

Abstract
RNA splicing plays a fundamental role in human biology. Its relevance in cancer is rapidly emerging as demonstrated by spliceosome mutations that determine the prognosis of patients with hematologic malignancies. We report studies using FD-895 and pladienolide-B in primary leukemia cells derived from patients with chronic lymphocytic leukemia and leukemia-lymphoma cell lines. We found that FD-895 and pladienolide-B induce an early pattern of mRNA intron retention – spliceosome modulation. This process was associated with apoptosis preferentially in cancer cells as compared to normal lymphocytes. The pro-apoptotic activity of these compounds was observed regardless of poor prognostic factors such as Del(17p), TP53 or SF3B1 mutations and was able to overcome the protective effect of culture conditions that resemble the tumor microenvironment. In addition, the activity of these compounds was observed not only in vitro but also in vivo using the A20 lymphoma murine model. Overall, these findings give evidence for the first time that spliceosome modulation is a valid target in chronic lymphocytic leukemia and provide an additional rationale for the development of spliceosome modulators for cancer therapy.
Introduction
Chronic lymphocytic leukemia (CLL) is the most common adult leukemia.1 Despite improvements in the survival of patients who are treated with chemoimmunotherapy,2 there is still no cure for this disease, except allogeneic bone marrow transplantation. High-risk patients, such as those with deletions in chromosome 17 - Del(17p) or TP53 mutations, generally fail to respond to chemotherapy and have a very poor prognosis.3 As such, there is a need for development of therapeutic agents that target novel pathways in CLL.4
Splicing, the removal of introns and joining of exons from nascent pre-mRNA, has gained attention as a target for cancer therapy given the distinct splicing patterns identified both in tumor cells and metastatic tumor populations.5,6 Recently, a series of studies identified heterozygous missense mutations in U2AF1 and splicing factor 3B subunit 1 (SF3B1) genes associated with myelodysplastic syndromes, and have shown that SF3B1 is frequently mutated in myelodysplastic syndromes,7,8 and CLL.9,10 This, combined with the identification of small molecules that target the spliceosome, motivated us to explore the application of these agents to CLL.
Identified in 1994,11 FD-895 was the first member of a large family of polyketides isolated from related strains of Streptomyces platensis, which includes pladienolide-B (PLAD-B) and pladienolide-D (PLAD-D) (Online Supplementary Figure S1).12 PLAD-B targets the splicing factor subunit SF3b and this interaction is postulated to be responsible for its mechanism of antitumor activity.13 Pladienolide-resistant clones from WiDr and DLD1 colorectal-cancer cell lines shared an identical mutation at Arg1074 (R1074H) in the SF3B1 gene suggesting that this mutation is critical for its anti-cancer activity via spliceosome modulation.14,15
Additional work has also identified other small molecules with splicing modulator activity including spliceostatin A, herboxidiene, isoginkgetin, and E7107, a compound that has been tested in phase I clinical studies in which it has shown clinical activity, albeit with unexpected visual toxicities.16 Additional data will be required to define the role of this and other related compounds as potential anti-cancer agents.
Here, we present studies using FD-895 and PLAD-B on primary leukemia cells derived from CLL patients and leukemia and lymphoma cell lines. We anticipate that these studies will provide the foundation for future development of pharmacologically-optimized spliceosome modulators.17
Methods
For in vivo study, PLAD-B was purchased from Santa Cruz Biotechnology (catalog # sc-391691). For in vitro studies, FD–895 was prepared by total synthesis in our laboratories,17 and PLAD–B was obtained as a gift from Merlion Pharmaceuticals. Fludarabine (F-ara-A) (catalog # F9813) and Bendamustine (catalog # B5437) were obtained from Sigma-Aldrich.
Peripheral blood mononuclear cells from CLL patients were obtained from the CLL Research Consortium tissue bank. After the diagnosis of CLL had been confirmed,18 patients provided written informed consent to collection of blood samples in a protocol approved by the Institutional Review Board of the University of California, San Diego (USCD) in accordance with the Declaration of Helsinki.19 The animal study protocol was approved by the Medical Experimental Animal Care Committee of UCSD.
Normal B cells (NBC) were purified from buffy coats of healthy volunteer donors. Dynabeads CD19 pan B (Life Technologies) and DETACHaBEAD CD19 (Life Technologies) were used to achieve more than 95% purity of the isolated NBC by flow cytometry analysis.
Additional techniques, methods and a list of polymerase chain reaction (PCR) primers (Online Supplementary Table S1) used in the study are provided in the Online Supplementary Information.
Results
FD-895 and PLAD-B induce intron retention – spliceosome modulation
Analysis of RNA-seq data from CLL cells and NBC showed differences in splicing efficiency between the two. In order to assess splicing efficiency quantitatively, the intron retention (IR) ratios were calculated for each gene by comparing the RNA-seq read density in exons, measured in fragments per kilobase per million fragments mapped, relative to the RNA-seq density of introns (Online Supplementary Table S2). Comparison of IR ratios showed that several genes have widespread changes in splicing efficiency, resulting in a basal increase of intron/exon RNA ratios in untreated CLL cells compared to NBC (1.4-fold average increase, Figure 1A and Online Supplementary Figure S2A, S2B). An average 71% of sequenced genes showed increased IR in CLL cells compared to NBC (Online Supplementary Table S3). Moreover, after incubation of CLL cells with FD-895 or PLAD-B there was an additional increase in IR ratios (2.2-fold average increase, Figure 1B, Online Supplementary Figures S2C, S2D, and 3A), but this was not observed after F-ara-A treatment (Figure 1C, 1D). We observed that treatment with FD-895 or PLAD-B induced IR more significantly in genes with the lowest basal IR suggesting a de novo spliceosome modulation induced by these compounds rather than exacerbation of an existing abnormal splicing process (Online Supplementary Figure S4). The DNAJB1 gene encodes a chaperone which is a member of the DnaJ or Hsp40 (heat shock protein 40 kD) family of proteins. DNAJB1 was one the genes that showed an increased IR ratio after treatment with FD-895 or PLAD-B (7-fold increase, Figure 1A–C) and because of that, this gene was selected for the validation studies described below.
