

Abstract
Asymptomatic prostate inflammation and prostate cancer have reached epidemic proportions among men in the developed world. Animal model studies implicate dietary carcinogens, such as the heterocyclic amines from over-cooked meats and sex steroid hormones, particularly estrogens, as candidate etiologies for prostate cancer. Each acts by causing epithelial cell damage, triggering an inflammatory response that can evolve into a chronic or recurrent condition. This milieu appears to spawn proliferative inflammatory atrophy (PIA) lesions, a type of focal atrophy that represents the earliest of prostate cancer precursor lesions. Rare PIA lesions contain cells which exhibit high c-Myc expression, shortened telomere segments, and epigenetic silencing of genes such as GSTP1, encoding the π-class glutathione S-transferase, all characteristic of prostatic intraepithelial neoplasia (PIN) and prostate cancer. Subsequent genetic changes, such as the gene translocations/deletions that generate fusion transcripts between androgen-regulated genes (such as TMPRSS2) and genes encoding ETS family transcription factors (such as ERG1), arise in PIN lesions and may promote invasiveness characteristic of prostatic adenocarcinoma cells. Lethal prostate cancers contain markedly corrupted genomes and epigenomes. Epigenetic silencing, which seems to arise in response to the inflamed microenvironment generated by dietary carcinogens and/or estrogens as part of an epigenetic “catastrophe” affecting hundreds of genes, persists to drive clonal evolution through metastatic dissemination. The cause of the initial epigenetic “catastrophe” has not been determined but likely involves defective chromatin structure maintenance by over-exuberant DNA methylation or histone modification. With dietary carcinogens and estrogens driving pro-carcinogenic inflammation in the developed world, it is tempting to speculate that dietary components associated with decreased prostate cancer risk, such as intake of fruits and vegetables, especially tomatoes and crucifers, might act to attenuate the ravages of the chronic or recurrent inflammatory processes. Specifically, nutritional agents might prevent PIA lesions or reduce the propensity of PIA lesions to suffer “catastrophic” epigenome corruption.
Keywords: Prostate, Proliferative inflammatory atrophy, Heterocyclic amines, Epigenetics, DNA methylation
Prostate cancer is the most common cancer diagnosed in men in the United States (US) and in Western Europe. Once rare in the rest of the world, the disease appears also to be on the rise throughout Asia and in many other developing nations. Diet and lifestyle, along with risk factors such as age, family history, and sex steroid hormones, have long been thought to contribute to prostatic carcinogenesis [57]. However, more recently, molecular pathology insights have indicted chronic or recurrent epithelial cell injury, accompanied by innate and adaptive inflammatory responses, in the early steps of prostate cancer development [18]. As a consequence, dietary components capable of inducing such injury, such as the heterocyclic amines created by over-cooking meats, loom large as candidate prostate carcinogens, while dietary components able to limit cell and genome damage and/or attenuate prostate inflammation, may protect against prostate cancer development. The mechanism(s) by which dietary components, inherited susceptibility, and sex steroid hormones cause epithelial damage and/or drive inflammatory processes that lead to cancer as men age, if better understood, could provide new opportunities for prostate cancer prevention, improved prostate cancer screening strategies, and perhaps even better prostate cancer treatment outcomes.
1 Proliferative Inflammatory Atrophy: A Lesion that Links Epithelial Injury to Prostate Cancer
In regions of the world with high prostate cancer incidence, prostate inflammation is essentially ubiquitous [18]. Though mostly asymptomatic, particularly if affecting the peripheral zone of the prostate where cancers arise, prostatitis has long been known to drive prostate cancer diagnoses independent of its propensity to cause the disease, because it tends to elevate serum prostate-specific antigen (PSA) levels. In the inflamed prostate, damage to the barrier function of the prostate epithelium stereotypically causes backflow of prostate secretions, including secreted proteins like PSA, into the prostate parenchyma and ultimately into the bloodstream. Because the detection of PSA in serum serves as the primary trigger of prostate biopsy for prostate cancer detection and diagnosis, prostate inflammation is responsible for a significant fraction of more than 30 million PSA tests, leading to more than a million prostate biopsies looking for cancer, performed in the United States each year [36]. Nonetheless, serum PSA elevations as early as age 40 years are associated with an increased risk of prostate cancer later in life [23, 24].
This tendency for prostate inflammation to elevate serum PSA has greatly undermined attempts to test causal associations between prostatitis and prostate cancer in population studies. Restricting cancer association studies to symptomatic prostatitis has not been very helpful either. Symptomatic prostatitis typically reflects inflammation of the transition zone near the urethra where benign prostatic hyperplasia (BPH) arises, leading to irritative voiding symptoms which prompt as many as 2 million physician visits in the United States each year, each accompanied by serum PSA tests [50]. As a consequence, apparent correlations between symptomatic prostatitis and prostate cancer in epidemiologic studies, which are numerous, have been frequently attributed to the bias that such men might be more likely subjected to prostate biopsy [19]. In addition, since symptomatic prostatitis does not reflect inflammation in the prostate peripheral zone where cancers arise, the condition presumably serves as a poor surrogate for pro-carcinogenic inflammatory processes. To circumvent these limitations and more directly test whether peripheral zone inflammation might be correlated with prostate cancer, an analysis of end-of-study prostate biopsies collected from placebo-treated subjects participating in the Prostate Cancer Prevention Trial of finasteride for asymptomatic men with serum PSA values <3 ng/mL was undertaken [83]. The results of this study, expected soon, should yield definitive insight into whether the presence of inflammation is directly associated with cancer within prostate glands.
Most of the inflammation in the prostate is a consequence of damage to the prostate epithelium, which can be caused by dietary carcinogens, estrogens, and inflammatory oxidants [18]. Microscopic examination of prostate tissues, particularly tissues from glands which contain prostate cancers, yields evidence of longstanding chronic or repeated tissue insults and innate immune responses. As an example, large numbers of corpora amylacea, microscopic laminated bodies containing calprotectin, myeloperoxidase, and α-defensins, from neutrophil granules, are often spread out through such prostate glands [71]. These bodies form as a consequence of neutrophil discharge during acute inflammation, and remain, like spent ammunition casings, even after the acute inflammatory process has subsided. Of interest, prostate tissues from rats prone to prostate inflammation and to prostate cancer also contain abundant corpora amylacea [32]. Characteristically, when the prostate epithelium suffers injury, focal atrophy lesions with dilated glandular lumens and immature epithelial cells appear [16, 17]. The epithelial cells act as if the normal secretory cell differentiation pathway has been abandoned in favor of a stress response with induced expression of α- and π-class glutathione S-transferases, cyclooxygenase-2, and other mediators of genome damage defense and cell survival [60, 86, 94]. Focal atrophy lesions, surrounded by corpora amylacea, mark prostate tissues that have suffered cell and tissue damage, and define a pro-carcinogenic milieu.
Proliferative inflammatory atrophy (PIA) lesions (Fig. 1), which comprise a subset of focal atrophy-type injury responses in the prostate, exhibit very high epithelial proliferation rates and infiltration by inflammatory cells [67]. This type of epithelial damage response is reminiscent of a number of other conditions in other organ sites, such as atrophic gastritis, hepatitis, and cirrhosis, that are known to lead to cancer development. Not surprisingly, PIA lesions also appear to be cancer precursor lesions, giving rise to prostatic intraepithelial neoplasia (PIN) or to prostate cancer directly [64]. The evidence that epithelial damage, leading to PIA, may initiate prostate cancer development, has accumulated over the past decade or so to include: (1) the detection of somatic genome and epigenome defects in PIA lesions that are identical to those seen in PIN and prostate cancer, (2) the visualization of direct morphological transitions between PIA and PIN and between PIA and prostate cancer, (3) the frequent appearance of somatic genetic and epigenetic alterations characteristic of PIN and cancer in PIA lesions, often to an intermediate degree between normal and neoplasia, (4) the greater prevalence of PIA lesions in prostate peripheral zone regions in areas of the world with high prostate cancer risk, and (5) the propensity for preclinical models of prostatic carcinogenesis, including those using dietary carcinogens, to first show PIA lesions before cancers arise [18].
