitans biofilms is lacking (Fine and Furgang 2002). From a clinical perspective, lactoferrin has been proposed as a general marker for inflammatory periodontal diseases (Friedman et al. 1983; Adonogianaki et al. 1993).
Localized Aggressive Periodontitis: Borderland between Caries and Periodontal Disease
A protease-sensitive, diffusible salivary protein with a molecular weight of <100,000 kDa and a basic isoelectric point that killed S. mutans was identified in whole saliva obtained from LAP subjects with minimal proximal decay (Fine et al. 2007). This unusual presentation of dental disease (aggressive periodontitis and minimal decay) provided a unique opportunity to study proximal caries resistance. In addition, data suggested that LAP saliva could have an effect on oral biofilm ecology. Furthermore, since this unusual clinical presentation was documented in adolescents of African American descent who had A. actinomycetemcomitans–associated LAP, a genetic basis for host-derived susceptibility could be inferred.
Comparison between caries-resistant and caries-susceptible individuals has been a focus of a great deal of interest (Anderson et al. 1982). However, most studies use DMFS (decayed, missing, filled, surfaces) as the clinical criteria, rarely do radiographic analysis, and fail to note the differences between occlusal and proximal decay (Azevedo et al. 2010). Using the LAP model, my colleagues and I set out to compare proximal decay in LAP subjects to matched healthy controls by means of radiographic analysis (Fine et al. 1984). Of 23 LAP subjects, 17 had no proximal decay, while 20 of 23 controls had 248 proximal lesions versus 19 proximal lesions for LAP subjects (P < 0.0001; Fine et al. 1984). Mixing occlusal and proximal decay obscured these differences. From a biological perspective, proximal caries-inducing bacteria such as S. mutans adhere tenaciously to smooth salivary-coated enamel surfaces, while bacteria that populate occlusal fissures are often nonadherent planktonic lactobacilli. In addition, the potential for saliva and/or crevice fluid to mollify the caries potential of bacteria deeply embedded into enamel pits and fissures is unlikely due to restricted access to fluid flow in these tortuous regions. By separating occlusal and proximal caries, interpretation of proximal caries resistance can be enhanced (Fine et al. 1984).
A Lactoferrin SNP and the Borderland
Caries
Caries result from a direct attack of the metabolic by-products of adherent bacteria that act as delivery devices for acid-induced demineralization of enamel. DNA analysis of the lactoferrin gene derived from an individual with LAP indicated that lactoferrin could kill S. mutans (Velliyagounder et al. 2003). Briefly, DNA obtained from an LAP subject showed sequence differences in lactoferrin at codon position 29 (or 47 depending on nomenclature) with AAA (lysine) at position 29 and AGA (arginine) in that position in the control subject. Recombinant human full-length lactoferrin of both variants was produced in an insect vector and the lysine (K) variant was shown to kill S. mutans in vitro by >1 log as compared to the arginine (R) variant (Velliyagounder et al. 2003; Appendix Fig.). The same lactoferrin lysine variant was related to reduced decay in a genetic association study of children in Brazil (Azevedo et al. 2010). While the Brazilian study appears to confirm the anti–S. mutans laboratory data reported above (Velliyagounder et al. 2003), note that occlusal and proximal caries were not separated in the Brazilian study (Azevedo et al. 2010).
Saliva collected from an LAP subject was passed through a S. mutans affinity column to isolate, capture, and identify the active anticaries fraction (Fine, Toruner, et al. 2013). The salivary protein that remained bound to the column containing S. mutans–coated beads was eluted and identified as lactoferrin by MALDI-TOF mass spectroscopy and confirmed by Western blot using human antibody to lactoferrin (Fig. 4). A clinical study was designed to determine whether periodontally healthy, non-LAP subjects homozygous for the lysine variant would have saliva with anti-mutans activity. The assumption was that those subjects homozygous for lysine would secrete lactoferrin with lysine in position 29, kill S. mutans, and show minimal smooth surface decay clinically. It was also assumed that those homozygous for arginine would secrete lactoferrin with arginine in amino acid position 29, which would have no effect on S. mutans and would show substantial levels of smooth surface decay clinically (Fine, Toruner, et al. 2013).
