

Abstract
NAD+ has emerged as a vital cofactor that can rewire metabolism, activate sirtuins and maintain mitochondrial fitness through mechanisms such as the mitochondrial unfolded protein response. This improved understanding of NAD+ metabolism revived interest in NAD+ boosting strategies to manage a wide spectrum of diseases, ranging from diabetes to cancer. In this review, we summarize how NAD+ metabolism links energy status with adaptive cellular and organismal responses and how this knowledge can be therapeutically exploited.
Keywords: NAD+ biosynthesis, NAD+ metabolism, NAD+ precursors, NAD+ therapeutics, energy signaling, mitochondrial function, Sirtuins, Poly(ADP-ribose) polymerases, Cyclic ADP-ribose synthases, Metabolic disease, Cancer, Neurodegenerative disease, Aging, Longevity
INTRODUCTION
The importance of nicotinamide adenine dinucleotide (NAD+) metabolism became apparent subsequent to the study of pellagra, a disease characterized by a darkly pigmented skin rash, dermatitis, diarrhea and dementia, later resulting in death (Sydenstricker, 1958). A century ago, pellagra was common in rural areas of Europe and became an epidemic in the southern United States (Sydenstricker, 1958). However, in 1914, Joseph Goldberger tested whether pellagra was caused by a dietary deficiency and discovered that substituting corn-based diets with milk, eggs and meat prevented and cured the condition (reprinted essay (Goldberger, 2006)). Later, Conrad Elvehjem found that a nicotinamide (NAM) enriched fraction from deproteinized liver and a sample of crystalline nicotinic acid (NA) cured pellagra (Elvehjem, 1940). NA and NAM, collectively termed niacin or vitamin B3, are now known as precursors for NAD+, an essential element for all cells (Bogan and Brenner, 2008; Chi and Sauve, 2013; Houtkooper et al., 2010a). Whereas pellagra remains endemic in underdeveloped countries (Seal et al., 2007), it is rare in developed countries, and mostly occurs in association with tuberculosis, malabsorption, alcoholism and eating disorders (Hegyi et al., 2004). Less severe niacin deficiencies are more difficult to detect and are linked with low metabolism, cold intolerance, and delayed brain development (Forbes and Duncan, 1961; Williams and Dunbar, 2014).
So, what is NAD+ and why is it important? NAD+ was originally described more than 100 years ago by Sir Arthur Harden and colleagues as a cofactor in fermentation (Harden and Young, 1906). Years later, another Nobel prize laureate, Hans von Euler-Chelpin, identified this factor as a nucleoside sugar phosphate (1940 Nobel lecture (Euler-Chelpin)). Yet it took a third Nobel laureate, Otto Warburg, to isolate NAD(P)+ and discover its key role for hydrogen transfer in biochemical reactions (Warburg et al., 1935). NAD+ and NADP+ perform similar redox functions within the cell, but the latter is more confined to biosynthetic pathways and redox protective roles (reviewed in (Ying, 2008)). Playing a vital role in energy metabolism within eukaryotic cells, NAD+ accepts hydride equivalents, to form reduced NADH, which furnishes reducing equivalents to the mitochondrial electron transport chain (ETC) to fuel oxidative phosphorylation. The roles of NAD+, however, have expanded beyond its role as a coenzyme, as NAD+ and its metabolites also act as degradation substrates for a wide range of enzymes, such as the sirtuins (Blander and Guarente, 2004; Haigis and Sinclair, 2010; Hall et al., 2013; Houtkooper et al., 2010a). Through these activities, NAD+ links cellular metabolism to changes in signaling and transcriptional events. Here, we give an overview of the current knowledge on NAD+ metabolism, including its biosynthesis, compartmentalization, degradation and actions as a signaling molecule.
1. NAD+: METABOLIC AND THERAPEUTIC INTERESTS
1.1 Food sources and bioavailability of NAD+
The daily requirements for NAD+ biosynthesis can be met with the consumption of less than 20 mg of niacin (Bogan and Brenner, 2008). Four major molecules have been described as the root substrates for different NAD+ biosynthetic pathways, i.e. the amino acid Tryptophan (Trp), NA, NAM and Nicotinamide Riboside (NR) (Figure 1A, B, D). However, intermediate compounds of these NAD+ biosynthetic pathways, such as nicotinamide mononucleotide (NMN), can also directly stimulate NAD+ synthesis. Vitamin B3 deficiency occurs on low protein diets or diets relying mostly on untreated maize. Interestingly, niacin is found in maize but is not bioavailable unless given an alkali treatment, a process used in Aztec and Mesoamerican times termed nixtamalization (Gwirtz and Garcia-Casal, 2014). In animal products, and probably in all uncooked foods, the NAD+ and NADP+ cellular content accounts for much of their dietary niacin content (Gross and Henderson, 1983), yet, as exemplified above with corn nixtamalization, their bioavailability might be affected by food processing or cooking.
Figure 1. NAD+ precursor metabolism and NAD+ consuming enzymes.
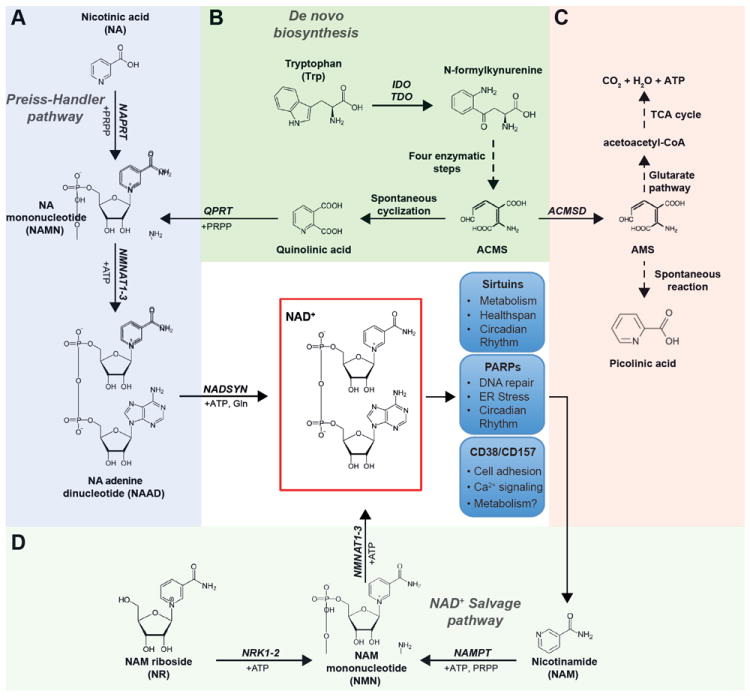
Tryptophan (Trp), nicotinic acid (NA), nicotinamide (NAM) and nicotinamide riboside (NR) are utilized through distinct metabolic pathways to form NAD+. A. NAD+ synthesis from NA, also known as the Preiss-Handler pathway, is initiated by the NA phosphoribosyltransferase (NAPRT), which uses phosphoribosyl pyrophosphate (PRPP) to form NAMN. Together with ATP, NAMN is then converted into NAAD by the NMN adenylyl transferase (NMNAT1-3) enzymes. Finally, NA adenine dinucleotide (NAAD) is transformed to NAD+ through an amidation reaction catalyzed by the NAD+ synthase (NADSYN) enzyme. B. The de novo biosynthesis of NAD+ from tryptophan (Trp) starts with the conversion of Trp to N-formylkynurenine by either indoleamine 2,3-dioxygenase (IDO) or tryptophan 2,3-dioxygenase (TDO). After four reaction steps, N-formylkynurenine can be subsequently converted to the unstable α-amino-β-carboxymuconate-ε-semialdehyde (ACMS), which can undergo nonenzymatic cyclization to quinolinic acid. The last step of the de novo biosynthesis component is comprised of the quinolinate phosphoribosyltransferase (QPRT)-catalyzed formation of NAMN, using PRPP as a co-substrate, which is converted to NAD+ via the remaining pathway described in panel A. C. ACMS can also be diverted away from NAD+ synthesis, by ACMS decarboxylase (ACMSD), to form α-amino-β-muconate-ε-semialdehyde (AMS) and can then be oxidized via the glutarate pathway and TCA cycle to CO2 and water, or nonenzymatically converted to picolinic acid. D. The synthesis of NAD+ from NAM or NR is more direct and relies on only 2 steps each. NAM is converted by the rate-limiting nicotinamide phosphoribosyltransferase (NAMPT) to form NMN, using PRPP as cosubstrate. NMN is also the product of phosphorylation of NR by the NR kinases (NRK1-2). The subsequent conversion of NMN to NAD+ is catalyzed by the NMNAT enzymes. The blue boxes depict the 3 families of NAD+ consuming enzymes and some of the key processes to which they have been linked. NMN, NAM mononucleotide; NAMN, NA mononucleotide; NAAD, NA adenine dinucleotide; NRK, NR kinase; NMNAT, NMN adenylyltransferase; NADSYN, NAD+ synthetase.
Bioavailability studies indicated that ingested NAD+ was primarily hydrolyzed in the small intestine by brush border cells (Baum et al., 1982; Gross and Henderson, 1983). As a first step, NAD+ is cleaved to NMN and 5’-AMP by a pyrophosphatase found either in intestinal secretions (Gross and Henderson, 1983) or in the brush border (Baum et al., 1982). Next NMN is rapidly hydrolyzed to NR, which in turn is more slowly converted into NAM (Gross and Henderson, 1983). NAM can also be formed directly by the cleavage of NAD+, obtaining ADP-ribose derivates as a side product (Gross and Henderson, 1983). The intestinal production of NAM from NAD+ or NR required the presence of intestinal cells, indicating that the enzymes for this process are membrane-bound or intracellular (Baum et al., 1982; Gross and Henderson, 1983). The direct perfusion with NAM, however, did not give rise to any of these species, indicating that NAM is the final degradation product and directly absorbed (Collins and Chaykin, 1972; Gross and Henderson, 1983; Henderson and Gross, 1979). In contrast, perfusion of the intestine with NA revealed a substantial cellular accumulation of labeled intermediates of the NAD+ biosynthetic pathway, including NAM, which suggest the presence of active NA metabolism in intestinal cells (Collins and Chaykin, 1972; Henderson and Gross, 1979). In line with this, blood concentrations of NA are relatively low (~100 nM), yet when pharmacologically primed (Jacobson et al., 1995; Tunaru et al., 2003), can increase and be rapidly converted to NAM by the liver (Collins and Chaykin, 1972). Strikingly, NAM levels in fasted human plasma are also too low to support NAD+ biosynthesis in cells (between 0.3 and 4 μM) (Hara et al., 2011; Jacobson et al., 1995). All of these results suggest that these NAD+ precursors are metabolized very quickly in mammalian blood and tissues.
1.2 Lipid lowering effect of niacin
NA attracted clinical attention for its cholesterol lowering actions (Altschul et al., 1955), and became the first drug used to treat dyslipidemia. Gram dosages of NA reduce plasma triglyceride and low-density lipoprotein (LDL) levels, while concomitantly increasing high-densitiy lipoproteins (HDL). However, the clinical use of NA has been limited by the fact that it induces cutaneous flushing, which compromises compliance (Birjmohun et al., 2005). This flushing does not derive from the ability of NA to drive NAD+ synthesis, but rather from the activation of a G-coupled receptor, GPR109A (Benyo et al., 2005). Given the low presence of NA in blood, the activation of this receptor is unlikely to be a native function of NA, but rather an effect from pharmacological dosing. It was also assumed that the beneficial effects of NA on plasma lipids are mediated via a receptor rather than a vitamin mechanism because of the high dose required (100-fold higher than that required to prevent pellagra) and the failure of NAM to provide similar benefits (Tunaru et al., 2003). Indeed, some evidence supports that GPR109A is necessary for NA to raise HDL cholesterol (Li et al., 2010; Tunaru et al., 2003). However, the absence of GRP109A expression in the liver (Soga et al., 2003; Tunaru et al., 2003; Wise et al., 2003), a central hub for HDL and LDL metabolism, also questions whether the effects of NA on blood lipids derive from GPR109A activation. Alternatively, strong evidence for the ability of NAD+ to enhance the activity of sirtuins provides a mechanism of action that also drives benefits on lipid homeostasis (Canto and Auwerx, 2012). In addition, sirtuin activity is inhibited by NAM (Anderson et al., 2003), which could explain why NAM failed to provide the benefits of NA, however, in some situations NAM treatments can have beneficial effects as discussed in Section 4.1. The intricate relationship between NAD+ and sirtuins will be discussed further in Section 3.2.
1.3 Introducing NAD+ as a metabolic regulator
The role of NAD+ as a coenzyme in most metabolic pathways suggests that NAD+ limitations could affect metabolic efficiency. Decreasing NAD+ levels could therefore prompt the development of many of the ailments associated with aging. Indeed, NAD+ levels can change during a number of physiological processes. Diverse lines of research on worms, rodents and human cellular models indicate that declining NAD+ levels are a hallmark for senescence (Braidy et al., 2011; Gomes et al., 2013; Khan et al., 2014; Massudi et al., 2012; Mouchiroud et al., 2013; Ramsey et al., 2008; Yoshino et al., 2011). Along a similar line, a reduction in muscle progenitor cell NAD+ content leads to a SIRT1-mediated metabolic switch that induces premature differentiation and a loss of regenerative capacity, reflecting a phenotype typical of aging muscle (Ryall et al., 2015). The link between metabolism and NAD+ is further solidified by observations that tissue NAD+ levels decrease with high fat diets (Bai et al., 2011b; Canto et al., 2012; Kraus et al., 2014; Pirinen et al., 2014; Yang et al., 2014; Yoshino et al., 2011). In contrast, NAD+ increases in mammalian cells and tissues in response to exercise (Canto et al., 2009; Canto et al., 2010; Costford et al., 2010) or calorie restriction (CR) (Chen et al., 2008), both of which are interventions associated with metabolic and age-related health benefits. In line with this, supplementation with NAD+ precursors has proven to enhance lifespan in budding yeast (Belenky et al., 2007) and worms (Mouchiroud et al., 2013). Also, in mammals, the enhancement of NAD+ levels has been linked with improved mitochondrial function under stress (Cerutti et al., 2014; Khan et al., 2014; Mouchiroud et al., 2013; Pirinen et al., 2014), leading to protection against dietary (Bai et al., 2011b; Canto et al., 2012) and age-related (Gomes et al., 2013; Yoshino et al., 2011) metabolic complications. Finally, hepatic NAD+ levels dynamically change in a circadian fashion (Asher et al., 2010; Nakahata et al., 2009; Ramsey et al., 2009), weaving an intricate relationship with nutritional states. Therefore, despite the classical misconception that intracellular NAD+ levels rarely change (Kaelin and McKnight, 2013), the evidence above unequivocally demonstrates the ability of NAD+ to respond dynamically to physiological stimuli. So, how do changes in NAD+ levels take place innately?
2. NAD+ SYNTHESIS AND SALVAGE, NEW WAYS TO BOOST NAD+
2.1 NAD+ biosynthesis and the discovery of new NAD+ precursors
NAD+ availability is determined by the relative rates of NAD+ biosynthesis and degradation. Ergo, the enhancement of NAD+ biosynthesis could provide a way to elevate NAD+ content. There are several known NAD+ precursors. First, dietary Trp can serve as an NAD+ precursor through an eight-step de novo pathway (Bender, 1983), which has been described in detail elsewhere (Houtkooper et al., 2010a); so we will only focus on some of its most interesting features (Figure 1A-D). The first and rate-limiting step in this path includes the conversion of Trp to N-formylkynurenine by either indoleamine 2,3-dioxygenase (IDO) or tryptophan 2,3-dioxygenase (TDO) (Figure 1B). These enzymes are strongly overexpressed in diverse cancers and the subsequent synthesis of kynurenines may act as potential second messengers in cancer immune tolerance (Stone and Darlington, 2002), possibly through binding to the aryl hydrocarbon receptor (AhR) (Bessede et al., 2014). An interesting branch point in the tryptophan catabolic pathway is the formation of the unstable α-amino-β-carboxymuconate-ε-semialdehyde (ACMS) (Bender, 1983). ACMS can be enzymatically converted to α-amino-β-muconate-ε-semialdehyde (AMS) by ACMS decarboxylase (ACMSD), leading to complete oxidation via the glutarate pathway and the tricarboxylic acid (TCA) cycle, or to the production of picolinic acid via a spontaneous reaction (Figure 1B, C)(Houtkooper et al., 2010a). Alternatively, ACMS can undergo spontaneous cyclization forming quinolinic acid, which subsequently serves as an NAD+ precursor (Bender, 1983). This latter nonenzymatic possibility seems to be only relevant when the metabolism of ACMS is limited in the cell. This might explain why, in general, Trp is considered a rather poor NAD+ precursor in vivo, as it will only be diverted to NAD+ synthesis when its supply exceeds the enzymatic capacity of ACMSD (Ikeda et al., 1965). In humans, diets ranging from 34mg to 86mg of Trp provide the equivalent of 1mg of Niacin (reviewed in (Horwitt et al., 1981)). Interestingly, the formation of NAD+ following Trp injections is further reduced in diabetic rats (Ikeda et al., 1965). When ACMSD capacity is surpassed, Trp-derived quinolinic acid is produced and used by quinolinate phosphoribosyltransferase (QPRT) to form NA mononucleotide (NAMN). NAMN is then converted to NA adenine dinucleotide (NAAD), using ATP, by the enzyme NMN adenylyltransferase (NMNAT) (Figure 1A) (Houtkooper et al., 2010a). This is a key enzyme for NAD+ synthesis in mammals, irrespective of the precursor used, since it is also needed for NAD+ salvage. Three NMNAT isoforms (NMNAT1-3) with different tissue and subcellular distributions have been described in mammals (Lau et al., 2009). NMNAT1 is a nuclear enzyme that is ubiquitously expressed, with its highest levels in skeletal muscle, heart, kidney, liver and pancreas, yet is almost undetectable in the brain (Emanuelli et al., 2001; Yalowitz et al., 2004). In contrast, NMNAT2 is mostly located in the cytosol and Golgi apparatus (Berger et al., 2005; Yalowitz et al., 2004). Finally, NMNAT3 is highly expressed in erythrocytes with a moderate expression in skeletal muscle and heart, and has been identified in both cytosolic and mitochondrial compartments, with cell/tissue specific subcellular localization patterns (Berger et al., 2005; Felici et al., 2013; Hikosaka et al., 2014; Zhang et al., 2003). The possible implications of the subcellular localization of NMNAT enzymes will be discussed in section 2.3. The last step in the primary biosynthesis of NAD+ includes the ATP-dependent amidation of NAAD by NAD+ synthase (NADSYN) using glutamine as a donor. NADSYN is mainly expressed in the small intestine, liver, kidney, and testis, where this pathway may be more relevant to NAD+ synthesis (Hara et al., 2003; Houtkooper et al., 2010a).
NAD+ can also be synthesized from metabolite recycling or the dietary uptake of other NAD+ precursors (Houtkooper et al., 2010a). NA can lead to NAD+ through the shorter, 3-step, Preiss-Handler pathway (Figure 1A). Here, NA is initially metabolized by the NA phosphoribosyltransferase (NAPRT) into NAMN, converging with the de novo pathway.
In mammals, NAM can also be an NAD+ precursor through its metabolism into NAM mononucleotide (NMN) by the rate-limiting enzyme nicotinamide phosphoribosyltransferase (NAMPT) (Figure 1D) (Revollo et al., 2004; Rongvaux et al., 2002). NMN can be then converted into NAD+ through a single additional reaction catalyzed by the NMNAT enzymes. NAM is also the product of NAD+ degradation by several enzyme families (see section 3). Consequently, NAMPT is key to not only metabolizing circulating NAM, but also to recycling intracellularly-produced NAM via the NAD+ salvage pathway. As a key enzyme, SNPs found in non-coding regions of human NAMPT are correlated with glucose and lipid metabolism alterations and type 2 diabetes, amongst other disease associations (Zhang et al., 2011).
Lastly, NR metabolism constitutes an additional path for NAD+ biosynthesis (Bieganowski and Brenner, 2004) (Figure 1D). NR is transported into cells by nucleoside transporters (Nikiforov et al., 2011) and is then phosphorylated by the NR kinases 1 and 2 (NRKs) (Bieganowski and Brenner, 2004), generating NMN. This phosphorylation step is a conserved feature in all eukaryotes (Bieganowski and Brenner, 2004), underscoring its evolutionary relevance. After the generation of NMN, NMNAT enzymes can then catalyze the formation of NAD+. While additional ways for NR metabolism have been described in yeast (Belenky et al., 2007), the phosphorylation by NRKs is still the only pathway described in mammalian cells for the transformation of NR into NAD+.
2.2. Whole body NAD+ transport
Despite Trp being the canonical NAD+ precursor, its action may be up to 60 times less efficient than NA (Institute of Medicine (US) Standing Committee on the Scientific Evaluation of Dietary Reference Intakes and its Panel on Folate, 1998), as Trp is also used for protein translation and other biosynthetic purposes. Indeed, the use of Trp as an NAD+ precursor would not be solely sufficient to support the physiological NAD+ requirements in mammals (Henderson, 1997). NA, in contrast, can act as a potent NAD+ precursor, primarily in liver and kidney where NAPRT demonstrates the highest activity levels (Hara et al., 2007). However, mammalian tissues are rarely exposed to NA, as its levels in blood are generally very low (Jacobson et al., 1995; Tunaru et al., 2003). Also, as discussed in section 1.1, most evidence suggests that NA might be quickly metabolized to NAM in the gut and the liver (Collins and Chaykin, 1972). However, the low plasma concentration of NAM (Hara et al., 2011) is ~1000-fold less than that required to increase NAD+ levels in cultured cells (Hara et al., 2007; Revollo et al., 2004).
These findings underscore the importance of naturally occurring NR as a possible alternative substrate for NAD+ biosynthesis (Bieganowski and Brenner, 2004). Supporting this idea, NR treatment enhances NAD+ levels in all mammalian cells tested (Canto et al., 2012; Yang et al., 2007b). Interestingly, different lines of evidence suggest that NR is the primary metabolite transported into the cell and metabolized into NAD+, even when cells are cultured in the presence NAD+ or NMN (Lu et al., 2009) (Figure 2). Thus by using specific inhibitors, it was suggested that free NAD+ or NMN in the medium are metabolized extracellularly to NR, which may be the final metabolite transported to cells for NAD+ biosynthesis (Nikiforov et al., 2011). Interestingly, milk extracts (whey vitamin fraction) rescue the survival of yeast cells defective for QNS1, a necessary enzyme for NA and NAM-triggered NAD+ biosynthesis in yeast (Bieganowski and Brenner, 2004). This extract, however, failed to rescue survival in NRK1-deficient yeast (Bieganowski and Brenner, 2004). The presence of significant NR levels in blood after oral intake is, however, not apparent, as classic reports generally indicate that radiolabelled NR is transformed into NAM at the brush border (Gross and Henderson, 1983). In this sense, it is intriguing that mammalian cultured cells commonly require almost millimolar NR concentrations in order to enhance NAD+ biosynthesis (Canto et al., 2012; Yang et al., 2007b), which is unlikely to be met in vivo. However, in some microorganisms, such as Haemophilus influenza, only the transport of NR across the membrane allows them to synthesize NAD+ and survive in the host bloodstream, as they are unable to use NA, NAM or the de novo pathway for this purpose (Cynamon et al., 1988; Herbert et al., 2003). This suggests that either NR or NMN is, in fact, available in the blood. This is partially corroborated by a report indicating that NMN might be present in the bloodstream at concentrations around 50 μM (Revollo et al., 2007). The presence of a circulating extracellular form of the NAMPT enzyme (eNAMPT), to convert NAM to NMN, supports this possibility (Revollo et al., 2007). Recent evidence indicates that eNAMPT activity in the plasma is required to safeguard hypothalamic NAD+ levels (Yoon et al., 2015). However, other labs have failed to detect NMN in plasma (Hara et al., 2011). In addition, the marginal presence of ATP and 5-phosphoribosyl 1-pyrophosphate (PRPP) in blood (Hara et al., 2011), both substrates for the reaction catalyzed by NAMPT, would impede the generation of NMN in the circulation. Furthermore, since NAM plasma levels are also low, it is difficult to substantiate significant NMN synthesis in the bloodstream (Hara et al., 2011).
