

Abstract
While multiple myeloma (MM) is almost invariably preceded by asymptomatic monoclonal gammopathy of undetermined significance (MGUS) and/or smoldering MM (SMM), the alterations of the bone marrow (BM) microenvironment that establish progression to symptomatic disease are circumstantial. Here we show that in Vk*MYC mice harboring oncogene-driven plasma cell proliferative disorder, disease appearance associated with substantial modifications of the BM microenvironment, including a progressive accumulation of both CD8+ and CD4+ T cells with a dominant T helper type 1 (Th1) response. Progression from asymptomatic to symptomatic MM was characterized by further BM accrual of T cells with reduced Th1 and persistently increased Th2 cytokine production, which associated with accumulation of CD206+Tie2+ macrophages, and increased pro-angiogenic cytokines and microvessel density (MVD). Notably, MVD was also increased at diagnosis in the BM of MGUS and SMM patients that subsequently progressed to MM when compared with MGUS and SMM that remained quiescent. These findings suggest a multistep pathogenic process in MM, in which the immune system may contribute to angiogenesis and disease progression. They also suggest initiating a large multicenter study to investigate MVD in asymptomatic patients as prognostic factor for the progression and outcome of this disease.
Keywords: angiogenesis, macrophages, multiple myeloma, smoldering multiple myeloma, T cells
Abbreviations: BM, bone marrow; BMb, BM biopsies; MGUS, monoclonal gammopathy of undetermined significance; MM, multiple myeloma; M-spike, monoclonal (M) protein; MVD, microvessel density; SMM, smoldering multiple myeloma; TAMs, M2 tumor associated macrophages; TEMs, Tie2-expressing macrophages
Introduction
Multiple myeloma (MM) is a B cell neoplasm characterized by the accumulation of clonal plasma cells within the BM, which in most cases is associated with monoclonal (M) protein (M-spike) in blood and/or urine. MM is also characterized by end-organ damage such as anemia, hypercalcemia, renal insufficiency and bone lesions.1 Often if not always, MM is preceded by MGUS, a pre-malignant condition without clinical symptoms of malignancy.2,3 In some patients, an intermediate condition defined as SMM, which is characterized by an M-spike of 3g/dL or more and BM containing ≥10% of plasma cells without evidence of end-organ damage related to the underlying plasma-cell disorder,4 anticipates MM. Despite recent promising advances in the treatment of MM,5 the disease remains incurable, and all patients eventually relapse and die for the disease.
Genetic studies have found copy number alterations at all stages, and lack of substantial genetic differences between MGUS and MM.6,7 Also, primary chromosomal translocations affecting the Ig locus are present at the MGUS phase of the disease, although additional aberrations, the so-called secondary translocations arise during disease progression.7 Thus, all together these data suggest that MM results from the progressive accumulation of epigenetic, genetic, transcriptional, and phenotypic changes within the transforming plasma cell.
While the BM microenvironment has a pivotal role in providing critical cell–cell interactions and autocrine and paracrine signaling to support malignant plasma cell growth and survival, its role in disease progression is only partially deciphered.8,9 This is particularly true for the immune response that in different solid tumors has been clearly shown to either eliminate transformed cells or promote their tumorigenic potential.8 Thus, the nature of the immune response during the early phases of MM could already have profound consequences in determining whether neoplastic plasma cells are eliminated or allowed to expand. This lack of knowledge has direct clinical implications. Indeed, while only 1% of MGUS patients per year will move forward to MM, and notwithstanding the risk to progress to MM in SMM patient increases to 10% per year, no reliable methods to predict disease progression are available. Several prognostic criteria have been proposed (e.g., the progressive increase in the M-spike in the first year of follow-up, the type of monoclonal immunoglobulin, or the serum free light chain ratio) with limited clinical impact,3 and attempts to treat MM precursor conditions have produced mixed results,3 which are likely due to the inability to select for patients that really need a treatment. A relevant exception has been one phase III trial in which specific biomarkers were implemented to focus on patients with the highest probability to have symptomatic MM.10
Thus, a better understanding of the changes to the complex interactions of malignant plasma cells and their surrounding immune cells in the BM milieu, especially at the early phases of disease, is eagerly needed to better define the risk of progression, and provide the framework for novel therapies targeting these interactions. To this aim, we have investigated the BM microenvironment during disease progression in Vk*MYC mice in which the activation of the transcription factor MYC, whose locus is found rearranged in half human MM tumors11 including SMM,12 occurs sporadically through the exploitation of the physiological somatic hypermutation process in germinal center B cells. Within a year, although with variable intensity, all mice develop a monoclonal plasmacytosis confined to the BM, a measurable serum M-spike, and progressively show typical end-organ damage.13 When compared with other existing mouse models of human MM, the Vk*MYC model appears to more fully recapitulate the human disease,14 and it has been already validated as a faithful model to predict single agent drug activity in human MM with a positive predictive value of 67% for clinical activity, and a negative predictive value of 88% for clinical inactivity,15
We have found in Vk*MYC mice that disease appearance was characterized by substantial modifications of the BM microenvironment, including a progressive accumulation of both CD8+ and CD4+ T cells with a dominant Th1 response. Instead, progression from asymptomatic to symptomatic MM in these mice associated with imbalance between Th1 and Th2 immune responses, accumulation of CD206+Tie2+ macrophages, and increased pro-angiogenic cytokines and MVD. Importantly, MVD was found significantly increased at diagnosis also in MGUS and SMM patients that, under monitoring, subsequently progressed to MM when compared with MGUS and SMM subjects who remained stable, thus suggesting that MVD at diagnosis might be a predictive marker of disease progression in MM.
Results
Modification of the BM immune infiltrate is an early event in Vk*MYC mice with M-spike
To identify immune correlates of disease progression in Vk*MYC mice, we conducted multi-parameter flow cytometry analyses in aging mice with M-spike at serum electrophoresis,13 and compared these findings with those obtained from age and sex-matched wild type (WT) littermates. The BM of Vk*MYC mice harboring an M-spike showed a progressive accumulation of CD3+ T cells that was independent of mouse age, and not present in WT mice (data not shown). This accumulation involved both CD8+ (Fig. 1A) and CD4+ T cells (Fig. 1B). Interestingly, a much higher frequency of CD8+ T cells and especially CD4+ T cells was evident in mice with an M-spike ≥6%, which corresponds to 3–5 g/dL of paraprotein (Figs. 1A and B). When these cells were assessed for intracellular cytokine production, the frequency of both CD8+ T cells producing IFNγ or TNFα, and CD4+ T cells producing IFNγ or IL2, all type-1 cytokines, was higher in the BM of Vk*MYC mice with an M-spike <6% than in WT mice (Figs. 1C and D, respectively), thus demonstrating that the BM microenvironment of Vk*MYC mice is the site of an inflammatory reaction with earlier homing of type-1 T cells. Interestingly, while the percentage of CD8+ IFNγ+ and CD4+ IL2+ type-1 T cells significantly dropped in Vk*MYC mice with an M-spike ≥6% when compared with mice with an M-spike <6%, the production of type-2 cytokines (i.e., IL4 and IL13; Fig. 1E) remained higher in all Vk*MYC than in WT mice, thus suggesting a tendency toward a type-2 immune response in more advanced phases of disease.
Figure 1.

