


Abstract
The functional characterization of miRNAs is still an open challenge. Here, we present DIANA-miRPath v3.0 (http://www.microrna.gr/miRPathv3) an online software suite dedicated to the assessment of miRNA regulatory roles and the identification of controlled pathways. The new miRPath web server renders possible the functional annotation of one or more miRNAs using standard (hypergeometric distributions), unbiased empirical distributions and/or meta-analysis statistics. DIANA-miRPath v3.0 database and functionality have been significantly extended to support all analyses for KEGG molecular pathways, as well as multiple slices of Gene Ontology (GO) in seven species (Homo sapiens, Mus musculus, Rattus norvegicus, Drosophila melanogaster, Caenorhabditis elegans, Gallus gallus and Danio rerio). Importantly, more than 600 000 experimentally supported miRNA targets from DIANA-TarBase v7.0 have been incorporated into the new schema. Users of DIANA-miRPath v3.0 can harness this wealth of information and substitute or combine the available in silico predicted targets from DIANA-microT-CDS and/or TargetScan v6.2 with high quality experimentally supported interactions. A unique feature of DIANA-miRPath v3.0 is its redesigned Reverse Search module, which enables users to identify and visualize miRNAs significantly controlling selected pathways or belonging to specific GO categories based on in silico or experimental data. DIANA-miRPath v3.0 is freely available to all users without any login requirement.
INTRODUCTION
microRNAs (miRNAs) are short (∼23nt) non-coding transcripts that act as potent post-transcriptional regulators of gene expression. miRNAs identify their target RNAs through sequence complementarity and guide the RNA-induced silencing complex (RISC) in order to induce cleavage, degradation and/or translation suppression in the case of protein coding genes (1). miRNAs exhibit a central regulatory role in animals and plants, controlling core biological processes and mechanisms. They are also actively researched as biomarkers and/or therapeutic targets for their involvement in numerous pathologies including cardiovascular diseases, pathogen infections, metabolic disorders and malignancies (2).
In silico miRNA target prediction algorithms have been proven invaluable tools for the elucidation of miRNA function. Currently available state-of-the-art implementations can identify miRNA:gene interactions in 3′ UTR as well as CDS regions, using complex physical models and/or machine learning approaches (2,3). However, even the most advanced methods still require experimental validation, since they exhibit a high number of false positive results. To this end, numerous low yield and high throughput wet lab techniques have been developed, that can be used to validate, explore and/or complement predicted results (4).
These approaches have revealed the complex functional roles of miRNAs. Each miRNA can control up to dozens of genes, while multiple miRNAs have been also shown to collaborate in targeting extensive cellular processes and molecular pathways (5,6). The high number of miRNAs (e.g. in Homo sapiens already exceed 2500) poses a significant bottleneck to the elucidation of their functional impact. Multiple targets have to be taken into account, which can be present in numerous pathways. The complexity of the problem increases when assessing the combinatorial effect of multiple miRNAs. A series of functional analysis web servers and packages have been developed, in order to assist in the assessment of the functional impact of miRNAs on biological processes and pathways (2). Some of the most commonly used applications, algorithms or methodologies include DIANA-miRPath (7), CORNA (8), miRTar (9), miTalos (10), the miRNA function module of StarBase (11) or an enrichment analysis using miRNA targets in DAVID (12).
The field is constantly evolving and surpassing impeding obstacles. However, a series of open problems still exists. A major hindrance is the lack of extensive experimentally validated miRNA:gene interaction datasets, which forces most available implementations to rely solely on in silico predicted interactions. As previously mentioned, even the most advanced miRNA target prediction algorithms exhibit high false positive rates (2). miRNA:gene interactions form the foundation of such implementations and biases present in the prediction algorithms can be subsequently introduced to the derived results. Until now there are no available implementations providing miRNA:gene interaction datasets on a scale comparable to in silico predictions. Recently, Bleazar et al. also challenged the standard enrichment statistics approach (one-sided Fisher's exact test), which is a fundamental aspect of such algorithms (13). In their work they have shown cases of bias existing in relevant enrichment analyses in Gene Ontology (GO) (14) and Kyoto Encyclopedia of Genes and Genomes (KEGG) pathways (15) using standard approaches. They have stressed the necessity for unbiased statistics that can derive biologically meaningful results and have proposed the use of empirical distributions, which bypasses the ‘preference’ of the hypergeometric test toward specific categories or pathways.