Fig. 1.
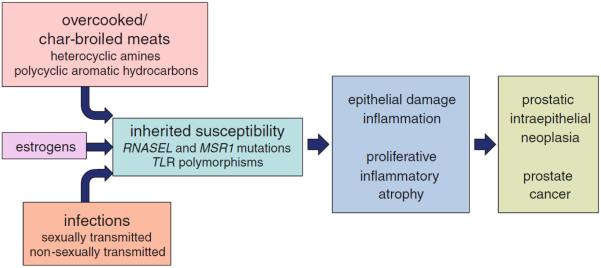
Etiological factors for prostate cancer. Prostatic carcinogenesis follows damage to the prostate epithelium, regenerative proliferation, and chronic/recurrent inflammation, leading to proliferative inflammatory atrophy lesions. These lesions sprout cells with c-Myc activation, telomere shortening, and epigenetic gene inactivation, which give rise to prostatic intraepithelial neoplasia and prostate cancer, carrying targeted gene rearrangements, mutations, and an extensively corrupted epigenome
Prostate damage, followed by a maladaptive innate immune response and tissue injury response, may be the pathway that connects environmental exposures, including the diet, to prostate cancer development. Certainly, both in population studies and in molecular pathology analyses, the association between prostate inflammation, PIA, and prostate cancer is clear. Yet, the mechanisms by which dietary habits, or other exposures, lead to prostate damage have not been fully elucidated.
2 The Diet: A Source of Carcinogens that can Damage the Prostate Epithelium and Cause PIA
Epidemiology studies of prostate cancer have strongly implicated the diet as a major modulator of prostate cancer risk. Prostate cancer incidence and mortality varies among different geographic regions, with high prostate cancer risk in the United States and in Europe and low prostate cancer risk in Asia, yet immigrants from low-risk regions to high-risk regions typically adopt higher prostate cancer risks, particularly with cultural assimilation [27, 73]. This likely reflects dietary differences: either dietary habits in high-risk regions promote prostate cancer, dietary habits in low-risk regions prevent prostate cancer, or both. When examined in greater detail, the most consistent dietary association for prostate cancer appears to be intake of red meats and/or animal fats [26, 43]. For red meats, cooking at high temperatures or char-broiling creates both heterocyclic aromatic amine and polycyclic aromatic hydrocarbon carcinogens [39, 44]. These cooking practices are also associated with an increased prostate cancer risk and may partially explain an increased propensity for prostate cancer development among African–American men versus Caucasian men in the United States [37, 82].
The best studied of these carcinogens for prostate cancer is 2-amino-1-methyl-6-phenylimidazopyridine (PhIP), the most abundant of the >20 heterocyclic amines that can appear in over-cooked meats [8, 85, 87]. In rat models, PhIP ingestion causes PIA and prostate cancer [74]. By itself, PhIP is non-toxic and non-mutagenic, but after activation, first to N–OH-PhIP by CYP1A1 or CYP1A2 in the liver or elsewhere and then to more reactive species in prostate epithelial cells by sulfotransferases or by a kinase/phosphatase, PhIP can cause marked prostate epithelial cell damage, elicit inflammatory responses, and form pro-mutagenic adducts with DNA [51]. Epithelial injury, accompanied by an inflammatory response, is a critical feature of PhIP prostate carcinogenesis in rats. Rats fed PhIP show genome mutations in ventral, dorsolateral, and anterior prostate lobes, but exhibit epithelial cell damage, inflammation, and PIA in only the ventral lobes [51]. For epidemiology studies, precise estimates of PhIP exposure from food frequency questionnaires, even with added questions about food preparation preferences, have proven difficult. This is likely because cooking practices can generate a wide variation of heterocyclic amine levels, though frequent intake of meat by men who prefer the meats to be well-done, pan-fried, or grilled, appears to be accompanied by an increased risk for prostate cancer [15]. More recent studies have suggested that germ line variants in PhIP-metabolizing enzymes, including cytochromes p450 enzymes, sulfotransferases, and UDP-glucuronide transferases, might affect prostate damage upon over-cooked meat consumption [40].
The influence of meat consumption may not be restricted to the initiation step of prostate cancer development. The natural history of human prostate cancer encompasses many years, with small prostate cancers appearing as early as age 20–30 years in the United States in autopsy studies, diagnoses of localized cancers seen at age 60–70 years, and death from prostate cancer occurring at age 70 years and older. This provides a large window of opportunity for chronic PhIP intake to influence the pathogenesis of prostate cancer. The rat models of PhIP prostate cancer development feature carcinogen exposure for a limited period of time after puberty, followed by observation for many months, isolating the action of PhIP on the initiation and early promotion steps of carcinogenesis [74]. However, during the pathogenesis of human prostate cancer, the most common somatic gene defect, epigenetic silencing of GSTP1, results in loss of π-class glutathione S-transferase (GST) expression, a phenotype that may be remarkably sensitive to PhIP-mediated cell and genome damage [56]. This change first appears in PIA lesions and persists in prostate carcinoma cells throughout metastatic dissemination [52, 91]. Thus, not only might PhIP cause the epithelial injury that leads to the formation of PIA lesions, but PhIP might also continue to inflict cell and genome damage for decades, driving ongoing malignant prostate cancer progression.
3 Non-Dietary Exposures that Cause Prostate Epithelial Damage, Prostate Inflammation, and PIA
Sex steroid hormones, infections, and inheritance have all been thought to influence prostate cancer development. Androgenic hormones, such as testosterone and dihydrotestosterone, along with a functioning androgen receptor, are needed for normal growth and development of all sex accessory glands, including the prostate and seminal vesicles, but there is little evidence that androgens cause prostate cancer per se. Androgen levels decline steadily throughout life in adult men, reaching a peak around age 21 years and falling thereafter, as prostate cancers begin to arise [66, 68]. In the United States, African-American men suffer more prostate cancers than Caucasians, despite similar age-adjusted androgen levels [66]. Also, in the prostates of adult men, androgen signaling is required for terminal differentiation to the columnar secretory epithelial cell phenotype, promoting the transcription and translation of genes like PSA and TMPRSS2 and driving the production of secretions for the ejaculate. By acting in this way, androgens tend to suppress epithelial cell proliferation.