Modifications of the T cell infiltrate in the BM of Vk*MYC mice during disease progression. (A and B) The frequency of CD8+ and CD4+ T cells was assessed in the BM of Vk*MYC mice and age-matched WT littermates by flow cytometry after staining with the indicated mAbs. Each dot represents an individual mouse. (C–E) Cells were also analyzed by intracellular cytokine production assay upon incubation with PMA and Ionomycin for 4 h at 37°C. BFA was added during the last 3 h of stimulation. Dead cells were excluded by live/dead staining. Aggregated data from five independent experiments are reported as mean ± SE. Statistical analyses (Student's t test): *p < 0.05; **p < 0.01; ***p < 0.001.
An asymptomatic phase that progresses to symptomatic MM can be identified in Vk*MYC mice
To better correlate the BM immune infiltrate with the disease status, plasma cells were quantified in the BM of Vk*MYC mice and age-and sex-matched littermates. As depicted in representative BM sections stained with an anti-IRF4/MUM1 mAb16 (Fig. 2A), and quantified in blind (Fig. 2B), plasma cells progressively accumulated in the BM of Vk*MYC mice, but not in WT mice. Indeed, Vk*MYC mice with an M-spike <6% (Fig. 2C) had an average plasma cell content of 30%, which was significantly higher than that found in WT mice (approximately 10%), but lower than the >50% of the nucleated cells (Fig. 2B) that could be found in the BM of mice with M-spike ≥6% (Fig. 2C).
Figure 2.

Both M-spike and BM plasma cells increase during disease progression in Vk*MYC mice. (A) Paraffin-embedded sections of the shinbone from Vk*MYC and sex-and age-matched WT littermates were analyzed by immunohistochemistry after staining with anti-IRF-4 mAb (40x magnification). (B) The number of BM plasma cells was also quantified in blind by an expert pathologist. (C) Mice were also assessed for percentage of serum M-spike by serum protein electrophoresis analyses. (D) Hemoglobin (Hgb) concentration, and (E) bone mineral density were assessed individually in the indicated groups of mice as described in the Materials and Methods section. Each dot represents an individual mouse. Data are reported as mean ± SE. Statistical analyses (Student's t test): *p < 0.05; **p < 0.01; ***p < 0.001.
As target organ damage is a defining criterion for MM, and Vk*MYC affected by MM also develop anemia, renal damage and bone lytic lesions,13 our cohort of Vk*MYC mice was investigated for symptomatic disease. Indeed, anemia (Fig. 2D) and a much more pronounced loss of bone mineral density (Fig. 2E) was evident in Vk*MYC mice with an M-spike ≥6%.
In summary, Vk*MYC mice with an M-spike <6%, while having by definition MYC alterations, were characterized by BM containing between 10% and 60% of plasma cells and no evidence of substantial end-organ damage attributable to the plasma cell disorder. Thus, for the sake of simplicity, Vk*MYC mice with an early disease characterized by M-spike <6% were hereafter defined as affected by SMM.
Progression from SMM to MM in Vk*Myc mice is heralded by an angiogenic switch
As rearrangement of MYC has been associated with progression to MM,13 and MM is also associated with increased BM angiogenesis,17 we hypothesized that also in Vk*MYC mice disease progression is characterized by an angiogenic switch. Thus, BM samples from Vk*MYC and WT mice were stained with an anti-CD31 mAb and vessels counted in blind. While we did not notice any difference in MVD when comparing samples from WT and SMM Vk*MYC mice, BM from MM Vk*MYC mice showed almost twice the amount of vessels (Fig. 3A). Of note, MVD strongly correlated with M-spike content (Fig. 3B), therefore suggesting a direct relationship between disease activity and paraprotein level in this model.
Figure 3.
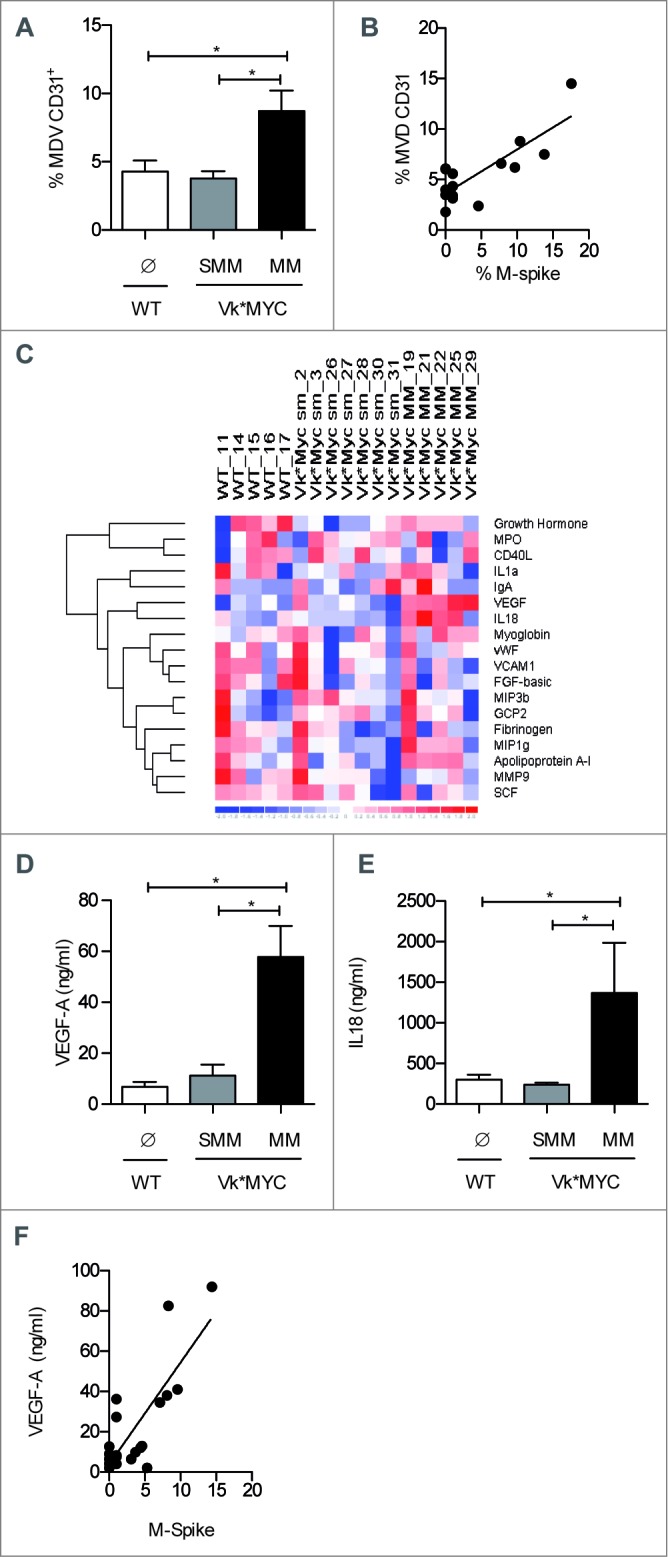
Increased BM angiogenesis characterizes the transition from SMM to MM in Vk*MYC mice. (A) Paraffin-embedded sections of the shinbone from Vk*MYC and sex-and age-matched WT littermates were stained with anti-CD31 mAb, and vessels were quantified by an expert pathologist. Data are reported as MVD for each shinbone. (B) Correlation plot of MVD and percentage of M-spike. Each dot represents an individual mouse. Statistical analyses (Linear Regression): p = 0.002 R2 = 0.65. (C) The indicated soluble molecules were quantified in the BM sera of the same groups of mice (n = 7/group) by Mouse Cytokine MAP B v 1.0. The Heat map shows the level of cytokines with values above 0, clustering using as distance metric 1-correlation, as linkage method average. (D and E) Data for the most relevant cytokines are also reported as mean ± SE, and are representative of two independent experiments. Statistical analyses (Student's t test): * p < 0.05. (F) Correlation plot for VEGF (pg/mL) and M-spike (%). Each dot represents an individual mouse. Statistical analyses (Linear Regression): p < 0.0001 R2 = 0.64.