Finally, the assessment of combined miRNA action is still a topic of active research, since miRNA functional analyses are often performed following miRNA differential expression experiments. However, the simple algebraic combination of miRNA targets can often result to sets comprising thousands of genes that hinder the derivation of accurate statistics. Furthermore, by forming a superset of target genes, the information that some genes are repressed by multiple miRNAs is lost. Such scenarios are common, especially when performing functional analyses of differentially expressed miRNAs in a Next Generation Sequencing (NGS) or microarray experiment. For instance, the 39 miRNA test set analyzed by Bleazard et al. —which is a realistic number of miRNAs in a differential analysis setting- comprised ∼10 000 gene targets, a number comparable to the ∼15 000 annotated genes in GO (13). This high number of targeted genes can reduce the biological significance of the functional analysis, since in the case of the example, two out of three genes in the GO gene universe are targeted. There is still a need for online implementations that can elegantly combine large numbers of miRNAs, while providing unbiased statistics.
In this manuscript, we present DIANA-miRPath v3.0, the new version of the DIANA-miRPath functional analysis online suite, which hosts numerous novel features, extensions and optimizations, including:
The use of predicted interactions derived from DIANA-microT-CDS (3) and TargetScan 6.2 (16), as well as more than 600 000 experimentally supported interactions derived from DIANA-TarBase v7.0 (17); enabling for the first time experimentally supported miRNA functional annotation on a similar scale to in silico predictions.
A new redesigned statistics engine that supports standard enrichment statistics (hypergeometric distributions), unbiased empirical distributions and/or meta-analysis statistics.
A significant extension to the annotation database, enabling DIANA-miRPath v3.0 users to not only identify miRNAs controlling molecular pathways but also to perform miRNA function annotation using GO or GOSlim terms (14), as well as to design publication-quality advanced visualizations.
A new Reverse Search Module with unprecedented flexibility that can assist in (re)-discovering miRNAs with not yet identified functions.
Support for seven model species: H. sapiens, Mus musculus, Rattus norvegicus, Drosophila melanogaster, Caenorhabditis elegans, Gallus gallus and Danio rerio.
The user interface has also been extended to support novel functionalities, while offering significantly more options than previous implementations. Its design, architecture and philosophy have been maintained, in order to minimize the learning curve of the transition to the new version for returning users. Specific details on the implemented algorithms, user interface and database schema are presented in detail in the following sections. Table 1 depicts a concise comparison between DIANA-miRPath v3.0 and relevant web servers.
Table 1. Comparison of included data, as well as basic and advanced features of online miRNA functional analysis tools.
