

Abstract
Chronic inflammation is a non-traditional risk factor for cardiovascular mortality in the chronic kidney disease (CKD) population. In recent years the gastrointestinal tract has emerged as a major instigator of systemic inflammation in CKD. Post-mortem studies previously discovered gut wall inflammation present throughout the digestive tract in chronic dialysis patients. In CKD animals, colon wall inflammation is associated with breakdown of the epithelial tight junction barrier (“leaky gut”) and translocation of bacterial DNA and endotoxin into the bloodstream. Gut bacterial DNA and endotoxin have also been detected in the serum from CKD and dialysis patients, whereby endotoxin levels increase with CKD stage and correlate with severity of systemic inflammation in the dialysis population. The CKD diet that is low in plant fiber and symbiotic organisms (in adherence with low potassium, low phosphorus intake) can alter the normal gut microbiome, leading to overgrowth of bacteria that produce uremic toxins such as cresyl and indoxyl molecules. The translocation of these toxins from the “leaky gut” into the bloodstream further promotes systemic inflammation, adverse cardiovascular outcomes and CKD progression. Data is lacking on optimal fiber and yogurt consumption in CKD that would favor growth of a more symbiotic microbiome while avoiding potassium and phosphorus overload. Prebiotic and probiotic formulations have shown promise in small clinical trials, in terms of lowering serum levels of uremic toxins and improving quality of life. The evidence points to a strong relationship between intestinal inflammation and adverse outcomes in CKD, and more trials investigating gut-targeted therapeutics are needed.
Keywords: Gut inflammation, CKD, tight junction barrier, microbiome, uremic toxins, chronic inflammation
Chronic kidney disease (CKD) is a state of accelerated cardiovascular disease, whereby the latter remains the #1 cause of death in this patient population [1]. Non-traditional risk factors contributing to cardiovascular pathology in CKD include chronic inflammation, oxidative stress, protein-energy wasting, disordered mineral metabolism, and deficiency of endogenous calcification inhibitors [2–4]. With respect to chronic inflammation, a multitude of dialysis and non-dialysis related factors likely play a role including infection, intravenous iron administration, blood-dialyzer interface, and preexisting heart failure [5,6]. Of note, despite technical innovations over recent decades such as biocompatible dialysis membranes, non-toxic equipment sterilization, less immunogenic iron preparations, and ultrapure dialysate, the phenomenon of systemic inflammation with its resultant oxidative stress has persisted in CKD patients [7].
Accumulating evidence over the recent years has spotlighted the gastrointestinal tract as a major source of chronic inflammation in CKD. Gut bacterial DNA fragments have been detected in the blood of both pre-dialysis CKD and chronic hemodialysis patients [8]. Using 16s ribosomal DNA amplification and pyrosequencing, Shi et al. detected bacteria (majority of gut origin) in the plasma of 12 of 52 chronic dialysis patients [9]. Presence of bacteria correlated with increased plasma C-reactive protein, interleukin-6, and D-lactate whereby the latter serves as a marker of gut permeability [9]. Endotoxin, derived from the cell wall of Gram-negative bacteria, is measurable in the blood of dialysis patients and correlates with severity of systemic inflammation in the absence of clinically detectable infection [10]. Indeed, levels of circulating endotoxin increase with severity of CKD stage and are most elevated in chronic hemodialysis and peritoneal dialysis patients [11,12].
Microbiota perturbation in CKD and production of uremic toxins
A symbiotic relationship with the gut microbiota is essential for regulation of local and systemic immunity [13,14], whereby the microbiota is an “outside-in” modifier that determines T-cell and natural killer cells subsets [15–17]. In the healthy gut, anaerobic Bacteroidetes and Firmicutes contribute >90% of bacterial species [18]. In accordance with decreasing oxygen tension, bacterial concentrations increase from the stomach (102–104 cells/mL) to the colon (>1012 cells/mL) [13]. The bacterial cells that make up the intestinal microbiota are also part of an important nutrient environment, providing energy-rich metabolites and vitamins to enterocytes [13]. Plant-derived polysaccharides or resistant starches transit intact to the colon where they are degraded by Bacteroides and fermented to release hydrogen, carbon dioxide, alcohol and short-chain fatty acids (acetate, butyrate, propionate and D-lactate) [13]. Colonic cells and bacterial compete for energy-rich short-chain fatty acids, and regulatory mechanisms are in place to ensure bacterial overgrowth does not occur which would deprive the intestinal cells of this nutrient source [13].