In search for a mechanistic explanation of the angiogenic switch, we measured several cytokines in BM serum of Vk*MYC and age-and sex-matched WT mice. As previously reported in MM patients,18,19 among the 18 cytokines explored (Fig. 3C), a strong, significant increase of VEGF-A and IL18, two angiogenesis-promoting cytokines,19,20 was evident in Vk*MYC mice at the phase of MM (Figs. 3D and E, respectively). Again, a correlation was found between peripheral blood M-spike and VEGF concentration in BM serum (Fig. 3F).
Having found that in the BM of Vk*MYC mice progressing to angiogenesis there were increased VEGF and frequency of T cells producing Th2 cytokines, such as IL4 and IL13 (Fig. 1E), both conditions favoring accumulation of alternatively activated/M2 tumor associated macrophages (TAMs21), we investigated the characteristics of BM-infiltrating CD11b+ F4/80+ TAMs. Indeed, CD11b+ F4/80+ TAMs appeared to preferentially accumulate in the BM of MM Vk*MYC mice (Fig. 4A), and the frequency of bona fide pro-angiogenic CD11b+ F4/80+Tie2+ macrophages (Tie2-expressing macrophages, TEMs22) were much higher than in BM of SMM mice (Fig. 4B). The frequency of BM TEMs well correlated with serum M-spike (Fig. 4C), thus suggesting a role for these cells in BM angiogenesis.
Figure 4.
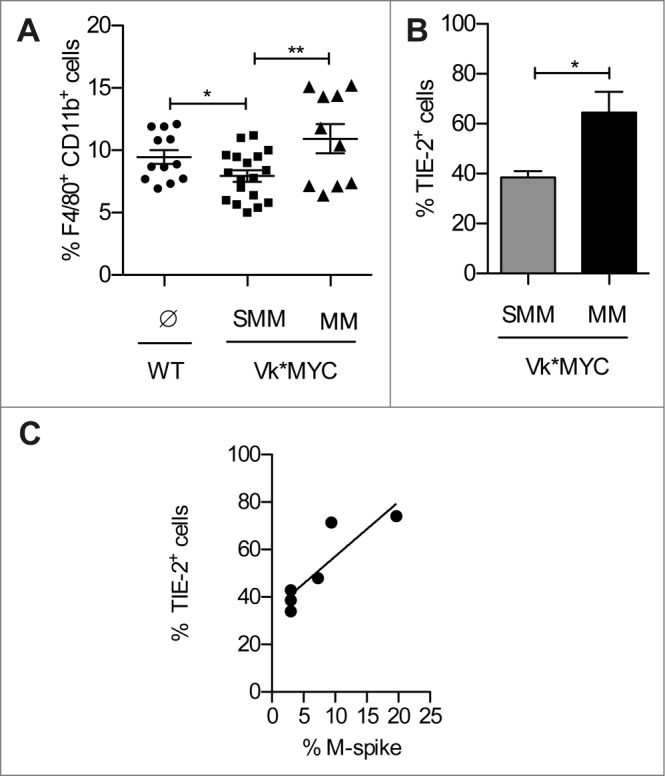
TEMs accumulate in the BM of Vk*MYC mice during disease progression. (A) The frequency of CD11b+ F4/80+ macrophages was assessed in the BM of Vk*MYC mice and age-matched WT littermates by flow cytometry after staining with the indicated mAbs. Dead cells were excluded by live/dead staining. Each dot represents an individual mouse. Data from three independent experiments were aggregated. (B) Frequency of TEMs within the CD11b+Tie-2+ cells. Data are reported as mean ± SE. Statistical analyses (Student's t test): * p < 0.05; ** p < 0.01. (C) Correlation plot for Tie-2+ TEMs (%) and M-spike (%). Statistical analyses (Linear Regression): p = 0.021 R2 = 0.76.
Increased MVD and angiogenic cytokines in the BM identify a subpopulation of asymptomatic patients that progress to symptomatic MM
Because of transgene expression, all Vk*MYC mice affected by SMM are bound to develop MM,13 which we have found is characterized by an angiogenic switch. This is substantially different from what occurs in MGUS/SMM patients, among which only a minority will move forward to MM. Hypothesizing that disease progression in Vk*MYC mice well recapitulates what occurs only in those MGUS and SMM patients that will progress to MM, we set out to investigate MVD in the BM of a cohort of MGUS and SMM patients that progressed to MM, and compared these data with those obtained from a cohort of MGUS and SMM patients that did not progress to MM in the same time lag (Table 1). To this aim, the microvessel area was investigated in blind on BM biopsies (BMb) obtained at the time of first diagnosis, and in the second BMb obtained 2–27 years later, and not earlier than 6 months before clinical and hematochemical signs of the overt progression in patients who progressed. Already at the first BMb, some MGUS and SMM showed increased micovessel area (Figs. 5A and B, respectively), and microvesssel area in the first BMb was significantly higher in those patients who at follow-up evolved to symptomatic MM when compared with those who persisted asymptomatic (Fig. 5C). As expected,23,24 patients who progressed to symptomatic MM showed at the second BMb a further increase in microvessel area (Fig. 5C).
Table 1.
Patients' data
| Pts | BMb | BJ | CRAB | ||||||
|---|---|---|---|---|---|---|---|---|---|
| SMM | Age | Type | ISS | at Diag | at Prog | at Diag | at Prog | at Diag | at Prog |
| 1 | 56 | IgG-k | I | 16% | 65% | NO | NO | NO | I, A, B |
| 2 | 48 | IgG-l | I | 3% | 24% | YES | YES | A | A, B |
| 3 | 74 | IgG-k | nd | 2% | 40% | NO | NO | NO | A, B |
| 4 | 64 | IgA-l | II | 16% | 75% | NO | YES | NO | A |
| 5 | 72 | IgG-k | II | 8% | 70% | NO | YES | A | A, B, R |
| 6 | 71 | IgA-k | YES | / | YES | / | NO | / | |
| 7 | 46 | IgA-k | NO | / | NO | / | NO | / | |
| 8 | 52 | IgG-l | NO | / | NO | / | NO | / | |
| MGUS | |||||||||
| 1 | 74 | IgA-k | II | 1% | 14% | NO | NO | NO | A, B, R |
| 2 | 69 | IgA-k | nd | 7% | 56% | NO | YES | NO | A, B |
| 3 | 74 | IgG-l | I | 1% | 8% | NO | NO | NO | B |
| 4 | 76 | IgG-k | nd | 2% | 25% | NO | NO | A | A, B |
| 5 | 72 | IgG-l | NO | / | NO | / | NO | / | |
| 6 | 63 | IgG-k | NO | / | NO | / | NO | / | |
| 7 | 65 | IgA-l | YES | / | YES | / | NO | / | |
| 8 | 51 | IgG-l | YES | / | YES | / | NO | / | |
| 9 | 44 | IgG-k | NO | / | NO | / | NO | / | |
| 10 | 66 | IgG-k | NO | / | NO | / | NO | / | |
Abbreviations: Pts, patients; BMb, BM biopsy; Diag, diagnosis; Prog, progression; BJ, Bence Jones; I, ipercalcemia; A, Anemia; B, bone lesions; R, renal insufficiency.