Figure 1.
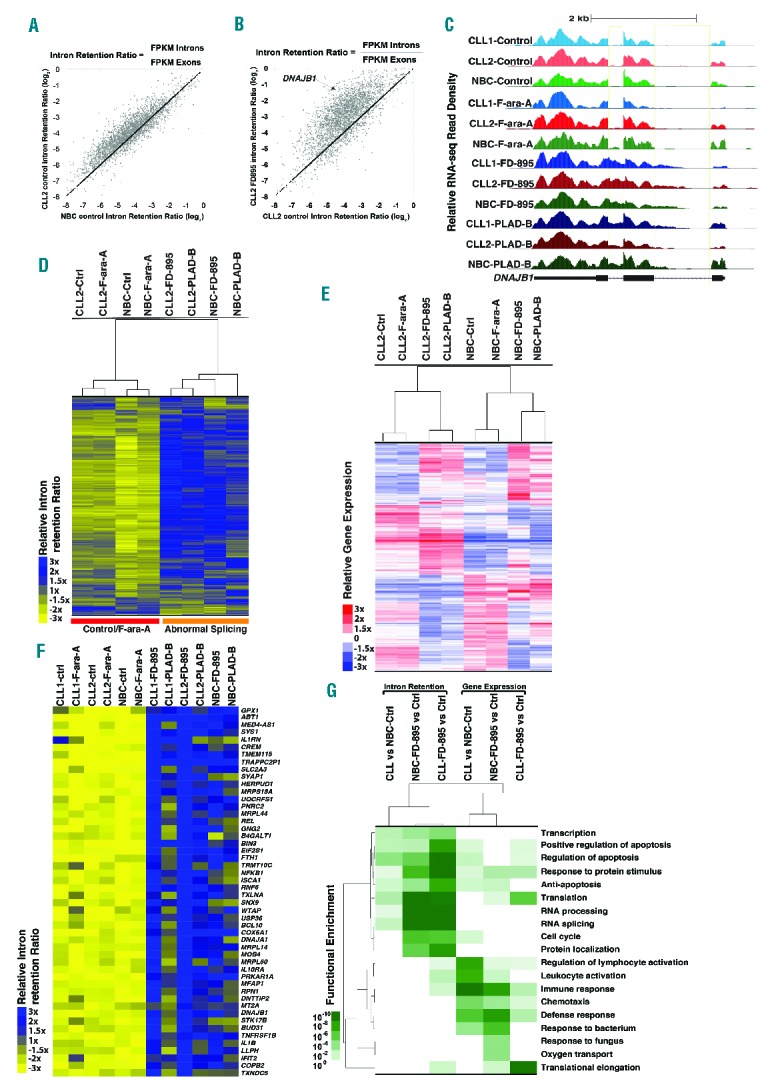
RNA sequencing analysis in samples treated with FD-895 and PLAD-B. RNA transcriptome analysis was conducted on RNA obtained from two separate CLL samples (wild-type for TP53 and SF3B1) and normal B cells from healthy controls. CLL and normal B cells were incubated with 100 nM FD-895, 100 nM PLAD-B or 10 μM F-ara-A for 2 h prior to harvesting. (A) Comparison of IR log2 ratios [fragments per kilobase per million fragments mapped (FPKM) intron / FPKM exon] in untreated CLL and untreated normal B cells. The black line represents the diagonal where IR ratios are equal in both samples. (B) Comparison of IR log2 ratios in CLL control and FD-895-treated samples. (C) RNA-seq read densities at the DNAJB1 locus, a sample gene with high IR pattern. FD-895- and PLAD-B-treated samples but not F-ara-A-treated or untreated controls showed a pattern of IR with accumulation of read densities in the regions expanding introns 1 and 2. (D) Hierarchical clustering for approximately 3,500 genes shown in the heat-map depicting the relative IR log2 ratios. (E) Gene expression across normal B cells and CLL samples treated with FD-895, PLAD-B and F-ara-A shown in the heat-map for approximately 4,000 genes. A similar clustering was observed with additional CLL and normal B-cell samples. (F) RNA-seq heat map showing 50 genes with a high level of IR after treatment with either FD-895 or PLAD-B. (G) Heat-map showing gene ontology pathway enrichment (P-values) using genes with greater than 3-fold increases in either their IR or mRNA expression ratios in CLL or normal B cells in untreated control samples compared with samples incubated with FD-895.
We performed an unsupervised cluster analysis of 3,500 highly expressed genes using IR ratios of CLL cell and NBC samples treated with FD-895, PLAD-B, or F-ara-A or untreated controls. We observed that IR ratios from untreated samples clustered together with F-ara-A samples, indicating minimal IR changes induced by F-ara-A regardless of the cell type (CLL or NBC). Distinctively, FD-895 and PLAD-B treated samples showed higher IR ratios compared to untreated controls or F-ara-A-treated samples (Figure 1D, Online Supplementary Figure S5). Additionally, we analyzed gene expression profiles of the same samples and we found that unlike IR ratios, gene expression profiles were segregated by cell type forming NBC and CLL clusters. Untreated cells and F-ara-A-treated samples clustered together while samples treated with FD-895 or PLAD-B showed similar gene expression profiles (Figure 1E).
We selected the top 50 genes that were affected by IR induced by FD-895 and found that the majority of those belong to pathways representing RNA splicing and gene regulation, signal transduction, endoplasmic reticulum stress and apoptosis (representative genes from those pathways include DNAJA1, DNAJB1, ABT1, NFKB1, GPX1, SLC2A3, HERPUD1, RBM4-RBM14, RBM8A, and WTAP) (Figure 1F, Online Supplementary Figure S5). There was a similar pathway distribution when the gene sampling was expanded to the top 900 targets affected by IR (Online Supplementary Figure S3B).