Simenhoff and colleagues in the 1970s performed intestinal intubation (endoscopy) in CKD and non-CKD individuals, and were the first to demonstrate markedly altered gut flora in CKD patients [19]. Further, they found that antibiotic treatment to alter composition of the microbiome favorably decreased serum levels of amine toxins and improved mentation [20]. The gut microbiome is markedly altered in CKD [21] whereby there is dominance of bacterial families that possess urease, uricase and indole and p-cresol-forming enzymes [22]. This may be related to the influx of circulating urea and other toxins into the gut lumen, favoring growth of bacteria that express urease. Further, CKD patients are often advised to adhere to diets that are low in fermentable plant fiber (low potassium diet) and poor in symbiont-rich cheese/yogurt (low phosphorus diet). This change in food substrate could further alter the bacterial composition while jeopardizing microbial nutrient production and thus affecting enterocyte health. End-stage kidney disease patients show decreased numbers of bacteria that are able to produce the short-chain fatty acid butyrate [22]. Gas chromatography studies have shown significantly altered exhaled breath gases in CKD rats [23] and dialysis patients [24] compared to healthy controls, further testament to the altered gut microbe composition.
In the presence of this disordered bacterial colonization, there is increased production of gut-derived uremic toxins such as indoxyl sulfate and p-cresyl sulfate [25]. These toxins induce pro-inflammatory responses and promote leukocyte stimulation [26,27], and have been associated with increased levels of inflammatory markers in stage 3–4 CKD patients, such as interleukin-6 and glutathione peroxidase [28,29]. In the 1990s, Niwa’s group demonstrated that indoxyl sulfate stimulated monocyte infiltration in the remnant kidney of 5/6-nephrectomized rats, thus stimulating production of transforming growth factor-beta 1 and progression of renal failure [30]. P-cresyl sulfate has also correlated with increased pulse-wave velocity indicative of vascular stiffening [28]. Recently, Hazen’s group demonstrated that plasma levels of gut microbial-derived trimethylamine-N-oxide (TMAO) correlated with increased 5-year mortality risk in CKD subjects after multivariate adjustment [31]. Dietary TMAO supplementation in animal models resulted in increased tubulointerstitial fibrosis and progressive renal dysfunction [31].
Gut epithelial barrier breakdown and translocation of bacterial toxins
Amazingly, the 1012 bacterial cells/mL in the colon lumen is separated from the host body by just a single-layer absorptive epithelium. In the healthy state, there is a mucosal barrier consisting of a mucus layer, defensins and lectins that shield the epithelium and immune system from direct contact with the microbiota [15,32].
Vaziri and colleagues discovered chronic inflammation throughout the gastrointestinal tract – extending from esophagus to large bowel – of chronic hemodialysis patients, on autopsy studies performed in the 1980s [33]. These inflammatory changes sometimes co-existed with peptic ulcer disease and ischemic lesions [33]. Permeability of the intestinal wall in CKD rats and in CKD patients was demonstrated in the early 1990s in studies utilizing polyethylene glycols of various molecular weights [34,35]. Since the time of these reports, there has been significant progress toward understanding the structure of the gut epithelial barrier, whereby the apical junctional complex is composed of transmembrane proteins including claudins (a group of at least 20 tissue-specific proteins), occludin, E-cadherin and cytoplasmic linker proteins such as zonula occludens [36]. Subsequently, rodent studies have confirmed that CKD is associated with depletion of gut epithelial tight junction proteins including occludin and claudin-1 [37,38]. This leaky barrier allowing translocation of gut bacterial DNA and uremic toxins could well explain the appearance of these noxious inflammatory stimuli in the blood of CKD and chronic dialysis patients [8–12,28]. As further confirmatory evidence, Wang and colleagues reported detection of colon bacterial DNA in the mesenteric lymph nodes, liver, spleen and blood of CKD rats [39].
To date, two mechanisms have been elucidated by which tight junction breakdown occurs (Figure 1). Urea diffuses from the blood into the gut lumen and is metabolized by gut bacterial urease to ammonia that is hydrolyzed into caustic ammonium hydroxide, which erodes the epithelial barrier [40,41]. This stimulates influx of inflammatory leukocytes, which triggers the second mechanism whereby local cytokine production induces retraction and endocytosis of the transcellular tight junction proteins (claudins and occludin) [42,43]. It is easy to see how these pathways can play into a vicious circuit that promotes chronic systemic inflammation.
Figure 1.