Of course, androgen signaling does play a significant role in the progression of established prostate cancers. The mechanism for this appears to involve the acquisition of somatic genome translocations and deletions which create fusion transcripts formed from androgen-regulated differentiation genes, such as TMPRSS2 and others, and oncogenes, such as ERG and ETV1 of the ETS family of transcription factors [84]. With these somatic genome defects, prostate cancer cells co-opt androgen signaling for the maintenance of a neoplastic phenotype. This may be the mechanistic basis for the frequent responses of advanced prostate cancers to androgen deprivation or to antiandrogens: interference with androgen signaling results in a reduction in the levels of TMPRSS2-ERG or other fusion transcripts in prostate cancer cells, attenuating cell growth and limiting cell survival. Remarkably, while androgens may act more to drive prostate cancer progression than to trigger prostate cancer initiation, new data have suggested that the initiation of androgen-target gene transcription might involve induction of DNA double-strand breaks by androgen receptor-associated TOP2B, a topoisomerase capable of resolving tangles in DNA, directed to sites in TMPRSS2 most often involved in translocations and deletions [28]. These breaks, which lead to TMPRSS2-ERG fusions, likely occur after the emergence of PIA lesions, perhaps driving PIN cells to become more invasive carcinoma cells. Topoisomerases prevent DNA tangling by catalyzing DNA breakage/rejoining reactions to permit strand passage. The enzymes are well-known to be sensitive to many compounds which can disrupt rejoining reactions and create recombinogenic DNA double-strand breaks. However, whether dietary components are responsible for any TOP2B-mediated DNA double-strand breaks during the pathogenesis of prostate cancer has not been reported.
Unlike androgens, estrogens may act to damage the prostate epithelium and promote the early steps of prostatic carcinogenesis. Breast cancers and prostate cancers are generally coincident throughout the world, leading to the hypothesis that estrogens might cause both diseases [14]. This contention is largely supported by rodent models, where estrogen exposures lead both to prostate inflammation and to prostatic cancer [59]. As an example, exposure of adult male Wistar rats to 17ß-estradiol results in prostate inflammation whether or not dihydrotestosterone is also given [55]. Male rodents given perinatal or neonatal estrogen exposures manifest prostatitis in adulthood [55, 75, 76]. Estrogens likely trigger inflammation in rodent prostates via induction of autoimmunity, as the condition can be induced in non-estrogen-treated rats via adoptive transfer of T-cells from adult male rats given 17ß-estradiol [69]. The mechanisms by which estrogens cause autoimmune prostate inflammation have not been fully elucidated, but may involve pituitary prolactin secretion, differential action of estrogenic hormones on estrogen receptor isoforms in the prostate, and/or reactive oxygen species generation by estrogen redox cycling. Also, the influence of diet habits on estrogen levels in men, largely produced in fatty tissues by aromatase action on androgens, has not been determined. Of note, however, in the United States, African-Americans, with higher prostate cancer risks, tend to have higher estrogen levels than Caucasians [66].
Infectious causes of prostate cancer have been more difficult to pin down. Many infectious agents have been detected in prostate tissue specimens or prostate secretions, but which of these actually cause prostate damage or trigger prostate inflammatory responses has not been systematically determined [70]. The best studied have been sexually transmitted infections, where gonorrhea and chlamydia have been reported to elevate serum PSA levels, an indicator of damage to the epithelial barrier function of the prostate, in at least 32 % of cases [78]. Remarkably, despite effective antibiotic treatment, PSA values can remain elevated after such infections for many months, providing evidence for chronic epithelial damage and dysfunction. This is consistent with population studies finding an increased prostate cancer risk with mild increases in serum PSA at early ages and/or with a history of sexually transmitted infections [23, 24, 79]. This complicates the search for infectious etiologies for prostate cancer in population cohort studies. If a prostate infection can damage the prostate epithelium in a young man, leading to PIA and chronic prostate inflammation even without persistent colonization as the man ages, then definitively establishing which infectious pathogen was responsible for this “hit-and-run” phenomenon will be very difficult. Nonetheless, direct inoculation of rodent prostates with bacteria or viruses triggers marked inflammatory responses and subsequent prostate cancer precursor lesions [21, 22].
Finally, some of the inherited susceptibility to prostate cancer development may be explained by genes encoding participants driving the activation and intensity of innate inflammatory responses. Two such genes, RNASEL and MSR1, appear responsible for some familial clusters of prostate cancer [11, 90]. RNASEL encodes a ribonuclease that participates in an interferon-inducible RNA destruction pathway activated in response to viral infection or other cellular damaging stress; MSR1 encodes subunits of a macrophage scavenger receptor that binds bacterial lipopolysaccharide and lipoteichoic acid. Diminished function of either protein in mice reduces the ability to fully clear various infections [80, 96]. In population studies, fairly consistent associations have been seen between prostate cancer and polymorphic variants of genes encoding toll-like receptors (TLRs), such as TLR4 and the cluster TLR1-TLR6-TLR10 [77, 95]. TLRs can bind a broad range of pathogens and/or damaged cell components, acting via NF-κB signaling to promote vigorous innate immune responses [12].
Even though estrogens and infections seem to be able to cause prostate epithelial damage which might lead to PIA and prostate cancer in the absence of dietary influences, each of these processes could be impacted by dietary habits common in high-risk prostate cancer regions of the world. Estrogen levels tend to be higher in men with increased fatty tissues. The microbiome, a source of infections or of colonization resistance to infections, varies widely with dietary practices. By influencing the propensity for estrogens and infections to inflict prostate damage, the diet can indirectly act to promote prostate cancer. Similarly, the degree of dietary heterocyclic amine-mediated prostate damage is likely to be subject to the same host genetic factors that regulate the intensity of host responses to prostate infections. RNASEL can degrade human RNA as well as viral RNA, leading to apoptosis [89]. MSR1 helps clear circulating oxidized serum lowdensity lipoproteins [41]. TLRs are activated by damaged human cell components [12]. Thus, the diet likely exerts direct and indirect effects on human prostate cancer development.
4 Inflammation, PIA, and the Molecular Pathogenesis of Prostate Cancer
Life-threatening human prostate cancer cells contain 3,866 mutations (20 non-silent coding mutations), 108 rearrangements, 5,408 regions with DNA hypermethylation, shortened telomere sequences, and activated c-Myc protein [7, 58, 92]. Notably, the somatic mutations do not seem to have singled out any common “driver” of prostatic carcinogenesis, nor hinted at any base change signature more consistent with one type of carcinogen versus another. Instead, mutations seem to accumulate over time in individual cancers, influenced by whether acquired mismatch repair gene abnormalities have appeared and/or whether pro-mutagenic treatments have been used. The more consistent somatic genetic defect is the translocations described above, particularly those involving gene targets of androgen signaling fused to cancer genes, such as TMPRSS2-ERG, which may be attributed to errors in initiation of transcription in response to androgen action leading to TOP2B-associated DNA double-strand breaks [28]. This somatic genome defect appears to occur in PIN lesions and likely underlies the invasiveness characteristic of carcinoma. Of all of the somatic changes in prostate cancer cells, the most consistent and earliest seem to involve epigenetic gene silencing, telomere shortening, and c-Myc induction [58].
Gene silencing, resulting from increased DNA methylation at gene regulatory sequences, is a candidate-initiating event for human prostate cancer. As an example, GSTP1, encoding the π-class GST, an oxidant and carcinogen detoxifying enzyme, has been found to be epigenetically silenced in some 5–10 % of PIA lesions, >70 % of PIN lesions, and almost all prostate cancers [9, 52, 58]. GSTP1 silencing has been attributed to de novo DNA hypermethylation at the 5′ regulatory region of the gene [45]. Of note, since such DNA methylation changes are potentially reversible, that is, the DNA sequence remains intact; persistent maintenance of GSTP1 inactivation throughout prostate cancer progression has served as a priori evidence for a selective growth or survival advantage during prostatic carcinogenesis. The mechanism for such a selective advantage has not been elucidated. Certainly, GSTP1 serves a caretaker gene function during carcinogenesis generally, as human prostate cancer cells devoid of GSTP1 better activate PhIP to cell and genome damaging species that cells with the enzyme, and mice carrying disrupted Gstp1/2 genes develop more skin tumors in response to topical carcinogens, and more intestinal tumors in the setting of relentless inflammation, than wild-type mice [30, 65]. A clue to a selected phenotype may be in mice carrying Pb-c-Myc transgenes that develop prostate cancer [33]. Preliminary data hint that loss of π-class GST function in these mice triggers accelerated prostatic carcinogenesis.