Using gene ontology (GO) functional enrichment analysis we found that incubation with FD-895 and PLAD-B induced IR mainly in pathways that are critical for RNA editing/processing, cell survival and cell cycle regulation (Figure 1G, Online Supplementary Figure S3B). Splicing abnormalities in these pathways were present as a baseline finding in untreated CLL cells compared with NBC, although with higher P values (Online Supplementary Figure S3C). We also analyzed the GO enrichment of differential gene expression induced by these compounds and found different pathways that were enhanced, including immune regulation and cell survival, while gene expression of the RNA processing and editing pathway was significantly down-regulated (Figure 1G).
Validation of FD-895 and PLAD-B induced intron retention/spliceosome modulation in chronic lymphocytic leukemia
Because IR is a process considered to be a surrogate marker of spliceosome modulation,20 and because the RNA-seq data suggested a process of IR, we decided to perform a series of validation studies in cells treated with FD-895 and PLAD-B under different conditions. For these experiments, we selected DNAJB1, a gene that has been used in previous studies,9 and one of the highest ranked genes showing an IR pattern (Figure 1B, 1C, and 1F).
CLL samples were incubated with a concentration gradient of FD-895, PLAD-B, bendamustine, or F-ara-A for 4 h (Figure 2A). After treatment, the levels of spliced and unspliced gene expression were evaluated by reverse transcriptase PCR and real-time quantitative PCR. While CLL cells treated with nanomolar concentrations of FD-895 or PLAD-B showed IR, this was not observed in samples treated with bendamustine or F-ara-A, even at supra-physiological concentrations (Figure 2A). FD-895 or PLAD-B did not induce IR in the housekeeping gene GAPDH (Figure 2A and Online Supplementary Figure S6A). IR was observed in cells treated with FD-895 or PLAD-B for all three genes examined, including: DNAJB1, RIOK3 (Figure 2B, Online Supplementary Figure S6B), and BRD2 (data not shown). The process of IR was time-dependent and occurred within 15 min of treatment (Figure 2B).
Figure 2.

Induction of intron retention / spliceosome modulation mediated by FD-895 and PLAD-B. (A) RT-PCR was used to access spliced (S) and unspliced (U) isoforms of DNAJB1 and GAPDH in CLL cells. (B) IR was evaluated by RT-PCR for DNAJB1, RIOK3 and GAPDH (used as loading control) in CLL cells after treatment. (C) Quantitative real-time-PCR analysis of CLL cells after treatment to evaluate levels of unspliced mRNA for DNAJB1. GAPDH was used for normalization. (D) Intron retention for DNAJB1 and (E) RIOK3 was evaluated by quantitative real-time-PCR in CLL and normal B cells using specific primers that allowed detection of intron-containing regions (Online Supplementary Table S1). The untreated controls (CLL cells or NBC) for DNAJB1 and RIOK3 were set to a value of 1. GAPDH was used as a control for normalization.
We quantified IR using real-time quantitative PCR and observed a time-dependent increase of unspliced DNAJB1 in samples treated with FD-895 or PLAD-B, but not with F-ara-A (Figure 2C). In addition, we found that FD-895 and PLAD-B induced ~4 fold increases in IR for both DNAJB1 and RIOK3 in CLL cells when compared to NBC (P<0.0001, Figure 2D, 2E).
The activity of FD-895 and PLAD-B in chronic lymphocytic leukemia cells is associated with regulation of alternative splicing
The RNA-seq data showed IR in the MCL-1 gene after treatment with FD-895 (Online Supplementary Figure S6C). We, therefore, investigated whether or not spliceosome modulation/IR by FD-895 and PLAD-B was associated with regulation of alternative splicing using MCL-1 and BCL-X, two genes involved in apoptosis, which are known to depend on alternative splicing for their functions, pro-apoptotic (short isoform) and anti-apoptotic (long isoform).21 We observed that treatment of CLL cells with FD-895 or PLAD-B induced expression of the short/pro-apoptotic alternatively spliced isoforms for both MCL-1 (Figure 3A) and BCL-X (Figure 3B). This process was time-dependent, occurring within 30 min of treatment. In contrast, there was no evidence of alternative splicing in NBC (Figure 3A, 3B). In addition, we did not observe IR or alternative splicing in GAPDH (Figure 3C and Online Supplementary Figure S6A).
Figure 3.
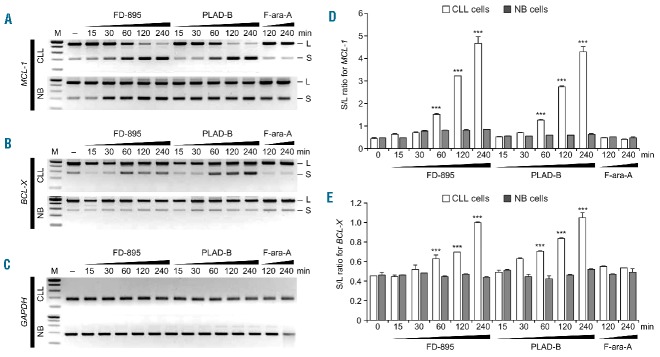
Regulation of alternative splicing after treatment with FD-895 or PLAD-B. (A–C) Normal B cells and CLL cells were treated with 100 nM FD-895, 100 nM PLAD-B or 10 μM F-ara-A for the indicated times. RT-PCR for (A) MCL-1, (B) BCL-X and (C) GAPDH was conducted. The RT-PCR generated two different products; a long or anti-apoptotic isoform (L) and a short or pro-apoptotic splice isoform (S). (D–E) The levels of the L and S isoforms were evaluated after incubation of 100 nM FD-895, 100 nM PLAD-B or 10 μM F-ara-A by densitometry of RT-PCR bands stained with ethidium bromide using Quantity One software (Bio-Rad). The S/L ratio was plotted as a mean of two separate experiments as shown in panel (D) for MCL-1 and (E) for BCL-X. Error bars represent SD and ***P<0.001.
Using gel electrophoresis and densitometric analysis we calculated the ratio of small to large (S/L) isoforms for MCL-1 and BCL-X, which is used as a marker for apoptosis.21 We found a higher S/L ratio (P<0.0001) for MCL-1 (Figure 3D) and BCL-X (Figure 3E) in CLL cells treated with FD-895 or PLAD-B, but there were no changes in NBC. S/L ratios remained unchanged in F-ara-A-treated cells.