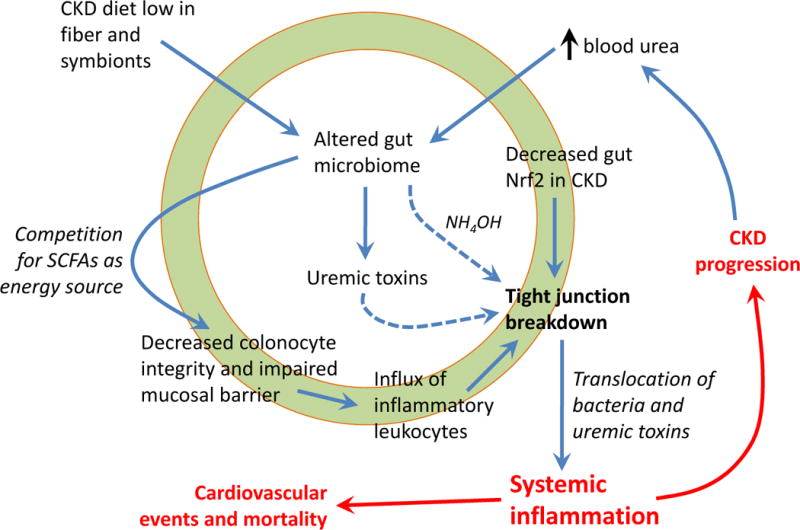
Pathways by which altered microbiome and chronic gut inflammation lead to systemic inflammation in chronic kidney disease (CKD). The low potassium and low phosphorus CKD diet leads to inadequate intake of plant fiber and symbiotic bacteria, and there is influx of urea from the blood circulation in the gut lumen. These factors promote expansion of bacteria that express urease, as well as overgrowth of bacteria that produce uremic toxins including p-cresyl and indoxyl sulfates. Urea is degraded by urease-producing bacteria to form ammonia that is hydrolyzed to caustic ammonium hydroxide (NH4OH) which causes degradation of tight junction proteins. Concurrently, the changed microbiome alters the symbiotic balance and competes with gut epithelial cells for short-chain fatty acids (SCFAs) for source of energy. This may impact colonocyte integrity and impair the protective mucosal barrier, with influx of inflammatory leukocytes and cytokine production. Gut inflammation further promotes epithelial barrier degradation via induction of endocytosis of tight junction proteins. Decreased tissue levels of the anti-inflammatory transcription factor Nrf2 (nuclear factor erythroid 2-related factor 2) further contributes to tight junction breakdown. Ultimately, there is translocation of bacteria, endotoxin and uremic toxins from the gut lumen into the circulation which drives systemic inflammation, a known non-traditional risk factor for cardiovascular mortality and CKD progression. Progression of kidney disease feeds into further elevation of blood urea levels, further propagating alterations in gut microbiome and forming a vicious circuit.
Our recent work showed that the anti-inflammatory transcription factor Nrf2 (nuclear factor erythroid 2-related factor 2) is suppressed in the gut of CKD rats, and treatment with an Nrf2 agonist not only decreased gut inflammation but also improved expression of tight junction proteins [44]. More research is needed to determine the clinical significance of the gut Nrf2 pathway as a novel therapeutic target.
Therapeutic avenues: pre- and pro-biotics, and anti-inflammatory interventions
It is well recognized that the CKD diet low in potassium and phosphorus affects gut microbial composition, and the potassium restriction in particular goes against the “heart healthy” high vegetables/fruit diet that is associated with less cardiovascular events in the general population [45]. However, there is clear data that higher serum potassium and phosphorus levels in the CKD and dialysis population correlate strongly with increased mortality risk [45]. Theoretically, judicious intake of vegetable fiber and symbiont-rich yogurt/cheeses (perhaps with concomitant use of potassium binding resins and phosphate binders) may result in a more balanced gut microbiome and thus improve inflammatory parameters, but randomized controlled trials are needed before new dietary guidelines can be proposed.
The realization that intestinal inflammation and microbiome disturbances are major contributors to chronic inflammation and adverse CKD outcomes has spurred gut-directed therapeutic investigations. Oral activated charcoal adsorbent was shown to partially restore expression of colonic tight junction proteins and resulted in lowering of plasma endotoxin and inflammatory markers in CKD rats, presumably via adsorbing urea and other luminal toxins [46]. However, randomized controlled trials in Japan and the U.S. failed to show beneficial effects in CKD patients in terms of slowing CKD progression [47,48]. Our recent report highlighting Nrf2 deficiency in the gut from CKD rodents demonstrated that treatment with a Nrf2 activator may improve expression of colon tight junction proteins, decrease colonic inflammation and lower blood urea and creatinine levels [44]. The potential benefit of modulating gut Nrf2 activity in patients with CKD remains to be explored.