Figure 4.

Isolation of active anti–Streptococcus mutans fraction from whole saliva from a subject with localized aggressive periodontitis. (A) Peak obtained when fraction 19 was eluted from saliva derived from a subject with localized aggressive periodontitis with salivary activity against S. mutans. Whole saliva was applied to S. mutans affinity column, and then fractions were collected after treatment with glycine hydrochloride for elution of fractions bound to S. mutans. (B) Activity of fraction 19 and its ability to prevent the growth of S. mutans by almost 2 logs as compared to phosphate buffered saline (PBS) control. Fraction 23 reduces growth by <1 log, and fraction 26 shows no reduction. (C) Fraction 19 was applied to a filter paper disc, which was then placed over a confluent inoculum of S. mutans grown on a mitis salivarius agar plate. Two days later, a zone of inhibition was seen around the filter disc coated with fraction 19 added to either PBS or normal saliva. Note that “normal” saliva without fraction 19 added has no anti–S. mutans activity. CFU, colony-forming unit; LAP, localized aggressive periodontitis.
Results indicated that all subjects with K29K genotype had activity against S. mutans that was blocked by pretreating saliva with antibody to human lactoferrin. In contrast, only 25% of subjects who had the R29R genotype had anti–S. mutans activity; however, this activity was not reduced by pretreatment with antibody to lactoferrin, suggesting that some other salivary factor was active in these subjects. Results also indicated that 87.5% of individuals with the K29K genotype had no smooth surface decay. Conversely, 70% subjects with the R29R genotype had multiple smooth surface lesions per individual (Fine, Toruner, et al. 2013).
To confirm and expand the understanding of the specificity of lactoferrin’s antibacterial activity, an 11-mer peptide (FQWQRNMRKVR) was prepared. The lysine variant had K in position 29, while the arginine variant had R in position 29 (Fig. 1). Each peptide was tested against a battery of Gram-positive, acid-producing, caries-promoting bacteria. All 5 strains of S. mutans analyzed were killed by >1 log by the lysine variant, whereas the arginine peptide had no activity above the phosphate buffered saline control (Table). Growth of other streptococcal species, including S. sobrinus and S. sanguinis, were also significantly reduced by the lysine peptide (Table). In addition, growth of Lactobacillus, Actinomyces, and Veillonella species was also reduced. Taken together, while the clinical data are limited in subject numbers, the information obtained suggests that the lysine variant of lactoferrin could directly affect growth of microbes related to proximal caries development and that those individuals with this variant have minimal proximal decay (Fine, Toruner, et al. 2013).
Table.
Effect of Lysine and Arginine Peptides on Mutans Streptococci and Other Bacteria.
| Bacterial Strain | Lysine Peptidea | Arginine Peptidea | PBS Controla | Growth Conditions |
|---|---|---|---|---|
| Actinomyces viscosus 43146 | −0.40 | −0.07 | 0.08 | ATCC medium 260, aerobic, 37°C |
| Streptococcus sanguinis 903 | −1.05 | −0.04 | −0.06 | ATCC medium 260, aerobic, 37°C |
| Streptococcus mitis 15914 | −0.39 | −0.04 | −0.03 | ATCC medium 260, aerobic, 37°C |
| Fusobacterium nucleatum 25586 | −0.46 | −0.45 | −0.02 | ATCC medium 1053, anaerobic, 37°C |
| Capnocytophaga sputigena 33612 | −0.59 | −0.60 | −0.02 | ATCC medium 1490, anaerobic, 37°C |
| Veillonella parvula 10790 | −0.58 | −0.47 | −0.06 | ATCC medium 1252, anaerobic, 37°C |
| Lactobacillus acidophilus 832 | −0.66 | −0.14 | −0.19 | ATCC medium 416, 5% CO2, 37°C |
| Streptococcus sanguinis 10556 | −0.56 | −0.15 | −0.12 | ATCC medium 260, aerobic, 37°C |
| Streptococcus gordonii 10558 | −0.52 | −0.06 | −0.10 | ATCC medium 260, aerobic, 37°C |
| Streptococcus mutans 25175 | −1.13 | −0.26 | −0.20 | ATCC medium 260, aerobic, 37°C |
| S. mutans 31383 | −1.08 | −0.16 | −0.12 | ATCC medium 260, aerobic, 37°C |
| S. mutans 33535 | −1.07 | −0.15 | −0.09 | ATCC medium 260, aerobic, 37°C |
| S. mutans 31441 | −1.03 | −0.14 | −0.16 | ATCC medium 260, aerobic, 37°C |
| S. mutans 33534 | −1.10 | −0.16 | −0.14 | ATCC medium 260, aerobic, 37°C |
| Streptococcus sobrinus 33340 | −1.16 | −0.08 | −0.02 | ATCC medium 260, aerobic, 37°C |
CFU, colony-forming unit; PBS, phosphate buffered saline.