Figure 2. Central nodes for cellular NAD+ metabolism.
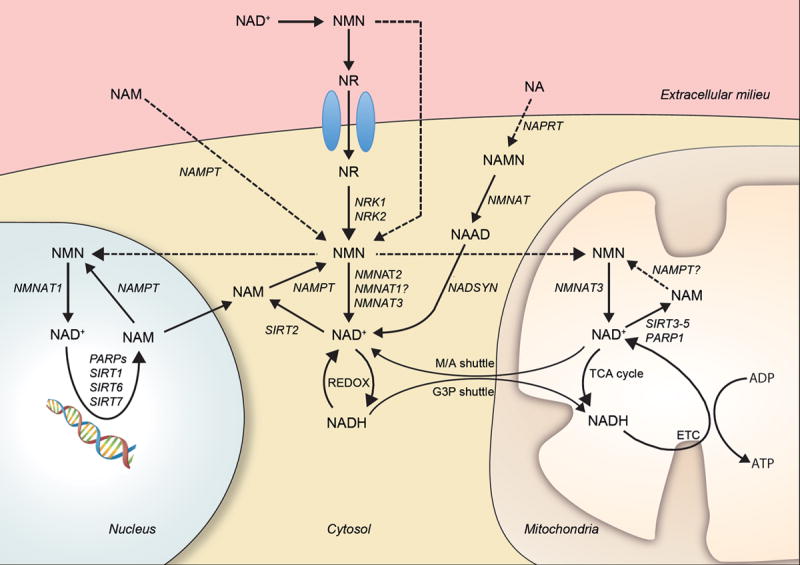
In normal circumstances, most NAD+ or NMN in blood is converted to NR, which enters the cell through specific transporters and is metabolized into NMN through NRK activity. Similarly, circulating NAM can be metabolized to NMN extracellulary by the extracellular NAMPT or enter the cell and be metabolized into NMN by the intracellular NAMPT. Extracellular NA can also enter the cell and be converted to NAD+ via a three-step reaction that is reliant on NAPRT, NMNAT and NADSYN. NMN and, possibly, NAM are potentially transported into the mitochondrial and nuclear compartments. In those compartments, NMN can lead to NAD+ synthesis via NMNAT activity. In each subcellular compartment, NAD+ and NADH equilibriums will be determined by their unique redox states. In the mitochondria, the electron transport chain is a major contributor to NADH oxidation into NAD+, coupling this action to ATP synthesis. In addition, the mitochondria and the cytosol can exchange redox equivalents through the malate/aspartate (M/A) and glyceraldehyde 3-phosphate (G3P) shuttles. In all compartments, the activity of NAD+ consuming enzymes, such as sirtuins or PARPs, lead to NAM production, which can be salvaged for NAD+ synthesis via NAMPT activity. Dashed arrows indicate pathways that need further validation.
All the above suggest that plasma levels of most NAD+ precursors are probably unable to systematically sustain high NAD+ production rates. Consequently, it seems that mammalian organisms largely rely on NAD+ salvage from intracellular NAM in order to maintain NAD+ pools. In fact, NAM is an end product of NAD+ consuming activities in the cell (i.e., sirtuins, poly(ADP-ribose) polymerases and cyclic ADP-ribose hydrolases) (Houtkooper et al., 2010a). Accordingly, mice lacking NAMPT are not viable (Revollo et al., 2007). This, however, does not rule out a limited contribution of circulating NR, NMN, NAM or Trp to NAD+ biosynthesis under basal conditions. However, further technical improvements will be needed, especially for NR, NMN and NAM determination, to precisely evaluate whether circulating precursors contribute to tissular NAD+ homeostasis.
2.3 Cell compartmentalization of NAD+
In general, intracellular NAD+ levels are maintained between 0.2 and 0.5 mM, depending on the cell type or tissue. However, NAD+ levels can change, up to ~2-fold, in response to diverse physiological stimuli. For example, NAD+ levels increase in response to energy stresses, such as glucose deprivation (Fulco et al., 2008), fasting (Canto et al., 2010; Rodgers et al., 2005), caloric restriction (Chen et al., 2008) and exercise (Canto et al., 2010; Costford et al., 2010), and fluctuate in a circadian fashion (Nakahata et al., 2009; Ramsey et al., 2009). So, where and how do these changes take place in the cell?
The presence of NMNATs in the nucleus, cytosol and mitochondria suggests that these compartments are fully capable to salvage NAD+ from NAM (Figure 2). NAD+ degrading enzymes, such as sirtuins, are also present in these compartments. In addition, the presence of different forms of NMNATs in each cellular compartment (e.g., NMNAT1 in the nucleus or NMNAT3 in the mitochondria/cytosol) suggests that NAD+ salvage is tailored according to compartment-specific metabolic needs. However, despite some evidence that NAMPT is localized to the mitochondria (Yang et al., 2007a), there is still some debate as to whether this is really the case (Pittelli et al., 2010). Therefore further experimental evidence is needed to confirm mitochondrial NAD+ salvage. Nonetheless, it is important to note that NAD+ is not evenly distributed in the cell. Most reports indicate that mitochondrial NAD+ content is ≥250 μM (Nakagawa et al., 2009; Yang et al., 2007a), while according to indirect estimations, nuclear NAD+ levels seem to be much lower, ~70 μM (Fjeld et al., 2003). To this effect, two-photon microscopy approaches have also been used to indirectly estimate NAD+ levels, confirming that NAD+ content in the nucleus is much lower than in the cytosol (Zhang et al., 2009). In addition, the different NAD+ pools can behave independently. As such, cells treated with methylmethane sulfonate, a genotoxic agent, can survive as long as mitochondrial NAD+ levels are maintained, irrespective of NAD+ depletion in other compartments (Yang et al., 2007a). Given that NAD+ or NADH cannot diffuse through membranes (van Roermund et al., 1995), the maintenance of NAD+ levels in each compartment is reliant on salvaging the NAM produced by NAD+-consuming enzymes (Figure 2). Alternatively, it can be derived from the intermediates NMN or NAMN, generated from NR metabolism or the Preiss-Handler pathway, respectively. It was recently shown that exogenous NAD+ can elevate mitochondrial NAD+ levels more than cytoplasmic levels, indicating that NAD+ precursors or intermediates traverse the mitochondrial membrane (Pittelli et al., 2011). Further, NR treatment was shown to enhance mitochondrial NAD+ levels in cultured cells and in mouse liver (Canto et al., 2012). However, the mitochondrial compartment lacks NRK activity to initiate NR conversion into NAD+ (Nikiforov et al., 2011). Hence, NR is likely converted to NMN in the cytosol and NMN may traverse the mitochondrial membrane to produce NAD+ via NMNATs (Figure 2) (Berger et al., 2005; Yang et al., 2007a). This way, both NMN and NAM might act as the main intracellular forms for regulating NAD+ levels between compartments.
The compartmentalization of NAD+ synthesis may have even more layers of complexity than once imagined. For example, NMNAT1 is recruited to target gene promoters by either the NAD+-consuming enzyme SIRT1 (Zhang et al., 2009) or PARP1 (Zhang et al., 2012), which suggests that NAD+ production is regulated at a sub-compartmental level during transcriptional regulation or DNA repair. These observations suggest that SIRT1 and PARP1 may compete for limiting amounts of localized NMNAT1-produced NAD+. Thus, despite estimations indicating that nuclear NAD+ levels are low, NAD+ maintenance is key for survival as testified by the fact that Nmnat1 knockout mice are embryonically lethal (Conforti et al., 2011).
3. THE ENZYMATIC USE OF NAD+
3.1 NAD+ and redox reactions in metabolism
While distinct, the cytosolic/nuclear and mitochondrial pools of NAD+ are interconnected by an intricate set of cellular redox processes. These NAD+ pools can modulate the activity of compartment specific metabolic pathways such as glycolysis in the cytoplasm and the TCA cycle/oxidative phosphorylation in the mitochondria.
In the cytoplasm, the conversion of glucose to pyruvate by glycolysis requires two NAD+ molecules per molecule of glucose. Following the conversion of glucose to two molecules of glyceraldehyde-3-phosphate (G3P), GAPDH (glyceraldehyde-3-phosphate dehydrogenase) reduces NAD+ to NADH to transform G3P into 1-3-biphosphoglycerate. Glycolysis will therefore net two NADH and two pyruvate molecules that can then be transported into to the mitochondrial matrix. Since the outer mitochondrial membrane is very porous, NADH is free to enter the intermembrane space. However, it is the reducing equivalent of NADH that is transported into the mitochondria, via either the malate-aspartate shuttle or the glycerol-3-phosphate shuttle of the inner mitochondrial membrane, rather than NADH itself (Figure 2) (McKenna et al., 2006). As discussed in section 2.3, cytoplasmic NAD+ levels cannot alter mitochondrial NAD+/NADH ratios directly since NAD+ is not permeable to the mitochondrial membrane (Barile et al., 1996). Therefore, changes in the cytoplasmic NAD+ pool do not acutely alter mitochondrial NAD+ levels (Pittelli et al., 2010; Yang et al., 2007a).
In the mitochondrion the TCA cycle reduces NAD+ molecules to produce multiple NADH molecules. Mitochondrial NADH, gained from glycolysis or the TCA cycle, are oxidized by Complex I (NADH:ubiquinone oxidoreductase) of the ETC. The subsequent two electrons gained by Complex I are relayed along ubiquinone (Coenzyme Q10), complex III (coenzyme Q - cytochrome c oxidoreductase), cytochrome c, and Complex IV (cytochrome c oxidase). In parallel to the oxidation of NADH to NAD+ by the ETC, the substrate succinate from the mitochondrial TCA cycle, provides additional electrons to ubiquinone in parallel with Complex I. Ultimately, the flow of electrons, generated from NADH and succinate, along the ETC is coupled to the pumping of protons from the mitochondrial matrix to the intermembrane space via Complex I, III and IV, creating a proton gradient. The proton gradient then provides the chemiosmotic gradient to couple the flux of protons back into the matrix via F0F1-ATP synthase with oxidative phosphorylation of ADP to ATP. Overall, the ETC reduces O2 to water and NADH to NAD+ for the purpose of generating ATP. As a result, mitochondrial NAD+ levels are 2-fold greater than the rest of the cell, as measured in mouse skeletal muscle (Pirinen et al., 2014) and 4-fold greater in mouse cardiac myocytes (Alano et al., 2007).
Since NAD+ levels within the cell can be limiting (Bai et al., 2011b; Pirinen et al., 2014; Pittelli et al., 2011), both glycolysis in the cytoplasm and the TCA cycle in the mitochondria can influence metabolic homeostasis by altering cytosolic/nuclear NAD+ and NADH levels. In addition, following DNA damage, NAD+ levels can drop low enough that glycolysis and substrate flux to the mitochondria is blocked leading to cell death, despite having an excess of available glucose (Alano et al., 2010; Benavente et al., 2009; Ying et al., 2005; Zhang et al., 2014). This finding highlights the need to understand the mechanisms interconnecting subcellular NAD+ pools, as their homeostasis and interaction is essential for the preservation of cell viability and ATP levels.
3.2 NAD+ consuming enzymes (I): Sirtuins
In mammals, there are seven sirtuin enzymes (SIRT1-SIRT7) based on the presence of a characteristic and evolutionarily conserved catalytic site, comprised of 275 amino acids (Haigis and Sinclair, 2010; Hall et al., 2013; Houtkooper et al., 2010a). Three sirtuins are located in the mitochondria (SIRT3-SIRT5), while SIRT1, SIRT6 and SIRT7 are predominantly located in the nucleus, and SIRT2 is found in the cytoplasm (Michishita et al., 2005; Verdin et al., 2010). However, some sirtuins, such as SIRT1 have been shown to shuttle in and out of the nucleus (Tanno et al., 2006).
Sirtuins use NAD+ as a cosubstrate to remove acetyl moieties from lysines on histones and proteins, releasing NAM and O-acetyl ADP-ribose (Houtkooper et al., 2010a). The consumption of NAD+ during deacetylation is what separates sirtuins, as type III lysine deactylases (KDACs), from type I, II and IV KDACs. SIRT1, SIRT2 and SIRT3 have strong deacetylase activity (Imai et al., 2000; North et al., 2003; Schwer et al., 2002; Vaziri et al., 2001), while SIRT4, SIRT5 and SIRT6 are weak in comparison. However, deacetylation is not the singular function of all sirtuins, as SIRT4 can act as a lipoamidase (Mathias et al., 2014) and, along with SIRT6, as an NAD+-dependent mono-ADP-ribosyltransferase (Haigis et al., 2006; Liszt et al., 2005). SIRT6 can also efficiently remove long-chain fatty acyl groups from lysine residues (Jiang et al., 2013), while SIRT5 has strong desuccinylase, demalonylase and deglutarylase enzymatic activities (Du et al., 2011; Tan et al., 2014). Finally, SIRT7 is an NAD+-dependent deacetylase with few known substrates, including p53 in vitro (Vakhrusheva et al., 2008), PAF53 in HeLa cells (Chen et al., 2013) and GABPβ1 in vivo (Ryu et al., 2014). Recent evidence indicates that long-chain deacylation is a general feature of all mammalian sirtuins (Feldman et al., 2013). For example, SIRT1, SIRT2 and SIRT3 can also act as effective decrotonylases (Bao et al., 2014; Feldman et al., 2013). The general deacylase activity of sirtuins, however, can differ in their preferential activity towards certain acyl chain lengths (Feldman et al., 2013).
Physiological roles of sirtuins
Generally, most sirtuins are activated during times of energy deficit and reduced carbohydrate energy sources, triggering cellular adaptations that improve metabolic efficiency (Figure 3A). For example, SIRT1 activity increases during exercise (Canto et al., 2009), CR (Chen et al., 2008), fasting (Canto et al., 2010; Rodgers et al., 2005) or low glucose availability (Fulco et al., 2008), all of which correlate with higher NAD+ levels. Due to space limitations, we will only briefly summarize the roles of sirtuins. For further information, we refer the reader to recent specialized reviews (Boutant and Canto, 2014; Chang and Guarente, 2013; Houtkooper et al., 2012).
Figure 3. The reciprocal relationship between SIRT1 and PARPs during NAD+ homeostasis and metabolic signaling in the cell.
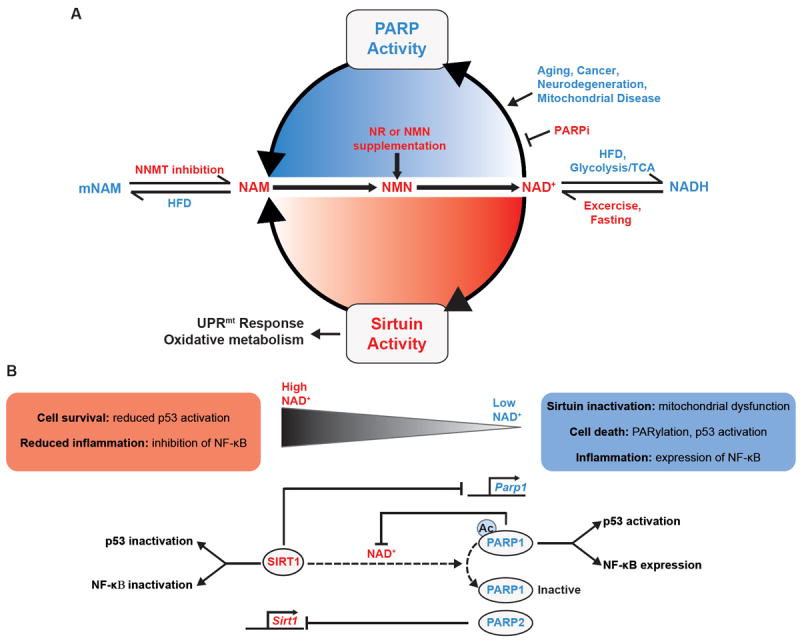
A. NAD+ is an essential coenzyme for sirtuin, PARP and CD38 activity, all of which metabolize NAD+ into NAM. Glycolysis and the TCA cycle also consume available NAD+ for the production of NADH, providing reducing equivalents for either lactate dehydrogenase (LDH) or the electron transport chain (ETC). Red font indicates environmental or physiological stimuli that activate sirtuins by increasing NAD+ while blue font indicates a reduction in NAD+, thereby diminishing sirtuin activity. NAM can be shunted away from NAD+ production following methylation by NNMT, a pathway activated by a HFD or with long term or high doses of NAM, which can favor the development of a fatty liver, due to reductions in available methyl groups. In contrast, NNMT depletion by NNMT-antisense oligonucleotides in animals, or mNAM supplementation in cells reduces NAM methylation. With a HFD, NAD+ can be reduced by elevating energy availability and NADH production, while exercise, fasting and CR reverses this process providing more NAD+ for sirtuin activation and protein deacetylation. NR supplementation or intraperitoneal NMN increases NAD+ availability via the NAD+ salvage pathway in mice. Ultimately, SIRT1 induces mitochondrial biogenesis, energy expenditure, antioxidant defenses, and lifespan extension by a mechanism that involves the mitochondrial unfolded protein response (UPRmt). PARPs consume NAD+, reducing SIRT1 activity, by increasing PARylation of DNA and proteins during aging, cancer, neurodegeneration, and mitochondrial diseases. B. SIRT1 negatively regulates PARP1 through the inhibition of transcription and possibly through deacetylation. Reciprocally, PARP1 inhibits SIRT1 by limiting NAD+ levels, while PARP2 directly inhibits SIRT1 transcription. Interestingly, PARP1 is required for the transcriptional co-activation of NF-κB, while SIRT1 inhibits NF-κB activity through the deacetylation of RelA/p65. In addition, PARP1 and SIRT1 oppositely regulate p53 nuclear accumulation and activation following cytotoxic stress. Since the Km of PARP1 for NAD+ is lower than that of SIRT1, as NAD+ levels drop following cell stress or senescence SIRT1 becomes less effective at regulating PARP1, and inhibiting inflammation or cell death through the inactivation of NF-κB and p53. Dashed arrows indicate pathways that need further validation.
Different nuclear sirtuin orthologs have been shown to influence lifespan in yeast, worms, flies and mice (Bauer et al., 2009; Boily et al., 2008; Kaeberlein et al., 1999; Kanfi et al., 2012; Rogina and Helfand, 2004; Tissenbaum and Guarente, 2001). Accordingly, the deacetylation of transcription factors, cofactors and histones by SIRT1 was shown to be important to enhance mitochondrial metabolism (Boily et al., 2008; Canto et al., 2009; Canto et al., 2010; Feige et al., 2008; Menzies et al., 2013; Price et al., 2012; Rodgers et al., 2005). Further evidence indicates that SIRT1 is key to linking nutrients to circadian rhythm (Asher et al., 2008; Chang and Guarente, 2013; Nakahata et al., 2008). The tight link between sirtuins and metabolism was reinforced by findings indicating that a moderate SIRT1 overexpression in mice could prevent metabolic and age-related complications, including insulin resistance, obesity and hepatic steatosis (Banks et al., 2008; Herranz et al., 2010; Pfluger et al., 2008). In addition, pharmacological SIRT1 activation protects against the lifespan reductions prompted by high-fat diets (Baur et al., 2006; Minor et al., 2011). Similarly, SIRT6 overexpression has been shown to increase mouse lifespan (Kanfi et al., 2012). Oppositely, loss of function models for SIRT1, SIRT3 and SIRT7 have been linked to a higher susceptibility to metabolic and age-related disease or reduced maximal lifespan (Boutant and Canto, 2014; Hirschey et al., 2011; Ryu et al., 2014; Vakhrusheva et al., 2008), while the absence of SIRT6 causes severe hypoglycemia, leading to mortality within the first month of life (Mostoslavsky et al., 2006; Zhong et al., 2010). Sirt2- and Sirt5-deficient mice, however, do not display an overt metabolic phenotype in the basal state (Beirowski et al., 2011; Bobrowska et al., 2012; Yu et al., 2013), while Sirt4 deficiency, in contrast to most sirtuins, enhances oxidative metabolism (Laurent et al., 2013).
Sirtuins as NAD+ sensors
Their ability to use NAD+ as a substrate led to speculation that sirtuins could act as metabolic sensors. The activity of sirtuins for a given intracellular NAD+ level is defined by the Michaelis constant, Km, for the reaction. This constant describes the NAD+ concentration when the reaction rate is half of the maximum during NAD+ excess. The estimated total intracellular content of NAD+ in mammals ranges from ~200 to ~500 μM (Bai et al., 2011b; Hong et al., 2014; Houtkooper et al., 2010a; Schmidt et al., 2004). The Km of SIRT1 for NAD+ has been reported to be in the range of 94-96 μM in mammals (Table 1) (Gerhart-Hines et al., 2011; Pacholec et al., 2010). The Km for NAD+, however, can differ very significantly between sirtuins. For example, the Km for NAD+ of SIRT2, SIRT3, SIRT4, SIRT5 and SIRT6 are reported as 83 μM (Borra et al., 2004), 880 μM (Hirschey et al., 2011), 35 μM (Laurent et al., 2013), 980 μM (Fischer et al., 2012) and 26 μM (Pan et al., 2011), respectively. The affinity of SIRT7 for NAD+ has not been reported to our knowledge. The above numbers help to classify sirtuins into two different categories. Firstly, there are sirtuins, such as SIRT2, 4 and 6, whose activity is unlikely to be rate-limited by NAD+, as NAD+ availability is considerably higher than their Km values. In contrast, there are other sirtuins, such as SIRT1, SIRT3 and SIRT5 whose Km for NAD+ falls within the range for physiological changes in NAD+. In this respect, it is important to note that SIRT1 is a nuclear enzyme, and NAD+ concentrations in the nucleus are below 100 μM, while NAD+ levels in the mitochondria can reach millimolar values, suggesting that NAD+ could limit SIRT3 and SIRT5 based on their Km values.
TABLE 1.
