

Abstract
Nearly all of the genetic material among cells within an organism is identical. However, single nucleotide variants (SNVs), indels, copy number variants (CNVs), and other structural variants (SVs) continually accumulate as cells divide during development. This process results in an organism composed of countless cells, each with its own unique personal genome. Thus, every human is undoubtedly mosaic. Mosaic mutations can go unnoticed, underlie genetic disease or normal human variation, and may be transmitted to the next generation as constitutional variants. Here, we review the influence of the developmental timing of mutations, the mechanisms by which they arise, methods for detecting mosaic variants, and the risk of passing these mutations on to the next generation.
Keywords: mosaicism, somatic mosaicism, post-zygotic mutation, transmission genetics, recurrence risk
Introduction
A fundamental principle of biology is that the DNA blueprint of a multicellular organism is identical among cells within the organism. The term mosaicism describes a violation of this principle that arises when specific cells within a developing organism mutate to develop two or more cell populations with distinct genotypes. Mosaicism stands in contrast to chimerism, the phenomenon of an individual being composed of the products of two or more fertilization events. Multiple lines of evidence suggest that mutation during mitosis is a common event, with DNA changes going uncorrected on average every two cell divisions to even multiple mutations per division [1–3]. Given the sheer number of mitoses, as many as 1016 that are required to generate an adult human composed of approximately 1014 cells [4], it is likely that cells exist within all of us harboring countless mutations that could potentially be causative of every human genetic disease. Yet almost none of these mutational events appear to affect organismal health. If detrimental to cellular function, mutant cells may be removed by apoptosis or immune surveillance. Otherwise, mutations may occur in a tissue where expression of the mutant gene is not relevant to function or disease, or mutant cells do not reach a proportion relevant to manifestation of a disease trait.
The exponential pace of cellular expansion during embryogenesis means that mutations occurring early during development have the most meaningful impact on the phenotype of an individual [5]. Mutations later in life, however, can transform cells with malignant potential, a process fundamental to cancer. This review focuses on non-oncologic phenotypes and processes. Conceptually, mosaicism can be categorized based on the distribution of mutant cells within the individual: somatic, germline and gonosomal (see glossary). Although molecular testing or observation of affected offspring can positively assign somatic, germline or combined mosaicism status, definitively ruling out mosaicism in either the somatic or germ cell compartment is challenging with current technology. Below, we explore how the timing, mechanisms, and developmental histories of post-zygotic mutations influence human health, disease, and potential risk of disease transmission.
Developmental Timing of Mutation
The precise timing during development when a mutation occurs strongly influences the distribution and phenotypic effects of mutant cells. In the most extreme case, a mutation can occur during the initial mitosis, resulting in approximately half of the cells in the individual harboring the new mutation (Figure 1A)—identical twins discordant for a dominant disorder caused by a new mutation might represent another manifestation of such a phenomenon [6]. Other signs of early embryologic mutational events may directly manifest in the observed phenotype. Individuals with congenital hemidysplasia with ichthyosiform erythroderma and limb defects (CHILD) syndrome exhibit striking demarcation of affected and unaffected tissue along the midline (Figure 2A) [7]. The exact time of left-right separation in humans is unknown [8]; in mice it appears to occur near the 8-cell stage [9]. If mutant cells arise before determination and are present on both sides of the left-right axis, tissues on both sides of the individual can be affected, potentially including one or both gonads (Figure 1B). By contrast, mutations occurring after the fate of the left and right sides of the embryo have become determined result in cells and phenotypes that are less likely to cross the midline (Figure 1C). In such individuals, only one gonad, if either, is likely to harbor mutations.
Figure 1.

The timing of post-zygotic mutation influences the distribution of mutant cells in the individual. A) Mutations that occur during the first mitosis result in approximately half of the individual being affected. Individuals with CHILD have been observed with this striking pattern (see Figure 2A). B) Mutations that occur before left-right determination can affect both sides of the individual, including one or both gonads. C) Mutations that arise after the determination of the two sides of the embryo can be confined to only one side of the individual. Only one gonad is likely to be effected. D) Mutations that occur after differentiation of primordial germ cells (PGCs) will be absence from somatic tissues. Thus, molecular investigations to detect such gonadal mosaicism must involve direct observation of germ cells. For males, this process is relatively straight forward, but for females it involves invasive biopsy of potentially both ovaries.
Figure 2.
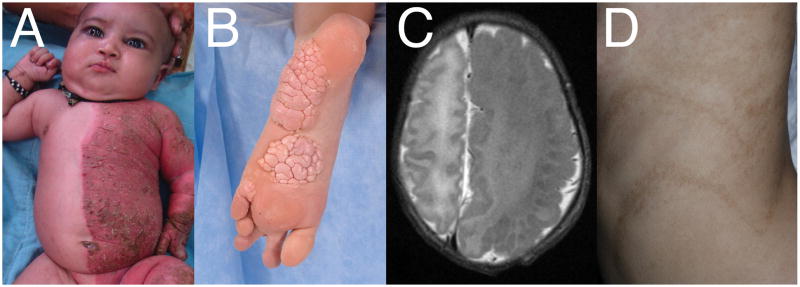
Phenotypic manifestations of mosaic mutations. A) Inflammatory nevus affecting the left side of the body of a 1-month-old individual with CHILD syndrome. Note the striking demarcation at the midline. Reproduced with permission from Chander et al. [7] B) Cerebriform connective tissue nevus on the plantar surface of the foot in an 11-year-old individual with Proteus syndrome. Reproduced with permission from Beachkofsky et al. [104]. C) Axial T2-weighted image showing markedly enlarged left cerebral hemisphere in a newborn with hemimegalencephaly. Reproduced with permission from Lang et al.[105]. D) Hyperpigmentation following lines of Blashko in an individual with linear and whorled nevoid hypermelanosis. Reproduced with permission from Molho-Pessach and Schaffer [106].
The observation of many individuals with affected tissues on both left and right sides suggests that left-right determination in humans may occur later during development or that migrational boundaries are less definitive. An alternative hypothesis is that earlier mitoses are more prone to mutation. Studies suggest that such a heightened mutagenic process or reduced repair and surveillance of genome instability may occur during very early development [10,11]. Up to 70% of human embryos display CNV or whole chromosomal aneuploidies in at least one blastomere during the first week of embryogenesis [11]. Transcription is not fully activated in humans until the 8-cell stage [12]; during this time, the embryo relies on maternally derived cytoplasmic and nuclear proteins while new transcription and translation machinery is being produced. A shortage of sufficient replication, error checking, or repair proteins may result in increased genomic instability and mutability.
Other embryologic milestones have considerable impact on the distribution of mutant cells in an individual. Primordial germ cells (PGCs), early cells that ultimately give rise to eggs or sperm, migrate out of the body of the embryo by the 24th day post-fertilization (Figure 1D) [13,14]. Later, PGCs migrate back into the gonadal ridge to populate the ovary or testis with germ cells. Mutations that occur before PGC differentiation, prior to ~15 mitotic divisions [15,16], can be present in both somatic and germ tissues. Mutations that occur after this key developmental period are confined to either the somatic or germinal lineage. Thus, testing for the timing of a mutation may provide insight into the abundance of mutant cells and, by extension, potential recurrence risk for that same mutation to be transmitted to multiple offspring [17].