GSTP1 is likely but one of many genes epigenetically silenced early during prostatic carcinogenesis, by some process tied to prostate inflammation and PIA [58]. Unfortunately, as of yet, the mechanism by which epigenetic silencing of any such genes occurs in the inflamed microenvironment of the prostate, or in other organs prone to cancer development, has remained elusive. Nonetheless, inflammation and epigenetic gene silencing appear to be major contributors to the earliest steps of epithelial carcinogenesis generally, with clear examples in inflammatory bowel disease, hepatitis, and gastritis in addition to PIA lesions in the prostate [81]. Presumably, the inflamed microenvironment promotes epigenetic gene silencing and aberrant DNA methylation by acting to influence the regulation of chromatin architecture, by interfering with fidelity of DNA methylation pattern preservation in such way as to promote over-methylation at certain gene sites, or by corrupting both chromatin structure and DNA methylation maintenance. In one mouse model study of intestinal tumorigenesis, new DNA methylation changes emerged in inflamed tissues at the loci of 250 genes, with 70 % of the genes known to be targeted by polycomb complexes for repression [29]. This observation nominates polycomb complex repression as a participant in the establishment of de novo DNA methylation changes in the setting of pro-neoplastic inflammation. In this type of mechanism, DNA methylation reinforces polycomb complex repression in some way to maintain epigenetic gene silencing. In support of a complementary type of mechanism, the inflammatory cytokine interleukin 1β was found to trigger gene silencing in certain cells by promoting nitric oxide generation, leading to an over-activation of DNA methyltransferases [31]. Which type of mechanism drives the epigenetic catastrophe in PIA lesions in the prostate is not clear. GSTP1, along with other stress-response genes, tends to be induced to high level expression in PIA cells, with rare PIA cells suffering loss of GSTP1 expression and de novo methylation [52]. Thus, epigenetic silencing, and DNA methylation, must occur despite transcriptional trans-activation.
In addition to DNA methylation, telomere shortening and over-expression of c-Myc protein consistently accompany human prostatic carcinogenesis [33, 49]. The telomeres of chromosomes, specialized structures containing ~2,000 repeats of the sequence 5′-TTAGGG-3′ maintained by the enzyme telomerase, act to prevent loss of DNA sequences which might otherwise occur with lagging-strand DNA synthesis during replication and to reduce illegitimate recombination [48]. Critically, short telomeres, reflecting replication in the absence of the enzyme telomerase or some sort of telomere sequence damage, are characteristic of PIN lesions and prostate cancer cells [49]. Of note, short telomere sequences have also been seen associated with hepatitis and inflammatory bowel disease [2, 38]. Perhaps, oxidants elaborated at sites of inflammation can damage and shorten telomere sequences in the prostate as well. c-Myc expression, also ubiquitous in human prostate cancer cells, can drive prostate tumorigenesis in mice: forced c-Myc expression in the mouse prostate leads to the appearance of neoplastic cells with increased nuclear and nucleolar size, with blunted differentiation, and with diminished expression of Nkx3.1, a prostate-specific homeodomain transcription factor and tumor suppressor [33]. Like telomere shortening, c-Myc over-expression may be influenced by an inflammatory milieu. Mice carrying defective Apc genes prone to intestinal tumorigenesis show c-Myc phosphorylation and stabilization in response to exposure to TLR ligands from the intestinal microflora [42].
PIA lesions, which are generated in response to cell and tissue damage accompanied by an induced inflammatory response, link exposures, like dietary carcinogens and estrogens, to prostate cancer. The earliest stereotypical molecular events, epigenetic gene silencing, telomere shortening, and c-Myc activation, arise in PIA lesions. However, the precise mechanisms by which these molecular accidents occur have not been elaborated. Each may have its origins, or at least be influenced, by either the damaging exposure, for example, a dietary carcinogen, or by the inflammatory response. In this way, the prostatic carcinogenesis may resemble exposure-driven cancer development in many organ sites.
5 Rational Interventions to Prevent Prostate Cancer
If the epidemic of prostate cancer in the developed world can be explained by exposures to dietary carcinogens and/or estrogens that lead to chronic prostate inflammation, rational prostate cancer prevention approaches should involve: (1) an avoidance of exposures, (2) an attenuation of prostate cell and tissue damage inflicted by carcinogens, and/or (3) a reduction in intensity or duration of inflammation in the prostate. Not surprisingly, epidemiologic and clinical trial evidence has emerged in support of each approach.
A reduction in dietary heterocyclic amine exposure could be accomplished not only by educating individuals to avoid eating over-cooked meats, but also by attempting to modify cooking practices at a population scale. Steaming, microwaving, and marinating are all known to produce less heterocyclic amines than pan-frying, char-broiling, and retaining pan-dripping [85]. Measuring heterocyclic amine content in marketed cooked foods, along with appropriate incentives, might promote safer cooking practices by restaurants and by grocers. Until this occurs, carcinogen-inflicted prostate damage may be able to be lessened even despite continued consumption of over-cooked meats. Because heterocyclic amine carcinogens, like PhIP, need to be activated by metabolism to trigger cell and genome damage, interference with bioactivation might present an attractive strategy to limit carcinogenesis. Diets rich in inducers of phase 2 metabolic enzyme expression, which activate the Keap1-Nrf2 pathway, both reduce carcinogen damage generally in animal models and lower prostate cancer risk in human epidemiology studies [1, 13, 20]. The foods with the highest levels of phase 2 enzyme inducers, such as the isothiocyanate sulforaphane, are the cruciferous vegetables, like broccoli, Brussels sprouts, cauliflower, and others. In a study of normal human volunteers, intake of cruciferous vegetables reduced PhIP adduction to DNA in response to a cooked meat meal [88]. A reduction in estrogen exposures may be more difficult to achieve. Estrogens are produced via aromatase action and androgens in fatty tissues, a worrisome accompaniment to the obesity epidemic arising in the United States and other developed countries. Also, African-American men have higher estrogen levels than Caucasian men, even though androgen levels are similar and have higher prostate cancer risk [66]. Perhaps, if obesity can be controlled, prostate cancer risk might fall.
Anti-inflammatory approaches to prostate cancer prevention have yielded mixed results. Chronic or recurrent inflammation can generate reactive oxygen and nitrogen species that can cause cell and tissue damage [3, 34]. For this reason, dietary antioxidants or anti-inflammatory agents should act to protect prostate cells from the ravages of ongoing injury even if PIA lesions have already emerged. Certainly, some of the dietary components consumed in more commonly or abundantly in Asia, such as soy, may have anti-inflammatory properties relevant to the prostate; in rat models, soy-rich diets have been shown to reduce prostate inflammation [72]. In addition, prostate cancer epidemiology focused on cohorts in the developed world strongly suggests that inadequate intake of any number of antioxidant micronutrients, including vitamin E, selenium, and lycopenes from tomatoes, results in increased prostate cancer risk [10, 25, 93]. However, a recent large clinical trial, the selenium and vitamin E cancer prevention trial (SELECT), revealed that supplementation with selenium alone (hazard ratio (HR) of 1.04 with 99 % confidence interval of 0.87–1.24), with vitamin E alone (HR of 1.13 with 99 % confidence interval of 0.95–1.35), or with the combination (HR of 1.05 with 99 % confidence interval of 0.88–1.25) did not reduce prostate cancer incidence [46]. Whether this trial truly targeted men with inadequate antioxidant intake, rather than over-supplementing men with adequate intake, has not yet been reported.