| miRPath | miRTar | miTalos | starbase - miRFunction | |
|---|---|---|---|---|
| Species | Human, mouse, rat, fly, worm, chicken, zebrafish | Human | Human, mouse | Human, mouse |
| miRNAs for human | 2,588 | 1,100 | 1,529 | 277 |
| In Silico Interactions | microT-CDS, TargetScan v6 | miRTar | TargetscanS, miRanda/mirSVR | TargetScan v6, PicTar, PITA, miRanda, RNA22 |
| Experimental Interactions | TarBase v7.0 | StarBase v2.0 | StarBase v2.0 | |
| Analysis using only Experimental Data | ✓ | |||
| Annotation | KEGG/GO, GO MF, GO BP, GO CC, GOSlim | KEGG | KEGG, NCI PID | KEGG/GO MF, GO CC, GO BP/BioCarta/Panther and others |
| Fisher's Exact Test | ✓ | ✓ | ✓ | ✓ |
| Unbiased Empirical Test | ✓ | |||
| Union of Targets | ✓ | ✓ | ✓ | |
| Intersection/Soft Intersection of Targets | ✓ | |||
| Pathways Meta-Analysis | ✓ | |||
| Visualization of Targets in Pathways | ✓ | ✓ | ✓ | |
| Clustering/Dendrograms | ✓ | |||
| miRNA functional Heatmaps | ✓ | |||
| Expression Filters | ✓ (free filter) | ✓ (Pre-calculated expression filters) | ✓ | |
| Pathogenic SNPs in miRNA binding Sites | ✓ | |||
| Reverse Search | ✓ | |||
| Version | v3.0 | Accessed (April 2015) | Accessed (April 2015) | v2.0 |
METHODS AND RESULTS
Database schema
DIANA-miRPath v3.0 database has been extended to support features such as microRNA nomenclature history (18), a novel miRNA/gene name suggestion mechanism, as well as analysis support for seven species (H. sapiens, M. musculus, R. norvegicus, D. melanogaster, C. elegans, G. gallus and D. rerio). The new database schema incorporates KEGG pathways, as well as GO and GOSlim annotations, enabling functional annotation of miRNAs and miRNA combinations using all datasets, or their subsets (GO cellular component, biological processes or molecular function). Gene and miRNA annotations are derived from Ensembl (19) and miRBase (20), respectively. Single nucleotide polymorphism locations and pathogenicity are derived from dbSNP (21).
miRNA:gene interactions are derived from the in silico miRNA target prediction algorithms: DIANA-microT-CDS and TargetScan 6.2, the latter in both Context+ and Conservation modes. DIANA-microT-CDS is the fifth version of the microT algorithm (3). It is a highly accurate target prediction algorithm trained against CLIP-Seq datasets, enabling target prediction in 3′ UTR and CDS mRNA regions. The user of DIANA-miRPath v3.0 can also utilize experimentally supported interactions from DIANA-TarBase v.7.0. TarBase v7.0 incorporates more than half a million experimentally supported miRNA:gene interactions derived from hundreds of publications and more than 150 CLIP-Seq libraries (17). The number of indexed interactions is 9–250-fold higher compared to any other manually curated database. The user of miRPath v3.0 can harness this wealth of information and substitute or combine in silico predicted targets with high quality experimentally validated interactions. Currently, this functionality is supported for H. sapiens and M. musculus and C. elegans, since most relevant wet-lab experiments correspond to these species. As more experimental data become available for other organisms in DIANA-TarBase, the experimentally supported functional analysis module will be further extended.
Algorithms and statistical methodologies
Implementation
DIANA-miRPath v3.0 interface has been implemented using PHP and JavaScript utilizing standardized web design patterns, like the Model-View-Controller model. Analysis algorithms are implemented in R. All miRNA:gene interactions, annotation and metadata are stored in a MySQL relational database. Database indexing and caching techniques were used to accelerate query processing. Extensive interim results and queries that are often requested by the analysis algorithms are pre-calculated and stored for future uses, in order to diminish result calculation time and enable an application-like user experience, even in the most sophisticated analysis settings.
Statistics
DIANA-miRPath v3.0 extends the Fisher's Exact Test, EASE score (22) and False Discovery Rate (23) methodologies that were available in the statistics engine of the second version (24), with the use of unbiased empirical distributions. DIANA-miRPath implements an adaptation of the sampling algorithm presented by Bleazard et al. (13). In brief, the algorithm samples without replacement from the set of all annotated miRNAs and extracts an empirical P-value, based on the proportion of simulations that produces an equal or greater KEGG/GO pathway/term overlap. The use of empirical distributions has been shown to change the scope of testing from gene level back to miRNA level and is robust against statistical biases present in GO or KEGG annotations (13).
DIANA-miRPath v3.0 extends this testing methodology with its meta-analysis statistics for the assessment of combined miRNA action. The meta-analysis algorithm enables the identification of pathways controlled by multiple miRNAs by examining each miRNA individually and subsequently combining the result probabilities and test statistics, as in meta-analysis studies (24). This approach avoids the aforementioned pitfalls, while promoting pathways which are targeted concurrently by multiple miRNAs.