Placental mosaicism
Another consideration in the origin and timing of mosaicism is the development of the placenta, which in mammals arises from cells derived from the embryo. In mice, a small minority of blastocyst cells ultimately give rise to the developing embryo and fetus; the remaining cells produce the placenta [18]. Our understanding of early human placental development is limited [19] but, conceptually, mutations that arise before placental determination may be confined to the placenta. The advent of prenatal testing of placental chorionic villi, as early as 10 weeks gestation, revealed that placental mosaicism occurs in 1–2% of samplings. Whether this mosaicism is the result of higher mutation rates early in development [11], an active segregation of mutant cells away from the embryo, or reduced selective pressure on the placenta versus development of the ultimate organism remains to be determined. Testing of amniotic fluid, primarily derived from the embryo and obtained later at amniocentesis, shows these mutations are confined to the placenta in the majority of cases [19]. Some cells from the placenta or developing fetus persist in the mother for decades [20], a phenomenon known as microchimerism, and should be distinguished from true mosaicism.
Major Molecular Classes of Mosaicism
Chromosomal Aneuploidy and Large-Scale Structural Abnormalities
Only trisomy of chromosomes 13, 18, 21 and X and monosomy of X are compatible with human life in the constitutional state. A broader range of aneuploidies has been observed in the mosaic state. These include trisomy 7, 8, 9, 12, 14, 15, 16, 17, 20 and 22 and monosomy 7, 18 and 21, each with varying prevalence, percentages of cells affected, and phenotypic features. Whole chromosomal aneuploidy occurs via nondisjunction with meiotic risk increasing with maternal age. Mosaic aneuploidy is the result of a combination of meiotic nondisjunction rescued by post-zygotic loss or copy of a whole chromosome, which can in turn lead to uniparental disomy (UPD) in the euploid cell line [21] or alternatively due to post-zygotic nondisjunction. Two other large-scale abnormalities that are observed in the mosaic state are ring chromosomes and isochromosomes. A notable mosaic abnormality involving isochromosomes is that of 12p, which causes Pallister-Killian syndrome (MIM#601803). Isochromosome 12p is only observed in the mosaic state, presumably because such an abnormality is lethal constitutionally.
Copy-number Variants and other Structural Variation
The techniques that elucidated the importance of constitutional CNVs to human health have increased sensitivity and revealed mosaicism for up to a few percent of all disease-associated mutations [21–24]. The observed frequency of mosaicism for disease-associated CNV is a balance between the rate of post-zygotic mutagenesis and milder mosaicism-associated phenotypes that evade clinical ascertainment. The vast majority of recurrent CNVs arise due to nonallelic homologous recombination (NAHR) between flanking low-copy repeats (LCRs). NAHR typically occurs during meiosis [25]. The apparent lack of paternal age effect on risk of classic genomic disorders such as Angelman (MIM#105830) and Smith-Magenis (MIM#182290) syndromes together with minimal bias in the parent of origin of recurrent CNVs [26] strengthen the hypothesis that most recurrent CNVs are of meiotic origin. Nonetheless, occasionally, CNVs that appear to be mediated by LCRs have been observed in the mosaic state [27]. The multi-copy nature of LCRs hampers detailed study of these mosaic CNVs. Thus, how many mosaic recurrent CNVs merely appear to be NAHR-mediated but are instead formed by other mechanisms remains unknown. Furthermore, the proportion of mosaic CNVs that actually result from post-zygotic mitotic recombination resulting in a loss of heterozygosity (LOH) rather than a post-zygotic new mutation has not been fully explored. Such mitotic LOH events, however, are known to occur in humans [28] and mice [29].
Nonrecurrent CNVs appear to form by two major mechanistic classes: replicative mechanisms and nonhomologous end joining (NHEJ). Although details of the proposed replicative mechanisms differ, common features emerge: long distance template switching, stalled replication (potentially resulting in collapsed forks generating one ended, double stranded DNA breaks), and stretches of short homology that facilitate template switching. Whichever mechanism of mutation transpires, because errors occur during DNA replication, mosaicism can arise. NHEJ, meanwhile, occurs throughout the cell cycle, and thus also results in mosaicism. Studies using a variety of techniques consistently find mosaic nonrecurrent CNVs in 0.5–5% of individuals with genomic disorders [21,23,30], underscoring the mitotic nature of these events.
Single Nucleotide Changes and Small Insertions and Deletions
Single nucleotide variants (SNVs) occur in six basic varieties: C-G➤T-A, T-A➤C-G (transitions) and C-G➤A-T, C-G➤G-C, T-A➤A-T, T-A➤G-C (transversions). Studies of post-zygotic mutations in tumors show that environmental stressors result in distinct mutational signatures [31–33]. A small study of mosaic SNVs in three phenotypically unremarkable individuals without cancer suggests that their distribution in healthy tissues are similar to mutations in cancer [34]. Thus, the environment an individual experiences in utero may influence the risk for specific types of mosaicism. A mutational class of similar scale to SNV is small (< ~100–200 bps) insertions and deletions (indels). Despite accounting for ~20% of disease causing mutations, the mechanisms that result in indels are more poorly understood. Conceivably, any polymerase can generate indels, but some specific human polymerases are experimentally observed to produce at least small indels at much higher rates [35]. Utilization of alternative polymerases due to damage or stress during development may influence the distribution of mosaic indels.
Trinucleotide and other repeat expansion and contraction
Expansions of tri- or rarely tetra- nucleotide repeats and other simple sequence repeats underlie at least 20 human genetic diseases [36]. In these diseases, wild-type length repeat alleles first expand to pre-mutation alleles, which in turn can rapidly expand to longer deleterious alleles. Repeats that occur in coding sequences tend to expand in the paternal germ line, suggesting a mitotic, replicative mechanism. Repeats that occur in non-coding regions, meanwhile, expand mostly in oogonia [37,38]. The molecular mechanism underlying this contrasting pattern is the focus of ongoing study. Nonetheless, somatic variation in both coding and non-coding trinucleotide repeat lengths has been observed. Somatic expansion of the coding trinucleotide repeat in the HD gene is associated with earlier age of onset of Huntington disease [39]. Monozygotic twins discordant for noncoding FMR1 repeat length have been reported [40], suggesting contraction also occurs mitotically. The extent to which somatic variation, and thus mosaicism, for nucleotide repeats influences disease expressivity and transmission is likely underappreciated.
Facioscapulohumeral muscular dystrophy (FSHD) is caused by contraction of a 3.3 kb repeat on chromosome 6, termed D4Z4 [41]. The exact mechanism of pathogenesis is unknown, but decreased methylation and repressive chromatin marks near a particular haplotype of the adjacent DUX4 gene are likely related [42,43]. One interesting feature of FSHD is that 20–40% of new mutations are post-zygotic and at relatively high mutant cell fractions [44,45]. Thus, the genome may be particularly susceptible to D4Z4 contraction early during embryogenesis. How many other loci and classes of mutation follow a similar pattern [11] remains a question of particular interest.
Autonomous mobile elements insertions
Despite original classification as “junk DNA,” the functional impact of the over half of the human genome that is comprised of repetitive elements, including mobile elements, is becoming clearer. For example, some mobile elements, particularly LINE-1, retain their intrinsic transposition capability and are able to move throughout the genome. Thus, during the life of an organism, elements are copied and pasted among the chromosomes through a replicative transposition mechanism [46], resulting in mosaicism. Recent analyses of adult cortical neurons reveal somatic mosaicism for mobile element insertions [47,48]. However, the scale of somatic retrotransposition events remains debated, with estimates varying widely from 0.6 [49] to 800 [50] insertions per neuron. Constitutional retrotransposition events can cause Mendelian disease by disrupting coding sequence, altering splicing, and even by exerting subtle positional effects [51]. Deleterious mosaic forms of the same lesions may also exist.