Dietary and anti-inflammatory drugs might provide another strategy for lowering prostate cancer risk [6]. Cyclooxygenase (COX) inhibitors, including aspirin and non-steroidal anti-inflammatory drugs (NSAIDs), have been associated with reduced incidence and mortality of many human cancers [5]. Unfortunately, these drugs also cause gastrointestinal bleeding and cardiovascular events, which even though rare, have limited widespread use for preventing cancer. The use of such drugs for prostate cancer prevention has proven even more problematic. The selective COX-2 inhibitors, including celecoxib and rofecoxib, seemed promising when used in rodent prostate cancer models, but have thus far not been found to be particularly effective in human trials [4, 53, 54]. One difference between the rodents and humans may be that PTGS2, which encodes human COX-2, is epigenetically silenced in almost all human prostate cancers, but not in any rodent prostate cancers [91, 94]. Despite this poor track record of success of COX inhibitors for prostate cancer risk reduction, epidemiology studies have detected a consistent inverse correlation between regular aspirin use and prostate cancer [35, 47, 61]. Whether this potential benefit can be attributed to non-selective COX inhibition or to some other anti-inflammatory property of the aspirin has not been resolved. The finding that statin drugs, which may also exhibit anti-inflammatory activity, are associated with lowered prostate cancer risks has also not been fully explained [62, 63].
6 Summary and Conclusions
In response to dietary carcinogens, excess estrogens, or both, chronic or recurrent prostate inflammation, induced by damage to the prostate epithelium, drives prostate carcinogenesis to epidemic rates in the developed world. This pathogenesis mechanism is revealed in the emergence of PIA, precursor lesions for PIN and prostate cancer. Cells in PIA lesions, attempting to regenerate the prostate epithelium despite ongoing inflammatory stresses, appear prone to suffer epigenome corruption, heralded by GSTP1 silencing and de novo DNA methylation, telomere shortening, and c-Myc activation. Activation of androgen signaling in such cells risks targeted translocations, caused by androgen recruitment of TOP2B to binding sites at the loci of prostate cell differentiation genes, which lead to fusion gene transcripts, such as TMPRSS2-ERG, that drive malignant prostate cancer progression. Careful attention, at a population scale, to dietary habits, with changing of cooking practices to limit carcinogen production, with increased intake of cruciferous vegetables to reduce carcinogen bioactivation, and with increased intake of anti-inflammatory and antioxidant micronutrients to attenuate prostate inflammation, might contribute to lowering the societal burden of prostate cancer.
Abbreviations
- PSA
Prostate-specific antigen
- BPH
Benign prostatic hyperplasia
- PIA
Proliferative inflammatory atrophy
- PIN
Prostatic intraepithelial neoplasia
- PhIP
Phenylimidazopyridine
- GST
Glutathione S-transferase
- TLRs
Toll-like receptors
- COX
Cyclooxygenase
- NSAIDs
Non-steroidal anti-inflammatory drugs
References
- 1.Ahn YH, Hwang Y, Liu H, et al. Electrophilic tuning of the chemoprotective natural product sulforaphane. Proc Natl Acad Sci USA. 2010;107:9590–9595. doi: 10.1073/pnas.1004104107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Aikata H, Takaishi H, Kawakami Y, et al. Telomere reduction in human liver tissues with age and chronic inflammation. Exp Cell Res. 2000;256:578–582. doi: 10.1006/excr.2000.4862. [DOI] [PubMed] [Google Scholar]
- 3.Ames BN, Gold LS, Willett WC. The causes and prevention of cancer. Proc Natl Acad Sci USA. 1995;92:5258–5265. doi: 10.1073/pnas.92.12.5258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Antonarakis ES, Heath EI, Walczak JR, et al. Phase II, randomized, placebo-controlled trial of neoadjuvant celecoxib in men with clinically localized prostate cancer: evaluation of drug-specific biomarkers. J Clin Oncol. 2009;27:4986–4993. doi: 10.1200/JCO.2009.21.9410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bardia A, Ebbert JO, Vierkant RA, et al. Association of aspirin and nonaspirin nonsteroidal anti-inflammatory drugs with cancer incidence and mortality. J Natl Cancer Inst. 2007;99:881–889. doi: 10.1093/jnci/djk200. [DOI] [PubMed] [Google Scholar]
- 6.Bardia A, Platz EA, Yegnasubramanian S, et al. Anti-inflammatory drugs, antioxidants, and prostate cancer prevention. Curr Opin Pharmacol. 2009;9:419–426. doi: 10.1016/j.coph.2009.06.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Berger MF, Lawrence MS, Demichelis F, et al. The genomic complexity of primary human prostate cancer. Nature. 2011;470:214–220. doi: 10.1038/nature09744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bordas M, Moyano E, Puignou L, et al. Formation and stability of heterocyclic amines in a meat flavour model system. Effect of temperature, time and precursors. J Chromatogr B Analyt Technol Biomed Life Sci. 2004;802:11–17. doi: 10.1016/j.jchromb.2003.09.024. [DOI] [PubMed] [Google Scholar]
- 9.Brooks JD, Weinstein M, Lin X, et al. CG island methylation changes near the GSTP1 gene in prostatic intraepithelial neoplasia (PIN) Cancer Epid Biom Prev. 1998;7:531–536. [PubMed] [Google Scholar]
- 10.Brooks JD, Metter EJ, Chan DW, et al. Plasma selenium level before diagnosis and the risk of prostate cancer development. J Urol. 2001;166:2034–2038. [PubMed] [Google Scholar]
- 11.Carpten J, Nupponen N, Isaacs S, et al. Germline mutations in the ribonuclease L gene in families showing linkage with HPC1. Nat Genet. 2002;30:181–184. doi: 10.1038/ng823. [DOI] [PubMed] [Google Scholar]
- 12.Caruso C, Balistreri CR, Candore G, et al. Polymorphisms of pro-inflammatory genes and prostate cancer risk: a pharmacogenomic approach. Cancer Immunol Immunother. 2009;58:1919–1933. doi: 10.1007/s00262-009-0658-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Cohen JH, Kristal AR, Stanford JL. Fruit and vegetable intakes and prostate cancer risk. J Natl Cancer Inst. 2000;92:61–68. doi: 10.1093/jnci/92.1.61. [DOI] [PubMed] [Google Scholar]
- 14.Coffey DS. Similarities of prostate and breast cancer: evolution, diet, and estrogens. Urology. 2001;57:31–38. doi: 10.1016/s0090-4295(00)00938-9. [DOI] [PubMed] [Google Scholar]
- 15.Cross AJ, Peters U, Kirsh VA, et al. A prospective study of meat and meat mutagens and prostate cancer risk. Cancer Res. 2005;65:11779–11784. doi: 10.1158/0008-5472.CAN-05-2191. [DOI] [PubMed] [Google Scholar]