Values in –log CFU/mL.
Periodontitis
Periodontal disease is a complex mucosal infection that is initiated by bacteria and perpetuated by the host response to the bacterial instigators (Curtis et al. 2011). While in the case of caries, it is feasible to suspect that the lactoferrin antimicrobial SNP of interest could directly attack caries-provoking bacteria, the SNP effect on periodontal disease is more nuanced. By reducing the Gram-positive inhabitants of the subgingival flora, the K29K lactoferrin SNP could suppress competing organisms and permit “pathogens” to overgrow; however, the K29K variant also can affect host modulation of the inflammatory response (Velliyagounder et al. 2003).
With respect to periodontal disease, a small subset of 16 LAP subjects was evaluated: 74% had lactoferrin with the K allele in amino acid position 29, while 26% of the LAP participants and their controls were K/R heterozygous and 74% of non-LAP controls were homozygous for the R allele. In addition, the same lysine variant stimulated higher levels of antimicrobial peptides of the hBD-2 variety in epithelial cells challenged with LPS as compared to the arginine variant. Thus, the K variant showed biological effects that could influence both bacterial and host-promoting factors (Velliyagounder et al. 2003).
Another larger study evaluated 124 African Americans (46 with aggressive periodontitis and 78 controls) and 208 Caucasians (77 aggressive periodontitis and 131 controls) for a SNP in this same N-terminal region of lactoferrin (Jordan et al. 2005). A single-point mutation was assessed causing a shift from threonine (T) to alanine (A) in amino acid position 11 in the LF-1 region of lactoferrin. Here, it was found that the African American aggressive periodontitis subjects were twice as likely to have the alanine shift as opposed to the threonine shift, while no such association was seen in Caucasian subjects. Since the switch from T/A at position 11 and from R/K at position 29 is in linkage disequilibrium (Keijser et al. 2008), these results support the data of Velliyagounder et al. (2003). Conversely, another study showed a SNP at amino acid position 29 where the arginine (R) variant was present in a group of individuals with aggressive periodontitis from Taiwan (Wu et al. 2009). Taken together, these data suggest that specific positional polymorphisms may be important in disease susceptibility but that the amino acid expressed by the polymorphism in that position as it relates to health outcomes may be dependent on genetic factors related to ethnicity. These ethnic distinctions that relate to disease susceptibility have been highlighted in a number of studies (Nibali et al. 2014).
In summary, it is worth noting that disease classification could be confounded by ethnicity-based genetic variants. Thus, in the case of LAP, clinical presentation in individuals of African descent could be similar to that seen in other ethnicities, but host-related microbial interactions could differ in content and quantity due to microbial transmission, diet, and/or host genetics (Haubek and Johansson 2014). With respect to lactoferrin and periodontal disease, one would expect the K or R lactoferrin SNP in position 29 to have a direct effect on a particular segment of the overall bacterial community. In addition, the SNP can indirectly affect the microflora by virtue of the influence of lactoferrin on the host cell response to these bacteria (Fig. 5). As such, both the direct and indirect effects of lactoferrin would apply to periodontal pathogenesis (Figs. 2, 3, 5). Undoubtedly, further work needs to be done in this area. When issues of clinical classification are resolved, there is no doubt that genetic variability will help disentangle microbial–host relationships with respect to disease status.
Figure 5.