As noted above, how best to manipulate the natural diet to engender a less pathogenic microbiome is unclear at this time. Another area of potential therapeutics is the use of refined prebiotics (nondigestible food ingredients that can stimulate growth and/or activity of beneficial gut bacteria) and probiotics (living organisms ingested via food or supplements that can improve the health of the host). Our group recently reported that feeding uremic rats the prebiotic amylose maize resistant starch, which reaches the colon undigested and is metabolized by bacteria to short-chain fatty acids, improved creatinine clearance and reduced kidney inflammation and fibrosis [49]. In a small trial with hemodialysis patients in Belgium, it was reported that oligofructose-inulin supplementation significantly reduced circulating p-cresyl sulfate levels [50]. A meta-analysis of controlled feeding trials found that fiber supplementation significantly decreased serum urea levels in a pooled cohort of 143 participants; the authors noted that trials examining harder end points are needed [51]. Probiotics have also shown some promise with the premise that correction of the intestinal microflora make-up can reduce production of uremic toxins. In two 1996 reports involving small cohorts of hemodialysis patients, treatment with Lactobacillus preparations decreased blood levels of uremic toxins [52,53] and improved nutritional status [52]. More recently, a multi-national crossover trial in patients with CKD stage 3 and 4 noted significant decrease in blood urea nitrogen and improved quality of life scores after treatment with a proprietary formulation of S. thermophilus, L. acidophilus and B. longum over 6 months [54]. It is not known how the interventions described above may have impacted gut tight junction barrier integrity, which would have required invasive testing.
Taking the next step given the promising effects of pre- and pro-biotic formulations, there are now efforts to combine both therapies into “synbiotic” preparations, and to examine whether this therapy results in decreased systemic inflammation and slowing of CKD progression. A small Japanese study reported that a synbiotic preparation reduced serum p-cresol levels and ameliorated constipation [55]. In attempts to fill the gap where high-quality intervention trials are lacking, Rossi and colleagues recently described the study design for the ongoing SYNERGY trial that is taking place in Australia and New Zealand [56]. This trial is recruiting stage 4–5 pre-dialysis CKD patients, and in placebo-controlled randomized cross-over design each patient will be subjected to a synbiotic therapy (a combination of Lactobacillus, Bifidobacteria and Streptococcus bacteria, with three complex fibers) or placebo ×6 weeks. While circulating indoxyl sulfate levels will be the primary outcome, the study will also examine systemic inflammation markers, proteinuria, kidney function and urinary kidney injury molecule-1, quality of life and stool microbiota. We note that the Streptococcus has high urease capacity, and given the abovementioned evidence pertaining to urea breakdown as a mechanism leading to leaky gut epithelial barrier [40,41], inclusion of this organism in the probiotic formulation may be deleterious. Nonetheless we applaud the rigorous study design of the SYNERGY trial and if promising data results, a longer intervention period examining hard outcomes such as cardiovascular events and mortality would be warranted.
Planning clinical trials around the gut microbiome and interpreting the results is certainly not straightforward. Factors that may contribute to variability in response to therapy across individuals include varying density of intestinal flora, genetic differences affecting host-microbe interactions, and different microbial composition. Indeed, it was reported in 2011 from analysis of fecal metagenomes from four countries that there are three discernable enterotypes of the human microbiome [57]. The degree of change in measured serum inflammatory markers that would translate into meaningful clinical outcomes is also unclear at this time.
Summary
Chronic inflammation is present throughout the gastrointestinal tract of uremic patients. Recent animal studies highlight breakdown of colon epithelial tight junctions, via activity of bacterial urease and local inflammation with cytokine production. The microbiota is altered due to the restrictive CKD diet and influx of urea from the circulation, favoring a microbial population that generates uremic toxins including cresyl and indoxyl molecules. The various pathways that converge and result in epithelial barrier breakdown are summarized in Figure 1. Ultimately, the damaged “leaky gut” allows translocation of bacteria and bacterial products into the systemic circulation, promoting chronic inflammation which drives adverse cardiovascular outcomes and CKD progression. Gut bacterial DNA fragments are detectable in the bloodstream from both CKD and dialysis patients, and correlate with severity of systemic inflammation. Serum levels of gut-derived indoxyl sulfate and p-cresyl sulfate toxins also correlate strongly with systemic inflammation, as well as vascular stiffness. The gut is a promising therapeutic target in CKD, whereby prebiotic and probiotic formulations have shown promise in small clinical trials in terms of lowering levels of uremic toxins and improving quality of life. Data is lacking on optimal dietary guidelines in CKD that would favor growth of a more symbiotic microbiome while avoiding potassium and phosphorus overload. Given that the evidence points to a key role for intestinal inflammation in CKD, more randomized clinical trials are needed to explore the benefits of gut-targeted therapeutics.