NAD+ Consuming Enzymes
| Enzyme | Km Value (μM) | References |
|---|---|---|
| SIRT1 | 94-96 | (Gerhart-Hines et al., 2011; Pacholec et al., 2010) |
| SIRT2 | 83 | (Borra et al., 2004) |
| SIRT3 | 880 | (Hirschey et al., 2011) |
| SIRT4 | 35 | (Laurent et al., 2013) |
| SIRT5 | 980 | (Fischer et al., 2012) |
| SIRT6 | 26 | (Pan et al., 2011) |
| SIRT7 | Unknown | |
| PARP1 | 50-97 | (Amé et al., 1999; Jiang et al., 2010; Mendoza-Alvarez and Alvarez-Gonzalez, 1993) |
| PARP2 | 130 | (Amé et al., 1999) |
| PARP4 (VPARP) | Unknown | |
| Tankyrase 1 (PARP5a) | 1125-1500 | (Jiang et al., 2010; Rippmann et al., 2002) |
| Tankyrase 2 (PARP5b) | Unknown | |
| CD38 | 15-25 | (Cakir-Kiefer et al., 2001; Fulco et al., 2008; Sauve et al., 1998) |
Only poly-ADP-Ribosylating PARPs were included in this table.
The above observations indicate that few sirtuins (i.e: SIRT1, SIRT3 and SIRT5) fulfill the key criteria to be considered NAD+ sensors. However, sirtuins are not exclusively regulated by NAD+. For example, NAM, the end-product of the sirtuin reaction, acts as a potent and general sirtuin deacetylase inhibitor. NAM was in fact shown to inhibit Sir2p, the yeast SIRT1 ortholog in a non-competitive manner with NAD+, with an IC50<50 μM (Anderson et al., 2003; Bitterman, 2002; Borra et al., 2004). Thus, sirtuin activity can potentially be differentially regulated by the cellular concentrations of both NAD+ and NAM.
NADH has also been proposed to act as an inhibitor of SIRT1, through competitive binding of the NAD+ pocket (Lin et al., 2004). Yet, the inhibition by NADH only occurs in the millimolar range, considerably above physiological NADH levels (Schmidt et al., 2004; Smith et al., 2009; Zhang et al., 2002). For example, intracellular concentrations of NADH in muscle cells range from 50 to 100 μM (Canto et al., 2012; Hong et al., 2014). Thus, based on the above findings the intracellular NAD+/NAM ratio may be a better predictor of sirtuin activity compared to the popularly used NAD+/NADH ratio.
3.3 NAD+ consuming enzymes (II): Poly(ADP-ribose) polymerases (PARPs)
Poly ADP-ribose (PAR) polymerases (PARPs) have been the center of intense focus due to their active role in DNA repair, inflammation and cell death but have now also been shown to influence circadian rhythm, neuronal function, endoplasmic reticulum stress and, metabolism, amongst other cellular pathways (reviewed in (Cantó et al., 2013; Gibson and Kraus, 2012; Kraus and Hottiger, 2013)). There are 17 different genes encoding PARP related proteins (Gibson and Kraus, 2012), but most research has so far focused on PARP1 and PARP2, which account for the vast majority of PARP activity in the cell (Canto et al., 2013). In general, PARP1 and PARP2 can be activated by DNA strand breaks endowing them with a role in the response to DNA damage (Amé et al., 1999; Benjamin and Gill, 1980; Gradwohl et al., 1990). However, PARPs can also be activated by interactions with the phosphorylated form of ERK to amplify ERK-mediated histone acetylation events (Cohen-Armon et al., 2007). Furthermore, PARPs are also activated by HSP70 during heat shock stress to alter nucleosome structure and by Trp tRNA synthetase (TrpRS) (Petesch and Lis, 2012; Sajish and Schimmel, 2015). Active PARP catalyzes the transfer of ADP-ribose subunits from NAD+ to protein acceptors, including different nuclear protein substrates, and even itself (a process called auto-poly-ADP-ribosylation), thus forming PAR chains (Kameshita et al., 1984). Classically, PARPs have been shown to play dual roles in the cell that can either result in the induction of cell death or DNA repair. PARP1, for instance, was shown to modify the effectiveness of the p53-mediated DNA damage response for different types of cytotoxic stress (Valenzuela et al., 2002). As a result, PARP inhibition can be an effective treatment for cancer (Bryant et al., 2005; Farmer et al., 2005; Fong et al., 2009), leading to the development of several potent PARP inhibitors as chemotherapeutic agents. From a purely metabolic angle, PARP1 activation has also been linked to a rapid reduction in the glycolytic rate. While this phenomenon has been classically linked to a reduction in NAD+ availability, recent evidence indicates that PARP1 might also directly PARylate hexokinase, leading to a reduction in hexokinase activity and the cellular glycolytic rate (Andrabi et al., 2014; Fouquerel et al., 2014). Indeed, the possible direct impact of PARP activities on metabolic enzymes will be a fascinating area of research for years to come.
The competition between PARPs and sirtuins for NAD+ as a metabolic determinant
Upon DNA damage, PARP enzymes utilize NAD+ to generate PAR polymers, yielding NAM as a reaction product. Excessive DNA damage dramatically reduces NAD+ levels (Berger, 1985), even down to 20-30% of their normal levels (Houtkooper et al., 2010a). In fact, the enzymatic properties of PARP1 indicate that it is an avid NAD+ consumer, with NAD+ increasing up to 2-fold in Parp1-KO mouse tissues (Bai and Canto, 2012). This, in turn, limits NAD+ availability for other nuclear enzymes such as SIRT1 (Figure 3A) (Bai et al., 2011b; Pillai et al., 2005; Qin et al., 2006; Rajamohan et al., 2009). In fact, the Km of PARP1 is in the ~50-59 μM range, unlike like that of PARP2 (Km 130 μM), dictating that NAD+ is rarely rate-limiting for PARP1 activity (Table 1) (Amé et al., 1999; Mendoza-Alvarez and Alvarez-Gonzalez, 1993). The lower affinity for, and consumption rate of, NAD+ by PARP2 is in agreement with the lack of change in NAD+ levels when PARP2 is knocked down in cultured cells (Bai et al., 2011a). Interestingly, however, Parp2 deficiency increased SIRT1 expression as a consequence of a direct negative regulatory effect on the SIRT1 promoter (Figure 3B) (Bai et al., 2011a). This further illustrates how PARP activity leads to SIRT1 inactivation, either by limiting NAD+ levels, in the case of PARP1 (Bai et al., 2011b), or by acting as a transcriptional repressor, in the case of PARP2 (Bai et al., 2011a).
The complexity of this pathway was heightened when SIRT1 was shown to directly inhibit PARP1 via its deacetylation (Figure 3B). Specifically, increased PAR activity was observed in Sirt1-KO cells treated with H2O2 (Kolthur-Seetharam et al., 2006), while the ability of SIRT1 to deacetylate PARP1 was confirmed by immunoprecipitation experiments (Rajamohan et al., 2009). Furthermore, SIRT1 also negatively regulates PARP1 transcription (Rajamohan et al., 2009). Illustrating the opposing roles of both enzymes, PARP1 is required for the transcriptional co-activation of NF-κB (Hassa et al., 2003), while SIRT1 inhibits NF-κB activity through the deacetylation of RelA/p65 (Yeung et al., 2004). Furthermore, PARP1 and SIRT1 have opposing effects on p53 nuclear accumulation and activation following cytotoxic stress (Figure 3B) (Langley et al., 2002; Luo et al., 2001; Valenzuela et al., 2002; Vaziri et al., 2001). Since the Km of SIRT1 for NAD+ is higher than that of PARP1, NAD+ levels can become so low following cell stress or senescence that SIRT1 no longer has the activity to keep PARP1 in check. This is supported by the fact that NAD+-repletion by expression of NAMPT can protect against PARP1 overexpression in a SIRT1-mediated manner (Pillai et al., 2005). Thus, it is likely that diverse cellular fates and metabolic decisions are closely regulated by the balance of the reciprocal regulation of SIRT1 and PARP1 activities, under the guidance of NAD+ levels (Figure 3B).
Recent work has further strengthened the hypothesis that PARP1 and SIRT1 have counterbalancing roles in metabolism and aging. For instance, PARP1 activity is enhanced with aging (Braidy et al., 2011; Mouchiroud et al., 2013) and high-caloric intake (Bai et al., 2011b), yet reduced upon nutrient scarcity (Bai et al., 2011b). Parp1 deletion in C57Bl/6 mice confers protection against diet-induced obesity (Bai et al., 2011b), Strikingly, Parp1-deficiency on a 129/SvImJ background has been reported to exacerbate high-fat diet-induced obesity (Devalaraja-Narashimha and Padanilam, 2010). In addition, some studies have shown that Parp1-deficiency can limit adipocyte function and size, leading to higher hepatic lipid accumulation (Erener et al., 2012). Despite these discrepancies, pharmacological PARP inhibition has consistently rendered protection against diet-induced obesity (Lehmann et al., 2015; Pirinen et al., 2014), possibly through an upregulation of SIRT1-dependent mitochondrial biogenesis and energy expenditure via the mitochondrial unfolded protein response (UPRmt; See section 4.3) (Pirinen et al., 2014). In addition, when the PARP1 worm homolog, pme-1, was knocked down in C. elegans, worms lived longer and maintained a more youthful phenotype at late adult stages (Mouchiroud et al., 2013). This was correlated to a marked increase in NAD+ availability, Sir2.1 activity and mitochondrial function that was linked to the activation of the UPRmt (Mouchiroud et al., 2013). Altogether, most studies certify that a reduction in PARP activity is beneficial against some aspects of metabolic disease.
Importantly, PARP inhibition might lead to higher NAD+ availability in a compartment specific fashion. In line with the predominant localization of PARP1 to the nucleus, reductions in PARP1 activity/expression markedly increases nucleo/cytoplasmic NAD+ levels and SIRT1 activity, yet does not alter mitochondrial NAD+ or SIRT3 activity (Bai et al., 2011b; Pirinen et al., 2014). However, this notion will need to be consolidated when further technical developments allow us to better directly measure NAD+ levels in a compartment-specific fashion, most notably in the nucleus.
To strengthen the hypothesis that PARPs can consume NAD+ to the point of impeding metabolism, the aryl hydrocarbon receptor (AHR) target gene, TiPARP (TCDD-inducible poly(ADP-ribose) polymerase or PARP7) was shown to increase PARylation of proteins, reducing NAD+ levels and SIRT1-mediated PGC-1α deacetylation in liver tissue (Diani-Moore et al., 2010). Furthermore, tankyrase 2 (PARP5b) knockout mice also have reduced fat pad and body weights, although no connection has yet been made to improvements in tissue NAD+ levels (Chiang et al., 2006). Altogether, these results suggest that ADP-ribosylation by several PARP family members can lead to metabolic dysfunction, suggesting that PARP inhibitors may have beneficial effects in this context.
3.4 NAD+ consuming enzymes (III): Cyclic ADP-ribose synthases
Cyclic ADP-ribose (cADPR), a secondary messenger implicated in Ca2+ signaling, cell cycle control and insulin signaling (Malavasi et al., 2008), is produced from NAD+ by cADPR synthases. The family of cADP-ribose synthases, including CD38 and its homolog CD157, were initially described as plasma membrane antigens on thymocytes and T lymphocytes. However, these ectoenzymes have also been found in non-lymphoid tissues, including muscle, liver and brain (Aksoy et al., 2006b; Quarona et al., 2013). In addition, recent topological studies have described the enzymatic activity of this transmembrane protein as both extra- and intra-cellular (Jackson and Bell, 1990; Lee, 2012; Zhao et al., 2012).
Mice deficient in Cd38 show significantly elevated levels of NAD+ (10-30-fold) in tissues such as liver, muscle, brain, and heart, with corresponding SIRT1 activation, confirming the role of CD38 as a major NAD+ consumer (Figure 3A) (Aksoy et al., 2006b; Barbosa et al., 2007). Conversely, cells overexpressing CD38 showed reductions in NAD+ levels and in the expression of proteins related to energy metabolism and antioxidant defense, as measured by quantitative proteomic analysis (Hu et al., 2014). Similar to Parp1-deficient mice, Cd38-KO animals were protected from diet-induced obesity, liver steatosis and glucose intolerance due to enhanced energy expenditure (Barbosa et al., 2007). In fact, the influence of Cd38-defiency on metabolism is so dramatic that despite having lower physical activity compared to WT animals, they still expend more total energy. One potential issue is that CD38-independent cADPR synthase and NAD+-glycohydrolase activity remained present in the developing brain of Cd38-KO mice (Ceni et al., 2003). Similarly, studies in heart (Kannt et al., 2012; Xie et al., 2005), skeletal muscle (Bacher et al., 2004) and kidney (Nam et al., 2006) also demonstrated that cADPR synthesis occurs independently of CD38 and CD157, suggesting the existence of other cADPR synthase family member(s). In further support of the existence of additional cADPR synthases, a small-molecule compound screen discovered two potent inhibitors, SAN2589 and SAN4825, that do not inhibit CD38, yet blunt cardiac cADPR synthase activity (Kannt et al., 2012). Despite observations that CD38 inhibition appears to enhance NAD+ levels, further work should clarify its cellular location and specific roles in various tissues to make it a viable therapeutic target.
4. RECENT ADVANCES IN NAD+-RELATED THERAPEUTICS
Although NA is effective to treat dyslipidemia (Altschul et al., 1955), due to its undesirable effects, niacin derivatives including acipimox and prolonged release forms, such as niaspan and enduracin, have largely replaced NA use in the clinical management of hyperlipidemia. The core of the hypothesis explaining the effectiveness of niacin rested in part on the activation of GPR109A in adipocytes, which apparently mediated the transient reduction of plasma free fatty acid (FFA) levels (Tunaru et al., 2003; Zhang et al., 2005). Yet, more recently, using both a mouse line deficient in Gpr109 and clinical trials with two GPR109 agonists it became clear that GPR109 did not mediate niacin’s lipid efficacy, thus questioning the GPR109-mediated FFA hypothesis (Lauring et al., 2012). This, in turn, gave strength to the possibility that the effects of niacin relied on the ability of NA or NAM to elevate NAD+ levels and activate the sirtuins (Canto and Auwerx, 2012). Beyond niacin, other NAD+ precursors, such as NMN and NR, are being considered as alternatives to niacin since they do not activate GPR109A receptors, yet still activate SIRT1 in mice (Canto et al., 2012). Similarly, the inhibition of PARP or CD38 activities has also proven to enhance NAD+ levels and sirtuin action (Figure 3A). Further disqualifying a GPR109-mediated effect and in support an NAD+-mediated metabolic response, in human type 2 diabetes patients, acipimox, increases muscle mitochondrial function, which is accompanied by a mitonuclear protein imbalance and the induction of UPRmt (see section 4.1 and 4.4), hallmarks of SIRT1, in lieu of GPR109, activation (van de Weijer et al., 2014). In the next section we will hence discuss the therapeutic targets, the prospective clinical indications, and the potential limitations for NAD+ boosting compounds that activate the sirtuins.
4.1 New perspectives in NAD+ therapeutics (I): Metabolic disease
Introducing new NAD+ precursors: NR and NMN
NR was recently demonstrated to have a surprisingly robust effect on systemic metabolism. First, dietary supplementation with NR protected against diet-induced obesity (Canto et al., 2012). NR treatment increased both intracellular and mitochondrial liver NAD+ levels, concomitant to an enhancement of SIRT1 as well as SIRT3 activities (Canto et al., 2012). As a result, there was a SIRT1-dependent increase in FOXO1 deacetylation, along with elevations in SOD2 expression, a FOXO1 target gene. Furthermore, in the mitochondrial compartment, NR led to the deacetylation of the well-established SIRT3 targets, SOD2 and NDUFA9. In line with the activation of SIRT1 and SIRT3 targets, mitochondrial content was higher in skeletal muscle and brown adipose tissue of NR-treated high fat fed animals, which increased the use of lipids as energy substrates, boosted energy expenditure, and improved insulin sensitivity (Canto et al., 2012). In alignment, impaired glucose tolerance and glucose-stimulated insulin secretion, induced by NAD+ shortages in NAMPT-deficient heterozygous animals, could be corrected by the administration of NMN (Revollo et al., 2007). Similarly, intraperitoneally administered NMN ameliorates glucose homeostasis in age- and diet-related insulin resistant states (Ramsey et al., 2008; Yoshino et al., 2011). Importantly, NMN reversed the loss of NAD+ levels observed in both circumstances. As with NR, NMN also safeguarded mitochondrial function in mice and improved age-related mitochondrial dysfunction (Gomes et al., 2013). Knockdown of the nuclear localized NMNAT1 attenuated the effect of NMN, consistent with the effect of NMN being driven by increases in NAD+ levels (Gomes et al., 2013). Furthermore, as NMNAT1 is located in the nucleus, the nuclear NAD+ pool may play a more dominant role for the induction of mitochondrial-encoded OXPHOS transcripts, potentially through alterations in SIRT1-directed HIF1α destabilization, leading to c-Myc activation of the nuclear-encoded mitochondrial factor TFAM (Gomes et al., 2013). Although these findings support the use of NR or NMN as a strategy for healthy aging, their efficacy in humans still needs testing. In fact, the dosages used for NR and NMN in mice, 400-500 mg/(kg*day), are high and potentially suboptimal for human application. Unlike NR, the use of NMN in mice has relied on intraperitoneal delivery, which could further complicate clinical use. Thus, the dosages, routes of administration and efficacy of NAD+ boosters need to be optimized for human use.
Rejuvenating old NAD+ precursors: the complexities around NAM
NAM, was first associated with diabetes when it was shown to protect against streptozotocin (STZ)-induced diabetes (Schein et al., 1967), which is accompanied by a robust reduction of NAD+ levels in pancreatic islet cells. NAM, but not NA, can recover this drop in NAD+ levels (Ho and Hashim, 1972). Later it was demonstrated that the NAD+ reduction induced by STZ was due to increased DNA damage, stimulating PARP1 activity (Yamamoto et al., 1981).
Unlike other NAD+ precursors, NAM has the capacity to exert end-product inhibition on SIRT1 deacetylase activity. However, long-term NAM treatment increases NAD+ levels via the NAD+ salvage pathway, which likely tips the balance of the NAD+/NAM ratio such that SIRT1 is activated. Despite NAM being suggested as a treatment for type 1 diabetes (Olmos et al., 2006), clinical trials failed to confirm this hypothesis (Cabrera-Rode et al., 2006; Gale et al., 2004). More recently OLETF rats, a rodent model of obesity and type 2 diabetes, exhibited profound metabolic improvements following NAM treatment (100mg/kg for 4 weeks). This treatment induced liver NAD+ levels, which were complimented by enhanced glucose control (Yang et al., 2014). However, some reports indicate that long term or high doses of NAM are detrimental, because they favor the development of a fatty liver, due to reductions in available methyl groups (Kang-Lee et al., 1983). For instance, NAM administration for 8 weeks (1-4g/kg) resulted in methyl-group deficiency, which is likely due to the conversion of NAM into 1-methyl-NAM (mNAM) by nicotinamide n-methyltransferase (NNMT) (Figure 3A). NNMT shunts NAM away from NAD+ using S-adenosylmethionine (SAM) as a methyl donor (Aksoy et al., 1994; Riederer et al., 2009). In line with this hypothesis, supplementation of methionine, a methyl group donor, prevented the formation of steatohepatosis caused by high doses of NAM (Kang-Lee et al., 1983).
Recently, NNMT expression was found to be negatively correlated with GLUT4, the insulin-responsive glucose transporter, in adipose tissue (Kraus et al., 2014). In adipose-specific Glut4-KO mice, Nnmt transcripts are increased, while they are reduced in adipose-specific Glut4-overexpressing mice Nnmt (Kraus et al., 2014). Similarly, Nnmt transcripts were increased in the WAT of ob/ob, db/db and high-fat fed mice, compared to lean insulin-sensitive controls (Kraus et al., 2014). In addition, tissue specific knockdown of Nnmt in WAT and liver, using antisense oligonucleotides, protected against diet-induced obesity by increasing the expression of Sirt1-target genes and energy expenditure. Accordingly, treating adipocytes with mNAM, which acts as an end-product inhibitor of NNMT (Aksoy et al., 1994), increased O2 consumption (Kraus et al., 2014). Coming from a totally different angle, a germline mouse model deficient in Maf1, a repressor of RNA polymerase III transcription of highly abundant cellular RNAs (Upadhya et al., 2002), also underscored the importance of NNMT in NAD+ homeostasis. Maf1-/- mice are resistant to obesity due to metabolic inefficiency as a consequence of futile tRNA production, which led to extreme reductions of NNMT levels, boosting NAM salvage to regenerate NAD+ (Bonhoure et al., 2015). In combination, these independent studies in widely different mouse models support that NNMT inhibition enhances NAD+-dependent SIRT1 activity and protects mice against obesity and type 2 diabetes. In a seemingly contradictory fashion, work in C. elegans has shown that NNMT and the methylation of NAM might actually be an integral part of the mechanism by which sirtuins provide health- and life-span benefits (Schmeisser et al., 2013). For this, NNMT-produced mNAM would act as a substrate to the ortholog of the mammalian aldehyde oxidase (AOx1), GAD-3, to generate hydrogen peroxide, which acts as a mitohormetic reactive oxygen species signal (Schmeisser et al., 2013). Taken together, however, NNMT activity seems strongly regulated in diverse metabolic contexts, and has a major impact on NAD+ homeostasis. The discrepancies in the current findings might arise from the distinct models used (i.e. worms vs. mice) and the amplitude of the mitohormetic response in different metabolic scenarios.
The potential for PARP inhibition in cell metabolism
The potential of PARP inhibition as a treatment for metabolic complications was first suggested by the observation that Parp1-KO mice were protected from STZ-induced β-cell death and dysfunction by maintaining NAD+ levels and therefore glucose tolerance (Masutani et al., 1999). Parp1-KO animals exhibit higher mitochondrial content, increased energy expenditure and protection against metabolic disease brought on by a high-fat diet (Bai et al., 2011b). Correspondingly, PARP inhibitors also prevent carbon tetrachloride-induced liver mitochondrial dysfunction and fibrosis (Mukhopadhyay et al., 2014), and diet-induced obesity in mice (Pirinen et al., 2014). Long-term treatment of up to 18 weeks with the dual PARP1 and PARP2 inhibitor, MRL-45696, was shown to enhance exercise capacity and muscle mitochondrial function in chow-diet fed mice (Cerutti et al., 2014; Pirinen et al., 2014). Both in worm and mouse models, the effect of PARP inhibition on mitochondrial function was linked with the activation of the UPRmt, as reflected by the induction of HSP60 and CLPP, two UPRmt biomarkers (Mouchiroud et al., 2013; Pirinen et al., 2014) (Figure 4). In fact, PARP inhibitors increased mitochondrial translation, without coordinate changes in cytosolic translation rates, thus leading to a mitonuclear protein imbalance (Pirinen et al., 2014), which on its turn triggers the UPRmt to maintain optimal mitochondrial function (Houtkooper et al., 2013). This finding is in line with the recent discovery that mitochondrially located PARP1 activity may PARylate and disrupt the interaction between key mitochondrial-specific DNA base excision repair (BER) enzymes, namely EXOG and DNA polymerase gamma (Polγ), and the mitochondrial DNA (mtDNA), hindering mitochondrial biogenesis and reducing mtDNA copy numbers (Szczesny et al., 2014).
Figure 4. Energy stress, NAD+-dependent UPRmt signaling and mitochondrial health.