Detection of Mosaicism
Cytogenetics and Fluorescent in situ Hybridization
Microscopic evaluation of banded chromosomes allowed the earliest molecular observations of mosaicism in human disease [52]. Banded chromosomes were also the first genome-wide test but have poor resolution (~5–10 Mb), limiting the types of lesions that can be detected. Fluorescent in situ hybridization (FISH) allows analysis of large numbers of interphase cells for CNVs as small as 50 kb. Identifying mosaicism, then, is an exercise in the number of cells visualized, but excluding even high levels of mosaicism requires counting more cells than is typically performed in diagnostics [53]. Identifying mosaicism for submicroscopic duplication CNVs by FISH can be particularly challenging owing to potentially confounding replication of the locus being interrogated during interphase [54].
Sanger Sequencing and Pyrosequencing
Sanger sequencing ignited the genomic era and elucidated many human genetic diseases caused by point mutations and indels. Sequencing DNA isolated from a mosaic tissue (see Box 1) offers the possibility of detecting a mutation. However, recent investigations underscore the limited detection abilities of Sanger sequencing with regards to mosaicism. Mosaicism for dominant alleles present in less than 25–35% or greater than 65–70% of cells can remain undetected [55]. Thus, the use of Sanger sequencing as the gold standard may need to be reconsidered. Pyrosequencing technology has the potential to offer better minimum detection limits (as low as 5%) with more quantitative results [56]. However, it can be impeded by sequence features of the target, specifically repetition of the same nucleotide. Special amplification techniques such as COLD-PCR can improve minimum detection limits [57], but assay design for arbitrary new mutations can be time consuming and quantitation is lost.
Box 1. Tissue Sampling.
A major obstacle to detecting mosaicism is testing a tissue sample that actually harbors the mutation. Most human genetic research and diagnostics are conducted on peripheral blood DNA because of ease of access and isolation. Unfortunately, blood cells are an unstable source of genetic material given multiple rounds of self-renewal during hematopoiesis. The diversity of the clonal lineages that give rise to circulating blood cells appears to decrease with age [91,92]. Thus, selective effects of mutations, negative or positive, can skew mutation frequency in blood compared to the rest of the organism. For example, isochromosome 12p is often undetectable in blood, but examination of cells of ectodermal origin frequently reveals the mosaic aneuploidy [93]. Culturing cell lines derived from individuals present similar challenges, not only because cells accumulate mutations as they undergo mitosis [94], but also because culture conditions exert selective forces. To overcome these limitations, other or multiple sources of primarily obtained DNA can be examined. Ectodermal tissues can be sampled from buccal brushings or hair root bulbs, mesodermal tissues are available from blood or saliva, while endodermal origin DNA is available from urothelial cells collected in urine samples. For male individuals, sperm samples may be particularly informative for recurrence risk assessment. Levels of mosaicism across tissues and body locations can show surprising variability, even within the same embryonic lineage [56]. This variation likely results from interplay between mutation timing, cell migration and determination during development, and tissue-specific cell-autonomous selective effects.
Array CGH and SNP Microarrays
Array comparative genomic hybridization (aCGH) and SNP microarray techniques ushered the switch of human and medical genetics from locus-specific to genome-wide assays. In addition to revealing the contribution of CNVs to human disease, they also led to appreciation of pathogenic mosaic CNVs. Under ideal conditions, CNV mosaicism can be identified by aCGH at levels as low as 10–20% [23,24,30]. SNP microarrays, meanwhile, measure the allele frequency of alternative alleles at polymorphic loci. Resolution is limited by availability of known polymorphic SNPs, but by searching for slight deviation from expected allele frequencies (0, 0.5, and 1), mosaicism for CNVs of modest size can be detected as low as 5% [58,59].
Massively Parallel Sequencing
Massively parallel sequencing revolutionized human genetic research and the practice of medical genetics and clinical genomics [60], allowing researchers and diagnosticians to test nearly the entire human genome with a single experiment. This power and convenience has allowed whole exome sequencing (WES) to become a front line clinical test [61,62]. These methods also dramatically altered the ability of researchers to observe mosaicism. Although the chemistries of sequencing approaches differ [63], the output is largely the same: digital sequence reads of DNA ranging in size from 50 bp to a few kb. Modestly low levels of mosaicism can be identified using general-purpose sequencing reagents [64]. But even using sophisticated Bayesian models to analyze sequence data [34], identifying mosaic calls among heterozygous variants and sequencing errors remains challenging. Despite limitations, standard exome sequencing has delineated the molecular underpinnings of some of the most recalcitrant mosaic disorders [65,66]. In general, though, the ability to detect mosaicism is proportional to the read depth of coverage or number of reads that are available covering a given base position.
Single-Cell Sequencing
The technologies described above utilize pooled DNA isolated from multiple cells; they produce data that reflect the overall average of the genomes of the constituent cells. By contrast, single-cell sequencing interrogates the personal genomes of individual cells. Single cells are isolated and their genomic DNA amplified by techniques such as multiplex displacement amplification (MDA) [67,68]. Massively parallel sequencing of the product together with computational algorithms to account for biases introduced during amplification [69] have allowed researchers to observe mosaicism at the cellular level. Recent single-cell sequencing studies reveal wide-spread mosaicism in apparently normal tissues [48,69,70]. Assessing the sensitivity and cost-effectiveness of single-cell sequencing to detect mosaicism for disease causing mutations, as well as overcoming technical challenges, is the focus of ongoing research.
Personalized Assays
If mosaicism is suspected at a specific locus or in a set of suspected genes, techniques can focus particular attention on variant alleles potentially occurring at these locations. Suspicion could arise from the individual’s phenotype or observation of a mutation in an offspring. For massively parallel sequencing, custom-capture reagents, which enrich the DNA library to be sequenced for template molecules of interest, allow for greater sensitivity. Capturing template from lists of candidate genes has pushed lower boundaries of detection [55]. Custom-capture reagents that multiplex private variations across clans [71] may be useful to explore very low-level mosaicism for variants identified in families in the future.
Structural variation, including deletion, duplication, translocation and inversions, brings DNA sequences that are normally disjointed into close proximity, generating novel breakpoint junctions (Figure 3A). If the fusion of a variant is known, it is often possible to design a simple PCR assay that amplifies DNA from a mutated allele but results in no or alternative amplification from the wild-type allele (Figure 3B). Under ideal conditions, these assays can detect mosaicism at ratios of 1:10,000 or even lower [17]. Such assays can be combined with “digital” PCR technology to add quantification to the detection of mosaicism [17].
Figure 3.

Personalized assays for detection of mosaicism. A) Structural variants including deletions, duplications and inversions result in two genomic loci that are normally located far apart coming close proximity. B) Researchers can design PCR primers that are capable of amplifying across the breakpoint of the SV. Genomic DNA from individuals harboring only normal alleles do not amplify the breakpoint junction. Meanwhile, individuals with mosaicism for the SV produce the junction because of exponential amplification from the rare allele. C) “Digital” droplet PCR improves detection of mosaicism by segregating wild-type and mutant alleles into individual droplets. Fluorescent probes specific for the mutant allele can anneal and are cleaved by DNA polymerase resulting in a fluorescent droplet. The number of positive fluorescent droplets is then detected by a droplet reader. D) Molecular inversion probes (MIPs) can isolate particular regions of interest for increased scrutiny. Linear probes are developed to anneal upstream and downstream of a target region. Polymerase and ligase fills in the gap to form circular DNA. Exonuclease treatment degrades linear genomic DNA. Further library preparation and massively parallel sequencing then assesses mosaicism.