FD-895 and PLAD-B induce early irreversible commitment to apoptosis in chronic lymphocytic leukemia cells within 2 hours of treatment
We cultured CLL cells in 100 nM FD-895 or 100 nM PLAD-B, and examined apoptosis over time. The minimum time for the irreversible development of apoptosis was determined by exposing the CLL cells to FD-895 or PLAD-B over increasing incubation times up to 48 h, followed by washing twice with media, and continuing to culture for a total of 48 h. We found that the effect of both FD-895 and PLAD-B is irreversible and at least 2 h of incubation was required to induce apoptosis (Figure 4A).
Figure 4.
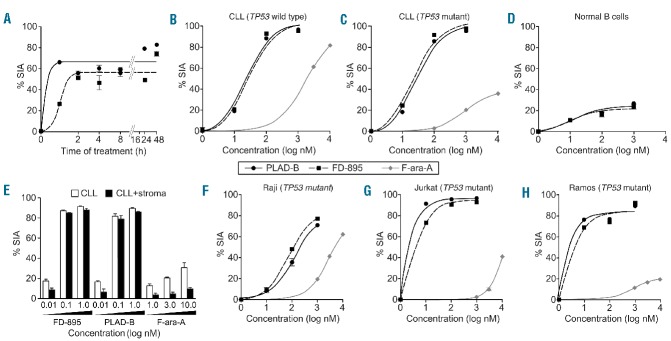
FD-895 or PLAD-B induced apoptosis in CLL cells but not in normal lymphocytes with TP53-independent activity. (A) Primary leukemia B cells alone from a CLL patient were treated with 100 nM FD-895 or 100 nM PLAD-B at time 0 and allowed to incubate for 0.5 h, to 48 h. After initial incubations, the cells were washed twice with fresh media to remove the remaining FD-895 or PLAD-B and then were cultured using media only for a total of 48 h. The dark black line indicates FD-895 and the light color line indicates PLAD-B. Apoptosis was measured by flow cytometry using a PI/DiOC6 assay. (B–D) Cells from different sources were evaluated for their level of apoptosis after incubation with FD-895, PLAD-B or F-ara-A for 48 h, including: (B) cells from a CLL patient with wild-type TP53 and sensitive to F-ara-A; (C) cells from a CLL patient with Del(17p) and TP53 mutation (TP53-mut), resistant to F-ara-A; and, (D) normal B cells from a healthy donor. (E) Primary CLL cells derived from a Del(17p) patient were treated alone or co-cultured with stroma-NK-tert cells with FD-895, PLAD-B, or F-ara-A for 48 h. Apoptosis was measured by flow cytometry using a CD19/CD5/DiOC6 assay. (F-H) In parallel, cell lines with different TP53 mutation status were incubated with FD-895, PLAD-B or F-ara-A: (F) Raji - TP53 mutant type; (G) Jurkat cells - TP53 mutant; (H) Ramos cells - TP53 mutant. After incubation, apoptosis was assessed by flow cytometry. These results are representative of at least three separate experiments. In order to discriminate the compound specific-induced apoptosis vs. background spontaneous cell death from in vitro culture conditions, we calculated the percentage of specific induced apoptosis (% SIA) using the following formula: % SIA = [(compound induced apoptosis − media only spontaneous apoptosis) / (100− media only spontaneous apoptosis)] × 100.
Determination of FD-895 and PLAD-B IC50 values and effect of co-culture conditions that resemble the tumor microenvironment
FD-895 and PLAD-B induced apoptosis in CLL cells at nanomolar concentrations with an IC50 in the range of 5.1-138.7 nM (Figure 4B, 4C). In contrast, NBC and normal T cells were resistant to the activity of both compounds, and the IC50 value was not achieved even when concentrations >1 μM were used (Figure 4D).
In order to assess the activity of FD-895 and PLAD-B under stringent conditions that resemble the anti-apoptotic tumor microenvironment,16 we incubated CLL cells with increasing concentrations of FD-895, PLAD-B (0.01–1 μM) and F-ara-A (1–10 μM) alone or with stroma-NK-tert cells. Stroma-NK-tert cells increased the viability of CLL cells and inhibited the activity of F-ara-A by more than 50%. However, FD-895 and PLAD-B were able to overcome the anti-apoptotic effect of the stroma cell support without causing direct cytopathic effects on stroma-NK-tert cells (Figure 4E).
FD-895 and PLAD-B selectively induce apoptosis in chronic lymphocytic leukemia cells but not in normal lymphocytes in a TP53-independent manner
We evaluated the anti-leukemia activity of FD-895 or PLAD-B in cells derived from CLL patients with Del(17p) and/or inactivating mutations in TP53. The samples were cultured alone or with stroma-NK-tert cells and harvested after treatment, at 48 h, and apoptosis was measured by flow cytometry. Cells derived from CLL patients with Del(17p) and/or TP53 mutations [% Del(17p) by fluorescence in situ hybridization, 66-99.5%] displayed resistance to F-ara-A, with IC50 values >10 μM (Figure 4C), while wild-type CLL samples underwent apoptosis when treated with F-ara-A at IC50 values of ~1 μM (Figure 4B). In contrast to the differential sensitivity to F-ara-A based on TP53 status, we observed that CLL samples underwent apoptosis with FD-895 or PLAD-B at IC50 values of 10–50 nM, regardless of the presence of Del(17p) or TP53 mutations (Figures 4B, 4C, Figure 5). Moreover, B and T lymphocytes from healthy volunteers were resistant to the pro-apoptotic activity of FD-895 or PLAD-B, and showed significantly lower levels of apoptosis after 48 h in culture compared with CLL cells (Figure 4D, Figure 5).
Figure 5.