Plasmacytosis (%) at BMb.
Figure 5.
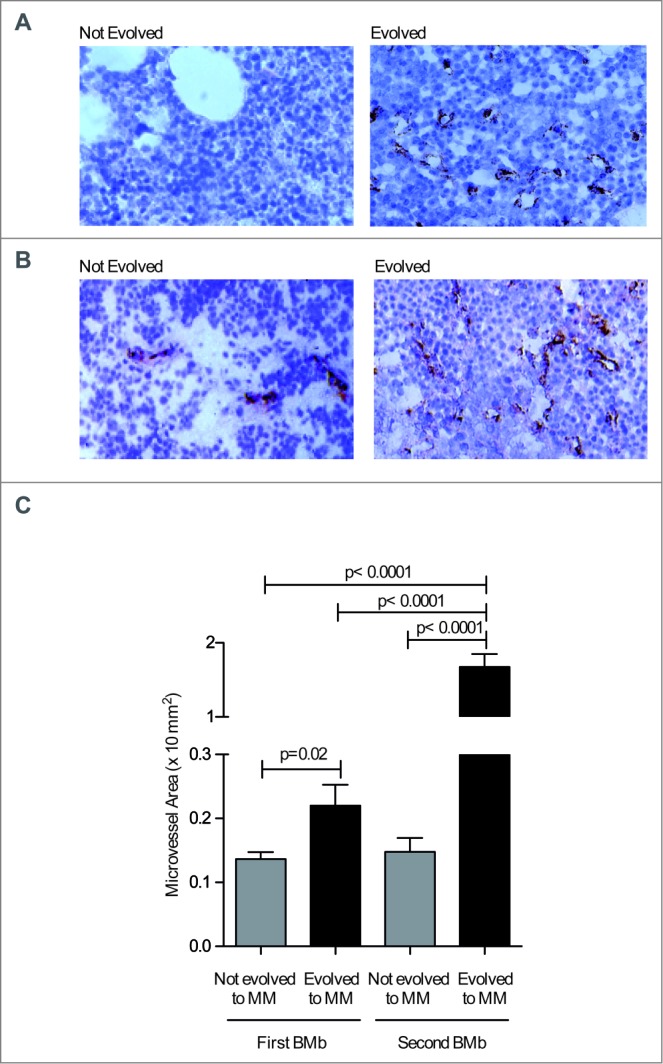
MVD is already increased in the BM of MGUS and SMM patients that subsequently progress to MM. (A and B) Paraffin-embedded sections of BMbs from MGUS (A) and SMM patients (B) were analyzed by immunohistochemistry after staining with anti-factorVIII-RA mAb (40x magnification). (C) Quantification of microvessel area as × 10−2 mm2 in the BM of MGUS and SMM patients at the time of first and second BMb. Data are reported as mean ± SE. Statistical analyses (Student's t test) are reported.
When we measured cytokines in the BM plasma of these patients at the time of first BMb, we found that levels of main angiogenic cytokines were significantly increased in evolved vs. not evolved patients (Table 2). Specifically, VEGF, FGF-2, ANG-2, HGF, and PDGF-BB were significantly increased, while no relevant variations or a trend to decrease were seen for TNFα, IL8, TIMP-1 and TIMP-2. There was no interrelation between the ANG-2, VEGF and PDGF-BB increase in a linear manner (rs = 0.53, p = 0.02, by Spearman correlation test). All together, these data suggest that a subgroup of asymptomatic patients affected by MGUS/SMM already harbor biological culprits of MM that predict progression to symptomatic disease.
Table 2.
BM plasma levels of angiogenic cytokines in patients with MGUS and SMM who evolved or not to MM
| Plasma levels | ||||
|---|---|---|---|---|
| Cytokines | Not evolved to MM | Evolved to MM | Variation | p |
| Mean ± SDa (range) | Mean ± SDa (range) | Mean ± SDa (range) | ||
| FGF-2 | 36,12 ± 10,71 | 128,8 ± 21,64 | 92,64 ± 19,88 | 0.03 |
| (16,34/70,84) | (60,34/163,8) | (32,88/147,5) | ||
| VEGF | 154,6 ± 7,292 | 232,1 ± 19,54 | 77,51 ± 20,86 | 0.03 |
| (138,3/170,9) | (174,4/280,1) | (3,510/141,8) | ||
| ANG2 | 92,71 ± 8,132 | 206,7 ± 45,12 | 113,9 ± 42,26 | 0.03 |
| (61,20/121,2) | (105,8/307,5) | (7,480/225,4) | ||
| HGF | 111,7 ± 51,71 | 655,1 ± 244,6 | 543,4 ± 233,8 | 0.03 |
| (17,26/272,9) | (31,08/1390) | (13,82/1372) | ||
| PDGF-BB | 809,0 ± 164,3 | 1572 ± 70,87 | 400,3 ± 162,6 | 0.03 |
| (325,2/1499) | (1387/1774) | (−111,6/930,0) | ||
| IL-8 | 11,23 ± 3,383 | 10,08 ± 2,171 | −1,145 ± 4,020 | ns |
| (3,660/18,79) | (4,780/16,51) | (−14,01/12,85) | ||
| TNF-α | 29,84 ± 8,944 | 36,90 ± 7,919 | 7,057 ± 11,95 | ns |
| (9,840/49,84) | (12,16/52,65) | (−37,68/42,81) | ||
| TIMP-1 | 6147 ±± 1221 | 4709 ± 437,3 | −1438 ± 995,1 | ns |
| (3990/10000) | (3651/6796) | (−5548/461,5) | ||
| TIMP-2 | 11437 ± 2398 | 7981 ± 166,9 | −3456 ± 2359 | ns |
| (7164/19000) | (7146/8154) | (−10854/981,9) | ||
Results are expressed as mean ± SD of two determinations per patient in the whole patients' group (significant p < 0.05 – Wilcoxon rank test).
pg/mL.
Discussion
Taking advantage of a reliable animal model that develops M-spike and invariably progresses to MM, we have identified and reported here relevant modifications of the BM microenvironment that were predictive of angiogenesis and progression to MM. More importantly, increased microvessel area allowed us to distinguish a subset of MGUS/SMM patients that progressed to MM. Thus, our data extend to this very relevant subset of patients previous findings demonstrating that angiogenesis is increased in MM patients when compared with MGUS and SMM patients,23 and high-grade angiogenesis or increased microvessel area are negative prognostic markers in MM patients.24,25 Interestingly, similar investigations have previously failed to either show a significant increase in BM angiogenesis in patients with MGUS when compared with healthy subjects, or to demonstrate that increased angiogenesis was a significant predictor of disease progression in MGUS and SMM patients.25 This was likely because the previous work relied on a relatively small number of unselected MGUS and SMM patients. Thus, potential informative results might have been diluted by the less relevant information gathered from the majority of MGUS and SMM that were unlikely to progress to MM.
Our findings also confirm the Vk*MYC as a primary model of human MM, and extend its potential use to investigate disease pathogenesis and progression.