Digital PCR technology can also identify mosaic SNVs and indels. Genomic DNA is partitioned into aqueous compartments such as droplets so that, on average, less than one target template is present in each. Fluorescent probes specific for the mutation anneal to mutant templates and are cleaved by DNA polymerase resulting in fluorescence (Figure 3C). Sensitivity and specificity are improved because each compartment, on average, contains only one type of target DNA—mutant or wild-type. Using this technique, post-zygotic mutations are identified at levels as low as 0.1% [72].
Molecular inversion probes (MIPs) can isolate particular regions of the genome harboring a suspected mosaic mutation. These linear DNA probes anneal both upstream and downstream of a region of interest (Figure 3D). DNA polymerase and ligase fills the gap between the MIP ends resulting in a circular molecule encoding the genotype of the target. Probes can be tiled and annealed to both plus and minus strands to increase sensitivity. Barcoded MIPs are subsequently multiplexed and subjected to massively parallel sequencing [73,74]. MIPs have been used successfully to identify mutant DNA at levels as low at 0.5% [74].
Phenotypic Manifestations of Mosaicism
Mosaic Presentations of Mendelian Disease
Mosaic presentations of Mendelian disease provide insight into the phenotypic effects of mosaicism. Haploinsufficiency of PAFAH1B1 underlies classic lissencephaly, a structural brain abnormality that results in smooth cortical surfaces. The resultant phenotype is severe cognitive impairment, refractory seizures and spastic quadriparesis. However, if PAFAH1B1 mutations occur post-zygotically, the phenotype can be much less severe [75]. A mother carrying a causative PAFAH1B1 mutation harbored by her son in ~25% of peripheral blood cells was reported with normal intellect and brain MRI but had seizures that were well controlled pharmacologically [76]. Interestingly, a mosaic PAFAH1B1 deletion has been observed adjacent to constitutional deletion of the nearby YWHAE gene within a single individual [77], exemplifying the complexity of mutational processes.
A wide array of Mendelian disorders have been observed in the mosaic state, including Duchene muscular dystrophy, hemophilia, Marfan, and ornithine transcarbamylase deficiency [5]. Some diseases, such as Cornelia de Lange syndrome, show markedly high rates of mosaicism, even across mutational mechanisms and genomic loci [78,79]. Rett syndrome provides another example of how mosaicism modulates Mendelian disease. Caused by mutations in the X-linked gene MECP2, Rett syndrome results in developmental regression, cognitive impairment, seizures and stereotypical hand movements in girls; mutations are lethal to hemizygous males. In rare cases, mosaicism for MECP2 mutations can result in classic Rett syndrome in boys, such as in a reported case with ~37% mosaicism for a truncating mutation [80]. Mosaicism for increased APP copy number has been identified in the brains of individuals with Alzheimer’s dementia [81], suggesting that mosaicism for mutations that cause Mendelian disease when present constitutionally may also contribute to sporadic disease.
Diseases that Manifest only in the Mosaic State
Much as MECP2 mutations are lethal for males, some mutations are lethal constitutionally regardless of sex. A poignant example is Proteus syndrome, which causes distorting overgrowth, bone abnormalities and cerebriform connective tissue nevi (Figure 2B). The overgrowth is caused by recurrent E17L mutations of the AKT1 gene [82]. No constitutional mutations have been detected, and there is no known intergenerational transmission. This striking syndrome was delineated by massively parallel sequencing of affected and unaffected tissues within individuals [65]. Similar approaches described a host of overgrowth syndromes caused by activating mutations of genes in similar pathways [83,84]. For example, activating mutations in PIK3CA, AKT3 and MTOR are associated with hemimegalencephaly [85], an overgrowth of one side of the brain (Figure 2C). Mutations were detected in as few as 8% of cells in affected brain [85], underscoring their powerful effect on trait manifestation and the challenge of detection. Many of these same mutations are found in tumors, presumably exerting the same growth effects to promote malignancy.
Normal Human Variation
“Normal phenotypic variation” between individuals, such as slight deviations of body symmetry and pigmentation, can often be noted by astute observers. These differences can be attributed to stochastic molecular processes, environmental factors, or physical events in utero or later in life. This variation may also be due in part to mosaicism. Individuals with linear and whorled nevoid hypermelanosis have sections of skin with increased pigmentation (Figure 2D). Careful genomic investigation of these areas can often identify chromosomal or CNV mosaicism that encompasses known pigmentation genes [86]. The majority of individuals have no extra-cutaneous manifestations. Other traits and imperfections that make us each unique may be influenced by somatic genome variation. Perhaps such high-level phenotypes as personality may be determined at least somewhat by mosaicism. The central nervous system (CNS) appears to be among the tissues most likely to experience mosaic aneuploidy [87]. Aneuploid neurons appear to be fully functional [88], but whether such genome variation is merely a byproduct of a mutable genome or an evolutionary answer to increase diversity in the CNS is less clear.
Paternal Bias of New Mutations
A significant majority of de novo SNVs [89] and nonrecurrent CNVs [26] arise on the chromosome inherited from an individual’s father. This bias shifts further towards paternal inheritance as a father’s age at conception of their child increases. A reasonable hypothesis for these observations is that most mutations occur during mitosis and that the paternal germ lineage is exposed to more mitoses and associated mitotic mutational risk. During embryogenesis, PGCs divide producing millions of germ cells that populate the primitive ovary or testis [90]. In females, primary oocytes become arrested in prophase of meiosis I in utero, thus ending exposure to additional mitotic errors. In males, starting at puberty, spermatogonia undergo mitosis every 16 days to produce more sperm precursors and self-renew [16]. As a man ages, his germ cells are continually exposed to more mitotic risk; an average sperm ejaculated from a 30-year-old male has experienced approximately 400 mitoses, an order of magnitude more mitoses than a typical oocyte [15,16]. Box 2 explores how these sexual dimorphisms may influence the transmission of post-zygotic mutations.
Box 2. Mosaicism in Transmission Genetics.
Constitutional mutations, the vast majority of which occur during meiosis, effectively have a single chance to be passed on to the next generation. However, mutations arise following DNA replication and failed repair, a process that occurs during mitosis as well as meiosis. Unless the meiotic mutation rate is many orders of magnitude higher than the mitotic rate, which is inconsistent with the paternal origin bias of new mutations [89], then many apparently de novo mutations generated more than one mutant gamete. Thus, mosaicism in one apparently healthy parent of a child with new constitutional mutations is likely common.
This hypothesis was tested in a cohort of 100 family trios with a simplex case of genetic disease associated with a genomic deletion. Four phenotypically unremarkable parents were identified with mosaicism for the same private CNV mutation [17]. This likely represents an underestimate, as only a single tissue type was tested. Although only genomic deletions were tested, other types of mosaic mutations likely exist in phenotypically unremarkable parents.
Thus, the idea of the extreme rarity of recurrence of de novo mutations must be reconsidered in light of our understanding of post-zygotic mutations. An important first consideration is the parent of origin of apparently de novo mutations. Mathematical analyses of the sexual dimorphisms of gametogenesis suggest that new mutations occurring on the maternal allele are more likely to be recurrently transmitted to offspring [95]. Although maternal origin mutations are a minority of all new mutations [89], they are more likely to have expanded and be shared across many germ cell precursors because the female germ line does not experience prolonged self-renewal. New X-linked recessive mutations provide a window into this process, because affected boys necessarily harbor the causative mutation on their mother’s X-chromosome. Indeed, apparently de novo X-linked recessive conditions show unexplained high levels of recurrence [95,96]. New mutations on paternal alleles are expected to recur less frequently, particularly for older fathers, because mutations likely arise during the self-renewal phase of spermatogenesis. Such mutations do not expand into large numbers of clonally related mutant sperm.