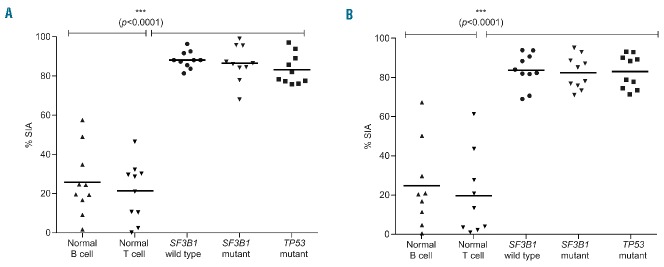
FD-895 and PLAD-B induce apoptosis in CLL cells independent of TP53 and SF3B1 mutational status. Primary leukemia B cells were obtained from CLL patients with wild-type SF3B1 (n=10), SF3B1 mutant (n=10), or Del(17p)/TP53 mutations (n=10). Normal B cells (n=10) and T lymphocytes (n=10) were obtained from healthy donors. Samples were independently incubated with (A) 100 nM FD-895 or (B) 100 nM PLAD-B for 48 h. After incubation, the samples were harvested and percentage of specific induced apoptosis (% SIA) was measured by flow cytometry. FD-895 and PLAD-B induced apoptosis in CLL cells regardless of Del(17p)/TP53 or SF3B1 mutation status, while normal B cells or T cells showed statistically-lower levels of apoptosis. The data show the results of samples analyzed in duplicate with the means and their respective SD.
Evaluation of the activity of FD-895 and PLAD-B across different human leukemia and lymphoma cell lines
In order to validate our observations in CLL, we studied the pro-apoptotic activity of FD-895 and PLAD-B in different cancer cell lines. Both FD-895 and PLAD-B induced apoptosis in TP53 mutant cell lines including Raji, Jurkat, and Ramos (Figure 4F, 4H)) with IC50 values in the nanomolar range (1–70 nM for FD-895 and 5.1–73.1 nM for PLAD-B).
FD-895 and PLAD-B induce apoptosis in chronic lymphocytic leukemia cells independently of TP53 and SF3B1 mutational status
We also tested whether treatment with FD-895 or PLAD-B could induce apoptosis in cells from CLL patients with wild-type or mutant SF3B1 (Online Supplementary Table S4). We found that treatment with 100 nM FD-895 (Figure 5A) or 100 nM PLAD-B (Figure 5B) induced cell death in CLL cells regardless of their SF3B1 mutational status with levels of apoptosis that were significantly higher compared to those of normal lymphocytes (Figure 5).
FD-895 and PLAD-B induce apoptosis via a caspase-dependent pathway
We performed a colorimetric-proteolytic assay to determine if FD-895 or PLAD-B induced apoptosis through a caspase-dependent mechanism. Treatment of CLL cells with FD-895 or PLAD-B induced activation of caspases 3, 6, 8, and 9 (Figure 6A). There was no caspase activation in NBC treated with either FD-895 or PLAD-B (Figure 6A). Caspase dependency was corroborated by co-culturing the cells with different compounds (F-ara-A, FD-895 or PLAD-B) in the presence of increasing concentrations of Z-VAD, a pan-caspase inhibitor. We observed that Z-VAD inhibited apoptosis induced by FD-895 and PLAD-B, suggesting that caspase activation is necessary for FD-895-and PLAD-B-induced apoptosis (Figure 6B).
Figure 6.

Effects of FD-895 and PLAD-B on modulation of caspases and apoptosis-associated proteins and in vivo effect on the A20 murine lymphoma BALB/c model. (A) CLL cells and normal B cells were assessed for caspase activation after treatment. Cells incubated with only media served as the control. (B) CLL cells were incubated with 100 nM FD-895, 100 nM PLAD-B, 10 μM F-ara-A, or 30 μM etoposide for 48 h either alone or in combination with different concentrations of Z-VAD (10 μM, 30 μM, or 90 μM). Following incubations, flow cytometry was performed using CD19/CD5/annexin-V to assess cell death. (C) CLL cells were incubated with 100 nM FD-895 or 100 nM PLAD-B for 6 h. Protein lysates from treated samples were analyzed by western blot using antibodies against PARP and Mcl-1. Cells incubated in media only were used as the negative control (−). (D) Induction of apoptosis in A20 cells after 48 h of treatment. (E) Tumor-bearing mice received intraperitoneal injections consecutively for 5 days with either vehicle, dexamethasone, PLAD-B low or high dose (3 mg/kg/day or 10 mg/kg/day). Tumor volume was measured over time. (F) Survival of mice treated as described above and followed for 35 days after injection; the error bars indicate SD. Survival was calculated using the Kaplan–Meier method.
Apoptosis induced by FD-895 or PLAD-B in chronic lymphocytic leukemia is associated with regulation of PARP and Mcl-1
We analyzed the levels of PARP and Mcl-1 by western blot analysis in CLL cells after treatment with 100 nM FD-895, 100 nM PLAD-B or 10 μM F-ara-A. CLL cells were treated for 6 h and 24 h for Mcl-1 and PARP, respectively. We observed increased levels of cleaved PARP (89 kDa) in CLL cells treated with FD-895, PLAD-B or the control F-ara-A. However, as observed in the Mcl-1 alternative splicing experiments, only samples treated with FD-895 or PLAD-B showed down-regulation of the anti-apoptotic isoform of Mcl-1 (48 kDa) (Figure 6C).