Data obtained in the Vk*MYC model suggest a role of the immune system in disease progression. It has been proposed that tumor cells activate the angiogenic process at some point by altering the balance between pro-and anti-angiogenic cytokines.26,27 While the mechanism by which neoplastic plasma cells impact on BM microenvironment is still poorly defined, our data suggest that a progressive loss of Th1 immune responses in favor of Th2 T cells and the accumulation of pro-angiogenic macrophages are key steps in the angiogenic switch. These data are in line with previous reports on a skew toward Th2 responses, including tumor-specific CD4+ T cells,28 in the peripheral blood of MM patients,29 and suggest that an imbalance in the Th1/Th2 ratio within the BM of MGUS and SMM patients might be predictive of progression to MM.
Even more instrumental to the angiogenic switch is the polarization of myeloid precursors to TEMs in the BM of Vk*MYC mice progressing to MM. TEMs are a minor and distinct fraction of tumor-infiltrating CD11b+ myeloid cells30 that preferentially locate in hypoxic perivascular areas, where they are likely induced to differentiate to Tie2+ MRC+ cells by vascular endothelial cells,31 and support angiogenesis through the angiopoietin2/Tie2 pathway.32 Unfortunately, no information is available to date on TEMs in MM patients, but in patients with active MM and in Vk*MYC mice, macrophages are an abundant component of the BM microenvironment (data reported herein and in33-35) where they support survival and proliferation of neoplastic plasma cells35-37 and associate with increased BM vascularity and poor prognosis.38 These macrophages are functionally, phenotypically, and morphologically different from those of patients with non-active disease and MGUS, and upon in vitro exposure to VEGF and FGF-2, they undergo a process of vascular mimicry, increasing the expression of Tie2 and VEGFR-1 molecules.39 Thus, investigating the characteristics of TAMs and TEMs in the BM of patients before they have developed symptomatic MM might help defining the pathogenic progression of the disease.
It has been proposed that MM evolves from pre-existing MGUS, and the angiogenic switch may be responsible, at least in part, for the progression of MGUS and SMM to MM.40 Our results support this hypothesis. Indeed, in Vk*MYC mice we found a direct progression from SMM to MM that associated with perturbation of the BM microenvironment and angiogenesis, and, more importantly, angiogenesis was already evident at the stage of MGUS or SMM in those patients that rapidly progressed to MM. All together, these results also suggest that disease in Vk*MYC mice mimics what occurs in a subset of patients that already at the phase of MGUS/SMM show biological culprits of MM (e.g., increased microvessel area). Thus, it should be assessed if MGUS/SMM patients with these characteristics at the BMb, even if asymptomatic, should be treated as having MM. This hypothesis deserves further experimental and clinical investigations.
A variety of angiogenic cytokines, including FGF-2, VEGF, HGF, PDGF-β and TNFα, promote angiogenesis.41 There is evidence that soluble angiogenic cytokines (i.e., FGF-2, VEGF, HGF and PDGF-β) are related to the tumor stage,17 can be measured in the patients' plasma or serum, rise with tumor progression and correlate with a more aggressive phenotype, while fall during response to treatment and long-term disease control. Accordingly, here we demonstrate that BM plasma levels of angiogenic cytokines increase significantly in patients with MGUS and SMM who evolve to MM indicating that these factors are involved in the “angiogenic switch” that characterizes the progression from MGUS/SMM to MM.
Taken together, these issues support the view that angiogenesis has a critical pathogenic role in MM.25 A much larger sample size is obviously needed to assess whether angiogenesis, independently from genetic properties of neoplastic plasma cells, could be an independent prognostic factor at the specific stage of MGUS/SMM. Results reported herein should prompt initiating a large multicenter study to address this medical need. As some MM patients have low-grade angiogenesis,25 other critical mechanisms of disease progression are likely involved in these patients, such as genetic and epigenetic alterations in cell proliferation and resistance to apoptosis.
Currently, the standard of care for patients with SMM has been observation until symptomatic disease occurs.42 Indeed, no interventional study in patients with SMM has shown improved overall survival in the treatment arm 43-47 except for one phase III trial in which patients with high-risk SMM were assigned to lenalinomide plus low-dose dexamethasone or to observation.10 Besides potential limitations of the study48 what distinguishes the latter from the others is a risk-stratified approach to patient selection. Indeed, Mateos and colleagues10 implemented specific biomarkers to focus their trial on patients with the highest probability to have symptomatic MM. The angiogenic switch might represent an additional biomarker of high-risk MGUS and SMM patients.
In conclusion, the availability of a primary animal model of SMM that invariably progresses to MM should help identifying both predictive markers of disease progression and drugs that avoid the switch to MM.
Materials and Methods
Animals
Wild type C57BL/6J mice (from Charles River Breeding Laboratories, Calco IT), and Vk*MYC mice13 were housed in a pathogen-free animal facility, and treated in accordance with the European Community Guidelines. Vk*MYC mice were screened by Real Time PCR in order to identify experimental Vk*MYC+/– animals. All in vivo experiments were approved by the Ethical Committee of Fondazione Centro San Raffaele (Milan IT).
Flow cytometry
Mouse bones were harvested and epiphyses were cut to collect BM. Single cell suspensions obtained from mouse femora were labeled with fluorochrome-conjugated monoclonal antibodies (mAbs, either from BD Bioscience, Buccinasco IT, Biolegend Europe, Uithoorn The Netherlands, or eBioscience Inc., Prodotti Gianni, Milan, IT) after neutralization of unspecific binding with FcR blocker (BD Biosciences), and acquired by BD LSR Fortessa™. Data were analyzed using the FlowJo software (TreeStar Inc., Ashland, OR, USA). Cells were also assessed for intracellular cytokine production after 4 h at 37°C of stimulation with Phorbol Myristate Acetate (PMA)/ionomycin. Brefeldin A (5 μg/mL) was added to the samples during the last 3 h of culture. After incubation, cells were washed and stained for surface markers 15 min at 4°C, fixed with 2% paraformaldehyde (PFA), and permeabilized with saponin (0.5% in PBS). Cells were then washed and stained for intracellular markers.
Serum protein electrophoresis
Mouse blood was periodically collected in Eppendorf by retro-orbital sampling. Sera were used undiluted and loaded on agarose gel (HYDRAGEL, SEBIA electrophoresis, Norcross, GA, USA). Electrophoresis was performed by the semi-automated multi-parameter HYDRASYS system SEBIA, and obtained gels were analyzed by the densitomer/scanner GELSCAN SEBIA and PHORESIS software for flat-bed scanner.
Peripheral quantitative computed tomography (pQCT)
The pQCT measurements were performed using a Stratec Research SA+ pQCT scanner (Stratec Medizintechnik GmbH, Pforzheim, D) with a voxel size of 70 μm and a scan speed of 3 mm/s. In order to orientate the long axes of the bones parallel to the image planes, the excised bone specimens were fixed with manufacturer-made plastic holders. The correct longitudinal positioning was determined by means of an initial “scout scan”. The scans were performed at the proximal metaphysis and at the diaphysis of the tibiae. The scans were analyzed with pQCT software 6.00B using contour mode 2 and peel mode 2 with a threshold of 350 mg/cm3 for the calculation of trabecular and total bone parameters and with a threshold of 710 mg/cm3 for cortical bone parameters.
Hemoglobin estimation
Venous blood samples were freshly obtained by retro-orbital sampling in BD Vacutainer® Blood Collection Tubes (BD Biosciences). Hemoglobin was estimated with an automated cell counter (System 9000; Serono–Baker Diagnostics, Lintech Components, NY, USA).