Acknowledgments
The work is supported by NIH-NIDDK grants K24-DK091419 (KKZ) and philanthropic grants from Mr. Harold Simmons and Mr. Louis Chang.
Footnotes
Potential Conflict of Interest:
KKZ has received honoraria from Abbott, Abbvie, Amgen, Genzyme/Sanofi, Otsuka, Shire, Vifor, and was the medical director of DaVita Harbor-UCLA/MFI in Long Beach, CA, during 2007–2012. Other authors have not declared any conflicts of interest.
References
- 1.Gansevoort RT, Correa-Rotter R, Hemmelgarn BR, et al. Chronic kidney disease and cardiovascular risk: epidemiology, mechanisms, and prevention. Lancet. 2013;382(9889):339–352. doi: 10.1016/S0140-6736(13)60595-4. [DOI] [PubMed] [Google Scholar]
- 2.Kalantar-Zadeh K, Ikizler TA, Block G, Avram MM, Kopple JD. Malnutrition-inflammation complex syndrome in dialysis patients: causes and consequences. Am J Kidney Dis. 2003;42(5):864–881. doi: 10.1016/j.ajkd.2003.07.016. [DOI] [PubMed] [Google Scholar]
- 3.Kendrick J, Chonchol MB. Nontraditional risk factors for cardiovascular disease in patients with chronic kidney disease. Nat Clin Pract Nephrol. 2008;4(12):672–681. doi: 10.1038/ncpneph0954. [DOI] [PubMed] [Google Scholar]
- 4.Lau WL, Ix JH. Clinical detection, risk factors, and cardiovascular consequences of medial arterial calcification: a pattern of vascular injury associated with aberrant mineral metabolism. Semin Nephrol. 2013;33(2):93–105. doi: 10.1016/j.semnephrol.2012.12.011. [DOI] [PubMed] [Google Scholar]
- 5.Carrero JJ, Stenvinkel P. Persistent inflammation as a catalyst for other risk factors in chronic kidney disease: a hypothesis proposal. Clin J Am Soc Nephrol. 2009;4(Suppl 1):S49–55. doi: 10.2215/CJN.02720409. [DOI] [PubMed] [Google Scholar]
- 6.Vaziri ND. Understanding iron: promoting its safe use in patients with chronic kidney failure treated by hemodialysis. Am J Kidney Dis. 2013;61(6):992–1000. doi: 10.1053/j.ajkd.2012.10.027. [DOI] [PubMed] [Google Scholar]
- 7.Carrero JJ, Stenvinkel P. Inflammation in end-stage renal disease–what have we learned in 10 years? Semin Dial. 2010;23(5):498–509. doi: 10.1111/j.1525-139X.2010.00784.x. [DOI] [PubMed] [Google Scholar]
- 8.Wang F, Jiang H, Shi K, Ren Y, Zhang P, Cheng S. Gut bacterial translocation is associated with microinflammation in end-stage renal disease patients. Nephrology (Carlton) 2012;17(8):733–738. doi: 10.1111/j.1440-1797.2012.01647.x. [DOI] [PubMed] [Google Scholar]
- 9.Shi K, Wang F, Jiang H, et al. Gut bacterial translocation may aggravate microinflammation in hemodialysis patients. Dig Dis Sci. 2014;59(9):2109–2117. doi: 10.1007/s10620-014-3202-7. [DOI] [PubMed] [Google Scholar]
- 10.Feroze U, Kalantar-Zadeh K, Sterling KA, et al. Examining associations of circulating endotoxin with nutritional status, inflammation, and mortality in hemodialysis patients. J Ren Nutr. 2012;22(3):317–326. doi: 10.1053/j.jrn.2011.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Szeto CC, Kwan BC, Chow KM, et al. Endotoxemia is related to systemic inflammation and atherosclerosis in peritoneal dialysis patients. Clin J Am Soc Nephrol. 2008;3(2):431–436. doi: 10.2215/CJN.03600807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.McIntyre CW, Harrison LE, Eldehni MT, et al. Circulating endotoxemia: a novel factor in systemic inflammation and cardiovascular disease in chronic kidney disease. Clin J Am Soc Nephrol. 2011;6(1):133–141. doi: 10.2215/CJN.04610510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Anders HJ, Andersen K, Stecher B. The intestinal microbiota, a leaky gut, and abnormal immunity in kidney disease. Kidney Int. 2013;83(6):1010–1016. doi: 10.1038/ki.2012.440. [DOI] [PubMed] [Google Scholar]