The statistics engine is highly customizable, depending on user needs. For instance, the user of DIANA-miRPath v3.0 can select between standard or robust statistics, combine results in gene (genes union or intersection) or pathways level (pathways union/intersection) in order to meet the specific requirements encountered in different research settings (e.g. exploratory versus focused functional analyses).
Reverse-search module
The new Reverse-Search Module enables users to perform powerful top-to-bottom queries: i.e. to identify miRNAs controlling specific pathways or those that belong to a specific GO category. DIANA-miRPath v3.0 users can select a pathway or GO category of interest from a suggestion-enabled free-text search box, as well the in silico prediction algorithm for the detection of miRNA targets (microT-CDS or TargetScan 6.2). The web server identifies miRNAs targeting the selected pathway and ranks them according to their enrichment P-values. DIANA-miRPath v3.0 returns a list of targeted genes per miRNA belonging to the selected pathway, as well as a visualization of the pathway with special notations on targeted genes. Importantly, the incorporation of TarBase v7.0 targets in miRPath v3.0 allows users to perform an extensive functional meta-analysis of numerous NGS datasets and publications, in order to identify miRNAs targeting the selected pathway or belonging to a specific GO category based on experimental findings.
User interface, results presentation and visualizations
The user interface has been enhanced to support standard or sophisticated analyses using KEGG pathways or GO/GOSlim categories in real-time (Figure 1). DIANA-miRPath v3.0 users can select miRNAs for analysis, the source of interactions (microT-CDS/Targetscan or TarBase), parameterize predicted interactions (prediction threshold, use of conservation), choose the merging algorithm (genes or pathways level) and the statistics engine (Hypergeometric or Empirical distributions). Users can also decide whether to use more conservative statistics (DAVID's EASE score (22)), as well as to control error rate due to multiple comparisons (Benjamini-Hochberg's FDR (23)). Furthermore, DIANA-miRPath enables the restriction of results and analyses only to an expressed mRNA subset.
Figure 1.
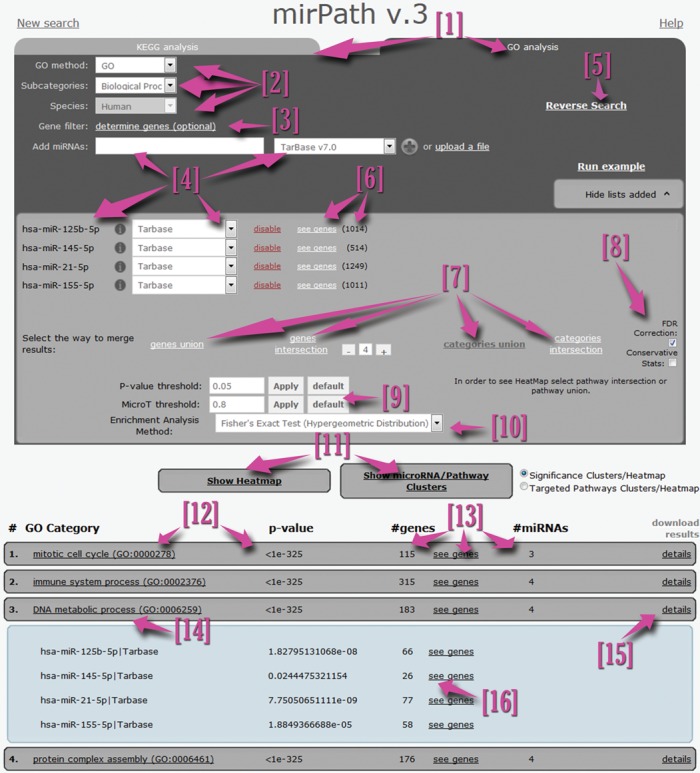
DIANA-miRPath v3.0 main user interface. Users can perform miRNA functional analyses in real-time using KEGG or Gene Ontology annotations [1]. Basic options [2] can be utilized to select the species or annotation subset to be included in the analysis. Users can restrain the interactions and statistics only to an expressed genes subset which can be uploaded using the optional Expressed genes filter menu [3]. Each miRNA and interactions dataset can be entered individually or by using a set up file [4]. Following the selection of miRNAs, further information regarding identified interactions and the targeted genes is presented in the miRNA matrix [6]. DIANA-miRPath v3.0 users can subsequently select the result merging algorithm [7], advanced statistics options [8], thresholds for predictions and p-values [9], as well as the main statistics engine [10]. Options for advanced visualizations are presented below [11]. The results pane [12] shows information regarding targeted pathways/enriched GO categories, p-values, as well as the number of miRNAs and genes present in each term [13]. All links are active and lead to further information [14]. The details pane [15] offers information for each miRNA, individual targets and p-values [16]. The Reverse Search module [5] is always accessible directly from the main user interface.