Proposed effect of lactoferrin single-nucleotide polymorphism (SNP) on dental diseases. Left side of panel (A) illustrates proposed direct effect of the lysine variant of lactoferrin on mutans streptococci and other Gram-positive acid-producing bacteria. This SNP also affects Candida species implicated in caries (data not shown). (B) Right side illustrates proposed effect of lysine variant on aggressive periodontitis. Here, both direct antibacterial and host-modulated immunity effects are seen. Lf, lactoferrin.
Conclusions and Future Directions
Over the last several years, it has become clearer that dental diseases involve a community of bacteria that move from health to disease by virtue of a shift in the composition of members of the resident flora (Hooper 2009; Hajishengallis 2014). The shifts can occur as a result of ecologic stimuli caused by numerous influences—physical (overhanging amalgams), chemical (antibiotic usage), or biological (genetic; Marsh 2003). In this review, it is proposed that a genetic variant in lactoferrin can provide the environmental impetus that could stabilize the maintenance and/or restoration of the resident microflora. While this review focuses on lactoferrin, it is more than likely that other salivary proteins singly or in combination affect the outcome of dental disease (Oppenheim et al. 2007). Constant exposure to antimicrobial elements in saliva and crevice fluid composed of substances such as lactoferrin, lysozyme, and lactoperoxidase with specific antimicrobial properties (i.e., kill S. mutans and other caries-associated microbes or not) can have an ecologic influence on biofilm composition and disease outcome. This scenario supports the ecologic plaque hypothesis and reinforces the concept that the host is a critical determinant with respect to microbial community composition (Marsh and Devine 2011).
This review presents a new way of looking at the constant exposure of the oral microflora to salivary elements with an emphasis on a specific genetically predetermined form of a salivary protein that can either protect or challenge community behavior. Protection (as in the case of caries) can come in the form of direct antimicrobial action on the disease-provoking members of the microflora (Fine, Toruner, et al. 2013). Alternatively, host-related components that interact with the biofilm community can produce an environment that can influence overall biofilm composition and stability unfavorably but indirectly and thus alter the clinical outcome (Nibali et al. 2014). Perhaps in the future, additional salivary protein variants will reveal other novel functional relationships with the clinical manifestation of oral diseases.
While this review suggests that host genetics can influence salivary protein expression—which can in turn influence microbial composition—many issues continue to need further resolution. Undoubtedly, other SNPs may coexist within lactoferrin and other prominent salivary proteins. Moreover, polymorphisms have been demonstrated in histatin, a salivary protein that can affect yeast (Helmerhorst and Oppenheim 2007). However, these issues aside, it was my intention to focus on how genetic variability in salivary lactoferrin can mitigate the outcome of dental disease.
Given the complexities depicted above and the limited database assessed, there is still sufficient evidence to indicate that the lysine SNP described in this review can have a direct effect on the microbial composition of dental plaque. Evidence indicates that lysine in amino acid position 29 in lactoferrin can kill mutans streptococci and associated acid-producing microbes and reduce proximal decay. Quantitation of acid-producing microbes that accumulate on proximal surfaces over time in subjects with this lactoferrin variant is still to be determined. Moreover, early evidence, based on the subjects studied thus far, suggests that individuals with the K29K genotype, for the most part regardless of race, will be less vulnerable to proximal decay that those with the R29R lactoferrin genotype. Neither genotype appears to affect occlusal decay (Fine, Toruner, et al. 2013). In addition, lactoferrin is related to periodontal vulnerability in African American adolescents, but the functional biology of this variant is still to be determined (Velliyagounder et al. 2003; Fine, Toruner, et al. 2013), and patient susceptibility in this case appears to be ethnicity dependent (Wu et al. 2009). Finally, this review illustrates the need for multicenter studies designed around specific genetic variants that show biological plausibility relative to disease pathogenesis in addition to genome-wide association studies. This strategy may make it possible for the emergence of new diagnostic and therapeutic approaches to dental diseases in the future.
Author Contributions
D.H. Fine, contributed to conception, design, data acquisition, analysis, and interpretation, drafted and critically revised the manuscript. The author gave final approval and agrees to be accountable for all aspects of the work.