- 14.Ramezani A, Raj DS. The gut microbiome, kidney disease, and targeted interventions. J Am Soc Nephrol. 2014;25(4):657–670. doi: 10.1681/ASN.2013080905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hooper LV, Littman DR, Macpherson AJ. Interactions between the microbiota and the immune system. Science. 2012;336(6086):1268–1273. doi: 10.1126/science.1223490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kranich J, Maslowski KM, Mackay CR. Commensal flora and the regulation of inflammatory and autoimmune responses. Semin Immunol. 2011;23(2):139–145. doi: 10.1016/j.smim.2011.01.011. [DOI] [PubMed] [Google Scholar]
- 17.Macpherson AJ, Harris NL. Interactions between commensal intestinal bacteria and the immune system. Nat Rev Immunol. 2004;4(6):478–485. doi: 10.1038/nri1373. [DOI] [PubMed] [Google Scholar]
- 18.Qin J, Li R, Raes J, et al. A human gut microbial gene catalogue established by metagenomic sequencing. Nature. 2010;464(7285):59–65. doi: 10.1038/nature08821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Simenhoff ML, Saukkonen JJ, Burke JF, Wesson LG, Schaedler RW, Gordon SJ. Bacterial populations of the small intestine in uremia. Nephron. 1978;22(1–3):63–68. doi: 10.1159/000181424. [DOI] [PubMed] [Google Scholar]
- 20.Simenhoff ML, Saukkonen JJ, Burke JF, Wesson LG, Schaedler RW. Amine metabolism and the small bowel in uraemia. Lancet. 1976;2(7990):818–821. doi: 10.1016/s0140-6736(76)91207-1. [DOI] [PubMed] [Google Scholar]
- 21.Vaziri ND, Wong J, Pahl M, et al. Chronic kidney disease alters intestinal microbial flora. Kidney Int. 2013;83(2):308–315. doi: 10.1038/ki.2012.345. [DOI] [PubMed] [Google Scholar]
- 22.Wong J, Piceno YM, Desantis TZ, Pahl M, Andersen GL, Vaziri ND. Expansion of urease- and uricase-containing, indole- and p-cresol-forming and contraction of short-chain fatty acid-producing intestinal microbiota in ESRD. Am J Nephrol. 2014;39(3):230–237. doi: 10.1159/000360010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Meinardi S, Jin KB, Barletta B, Blake DR, Vaziri ND. Exhaled breath and fecal volatile organic biomarkers of chronic kidney disease. Biochim Biophys Acta. 2013;1830(3):2531–2537. doi: 10.1016/j.bbagen.2012.12.006. [DOI] [PubMed] [Google Scholar]
- 24.Lee HJ, Pahl MV, Vaziri ND, Blake DR. Effect of hemodialysis and diet on the exhaled breath methanol concentration in patients with ESRD. J Ren Nutr. 2012;22(3):357–364. doi: 10.1053/j.jrn.2011.07.003. [DOI] [PubMed] [Google Scholar]
- 25.Evenepoel P, Meijers BK, Bammens BR, Verbeke K. Uremic toxins originating from colonic microbial metabolism. Kidney Int Suppl. 2009(114):S12–19. doi: 10.1038/ki.2009.402. [DOI] [PubMed] [Google Scholar]
- 26.Motojima M, Hosokawa A, Yamato H, Muraki T, Yoshioka T. Uremic toxins of organic anions up-regulate PAI-1 expression by induction of NF-kappaB and free radical in proximal tubular cells. Kidney Int. 2003;63(5):1671–1680. doi: 10.1046/j.1523-1755.2003.00906.x. [DOI] [PubMed] [Google Scholar]
- 27.Schepers E, Meert N, Glorieux G, Goeman J, Van der Eycken J, Vanholder R. Pcresylsulphate, the main in vivo metabolite of p-cresol, activates leucocyte free radical production. Nephrol Dial Transplant. 2007;22(2):592–596. doi: 10.1093/ndt/gfl584. [DOI] [PubMed] [Google Scholar]
- 28.Rossi M, Campbell KL, Johnson DW, et al. Protein-bound uremic toxins, inflammation and oxidative stress: a cross-sectional study in stage 3–4 chronic kidney disease. Arch Med Res. 2014;45(4):309–317. doi: 10.1016/j.arcmed.2014.04.002. [DOI] [PubMed] [Google Scholar]
- 29.Moradi H, Sica DA, Kalantar-Zadeh K. Cardiovascular burden associated with uremic toxins in patients with chronic kidney disease. Am J Nephrol. 2013;38(2):136–148. doi: 10.1159/000351758. [DOI] [PubMed] [Google Scholar]