The web server returns the outcome of the analysis in real-time. The user interface caters results in text format (targeted gene lists, enriched pathways and P-values), figures (pathway diagrams with marked targeted genes) and meta-data (miRNA/gene information, GO descriptions, hierarchy/ancestor diagrams), as well as active links to relevant database entries from miRBase, KEGG, Ensembl, GONuts (25), AmiGO (26) and QuickGO (27). It incorporates active links for miRNA, gene and functional related terms. Further information and metadata including miRNA MeSH term tag clouds, KEGG pathway graphs, interactive GO Visualizations from OLSVis (28) and graphs, are also supported. miRNA target lists have now been extended with prediction scores and specific notations for experimental support in cases of in silico predictions, while TarBase targets are accompanied with a description of the utilized validation method. DIANA-miRPath v3.0 acts also as a miRNA research hub, enabling users to extend their queries to other DIANA tools. For instance, these tools can be used to identify all predicted/experimentally supported targets for a specific miRNA in mRNAs or lncRNAs using microT, TarBase and lncBase (29), respectively. The user of miRPath v3.0 can also utilize the SNP module to identify pathogenic SNPs on in silico predicted binding sites.
The new visualization module is now able to support the creation of publication-quality miRNA/pathway/GO/GOSlim dendrograms and miRNA versus GO/GOSlim/KEGG entries heatmaps (Figure 2). The latter functionalities are powerful exploratory tools, enabling the identification of miRNAs belonging to similar functional categories or the identification of categories/pathways characterized or controlled by similar miRNAs. The interface of the Reverse Search module has also been redesigned and extended with novel functionalities (Figure 3). The new version provides enrichment statistics, as well as targeting visualizations, in order to assist in the assessment of miRNA function in selected pathways or GO categories. A detailed use-case example can be found in the provided Supporting material.
Figure 2.
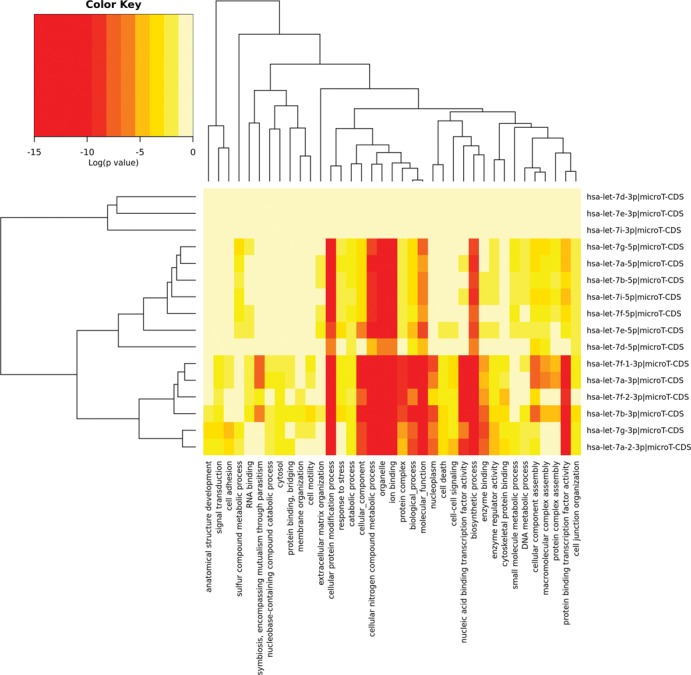
A miRNA versus GOSlim categories heatmap created directly from the DIANA-miRPath v3.0 interface. The heatmap depicts the level of enrichment in GO categories of various let-7 family members in Homo sapiens. The heatmap enables the identification of miRNA subclasses or GO terms that characterize similar miRNAs, since they are clustered together.