Somatic mosaicism also provides insight into mutation timing. To be present in somatic tissues and transmitted to an offspring, mutations must have arisen before the differentiation of the germ cells (Figure 1). Mathematical modeling suggests that because somatically mosaic mutations necessarily occurred earlier, average somatically mosaic parents are at two to three orders of magnitude higher recurrence risk than parents with only germline mutations [95]. Moreover, despite asymmetry in the parent of origin of new mutations, early embryogenesis events are similar between sexes, and thus mosaic mothers and fathers are predicted to be at roughly equivalent risks.
Implications for counseling
Our current limited ability to intervene in the natural history of many genetic diseases means that recurrence risk counseling is among the most effective interventions that can be provided in medical genetics. Many couples with a child severely affected with a simplex genetic disease prefer to avoid the added hardship of another affected child. Empiric recurrence risk estimates are only available for the most prevalent and well-studied diseases. More in-depth understanding of mosaicism, including the use of mathematical models (see Box 3), together with more widespread screening for mosaicism may improve recurrence risk estimation. For couples interested in having additional children, determining the parent of origin of a new mutation my serve to reassure or trigger additional studies. Similarly, testing with one or more of the sensitive, personalized molecular assays discussed above may identify couples with a somatically mosaic parent where preimplantation or prenatal diagnostics could potentially be made available.
Box 3. Mathematical Models of Mosaicism.
Understanding the implications of mosaicism and recurrence risk is challenging without a formal model; the large numbers of cells involved and subtle selective forces at work are difficult to conceptualize. An effective mathematical model of mosaicism should consider the emergence, persistence, and sampling properties of mosaic variation in clonal lineages arising during mitosis. Some of the earliest mathematical analyses of mosaicism and recurrence risk consisted of simple exponential expansion (Figure IA) [97]. Although simplistic, the importance of this formalism should not be understated. Branching process models, which explicitly consider systems of reproducing particles organized into lineages of ancestral cells and their offspring, are more advanced approaches. Multi-type Galton-Watson (GW) processes permit modeling of mosaicism in a finite type space (for example wildtype, mutant a, mutant b, etc.) in a discrete number of cell generations (Figure IB).
One of the earliest GW models to capture the dynamics of mosaicism was introduced to consider deletions of the mitochondrial genome [98]. Subsequent work determined the rate of accumulation of mutants, the expected proportion of mutants at each cell division and the variance of that proportion [99]. More recently, the model was extended to apply to analysis of mosaicism in transmission genetics and specifically modeled sexual dimorphism in gametogenesis [17,95]. The framework of the model allows for predictions that are intuitively reasonable, but difficult to arrive at without the mathematical formalism because of the complex interplay between different factors. These factors can be explored online at http://www.recurrencerisk.org.
A more complex extension of branching process models are continuous time branching processes [100,101]. These continuous time models allow cell division times to vary (Figure IC). Such continuous time models can incorporate more sources of variability for the study of mosaicism and enable a more complete framework to approach biological reality. These models require specification of the cell life length distribution in addition to the mutation parameters discussed above. The analysis of continuous time models, however, is more mathematically challenging. Biological studies using continuous time models have been applied for the analysis of passenger and driver mutations in cancer [102] and modeling variation in cell life-lengths among oligodendrocytes [103].
The application of continuous time models to mosaicism may prove fruitful in the near future. However, even these advanced models fail to account for bottlenecks and density dependent growth that occur during cell fate determination. Such a model would rely on a detailed knowledge of human embryogenesis. Therefore, to fully appreciate the process of mutations expanding through clonal lineages and being transmitted, we will need to improve not only our understanding of the earliest stages of human development in utero but also investigate novel stochastic process frameworks.
Figure I.
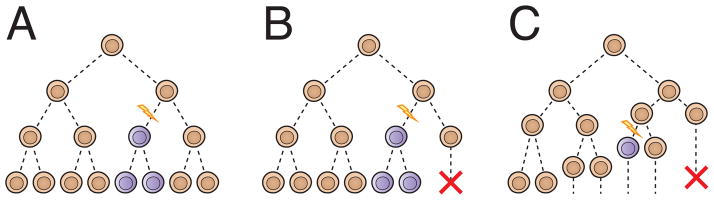
Use of stochastic process models to study mosaicism. A) A simple exponential model of the emergence of mutations. All cells divide into exactly two cells during each of a predetermined number of mitoses. Mutations are equally likely to occur during each division. B) Galton-Watson model. Cells may divide, give rise to one mutant and one wildtype or expire without dividing during each of a predetermined number of divisions. Fitness can vary with genotype and mutation rate can vary over the process of development. Two or more such models can be combined to model sexual dimorphisms. C) Continuous time branching process model. Cells undergo mitoses after a time determined by a distribution. Thus any two cells may have experienced varying numbers of divisions.
Concluding Remarks
The explosion of massively parallel sequencing data and the difficulty of variant interpretation often raises a rhetorical question, “we don’t even understand constitutional variation, do we really have to worry about the mosaic genome?” Recent studies have shown that mutations that exist only in the mosaic state can have profound effects on an individual’s phenotype. Moreover, studies of random new mutations reveal that a considerable proportion of these changes occurred in the previous generation as post-zygotic events. Thus, considering mitotic mutations is vital to a full view of human genetics. Improved technologies for identifying mosaicism against the noise of the average DNA content of an individual will hopefully allow us to more fully appreciate the vast genetic variation within our own organismal and cellular personal genomes and how these variations influence our lives.
Highlights.
Post-zygotic mutation is a common occurrence.
The developmental stage and timing of new mutations influences phenotypic effects and chance of transmission.
All major classes of mutations are observed in the mosaic state.
Mathematical modeling of mosaicism can inform estimates of recurrence risk for new mutations.
Acknowledgments
We thank Christine R. Beck and Priya Sivaramakrishnan for critical review of the manuscript. IMC is a fellow of the BCM Medical Scientist Training Program (T32 GM007330) and was supported by a fellowship from the National Institute of Neurological Disorders and Stroke (F31 NS083159). This work was also supported in part by grants from the Intellectual and Developmental Disabilities Research Center (P30 HD024064), the Baylor-Hopkins Center for Mendelian Genomics (U54 HG006542), and the National Institute of Neurological Disorders and Stroke (R01 NS058529) to JRL; and from the National Heart Blood and Lung Institute (R01 HL101975) and Polish Ministry of Science and Higher Education (R13-0005-04/2008) to PS.