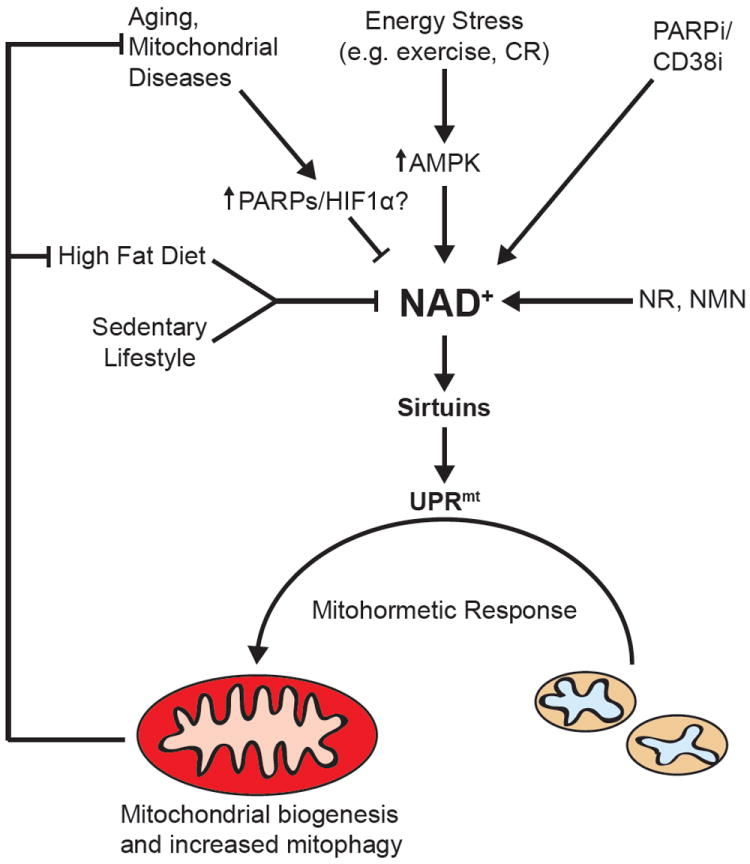
The aging process and associated metabolic diseases, including obesity and mitochondrial diseases can be improved in mice and C. elegans using NAD+ boosters or PARP/CD38 inhibitors (PARPi/CD38i) in much the same way as has been demonstrated by calorie restriction (CR). Part of the metabolic decline during aging is due to a PARP-directed reduction in NAD+ levels, attenuating SIRT1 and FOXOA3 activities and leading to the activation of HIF1α and an increased reliance on glycolysis. Recently, a mechanism has been proposed for these NAD+-mediated improvements that include the induction of UPRmt, which is triggered by SIRT1 and SIRT3 induced mitochondrial biogenesis, creating an imbalance in mitochondrial- versus nuclear-encoded mitochondrial proteins. This mitonuclear imbalance activates the UPRmt, a retrograde signal that induces a mitohormetic and adaptive nuclear response, ultimately repairing and improving mitochondrial function. These mitohormetic signals can attenuate the impact of aging, mitochondrial diseases or a high-fat diet on metabolism.
While the above observations set the stage for PARP inhibition to treat complex human metabolic diseases, it is important that inhibitors are selective for PARP1 and do not affect other members of the PARP family. For instance, although Parp2-KO mice were protected from diet-induced obesity, they were glucose intolerant due to defective pancreatic function (Bai et al., 2011a). In this sense, albeit diverse and highly efficient PARP inhibitors exist and are currently used in humans for anti-cancer therapy (Curtin and Szabo, 2013), none of them are selective for PARP1. Furthermore, since several of the PARPs play key roles in DNA damage repair upon genotoxic stress (Curtin and Szabo, 2013), further work must also ensure the long-term safety of selective PARP1 inhibition to treat metabolic diseases.
Inhibition of cADP-Ribose synthases improves metabolism
As described above, cADP-Ribose synthases, such as CD38, are primary NADases in mammalian tissues with a strong impact on SIRT1 activity (Aksoy et al., 2006a; Escande et al., 2010). This led to the hypothesis that CD38 inhibition (and subsequent increases in NAD+ levels) could be applied to treat metabolic disorders. In line with this, mice lacking CD38 are protected against diet-induced metabolic disease (Barbosa et al., 2007). Some natural flavonoids, such as quercetin, apigenin, luteolinidin, kuromanin and luteolin, were found to inhibit CD38 in the low micromolar range (Escande et al., 2013; Kellenberger et al., 2011). Accordingly, quercetin and apigenin increased liver NAD+ levels and SIRT1 activity resulting in improved glucose homeostasis and fatty acid oxidation in the liver of these mice (Escande et al., 2013). However, the recent development of potent thiazoloquin(az)olinone inhibitors for CD38, which can enhance NAD+ levels in multiple tissues, may prove to be effective for the design of future therapies (Haffner et al., 2015). Yet, as discussed in section 3.4, there remain several issues that require further work before CD38 inhibitors can be recommended to treat metabolic dysfunction. First, it is not entirely clear that CD38 is the main cADP-ribose synthase enzyme, therefore potentially compromising the efficacy of CD38 inhibitors for clinical use. Second, despite evidence indicating that it might also exist in nuclear and mitochondrial fractions (Aksoy et al., 2006a), CD38 activity is highest on the extracellular side of the plasma membrane, where NAD+ levels are generally very low (De Flora et al., 1997). Finally, the increase in NAD+ observed in Cd38-deficient mice is ~30 fold, while most other strategies described to date lead to a ~2-fold increase in NAD+ at best. The massive effect of CD38 on NAD+ levels could therefore be indicative for major alterations in additional NAD+-utilizing metabolic pathways.
4.2 New perspectives in NAD+ therapeutics (II): Neurodegenerative disease
Although the elimination of neurons by axonal degradation plays a role in normal nervous system development, aberrant neuronal cell death is typical of insults such as trauma, chemical toxicity, or of aging and neurodegenerative disorders such as Parkinson’s disease, Alzheimer’s disease and amyotrophic lateral sclerosis (for review see (Wang et al., 2012)).
Controversial links between NMNAT and neurodegenerative phenotypes
Axon degradation had originally been assumed to be a passive process. However, this view changed with the characterization of the naturally occurring Wallerian degeneration slow (WldS) dominant mutation (Conforti et al., 2000). Rodent carriers of this mutation displayed a dramatic reduction in axonal degeneration in both central and peripheral neurons. The WldS mutant protein is a chimeric protein composed of the complete sequence of NMNAT1 fused to the ubiquitination factor E4B at the N-terminus (Conforti et al., 2000; Mack et al., 2001). Efforts from diverse labs have since confirmed that it is the NMNAT enzymatic activity that is required to delay axon degeneration (Araki et al., 2004; Conforti et al., 2009; Gilley and Coleman, 2010; Llopis, 2000; Sasaki et al., 2009; Yahata et al., 2009; Yan et al., 2010), probably by promoting an increase in NAD+-directed SIRT1 activity (Araki et al., 2004). Interestingly, WldS mutant mice exhibit enhanced insulin secretion from isolated islets with an improvement in glucose homeostasis, also via an NAD+-directed activation of SIRT1 (Wu et al., 2011). Another study specifically demonstrated that it is the cytosolic distribution of NMNAT proteins that is crucial for slowing Wallerian degeneration (Sasaki et al., 2009). Further work should define changes in nuclear and cytosolic NAD+ levels, as most studies measure NAD+ in whole brain lysate, the outcome of which is confounded by the high level of NAD+ found in neuronal mitochondria.
NAD+ precursors protect against neurodegenerative disease
Following the injury of neurons there is an induction of multiple transcripts for NAD+ biosynthetic enzymes, including a more than 20-fold increase in NRK2, which catalyzes the synthesis of NAD+ from NR, suggesting a compensatory response to elevate NAD+ levels (Sasaki et al., 2006). In line with this, the pretreatment of neurons with either high levels of NAD+ in cell culture, or precursors such as NMN or NR, protects against axonal degeneration following axotomy, hearing loss caused by excess manganese toxicity or even noise-induced hearing loss in mice (Brown et al., 2014; Gerdts et al., 2015; Sasaki et al., 2006; Wang et al., 2014b). Similarly, rodent studies have demonstrated that pharmacological doses of NAM increases NAD+ biosynthesis and provides protection against ischemia (Klaidman et al., 2003; Sadanaga-Akiyoshi et al., 2003), fetal alcohol induced neurodegeneration (Ieraci and Herrera, 2006) and fetal ischemic brain injuries (Feng et al., 2006) by preventing NAD+ depletion. Further supporting the role of NAD+ in neuroprotection, a high-throughput screen identified an aminopropyl carbazole chemical P7C3 (Pieper et al., 2010), which was only recently discovered to be a pharmacological activator of NAMPT (Wang et al., 2014a), but had previously been shown to possess neuroprotective activity in models of traumatic brain injury (Yin et al., 2014), Parkinson’s disease (De Jesús-Cortés et al., 2012) and amyotrophic lateral sclerosis (Tesla et al., 2012). Increasing the activity of existing NAMPT using similar pharmacological approaches may therefore improve NAD+ depletion in aged animals, exhibiting reduced NAMPT and impairments in neural stem/progenitor cell self-renewal and differentiation, a treatment phenomenon already demonstrated using NMN on aging mice (Stein and Imai, 2014). In another model of neuronal degeneration, raised NAD+ levels after CR attenuated increases in Alzheimer disease (AD)-type β-amyloid content, in a rodent model of AD (Qin et al., 2006). NAM was also able to improve β-amyloid peptide (1–42)-induced oxidative damage and therefore protect against neurodegeneration (Turunc Bayrakdar et al., 2014a; Turunc Bayrakdar et al., 2014b). Similarly, exposing neuronal cells to toxic prion proteins to model protein misfolding in Alzheimer’s and Parkinson’s disease induced NAD+ depletion that was improved with exogenous NAD+ or NAM (Zhou et al., 2015). Additionally, NR has been shown to improve the AD phenotype via PGC-1α-mediated β-secretase (BACE1) degradation and the induction of mitochondrial biogenesis (Gong et al., 2013).
Maintaining NAD+ levels seems to, hence, sustain basal metabolic function and health in neurons. Furthermore, based on the preliminary evidence above, NR might have a privileged position among different NAD+ precursors in the prevention of neurodegeneration, as the effect of NR may be enhanced by the increase in NRK2 during axonal damage.
The role of PARPs in neurodegeneration
The depletion of NAD+ in neurodegeneration has been generally attributed to the activation of PARP enzymes. Well known neurodegenerative DNA repair disorders include ataxiatelangiectasia (AT), Cockayne syndrome (CS) and xeroderma pigmentosum group A (XPA), all of which demonstrate mitochondrial dysregulation due to SIRT1 inhibition and a reduction in mitophagy, the process of autophagic clearance of defective mitochondria (Fang et al., 2014). The reduction in SIRT1 activity and mitophagy in XPA-, CSB- and ATM-deficient cells can be attributed to the aberrant activation of PARP1, as reflected by the ability of PARP inhibitor AZD2281 (olaparib) to rescue the mitochondrial defect in cells and to extend the lifespan of xpa-1 mutant worms (Fang et al., 2014). In extension of these findings, using NR or a PARP inhibitor both improved the phenotype of a mouse model of Cockayne Syndrome group B (CSB), an accelerated aging disorder featuring the disinhibition of PARP activity by CSB protein, through SIRT1-mediated improvements of metabolic, mitochondrial, and transcriptional alterations (Scheibye-Knudsen et al., 2014). Similarly, augmented PARylation in the Csa-/-/Xpa-/- (CX) mouse model of cerebellar ataxia was reduced upon NR treatment, which improved NAD+ levels, SIRT1 activity, and mitochondrial function (Fang et al., 2014). Both interventions using NAD+ precursors or PARP inhibition could hence be helpful to improve neurodegenerative phenotypes.
4.3 New perspectives in NAD+ therapeutics (III): cancer and cell fate
Genomic stress is the root of all cancers and so maintaining genome integrity is an essential tool for the prevention of cancer. The fact that PARPs and several sirtuin enzymes are key for genomic maintenance suggests that the regulation of NAD+ could have an impact on cancer susceptibility and development (Canto et al., 2013). As protectors of genomic stability, PARPs can potentially play a multifunctional role in various cancer-related processes, including DNA repair, recombination, cell proliferation or death. In general, PARP activity protects cancer cells, especially those with high genome instability, from cellular death. Thus, PARP inhibitors are currently being clinically studied for the treatment of cancers that result from dysfunctional homologous DNA recombination repair (Curtin and Szabo, 2013). While a role for SIRT1 in cancer has been controversial, transgenic animal models demonstrate that SIRT1 protects against age-related carcinomas and sarcomas, but not lymphomas (Herranz et al., 2010). Given that PARP activity is rarely affected by physiological changes in NAD+ availability, it would be intuitive to think that most effects derived from fluctuations in NAD+ levels might crystalize into expected outcomes of modulating the activity of some sirtuins, such as SIRT1 (i.e.: protection against cancer). In this sense, it has been demonstrated that niacin supplementation can decrease the development of skin cancer (Gensler et al., 1999), while NR can both reduce the incidence of cancer and have a therapeutic effect on fully formed tumors, in a genetic mouse model for liver cancer (Tummala et al., 2014). Conversely, niacin deficiency can enhance cancer susceptibility, indicating that cellular NAD+ levels are inversely related to the incidence of cancer (Benavente et al., 2012; Jacobson, 1993). In another approach, some evidence suggests that the protective effects of niacin against the development of skin cancer are due to elevation in both PARPs and SIRT1 activity (Benavente et al., 2012). Firstly, niacin can effectively restore NAD+ levels and Poly(ADP)-ribosylated proteins in keratinocytes following photodamage, indicating an increase in the activity of PARPs. Secondly, SIRT1, also related to DNA repair and maintaining genomic stability and nucleotide excision repair pathways (Fan and Luo, 2010; Wang et al., 2008), exhibited increased activity in this same model as evidenced by increased protein deacetylation. However, the balance of SIRT1 and PARP activities upon genotoxic stress is further complicated by the fact that SIRT1 may reduce the expression or be a direct inhibitor of PARP1 (Kolthur-Seetharam et al., 2006; Rajamohan et al., 2009) and PARP2 is a negative regulator of SIRT1 expression (Bai et al., 2011a). This emphasizes the potential for an NAD+-dependent failsafe mechanism that can decide to elicit either repair or apoptosis depending on the severity of the cellular genotoxic insult through the manipulation and balance of SIRT1 and PARP activity levels. Furthermore, SIRT6, which, as described above, most likely does not act as an NAD+ sensor (see section 3.2), can activate PARP1 to stimulate highly efficient double-strand break repair, but only in response to oxidative stress-induced DNA damage (Mao et al., 2011). In addition, SIRT3 and SIRT5 have both been shown to play roles as either tumor suppressors or oncogenes depending on the cellular and molecular context, while SIRT4 acts as a tumor repressor due to its repression of glutamine metabolism, a process that is essential during rapid cell proliferation, as is seen in cancer (reviewed in (Kumar and Lombard, 2015)).
In a more speculative territory, it should also be highlighted that higher NAD+ levels, either through PARP inhibition or NAD+ precursor supplementation, rewires metabolism and enhances oxidative vs. glycolytic metabolism. Most cancer cells rely indisputably on glycolytic metabolism. Therefore, the metabolic remodeling by enhanced NAD+ levels could constitute a complementary mechanism to slow-down cancer progression or initiate cell death. However, NAD+ depletion might also inhibit growth of several cancers. NAMPT has been found to be overexpressed in several types of tumors and its expression is associated to tumor progression (Bi et al., 2011; Hasmann and Schemainda, 2003; Van Beijnum et al., 2002; Wang et al., 2011). Consequently, several studies showed that the NAD+ depletion triggered by the down-regulation of NAMPT activity can reduce tumor cell growth and sensitize cells to chemotoxic agents (Bi et al., 2011; Hasmann and Schemainda, 2003; Wang et al., 2011; Watson et al., 2009). One must keep in mind that in most cancer cells, PARPs are activated due to DNA damage and genome instability, leading to NAD+ depletion in cancer cells (Garten et al., 2009). As a result, the down-regulation of NAMPT sensitizes cancer cells to DNA damaging agents and apoptosis. In addition, NAD+ depletion also impairs glycolytic capacity in tumor cells (Bai and Canto, 2012). Altogether, the above data suggest that both NAD+ boosting and depletion can impact on tumor development and progression depending on the type and the metabolic properties of the tumor.
4.4 New perspectives in NAD+ therapeutics (IV): Aging
We have only recently started to understand the key pathways involved in defining lifespan. Much of this insight came from studies on CR, the most consistent intervention that extends longevity (as reviewed in (Canto and Auwerx, 2009)). Despite some evidence disputing the link between sirtuins and the longevity effects of CR, or longevity in general (Burnett et al., 2011; Jiang et al., 2000; Lamming et al., 2005), most data agree that sirtuin activation in mammals delays the onset of age-related degenerative processes (Herranz et al., 2010; Kanfi et al., 2012; Pearson et al., 2008; Satoh et al., 2013), and that defective sirtuin activity impairs some of the metabolic and physiological benefits triggered by CR (Boily et al., 2008; Hallows et al., 2011; Mercken et al., 2013; Someya et al., 2010). Of note, SIRT1 and SIRT3 have so far been the sirtuins most tightly linked to adaptations during CR. As discussed in section 3.1, the enzymatic characteristics of these sirtuins, but not of other sirtuins, may allow them to act as predominant NAD+ sensors, enabling them to monitor changes in nutrient availability.
During aging, reductions in NAD+ have been consistently observed in worms, diverse rodent tissues – including liver, pancreas, kidney, skeletal muscle, heart and white adipose – and in human skin samples (Braidy et al., 2011; Gomes et al., 2013; Khan et al., 2014; Massudi et al., 2012; Mouchiroud et al., 2013; Yoshino et al., 2011). Several hypotheses can explain the reductions in NAD+ levels during aging. The first relies on reductions in NAMPT expression with age, which may be in part due to the dysregulation of the circadian rhythm and therefore the CLOCK/BMAL-regulation of NAMPT (Nakahata et al., 2009). Another possible explanation for this lies in the higher PARP activity (due to cumulative DNA damage or alternative pathways of PARP activation, such as inflammatory or metabolic stress) observed in old worms and tissues from aged mice (Braidy et al., 2011; Mouchiroud et al., 2013). Supporting this possibility, blocking PARP activity is enough to recover NAD+ levels in aged organisms (Mouchiroud et al., 2013). The age-related reduction in NAD+, in turn, compromises mitochondrial function, which can be recovered via PARP inhibition or NAD+ precursor supplementation (Gomes et al., 2013; Mouchiroud et al., 2013). These observations are in line with the protection from metabolic dysfunction and disease in mice with genetically or pharmacologically-triggered deficiencies in PARP activity (Bai et al., 2011b; Pirinen et al., 2014) or in mice treated with NR (Canto et al., 2012) or NMN (Yoshino et al., 2011). As found in natural aging, significant reductions in skeletal muscle NAD+ occur in Deletor mice, with a mutation in the mitochondrial replicative helicase Twinkle, resulting in the accumulation of damage and a progressive muscle myopathy (Khan et al., 2014). Treatment of Deletor mice with NR delayed early- and late-stage disease progression by increasing mitochondrial biogenesis in skeletal muscle and brown adipose tissue while preventing mtDNA damage (Khan et al., 2014). In line with this, NR and PARP inhibition also improve the respiratory chain defect and exercise intolerance in Sco2-knockout/knockin mice, another model for mitochondrial disease (Cerutti et al., 2014). Altogether, the above data demonstrate that NAD+ supplementation maintains mitochondrial function, not only upon age-related decline but also in genetically-determined mitochondrial diseases that are known to accelerate the aging process (Figure 4).
The induction of UPRmt by NAD+ as a mechanism to enhance longevity
The ability of NAD+ to induce a mitonuclear protein imbalance could provide a key link between NAD+ and mitochondrial function. Mitonuclear protein imbalance can be defined as a stoichiometric difference between nuclear and mitochondrial-encoded respiratory subunit proteins (Houtkooper et al., 2013), an effect known to promote longevity in worms (Durieux et al., 2011; Houtkooper et al., 2013). Forced expression of genes regulating UPRmt, such as Hsp60 paralogs, in Drosophila slows age-dependent mitochondrial and muscle dysfunction, due to the compensatory actions of UPRmt signaling (Owusu-Ansah et al., 2013). Likewise, the activation of UPRmt by targeting mitochondrial ribosomal protein (Mrp) translation, using a knockdown of ribosomal proteins or antibiotics that specifically inhibit mitochondrial translation, increases longevity in C. elegans (Houtkooper et al., 2013). Strikingly, even subtle changes in expression levels of the Mrp’s in the BXD mouse genetic reference population have robust effects on mouse lifespan (Houtkooper et al., 2010b; Wu et al., 2014). Similarly, both NR and PARP inhibitors increased C. elegans lifespan, via an induction of the UPRmt by Sir-2.1, the worm SIRT1 ortholog (Mouchiroud et al., 2013). Part of the metabolic decline during aging is due to a PARP-directed decay in NAD+ levels, leading to reduced SIRT1, or Sir-2.1, activity and subsequent reduction in FOXO3A/Daf-16 activity and anti-oxidant defense (Mouchiroud et al., 2013) and increased HIF1α-led glycolytic reliance (Gomes et al., 2013). Furthermore, Sir-2.1-directed longevity is blunted in worms deficient in ubl-5 (Mouchiroud et al., 2013), an essential component for UPRmt-directed communication from the mitochondria to the nucleus (Durieux et al., 2011), solidifying the necessity of UPRmt induction for NAD+-induced longevity. In this sense, the decline in NAD+ observed with aging, deregulates the mitonuclear protein balance and respiratory function, which accelerates the aging process. Importantly, mitochondrial dysfunction in aged mice or worms could be recovered following NMN or NR treatment (Gomes et al., 2013; Mouchiroud et al., 2013). The above results highlight how the UPRmt triggers an adaptive mitohormetic response as long as the cell is properly furnished with NAD+. However, in situations of limited NAD+ availability, SIRT1 fails to drive mitohormesis and, hence, mitochondria remain dysfunctional. As a whole, the above demonstrates a tight feedback loop between the mitonuclear protein imbalance and UPRmt on the one hand and NAD+ metabolism and SIRT1 activity on the other. In summary, this assigns a crucial role for NAD+ to synchronize the nuclear and mitochondrial genomes
The identification of UPRmt as a main mechanism by which NAD+ levels modulate mitochondrial fitness constitutes a major leap forward in our understanding of the molecular mechanisms driving a healthy lifespan. Importantly, UPRmt is not just the principle mechanism by which NAD+ affects longevity, but is also a key mode of action for other well- studied longevity compounds, such as rapamycin and resveratrol (Houtkooper et al., 2013).