Figure 3.
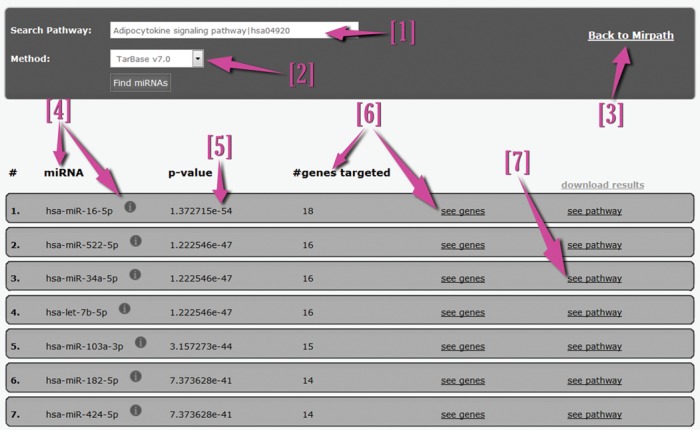
The new Reverse Search module. From the new interface, users can select the pathway/GO category of interest [1] and the interactions database [2]. The Reverse search module presents miRNAs [4], enrichment P-values [5] and targeted genes [6]. The links and icons of miRNAs and genes are active and lead to further information and metadata. The pathway visualization link [7] leads to a KEGG pathway map depicting the selected pathway, as well as the miRNA targets with a special color notation.
CONCLUSION
As we learn more about miRNA:gene interactions, the in silico analysis tools and applications mature and grow, in order to support more demanding research scenarios. The third version of DIANA-miRPath combines leading state of the art target prediction algorithms, with the most extensive manually curated miRNA:gene interaction dataset to date, in order to chart the miRNA target search space. The new user interface enables extensive parameterization and tailor-made analyses, with selection options spanning from prediction thresholds to settings deep under the hood of the statistics engine. The latter has been significantly redesigned, in order to support novel statistics methodologies that are based on empirical distributions and do not suffer from the biases observed in standard approaches.
We believe that the new miRPath version is designed to accommodate diverse research needs that require accurate functional characterization of one or more microRNAs. The incorporation of KEGG pathways and multiple gene ontologies, as well as numerous links to DIANA and external tools or databases, meta-data, SNP analysis modules, clustering algorithms and advanced visualizations, render DIANA-miRPath v3.0 as an one-stop hub for miRNA research projects. Importantly, the new Reverse Search module can be used to identify novel mechanisms and research targets by meta-analyzing in silico predictions or vast experimental datasets. This unique feature enables miRNA functional analysis tools to be also utilized as a first exploratory research step, as well as a companion along a research endeavor.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
‘TOM’ (4580), ‘ARISTEIA’ Action of the ‘OPERATIONAL PROGRAMME EDUCATION AND LIFELONG LEARNING’; European Social Fund (ESF) and National Resources; John S. Latsis Public Benefit Foundation; ‘MEDA’ (MIS 448842), Development Grants For Research Institutions – KRIPIS, General Secretariat for Research and Technology, Ministry of Education, Greece; European Regional Development Fund.
Conflict of interest statement. None declared.