- 16.De Marzo AM, Marchi VL, Epstein JI, et al. Proliferative inflammatory atrophy of the prostate: implications for prostatic carcinogenesis. Am J Pathol. 1999;155:1985–1992. doi: 10.1016/S0002-9440(10)65517-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.De Marzo AM, Platz EA, Epstein JI, et al. A working group classification of focal prostate atrophy lesions. Am J Surg Pathol. 2006;30:1281–1291. doi: 10.1097/01.pas.0000213289.50660.be. [DOI] [PubMed] [Google Scholar]
- 18.De Marzo AM, Platz EA, Sutcliffe S, et al. Inflammation in prostate carcinogenesis. Nat Rev Cancer. 2007;7:256–269. doi: 10.1038/nrc2090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Dennis LK, Lynch CF, Torner JC. Epidemiologic association between prostatitis and prostate cancer. Urology. 2002;60:78–83. doi: 10.1016/s0090-4295(02)01637-0. [DOI] [PubMed] [Google Scholar]
- 20.Dinkova-Kostova AT, Talalay P. Direct and indirect antioxidant properties of inducers of cytoprotective proteins. Mol Nutr Food Res. 2008;52(Suppl 1):S128–S138. doi: 10.1002/mnfr.200700195. [DOI] [PubMed] [Google Scholar]
- 21.Elkahwaji JE, Zhong W, Hopkins WJ, et al. Chronic bacterial infection and inflammation incite reactive hyperplasia in a mouse model of chronic prostatitis. Prostate. 2007;67:14–21. doi: 10.1002/pros.20445. [DOI] [PubMed] [Google Scholar]
- 22.Elkahwaji JE, Hauke RJ, Brawner CM. Chronic bacterial inflammation induces prostatic intraepithelial neoplasia in mouse prostate. Br J Cancer. 2009;101:1740–1748. doi: 10.1038/sj.bjc.6605370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Fang J, Metter EJ, Landis P, et al. Low levels of prostate-specific antigen predict long-term risk of prostate cancer: results from the Baltimore longitudinal study of aging. Urology. 2001;58:411–416. doi: 10.1016/s0090-4295(01)01304-8. [DOI] [PubMed] [Google Scholar]
- 24.Gann PH, Hennekens CH, Stampfer MJ. A prospective evaluation of plasma prostate-specific antigen for detection of prostatic cancer. JAMA. 1995;273:289–294. [PubMed] [Google Scholar]
- 25.Gann PH, Ma J, Giovannucci E, et al. Lower prostate cancer risk in men with elevated plasma lycopene levels: results of a prospective analysis. Cancer Res. 1999;59:1225–1230. [PubMed] [Google Scholar]
- 26.Giovannucci E, Rimm EB, Colditz GA, et al. A prospective study of dietary fat and risk of prostate cancer [see comments] J Natl Cancer Inst. 1993;85:1571–1579. doi: 10.1093/jnci/85.19.1571. [DOI] [PubMed] [Google Scholar]
- 27.Haenszel W, Kurihara M. Studies of Japanese migrants. I. Mortality from cancer and other diseases among Japanese in the United States. J Natl Cancer Inst. 1968;40:43–68. [PubMed] [Google Scholar]
- 28.Haffner MC, Aryee MJ, Toubaji A, et al. Androgen-induced TOP2B-mediated double-strand breaks and prostate cancer gene rearrangements. Nat Genet. 2010;42:668–675. doi: 10.1038/ng.613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hahn MA, Hahn T, Lee DH, et al. Methylation of polycomb target genes in intestinal cancer is mediated by inflammation. Cancer Res. 2008;68:10280–10289. doi: 10.1158/0008-5472.CAN-08-1957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Henderson CJ, Smith AG, Ure J, et al. Increased skin tumorigenesis in mice lacking pi class glutathione S- transferases. Proc Natl Acad Sci USA. 1998;95:5275–5280. doi: 10.1073/pnas.95.9.5275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hmadcha A, Bedoya FJ, Sobrino F, et al. Methylation-dependent gene silencing induced by interleukin 1beta via nitric oxide production. J Exp Med. 1999;190:1595–1604. doi: 10.1084/jem.190.11.1595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Isaacs JT. The aging ACI/Seg versus Copenhagen male rat as a model system for the study of prostatic carcinogenesis. Cancer Res. 1984;44:5785–5796. [PubMed] [Google Scholar]
- 33.Iwata T, Schultz D, Hicks J, et al. MYC overexpression induces prostatic intraepithelial neoplasia and loss of Nkx3.1 in mouse luminal epithelial cells. PLoS ONE. 2010;5:e9427. doi: 10.1371/journal.pone.0009427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Jackson AL, Loeb LA. The contribution of endogenous sources of DNA damage to the multiple mutations in cancer. Mutat Res. 2001;477:7–21. doi: 10.1016/s0027-5107(01)00091-4. [DOI] [PubMed] [Google Scholar]
- 35.Jacobs EJ, Rodriguez C, Mondul AM, et al. A large cohort study of aspirin and other nonsteroidal anti-inflammatory drugs and prostate cancer incidence. J Natl Cancer Inst. 2005;97:975–980. doi: 10.1093/jnci/dji173. [DOI] [PubMed] [Google Scholar]
- 36.Jemal A, Siegel R, Ward E, Hao Y, Xu J, Thun MJ. Cancer statistics, 2009. CA Cancer J Clin. 2009;59:225–249. doi: 10.3322/caac.20006. [DOI] [PubMed] [Google Scholar]
- 37.Kidd LC, Stillwell WG, Yu MC, et al. Urinary excretion of 2-amino-1-methyl-6-phenylimidazo[4,5-b]pyridine (PhIP) in White, African–American, and Asian–American men in Los Angeles county. Cancer Epidemiol Biomarkers Prevent. 1999;8:439–445. [PubMed] [Google Scholar]
- 38.Kinouchi Y, Hiwatashi N, Chida M, et al. Telomere shortening in the colonic mucosa of patients with ulcerative colitis. J Gastroenterol. 1998;33:343–348. doi: 10.1007/s005350050094. [DOI] [PubMed] [Google Scholar]
- 39.Knize MG, Salmon CP, Mehta SS, et al. Analysis of cooked muscle meats for heterocyclic aromatic amine carcinogens. Mutat Res. 1997;376:129–134. doi: 10.1016/s0027-5107(97)00035-3. [DOI] [PubMed] [Google Scholar]
- 40.Koutros S, Berndt SI, Sinha R, et al. Xenobiotic metabolizing gene variants, dietary heterocyclic amine intake, and risk of prostate cancer. Cancer Res. 2009;69:1877–1884. doi: 10.1158/0008-5472.CAN-08-2447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Krieger M, Herz J. Structures and functions of multiligand lipoprotein receptors: macrophage scavenger receptors and LDL receptor-related protein (LRP) Annu Rev Biochem. 1994;63:601–637. doi: 10.1146/annurev.bi.63.070194.003125. [DOI] [PubMed] [Google Scholar]
- 42.Lee SH, Hu LL, Gonzalez-Navajas J, et al. ERK activation drives intestinal tumorigenesis in Apc(min/+) mice. Nat Med. 2010;16:665–670. doi: 10.1038/nm.2143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Le Marchand L, Kolonel LN, Wilkens LR, et al. Animal fat consumption and prostate cancer: a prospective study in Hawaii. Epidemiology. 1994;5:276–282. doi: 10.1097/00001648-199405000-00004. [DOI] [PubMed] [Google Scholar]