CONCLUSIONS
The association between metabolism, health and lifespan have long been proposed based on similarities, between metabolic dysfunction and disease (e.g. obesity, diabetes, neurodegeneration, cancer) and the aging process. Only recently have these processes been linked so tightly by multiple proteins, including the sirtuins and PARPs, all of which are tightly controlled by the regulation and subcellular balance of the metabolite NAD+. As such, we have never been so close to solving the ancient question of how we age and what we can do to slow this process, while simultaneously not compromising on our quality of life. Despite these insights, several aspects of NAD+ metabolism remain obscure. On one side, the complex detection and quantification of NAD+ metabolites and fluxes has not yet allowed us to obtain a clear picture of how different NAD+ precursors are metabolized to feed cells and tissues. We also speculate that additional proteins controlling the supply or salvage of NAD+, along with proteins that are controlled by NAD+ levels, will be identified. Furthermore, the potential preventive and therapeutic use of NAD+ boosting strategies requires an assessment of the bioavailability and effectiveness of various precursor doses in human therapy. In addition, new NAD+ boosters are welcomed since the side effects of niacin generally lead to poor compliance, despite its known efficacy in a myriad of diseases. Therefore, the dosing and safety of these new NAD+ boosters (e.g. NAD+ precursors, CD38 inhibitors and PARP inhibitors) must be thoroughly assessed to translate these exciting insights into NAD+ biology towards human relevance.
Highlights.
Adaptive cellular metabolism relies on NAD+ to mediate energy signaling
NAD+ therapeutics is showing its potential to treat disease
Metabolic syndrome, cancer and aging all involve NAD+ signaling
Acknowledgments
We would like to kindly thank Yoh Terada for his helpful advice during the creation of this review. KJM is the recipient of a Heart and Stroke Foundation of Canada research fellowship award. CC is an employee of the Nestlé Institute of Health Sciences S.A. JA is the Nestlé Chair in Energy Metabolism. Work in the laboratory is supported by the École Polytechnique Fédérale de Lausanne, the National Institutes of Health (R01AG043930), the Swiss National Science Foundation (31003A-124713), and Systems X (51RTP0-151019).
Footnotes
AUTHOR CONTRIBUTIONS
CC and KJM both designed the outline and generated substantial text. JA contributed to discussion of the ideas that make up this review and provided edits to all sections.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Aksoy P, Escande C, White TA, Thompson M, Soares S, Benech JC, Chini EN. Regulation of SIRT 1 mediated NAD dependent deacetylation: a novel role for the multifunctional enzyme CD38. Biochemical and biophysical research communications. 2006a;349:353–359. doi: 10.1016/j.bbrc.2006.08.066. [DOI] [PubMed] [Google Scholar]
- Aksoy P, White TA, Thompson M, Chini EN. Regulation of intracellular levels of NAD: a novel role for CD38. Biochemical and biophysical research communications. 2006b;345:1386–1392. doi: 10.1016/j.bbrc.2006.05.042. [DOI] [PubMed] [Google Scholar]
- Aksoy S, Szumlanski CL, Weinshilboum RM. Human liver nicotinamide N-methyltransferase cDNA cloning, expression, and biochemical characterization. The Journal of biological chemistry. 1994;269:14835–14840. [PubMed] [Google Scholar]
- Alano CC, Garnier P, Ying W, Higashi Y, Kauppinen TM, Swanson RA. NAD+ depletion is necessary and sufficient for poly(ADP-ribose) polymerase-1-mediated neuronal death. The Journal of neuroscience : the official journal of the Society for Neuroscience. 2010;30:2967–2978. doi: 10.1523/JNEUROSCI.5552-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alano CC, Tran A, Tao R, Ying W, Karliner JS, Swanson RA. Differences among cell types in NAD(+) compartmentalization: a comparison of neurons, astrocytes, and cardiac myocytes. Journal of neuroscience research. 2007;85:3378–3385. doi: 10.1002/jnr.21479. [DOI] [PubMed] [Google Scholar]
- Altschul R, Hoffer A, Stephen JD. Influence of nicotinic acid on serum cholesterol in man. Archives of biochemistry and biophysics. 1955;54:558–559. doi: 10.1016/0003-9861(55)90070-9. [DOI] [PubMed] [Google Scholar]
- Amé JC, Rolli V, Schreiber V, Niedergang C, Apiou F, Decker P, Muller S, Höger T, Ménissier-de Murcia J, de Murcia G. PARP-2, A novel mammalian DNA damage-dependent poly(ADP-ribose) polymerase. The Journal of biological chemistry. 1999;274:17860–17868. doi: 10.1074/jbc.274.25.17860. [DOI] [PubMed] [Google Scholar]
- Anderson RM, Bitterman KJ, Wood JG, Medvedik O, Sinclair DA. Nicotinamide and PNC1 govern lifespan extension by calorie restriction in Saccharomyces cerevisiae. Nature. 2003;423:181–185. doi: 10.1038/nature01578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andrabi SA, Umanah GKE, Chang C, Stevens DA, Karuppagounder SS, Gagné J-P, Poirier GG, Dawson VL, Dawson TM. Poly(ADP-ribose) polymerase-dependent energy depletion occurs through inhibition of glycolysis. Proceedings of the National Academy of Sciences. 2014;111:10209–10214. doi: 10.1073/pnas.1405158111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Araki T, Sasaki Y, Milbrandt J. Increased nuclear NAD biosynthesis and SIRT1 activation prevent axonal degeneration. Science (New York, N Y) 2004;305:1010–1013. doi: 10.1126/science.1098014. [DOI] [PubMed] [Google Scholar]
- Asher G, Gatfield D, Stratmann M, Reinke H, Dibner C, Kreppel F, Mostoslavsky R, Alt FW, Schibler U. SIRT1 regulates circadian clock gene expression through PER2 deacetylation. Cell. 2008;134:317–328. doi: 10.1016/j.cell.2008.06.050. [DOI] [PubMed] [Google Scholar]
- Asher G, Reinke H, Altmeyer M, Gutierrez-Arcelus M, Hottiger MO, Schibler U. Poly(ADP-ribose) polymerase 1 participates in the phase entrainment of circadian clocks to feeding. Cell. 2010;142:943–953. doi: 10.1016/j.cell.2010.08.016. [DOI] [PubMed] [Google Scholar]
- Bacher I, Zidar A, Kratzel M, Hohenegger M. Channelling of substrate promiscuity of the skeletal-muscle ADP-ribosyl cyclase isoform. The Biochemical journal. 2004;381:147–154. doi: 10.1042/BJ20031977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bai P, Canto C. The role of PARP-1 and PARP-2 enzymes in metabolic regulation and disease. Cell Metab. 2012;16:290–295. doi: 10.1016/j.cmet.2012.06.016. [DOI] [PubMed] [Google Scholar]
- Bai P, Canto C, Brunyanszki A, Huber A, Szanto M, Cen Y, Yamamoto H, Houten SM, Kiss B, Oudart H, et al. PARP-2 regulates SIRT1 expression and whole-body energy expenditure. Cell Metab. 2011a;13:450–460. doi: 10.1016/j.cmet.2011.03.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bai P, Canto C, Oudart H, Brunyanszki A, Cen Y, Thomas C, Yamamoto H, Huber A, Kiss B, Houtkooper RH, et al. PARP-1 inhibition increases mitochondrial metabolism through SIRT1 activation. Cell Metab. 2011b;13:461–468. doi: 10.1016/j.cmet.2011.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Banks AS, Kon N, Knight C, Matsumoto M, Gutierrez-Juarez R, Rossetti L, Gu W, Accili D. SirT1 gain of function increases energy efficiency and prevents diabetes in mice. Cell Metab. 2008;8:333–341. doi: 10.1016/j.cmet.2008.08.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bao X, Wang Y, Li X, Li X-M, Liu Z, Yang T, Wong CF, Zhang J, Hao Q, Li XD. Identification of ‘erasers’ for lysine crotonylated histone marks using a chemical proteomics approach. eLife. 2014;3 doi: 10.7554/eLife.02999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barbosa MTP, Soares SM, Novak CM, Sinclair D, Levine JA, Aksoy P, Chini EN. The enzyme CD38 (a NAD glycohydrolase, EC 3.2.2.5) is necessary for the development of diet-induced obesity. FASEB journal : official publication of the Federation of American Societies for Experimental Biology. 2007;21:3629–3639. doi: 10.1096/fj.07-8290com. [DOI] [PubMed] [Google Scholar]
- Barile M, Passarella S, Danese G, Quagliariello E. Rat liver mitochondria can synthesize nicotinamide adenine dinucleotide from nicotinamide mononucleotide and ATP via a putative matrix nicotinamide mononucleotide adenylyltransferase. Biochemistry and molecular biology international. 1996;38:297–306. [PubMed] [Google Scholar]
- Bauer JH, Morris SNS, Chang C, Flatt T, Wood JG, Helfand SL. dSir2 and Dmp53 interact to mediate aspects of CR-dependent lifespan extension in D. melanogaster Aging. 2009;1:38–48. doi: 10.18632/aging.100001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baum CL, Selhub J, Rosenberg IH. The hydrolysis of nicotinamide adenine nucleotide by brush border membranes of rat intestine. Biochem J. 1982;204:203–207. doi: 10.1042/bj2040203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baur JA, Pearson KJ, Price NL, Jamieson HA, Lerin C, Kalra A, Prabhu VV, Allard JS, Lopez-Lluch G, Lewis K, et al. Resveratrol improves health and survival of mice on a high-calorie diet. Nature. 2006;444:337–342. doi: 10.1038/nature05354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beirowski B, Gustin J, Armour SM, Yamamoto H, Viader A, North BJ, Michan S, Baloh RH, Golden JP, Schmidt RE, et al. Sir-two-homolog 2 (Sirt2) modulates peripheral myelination through polarity protein Par-3/atypical protein kinase C (aPKC) signaling. Proceedings of the National Academy of Sciences. 2011;108:E952–961. doi: 10.1073/pnas.1104969108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Belenky P, Racette FG, Bogan KL, McClure JM, Smith JS, Brenner C. Nicotinamide riboside promotes Sir2 silencing and extends lifespan via Nrk and Urh1/Pnp1/Meu1 pathways to NAD+ Cell. 2007;129:473–484. doi: 10.1016/j.cell.2007.03.024. [DOI] [PubMed] [Google Scholar]
- Benavente CA, Jacobson MK, Jacobson EL. NAD in skin: therapeutic approaches for niacin. Current pharmaceutical design. 2009;15:29–38. doi: 10.2174/138161209787185760. [DOI] [PubMed] [Google Scholar]
- Benavente CA, Schnell SA, Jacobson EL. Effects of niacin restriction on sirtuin and PARP responses to photodamage in human skin. PloS one. 2012;7:e42276. doi: 10.1371/journal.pone.0042276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bender DA. Biochemistry of tryptophan in health and disease. Mol Aspects Med. 1983;6:101–197. doi: 10.1016/0098-2997(83)90005-5. [DOI] [PubMed] [Google Scholar]
- Benjamin RC, Gill DM. Poly(ADP-ribose) synthesis in vitro programmed by damaged DNA. A comparison of DNA molecules containing different types of strand breaks. The Journal of biological chemistry. 1980;255:10502–10508. [PubMed] [Google Scholar]
- Benyo Z, Gille A, Kero J, Csiky M, Suchankova MC, Nusing RM, Moers A, Pfeffer K, Offermanns S. GPR109A (PUMA-G/HM74A) mediates nicotinic acid-induced flushing. J Clin Invest. 2005;115:3634–3640. doi: 10.1172/JCI23626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berger F, Lau C, Dahlmann M, Ziegler M. Subcellular compartmentation and differential catalytic properties of the three human nicotinamide mononucleotide adenylyltransferase isoforms. The Journal of biological chemistry. 2005;280:36334–36341. doi: 10.1074/jbc.M508660200. [DOI] [PubMed] [Google Scholar]
- Berger NA. Poly(ADP-ribose) in the cellular response to DNA damage. Radiation research. 1985;101:4–15. [PubMed] [Google Scholar]
- Bessede A, Gargaro M, Pallotta MT, Matino D, Servillo G, Brunacci C, Bicciato S, Mazza EMC, Macchiarulo A, Vacca C, et al. Aryl hydrocarbon receptor control of a disease tolerance defence pathway. Nature. 2014;511:184–190. doi: 10.1038/nature13323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bi T-Q, Che X-M, Liao X-H, Zhang D-J, Long H-L, Li H-J, Zhao W. Overexpression of Nampt in gastric cancer and chemopotentiating effects of the Nampt inhibitor FK866 in combination with fluorouracil. Oncology reports. 2011;26:1251–1257. doi: 10.3892/or.2011.1378. [DOI] [PubMed] [Google Scholar]
- Bieganowski P, Brenner C. Discoveries of nicotinamide riboside as a nutrient and conserved NRK genes establish a Preiss-Handler independent route to NAD+ in fungi and humans. Cell. 2004;117:495–502. doi: 10.1016/s0092-8674(04)00416-7. [DOI] [PubMed] [Google Scholar]
- Birjmohun RS, Hutten BA, Kastelein JJ, Stroes ES. Efficacy and safety of high-density lipoprotein cholesterol-increasing compounds: a meta-analysis of randomized controlled trials. J Am Coll Cardiol. 2005;45:185–197. doi: 10.1016/j.jacc.2004.10.031. [DOI] [PubMed] [Google Scholar]
- Bitterman KJ. Inhibition of Silencing and Accelerated Aging by Nicotinamide, a Putative Negative Regulator of Yeast Sir2 and Human SIRT1. Journal of Biological Chemistry. 2002;277:45099–45107. doi: 10.1074/jbc.M205670200. [DOI] [PubMed] [Google Scholar]
- Blander G, Guarente L. The Sir2 family of protein deacetylases. Annual review of biochemistry. 2004;73:417–435. doi: 10.1146/annurev.biochem.73.011303.073651. [DOI] [PubMed] [Google Scholar]
- Bobrowska A, Donmez G, Weiss A, Guarente L, Bates G. SIRT2 ablation has no effect on tubulin acetylation in brain, cholesterol biosynthesis or the progression of Huntington’s disease phenotypes in vivo. PLoS One. 2012;7:e34805. doi: 10.1371/journal.pone.0034805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bogan KL, Brenner C. Nicotinic acid, nicotinamide, and nicotinamide riboside: a molecular evaluation of NAD+ precursor vitamins in human nutrition. Annual review of nutrition. 2008;28:115–130. doi: 10.1146/annurev.nutr.28.061807.155443. [DOI] [PubMed] [Google Scholar]
- Boily G, Seifert EL, Bevilacqua L, He XH, Sabourin G, Estey C, Moffat C, Crawford S, Saliba S, Jardine K, et al. SirT1 regulates energy metabolism and response to caloric restriction in mice. PloS one. 2008;3:e1759. doi: 10.1371/journal.pone.0001759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonhoure N, Byrnes A, Moir RD, Hodroj W, Preitner F, Praz V, Marcelin G, Chua SC, Jr, Martinez-Lopez N, Singh R, et al. Loss of the RNA polymerase III repressor MAF1 confers obesity resistance. Genes dev. 2015;29:934–937. doi: 10.1101/gad.258350.115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borra MT, Langer MR, Slama JT, Denu JM. Substrate specificity and kinetic mechanism of the Sir2 family of NAD+-dependent histone/protein deacetylases. Biochemistry. 2004;43:9877–9887. doi: 10.1021/bi049592e. [DOI] [PubMed] [Google Scholar]
- Boutant M, Canto C. SIRT1 metabolic actions: Integrating recent advances from mouse models. Molecular metabolism. 2014;3:5–18. doi: 10.1016/j.molmet.2013.10.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Braidy N, Guillemin GJ, Mansour H, Chan-Ling T, Poljak A, Grant R. Age related changes in NAD+ metabolism oxidative stress and Sirt1 activity in wistar rats. PloS one. 2011;6:e19194. doi: 10.1371/journal.pone.0019194. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Brown KD, Maqsood S, Huang J-Y, Pan Y, Harkcom W, Li W, Sauve A, Verdin E, Jaffrey SR. Activation of SIRT3 by the NAD(+) Precursor Nicotinamide Riboside Protects from Noise-Induced Hearing Loss. Cell metabolism. 2014;20:1059–1068. doi: 10.1016/j.cmet.2014.11.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bryant HE, Schultz N, Thomas HD, Parker KM, Flower D, Lopez E, Kyle S, Meuth M, Curtin NJ, Helleday T. Specific killing of BRCA2-deficient tumours with inhibitors of poly(ADP-ribose) polymerase. Nature. 2005;434:913–917. doi: 10.1038/nature03443. [DOI] [PubMed] [Google Scholar]
- Burnett C, Valentini S, Cabreiro F, Goss M, Somogyvári M, Piper MD, Hoddinott M, Sutphin GL, Leko V, McElwee JJ, et al. Absence of effects of Sir2 overexpression on lifespan in C elegans and Drosophila. Nature. 2011;477:482–485. doi: 10.1038/nature10296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cabrera-Rode E, Molina G, Arranz C, Vera M, González P, Suárez R, Prieto M, Padrón S, León R, Tillan J, et al. Effect of standard nicotinamide in the prevention of type 1 diabetes in first degree relatives of persons with type 1 diabetes. Autoimmunity. 2006;39:333–340. doi: 10.1080/08916930600738383. [DOI] [PubMed] [Google Scholar]
- Cakir-Kiefer C, Muller-Steffner H, Oppenheimer N, Schuber F. Kinetic competence of the cADP-ribose-CD38 complex as an intermediate in the CD38/NAD+ glycohydrolase-catalysed reactions: implication for CD38 signalling. The Biochemical journal. 2001;358:399–406. doi: 10.1042/0264-6021:3580399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Canto C, Auwerx J. Caloric restriction, SIRT1 and longevity. Trends Endocrinol Metab. 2009;20:325–331. doi: 10.1016/j.tem.2009.03.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Canto C, Auwerx J. Targeting sirtuin 1 to improve metabolism: all you need is NAD(+)? Pharmacol Rev. 2012;64:166–187. doi: 10.1124/pr.110.003905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Canto C, Gerhart-Hines Z, Feige JN, Lagouge M, Noriega L, Milne JC, Elliott PJ, Puigserver P, Auwerx J. AMPK regulates energy expenditure by modulating NAD+ metabolism and SIRT1 activity. Nature. 2009;458:1056–1060. doi: 10.1038/nature07813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Canto C, Houtkooper RH, Pirinen E, Youn DY, Oosterveer MH, Cen Y, Fernandez-Marcos PJ, Yamamoto H, Andreux PA, Cettour-Rose P, et al. The NAD(+) precursor nicotinamide riboside enhances oxidative metabolism and protects against high-fat diet-induced obesity. Cell Metab. 2012;15:838–847. doi: 10.1016/j.cmet.2012.04.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Canto C, Jiang LQ, Deshmukh AS, Mataki C, Coste A, Lagouge M, Zierath JR, Auwerx J. Interdependence of AMPK and SIRT1 for metabolic adaptation to fasting and exercise in skeletal muscle. Cell Metab. 2010;11:213–219. doi: 10.1016/j.cmet.2010.02.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Canto C, Sauve AA, Bai P. Crosstalk between poly(ADP-ribose) polymerase and sirtuin enzymes. Mol Aspects Med. 2013;34:1168–1201. doi: 10.1016/j.mam.2013.01.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cantó C, Sauve AA, Bai P. Crosstalk between poly(ADP-ribose) polymerase and sirtuin enzymes. Molecular aspects of medicine. 2013;34:1168–1201. doi: 10.1016/j.mam.2013.01.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ceni C, Muller-Steffner H, Lund F, Pochon N, Schweitzer A, De Waard M, Schuber F, Villaz M, Moutin MJ. Evidence for an intracellular ADP-ribosyl cyclase/NAD(+)-glycohydrolase in brain from CD38-deficient mice. The Journal of biological chemistry. 2003;278:40670–40678. doi: 10.1074/jbc.M301196200. [DOI] [PubMed] [Google Scholar]
- Cerutti R, Pirinen E, Lamperti C, Marchet S, Sauve AA, Li W, Leoni V, Schon EA, Dantzer F, Auwerx J, et al. NAD(+)-Dependent Activation of Sirt1 Corrects the Phenotype in a Mouse Model of Mitochondrial Disease. Cell metabolism. 2014;19:1042–1049. doi: 10.1016/j.cmet.2014.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang H-C, Guarente L. SIRT1 mediates central circadian control in the SCN by a mechanism that decays with aging. Cell. 2013;153:1448–1460. doi: 10.1016/j.cell.2013.05.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen D, Bruno J, Easlon E, Lin SJ, Cheng HL, Alt FW, Guarente L. Tissue-specific regulation of SIRT1 by calorie restriction. Genes Dev. 2008;22:1753–1757. doi: 10.1101/gad.1650608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen S, Seiler J, Santiago-Reichelt M, Felbel K, Grummt I, Voit R. Repression of RNA polymerase I upon stress is caused by inhibition of RNA-dependent deacetylation of PAF53 by SIRT7. Molecular cell. 2013;52:303–313. doi: 10.1016/j.molcel.2013.10.010. [DOI] [PubMed] [Google Scholar]
- Chi Y, Sauve AA. Nicotinamide riboside, a trace nutrient in foods, is a vitamin B3 with effects on energy metabolism and neuroprotection. Current Opinion in Clinical Nutrition and Metabolic Care. 2013;16:657–661. doi: 10.1097/MCO.0b013e32836510c0. [DOI] [PubMed] [Google Scholar]
- Chiang YJ, Nguyen M-L, Gurunathan S, Kaminker P, Tessarollo L, Campisi J, Hodes RJ. Generation and characterization of telomere length maintenance in tankyrase 2-deficient mice. Molecular and cellular biology. 2006;26:2037–2043. doi: 10.1128/MCB.26.6.2037-2043.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cohen-Armon M, Visochek L, Rozensal D, Kalal A, Geistrikh I, Klein R, Bendetz-Nezer S, Yao Z, Seger R. DNA-independent PARP-1 activation by phosphorylated ERK2 increases Elk1 activity: a link to histone acetylation. Molecular cell. 2007;25:297–308. doi: 10.1016/j.molcel.2006.12.012. [DOI] [PubMed] [Google Scholar]
- Collins PB, Chaykin S. The management of nicotinamide and nicotinic acid in the mouse. The Journal of biological chemistry. 1972;247:778–783. [PubMed] [Google Scholar]
- Conforti L, Janeckova L, Wagner D, Mazzola F, Cialabrini L, Di Stefano M, Orsomando G, Magni G, Bendotti C, Smyth N, et al. Reducing expression of NAD+ synthesizing enzyme NMNAT1 does not affect the rate of Wallerian degeneration. The FEBS journal. 2011;278:2666–2679. doi: 10.1111/j.1742-4658.2011.08193.x. [DOI] [PubMed] [Google Scholar]