REFERENCES
- 1.Huntzinger E., Izaurralde E. Gene silencing by microRNAs: contributions of translational repression and mRNA decay. Nat. Rev. Genet. 2011;12:99–110. doi: 10.1038/nrg2936. [DOI] [PubMed] [Google Scholar]
- 2.Vlachos I.S., Hatzigeorgiou A.G. Online resources for miRNA analysis. Clin. Biochem. 2013;46:879–900. doi: 10.1016/j.clinbiochem.2013.03.006. [DOI] [PubMed] [Google Scholar]
- 3.Paraskevopoulou M.D., Georgakilas G., Kostoulas N., Vlachos I.S., Vergoulis T., Reczko M., Filippidis C., Dalamagas T., Hatzigeorgiou A.G. DIANA-microT web server v5.0: service integration into miRNA functional analysis workflows. Nucleic Acids Res. 2013;41:W169–W173. doi: 10.1093/nar/gkt393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Thomson D.W., Bracken C.P., Goodall G.J. Experimental strategies for microRNA target identification. Nucleic Acids Res. 2011;39:6845–6853. doi: 10.1093/nar/gkr330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Olive V., Jiang I., He L. mir-17–92, a cluster of miRNAs in the midst of the cancer network. Int. J. Biochem. Cell Biol. 2010;42:1348–1354. doi: 10.1016/j.biocel.2010.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Peter M.E. Targeting of mRNAs by multiple miRNAs: the next step. Oncogene. 2010;29:2161–2164. doi: 10.1038/onc.2010.59. [DOI] [PubMed] [Google Scholar]
- 7.Papadopoulos G.L., Alexiou P., Maragkakis M., Reczko M., Hatzigeorgiou A.G. DIANA-mirPath: Integrating human and mouse microRNAs in pathways. Bioinformatics. 2009;25:1991–1993. doi: 10.1093/bioinformatics/btp299. [DOI] [PubMed] [Google Scholar]
- 8.Wu X., Watson M. CORNA: testing gene lists for regulation by microRNAs. Bioinformatics. 2009;25:832–833. doi: 10.1093/bioinformatics/btp059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hsu J.B., Chiu C.M., Hsu S.D., Huang W.Y., Chien C.H., Lee T.Y., Huang H.D. miRTar: an integrated system for identifying miRNA-target interactions in human. BMC Bioinformatics. 2011;12:300. doi: 10.1186/1471-2105-12-300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kowarsch A., Preusse M., Marr C., Theis F.J. miTALOS: analyzing the tissue-specific regulation of signaling pathways by human and mouse microRNAs. RNA. 2011;17:809–819. doi: 10.1261/rna.2474511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Li J.-H., Liu S., Zhou H., Qu L.-H., Yang J.-H. starBase v2.0: decoding miRNA-ceRNA, miRNA-ncRNA and protein–RNA interaction networks from large-scale CLIP-Seq data. Nucleic Acids Res. 2014;42:D92–D97. doi: 10.1093/nar/gkt1248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Huang da W., Sherman B.T., Lempicki R.A. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat. Protoc. 2009;4:44–57. doi: 10.1038/nprot.2008.211. [DOI] [PubMed] [Google Scholar]
- 13.Bleazard T., Lamb J.A., Griffiths-Jones S. Bias in microRNA functional enrichment analysis. Bioinformatics. 2015 doi: 10.1093/bioinformatics/btv023. doi:10.1093/bioinformatics/btv023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ashburner M., Ball C.A., Blake J.A., Botstein D., Butler H., Cherry J.M., Davis A.P., Dolinski K., Dwight S.S., Eppig J.T., et al. Gene ontology: tool for the unification of biology. The Gene Ontology Consortium. Nat. Genet. 2000;25:25–29. doi: 10.1038/75556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kanehisa M., Goto S., Sato Y., Kawashima M., Furumichi M., Tanabe M. Data, information, knowledge and principle: back to metabolism in KEGG. Nucleic Acids Res. 2014;42:D199–205. doi: 10.1093/nar/gkt1076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Garcia D.M., Baek D., Shin C., Bell G.W., Grimson A., Bartel D.P. Weak seed-pairing stability and high target-site abundance decrease the proficiency of lsy-6 and other microRNAs. Nat. Struct. Mol. Biol. 2011;18:1139–1146. doi: 10.1038/nsmb.2115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Vlachos I.S., Paraskevopoulou M.D., Karagkouni D., Georgakilas G., Vergoulis T., Kanellos I., Anastasopoulos I.