Induction of intron retention and apoptosis correlates with tumor regression in vivo
FD-895 and PLAD-B both induce apoptosis and IR at nanomolar concentrations in a wide range of cell lines including A20 murine lymphoma (Figure 6D). To assess whether the in vitro activity of these compounds correlates with antitumor activity in vivo, we treated BALB/c mice bearing subcutaneous A20 lymphoma tumors with intraperiotoneal injections of PLAD-B for 5 consecutive days. We observed tumor regression and improved survival (P<0.001) in the group of mice treated with PLAD-B. There was a dose-dependent effect that favored higher doses of this compound (3 mg/Kg/day versus 10 mg/Kg/day). In contrast, mice injected with vehicle or dexamethasone control continued to show tumor progression (Figure 6E). After 35 days of follow up, none of the vehicle- or dexamethasone control-treated mice had survived while 33% (low dose) and 83% (high dose) of PLAD-B-treated mice were still alive (Figure 6F). None of the mice treated with PLAD-B showed evidence of toxicity, behavioral changes, diarrhea, rough coat, withdrawal, weight loss or gross visual impairment.22
Discussion
Gene expression consists of several steps, including transcription, pre-mRNA processing (capping, splicing, and poly-adenylation), mRNA surveillance, and mRNA export. These steps are extensively coupled to form ‘gene expression factories’,23 whose modulation, if selective, offers new avenues into cancer therapy. Among these events, the regulation of mRNA maturation by the spliceosome has gained significant recognition as a potential therapeutic target due to the recent discovery of mutations in genes associated with the spliceosome. These include mutations in U2AF35, ZRSR2, SRSF2 and SF3B1, which have been reported in solid tumors as well as hematologic malignancies including CLL and myelodysplastic syndromes.9,10,24
In CLL, SF3B1 mutations are found in ~10–15% of cases and constitute an independent prognostic factor associated with rapid progression and short survival with a frequency that increases after exposure to chemotherapy, suggesting clonal selection of SF3B1 mutant cells after treatment.9,10,25 SF3B1 is upregulated in CLL cells compared with NBC, possibly due to epigenetic regulation through hypomethylation.26 In addition, in silico data strongly suggest that hot spot mutations of SF3B1 in CLL are in fact oncogenic gain-of-function mutations conferring SF3B1 attributes of a proto-oncogene.27 It is, therefore, plausible that the expression of mutated SF3B1 could result in “hyper-activation” of the spliceosome system leading to the generation of oncogenic/leukemogenic alternatively spliced mRNA isoforms that participate in cell survival, proliferation and possibly chemoresistance. Although the causative link between SF3B1 mutations and CLL pathogenesis remains unclear, recent findings suggest that SF3B1 mutations might be linked not only to deregulation of the spliceosome but also to genomic instability and epi-genetic alterations.28
In this project, we studied FD-895 and PLAD-B, two polyketides, whose biological activity has been associated with the ability to modulate splicing by targeting SF3b.15 We evaluated the effects of these two compounds in primary leukemia cells from CLL patients as well as normal lymphocytes and leukemia-lymphoma cells lines.
RNA-seq transcriptome analysis allowed us to analyze the effect of FD-895 and PLAD-B from a global perspective. We used fragments per kilobase per million fragments mapped intron/exon ratios as a surrogate marker of IR/spliceosome modulation.13,29 By calculating IR ratios, we can estimate the relative efficiency of splicing for each gene and indirectly assess the functional status of the spliceosome system.
Our initial experiments showed an increased pattern of IR in untreated CLL cells compared with NBC. This was somewhat unexpected, although amplified IR has been recently reported in solid tumors including lung and breast cancers.30,31 To our knowledge, this is the first time that a basal increase in IR has been reported in CLL or hematologic malignancies. This suggests that baseline abnormalities that involve the RNA processing/spliceosome system could be implicated in CLL leukemogenesis.
We observed that FD-895 and PLAD-B very rapidly induced a generalized process of IR in which the majority of genes (>82%) showed intronic sequences after incubation with this agent but no IR in a housekeeping gene (GAPDH) or intronless genes. The majority of genes (25%) with the highest levels of IR belonged to the gene regulation/RNA splicing pathway. This strongly suggests that the IR effect mediated by FD-895 and PLAD-B involves a broad process that is not stochastic in nature and that certain pathways, mainly RNA processing/editing, are highly sensitive to the effect of these compounds and may provide an explanation for their mechanism of action. In addition, genes with a high basal IR ratio showed the lowest rates of IR increase after treatment. Contrariwise, genes that initially had low levels of IR were the ones that showed the highest post-treatment increase in IR (Online Supplementary Figure S4). Overall, this suggests that the activity of the compounds used in our experiments induced a de novo pattern of spliceosome inhibition more than exacerbation of an existing abnormal splicing process.
Our data show that structurally similar macrolides (PLAD-B and FD-895) have common potent pro-apoptotic activity at nanomolar concentrations in vitro and in vivo. FD-895 and PLAD-B induced early and irreversible commitment to apoptosis within 2 h of treatment (Figure 4A). These nanomolar concentrations compare favorably with pharmacokinetic data of E7107, a compound used in clinical trials.16 In addition, these polyketides not only produced effects in vitro but also demonstrated encouraging clinical activity in vivo using the A20 lymphoma mouse model. Overall, this suggests the potential applicability of these compounds in cancer therapy.
We found a striking difference between the activity of FD-895 and PLAD-B compared with chemotherapy agents such as bendamustine and F-ara-A. Even though CLL cells ultimately die after incubation with these latter, we never observed evidence of IR induced by these agents. In contrast, a hallmark of the apoptosis induced by FD-895 and PLAD-B was early induction of IR. IR after incubation with FD-895 and PLAD-B was not only very rapid, occurring within minutes of incubation, but also took place at nanomolar concentrations and was at least 50% to 75% higher in CLL cells than in NBC. Overall, these findings strongly suggest that the mechanism of action of FD-895 and PLAD-B is mediated, at least in part, by IR/spliceosome modulation.
The pro-apoptotic activity of FD-895 and PLAD-B was observed in all malignant cells tested (CLL, Ramos, Jurkat, Raji, A20 mouse lymphoma cell line), but it was much lower in normal T and B lymphocytes suggesting that malignant cells may be more dependent on the spliceosome system for their survival. This finding shows a therapeutic window that could facilitate future clinical development of this class of compounds.
Not only did we observe IR induced by FD-895 and PLAD-B but we also found that these compounds have the ability to regulate alternative splicing. We demonstrated this by using apoptosis-related genes (MCL-1, BCL-X), which are known to be dependent on alternative splicing for their function.32 After incubation with FD-895 and PLAD-B we observed a change of isoform ratios that favor the expression of pro-apoptotic isoforms. This was not observed with F-ara-A or bendamustine. It is very likely that the modulation of alternative splicing induced by these compounds is broader and may involve other genes critical for cancer cell survival, proliferation, etc. Importantly, we did not observe alternative splicing induced by FD-895 or PLAD-B in normal lymphocytes suggesting that this is probably a very important pro-apoptotic mechanism of action that explains the differential sensitivity observed in CLL cells compared to NBC.