- Conforti L, Tarlton A, Mack TG, Mi W, Buckmaster EA, Wagner D, Perry VH, Coleman MP. A Ufd2/D4Cole1e chimeric protein and overexpression of Rbp7 in the slow Wallerian degeneration (WldS) mouse. Proceedings of the National Academy of Sciences of the United States of America. 2000;97:11377–11382. doi: 10.1073/pnas.97.21.11377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conforti L, Wilbrey A, Morreale G, Janeckova L, Beirowski B, Adalbert R, Mazzola F, Di Stefano M, Hartley R, Babetto E, et al. Wld S protein requires Nmnat activity and a short N-terminal sequence to protect axons in mice. The Journal of cell biology. 2009;184:491–500. doi: 10.1083/jcb.200807175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Costford SR, Bajpeyi S, Pasarica M, Albarado DC, Thomas SC, Xie H, Church TS, Jubrias SA, Conley KE, Smith SR. Skeletal muscle NAMPT is induced by exercise in humans. AJP: Endocrinology and Metabolism. 2010;298:E117–126. doi: 10.1152/ajpendo.00318.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Curtin NJ, Szabo C. Therapeutic applications of PARP inhibitors: anticancer therapy and beyond. Molecular aspects of medicine. 2013;34:1217–1256. doi: 10.1016/j.mam.2013.01.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cynamon MH, Sorg TB, Patapow A. Utilization and metabolism of NAD by Haemophilus parainfluenzae. Journal of general microbiology. 1988;134:2789–2799. doi: 10.1099/00221287-134-10-2789. [DOI] [PubMed] [Google Scholar]
- De Flora A, Guida L, Franco L, Zocchi E. The CD38/cyclic ADP-ribose system: a topological paradox. The international journal of biochemistry & cell biology. 1997;29:1149–1166. doi: 10.1016/s1357-2725(97)00062-9. [DOI] [PubMed] [Google Scholar]
- De Jesús-Cortés H, Xu P, Drawbridge J, Estill SJ, Huntington P, Tran S, Britt J, Tesla R, Morlock L, Naidoo J, et al. Neuroprotective efficacy of aminopropyl carbazoles in a mouse model of Parkinson disease. Proceedings of the National Academy of Sciences. 2012;109:17010–17015. doi: 10.1073/pnas.1213956109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Devalaraja-Narashimha K, Padanilam BJ. PARP1 deficiency exacerbates diet-induced obesity in mice. The Journal of endocrinology. 2010;205:242–251. doi: 10.1677/JOE-09-0402. [DOI] [PubMed] [Google Scholar]
- Diani-Moore S, Ram P, Li X, Mondal P, Youn DY, Sauve AA, Rifkind AB. Identification of the aryl hydrocarbon receptor target gene TiPARP as a mediator of suppression of hepatic gluconeogenesis by 2,3,7,8-tetrachlorodibenzo-p-dioxin and of nicotinamide as a corrective agent for this effect. Journal of Biological Chemistry. 2010;285:38801–38810. doi: 10.1074/jbc.M110.131573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Du J, Zhou Y, Su X, Yu JJ, Khan S, Jiang H, Kim J, Woo J, Kim JH, Choi BH, et al. Sirt5 is a NAD-dependent protein lysine demalonylase and desuccinylase. Science (New York, N Y) 2011;334:806–809. doi: 10.1126/science.1207861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Durieux J, Wolff S, Dillin A. The cell-non-autonomous nature of electron transport chain-mediated longevity. Cell. 2011;144:79–91. doi: 10.1016/j.cell.2010.12.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elvehjem CA. The Biological Significance of Nicotinic Acid: Harvey Lecture, November 16, 1939. Bulletin of the New York Academy of Medicine. 1940;16:173–189. [PMC free article] [PubMed] [Google Scholar]
- Emanuelli M, Carnevali F, Saccucci F, Pierella F, Amici A, Raffaelli N, Magni G. Molecular cloning, chromosomal localization, tissue mRNA levels, bacterial expression, and enzymatic properties of human NMN adenylyltransferase. J Biol Chem. 2001;276:406–412. doi: 10.1074/jbc.M008700200. [DOI] [PubMed] [Google Scholar]
- Erener S, Mirsaidi A, Hesse M, Tiaden AN, Ellingsgaard H, Kostadinova R, Donath MY, Richards PJ, Hottiger MO. ARTD1 deletion causes increased hepatic lipid accumulation in mice fed a high-fat diet and impairs adipocyte function and differentiation. FASEB J. 2012;26:2631–2638. doi: 10.1096/fj.11-200212. [DOI] [PubMed] [Google Scholar]
- Escande C, Chini CC, Nin V, Dykhouse KM, Novak CM, Levine J, van Deursen J, Gores GJ, Chen J, Lou Z, et al. Deleted in breast cancer-1 regulates SIRT1 activity and contributes to high-fat diet-induced liver steatosis in mice. J Clin Invest. 2010;120:545–558. doi: 10.1172/JCI39319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Escande C, Nin V, Price NL, Capellini V, Gomes AP, Barbosa MT, O’Neil L, White TA, Sinclair DA, Chini EN. Flavonoid apigenin is an inhibitor of the NAD+ ase CD38: implications for cellular NAD+ metabolism, protein acetylation, and treatment of metabolic syndrome. Diabetes. 2013;62:1084–1093. doi: 10.2337/db12-1139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Euler-Chelpin Hv. Nobel Lecture: Fermentation of Sugars and Fermentative Enzymes. 1929 ( http://www.nobelprize.org/nobel_prizes/chemistry/laureates/1929/euler-chelpin-lecture.html:TheNobelFoundation)
- Fan W, Luo J. SIRT1 regulates UV-induced DNA repair through deacetylating XPA. Molecular cell. 2010;39:247–258. doi: 10.1016/j.molcel.2010.07.006. [DOI] [PubMed] [Google Scholar]
- Fang EF, Scheibye-Knudsen M, Brace LE, Kassahun H, SenGupta T, Nilsen H, Mitchell JR, Croteau DL, Bohr VA. Defective mitophagy in XPA via PARP-1 hyperactivation and NAD(+)/SIRT1 reduction. Cell. 2014;157:882–896. doi: 10.1016/j.cell.2014.03.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farmer H, McCabe N, Lord CJ, Tutt ANJ, Johnson DA, Richardson TB, Santarosa M, Dillon KJ, Hickson I, Knights C, et al. Targeting the DNA repair defect in BRCA mutant cells as a therapeutic strategy. Nature. 2005;434:917–921. doi: 10.1038/nature03445. [DOI] [PubMed] [Google Scholar]
- Feige JN, Lagouge M, Canto C, Strehle A, Houten SM, Milne JC, Lambert PD, Mataki C, Elliott PJ, Auwerx J. Specific SIRT1 activation mimics low energy levels and protects against diet-induced metabolic disorders by enhancing fat oxidation. Cell Metab. 2008;8:347–358. doi: 10.1016/j.cmet.2008.08.017. [DOI] [PubMed] [Google Scholar]
- Feldman JL, Baeza J, Denu JM. Activation of the protein deacetylase SIRT6 by long-chain fatty acids and widespread deacylation by mammalian sirtuins. Journal of Biological Chemistry. 2013;288:31350–31356. doi: 10.1074/jbc.C113.511261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Felici R, Lapucci A, Ramazzotti M, Chiarugi A. Insight into molecular and functional properties of NMNAT3 reveals new hints of NAD homeostasis within human mitochondria. PloS one. 2013;8:e76938. doi: 10.1371/journal.pone.0076938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feng Y, Paul IA, LeBlanc MH. Nicotinamide reduces hypoxic ischemic brain injury in the newborn rat. Brain research bulletin. 2006;69:117–122. doi: 10.1016/j.brainresbull.2005.11.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fischer F, Gertz M, Suenkel B, Lakshminarasimhan M, Schutkowski M, Steegborn C. Sirt5 deacylation activities show differential sensitivities to nicotinamide inhibition. PLoS One. 2012;7:e45098. doi: 10.1371/journal.pone.0045098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fjeld CC, Birdsong WT, Goodman RH. Differential binding of NAD+ and NADH allows the transcriptional corepressor carboxyl-terminal binding protein to serve as a metabolic sensor. Proc Natl Acad Sci U S A. 2003;100:9202–9207. doi: 10.1073/pnas.1633591100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fong PC, Boss DS, Yap TA, Tutt A, Wu P, Mergui-Roelvink M, Mortimer P, Swaisland H, Lau A, O’Connor MJ, et al. Inhibition of poly(ADP-ribose) polymerase in tumors from BRCA mutation carriers. The New England journal of medicine. 2009;361:123–134. doi: 10.1056/NEJMoa0900212. [DOI] [PubMed] [Google Scholar]
- Forbes JC, Duncan GM. Effect of a tryptophan-niacin deficient diet on the adrenal response of rats exposed to cold and alcohol intoxication. Quarterly journal of studies on alcohol. 1961;22:254–260. [PubMed] [Google Scholar]
- Fouquerel E, Goellner EM, Yu Z, Gagné J-P, Barbi de Moura M, Feinstein T, Wheeler D, Redpath P, Li J, Romero G, et al. ARTD1/PARP1 negatively regulates glycolysis by inhibiting hexokinase 1 independent of NAD+ depletion. Cell reports. 2014;8:1819–1831. doi: 10.1016/j.celrep.2014.08.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fulco M, Cen Y, Zhao P, Hoffman EP, McBurney MW, Sauve AA, Sartorelli V. Glucose restriction inhibits skeletal myoblast differentiation by activating SIRT1 through AMPK-mediated regulation of Nampt. Developmental cell. 2008;14:661–673. doi: 10.1016/j.devcel.2008.02.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gale EAM, Bingley P, Emmett CL, Collier T Group, E.N.D.I.T.E. European Nicotinamide Diabetes Intervention Trial (ENDIT): a randomised controlled trial of intervention before the onset of type 1 diabetes. Lancet. 2004;363:925–931. doi: 10.1016/S0140-6736(04)15786-3. [DOI] [PubMed] [Google Scholar]
- Garten A, Petzold S, Körner A, Imai S-i, Kiess W. Nampt: linking NAD biology, metabolism and cancer. Trends in endocrinology and metabolism: TEM. 2009;20:130–138. doi: 10.1016/j.tem.2008.10.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gensler HL, Williams T, Huang AC, Jacobson EL. Oral niacin prevents photocarcinogenesis and photoimmunosuppression in mice. Nutrition and cancer. 1999;34:36–41. doi: 10.1207/S15327914NC340105. [DOI] [PubMed] [Google Scholar]
- Gerdts J, Brace EJ, Sasaki Y, DiAntonio A, Milbrandt J. Neurobiology. SARM1 activation triggers axon degeneration locally via NAD+destruction. Science (New York, N Y) 2015;348:453–457. doi: 10.1126/science.1258366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerhart-Hines Z, Dominy JE, Jr, Blättler SM, Jedrychowski MP, Banks AS, Lim J-H, Chim H, Gygi SP, Puigserver P. The cAMP/PKA Pathway Rapidly Activates SIRT1 to Promote Fatty Acid Oxidation Independently of Changes in NAD+ Molecular cell. 2011;44:851–863. doi: 10.1016/j.molcel.2011.12.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gibson BA, Kraus WL. New insights into the molecular and cellular functions of poly(ADP-ribose) and PARPs. Nature reviews Molecular cell biology. 2012;13:411–424. doi: 10.1038/nrm3376. [DOI] [PubMed] [Google Scholar]
- Gilley J, Coleman MP. Endogenous Nmnat2 is an essential survival factor for maintenance of healthy axons. PLoS biology. 2010;8:e1000300. doi: 10.1371/journal.pbio.1000300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldberger J. The etiology of pellagra. 1914. Public health reports. 2006;121(1 Supp l):77–79. discussion 76. [PubMed] [Google Scholar]
- Gomes AP, Price NL, Ling AJY, Moslehi JJ, Montgomery MK, Rajman L, White JP, Teodoro JS, Wrann CD, Hubbard BP, et al. Declining NAD(+) induces a pseudohypoxic state disrupting nuclear-mitochondrial communication during aging. Cell. 2013;155:1624–1638. doi: 10.1016/j.cell.2013.11.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gong B, Pan Y, Vempati P, Zhao W, Knable L, Ho L, Wang J, Sastre M, Ono K, Sauve AA, et al. Nicotinamide riboside restores cognition through an upregulation of proliferator-activated receptor-γ coactivator 1α regulated β-secretase 1 degradation and mitochondrial gene expression in Alzheimer’s mouse models. Neurobiology of Aging. 2013;34:1581–1588. doi: 10.1016/j.neurobiolaging.2012.12.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gradwohl G, Ménissier de Murcia JM, Molinete M, Simonin F, Koken M, Hoeijmakers JH, de Murcia G. The second zinc-finger domain of poly(ADP-ribose) polymerase determines specificity for single-stranded breaks in DNA. Proceedings of the National Academy of Sciences of the United States of America. 1990;87:2990–2994. doi: 10.1073/pnas.87.8.2990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gross CJ, Henderson LM. Digestion and absorption of NAD by the small intestine of the rat. J Nutr. 1983;113:412–420. doi: 10.1093/jn/113.2.412. [DOI] [PubMed] [Google Scholar]
- Gwirtz JA, Garcia-Casal MN. Processing maize flour and corn meal food products. Annals of the New York Academy of Sciences. 2014;1312:66–75. doi: 10.1111/nyas.12299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haffner CD, Becherer JD, Boros EE, Cadilla R, Carpenter T, Cowan D, Deaton DN, Guo Y, Harrington W, Henke BR, et al. Discovery, Synthesis, and Biological Evaluation of Thiazoloquin(az)olin(on)es as Potent CD38 Inhibitors. Journal of medicinal chemistry. 2015;58:3548–3571. doi: 10.1021/jm502009h. [DOI] [PubMed] [Google Scholar]
- Haigis MC, Mostoslavsky R, Haigis KM, Fahie K, Christodoulou DC, Murphy AJ, Valenzuela DM, Yancopoulos GD, Karow M, Blander G, et al. SIRT4 inhibits glutamate dehydrogenase and opposes the effects of calorie restriction in pancreatic beta cells. Cell. 2006;126:941–954. doi: 10.1016/j.cell.2006.06.057. [DOI] [PubMed] [Google Scholar]
- Haigis MC, Sinclair DA. Mammalian sirtuins: biological insights and disease relevance. Annual review of pathology. 2010;5:253–295. doi: 10.1146/annurev.pathol.4.110807.092250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hall JA, Dominy JE, Lee Y, Puigserver P. The sirtuin family’s role in aging and age-associated pathologies. J Clin Invest. 2013;123:973–979. doi: 10.1172/JCI64094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hallows WC, Yu W, Smith BC, Devries MK, Ellinger JJ, Someya S, Shortreed MR, Prolla T, Markley JL, Smith LM, et al. Sirt3 promotes the urea cycle and fatty acid oxidation during dietary restriction. Mol Cell. 2011;41:139–149. doi: 10.1016/j.molcel.2011.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hara N, Yamada K, Shibata T, Osago H, Hashimoto T, Tsuchiya M. Elevation of cellular NAD levels by nicotinic acid and involvement of nicotinic acid phosphoribosyltransferase in human cells. The Journal of biological chemistry. 2007;282:24574–24582. doi: 10.1074/jbc.M610357200. [DOI] [PubMed] [Google Scholar]
- Hara N, Yamada K, Shibata T, Osago H, Tsuchiya M. Nicotinamide phosphoribosyltransferase/visfatin does not catalyze nicotinamide mononucleotide formation in blood plasma. PLoS One. 2011;6:e22781. doi: 10.1371/journal.pone.0022781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hara N, Yamada K, Terashima M, Osago H, Shimoyama M, Tsuchiya M. Molecular identification of human glutamine- and ammonia-dependent NAD synthetases. Carbon-nitrogen hydrolase domain confers glutamine dependency. J Biol Chem. 2003;278:10914–10921. doi: 10.1074/jbc.M209203200. [DOI] [PubMed] [Google Scholar]
- Harden A, Young WJ. The Alcoholic Ferment of Yeast-Juice. Part II.--The Conferment of Yeast-Juice. Proceedings of the Royal Society of London Series B, Containing Papers of a Biological Character. 1906;78:369–375. [Google Scholar]
- Hasmann M, Schemainda I. FK866, a highly specific noncompetitive inhibitor of nicotinamide phosphoribosyltransferase, represents a novel mechanism for induction of tumor cell apoptosis. Cancer research. 2003;63:7436–7442. [PubMed] [Google Scholar]
- Hassa PO, Buerki C, Lombardi C, Imhof R, Hottiger MO. Transcriptional coactivation of nuclear factor-kappaB-dependent gene expression by p300 is regulated by poly(ADP)-ribose polymerase-1. The Journal of biological chemistry. 2003;278:45145–45153. doi: 10.1074/jbc.M307957200. [DOI] [PubMed] [Google Scholar]
- Hegyi J, Schwartz RA, Hegyi V. Pellagra: dermatitis, dementia, and diarrhea. International journal of dermatology. 2004;43:1–5. doi: 10.1111/j.1365-4632.2004.01959.x. [DOI] [PubMed] [Google Scholar]
- Henderson LM. Tryptophan’s role as a vitamin precursor (Krehl et al., 1945) J Nutr. 1997;127:1043S–1045S. [PubMed] [Google Scholar]
- Henderson LM, Gross CJ. Metabolism of niacin and niacinamide in perfused rat intestine. The Journal of nutrition. 1979;109:654–662. doi: 10.1093/jn/109.4.654. [DOI] [PubMed] [Google Scholar]
- Herbert M, Sauer E, Smethurst G, Kraiss A, Hilpert AK, Reidl J. Nicotinamide ribosyl uptake mutants in Haemophilus influenzae. Infection and immunity. 2003;71:5398–5401. doi: 10.1128/IAI.71.9.5398-5401.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herranz D, Munoz-Martin M, Canamero M, Mulero F, Martinez-Pastor B, Fernandez-Capetillo O, Serrano M. Sirt1 improves healthy ageing and protects from metabolic syndrome-associated cancer. Nat Commun. 2010;1:3. doi: 10.1038/ncomms1001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hikosaka K, Ikutani M, Shito M, Kazuma K, Gulshan M, Nagai Y, Takatsu K, Konno K, Tobe K, Kanno H, et al. Deficiency of nicotinamide mononucleotide adenylyltransferase 3 (nmnat3) causes hemolytic anemia by altering the glycolytic flow in mature erythrocytes. Journal of Biological Chemistry. 2014;289:14796–14811. doi: 10.1074/jbc.M114.554378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirschey MD, Shimazu T, Jing E, Grueter CA, Collins AM, Aouizerat B, Stancakova A, Goetzman E, Lam MM, Schwer B, et al. SIRT3 deficiency and mitochondrial protein hyperacetylation accelerate the development of the metabolic syndrome. Mol Cell. 2011;44:177–190. doi: 10.1016/j.molcel.2011.07.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ho CK, Hashim SA. Pyridine nucleotide depletion in pancreatic islets associated with streptozotocin-induced diabetes. Diabetes. 1972;21:789–793. doi: 10.2337/diab.21.7.789. [DOI] [PubMed] [Google Scholar]
- Hong J, Kim B-W, Choo H-J, Park J-J, Yi J-S, Yu D-M, Lee H, Yoon G-S, Lee J-S, Ko Y-G. Mitochondrial complex I deficiency enhances skeletal myogenesis but impairs insulin signaling through SIRT1 inactivation. Journal of Biological Chemistry. 2014 doi: 10.1074/jbc.M114.560078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horwitt MK, Harper AE, Henderson LM. Niacin-tryptophan relationships for evaluating niacin equivalents. The American journal of clinical nutrition. 1981;34:423–427. doi: 10.1093/ajcn/34.3.423. [DOI] [PubMed] [Google Scholar]
- Houtkooper RH, Canto C, Wanders RJ, Auwerx J. The secret life of NAD+: an old metabolite controlling new metabolic signaling pathways. Endocr Rev. 2010a;31:194–223. doi: 10.1210/er.2009-0026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Houtkooper RH, Mouchiroud L, Ryu D, Moullan N, Katsyuba E, Knott G, Williams RW, Auwerx J. Mitonuclear protein imbalance as a conserved longevity mechanism. Nature. 2013;497:451–457. doi: 10.1038/nature12188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Houtkooper RH, Pirinen E, Auwerx J. Sirtuins as regulators of metabolism and healthspan. Nature reviews Molecular cell biology. 2012;13:225–238. doi: 10.1038/nrm3293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Houtkooper RH, Williams RW, Auwerx J. Metabolic networks of longevity. Cell. 2010b;142:9–14. doi: 10.1016/j.cell.2010.06.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu Y, Wang H, Wang Q, Deng H. Overexpression of CD38 decreases cellular NAD levels and alters the expression of proteins involved in energy metabolism and antioxidant defense. Journal of proteome research. 2014;13:786–795. doi: 10.1021/pr4010597. [DOI] [PubMed] [Google Scholar]
- Ieraci A, Herrera DG. Nicotinamide protects against ethanol-induced apoptotic neurodegeneration in the developing mouse brain. PLoS medicine. 2006;3:e101. doi: 10.1371/journal.pmed.0030101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ikeda M, Tsui H, S N, Ichiyama A, Nishizuka Y, Hayaishi O. Studies on the Biosynthesis of Nicotinamide Adenine Dinucleotide. II. A ROLE OF PICOLINIC CARBOXYLASE IN THE BIOSYNTHESIS OF NICOTINAMIDE ADENINE DINUCLEOTIDE FROM TRYPTOPHAN IN MAMMALS. The Journal of biological chemistry. 1965;240:1395–1401. [PubMed] [Google Scholar]
- Imai S, Armstrong CM, Kaeberlein M, Guarente L. Transcriptional silencing and longevity protein Sir2 is an NAD-dependent histone deacetylase. Nature. 2000;403:795–800. doi: 10.1038/35001622. [DOI] [PubMed] [Google Scholar]
- Institute of Medicine (US) Standing Committee on the Scientific Evaluation of Dietary Reference Intakes and its Panel on Folate, O.B.V., and Choline. Dietary Reference Intakes for Thiamin, Riboflavin, Niacin, Vitamin B6, Folate, Vitamin B12, Pantothenic Acid, Biotin, and Choline. Washington (DC): National Academies Press (US); 1998. [PubMed] [Google Scholar]
- Jackson DG, Bell JI. Isolation of a cDNA encoding the human CD38 (T10) molecule, a cell surface glycoprotein with an unusual discontinuous pattern of expression during lymphocyte differentiation. Journal of immunology (Baltimore, Md : 1950) 1990;144:2811–2815. [PubMed] [Google Scholar]
- Jacobson EL. Niacin deficiency and cancer in women. Journal of the American College of Nutrition. 1993;12:412–416. doi: 10.1080/07315724.1993.10718330. [DOI] [PubMed] [Google Scholar]
- Jacobson EL, Dame AJ, Pyrek JS, Jacobson MK. Evaluating the role of niacin in human carcinogenesis. Biochimie. 1995;77:394–398. doi: 10.1016/0300-9084(96)88152-1. [DOI] [PubMed] [Google Scholar]