-L., Maniou S., Karathanou K., Kalfakakou D. DIANA-TarBase v7.0: indexing more than half a million experimentally supported miRNA:mRNA interactions. Nucleic Acids Res. 2015;43:D153–D159. doi: 10.1093/nar/gku1215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Vergoulis T., Kanellos I., Kostoulas N., Georgakilas G., Sellis T., Hatzigeorgiou A., Dalamagas T. mirPub: a database for searching microRNA publications. Bioinformatics. 2014 doi: 10.1093/bioinformatics/btu819. doi:10.1093/bioinformatics/btu819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Cunningham F., Amode M.R., Barrell D., Beal K., Billis K., Brent S., Carvalho-Silva D., Clapham P., Coates G., Fitzgerald S., et al. Ensembl 2015. Nucleic Acids Res. 2015;43:D662–D669. doi: 10.1093/nar/gku1010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kozomara A., Griffiths-Jones S. miRBase: annotating high confidence microRNAs using deep sequencing data. Nucleic Acids Res. 2014;42:D68–D73. doi: 10.1093/nar/gkt1181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Sherry S.T., Ward M.H., Kholodov M., Baker J., Phan L., Smigielski E.M., Sirotkin K. dbSNP: the NCBI database of genetic variation. Nucleic Acids Res. 2001;29:308–311. doi: 10.1093/nar/29.1.308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hosack D.A., Dennis G. Jr, Sherman B.T., Lane H.C., Lempicki R.A. Identifying biological themes within lists of genes with EASE. Genome Biol. 2003;4:R70. doi: 10.1186/gb-2003-4-10-r70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Benjamini Y., Hochberg Y. Controlling the False Discovery Rate: a practical and powerful approach to multiple testing. J. Roy. Stat. Soc. Ser. B (Methodological) 1995;57:289–300. [Google Scholar]
- 24.Vlachos I.S., Kostoulas N., Vergoulis T., Georgakilas G., Reczko M., Maragkakis M., Paraskevopoulou M.D., Prionidis K., Dalamagas T., Hatzigeorgiou A.G. DIANA miRPath v.2.0: investigating the combinatorial effect of microRNAs in pathways. Nucleic Acids Res. 2012;40:W498–W504. doi: 10.1093/nar/gks494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Renfro D.P., McIntosh B.K., Venkatraman A., Siegele D.A., Hu J.C. GONUTS: the Gene Ontology Normal Usage Tracking System. Nucleic Acids Res. 2011;40:D1262–D1269. doi: 10.1093/nar/gkr907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Carbon S., Ireland A., Mungall C.J., Shu S., Marshall B., Lewis S., Ami G.O.H., Web Presence Working, G. AmiGO: online access to ontology and annotation data. Bioinformatics. 2009;25:288–289. doi: 10.1093/bioinformatics/btn615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Binns D., Dimmer E., Huntley R., Barrell D., O'Donovan C., Apweiler R. QuickGO: a web-based tool for Gene Ontology searching. Bioinformatics. 2009;25:3045–3046. doi: 10.1093/bioinformatics/btp536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Vercruysse S., Venkatesan A., Kuiper M. OLSVis: an animated, interactive visual browser for bio-ontologies. BMC Bioinformatics. 2012;13:116. doi: 10.1186/1471-2105-13-116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Paraskevopoulou M.D., Georgakilas G., Kostoulas N., Reczko M., Maragkakis M., Dalamagas T.M., Hatzigeorgiou A.G. DIANA-LncBase: experimentally verified and computationally predicted microRNA targets on long non-coding RNAs. Nucleic Acids Res. 2013;41:D239–D245. doi: 10.1093/nar/gks1246. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.