- 30.Miyazaki T, Ise M, Hirata M, et al. Indoxyl sulfate stimulates renal synthesis of transforming growth factor-beta 1 and progression of renal failure. Kidney Int Suppl. 1997;63:S211–214. [PubMed] [Google Scholar]
- 31.Tang WH, Wang Z, Kennedy DJ, et al. Gut Microbiota-Dependent Trimethylamine N-Oxide (TMAO) Pathway Contributes to Both Development of Renal Insufficiency and Mortality Risk in Chronic Kidney Disease. Circ Res. 2015;116(3):448–455. doi: 10.1161/CIRCRESAHA.116.305360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Lathrop SK, Bloom SM, Rao SM, et al. Peripheral education of the immune system by colonic commensal microbiota. Nature. 2011;478(7368):250–254. doi: 10.1038/nature10434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Vaziri ND, Dure-Smith B, Miller R, Mirahmadi MK. Pathology of gastrointestinal tract in chronic hemodialysis patients: an autopsy study of 78 cases. Am J Gastroenterol. 1985;80(8):608–611. [PubMed] [Google Scholar]
- 34.Magnusson M, Magnusson KE, Sundqvist T, Denneberg T. Increased intestinal permeability to differently sized polyethylene glycols in uremic rats: effects of low- and high-protein diets. Nephron. 1990;56(3):306–311. doi: 10.1159/000186158. [DOI] [PubMed] [Google Scholar]
- 35.Magnusson M, Magnusson KE, Sundqvist T, Denneberg T. Impaired intestinal barrier function measured by differently sized polyethylene glycols in patients with chronic renal failure. Gut. 1991;32(7):754–759. doi: 10.1136/gut.32.7.754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Laukoetter MG, Bruewer M, Nusrat A. Regulation of the intestinal epithelial barrier by the apical junctional complex. Curr Opin Gastroenterol. 2006;22(2):85–89. doi: 10.1097/01.mog.0000203864.48255.4f. [DOI] [PubMed] [Google Scholar]
- 37.Vaziri ND, Yuan J, Rahimi A, Ni Z, Said H, Subramanian VS. Disintegration of colonic epithelial tight junction in uremia: a likely cause of CKD-associated inflammation. Nephrol Dial Transplant. 2012;27(7):2686–2693. doi: 10.1093/ndt/gfr624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Vaziri ND, Yuan J, Nazertehrani S, Ni Z, Liu S. Chronic kidney disease causes disruption of gastric and small intestinal epithelial tight junction. Am J Nephrol. 2013;38(2):99–103. doi: 10.1159/000353764. [DOI] [PubMed] [Google Scholar]
- 39.Wang F, Zhang P, Jiang H, Cheng S. Gut bacterial translocation contributes to microinflammation in experimental uremia. Dig Dis Sci. 2012;57(11):2856–2862. doi: 10.1007/s10620-012-2242-0. [DOI] [PubMed] [Google Scholar]
- 40.Vaziri ND, Goshtasbi N, Yuan J, et al. Uremic plasma impairs barrier function and depletes the tight junction protein constituents of intestinal epithelium. Am J Nephrol. 2012;36(5):438–443. doi: 10.1159/000343886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Vaziri ND, Yuan J, Norris K. Role of urea in intestinal barrier dysfunction and disruption of epithelial tight junction in chronic kidney disease. Am J Nephrol. 2013;37(1):1–6. doi: 10.1159/000345969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Nusrat A, Turner JR, Madara JL. Molecular physiology and pathophysiology of tight junctions. IV. Regulation of tight junctions by extracellular stimuli: nutrients, cytokines, and immune cells. Am J Physiol Gastrointest Liver Physiol. 2000;279(5):G851–857. doi: 10.1152/ajpgi.2000.279.5.G851. [DOI] [PubMed] [Google Scholar]
- 43.Al-Sadi R, Boivin M, Ma T. Mechanism of cytokine modulation of epithelial tight junction barrier. Front Biosci (Landmark Ed) 2009;14:2765–2778. doi: 10.2741/3413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Lau WL, Liu SM, Pahlevan S, et al. Role of Nrf2 Dysfunction in Uremia-Associated Intestinal Inflammation and Epithelial Barrier Disruption. Dig Dis Sci. 2014 doi: 10.1007/s10620-014-3428-4. [DOI] [PubMed] [Google Scholar]