- Jiang H, Khan S, Wang Y, Charron G, He B, Sebastian C, Du J, Kim R, Ge E, Mostoslavsky R. SIRT6 regulates TNF-α secretion through hydrolysis of long-chain fatty acyl lysine. Nature. 2013;496:110–113. doi: 10.1038/nature12038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang H, Kim JH, Frizzell KM, Kraus WL, Lin H. Clickable NAD analogues for labeling substrate proteins of poly(ADP-ribose) polymerases. Journal of the American Chemical Society. 2010;132:9363–9372. doi: 10.1021/ja101588r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang JC, Jaruga E, Repnevskaya MV, Jazwinski SM. An intervention resembling caloric restriction prolongs life span and retards aging in yeast. FASEB journal : official publication of the Federation of American Societies for Experimental Biology. 2000;14:2135–2137. doi: 10.1096/fj.00-0242fje. [DOI] [PubMed] [Google Scholar]
- Kaeberlein M, McVey M, Guarente L. The SIR2/3/4 complex and SIR2 alone promote longevity in Saccharomyces cerevisiae by two different mechanisms. Genes & development. 1999;13:2570–2580. doi: 10.1101/gad.13.19.2570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaelin WG, McKnight SL. Influence of metabolism on epigenetics and disease. Cell. 2013;153:56–69. doi: 10.1016/j.cell.2013.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kameshita I, Matsuda Z, Taniguchi T, Shizuta Y. Poly (ADP-Ribose) synthetase. Separation and identification of three proteolytic fragments as the substrate-binding domain, the DNA-binding domain, and the automodification domain. The Journal of biological chemistry. 1984;259:4770–4776. [PubMed] [Google Scholar]
- Kanfi Y, Naiman S, Amir G, Peshti V, Zinman G, Nahum L, Bar-Joseph Z, Cohen HY. The sirtuin SIRT6 regulates lifespan in male mice. Nature. 2012;483:218–221. doi: 10.1038/nature10815. [DOI] [PubMed] [Google Scholar]
- Kang-Lee YA, McKee RW, Wright SM, Swendseid ME, Jenden DJ, Jope RS. Metabolic effects of nicotinamide administration in rats. The Journal of nutrition. 1983;113:215–221. doi: 10.1093/jn/113.2.215. [DOI] [PubMed] [Google Scholar]
- Kannt A, Sicka K, Kroll K, Kadereit D, Gögelein H. Selective inhibitors of cardiac ADPR cyclase as novel anti-arrhythmic compounds. Naunyn-Schmiedeberg’s archives of pharmacology. 2012;385:717–727. doi: 10.1007/s00210-012-0750-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kellenberger E, Kuhn I, Schuber F, Muller-Steffner H. Flavonoids as inhibitors of human CD38. Bioorganic & medicinal chemistry letters. 2011;21:3939–3942. doi: 10.1016/j.bmcl.2011.05.022. [DOI] [PubMed] [Google Scholar]
- Khan NA, Auranen M, Paetau I, Pirinen E, Euro L, Forsstrom S, Pasila L, Velagapudi V, Carroll CJ, Auwerx J, et al. Effective treatment of mitochondrial myopathy by nicotinamide riboside, a vitamin B3. EMBO Mol Med. 2014 doi: 10.1002/emmm.201403943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klaidman L, Morales M, Kem S, Yang J, Chang M-L, Adams JD. Nicotinamide offers multiple protective mechanisms in stroke as a precursor for NAD+, as a PARP inhibitor and by partial restoration of mitochondrial function. Pharmacology. 2003;69:150–157. doi: 10.1159/000072668. [DOI] [PubMed] [Google Scholar]
- Kolthur-Seetharam U, Dantzer F, McBurney MW, de Murcia G, Sassone-Corsi P. Control of AIF-mediated cell death by the functional interplay of SIRT1 and PARP-1 in response to DNA damage. Cell cycle (Georgetown, Tex) 2006;5:873–877. doi: 10.4161/cc.5.8.2690. [DOI] [PubMed] [Google Scholar]
- Kraus D, Yang Q, Kong D, Banks AS, Zhang L, Rodgers JT, Pirinen E, Pulinilkunnil TC, Gong F, Wang YC, et al. Nicotinamide N-methyltransferase knockdown protects against diet-induced obesity. Nature. 2014;508:258–262. doi: 10.1038/nature13198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kraus WL, Hottiger MO. PARP-1 and gene regulation: progress and puzzles. Molecular aspects of medicine. 2013;34:1109–1123. doi: 10.1016/j.mam.2013.01.005. [DOI] [PubMed] [Google Scholar]
- Kumar S, Lombard DB. Mitochondrial sirtuins and their relationships with metabolic disease and cancer. Antioxidants & Redox Signaling. 2015;22:1060–1077. doi: 10.1089/ars.2014.6213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lamming DW, Latorre-Esteves M, Medvedik O, Wong SN, Tsang FA, Wang C, Lin S-J, Sinclair DA. HST2 mediates SIR2-independent life-span extension by calorie restriction. Science (New York, N Y) 2005;309:1861–1864. doi: 10.1126/science.1113611. [DOI] [PubMed] [Google Scholar]
- Langley E, Pearson M, Faretta M, Bauer U-M, Frye RA, Minucci S, Pelicci PG, Kouzarides T. Human SIR2 deacetylates p53 and antagonizes PML/p53-induced cellular senescence. The EMBO Journal. 2002;21:2383–2396. doi: 10.1093/emboj/21.10.2383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lau C, Niere M, Ziegler M. The NMN/NaMN adenylyltransferase (NMNAT) protein family. Frontiers in bioscience. 2009;14:410–431. doi: 10.2741/3252. [DOI] [PubMed] [Google Scholar]
- Laurent G, German NJ, Saha AK, de Boer VC, Davies M, Koves TR, Dephoure N, Fischer F, Boanca G, Vaitheesvaran B, et al. SIRT4 coordinates the balance between lipid synthesis and catabolism by repressing malonyl CoA decarboxylase. Mol Cell. 2013;50:686–698. doi: 10.1016/j.molcel.2013.05.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lauring B, Taggart AKP, Tata JR, Dunbar R, Caro L, Cheng K, Chin J, Colletti SL, Cote J, Khalilieh S, et al. Niacin lipid efficacy is independent of both the niacin receptor GPR109A and free fatty acid suppression. Sci Transl Med. 2012;4:148ra115. doi: 10.1126/scitranslmed.3003877. [DOI] [PubMed] [Google Scholar]
- Lee HC. Cyclic ADP-ribose and nicotinic acid adenine dinucleotide phosphate (NAADP) as messengers for calcium mobilization. Journal of Biological Chemistry. 2012;287:31633–31640. doi: 10.1074/jbc.R112.349464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lehmann M, Pirinen E, Mirsaidi A, Kunze FA, Richards PJ, Auwerx J, Hottiger MO. ARTD1-induced poly-ADP-ribose formation enhances PPAR ligand binding and co-factor exchange. Nucleic Acids Research. 2015;43:129–142. doi: 10.1093/nar/gku1260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li X, Millar JS, Brownell N, Briand F, Rader DJ. Modulation of HDL metabolism by the niacin receptor GPR109A in mouse hepatocytes. Biochem Pharmacol. 2010;80:1450–1457. doi: 10.1016/j.bcp.2010.07.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin S-J, Ford E, Haigis M, Liszt G, Guarente L. Calorie restriction extends yeast life span by lowering the level of NADH. Genes & development. 2004;18:12–16. doi: 10.1101/gad.1164804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liszt G, Ford E, Kurtev M, Guarente L. Mouse Sir2 homolog SIRT6 is a nuclear ADP-ribosyltransferase. The Journal of biological chemistry. 2005;280:21313–21320. doi: 10.1074/jbc.M413296200. [DOI] [PubMed] [Google Scholar]
- Llopis J, Westin S, Ricote M, Wang Z, Cho CY, Kurokawa R, Mullen TM, Rose DW, Rosenfeld MG, Tsien RY, Glass CK, Wang J. Ligand-dependent interactions of coactivators steroid receptor coactivator-1 and peroxisome proliferator-activated receptor binding protein with nuclear hormone receptors can be imaged in live cells and are required for transcription. Proceedings of the National Academy of Sciences of the United States of America. 2000;97:4363–4368. doi: 10.1073/pnas.97.8.4363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu S-P, Kato M, Lin S-J. Assimilation of endogenous nicotinamide riboside is essential for calorie restriction-mediated life span extension in Saccharomyces cerevisiae. Journal of Biological Chemistry. 2009;284:17110–17119. doi: 10.1074/jbc.M109.004010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo J, Nikolaev AY, Imai S, Chen D, Su F, Shiloh A, Guarente L, Gu W. Negative control of p53 by Sir2alpha promotes cell survival under stress. Cell. 2001;107:137–148. doi: 10.1016/s0092-8674(01)00524-4. [DOI] [PubMed] [Google Scholar]
- Mack TG, Reiner M, Beirowski B, Mi W, Emanuelli M, Wagner D, Thomson D, Gillingwater T, Court F, Conforti L, et al. Wallerian degeneration of injured axons and synapses is delayed by a Ube4b/Nmnat chimeric gene. Nature neuroscience. 2001;4:1199–1206. doi: 10.1038/nn770. [DOI] [PubMed] [Google Scholar]
- Malavasi F, Deaglio S, Funaro A, Ferrero E, Horenstein AL, Ortolan E, Vaisitti T, Aydin S. Evolution and function of the ADP ribosyl cyclase/CD38 gene family in physiology and pathology. Physiological reviews. 2008;88:841–886. doi: 10.1152/physrev.00035.2007. [DOI] [PubMed] [Google Scholar]
- Mao Z, Hine C, Tian X, Van Meter M, Au M, Vaidya A, Seluanov A, Gorbunova V. SIRT6 promotes DNA repair under stress by activating PARP1. Science (New York, N Y) 2011;332:1443–1446. doi: 10.1126/science.1202723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Massudi H, Grant R, Braidy N, Guest J, Farnsworth B, Guillemin GJ. Age-associated changes in oxidative stress and NAD+ metabolism in human tissue. PloS one. 2012;7:e42357. doi: 10.1371/journal.pone.0042357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Masutani M, Suzuki H, Kamada N, Watanabe M, Ueda O, Nozaki T, Jishage K, Watanabe T, Sugimoto T, Nakagama H, et al. Poly(ADP-ribose) polymerase gene disruption conferred mice resistant to streptozotocin-induced diabetes. Proceedings of the National Academy of Sciences of the United States of America. 1999;96:2301–2304. doi: 10.1073/pnas.96.5.2301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mathias RA, Greco TM, Oberstein A, Budayeva HG, Chakrabarti R, Rowland EA, Kang Y, Shenk T, Cristea IM. Sirtuin 4 is a lipoamidase regulating pyruvate dehydrogenase complex activity. Cell. 2014;159:1615–1625. doi: 10.1016/j.cell.2014.11.046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKenna MC, Waagepetersen HS, Schousboe A, Sonnewald U. Neuronal and astrocytic shuttle mechanisms for cytosolic-mitochondrial transfer of reducing equivalents: current evidence and pharmacological tools. Biochemical pharmacology. 2006;71:399–407. doi: 10.1016/j.bcp.2005.10.011. [DOI] [PubMed] [Google Scholar]
- Mendoza-Alvarez H, Alvarez-Gonzalez R. Poly(ADP-ribose) polymerase is a catalytic dimer and the automodification reaction is intermolecular. The Journal of biological chemistry. 1993;268:22575–22580. [PubMed] [Google Scholar]
- Menzies KJ, Singh K, Saleem A, Hood DA. Sirtuin 1-mediated Effects of Exercise and Resveratrol on Mitochondrial Biogenesis. Journal of Biological Chemistry. 2013;288:6968–6979. doi: 10.1074/jbc.M112.431155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mercken EM, Hu J, Krzysik-Walker S, Wei M, Li Y, McBurney MW, de Cabo R, Longo VD. SIRT1 but not its increased expression is essential for lifespan extension in caloric restricted mice. Aging Cell. 2013 doi: 10.1111/acel.12151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Michishita E, Park JY, Burneskis JM, Barrett JC, Horikawa I. Evolutionarily conserved and nonconserved cellular localizations and functions of human SIRT proteins. Molecular biology of the cell. 2005;16:4623–4635. doi: 10.1091/mbc.E05-01-0033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Minor RK, Baur JA, Gomes AP, Ward TM, Csiszar A, Mercken EM, Abdelmohsen K, Shin YK, Canto C, Scheibye-Knudsen M, et al. SRT1720 improves survival and healthspan of obese mice. Sci Rep. 2011;1:70. doi: 10.1038/srep00070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mostoslavsky R, Chua KF, Lombard DB, Pang WW, Fischer MR, Gellon L, Liu P, Mostoslavsky G, Franco S, Murphy MM, et al. Genomic instability and aging-like phenotype in the absence of mammalian SIRT6. Cell. 2006;124:315–329. doi: 10.1016/j.cell.2005.11.044. [DOI] [PubMed] [Google Scholar]
- Mouchiroud L, Houtkooper RH, Moullan N, Katsyuba E, Ryu D, Cantó C, Mottis A, Jo Y-S, Viswanathan M, Schoonjans K, et al. The NAD(+)/Sirtuin Pathway Modulates Longevity through Activation of Mitochondrial UPR and FOXO Signaling. Cell. 2013;154:430–441. doi: 10.1016/j.cell.2013.06.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mukhopadhyay P, Rajesh M, Cao Z, Horváth B, Park O, Wang H, Erdelyi K, Holovac E, Wang Y, Liaudet L, et al. Poly (ADP-ribose) polymerase-1 is a key mediator of liver inflammation and fibrosis. Hepatology (Baltimore, Md) 2014;59:1998–2009. doi: 10.1002/hep.26763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakagawa T, Lomb DJ, Haigis MC, Guarente L. SIRT5 Deacetylates carbamoyl phosphate synthetase 1 and regulates the urea cycle. Cell. 2009;137:560–570. doi: 10.1016/j.cell.2009.02.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakahata Y, Kaluzova M, Grimaldi B, Sahar S, Hirayama J, Chen D, Guarente LP, Sassone-Corsi P. The NAD+-dependent deacetylase SIRT1 modulates CLOCK-mediated chromatin remodeling and circadian control. Cell. 2008;134:329–340. doi: 10.1016/j.cell.2008.07.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakahata Y, Sahar S, Astarita G, Kaluzova M, Sassone-Corsi P. Circadian control of the NAD+ salvage pathway by CLOCK-SIRT1. Science. 2009;324:654–657. doi: 10.1126/science.1170803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nam T-S, Choi SH, Rah S-Y, Kim S-Y, Jang W, Im M-J, Kwon HJ, Kim U-H. Discovery of a small-molecule inhibitor for kidney ADP-ribosyl cyclase: Implication for intracellular calcium signal mediated by cyclic ADP-ribose. Experimental & molecular medicine. 2006;38:718–726. doi: 10.1038/emm.2006.84. [DOI] [PubMed] [Google Scholar]
- Nikiforov A, Dolle C, Niere M, Ziegler M. Pathways and subcellular compartmentation of NAD biosynthesis in human cells: from entry of extracellular precursors to mitochondrial NAD generation. J Biol Chem. 2011;286:21767–21778. doi: 10.1074/jbc.M110.213298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- North BJ, Marshall BL, Borra MT, Denu JM, Verdin E. The human Sir2 ortholog, SIRT2, is an NAD+-dependent tubulin deacetylase. Molecular cell. 2003;11:437–444. doi: 10.1016/s1097-2765(03)00038-8. [DOI] [PubMed] [Google Scholar]
- Olmos PR, Hodgson MI, Maiz A, Manrique M, De Valdés MD, Foncea R, Acosta AM, Emmerich MV, Velasco S, Muñiz OP, et al. Nicotinamide protected first-phase insulin response (FPIR) and prevented clinical disease in first-degree relatives of type-1 diabetics. Diabetes research and clinical practice. 2006;71:320–333. doi: 10.1016/j.diabres.2005.07.009. [DOI] [PubMed] [Google Scholar]
- Owusu-Ansah E, Song W, Perrimon N. Muscle mitohormesis promotes longevity via systemic repression of insulin signaling. Cell. 2013;155:699–712. doi: 10.1016/j.cell.2013.09.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pacholec M, Bleasdale JE, Chrunyk B, Cunningham D, Flynn D, Garofalo RS, Griffith D, Griffor M, Loulakis P, Pabst B, et al. SRT1720, SRT2183, SRT1460, and resveratrol are not direct activators of SIRT1. Journal of Biological Chemistry. 2010;285:8340–8351. doi: 10.1074/jbc.M109.088682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pan PW, Feldman JL, Devries MK, Dong A, Edwards AM, Denu JM. Structure and biochemical functions of SIRT6. J Biol Chem. 2011;286:14575–14587. doi: 10.1074/jbc.M111.218990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pearson KJ, Baur JA, Lewis KN, Peshkin L, Price NL, Labinskyy N, Swindell WR, Kamara D, Minor RK, Perez E, et al. Resveratrol Delays Age-Related Deterioration and Mimics Transcriptional Aspects of Dietary Restriction without Extending Life Span. Cell metabolism. 2008;8:157–168. doi: 10.1016/j.cmet.2008.06.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petesch SJ, Lis JT. Activator-induced spread of poly(ADP-ribose) polymerase promotes nucleosome loss at Hsp70. Molecular cell. 2012;45:64–74. doi: 10.1016/j.molcel.2011.11.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pfluger PT, Herranz D, Velasco-Miguel S, Serrano M, Tschöp MH. Sirt1 protects against high-fat diet-induced metabolic damage. Proceedings of the National Academy of Sciences. 2008;105:9793–9798. doi: 10.1073/pnas.0802917105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pieper AA, Xie S, Capota E, Estill SJ, Zhong J, Long JM, Becker GL, Huntington P, Goldman SE, Shen C-H, et al. Discovery of a proneurogenic, neuroprotective chemical. Cell. 2010;142:39–51. doi: 10.1016/j.cell.2010.06.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pillai JB, Isbatan A, Imai S-i, Gupta MP. Poly(ADP-ribose) polymerase-1-dependent cardiac myocyte cell death during heart failure is mediated by NAD+ depletion and reduced Sir2alpha deacetylase activity. The Journal of biological chemistry. 2005;280:43121–43130. doi: 10.1074/jbc.M506162200. [DOI] [PubMed] [Google Scholar]
- Pirinen E, Canto C, Jo YS, Morato L, Zhang H, Menzies KJ, Williams EG, Mouchiroud L, Moullan N, Hagberg C, et al. Pharmacological Inhibition of poly(ADP-ribose) polymerases improves fitness and mitochondrial function in skeletal muscle. Cell Metab. 2014;19:1034–1041. doi: 10.1016/j.cmet.2014.04.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pittelli M, Felici R, Pitozzi V, Giovannelli L, Bigagli E, Cialdai F, Romano G, Moroni F, Chiarugi A. Pharmacological effects of exogenous NAD on mitochondrial bioenergetics, DNA repair, and apoptosis. Molecular pharmacology. 2011;80:1136–1146. doi: 10.1124/mol.111.073916. [DOI] [PubMed] [Google Scholar]
- Pittelli M, Formentini L, Faraco G, Lapucci A, Rapizzi E, Cialdai F, Romano G, Moneti G, Moroni F, Chiarugi A. Inhibition of nicotinamide phosphoribosyltransferase: cellular bioenergetics reveals a mitochondrial insensitive NAD pool. Journal of Biological Chemistry. 2010;285:34106–34114. doi: 10.1074/jbc.M110.136739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Price NL, Gomes AP, Ling AJY, Duarte FV, Martin-Montalvo A, North BJ, Agarwal B, Ye L, Ramadori G, Teodoro JS, et al. SIRT1 Is Required for AMPK Activation and the Beneficial Effects of Resveratrol on Mitochondrial Function. Cell metabolism. 2012;15:675–690. doi: 10.1016/j.cmet.2012.04.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qin W, Yang T, Ho L, Zhao Z, Wang J, Chen L, Zhao W, Thiyagarajan M, MacGrogan D, Rodgers JT, et al. Neuronal SIRT1 activation as a novel mechanism underlying the prevention of Alzheimer disease amyloid neuropathology by calorie restriction. The Journal of biological chemistry. 2006;281:21745–21754. doi: 10.1074/jbc.M602909200. [DOI] [PubMed] [Google Scholar]
- Quarona V, Zaccarello G, Chillemi A, Brunetti E, Singh VK, Ferrero E, Funaro A, Horenstein AL, Malavasi F. CD38 and CD157: a long journey from activation markers to multifunctional molecules. Cytometry Part B, Clinical cytometry. 2013;84:207–217. doi: 10.1002/cyto.b.21092. [DOI] [PubMed] [Google Scholar]
- Rajamohan SB, Pillai VB, Gupta M, Sundaresan NR, Birukov KG, Samant S, Hottiger MO, Gupta MP. SIRT1 promotes cell survival under stress by deacetylation-dependent deactivation of poly(ADP-ribose) polymerase 1. Molecular and cellular biology. 2009;29:4116–4129. doi: 10.1128/MCB.00121-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramsey KM, Mills KF, Satoh A, Imai S. Age-associated loss of Sirt1-mediated enhancement of glucose-stimulated insulin secretion in beta cell-specific Sirt1-overexpressing (BESTO) mice. Aging Cell. 2008;7:78–88. doi: 10.1111/j.1474-9726.2007.00355.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramsey KM, Yoshino J, Brace CS, Abrassart D, Kobayashi Y, Marcheva B, Hong HK, Chong JL, Buhr ED, Lee C, et al. Circadian clock feedback cycle through NAMPT-mediated NAD+ biosynthesis. Science. 2009;324:651–654. doi: 10.1126/science.1171641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Revollo JR, Grimm AA, Imai S-i. The NAD biosynthesis pathway mediated by nicotinamide phosphoribosyltransferase regulates Sir2 activity in mammalian cells. The Journal of biological chemistry. 2004;279:50754–50763. doi: 10.1074/jbc.M408388200. [DOI] [PubMed] [Google Scholar]
- Revollo JR, Korner A, Mills KF, Satoh A, Wang T, Garten A, Dasgupta B, Sasaki Y, Wolberger C, Townsend RR, et al. Nampt/PBEF/Visfatin regulates insulin secretion in beta cells as a systemic NAD biosynthetic enzyme. Cell Metab. 2007;6:363–375. doi: 10.1016/j.cmet.2007.09.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riederer M, Erwa W, Zimmermann R, Frank S, Zechner R. Adipose tissue as a source of nicotinamide N-methyltransferase and homocysteine. Atherosclerosis. 2009;204:412–417. doi: 10.1016/j.atherosclerosis.2008.09.015. [DOI] [PubMed] [Google Scholar]
- Rippmann JF, Damm K, Schnapp A. Functional characterization of the poly(ADP-ribose) polymerase activity of tankyrase 1 a potential regulator of telomere length. Journal of molecular biology. 2002;323:217–224. doi: 10.1016/s0022-2836(02)00946-4. [DOI] [PubMed] [Google Scholar]
- Rodgers JT, Lerin C, Haas W, Gygi SP, Spiegelman BM, Puigserver P. Nutrient control of glucose homeostasis through a complex of PGC-1alpha and SIRT1. Nature. 2005;434:113–118. doi: 10.1038/nature03354. [DOI] [PubMed] [Google Scholar]