Glossary
- Somatic mosaicism
Genetic variation that is present in the genomes of cells that make up the body of the organism and do not contribute to gametes produced by the individual
- Gonadal mosaicism
Genetic variation that is present in the genomes of cells that specifically contribute to the gametes
- Gonosomal mosaicism
Genetic variation that is present in the genomes of both somatic cells and germline cells
- Constitutional variation
Genetic variation that is present in the genome of every (or the vast majority of every) cell in an individual
- Post-zygotic mutation
A mutation that occurs after the fertilization of the ovum by the sperm
- Simplex genetic disease
The first occurrence of a genetic disease in a family, which may be due to a de novo mutation, recessive inheritance in a small sibship, or an inherited dominant allele with reduced penetrance
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Seshadri R, et al. Mutation rate of normal and malignant human lymphocytes. Cancer Res. 1987;47:407–409. [PubMed] [Google Scholar]
- 2.Gong Y, et al. The influence of premeiotic clusters of mutation on indirect estimations of mutation rate. Human Heredity. 2006 doi: 10.1159/000089955. [DOI] [PubMed] [Google Scholar]
- 3.Behjati S, et al. Genome sequencing of normal cells reveals developmental lineages and mutational processes. Nature. 2014;513:422–425. doi: 10.1038/nature13448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Iourov IY, et al. Somatic genome variations in health and disease. Curr Genomics. 2010;11:387–396. doi: 10.2174/138920210793176065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Erickson RP. Somatic gene mutation and human disease other than cancer: an update. Mutat Res. 2010;705:96–106. doi: 10.1016/j.mrrev.2010.04.002. [DOI] [PubMed] [Google Scholar]
- 6.Vadlamudi L, et al. Timing of de novo mutagenesis--a twin study of sodium-channel mutations. N Engl J Med. 2010;363:1335–1340. doi: 10.1056/NEJMoa0910752. [DOI] [PubMed] [Google Scholar]
- 7.Chander R, et al. CHILD syndrome with thrombocytosis and congenital dislocation of hip: A case report from India. Dermatol Online J. 2010;16:6. [PubMed] [Google Scholar]
- 8.Ma K. Embryonic left-right separation mechanism allows confinement of mutation-induced phenotypes to one lateral body half of bilaterians. Am J Med Genet A. 2013;161:3095–3114. doi: 10.1002/ajmg.a.36188. [DOI] [PubMed] [Google Scholar]
- 9.Gardner RL. Normal bias in the direction of fetal rotation depends on blastomere composition during early cleavage in the mouse. PLoS ONE. 2010;5:e9610. doi: 10.1371/journal.pone.0009610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Russell LB, Russell WL. Spontaneous mutations recovered as mosaics in the mouse specific-locus test. Proc Natl Acad Sci USA. 1996;93:13072–13077. doi: 10.1073/pnas.93.23.13072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Vanneste E, et al. Chromosome instability is common in human cleavage-stage embryos. Nat Med. 2009;15:577–583. doi: 10.1038/nm.1924. [DOI] [PubMed] [Google Scholar]
- 12.Braude P, et al. Human gene expression first occurs between the four- and eight-cell stages of preimplantation development. Nature. 1988;332:459–461. doi: 10.1038/332459a0. [DOI] [PubMed] [Google Scholar]
- 13.Fujimoto T, et al. The origin, migration and fine morphology of human primordial germ cells. Anat Rec. 1977;188:315–330. doi: 10.1002/ar.1091880305. [DOI] [PubMed] [Google Scholar]
- 14.Marques-Mari AI, et al. Differentiation of germ cells and gametes from stem cells. Hum Reprod Update. 2009;15:379–390. doi: 10.1093/humupd/dmp001. [DOI] [PubMed] [Google Scholar]
- 15.Drost JB, Lee WR. Biological basis of germline mutation: comparisons of spontaneous germline mutation rates among drosophila, mouse, and human. Environ Mol Mutagen. 1995;25:48–64. doi: 10.1002/em.2850250609. [DOI] [PubMed] [Google Scholar]
- 16.Qin J, et al. The molecular anatomy of spontaneous germline mutations in human testes. PLoS Biol. 2007;5:e224. doi: 10.1371/journal.pbio.0050224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Campbell IM, et al. Parental Somatic Mosaicism Is Underrecognized and Influences Recurrence Risk of Genomic Disorders. Am J Hum Genet. 2014;95:173–182. doi: 10.1016/j.ajhg.2014.07.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Markert CL, Petters RM. Manufactured hexaparental mice show that adults are derived from three embyronic cells. Science. 1978;202:56–58. doi: 10.1126/science.694518. [DOI] [PubMed] [Google Scholar]
- 19.Kalousek DK, Vekemans M. Confined placental mosaicism. J Med Genet. 1996;33:529–533. doi: 10.1136/jmg.33.7.529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Bianchi DW, et al. Male fetal progenitor cells persist in maternal blood for as long as 27 years postpartum. Proc Natl Acad Sci USA. 1996;93:705–708. doi: 10.1073/pnas.93.2.705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Conlin LK, et al. Mechanisms of mosaicism, chimerism and uniparental disomy identified by single nucleotide polymorphism array analysis. Hum Mol Genet. 2010;19:1263–1275. doi: 10.1093/hmg/ddq003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Cheung SW, et al. Microarray-based CGH detects chromosomal mosaicism not revealed by conventional cytogenetics. Am J Med Genet A. 2007;143:1679–1686. doi: 10.1002/ajmg.a.31740. [DOI] [PubMed] [Google Scholar]
- 23.Pham J, et al. Somatic mosaicism detected by exon-targeted, high-resolution aCGH in 10 362 consecutive cases. Eur J Hum Genet. 2014;22:969–978. doi: 10.1038/ejhg.2013.285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Boone PM, et al. Detection of clinically relevant exonic copy-number changes by array CGH. Hum Mutat. 2010;31:1326–1342. doi: 10.1002/humu.21360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Turner DJ, et al. Germline rates of de novo meiotic deletions and duplications causing several genomic disorders. Nat Genet. 2008;40:90–95. doi: 10.1038/ng.2007.40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hehir-Kwa JY, et al. De novo copy number variants associated with intellectual disability have a paternal origin and age bias. J Med Genet. 2011;48:776–778. doi: 10.1136/jmedgenet-2011-100147. [DOI] [PubMed] [Google Scholar]
- 27.Messiaen L, et al. Mosaic type-1 NF1 microdeletions as a cause of both generalized and segmental neurofibromatosis type-1 (NF1) Hum Mutat. 2011;32:213–219. doi: 10.1002/humu.21418. [DOI] [PubMed] [Google Scholar]
- 28.Choate KA, et al. Mitotic recombination in patients with ichthyosis causes reversion of dominant mutations in KRT10. Science. 2010;330:94–97. doi: 10.1126/science.1192280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Shao C, et al. Mitotic recombination is suppressed by chromosomal divergence in hybrids of distantly related mouse strains. Nat Genet. 2001;28:169–172. doi: 10.1038/88897. [DOI] [PubMed] [Google Scholar]
- 30.Ballif BC, et al. Detection of low-level mosaicism by array CGH in routine diagnostic specimens. Am J Med Genet A. 2006;140:2757–2767. doi: 10.1002/ajmg.a.31539. [DOI] [PubMed] [Google Scholar]
- 31.Pleasance ED, et al. A small-cell lung cancer genome with complex signatures of tobacco exposure. Nature. 2010;463:184–190. doi: 10.1038/nature08629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Pfeifer GP, et al. Mutations induced by ultraviolet light. Mutat Res. 2005;571:19–31. doi: 10.1016/j.mrfmmm.2004.06.057. [DOI] [PubMed] [Google Scholar]