Supplementary Material
Acknowledgments
The author acknowledges that he has a patent through Rutgers University using a derivative of the peptides described in this review. The patent is owned by Rutgers University and has not been licensed, and no remuneration is attributed to the inventor or Rutgers at this moment. The author acknowledges that references to work that may have had a direct association with this review may have been omitted due to limitations imposed by journal guidelines.
Footnotes
Support for research presented in this review was provided to D.H.F. from the National Institutes of Health, National Institute of Dental and Craniofacial Research (DE-016306, DE-016474, DE-017968).
The author declares no potential conflicts of interest with respect to the authorship and/or publication of this article.
A supplemental appendix to this article is published electronically only at http://jdr.sagepub.com/supplemental.
References
- Abreu MT, Vora P, Faure E, Thomas LS, Arnold ET, Arditi M. 2001. Decreased expression of Toll-like receptor-4 and MD-2 correlates with intestinal epithelial cell protection against dysregulated proinflammatory gene expression in response to bacterial lipopolysaccharide. J Immunol. 167(3):1609–1616. [DOI] [PubMed] [Google Scholar]
- Actor JK, Hwang SA, Kruzel ML. 2009. Lactoferrin as a natural immune modulator. Curr Pharm Des. 15(17):1956–1973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Adonogianaki E, Moughal NA, Kinane DF. 1993. Lactoferrin in the gingival crevice as a marker of polymorphonuclear leucocytes in periodontal diseases. J Clin Periodontol. 20(1):26–31. [DOI] [PubMed] [Google Scholar]
- Alugupalli KR, Kalfas S. 1995. Inhibitory effect of lactoferrin on the adhesion of Actinobacillus actinomycetemcomitans and Prevotella intermedia to fibroblasts and epithelial cells. APMIS. 103(2):154–160. [PubMed] [Google Scholar]
- Anderson LC, Lamberts BL, Bruton WF. 1982. Salivary protein polymorphisms in caries-free and caries-active adults. J Dent Res. 61(2):393–396. [DOI] [PubMed] [Google Scholar]
- Appelmelk BJ, An YQ, Geerts M, Thijs BG, de Boer HA, MacLaren DM, de Graaff J, Nuijens JH. 1994. Lactoferrin is a lipid A-binding protein. Infect Immun. 62(6):2628–2632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arnold RR, Cole MF, McGhee JR. 1977. A bactericidal effect for human lactoferrin. Science. 197(4300):263–265. [DOI] [PubMed] [Google Scholar]
- Arnold RR, Russell JE, Champion WJ, Gauthier JJ. 1981. Bactericidal activity of human lactoferrin: influence of physical conditions and metabolic state of the target microorganism. Infect Immun. 32(2):655–660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Azevedo LF, Pecharki GD, Brancher JA, Cordeiro CA, Jr, Medeiros KG, Antunes AA, Arruda ES, Werneck RI, de Azevedo LR, Mazur RF, et al. 2010. Analysis of the association between lactotransferrin (LTF) gene polymorphism and dental caries. J Appl Oral Sci. 18(2):166–170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baker EN, Baker HM. 2009. A structural framework for understanding the multifunctional character of lactoferrin. Biochimie. 91(1):3–10. [DOI] [PubMed] [Google Scholar]
- Baveye S, Elass E, Fernig DG, Blanquart C, Mazurier J, Legrand D. 2000. Human lactoferrin interacts with soluble CD14 and inhibits expression of endothelial adhesion molecules, E-selectin and ICAM-1, induced by the CD14-lipopolysaccharide complex. Infect Immun. 68(12):6519–6525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Curtis MA, Zenobia C, Darveau RP. 2011. The relationship of the oral microbiotia to periodontal health and disease. Cell Host Microbe. 10(4):302–326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dreizen S, Brown LR. (1976). Xerostomia and dental caries. In: Stiles HM, Loesche WJ, O’Brien TC. editors. Microbial aspects of dental caries: proceedings of a workshop on microbial aspects of dental caries, June 21–24, 1976, St Simons Island, Georgia Washington (DC): Information Retrieval; p. 263–274. [Google Scholar]