Immunohistochemistry (IHC)
Vk*MYC mouse bones were formalin fixed and decalcified in Fix Decal (Pro-Eco) overnight. Bones were then paraffin-embedded, and 4-μm sectioned. Sections were stained with primary antibodies: anti-CD31 rabbit mAb (Neomarkers, Fremont, CA, USA; diluted 1:200 over-night) or with IRF4/MUM1 mouse mAb (Santa Cruz Biotechnology, CA, USA, diluted 1:500 for 1 h). Samples were washed and incubated with biotinylated horseradish peroxidase (HRP)-labeled secondary antibodies used 1:2 for 30 min (BioCare, Birmingham, UK). Slides were then incubated for 5 min with the chromogen 3,3′ – diaminobenzidine (DAB), which is converted in an insoluble brown precipitate by the HRP. After washing, slides were contrasted with Mayer-Hematoxylin (BioOptica, Milan, IT) and mounted with a cover glass. All sections were blindly evaluated by an expert hematopathologist.
Patients
A total of 18 patients who fulfilled the International Myeloma Working Group (IMWG) diagnostic criteria for SMM – 8 patients and MGUS – 10 patients49 who progressed (9 patients) or not (9 patients) to symptomatic MM were studied. The ethics committee of the University Medical School of Bari (IT) approved the study and all patients gave their informed consent.
Measurement of BM angiogenesis
Blood vessels were detected in 6-μm sections of 4% PFA fixed paraffin embedded biopsies by staining endothelial cells with the anti-factor VIII related antigen (FVIII-RA) mAb (Dako, Glostrup, Denmark) and a three-layer biotin-avidin-peroxidase system. Angiogenesis was measured as microvessel area without knowledge of the clinical diagnosis. Briefly, four to six 250x fields covering each of two sections per biopsy were examined with a superimposed 484-point square reticulum (12.5 × 10−2 mm2) for the presence of microvessels (capillaries and small venules). These were identified as endothelial cells, either single or clustered in nests or tubes, and clearly separated from one another, and either without or with a lumen (not exceeding 10 μm in diameter). A planimetric point count method with slight modifications for the computed image analysis (Zeiss KS300, Zeiss, Oberkochen, D) was applied to measure the microvessel area within the cellular area (reticulum area minus dense connective tissue, fat, bone lamellae, necrosis, and hemorrhage areas). Values are expressed as mean ± 1 standard deviation (SD) per patient, and groups of patients.
Cytokine quantification in mice BM serum samples
Cytokines were quantified by the Myriad RBD™ multiplex immunoassay (Myriad RBD, Austin, TX, USA). The sera were 1:10 diluted with PBS, and stored at –80˚C until sending to Myriad RBD for cytokine quantification.
BM plasma collection and storage and enzyme-linked immunosorbent assay (ELISA) in patients
BM plasma samples were collected in not progressed MGUS and SMM patients and in those who progressed to MM at least 6 months before clinical and hematochemical signs of the overt progression. BM aspirates were processed immediately after harvesting by centrifugation at 1500 rpm for 10 min and the plasma stored at –80°C until use. Angiogenic cytokines (FGF-2, VEGF, ANG-2, HGF, PDGF-BB, IL8. TNFα, TIMP-1, TIMP-2) were measured using the “Q-Plex Human Angiogenesis for the chemiluminescent Platform” (Quansys Biosciences, Logan, UT, USA) and the Q-View Software.
Statistical analyses
These were performed using the Student's t test. Values were considered statistically significant for p < 0.05. For all tests symbols mean: * p < 0.05; ** p < 0.01; *** pP < 0.001. Non-parametric tests were applied because variables were not normally distributed using the SPSS statistical software. The Wilcoxon signed rank test was employed to compare pre-and post-progression cytokine serum levels. The Spearman rank correlation test was used to evaluate the correlation between the cytokine variations.
Acknowledgments
We thank Martina Rocchi and Anna Innocenzi for histopathological technical support.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Funding
The work was supported by Associazione Italiana per la Ricerca sul Cancro, AIRC 5 × 1000 Molecular Clinical Oncology Special Program, Milan, IT (grant no. 9965 to M. Bellone, M. Ponzoni, G. Tonon and A. Vacca), the European Commission's Seventh Framework Program (EU FPT7) under grant agreement no. 278706 (OVERMyR; to A.Vacca), and grants from MIUR PRIN 2010NECHBX (to A. Vacca). Arianna Calcinotto was awarded a fellowship from AIRC/FIRC and conducted this study in partial fulfillment of her Ph.D. at San Raffaele University.
Authors' Contributions
AC designed the research, performed experiments, analyzed the data, designed the figures, and wrote the manuscript; MP analyzed BM samples and reviewed the manuscript; RR and AV provided patient samples, cytokine measurement and BM immunohistochemistry and reviewed the manuscript. MG, EC and IV performed experiments. MTSB took care of the Vk*MYC colony. MC and LB provided the first Vk*MYC mice and reviewed the manuscript; AR supervised the work of IV and reviewed the manuscript; GT analyzed data related to BM cytokines and reviewed the manuscript; MB designed the research, analyzed the data, and wrote the manuscript.
Disclaimer
The study involving human biopsy samples was conducted in accordance with the Declaration of Helsinki and approved by the local ethics committee of the University of Bari. Patients gave written consent for the sample collection.
References
- 1.Anderson KC, Carrasco RD. Pathogenesis of myeloma. Annu Rev Pathol 2011; 6:249-74; PMID:; http://dx.doi.org/ 10.1146/annurev-pathol-011110-130249 [DOI] [PubMed] [Google Scholar]
- 2.Kyle RA. Monoclonal gammopathy of undetermined significance. Natural history in 241 cases. Am J Med 1978; 64:814-26; PMID:; http://dx.doi.org/ 10.1016/0002-9343(78)90522-3 [DOI] [PubMed] [Google Scholar]
- 3.Agarwal A, Ghobrial IM. Monoclonal gammopathy of undetermined significance and smoldering multiple myeloma: a review of the current understanding of epidemiology, biology, risk stratification, and management of myeloma precursor disease. Clin Cancer Res 2013; 19:985-94; PMID:; http://dx.doi.org/ 10.1158/1078-0432.CCR-12-2922 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Rajkumar SV, Larson D, Kyle RA. Diagnosis of smoldering multiple myeloma. N Engl J Med 2011; 365:474-5; PMID:; http://dx.doi.org/ 10.1056/NEJMc1106428 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Mahindra A, Laubach J, Raje N, Munshi N, Richardson PG, Anderson K. Latest advances and current challenges in the treatment of multiple myeloma. Nat Rev Clin Oncol 2012; 9:135-43; PMID:; http://dx.doi.org/ 10.1038/nrclinonc.2012.15 [DOI] [PubMed] [Google Scholar]