- 33.Helleday T, et al. Mechanisms underlying mutational signatures in human cancers. Nat Rev Genet. 2014;15:585–598. doi: 10.1038/nrg3729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Huang AY, et al. Postzygotic single-nucleotide mosaicisms in whole-genome sequences of clinically unremarkable individuals. Cell Res. 2014 doi: 10.1038/cr.2014.131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Hile SE, et al. Beyond translesion synthesis: polymerase κ fidelity as a potential determinant of microsatellite stability. Nucleic Acids Res. 2012;40:1636–1647. doi: 10.1093/nar/gkr889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.McMurray CT. Mechanisms of trinucleotide repeat instability during human development. Nat Rev Genet. 2010;11:786–799. doi: 10.1038/nrg2828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Rifé M, et al. Analysis of CGG variation through 642 meioses in Fragile X families. Mol Hum Reprod. 2004;10:773–776. doi: 10.1093/molehr/gah102. [DOI] [PubMed] [Google Scholar]
- 38.McMurray JJV. Clinical practice. Systolic heart failure. N Engl J Med. 2009;362:228–238. doi: 10.1056/NEJMcp0909392. [DOI] [PubMed] [Google Scholar]
- 39.Swami M, et al. Somatic expansion of the Huntington’s disease CAG repeat in the brain is associated with an earlier age of disease onset. Hum Mol Genet. 2009;18:3039–3047. doi: 10.1093/hmg/ddp242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Helderman-van den Enden AT, et al. Monozygotic twin brothers with the fragile X syndrome: different CGG repeats and different mental capacities. J Med Genet. 1999;36:253–257. [PMC free article] [PubMed] [Google Scholar]
- 41.Lemmers RJLF, et al. A unifying genetic model for facioscapulohumeral muscular dystrophy. Science. 2010;329:1650–1653. doi: 10.1126/science.1189044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Lemmers RJLF, et al. Digenic inheritance of an SMCHD1 mutation and an FSHD-permissive D4Z4 allele causes facioscapulohumeral muscular dystrophy type 2. Nat Genet. 2012;44:1370–1374. doi: 10.1038/ng.2454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Lupski JR. Digenic inheritance and Mendelian disease. Nat Genet. 2012;44:1291–1292. doi: 10.1038/ng.2479. [DOI] [PubMed] [Google Scholar]
- 44.van der Maarel SM, et al. De novo facioscapulohumeral muscular dystrophy: frequent somatic mosaicism, sex-dependent phenotype, and the role of mitotic transchromosomal repeat interaction between chromosomes 4 and 10. The American Journal of Human Genetics. 2000;66:26–35. doi: 10.1086/302730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Lemmers RJLF, et al. Somatic mosaicism in FSHD often goes undetected. Ann Neurol. 2004;55:845–850. doi: 10.1002/ana.20106. [DOI] [PubMed] [Google Scholar]
- 46.Shapiro JA. Molecular model for the transposition and replication of bacteriophage Mu and other transposable elements. Proc Natl Acad Sci USA. 1979;76:1933–1937. doi: 10.1073/pnas.76.4.1933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Erwin JA, et al. Mobile DNA elements in the generation of diversity and complexity in the brain. Nat Rev Neurosci. 2014;15:497–506. doi: 10.1038/nrn3730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Evrony GD, et al. Cell lineage analysis in human brain using endogenous retroelements. Neuron. 2015;85:49–59. doi: 10.1016/j.neuron.2014.12.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Evrony GD, et al. Single-neuron sequencing analysis of L1 retrotransposition and somatic mutation in the human brain. Cell. 2012;151:483–496. doi: 10.1016/j.cell.2012.09.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Coufal NG, et al. L1 retrotransposition in human neural progenitor cells. Nature. 2009;460:1127–1131. doi: 10.1038/nature08248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Beck CR, et al. LINE-1 elements in structural variation and disease. Annu Rev Genomics Hum Genet. 2011;12:187–215. doi: 10.1146/annurev-genom-082509-141802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Hirschhorn K, et al. Human intersex with chromosome mosaicism of type XY/XO. Report of a case. N Engl J Med. 1960;263:1044–1048. doi: 10.1056/NEJM196011242632102. [DOI] [PubMed] [Google Scholar]
- 53.Hook EB. Exclusion of chromosomal mosaicism: tables of 90%, 95% and 99% confidence limits and comments on use. Am J Hum Genet. 1977;29:94–97. [PMC free article] [PubMed] [Google Scholar]
- 54.Shaffer LG, et al. Diagnosis of CMT1A duplications and HNPP deletions by interphase FISH: implications for testing in the cytogenetics laboratory. Am J Med Genet. 1997;69:325–331. [PubMed] [Google Scholar]
- 55.Jamuar SS, et al. Somatic mutations in cerebral cortical malformations. N Engl J Med. 2014;371:733–743. doi: 10.1056/NEJMoa1314432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Goriely A, et al. Germline and somatic mosaicism for FGFR2 mutation in the mother of a child with Crouzon syndrome: Implications for genetic testing in “paternal age-effect” syndromes. Am J Med Genet A. 2010;152:2067–2073. doi: 10.1002/ajmg.a.33513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Li J, et al. Replacing PCR with COLD-PCR enriches variant DNA sequences and redefines the sensitivity of genetic testing. Nat Med. 2008;14:579–584. doi: 10.1038/nm1708. [DOI] [PubMed] [Google Scholar]
- 58.Conlin LK, et al. Utility of SNP arrays in detecting, quantifying, and determining meiotic origin of tetrasomy 12p in blood from individuals with Pallister-Killian syndrome. Am J Med Genet A. 2012;158:3046–3053. doi: 10.1002/ajmg.a.35726. [DOI] [PubMed] [Google Scholar]
- 59.González JR, et al. A fast and accurate method to detect allelic genomic imbalances underlying mosaic rearrangements using SNP array data. BMC Bioinformatics. 2011;12:166. doi: 10.1186/1471-2105-12-166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Gonzaga-Jauregui C, et al. Human genome sequencing in health and disease. Annu Rev Med. 2012;63:35–61. doi: 10.1146/annurev-med-051010-162644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Yang Y, et al. Clinical whole-exome sequencing for the diagnosis of mendelian disorders. N Engl J Med. 2013;369:1502–1511. doi: 10.1056/NEJMoa1306555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Yang Y, et al. Molecular findings among patients referred for clinical whole-exome sequencing. JAMA. 2014;312:1870–1879. doi: 10.1001/jama.2014.14601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Mardis ER. Next-generation sequencing platforms. Annu Rev Anal Chem (Palo Alto Calif) 2013;6:287–303. doi: 10.1146/annurev-anchem-062012-092628. [DOI] [PubMed] [Google Scholar]
- 64.Pagnamenta AT, et al. Exome sequencing can detect pathogenic mosaic mutations present at low allele frequencies. J Hum Genet. 2012;57:70–72. doi: 10.1038/jhg.2011.128. [DOI] [PubMed] [Google Scholar]