- 45.Kalantar-Zadeh K, Tortorici AR, Chen JL, et al. Dietary restrictions in dialysis patients: is there anything left to eat? Semin Dial. 2015;28(2):159–168. doi: 10.1111/sdi.12348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Vaziri ND, Yuan J, Khazaeli M, Masuda Y, Ichii H, Liu S. Oral activated charcoal adsorbent (AST-120) ameliorates chronic kidney disease-induced intestinal epithelial barrier disruption. Am J Nephrol. 2013;37(6):518–525. doi: 10.1159/000351171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Akizawa T, Asano Y, Morita S, et al. Effect of a carbonaceous oral adsorbent on the progression of CKD: a multicenter, randomized, controlled trial. Am J Kidney Dis. 2009;54(3):459–467. doi: 10.1053/j.ajkd.2009.05.011. [DOI] [PubMed] [Google Scholar]
- 48.Schulman G, Berl T, Beck GJ, et al. Randomized Placebo-Controlled EPPIC Trials of AST-120 in CKD. J Am Soc Nephrol. 2014 doi: 10.1681/ASN.2014010042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Vaziri ND, Liu SM, Lau WL, et al. High Amylose Resistant Starch Diet Ameliorates Oxidative Stress, Inflammation, and Progression of Chronic Kidney Disease. PLoS One. 2014;9(12):e114881. doi: 10.1371/journal.pone.0114881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Meijers BK, De Preter V, Verbeke K, Vanrenterghem Y, Evenepoel P. p-Cresyl sulfate serum concentrations in haemodialysis patients are reduced by the prebiotic oligofructose-enriched inulin. Nephrol Dial Transplant. 2010;25(1):219–224. doi: 10.1093/ndt/gfp414. [DOI] [PubMed] [Google Scholar]
- 51.Chiavaroli L, Mirrahimi A, Sievenpiper JL, Jenkins DJ, Darling PB. Dietary fiber effects in chronic kidney disease: a systematic review and meta-analysis of controlled feeding trials. Eur J Clin Nutr. 2014 doi: 10.1038/ejcn.2014.237. [DOI] [PubMed] [Google Scholar]
- 52.Simenhoff ML, Dunn SR, Zollner GP, et al. Biomodulation of the toxic and nutritional effects of small bowel bacterial overgrowth in end-stage kidney disease using freeze-dried Lactobacillus acidophilus. Miner Electrolyte Metab. 1996;22(1–3):92–96. [PubMed] [Google Scholar]
- 53.Hida M, Aiba Y, Sawamura S, Suzuki N, Satoh T, Koga Y. Inhibition of the accumulation of uremic toxins in the blood and their precursors in the feces after oral administration of Lebenin, a lactic acid bacteria preparation, to uremic patients undergoing hemodialysis. Nephron. 1996;74(2):349–355. doi: 10.1159/000189334. [DOI] [PubMed] [Google Scholar]
- 54.Ranganathan N, Ranganathan P, Friedman EA, et al. Pilot study of probiotic dietary supplementation for promoting healthy kidney function in patients with chronic kidney disease. Adv Ther. 2010;27(9):634–647. doi: 10.1007/s12325-010-0059-9. [DOI] [PubMed] [Google Scholar]
- 55.Nakabayashi I, Nakamura M, Kawakami K, et al. Effects of synbiotic treatment on serum level of p-cresol in haemodialysis patients: a preliminary study. Nephrol Dial Transplant. 2011;26(3):1094–1098. doi: 10.1093/ndt/gfq624. [DOI] [PubMed] [Google Scholar]
- 56.Rossi M, Johnson DW, Morrison M, et al. SYNbiotics Easing Renal failure by improving Gut microbiologY (SYNERGY): a protocol of placebo-controlled randomised cross-over trial. BMC Nephrol. 2014;15:106. doi: 10.1186/1471-2369-15-106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Arumugam M, Raes J, Pelletier E, et al. Enterotypes of the human gut microbiome. Nature. 2011;473(7346):174–180. doi: 10.1038/nature09944. [DOI] [PMC free article] [PubMed] [Google Scholar]