In our experiments we used an in vitro model of stromal cell support that mimics conditions present in the tumor microenvironment. This is important, as the tumor microenvironment has been shown to provide anti-apoptotic protection to cancer cells, including CLL, and could be responsible for chemoresistance as well as persistence of minimal residual disease.33 Under these conditions, we showed that the pro-apoptotic activity of FD-895 and PLAD-B overcomes the protective effect of the microenvironment, suggesting that spliceosome modulators may have potential applications in refractory cancer and eradication of minimal residual disease.
The pro-apoptotic activity of FD-895 and PLAD-B was independent of Del(17p) - TP53 or SF3B1 mutations found in CLL. Both of these compounds displayed com parable activity in cells from CLL patients as well as cell lines with these genetic abnormalities and resistance to F-ara-A (known for its p53-dependent cytotoxicity). The fact that the activity of FD-895 and PLAD-B is independent of p53 opens significant opportunities for the development of therapies for refractory malignancies in which the prevalence of p53 dysfunction is significantly high.34 In addition, the finding that the pro-apoptotic activity of these agents was independent of SF3B1 mutational status was somewhat unexpected, mainly because in vitro data have shown that SF3B1 mutations may confer resistance to this kind of compound.15 There are various potential explanations for this observation. First, all the mutations described in CLL patients (including the patients tested at UCSD) cluster in the heat repeat region 2-6 of the SF3b1 protein.24,35 On the other hand, the R1074H mutation that confers resistance to PLAD-B is located in a distant domain (9th heat repeat region, Online Supplementary Figure S7). This highlights functional differences of these mutations and their impact on FD-895 and PLAD-B activity. Second, the activity of PLAD-B and FD-895 may be more complex than simply binding to SF3B1 as these compounds can also target other members of this protein family including SF3B1, SF3B3, SF3B4, and SF3B2 and potentially other proteins of the spliceosome system. Third, the fact that all CLL samples tested were sensitive to FD-895 and PLAD-B, regardless of SF3B1 mutation status, highlights the importance of this pathway in CLL and the potential relevance of a more global spliceosome deregulation in cancer.
We found that the pro-apoptotic activity of FD-895 and PLAD-B in CLL cells was caspase-dependent (with activation of the intrinsic and extrinsic pathways) and correlated with changes in apoptosis-related proteins including poly (ADP-ribose) polymerase (PARP) cleavage and Mcl-1 (long isoform) down-regulation. It is possible that the IR and alternative splicing induced by FD-895 and PLAD-B tips over the strong anti-apoptotic protein balance that favors survival in CLL and other malignancies.36 This could have potential implications in terms of synergistic activity of these compounds in combination with other anti-cancer agents.
Together, our studies provide a solid foundation for further therapeutic development of this class of compounds. We have shown for the first time that FD-895 and PLAD-B induce a global pattern of IR/spliceosome modulation and regulation of alternative RNA splicing at low nanomolar concentrations, and that the functional consequence of this process leads to apoptosis in vitro and in vivo preferentially in cancer cells. This constitutes evidence that the spliceosome system is a relevant target in leukemia and lymphoma and potentially other cancers.
Our current efforts using key structure-activity relationships are focused on adapting this knowledge to further guide analog discovery and synthesis,17 with the ultimate goal of developing novel spliceosome modulators for cancer therapy.
Footnotes
The online version of this article has a Supplementary Appendix
Funding
The authors would like to thank the following organizations for their grant support: Lymphoma Research Foundation (LRF, grant #285871) to JEC and MDB, the American Cancer Society (RSG–06–011–01–CDD) to MDB, the National Institutes of Health (R01-GM086225) to MDB, the National Institutes of Health (PO1-CA081534)-CLL Research Consortium Grant to TJK, and JEC, the UC San Diego Foundation Blood Cancer Research Fund (to TJK), the Bennett Family Foundation (to JEC), and support provided by Moores Cancer Center-UCSD for open access publication.
Authorship and Disclosures
Information on authorship, contributions, and financial & other disclosures was provided by the authors and is available with the online version of this article at www.haematologica.org.
References
- 1.Rai KR. Pathophysiologic mechanisms of chronic lymphocytic leukemia and their application to therapy. Exp Hematol. 2007;35(4 Suppl 1):134–136. [DOI] [PubMed] [Google Scholar]
- 2.Hallek M, Fischer K, Fingerle-Rowson G, et al. Addition of rituximab to fludarabine and cyclophosphamide in patients with chronic lymphocytic leukaemia: a randomised, open-label, phase 3 trial. Lancet. 2010;376(9747):1164–1174. [DOI] [PubMed] [Google Scholar]
- 3.Castro JE. Treatment of patients with chronic lymphocytic leukemia with 17p deletion: the saga continues. Leuk Lymphoma. 2012;53(2):179–180. [DOI] [PubMed] [Google Scholar]
- 4.Hallek M. Signaling the end of chronic lymphocytic leukemia: new frontline treatment strategies. Blood. 2013;122(23):3723–3734. [DOI] [PubMed] [Google Scholar]
- 5.Kornblihtt AR, Schor IE, Alló M, Dujardin G, Petrillo E, Muñoz MJ. Alternative splicing: a pivotal step between eukaryotic transcription and translation. Nat Rev Mol Cell Biol. 2013;14(3):153–165. [DOI] [PubMed] [Google Scholar]
- 6.Kim E, Goren A, Ast G. Insights into the connection between cancer and alternative splicing. Trends Genet. 2008;24(1):7–10. [DOI] [PubMed] [Google Scholar]