- Elass-Rochard E, Roseanu A, Legrand D, Trif M, Salmon V, Motas C, Montreuil J, Spik G. 1995. Lactoferrin-lipopolysaccharide interaction: involvement of the 28–34 loop region of human lactoferrin in the high-affinity binding to Escherichia coli 055B5 lipopolysaccharide. Biochem J. 312(Pt 3):839–845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farnaud S, Evans RW. 2003. Lactoferrin: a multifunctional protein with antimicrobial properties. Mol Immunol. 40(7):395–405. [DOI] [PubMed] [Google Scholar]
- Fine DH, Furgang D. 2002. Lactoferrin iron levels affect attachment of Actinobacillus actinomycetemcomitans to buccal epithelial cells. J Periodontol. 73(6):616–623. [DOI] [PubMed] [Google Scholar]
- Fine DH, Furgang D, Beydouin F. 2002. Lactoferrin iron levels are reduced in saliva of patients with localized aggressive periodontitis. J Periodontol. 73(6):624–630. [DOI] [PubMed] [Google Scholar]
- Fine DH, Furgang D, Goldman D. 2007. Saliva from subjects harboring Actinobacillus actinomycetemcomitans kills Streptococcus mutans in vitro. J Periodontol. 78(3):518–526. [DOI] [PubMed] [Google Scholar]
- Fine DH, Furgang D, McKiernan M, Rubin M. 2013. Can salivary activity predict periodontal breakdown in A. actinomycetemcomitans infected adolescents? Arch Oral Biol. 58(6):611–620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fine DH, Goldberg D, Karol R. 1984. Caries levels in patients with juvenile periodontitis. J Periodontol. 55(4):242–246. [DOI] [PubMed] [Google Scholar]
- Fine DH, Toruner GA, Velliyagounder K, Sampathkumar V, Godboley D, Furgang D. 2013. A lactotransferrin single nucleotide polymorphism demonstrates biological activity that can reduce susceptibility to caries. Infect Immun. 81(5):1596–15605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Friedman SA, Mandel ID, Herrera MS. 1983. Lysozyme and lactoferrin quantitation in the crevicular fluid. J Periodontol. 54(6):347–350. [DOI] [PubMed] [Google Scholar]
- Glimvall P, Wickstrom C, Jansson H. 2012. Elevated levels of salivary lactoferrin, a marker for chronic periodontitis? J Periodontal Res. 47(5):655–660. [DOI] [PubMed] [Google Scholar]
- Gorr SU, Abdolhosseini M. 2011. Antimicrobial peptides and periodontal disease. J Clin Periodontol 38(Suppl 11):126–141. [DOI] [PubMed] [Google Scholar]
- Griffen AL, Beall CJ, Campbell JH, Firestone ND, Kumar PS, Yang ZK, Podar M, Leys EJ. 2012. Distinct and complex bacterial profiles in human periodontitis and health revealed by 16S pyrosequencing. ISME J. 6(6):1176–1185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gross EL, Beall CJ, Kutsch SR, Firestone ND, Leys EJ, Griffen AL. 2012. Beyond Streptococcus mutans: dental caries onset linked to multiple species by 16S rRNA community analysis. PloS One. 7(10):e47722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guillen C, McInnes IB, Vaughan D, Speekenbrink AB, Brock JH. 2000. The effects of local administration of lactoferrin on inflammation in murine autoimmune and infectious arthritis. Arthritis Rheum. 43(9):2073–2080. [DOI] [PubMed] [Google Scholar]