- 44.Lijinsky W, Shubik P. Benzo(a)pyrene and other polynuclear hydrocarbons in charcoal-broiled meat. Science. 1964;145:53–55. doi: 10.1126/science.145.3627.53. [DOI] [PubMed] [Google Scholar]
- 45.Lin X, Tascilar M, Lee W-H, et al. GSTP1 CpG island hypermethylation is responsible for the absence of GSTP1 expression in human prostate cancer cells. Am J Pathol. 2001;159:1815–1826. doi: 10.1016/S0002-9440(10)63028-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Lippman SM, Klein EA, Goodman PJ, et al. Effect of selenium and vitamin E on risk of prostate cancer and other cancers: the selenium and vitamin E cancer prevention trial (SELECT) JAMA. 2009;301:39–51. doi: 10.1001/jama.2008.864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Mahmud S, Franco E, Aprikian A. Prostate cancer and use of nonsteroidal anti-inflammatory drugs: systematic review and meta-analysis. Br J Cancer. 2004;90:93–99. doi: 10.1038/sj.bjc.6601416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Maser RS, DePinho RA. Connecting chromosomes, crisis, and cancer. Science. 2002;297:565–569. doi: 10.1126/science.297.5581.565. [DOI] [PubMed] [Google Scholar]
- 49.Meeker AK, Hicks JL, Platz EA, et al. Telomere shortening is an early somatic DNA alteration in human prostate tumorigenesis. Cancer Res. 2002;62:6405–6409. [PubMed] [Google Scholar]
- 50.Murphy AB, Macejko A, Taylor A, et al. Chronic prostatitis: management strategies. Drugs. 2009;69:71–84. doi: 10.2165/00003495-200969010-00005. [DOI] [PubMed] [Google Scholar]
- 51.Nakai Y, Nelson WG, De Marzo AM. The dietary charred meat carcinogen 2-amino-1-methyl-6-phenylimidazo[4,5-b]pyridine acts as both a tumor initiator and promoter in the rat ventral prostate. Cancer Res. 2007;67:1378–1384. doi: 10.1158/0008-5472.CAN-06-1336. [DOI] [PubMed] [Google Scholar]
- 52.Nakayama M, Bennett CJ, Hicks JL, et al. Hypermethylation of the human glutathione S-transferase-pi gene (GSTP1) CpG island is present in a subset of proliferative inflammatory atrophy lesions but not in normal or hyperplastic epithelium of the prostate: a detailed study using laser-capture microdissection. Am J Pathol. 2003;163:923–933. doi: 10.1016/s0002-9440(10)63452-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Narayanan BA, Condon MS, Bosland MC, et al. Suppression of N-methyl-N-nitrosourea/testosterone-induced rat prostate cancer growth by celecoxib: effects on cyclooxygenase-2, cell cycle regulation, and apoptosis mechanism(s) Clin Cancer Res. 2003;9:3503–3513. [PubMed] [Google Scholar]
- 54.Narayanan BA, Narayanan NK, Pittman B, et al. Regression of mouse prostatic intraepithelial neoplasia by nonsteroidal anti-inflammatory drugs in the transgenic adenocarcinoma mouse prostate model. Clin Cancer Res. 2004;10:7727–7737. doi: 10.1158/1078-0432.CCR-04-0732. [DOI] [PubMed] [Google Scholar]
- 55.Naslund MJ, Strandberg JD, Coffey DS. The role of androgens and estrogens in the pathogenesis of experimental nonbacterial prostatitis. J Urol. 1988;140:1049–1053. doi: 10.1016/s0022-5347(17)41924-0. [DOI] [PubMed] [Google Scholar]
- 56.Nelson CP, Kidd LC, Sauvageot J, et al. Protection against 2-hydroxyamino-1-methyl-6-phenylimidazo[4,5- b]pyridine cytotoxicity and DNA adduct formation in human prostate by glutathione S-transferase P1. Cancer Res. 2001;61:103–109. [PubMed] [Google Scholar]
- 57.Nelson WG, De Marzo AM, Isaacs WB. Prostate cancer. N Engl J Med. 2003;349:366–381. doi: 10.1056/NEJMra021562. [DOI] [PubMed] [Google Scholar]
- 58.Nelson WG, De Marzo AM, Yegnasubramanian S. Epigenetic alterations in human prostate cancers. Endocrinology. 2009;150:3991–4002. doi: 10.1210/en.2009-0573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Palapattu GS, Sutcliffe S, Bastian PJ, et al. Prostate carcinogenesis and inflammation: emerging insights. Carcinogenesis. 2005;26:1170–1181. doi: 10.1093/carcin/bgh317. [DOI] [PubMed] [Google Scholar]
- 60.Parsons JK, Nelson CP, Gage WR, et al. GSTA1 expression in normal, preneoplastic, and neoplastic human prostate tissue. Prostate. 2001;49:30–37. doi: 10.1002/pros.1115. [DOI] [PubMed] [Google Scholar]
- 61.Platz EA, Rohrmann S, Pearson JD, et al. Nonsteroidal anti-inflammatory drugs and risk of prostate cancer in the Baltimore longitudinal study of aging. Cancer Epidemiol Biomarkers Prev. 2005;14:390–396. doi: 10.1158/1055-9965.EPI-04-0532. [DOI] [PubMed] [Google Scholar]
- 62.Platz EA, Leitzmann MF, Visvanathan K, et al. Statin drugs and risk of advanced prostate cancer. J Natl Cancer Inst. 2006;98:1819–1825. doi: 10.1093/jnci/djj499. [DOI] [PubMed] [Google Scholar]
- 63.Platz EA, Till C, Goodman PJ, et al. Men with low serum cholesterol have a lower risk of high-grade prostate cancer in the placebo arm of the prostate cancer prevention trial. Cancer Epidemiol Biomarkers Prev. 2009;18:2807–2813. doi: 10.1158/1055-9965.EPI-09-0472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Putzi MJ, De Marzo AM. Morphologic transitions between proliferative inflammatory atrophy and high-grade prostatic intraepithelial neoplasia. Urology. 2000;56:828–832. doi: 10.1016/s0090-4295(00)00776-7. [DOI] [PubMed] [Google Scholar]
- 65.Ritchie KJ, Walsh S, Sansom OJ, et al. Markedly enhanced colon tumorigenesis in ApcMin mice lacking glutathione S-transferase Pi. Proc Natl Acad Sci USA. 2009;106:20859–20864. doi: 10.1073/pnas.0911351106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Rohrmann S, Nelson WG, Rifai N, et al. Serum estrogen, but not testosterone, levels differ between black and white men in a nationally representative sample of Americans. J Clin Endocrinol Metab. 2007;92:2519–2525. doi: 10.1210/jc.2007-0028. [DOI] [PubMed] [Google Scholar]
- 67.Ruska KM, Sauvageot J, Epstein JI. Histology and cellular kinetics of prostatic atrophy. Am J Surg Pathol. 1998;22:1073–1077. doi: 10.1097/00000478-199809000-00005. [DOI] [PubMed] [Google Scholar]
- 68.Sakr WA, Grignon DJ, Crissman JD, et al. High grade prostatic intraepithelial neoplasia (HGPIN) and prostatic adenocarcinoma between the ages of 20–69: an autopsy study of 249 cases. In Vivo. 1994;8:439–443. [PubMed] [Google Scholar]
- 69.Seethalakshmi L, Bala RS, Malhotra RK, et al. 17 beta-estradiol induced prostatitis in the rat is an autoimmune disease. J Urol. 1996;156:1838–1842. [PubMed] [Google Scholar]
- 70.Sfanos KS, Sauvageot J, Fedor HL, et al. A molecular analysis of prokaryotic and viral DNA sequences in prostate tissue from patients with prostate cancer indicates the presence of multiple and diverse microorganisms. Prostate. 2008;68:306–320. doi: 10.1002/pros.20680. [DOI] [PubMed] [Google Scholar]