- 65.Lindhurst MJ, et al. A mosaic activating mutation in AKT1 associated with the Proteus syndrome. N Engl J Med. 2011;365:611–619. doi: 10.1056/NEJMoa1104017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Kurek KC, et al. Somatic mosaic activating mutations in PIK3CA cause CLOVES syndrome. Am J Hum Genet. 2012;90:1108–1115. doi: 10.1016/j.ajhg.2012.05.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Dean FB, et al. Comprehensive human genome amplification using multiple displacement amplification. Proc Natl Acad Sci USA. 2002;99:5261–5266. doi: 10.1073/pnas.082089499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Gole J, et al. Massively parallel polymerase cloning and genome sequencing of single cells using nanoliter microwells. Nat Biotechnol. 2013;31:1126–1132. doi: 10.1038/nbt.2720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Cai X, et al. Single-cell, genome-wide sequencing identifies clonal somatic copy-number variation in the human brain. Cell Rep. 2014;8:1280–1289. doi: 10.1016/j.celrep.2014.07.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.McConnell MJ, et al. Mosaic copy number variation in human neurons. Science. 2013;342:632–637. doi: 10.1126/science.1243472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Lupski JR, et al. Clan genomics and the complex architecture of human disease. Cell. 2011;147:32–43. doi: 10.1016/j.cell.2011.09.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Johnson BE, et al. Mutational analysis reveals the origin and therapy-driven evolution of recurrent glioma. Science. 2014;343:189–193. doi: 10.1126/science.1239947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.O’Roak BJ, et al. Multiplex targeted sequencing identifies recurrently mutated genes in autism spectrum disorders. Science. 2012;338:1619–1622. doi: 10.1126/science.1227764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Hiatt JB, et al. Single molecule molecular inversion probes for targeted, high-accuracy detection of low-frequency variation. Genome Res. 2013;23:843–854. doi: 10.1101/gr.147686.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Sicca F, et al. Mosaic mutations of the LIS1 gene cause subcortical band heterotopia. Neurology. 2003;61:1042–1046. doi: 10.1212/wnl.61.8.1042. [DOI] [PubMed] [Google Scholar]
- 76.Mineyko A, et al. A novel missense mutation in LIS1 in a child with subcortical band heterotopia and pachygyria inherited from his mildly affected mother with somatic mosaicism. J Child Neurol. 2010;25:738–741. doi: 10.1177/0883073809343312. [DOI] [PubMed] [Google Scholar]
- 77.Nagamani SCS, et al. Microdeletions including YWHAE in the Miller-Dieker syndrome region on chromosome 17p13.3 result in facial dysmorphisms, growth restriction, and cognitive impairment. J Med Genet. 2009;46:825–833. doi: 10.1136/jmg.2009.067637. [DOI] [PubMed] [Google Scholar]
- 78.Huisman SA, et al. High rate of mosaicism in individuals with Cornelia de Lange syndrome. J Med Genet. 2013;50:339–344. doi: 10.1136/jmedgenet-2012-101477. [DOI] [PubMed] [Google Scholar]
- 79.Ansari M, et al. Genetic heterogeneity in Cornelia de Lange syndrome (CdLS) and CdLS-like phenotypes with observed and predicted levels of mosaicism. J Med Genet. 2014;51:659–668. doi: 10.1136/jmedgenet-2014-102573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Topcu M, et al. Somatic mosaicism for a MECP2 mutation associated with classic Rett syndrome in a boy. Eur J Hum Genet. 2002;10:77–81. doi: 10.1038/sj.ejhg.5200745. [DOI] [PubMed] [Google Scholar]
- 81.Bushman DM, et al. Genomic mosaicism with increased amyloid precursor protein (APP) gene copy number in single neurons from sporadic Alzheimer’s disease brains. Elife. 2015;4 doi: 10.7554/eLife.05116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Cohen MM. Proteus syndrome review: molecular, clinical, and pathologic features. Clinical Genetics. 2014;85:111–119. doi: 10.1111/cge.12266. [DOI] [PubMed] [Google Scholar]
- 83.Poduri A, et al. Somatic activation of AKT3 causes hemispheric developmental brain malformations. Neuron. 2012;74:41–48. doi: 10.1016/j.neuron.2012.03.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Lindhurst MJ, et al. Mosaic overgrowth with fibroadipose hyperplasia is caused by somatic activating mutations in PIK3CA. Nat Genet. 2012;44:928–933. doi: 10.1038/ng.2332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Lee JH, et al. De novo somatic mutations in components of the PI3K-AKT3-mTOR pathway cause hemimegalencephaly. Nat Genet. 2012;44:941–945. doi: 10.1038/ng.2329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Lombillo VA, Sybert VP. Mosaicism in cutaneous pigmentation. Curr Opin Pediatr. 2005;17:494–500. doi: 10.1097/01.mop.0000171319.84053.45. [DOI] [PubMed] [Google Scholar]
- 87.Bushman DM, Chun J. The genomically mosaic brain: aneuploidy and more in neural diversity and disease. Semin Cell Dev Biol. 2013;24:357–369. doi: 10.1016/j.semcdb.2013.02.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Pack SD, et al. Individual adult human neurons display aneuploidy: detection by fluorescence in situ hybridization and single neuron PCR. Cell Cycle. 2005;4:1758–1760. doi: 10.4161/cc.4.12.2153. [DOI] [PubMed] [Google Scholar]
- 89.Kong A, et al. Rate of de novo mutations and the importance of father’s age to disease risk. Nature. 2012;488:471–475. doi: 10.1038/nature11396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Mamsen LS, et al. Germ cell numbers in human embryonic and fetal gonads during the first two trimesters of pregnancy: analysis of six published studies. Hum Reprod. 2011;26:2140–2145. doi: 10.1093/humrep/der149. [DOI] [PubMed] [Google Scholar]
- 91.Busque L, et al. Nonrandom X-inactivation patterns in normal females: lyonization ratios vary with age. Blood. 1996;88:59–65. [PubMed] [Google Scholar]
- 92.Jaiswal S, et al. Age-related clonal hematopoiesis associated with adverse outcomes. N Engl J Med. 2014;371:2488–2498. doi: 10.1056/NEJMoa1408617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Jamuar S, et al. Clinical and radiological findings in Pallister-Killian syndrome. European Journal of Medical Genetics. 2012;55:167–172. doi: 10.1016/j.ejmg.2012.01.019. [DOI] [PubMed] [Google Scholar]
- 94.Liu P, et al. Passage Number is a Major Contributor to Genomic Structural Variations in Mouse iPSCs. Stem Cells. 2014;32:2657–2667. doi: 10.1002/stem.1779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Campbell IM, et al. Parent of Origin, Mosaicism, and Recurrence Risk: Probabilistic Modeling Explains the Broken Symmetry of Transmission Genetics. Am J Hum Genet. 2014 doi: 10.1016/j.ajhg.2014.08.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Helderman-van den Enden ATJM, et al. Recurrence risk due to germ line mosaicism: Duchenne and Becker muscular dystrophy. Clinical Genetics. 2009;75:465–472. doi: 10.1111/j.1399-0004.2009.01173.x. [DOI] [PubMed] [Google Scholar]
- 97.Hartl DL. Recurrence risks for germinal mosaics. The American Journal of Human Genetics. 1971;23:124–134. [PMC free article] [PubMed] [Google Scholar]
- 98.Shenkar R, et al. The mutation rate of the human mtDNA deletion mtDNA4977. The American Journal of Human Genetics. 1996;59:772–780. [PMC free article] [PubMed] [Google Scholar]
- 99.Olofsson P, Shaw CA. Exact sampling formulas for multi-type Galton-Watson processes. J Math Biol. 2002;45:279–293. doi: 10.1007/s002850200148. [DOI] [PubMed] [Google Scholar]
- 100.Athreya KB, Ney PE. Branching Process. Springer; 1972. [Google Scholar]
- 101.Kimmel M, Axelrod DE. Branching Processes in Biology. Springer Science & Business Media; 2002. [Google Scholar]
- 102.Bauer B, et al. Cancer initiation with epistatic interactions between driver and passenger mutations. J Theor Biol. 2014;358:52–60. doi: 10.1016/j.jtbi.2014.05.018. [DOI] [PubMed] [Google Scholar]
- 103.Hyrien O, et al. Stochastic modeling of oligodendrocyte generation in cell culture: model validation with time-lapse data. Theor Biol Med Model. 2006;3:21. doi: 10.1186/1742-4682-3-21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Beachkofsky TM, et al. Progressive overgrowth of the cerebriform connective tissue nevus in patients with Proteus syndrome. J Am Acad Dermatol. 2010;63:799–804. doi: 10.1016/j.jaad.2009.12.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Lang SS, et al. Prenatal diagnosis of hemimegalencephaly. World Neurosurg. 2014;82:241, e5–8. doi: 10.1016/j.wneu.2013.09.028. [DOI] [PubMed] [Google Scholar]
- 106.Molho-Pessach V, Schaffer JV. Blaschko lines and other patterns of cutaneous mosaicism. Clin Dermatol. 2011;29:205–225. doi: 10.1016/j.clindermatol.2010.09.012. [DOI] [PubMed] [Google Scholar]