- Hajishengallis G. 2014. Immunomicrobial pathogenesis of periodontitis: keystones, pathobionts, and host response. Trends Immunol. 35(1):3–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haubek D, Johansson A. 2014. Pathogenicity of the highly leukotoxic JP2 clone of Aggregatibacter actinomycetemcomitans and its geographic dissemination and role in aggressive periodontitis. J Oral Microbiol [epub ahead of print 14 Aug 2014]. doi: 10.3402/jom.v6.23980 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Helmerhorst EJ, Oppenheim FG. 2007. Saliva: a dynamic proteome. J Dent Res. 86(8):680–693. [DOI] [PubMed] [Google Scholar]
- Hooper LV. 2009. Do symbiotic bacteria subvert host immunity? Nat Rev Microbiol. 7(5):367–374. [DOI] [PubMed] [Google Scholar]
- Jordan WJ, Eskdale J, Lennon GP, Pestoff R, Wu L, Fine DH, Gallagher G. 2005. A non-conservative, coding single-nucleotide polymorphism in the N-terminal region of lactoferrin is associated with aggressive periodontitis in an African-American, but not a Caucasian population. Genes Immun. 6(7):632–635. [DOI] [PubMed] [Google Scholar]
- Kawai T, Akira S. 2005. Pathogen recognition with Toll-like receptors. Curr Opin Immunol. 17(4):338–344. [DOI] [PubMed] [Google Scholar]
- Keijser S, Jager MJ, Dogterom-Ballering HC, Schoonderwoerd DT, de Keizer RJ, Krose CJ, Houwing-Duistermaat JJ, van der Plas MJ, van Dissel JT, Nibbering PH. 2008. Lactoferrin Glu561Asp polymorphism is associated with susceptibility to herpes simplex keratitis. Exp Eye Res. 86(1):105–109. [DOI] [PubMed] [Google Scholar]
- Legrand D, Elass E, Carpentier M, Mazurier J. 2005. Lactoferrin: a modulator of immune and inflammatory responses. Cell Mol Life Sci. 62(22):2549–2559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Legrand D, Elass E, Carpentier M, Mazurier J. 2006. Interactions of lactoferrin with cells involved in immune function. Biochem Cell Bio. 84(3):282–290. [DOI] [PubMed] [Google Scholar]
- Mandel ID. 1987. The functions of saliva. J Dent Res. 66(Spec No):623–627. [DOI] [PubMed] [Google Scholar]
- Marsh PD. 2003. Are dental diseases examples of ecological catastrophes? Microbiol. 149(Pt 2):279–294. [DOI] [PubMed] [Google Scholar]
- Marsh PD, Devine DA. 2011. How is the development of dental biofilms influenced by the host? J Clin Periodontol. 38(Suppl 11):28–35. [DOI] [PubMed] [Google Scholar]
- Miyake K. 2004. Innate recognition of lipopolysaccharide by Toll-like receptor 4-MD-2. Trends Microbiol. 12(4):186–192. [DOI] [PubMed] [Google Scholar]
- Mohamed JA, DuPont HL, Flores J, Palur H, Nair P, Jiang ZD, Guo D, Belkind-Gerson J, Okhuysen PC. 2011. Single nucleotide polymorphisms in the promoter of the gene encoding the lipopolysaccharide receptor CD14 are associated with bacterial diarrhea in US and Canadian travelers to Mexico. Clin Infect Dis. 52(11):1332–1341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mohamed JA, DuPont HL, Jiang ZD, Belkind-Gerson J, Figueroa JF, Armitige LY, Tsai A, Nair P, Martinez-Sandoval FJ, Guo DC, et al. 2007. A novel single-nucleotide polymorphism in the lactoferrin gene is associated with susceptibility to diarrhea in North American travelers to Mexico. Clin Infect Dis. 44(7):945–952. [DOI] [PubMed] [Google Scholar]
- Naidu SS, Svensson U, Kishore AR, Naidu AS. 1993. Relationship between antibacterial activity and porin binding of lactoferrin in Escherichia coli and Salmonella typhimurium. Antimicrob Agents Chemother. 37(2):240–245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nibali L, Henderson B, Sadiq ST, Donos N. 2014. Genetic dysbiosis: the role of microbial insults in chronic inflammatory diseases [epub ahead of print 25 Feb 2014]. J Oral Microbiol. doi: 10.3402/jom.v6.22962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nibbering PH, Ravensbergen E, Welling MM, van Berkel LA, van Berkel PH, Pauwels EK, Nuijens JH. 2001. Human lactoferrin and peptides derived from its N terminus are highly effective against infections with antibiotic-resistant bacteria. Infect Immun. 69(3):1469–1476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Odell EW, Sarra R, Foxworthy M, Chapple DS, Evans RW. 1996. Antibacterial activity of peptides homologous to a loop region in human lactoferrin. FEBS Lett. 382(1–2):175–178. [DOI] [PubMed] [Google Scholar]