- 71.Sfanos KS, Wilson BA, De Marzo AM, et al. Acute inflammatory proteins constitute the organic matrix of prostatic corpora amylacea and calculi in men with prostate cancer. Proc Natl Acad Sci USA. 2009;106:3443–3448. doi: 10.1073/pnas.0810473106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Sharma OP, Adlercreutz H, Strandberg JD, et al. Soy of dietary source plays a preventive role against the pathogenesis of prostatitis in rats. J Steroid Biochem Mol Biol. 1992;43:557–564. doi: 10.1016/0960-0760(92)90244-d. [DOI] [PubMed] [Google Scholar]
- 73.Shimizu H, Ross RK, Bernstein L, et al. Cancers of the prostate and breast among Japanese and white immigrants in Los Angeles County. Br J Cancer. 1991;63:963–966. doi: 10.1038/bjc.1991.210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Shirai T, Sano M, Tamano S, et al. The prostate: a target for carcinogenicity of 2-amino-1-methyl-6-phenylimidazo[4,5-b]pyridine (PhIP) derived from cooked foods. Cancer Res. 1997;57:195–198. [PubMed] [Google Scholar]
- 75.Stoker TE, Robinette CL, Britt BH, et al. Prepubertal exposure to compounds that increase prolactin secretion in the male rat: effects on the adult prostate. Biol Reprod. 1999;61:1636–1643. doi: 10.1095/biolreprod61.6.1636. [DOI] [PubMed] [Google Scholar]
- 76.Stoker TE, Robinette CL, Cooper RL. Perinatal exposure to estrogenic compounds and the subsequent effects on the prostate of the adult rat: evaluation of inflammation in the ventral and lateral lobes. Reprod Toxicol. 1999;13:463–472. doi: 10.1016/s0890-6238(99)00049-0. [DOI] [PubMed] [Google Scholar]
- 77.Sun J, Wiklund F, Zheng SL, et al. Sequence variants in Toll-like receptor gene cluster (TLR6-TLR1-TLR10) and prostate cancer risk. J Natl Cancer Inst. 2005;97:525–532. doi: 10.1093/jnci/dji070. [DOI] [PubMed] [Google Scholar]
- 78.Sutcliffe S, Zenilman JM, Ghanem KG, et al. Sexually transmitted infections and prostatic inflammation/cell damage as measured by serum prostate specific antigen concentration. J Urol. 2006;175:1937–1942. doi: 10.1016/S0022-5347(05)00892-X. [DOI] [PubMed] [Google Scholar]
- 79.Sutcliffe S, Platz EA. Inflammation and prostate cancer: a focus on infections. Curr Urol Rep. 2008;9:243–249. doi: 10.1007/s11934-008-0042-z. [DOI] [PubMed] [Google Scholar]
- 80.Suzuki H, Kurihara Y, Takeya M, et al. A role for macrophage scavenger receptors in atherosclerosis and susceptibility to infection. Nature. 1997;386:292–296. doi: 10.1038/386292a0. [DOI] [PubMed] [Google Scholar]
- 81.Suzuki H, Toyota M, Kondo Y, et al. Inflammation-related aberrant patterns of DNA methylation: detection and role in epigenetic deregulation of cancer cell transcriptome. Methods Mol Biol. 2009;512:55–69. doi: 10.1007/978-1-60327-530-9_5. [DOI] [PubMed] [Google Scholar]
- 82.Tang D, Liu JJ, Bock CH, et al. Racial differences in clinical and pathological associations with PhIP-DNA adducts in prostate. Int J Cancer. 2007;121:1319–1324. doi: 10.1002/ijc.22806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Thompson IM, Goodman PJ, Tangen CM, et al. The influence of finasteride on the development of prostate cancer. N Engl J Med. 2003;349:215–224. doi: 10.1056/NEJMoa030660. [DOI] [PubMed] [Google Scholar]
- 84.Tomlins SA, Rhodes DR, Perner S, et al. Recurrent fusion of TMPRSS2 and ETS transcription factor genes in prostate cancer. Science. 2005;310:644–648. doi: 10.1126/science.1117679. [DOI] [PubMed] [Google Scholar]
- 85.Turesky RJ. Formation and biochemistry of carcinogenic heterocyclic aromatic amines in cooked meats. Toxicol Lett. 2007;168:219–227. doi: 10.1016/j.toxlet.2006.10.018. [DOI] [PubMed] [Google Scholar]
- 86.van Leenders GJ, Gage WR, Hicks JL, et al. Intermediate cells in human prostate epithelium are enriched in proliferative inflammatory atrophy. Am J Pathol. 2003;162:1529–1537. doi: 10.1016/S0002-9440(10)64286-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Wakabayashi K, Ushiyama H, Takahashi M, et al. Exposure to heterocyclic amines. Environ Health Perspect. 1993;99:129–134. doi: 10.1289/ehp.9399129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Walters DG, Young PJ, Agus C, et al. Cruciferous vegetable consumption alters the metabolism of the dietary carcinogen 2-amino-1-methyl-6-phenylimidazo[4,5-b]pyridine (PhIP) in humans. Carcinogenesis. 2004;25:1659–1669. doi: 10.1093/carcin/bgh164. [DOI] [PubMed] [Google Scholar]
- 89.Xiang Y, Wang Z, Murakami J, et al. Effects of RNase L mutations associated with prostate cancer on apoptosis induced by 2′, 5′-oligoadenylates. Cancer Res. 2003;63:6795–6801. [PubMed] [Google Scholar]
- 90.Xu J, Zheng SL, Komiya A, et al. Germline mutations and sequence variants of the macrophage scavenger receptor 1 gene are associated with prostate cancer risk. Nat Genet. 2002;32:321–325. doi: 10.1038/ng994. [DOI] [PubMed] [Google Scholar]
- 91.Yegnasubramanian S, Kowalski J, Gonzalgo ML, et al. Hypermethylation of CpG islands in primary and metastatic human prostate cancer. Cancer Res. 2004;64:1975–1986. doi: 10.1158/0008-5472.can-03-3972. [DOI] [PubMed] [Google Scholar]
- 92.Yegnasubramanian S, Wu Z, Haffner MC, et al. Chromosome-wide mapping of DNA methylation patterns in normal and malignant prostate cells reveals pervasive methylation of gene-associated and conserved intergenic sequences. BMC Genomics. 2011;12:213. doi: 10.1186/1471-2164-12-313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Yoshizawa K, Willett WC, Morris SJ, et al. Study of prediagnostic selenium level in toenails and the risk of advanced prostate cancer. J Natl Cancer Inst. 1998;90:1219–1224. doi: 10.1093/jnci/90.16.1219. [DOI] [PubMed] [Google Scholar]
- 94.Zha S, Gage WR, Sauvageot J, et al. Cyclooxygenase-2 is up-regulated in proliferative inflammatory atrophy of the prostate, but not in prostate carcinoma. Cancer Res. 2001;61:8617–8623. [PubMed] [Google Scholar]
- 95.Zheng SL, Augustsson-Balter K, Chang B, et al. Sequence variants of toll-like receptor 4 are associated with prostate cancer risk: results from the cancer prostate in Sweden study. Cancer Res. 2004;64:2918–2922. doi: 10.1158/0008-5472.can-03-3280. [DOI] [PubMed] [Google Scholar]
- 96.Zhou A, Paranjape J, Brown TL, et al. Interferon action and apoptosis are defective in mice devoid of 2′,5′- oligoadenylate-dependent RNase L. EMBO J. 1997;16:6355–6363. doi: 10.1093/emboj/16.21.6355. [DOI] [PMC free article] [PubMed] [Google Scholar]