- 6.Bergsagel PL, Kuehl WM. Degree of focal immunoglobulin heavy chain locus deletion as a measure of B-cell tumor purity. Leukemia 2013; 27:2067-8; PMID:; http://dx.doi.org/ 10.1038/leu.2013.139 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bergsagel PL, Kuehl WM, Zhan F, Sawyer J, Barlogie B, Shaughnessy J Jr. Cyclin D dysregulation: an early and unifying pathogenic event in multiple myeloma. Blood 2005; 106:296-303; PMID:; http://dx.doi.org/ 10.1182/blood-2005-01-0034 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Vesely MD, Kershaw MH, Schreiber RD, Smyth MJ. Natural innate and adaptive immunity to cancer. Annu Rev Immunol 2011; 29:235-71; PMID:; http://dx.doi.org/ 10.1146/annurev-immunol-031210-101324 [DOI] [PubMed] [Google Scholar]
- 9.Palumbo A, Anderson K. Multiple myeloma. N Engl J Med 2011; 364:1046-60; PMID:; http://dx.doi.org/ 10.1056/NEJMra1011442 [DOI] [PubMed] [Google Scholar]
- 10.Mateos MV, Hernandez MT, Giraldo P, de la Rubia J, de Arriba F, Lopez Corral L, Rosinol L, Paiva B, Palomera L, Bargay J et al.. Lenalidomide plus dexamethasone for high-risk smoldering multiple myeloma. N Engl J Med 2013; 369:438-47; PMID:; http://dx.doi.org/ 10.1056/NEJMoa1300439 [DOI] [PubMed] [Google Scholar]
- 11.Shou Y, Martelli ML, Gabrea A, Qi Y, Brents LA, Roschke A, Dewald G, Kirsch IR, Bergsagel PL, Kuehl WM. Diverse karyotypic abnormalities of the c-myc locus associated with c-myc dysregulation and tumor progression in multiple myeloma. Proc Natl Acad Sci USA 2000; 97:228-33; PMID:; http://dx.doi.org/ 10.1073/pnas.97.1.228 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Affer M, Chesi M, Chen WD, Keats JJ, Demchenko YN, Tamizhmani K, Garbitt VM, Riggs DL, Brents LA, Roschke AV et al.. Promiscuous MYC locus rearrangements hijack enhancers but mostly super-enhancers to dysregulate MYC expression in multiple myeloma. Leukemia 2014; 28:1725-35; PMID:; http://dx.doi.org/ 10.1038/leu.2014.70 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Chesi M, Robbiani DF, Sebag M, Chng WJ, Affer M, Tiedemann R, Valdez R, Palmer SE, Haas SS, Stewart AK et al.. AID-dependent activation of a MYC transgene induces multiple myeloma in a conditional mouse model of post-germinal center malignancies. Cancer Cell 2008; 13:167-80; PMID:; http://dx.doi.org/ 10.1016/j.ccr.2008.01.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kuehl WM. Modeling multiple myeloma by AID-dependent conditional activation of MYC. Cancer Cell 2008; 13:85-7; PMID:; http://dx.doi.org/ 10.1016/j.ccr.2008.01.022 [DOI] [PubMed] [Google Scholar]
- 15.Chesi M, Bergsagel PL. Molecular pathogenesis of multiple myeloma: basic and clinical updates. Int J Hematol 2013; 97:313-23; PMID:; http://dx.doi.org/ 10.1007/s12185-013-1291-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Falini B, Fizzotti M, Pucciarini A, Bigerna B, Marafioti T, Gambacorta M, Pacini R, Alunni C, Natali-Tanci L, Ugolini B et al.. A monoclonal antibody (MUM1p) detects expression of the MUM1/IRF4 protein in a subset of germinal center B cells, plasma cells, and activated T cells. Blood 2000; 95:2084-92; PMID: [PubMed] [Google Scholar]
- 17.Folkman J. Angiogenesis-dependent diseases. Semin Oncol 2001; 28:536-42; PMID:; http://dx.doi.org/ 10.1016/S0093-7754(01)90021-1 [DOI] [PubMed] [Google Scholar]
- 18.Di Raimondo F, Azzaro MP, Palumbo G, Bagnato S, Giustolisi G, Floridia P, Sortino G, Giustolisi R. Angiogenic factors in multiple myeloma: higher levels in bone marrow than in peripheral blood. Haematologica 2000; 85:800-5; PMID: [PubMed] [Google Scholar]
- 19.Alexandrakis MG, Passam FH, Sfiridaki K, Moschandrea J, Pappa C, Liapi D, Petreli E, Roussou P, Kyriakou DS. Interleukin-18 in multiple myeloma patients: serum levels in relation to response to treatment and survival. Leuk Res 2004; 28:259-66; PMID:; http://dx.doi.org/ 10.1016/S0145-2126(03)00261-3 [DOI] [PubMed] [Google Scholar]
- 20.Leung DW, Cachianes G, Kuang WJ, Goeddel DV, Ferrara N. Vascular endothelial growth factor is a secreted angiogenic mitogen. Science 1989; 246:1306-9; PMID:; http://dx.doi.org/ 10.1126/science.2479986 [DOI] [PubMed] [Google Scholar]
- 21.Gordon S, Martinez FO. Alternative activation of macrophages: mechanism and functions. Immunity 2010; 32:593-604; PMID:; http://dx.doi.org/ 10.1016/j.immuni.2010.05.007 [DOI] [PubMed] [Google Scholar]
- 22.De Palma M, Murdoch C, Venneri MA, Naldini L, Lewis CE. Tie2-expressing monocytes: regulation of tumor angiogenesis and therapeutic implications. Trends Immunol 2007; 28:519-24; PMID:; http://dx.doi.org/ 10.1016/j.it.2007.09.004 [DOI] [PubMed] [Google Scholar]
- 23.Vacca A, Ribatti D, Presta M, Minischetti M, Iurlaro M, Ria R, Albini A, Bussolino F, Dammacco F. Bone marrow neovascularization, plasma cell angiogenic potential, and matrix metalloproteinase-2 secretion parallel progression of human multiple myeloma. Blood 1999; 93:3064-73; PMID: [PubMed] [Google Scholar]
- 24.Rajkumar SV, Leong T, Roche PC, Fonseca R, Dispenzieri A, Lacy MQ, Lust JA, Witzig TE, Kyle RA, Gertz MA et al.. Prognostic value of bone marrow angiogenesis in multiple myeloma. Clin Cancer Res 2000; 6:3111-6; PMID: [PubMed] [Google Scholar]
- 25.Rajkumar SV, Mesa RA, Fonseca R, Schroeder G, Plevak MF, Dispenzieri A, Lacy MQ, Lust JA, Witzig TE, Gertz MA et al.. Bone marrow angiogenesis in 400 patients with monoclonal gammopathy of undetermined significance, multiple myeloma, and primary amyloidosis. Clin Cancer Res 2002; 8:2210-6; PMID: [PubMed] [Google Scholar]
- 26.Folkman J. Angiogenesis in cancer, vascular, rheumatoid and other disease. Nat Med 1995; 1:27-31; PMID:; http://dx.doi.org/ 10.1038/nm0195-27 [DOI] [PubMed] [Google Scholar]
- 27.Bhutani M, Turkbey B, Tan E, Kemp TJ, Pinto LA, Berg AR, Korde N, Minter AR, Weiss BM, Mena E et al.. Bone marrow angiogenesis in myeloma and its precursor disease: a prospective clinical trial. Leukemia 2014; 28:413-6; PMID:; http://dx.doi.org/ 10.1038/leu.2013.268 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ostad M, Andersson M, Gruber A, Sundblad A. Expansion of immunoglobulin autoreactive T-helper cells in multiple myeloma. Blood 2008; 111:2725-32; PMID:; http://dx.doi.org/ 10.1182/blood-2006-11-056242 [DOI] [PubMed] [Google Scholar]