- 7.Visconte V, Tabarroki A, Rogers HJ, et al. SF3B1 mutations are infrequently found in non-myelodysplastic bone marrow failure syndromes and mast cell diseases but, if present, are associated with the ring sideroblast phenotype. Haematologica. 2013;98(9):e105–107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Visconte V, Rogers HJ, Singh J, et al. SF3B1 haploinsufficiency leads to formation of ring sideroblasts in myelodysplastic syndromes. Blood. 2012;120(16):3173–3186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Wang L, Lawrence MS, Wan Y, et al. SF3B1 and other novel cancer genes in chronic lymphocytic leukemia. N Engl J Med. 2011;365(26):2497–2506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Landau DA, Carter SL, Stojanov P, et al. Evolution and impact of subclonal mutations in chronic lymphocytic leukemia. Cell. 2013;152(4):714–726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Seki-Asano M, Okazaki T, Yamagishi M, et al. Isolation and characterization of a new 12-membered macrolide FD-895. J Antibiot (Tokyo). 1994;47(12):1395–1401. [DOI] [PubMed] [Google Scholar]
- 12.Asai N, Kotake Y, Niijima J, Fukuda Y, Uehara T, Sakai T. Stereochemistry of pladienolide B. J Antibiot (Tokyo). 2007;60(6): 364–369. [DOI] [PubMed] [Google Scholar]
- 13.Kotake Y, Sagane K, Owa T, et al. Splicing factor SF3b as a target of the antitumor natural product pladienolide. Nat Chem Biol. 2007;3(9):570–575. [DOI] [PubMed] [Google Scholar]
- 14.Webb TR, Joyner AS, Potter PM. The development and application of small molecule modulators of SF3b as therapeutic agents for cancer. Drug Discov Today. 2013;18(1-2):43–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Yokoi A, Kotake Y, Takahashi K, et al. Biological validation that SF3b is a target of the antitumor macrolide pladienolide. FEBS J. 2011;278(24):4870–4880. [DOI] [PubMed] [Google Scholar]
- 16.Eskens FA, Ramos FJ, Burger H, et al. Phase I pharmacokinetic and pharmacodynamic study of the first-in-class spliceosome inhibitor E7107 in patients with advanced solid tumors. Clin Cancer Res. 2013;19(22): 6296–6304. [DOI] [PubMed] [Google Scholar]
- 17.Villa R, Kashyap MK, Kumar D, et al. Stabilized cyclopropane analogs of the splicing inhibitor FD-895. J Med Chem. 2013;56(17):6576–6582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Matutes E, Owusu-Ankomah K, Morilla R, et al. The immunological profile of B-cell disorders and proposal of a scoring system for the diagnosis of CLL. Leukemia. 1994;8(10):1640–1645. [PubMed] [Google Scholar]
- 19.WMA Declaration of Helsinki - Ethical Principles for Medical Research Involving Human Subjects. [http://www.wma.net/en/30publications/10policies/b3/index.html%5D. [PubMed]
- 20.Landau DA, Wu CJ. Chronic lymphocytic leukemia: molecular heterogeneity revealed by high-throughput genomics. Genome Med. 2013;5(5):47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Miura K, Fujibuchi W, Unno M. Splice variants in apoptotic pathway. Exp Oncol. 2012;34(3):212–217. [PubMed] [Google Scholar]
- 22.Fox MW. The visual cliff test for the study of visual depth perception in the mouse. Anim Behav. 1965;13(2):232–233. [DOI] [PubMed] [Google Scholar]
- 23.Proudfoot NJ, Furger A, Dye MJ. Integrating mRNA processing with transcription. Cell. 2002;108(4):501–512. [DOI] [PubMed] [Google Scholar]
- 24.Schwaederlé M, Ghia E, Rassenti LZ, et al. Subclonal evolution involving SF3B1 mutations in chronic lymphocytic leukemia. Leukemia. 2013;27(5):1214–1217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Quesada V, Ramsay AJ, Lopez-Otin C. Chronic lymphocytic leukemia with SF3B1 mutation. N Engl J Med. 2012;366(26):2530. [DOI] [PubMed] [Google Scholar]
- 26.Rossi D, Bruscaggin A, Spina V, et al. Mutations of the SF3B1 splicing factor in chronic lymphocytic leukemia: association with progression and fludarabine-refractoriness. Blood. 2011;118(26):6904–6908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wu X, Tschumper RC, Jelinek DF. Genetic characterization of SF3B1 mutations in single chronic lymphocytic leukemia cells. Leukemia. 2013;27(11):2264–2267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ramsay AJ, Rodríguez D, Villamor N, et al. Frequent somatic mutations in components of the RNA processing machinery in chronic lymphocytic leukemia. Leukemia. 2013;27(7):1600–1603. [DOI] [PubMed] [Google Scholar]
- 29.Cvitkovic I, Jurica MS. Spliceosome database: a tool for tracking components of the spliceosome. Nucleic Acids Res. 2013;41(Database issue):D132–141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Zhang Q, Li H, Jin H, Tan H, Zhang J, Sheng S. The global landscape of intron retentions in lung adenocarcinoma. BMC Med Genomics. 2014;7:15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Eswaran J, Horvath A, Godbole S, et al. RNA sequencing of cancer reveals novel splicing alterations. Sci Rep. 2013;3:1689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Gao Y, Koide K. Chemical perturbation of Mcl-1 pre-mRNA splicing to induce apoptosis in cancer cells. ACS Chem Biol. 2013;8(5):895–900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Burger JA, Tsukada N, Burger M, Zvaifler NJ, Dell’Aquila M, Kipps TJ. Blood-derived nurse-like cells protect chronic lymphocytic leukemia B cells from spontaneous apoptosis through stromal cell-derived factor-1. Blood. 2000;96(8):2655–2663. [PubMed] [Google Scholar]
- 34.te Raa GD, Malcikova J, Pospisilova S, et al. Overview of available p53 function tests in relation to TP53 and ATM gene alterations and chemoresistance in chronic lymphocytic leukemia. Leuk Lymphoma. 2013;54(8): 1849–1853. [DOI] [PubMed] [Google Scholar]
- 35.Oscier DG, Rose-Zerilli MJ, Winkelmann N, et al. The clinical significance of NOTCH1 and SF3B1 mutations in the UK LRF CLL4 trial. Blood. 2013;121(3):468–475. [DOI] [PubMed] [Google Scholar]
- 36.Chu P, Deforce D, Pedersen IM, et al. Latent sensitivity to Fas-mediated apoptosis after CD40 ligation may explain activity of CD154 gene therapy in chronic lymphocytic leukemia. Proc Natl Acad Sci USA. 2002;99(6):3854–3859. [DOI] [PMC free article] [PubMed] [Google Scholar]