- Oppenheim FG, Salih E, Siqueira WL, Zhang W, Helmerhorst EJ. 2007. Salivary proteome and its genetic polymorphisms. Ann NY Acad of Sci. 1098:22–50. [DOI] [PubMed] [Google Scholar]
- Ren L, Leung WK, Darveau RP, Jin L. 2005. The expression profile of lipopolysaccharide-binding protein, membrane-bound CD14, and toll-like receptors 2 and 4 in chronic periodontitis. J Periodontol. 76(11):1950–1959. [DOI] [PubMed] [Google Scholar]
- Rosenfeld Y, Papo N, Shai Y. 2006. Endotoxin (lipopolysaccharide) neutralization by innate immunity host-defense peptides. J Biol Chem. 281(3):1636–1643. [DOI] [PubMed] [Google Scholar]
- Rudney JD, Hickey KL, Ji Z. 1999. Cumulative correlations of lysozyme, lactoferrin, peroxidase, S-IgA, amylase, and total protein concentrations with adherence of oral viridans streptococci to microplates coated with human saliva. J Dent Res. 78(3):759–768. [DOI] [PubMed] [Google Scholar]
- Scannapieco FA. 1994. Saliva-bacterium interactions in oral microbial ecology. Crit Rev Oral Biol Med 5(3–4):203–248. [DOI] [PubMed] [Google Scholar]
- Scott MG, Vreugdenhil AC, Buurman MA, Hancock RE, Gold MR. 2000. Cutting edge: cationic antimicrobial peptides block the binding of lipopolysaccharide (LPS) to LPS binding protein. J Immunol. 164(2):549–553. [DOI] [PubMed] [Google Scholar]
- Sidhu R, Wilson P, Wright A, Yau CW, D’Cruz FA, Foye L, Morley S, Lobo AJ, McAlindon ME, Sanders DS. 2010. Faecal lactoferrin: a novel test to differentiate between the irritable and inflamed bowel? Aliment Pharmacol Ther. 31(12):1365–1370. [DOI] [PubMed] [Google Scholar]
- Sikorska MH, Mielnik-Blaszczak M, Kapeć E. 2002. The relationship between the levels of SigA, lactoferrin and alpha(1) proteinase inhibitor in saliva and permanent dentition caries in 15-year-olds. Oral Microbiol Immunol. 17(5):272–276. [DOI] [PubMed] [Google Scholar]
- Singh PK, Parsek MR, Greenberg EP, Welsh MJ. 2002. A component of innate immunity prevents bacterial biofilm development. Nature. 417(6888):552–555. [DOI] [PubMed] [Google Scholar]
- Velliyagounder K, Kaplan JB, Furgang D, Legarda D, Diamond G, Parkin RE, Fine DH. 2003. One of two human lactoferrin variants exhibits increased antibacterial and transcriptional activation activities and is associated with localized juvenile periodontitis. Infect Immun. 71(11):6141–6147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Velusamy SK, Ganeshnarayan K, Markowitz K, Schreiner H, Furgang D, Fine DH, Velliyagounder K. 2013. Lactoferrin knockout mice demonstrates greater susceptibility to Aggregatibacter actinomycetemcomitans–induced periodontal disease. J Periodontol. 84(11):1690–1701. [DOI] [PubMed] [Google Scholar]
- Visintin A, Iliev DB, Monks BG, Halmen KA, Golenbock DT. 2006. MD-2. Immunobiology. 211(6–8):437–447. [DOI] [PubMed] [Google Scholar]
- Wu YM, Juo SH, Ho YP, Ho KY, Yang YH, Tsai CC. 2009. Association between lactoferrin gene polymorphisms and aggressive periodontitis among Taiwanese patients. J Periodontal Res. 44(3):418–424. [DOI] [PubMed] [Google Scholar]
- Wurfel MM, Hailman E, Wright SD. 1995. Soluble CD14 acts as a shuttle in the neutralization of lipopolysaccharide (LPS) by LPS-binding protein and reconstituted high density lipoprotein. J Exp Med. 181(5):1743–1754. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.