- 29.Sharma A, Khan R, Joshi S, Kumar L, Sharma M. Dysregulation in T helper 1/T helper 2 cytokine ratios in patients with multiple myeloma. Leuk Lymphoma 2010; 51:920-7; PMID:; http://dx.doi.org/ 10.3109/10428191003699563 [DOI] [PubMed] [Google Scholar]
- 30.Pucci F, Venneri MA, Biziato D, Nonis A, Moi D, Sica A, Di Serio C, Naldini L, De Palma M. A distinguishing gene signature shared by tumor-infiltrating Tie2-expressing monocytes, blood “resident” monocytes, and embryonic macrophages suggests common functions and developmental relationships. Blood 2009; 114:901-14; PMID:; http://dx.doi.org/ 10.1182/blood-2009-01-200931 [DOI] [PubMed] [Google Scholar]
- 31.He H, Xu J, Warren CM, Duan D, Li X, Wu L, Iruela-Arispe ML. Endothelial cells provide an instructive niche for the differentiation and functional polarization of M2-like macrophages. Blood 2012; 120:3152-62; PMID:; http://dx.doi.org/ 10.1182/blood-2012-04-422758 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Mazzieri R, Pucci F, Moi D, Zonari E, Ranghetti A, Berti A, Politi LS, Gentner B, Brown JL, Naldini L et al.. Targeting the ANG2/TIE2 axis inhibits tumor growth and metastasis by impairing angiogenesis and disabling rebounds of proangiogenic myeloid cells. Cancer Cell 2011; 19:512-26; PMID:; http://dx.doi.org/ 10.1016/j.ccr.2011.02.005 [DOI] [PubMed] [Google Scholar]
- 33.Zheng Y, Cai Z, Wang S, Zhang X, Qian J, Hong S, Li H, Wang M, Yang J, Yi Q. Macrophages are an abundant component of myeloma microenvironment and protect myeloma cells from chemotherapy drug-induced apoptosis. Blood 2009; 114:3625-8; PMID:; http://dx.doi.org/ 10.1182/blood-2009-05-220285 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ribatti D, Nico B, Vacca A. Importance of the bone marrow microenvironment in inducing the angiogenic response in multiple myeloma. Oncogene 2006; 25:4257-66; PMID:; http://dx.doi.org/ 10.1038/sj.onc.1209456 [DOI] [PubMed] [Google Scholar]
- 35.Hope C, Ollar SJ, Heninger E, Hebron E, Jensen JL, Kim J, Maroulakou I, Miyamoto S, Leith C, Yang DT et al.. TPL2 kinase regulates the inflammatory milieu of the myeloma niche. Blood 2014; 123:3305-15; PMID:; http://dx.doi.org/ 10.1182/blood-2014-02-554071 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kim J, Denu RA, Dollar BA, Escalante LE, Kuether JP, Callander NS, Asimakopoulos F, Hematti P. Macrophages and mesenchymal stromal cells support survival and proliferation of multiple myeloma cells. Br J Haematol 2012; 158:336-46; PMID:; http://dx.doi.org/ 10.1111/j.1365-2141.2012.09154.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ribatti D. Mast cells and macrophages exert beneficial and detrimental effects on tumor progression and angiogenesis. Immunol Lett 2013; 152:83-8; PMID:; http://dx.doi.org/ 10.1016/j.imlet.2013.05.003 [DOI] [PubMed] [Google Scholar]
- 38.Berardi S, Ria R, Reale A, De Luisi A, Catacchio I, Moschetta M, Vacca A. Multiple myeloma macrophages: pivotal players in the tumor microenvironment. J Oncol 2013; 2013:183602; PMID:; http://dx.doi.org/ 10.1155/2013/183602 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Scavelli C, Nico B, Cirulli T, Ria R, Di Pietro G, Mangieri D, Bacigalupo A, Mangialardi G, Coluccia AML, Caravita T et al.. Vasculogenic mimicry by bone marrow macrophages in patients with multiple myeloma. Oncogene 2007; 27:663-74; PMID:; http://dx.doi.org/ 10.1038/sj.onc.1210691 [DOI] [PubMed] [Google Scholar]
- 40.Rajkumar SV, Witzig TE. A review of angiogenesis and antiangiogenic therapy with thalidomide in multiple myeloma. Cancer Treat Rev 2000; 26:351-62; PMID:; http://dx.doi.org/ 10.1053/ctrv.2000.0188 [DOI] [PubMed] [Google Scholar]
- 41.Carmeliet P, Moons L, Luttun A, Vincenti V, Compernolle V, De Mol M, Wu Y, Bono F, Devy L, Beck H et al.. Synergism between vascular endothelial growth factor and placental growth factor contributes to angiogenesis and plasma extravasation in pathological conditions. Nat Med 2001; 7:575-83; PMID:; http://dx.doi.org/ 10.1038/87904 [DOI] [PubMed] [Google Scholar]
- 42.Korde N, Kristinsson SY, Landgren O. Monoclonal gammopathy of undetermined significance (MGUS) and smoldering multiple myeloma (SMM): novel biological insights and development of early treatment strategies. Blood 2011; 117:5573-81; PMID:; http://dx.doi.org/ 10.1182/blood-2011-01-270140 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Rajkumar SV, Dispenzieri A, Fonseca R, Lacy MQ, Geyer S, Lust JA, Kyle RA, Greipp PR, Gertz MA, Witzig TE. Thalidomide for previously untreated indolent or smoldering multiple myeloma. Leukemia 2001; 15:1274-6; PMID:; http://dx.doi.org/ 10.1038/sj.leu.2402183 [DOI] [PubMed] [Google Scholar]
- 44.Martin A, Garcia-Sanz R, Hernandez J, Blade J, Suquia B, Fernandez-Calvo J, Gonzalez M, Mateo G, Orfao A, San Miguel JF. Pamidronate induces bone formation in patients with smouldering or indolent myeloma, with no significant anti-tumour effect. Br J Haematol 2002; 118:239-42; PMID:; http://dx.doi.org/ 10.1046/j.1365-2141.2002.03549.x [DOI] [PubMed] [Google Scholar]
- 45.Weber D, Rankin K, Gavino M, Delasalle K, Alexanian R. Thalidomide alone or with dexamethasone for previously untreated multiple myeloma. J Clin Oncol 2003; 21:16-9; PMID:; http://dx.doi.org/ 10.1200/JCO.2003.03.139 [DOI] [PubMed] [Google Scholar]
- 46.Musto P, Petrucci MT, Bringhen S, Guglielmelli T, Caravita T, Bongarzoni V, Andriani A, D'Arena G, Balleari E, Pietrantuono G et al.. A multicenter, randomized clinical trial comparing zoledronic acid versus observation in patients with asymptomatic myeloma. Cancer 2008; 113:1588-95; PMID:; http://dx.doi.org/ 10.1002/cncr.23783 [DOI] [PubMed] [Google Scholar]
- 47.Detweiler-Short K, Hayman S, Gertz MA, Lacy MQ, Dispenzieri A, Kumar S, Zeldenrust SR, Russell SJ, Lust JA, Kyle RA et al.. Long-term results of single-agent thalidomide as initial therapy for asymptomatic (smoldering or indolent) myeloma. Am J Hematol 2010; 85:737-40; PMID:; http://dx.doi.org/ 10.1002/ajh.21821 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Rajkumar SV. Multiple myeloma: 2013 update on diagnosis, risk-stratification, and management. Am J Hematol 2013; 88:226-35; PMID:; http://dx.doi.org/ 10.1002/ajh.23390 [DOI] [PubMed] [Google Scholar]
- 49.International Myeloma Working Group. Criteria for the classification of monoclonal gammopathies, multiple myeloma and related disorders: a report of the International Myeloma Working Group. Br J Haematol. 2003. Jun; 121(5):749-57; PMID:; http://dx.doi.org/ 10.1046/j.1365-2141.2003.04355.x [DOI] [PubMed] [Google Scholar]