- Rogina B, Helfand SL. Sir2 mediates longevity in the fly through a pathway related to calorie restriction. Proceedings of the National Academy of Sciences of the United States of America. 2004;101:15998–16003. doi: 10.1073/pnas.0404184101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rongvaux A, Shea RJ, Mulks MH, Gigot D, Urbain J, Leo O, Andris F. Pre-B-cell colony-enhancing factor, whose expression is up-regulated in activated lymphocytes, is a nicotinamide phosphoribosyltransferase, a cytosolic enzyme involved in NAD biosynthesis. European journal of immunology. 2002;32:3225–3234. doi: 10.1002/1521-4141(200211)32:11<3225::AID-IMMU3225>3.0.CO;2-L. [DOI] [PubMed] [Google Scholar]
- Ryall JG, Dell’Orso S, Derfoul A, Juan A, Zare H, Feng X, Clermont D, Koulnis M, Gutierrez-Cruz G, Fulco M, et al. The NAD+-Dependent SIRT1 Deacetylase Translates a Metabolic Switch into Regulatory Epigenetics in Skeletal Muscle Stem Cells. STEM. 2015:1–14. doi: 10.1016/j.stem.2014.12.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ryu D, Jo YS, Lo Sasso G, Stein S, Zhang H, Perino A, Lee JU, Zeviani M, Romand R, Hottiger MO, et al. A SIRT7-Dependent Acetylation Switch of GABPbeta1 Controls Mitochondrial Function. Cell Metab. 2014 doi: 10.1016/j.cmet.2014.08.001. [DOI] [PubMed] [Google Scholar]
- Sadanaga-Akiyoshi F, Yao H, Tanuma S-i, Nakahara T, Hong JS, Ibayashi S, Uchimura H, Fujishima M. Nicotinamide attenuates focal ischemic brain injury in rats: with special reference to changes in nicotinamide and NAD+ levels in ischemic core and penumbra. Neurochemical research. 2003;28:1227–1234. doi: 10.1023/a:1024236614015. [DOI] [PubMed] [Google Scholar]
- Sajish M, Schimmel P. A human tRNA synthetase is a potent PARP1-activating effector target for resveratrol. Nature. 2015;519:370–373. doi: 10.1038/nature14028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sasaki Y, Araki T, Milbrandt J. Stimulation of nicotinamide adenine dinucleotide biosynthetic pathways delays axonal degeneration after axotomy. The Journal of neuroscience : the official journal of the Society for Neuroscience. 2006;26:8484–8491. doi: 10.1523/JNEUROSCI.2320-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sasaki Y, Vohra BPS, Lund FE, Milbrandt J. Nicotinamide mononucleotide adenylyl transferase-mediated axonal protection requires enzymatic activity but not increased levels of neuronal nicotinamide adenine dinucleotide. The Journal of neuroscience : the official journal of the Society for Neuroscience. 2009;29:5525–5535. doi: 10.1523/JNEUROSCI.5469-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Satoh A, Brace CS, Rensing N, Cliften P, Wozniak DF, Herzog ED, Yamada KA, Imai S-i. Sirt1 extends life span and delays aging in mice through the regulation of Nk2 homeobox 1 in the DMH and LH. Cell metabolism. 2013;18:416–430. doi: 10.1016/j.cmet.2013.07.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sauve AA, Munshi C, Lee HC, Schramm VL. The reaction mechanism for CD38. A single intermediate is responsible for cyclization, hydrolysis, and base-exchange chemistries. Biochemistry. 1998;37:13239–13249. doi: 10.1021/bi981248s. [DOI] [PubMed] [Google Scholar]
- Scheibye-Knudsen M, Fang EF, Croteau DL, Bohr VA. Contribution of defective mitophagy to the neurodegeneration in DNA repair-deficient disorders. Autophagy. 2014;10:1468–1469. doi: 10.4161/auto.29321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schein PS, Cooney DA, Vernon ML. The use of nicotinamide to modify the toxicity of streptozotocin diabetes without loss of antitumor activity. Cancer research. 1967;27:2324–2332. [PubMed] [Google Scholar]
- Schmeisser K, Mansfeld J, Kuhlow D, Weimer S, Priebe S, Heiland I, Birringer M, Groth M, Segref A, Kanfi Y, et al. Role of sirtuins in lifespan regulation is linked to methylation of nicotinamide. Nat Chem Biol. 2013 doi: 10.1038/nchembio.1352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmidt MT, Smith BC, Jackson MD, Denu JM. Coenzyme specificity of Sir2 protein deacetylases: implications for physiological regulation. The Journal of biological chemistry. 2004;279:40122–40129. doi: 10.1074/jbc.M407484200. [DOI] [PubMed] [Google Scholar]
- Schwer B, North BJ, Frye RA, Ott M, Verdin E. The human silent information regulator (Sir)2 homologue hSIRT3 is a mitochondrial nicotinamide adenine dinucleotide-dependent deacetylase. The Journal of cell biology. 2002;158:647–657. doi: 10.1083/jcb.200205057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seal AJ, Creeke PI, Dibari F, Cheung E, Kyroussis E, Semedo P, van den Briel T. Low and deficient niacin status and pellagra are endemic in postwar Angola. The American journal of clinical nutrition. 2007;85:218–224. doi: 10.1093/ajcn/85.1.218. [DOI] [PubMed] [Google Scholar]
- Smith BC, Hallows WC, Denu JM. A continuous microplate assay for sirtuins and nicotinamide-producing enzymes. Analytical biochemistry. 2009;394:101–109. doi: 10.1016/j.ab.2009.07.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soga T, Kamohara M, Takasaki J, Matsumoto S, Saito T, Ohishi T, Hiyama H, Matsuo A, Matsushime H, Furuichi K. Molecular identification of nicotinic acid receptor. Biochem Biophys Res Commun. 2003;303:364–369. doi: 10.1016/s0006-291x(03)00342-5. [DOI] [PubMed] [Google Scholar]
- Someya S, Yu W, Hallows WC, Xu J, Vann JM, Leeuwenburgh C, Tanokura M, Denu JM, Prolla TA. Sirt3 mediates reduction of oxidative damage and prevention of age-related hearing loss under caloric restriction. Cell. 2010;143:802–812. doi: 10.1016/j.cell.2010.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stein LR, Imai S-i. Specific ablation of Nampt in adult neural stem cells recapitulates their functional defects during aging. The EMBO Journal. 2014;33:1321–1340. doi: 10.1002/embj.201386917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stone TW, Darlington LG. Endogenous kynurenines as targets for drug discovery and development. Nature reviews Drug discovery. 2002;1:609–620. doi: 10.1038/nrd870. [DOI] [PubMed] [Google Scholar]
- Sydenstricker VP. The history of pellagra, its recognition as a disorder of nutrition and its conquest. The American journal of clinical nutrition. 1958;6:409–414. doi: 10.1093/ajcn/6.4.409. [DOI] [PubMed] [Google Scholar]
- Szczesny B, Brunyánszki A, Olah G, Mitra S, Szabo C. Opposing roles of mitochondrial and nuclear PARP1 in the regulation of mitochondrial and nuclear DNA integrity: implications for the regulation of mitochondrial function. Nucleic Acids Research. 2014;42:13161–13173. doi: 10.1093/nar/gku1089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tan M, Peng C, Anderson KA, Chhoy P, Xie Z, Dai L, Park J, Chen Y, Huang H, Zhang Y, et al. Lysine glutarylation is a protein posttranslational modification regulated by SIRT5. Cell Metab. 2014;19:605–617. doi: 10.1016/j.cmet.2014.03.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanno M, Sakamoto J, Miura T, Shimamoto K, Horio Y. Nucleocytoplasmic Shuttling of the NAD+-dependent Histone Deacetylase SIRT1. Journal of Biological Chemistry. 2006;282:6823–6832. doi: 10.1074/jbc.M609554200. [DOI] [PubMed] [Google Scholar]
- Tesla R, Wolf HP, Xu P, Drawbridge J, Estill SJ, Huntington P, McDaniel L, Knobbe W, Burket A, Tran S, et al. Neuroprotective efficacy of aminopropyl carbazoles in a mouse model of amyotrophic lateral sclerosis. Proceedings of the National Academy of Sciences. 2012;109:17016–17021. doi: 10.1073/pnas.1213960109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tissenbaum HA, Guarente L. Increased dosage of a sir-2 gene extends lifespan in Caenorhabditis elegans. Nature. 2001;410:227–230. doi: 10.1038/35065638. [DOI] [PubMed] [Google Scholar]
- Tummala KS, Gomes AL, Yilmaz M, Graña O, Bakiri L, Ruppen I, Ximénez-Embún P, Sheshappanavar V, Rodriguez-Justo M, Pisano DG, et al. Inhibition of De Novo NAD(+) Synthesis by Oncogenic URI Causes Liver Tumorigenesis through DNA Damage. Cancer cell. 2014;26:826–839. doi: 10.1016/j.ccell.2014.10.002. [DOI] [PubMed] [Google Scholar]
- Tunaru S, Kero J, Schaub A, Wufka C, Blaukat A, Pfeffer K, Offermanns S. PUMA-G and HM74 are receptors for nicotinic acid and mediate its anti-lipolytic effect. Nature medicine. 2003;9:352–355. doi: 10.1038/nm824. [DOI] [PubMed] [Google Scholar]
- TuruncBayrakdar E, Armagan G, Uyanikgil Y, Kanit L, Koylu E, Yalcin A. Ex vivo protective effects of nicotinamide and 3-aminobenzamide on rat synaptosomes treated with Aβ(1-42) Cell biochemistry and function. 2014a doi: 10.1002/cbf.3049. [DOI] [PubMed] [Google Scholar]
- Turunc Bayrakdar E, Uyanikgil Y, Kanit L, Koylu E, Yalcin A. Nicotinamide treatment reduces the levels of oxidative stress, apoptosis, and PARP-1 activity in Aβ(1-42)-induced rat model of Alzheimer’s disease. Free radical research. 2014b;48:146–158. doi: 10.3109/10715762.2013.857018. [DOI] [PubMed] [Google Scholar]
- Upadhya R, Lee J, Willis IM. Maf1 is an essential mediator of diverse signals that repress RNA polymerase III transcription. Molecular cell. 2002;10:1489–1494. doi: 10.1016/s1097-2765(02)00787-6. [DOI] [PubMed] [Google Scholar]
- Vakhrusheva O, Smolka C, Gajawada P, Kostin S, Boettger T, Kubin T, Braun T, Bober E. Sirt7 increases stress resistance of cardiomyocytes and prevents apoptosis and inflammatory cardiomyopathy in mice. Circulation Research. 2008;102:703–710. doi: 10.1161/CIRCRESAHA.107.164558. [DOI] [PubMed] [Google Scholar]
- Valenzuela MT, Guerrero R, Núñez MI, Ruiz De Almodóvar JM, Sarker M, de Murcia G, Oliver FJ. PARP-1 modifies the effectiveness of p53-mediated DNA damage response. Oncogene. 2002;21:1108–1116. doi: 10.1038/sj.onc.1205169. [DOI] [PubMed] [Google Scholar]
- Van Beijnum JR, Moerkerk PTM, Gerbers AJ, De Bruïne AP, Arends J-W, Hoogenboom HR, Hufton SE. Target validation for genomics using peptide-specific phage antibodies: a study of five gene products overexpressed in colorectal cancer. International Journal of Cancer. 2002;101:118–127. doi: 10.1002/ijc.10584. [DOI] [PubMed] [Google Scholar]
- van de Weijer T, Phielix E, Bilet L, Williams EG, Ropelle ER, Bierwagen A, Livingstone R, Nowotny P, Sparks LM, Paglialunga S, et al. Evidence for a direct effect of the NAD+ precursor Acipimox on muscle mitochondrial function in humans. Diabetes. 2014 doi: 10.2337/db14-0667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Roermund CW, Elgersma Y, Singh N, Wanders RJ, Tabak HF. The membrane of peroxisomes in Saccharomyces cerevisiae is impermeable to NAD(H) and acetyl-CoA under in vivo conditions. EMBO J. 1995;14:3480–3486. doi: 10.1002/j.1460-2075.1995.tb07354.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vaziri H, Dessain SK, Ng Eaton E, Imai SI, Frye RA, Pandita TK, Guarente L, Weinberg RA. hSIR2(SIRT1) functions as an NAD-dependent p53 deacetylase. Cell. 2001;107:149–159. doi: 10.1016/s0092-8674(01)00527-x. [DOI] [PubMed] [Google Scholar]
- Verdin E, Hirschey MD, Finley LWS, Haigis MC. Sirtuin regulation of mitochondria: energy production, apoptosis, and signaling. Trends in biochemical sciences. 2010;35:669–675. doi: 10.1016/j.tibs.2010.07.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang B, Hasan MK, Alvarado E, Yuan H, Wu H, Chen WY. NAMPT overexpression in prostate cancer and its contribution to tumor cell survival and stress response. Oncogene. 2011;30:907–921. doi: 10.1038/onc.2010.468. [DOI] [PubMed] [Google Scholar]
- Wang G, Han T, Nijhawan D, Theodoropoulos P, Naidoo J, Yadavalli S, Mirzaei H, Pieper AA, Ready JM, McKnight SL. P7C3 neuroprotective chemicals function by activating the rate-limiting enzyme in NAD salvage. Cell. 2014a;158:1324–1334. doi: 10.1016/j.cell.2014.07.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang JT, Medress ZA, Barres BA. Axon degeneration: molecular mechanisms of a self-destruction pathway. The Journal of cell biology. 2012;196:7–18. doi: 10.1083/jcb.201108111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang L, Ding D, Salvi R, Roth JA. Nicotinamide adenine dinucleotide prevents neuroaxonal degeneration induced by manganese in cochlear organotypic cultures. Neurotoxicology. 2014b;40:65–74. doi: 10.1016/j.neuro.2013.11.007. [DOI] [PubMed] [Google Scholar]
- Wang R-H, Sengupta K, Li C, Kim H-S, Cao L, Xiao C, Kim S, Xu X, Zheng Y, Chilton B, et al. Impaired DNA damage response, genome instability, and tumorigenesis in SIRT1 mutant mice. Cancer cell. 2008;14:312–323. doi: 10.1016/j.ccr.2008.09.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Warburg O, Christian W, Griese A. Hydrogen-transferring co-enzyme, its composition and mode of functioning. Biochemische Zeitschrift. 1935;280:157–205. [Google Scholar]
- Watson M, Roulston A, Bélec L, Billot X, Marcellus R, Bédard D, Bernier C, Branchaud S, Chan H, Dairi K, et al. The small molecule GMX1778 is a potent inhibitor of NAD+ biosynthesis: strategy for enhanced therapy in nicotinic acid phosphoribosyltransferase 1-deficient tumors. Molecular and cellular biology. 2009;29:5872–5888. doi: 10.1128/MCB.00112-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams AC, Dunbar RIM. Big brains, meat, tuberculosis and the nicotinamide switches: co-evolutionary relationships with modern repercussions on longevity and disease? Medical hypotheses. 2014;83:79–87. doi: 10.1016/j.mehy.2014.04.003. [DOI] [PubMed] [Google Scholar]
- Wise A, Foord SM, Fraser NJ, Barnes AA, Elshourbagy N, Eilert M, Ignar DM, Murdock PR, Steplewski K, Green A, et al. Molecular identification of high and low affinity receptors for nicotinic acid. J Biol Chem. 2003;278:9869–9874. doi: 10.1074/jbc.M210695200. [DOI] [PubMed] [Google Scholar]
- Wu J, Zhang F, Yan M, Wu D, Yu Q, Zhang Y, Zhou B, McBurney MW, Zhai Q. WldS enhances insulin transcription and secretion via a SIRT1-dependent pathway and improves glucose homeostasis. Diabetes. 2011;60:3197–3207. doi: 10.2337/db11-0232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu Y, Williams EG, Dubuis S, Mottis A, Jovaisaite V, Houten SM, Argmann CA, Faridi P, Wolski W, Kutalik Z, et al. Multilayered genetic and omics dissection of mitochondrial activity in a mouse reference population. Cell. 2014;158:1415–1430. doi: 10.1016/j.cell.2014.07.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie G-H, Rah S-Y, Kim SJ, Nam T-S, Ha K-C, Chae S-W, Im M-J, Kim U-H. ADP-ribosyl cyclase couples to cyclic AMP signaling in the cardiomyocytes. Biochemical and biophysical research communications. 2005;330:1290–1298. doi: 10.1016/j.bbrc.2005.03.114. [DOI] [PubMed] [Google Scholar]
- Yahata N, Yuasa S, Araki T. Nicotinamide mononucleotide adenylyltransferase expression in mitochondrial matrix delays Wallerian degeneration. The Journal of neuroscience : the official journal of the Society for Neuroscience. 2009;29:6276–6284. doi: 10.1523/JNEUROSCI.4304-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yalowitz JA, Xiao S, Biju MP, Antony AC, Cummings OW, Deeg MA, Jayaram HN. Characterization of human brain nicotinamide 5’-mononucleotide adenylyltransferase-2 and expression in human pancreas. Biochem J. 2004;377:317–326. doi: 10.1042/BJ20030518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamamoto H, Uchigata Y, Okamoto H. Streptozotocin and alloxan induce DNA strand breaks and poly(ADP-ribose) synthetase in pancreatic islets. Nature. 1981;294:284–286. doi: 10.1038/294284a0. [DOI] [PubMed] [Google Scholar]
- Yan T, Feng Y, Zheng J, Ge X, Zhang Y, Wu D, Zhao J, Zhai Q. Nmnat2 delays axon degeneration in superior cervical ganglia dependent on its NAD synthesis activity. Neurochemistry international. 2010;56:101–106. doi: 10.1016/j.neuint.2009.09.007. [DOI] [PubMed] [Google Scholar]
- Yang H, Yang T, Baur JA, Perez E, Matsui T, Carmona JJ, Lamming DW, Souza-Pinto NC, Bohr VA, Rosenzweig A, et al. Nutrient-sensitive mitochondrial NAD+ levels dictate cell survival. Cell. 2007a;130:1095–1107. doi: 10.1016/j.cell.2007.07.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang SJ, Choi JM, Kim L, Park SE, Rhee EJ, Lee WY, Oh KW, Park SW, Park C-Y. Nicotinamide improves glucose metabolism and affects the hepatic NAD-sirtuin pathway in a rodent model of obesity and type 2 diabetes. The Journal of nutritional biochemistry. 2014;25:66–72. doi: 10.1016/j.jnutbio.2013.09.004. [DOI] [PubMed] [Google Scholar]
- Yang T, Chan NY, Sauve AA. Syntheses of nicotinamide riboside and derivatives: effective agents for increasing nicotinamide adenine dinucleotide concentrations in mammalian cells. J Med Chem. 2007b;50:6458–6461. doi: 10.1021/jm701001c. [DOI] [PubMed] [Google Scholar]
- Yeung F, Hoberg JE, Ramsey CS, Keller MD, Jones DR, Frye RA, Mayo MW. Modulation of NF-kappaB-dependent transcription and cell survival by the SIRT1 deacetylase. The EMBO Journal. 2004;23:2369–2380. doi: 10.1038/sj.emboj.7600244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yin TC, Britt JK, De Jesús-Cortés H, Lu Y, Genova RM, Khan MZ, Voorhees JR, Shao J, Katzman AC, Huntington PJ, et al. P7C3 Neuroprotective Chemicals Block Axonal Degeneration and Preserve Function after Traumatic Brain Injury. Cell reports. 2014 doi: 10.1016/j.celrep.2014.08.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ying W. NAD+/NADH and NADP+/NADPH in cellular functions and cell death: regulation and biological consequences. Antioxidants & Redox Signaling. 2008;10:179–206. doi: 10.1089/ars.2007.1672. [DOI] [PubMed] [Google Scholar]
- Ying W, Alano CC, Garnier P, Swanson RA. NAD+ as a metabolic link between DNA damage and cell death. Journal of neuroscience research. 2005;79:216–223. doi: 10.1002/jnr.20289. [DOI] [PubMed] [Google Scholar]
- Yoon MJ, Yoshida M, Johnson S, Takikawa A, Usui I, Tobe K, Nakagawa T, Yoshino J, Imai S-i. SIRT1-Mediated eNAMPT Secretion from Adipose Tissue Regulates Hypothalamic NAD(+) and Function in Mice. Cell metabolism. 2015 doi: 10.1016/j.cmet.2015.04.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshino J, Mills KF, Yoon MJ, Imai S. Nicotinamide mononucleotide, a key NAD(+) intermediate, treats the pathophysiology of diet- and age-induced diabetes in mice. Cell Metab. 2011;14:528–536. doi: 10.1016/j.cmet.2011.08.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu J, Sadhukhan S, Noriega LG, Moullan N, He B, Weiss RS, Lin H, Schoonjans K, Auwerx J. Metabolic characterization of a Sirt5 deficient mouse model. Scientific reports. 2013;3:2806. doi: 10.1038/srep02806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang F, Xie R, Munoz FM, Lau SS, Monks TJ. PARP-1 Hyperactivation and Reciprocal Elevations in Intracellular Ca2+ During ROS-Induced Nonapoptotic Cell Death. Toxicological sciences : an official journal of the Society of Toxicology. 2014;140:118–134. doi: 10.1093/toxsci/kfu073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang LQ, Heruth DP, Ye SQ. Nicotinamide Phosphoribosyltransferase in Human Diseases. Journal of bioanalysis & biomedicine. 2011;3:13–25. doi: 10.4172/1948-593X.1000038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Q, Piston DW, Goodman RH. Regulation of corepressor function by nuclear NADH. Science (New York, N Y) 2002;295:1895–1897. doi: 10.1126/science.1069300. [DOI] [PubMed] [Google Scholar]
- Zhang T, Berrocal JG, Frizzell KM, Gamble MJ, DuMond ME, Krishnakumar R, Yang T, Sauve AA, Kraus WL. Enzymes in the NAD+ salvage pathway regulate SIRT1 activity at target gene promoters. J Biol Chem. 2009;284:20408–20417. doi: 10.1074/jbc.M109.016469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang T, Berrocal JG, Yao J, DuMond ME, Krishnakumar R, Ruhl DD, Ryu KW, Gamble MJ, Kraus WL. Regulation of Poly(ADP-ribose) Polymerase-1-dependent Gene Expression through Promoter-directed Recruitment of a Nuclear NAD(+) Synthase. The Journal of biological chemistry. 2012;287:12405–12416. doi: 10.1074/jbc.M111.304469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang X, Kurnasov OV, Karthikeyan S, Grishin NV, Osterman AL, Zhang H. Structural characterization of a human cytosolic NMN/NaMN adenylyltransferase and implication in human NAD biosynthesis. J Biol Chem. 2003;278:13503–13511. doi: 10.1074/jbc.M300073200. [DOI] [PubMed] [Google Scholar]
- Zhang Y, Schmidt RJ, Foxworthy P, Emkey R, Oler JK, Large TH, Wang H, Su EW, Mosior MK, Eacho PI, et al. Niacin mediates lipolysis in adipose tissue through its G-protein coupled receptor HM74A. Biochemical and biophysical research communications. 2005;334:729–732. doi: 10.1016/j.bbrc.2005.06.141. [DOI] [PubMed] [Google Scholar]
- Zhao YJ, Lam CMC, Lee HC. The membrane-bound enzyme CD38 exists in two opposing orientations. Science Signaling. 2012;5:ra67. doi: 10.1126/scisignal.2002700. [DOI] [PubMed] [Google Scholar]
- Zhong L, D’Urso A, Toiber D, Sebastian C, Henry RE, Vadysirisack DD, Guimaraes A, Marinelli B, Wikstrom JD, Nir T, et al. The histone deacetylase Sirt6 regulates glucose homeostasis via Hif1alpha. Cell. 2010;140:280–293. doi: 10.1016/j.cell.2009.12.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou M, Ottenberg G, Sferrazza GF, Hubbs C, Fallahi M, Rumbaugh G, Brantley AF, Lasmézas CI. Neuronal death induced by misfolded prion protein is due to NAD+ depletion and can be relieved in vitro and in vivo by NAD+ replenishment. Brain : a journal of neurology. 2015;138:992–1008. doi: 10.1093/brain/awv002. [DOI] [PMC free article] [PubMed] [Google Scholar]