

Abstract
The 5´-AMP-activated protein kinase (AMPK) is a key regulator of cellular metabolism and energy homeostasis in mammalian tissues. Metabolic adaptation is a critical step in ensuring cell survival during metabolic stress. Because of its critical role in the regulation of glucose homeostasis and carbohydrate, lipid, and protein metabolism, AMPK is involved in many human diseases, including cancers. Although AMPK signaling was originally characterized as a tumor-suppressive signaling pathway, several lines of evidence suggest that AMPK plays a much broader role and cannot simply be defined as either an oncogenic regulator or tumor suppressor. Notably, several recent studies demonstrated that the antitumorigenic effects of many indirect AMPK activators, such as metformin, do not depend on AMPK. Conversely, activation of AMPK induces the progression of cancers, emphasizing its oncogenic effect. Bladder cancer can be divided into two groups: non–muscle-invasive bladder cancer (NMIBC) and muscle-invasive bladder cancer (MIBC). The molecular mechanisms underlying these two types of cancer are distinct: NMIBC is associated with activation of the Ras pathway, whereas MIBC is characterized by loss of major tumor suppressors. Importantly, both pathways are connected to the mammalian target of rapamycin (mTOR) pathway. In addition, our recent metabolomic findings suggest that β-oxidation of fatty acids is an important factor in the development of bladder cancer. Both mTOR and β-oxidation are tightly associated with the AMPK pathway. Here, I summarize and discuss the recent findings on the two distinct roles of AMPK in cancer, as well as the relationship between bladder cancer and AMPK.
Keywords: AMP-Activated Protein Kinases, Urinary Bladder Neoplasms, Metabolism
INTRODUCTION
5´-AMP-activated protein kinase (AMPK), a key regulator of cellular energy homeostasis, is activated in response to various metabolic stresses including starvation, hypoxia, and ischemia. AMPK is a heterotrimeric enzyme, which is activated by phosphorylation of Thr172 on its α subunit. Several upstream kinases can modify Thr172, including liver kinase B1 (LKB1), calcium/calmodulin-dependent protein kinase kinase 2 (CaMKK2), and transforming growth factor β activated kinase-1 (Fig. 1). AMPK is considered to be a metabolic “master switch” that regulates intracellular processes such as the cellular uptake of glucose, β-oxidation of fatty acids, biogenesis of glucose transporter 4, and mitochondrial biogenesis [1-4].
Fig. 1.
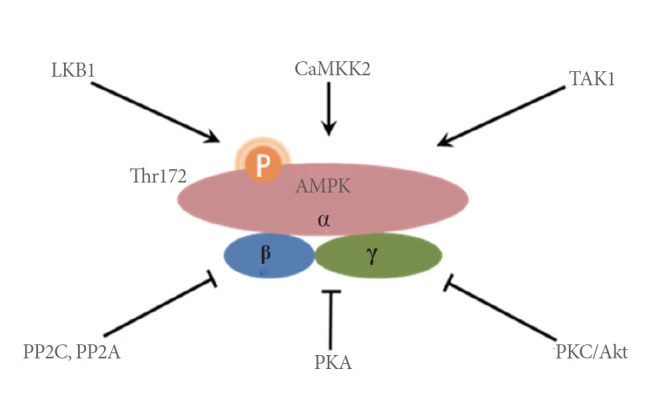
Activation and inactivation of AMPK. AMPK is a heterotrimeric enzyme activated by phosphorylation of Thr172 on its α subunit. Several upstream kinases can modify Thr172, including LKB1, CaMKK2, and TAK1. The activity of AMPK can be reduced by PP2A and PP2C, which mediate the dephosphorylation of phospho-Thr172. PKC and Akt also phosphorylate Ser485/491 of the α chain, which in turn decreases AMPK activity. Additionally, PKA-dependent modification of Ser173 diminishes AMPK activity. AMPK, 5´-AMP-activated protein kinase; LKB1, liver kinase B1; CaMKK2, calcium/calmodulin-dependent protein kinase kinase 2; TAK1, transforming growth factor β activated kinase-1; PP2C/A, protein phosphatase 2C/A; PKA/C, protein kinase A/C.
Upon activation, AMPK increases cellular energy levels by inhibiting anabolic energy-consuming pathways (including fatty acid synthesis and protein synthesis) and stimulating energy-producing catabolic pathways (including fatty acid oxidation and glucose transport) (Fig. 2). Because it is a major regulator of both lipid and glucose metabolism, AMPK has been considered as a potential therapeutic target for the treatment of diabetes, obesity, and cancer. Recently, however, many reports show that AMPK actually plays a critical role in tumor survival and growth [5-8]. The metabolism reprogramming triggered by the activation of AMPK is important for the survival of cells in the tumor microenvironment. Thus, it would not be surprising if, in some cases, AMPK acts as a tumor promoter.
Fig. 2.
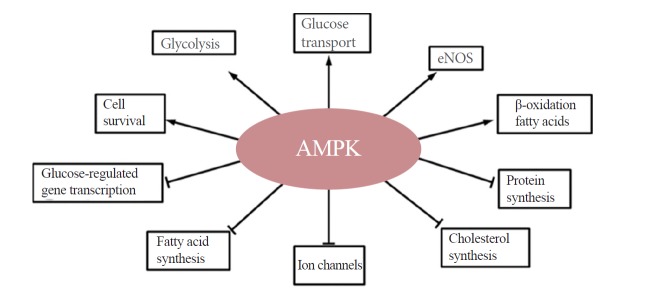
Function of AMPK. Arrows denote activation, and horizontal bars indicate inhibition. AMPK, 5´-AMP-activated protein kinase; eNOS, endothelial nitric oxide synthase 3.
AMPK AND THE WARBURG EFFECT
In contrast to normal cells, which produce energy through a relatively low rate of glycolysis and oxidation of pyruvate in the mitochondria, the energy production of most cancer cells relies on a high rate of glycolysis followed by lactic acid fermentation in the cytosol; this phenomenon is known as the Warburg effect [9-11]. Rapidly growing cancer cells achieve glycolytic rates that are up to 200 folds higher than those of normal cells, even when oxygen is plentiful. The Warburg effect may reflect adaptation to low-oxygen environments within tumors, mitochondrial damage, or oncogene-mediated shutdown of mitochondria, which would otherwise induce apoptosis in cancerous cells. On the other hand, the effect might also be associated with rapid cell proliferation. Because glycolysis provides most of the building blocks required for cell proliferation, cancer cells may need to activate glycolysis despite the availability of oxygen [12] (Fig. 3).
Fig. 3.
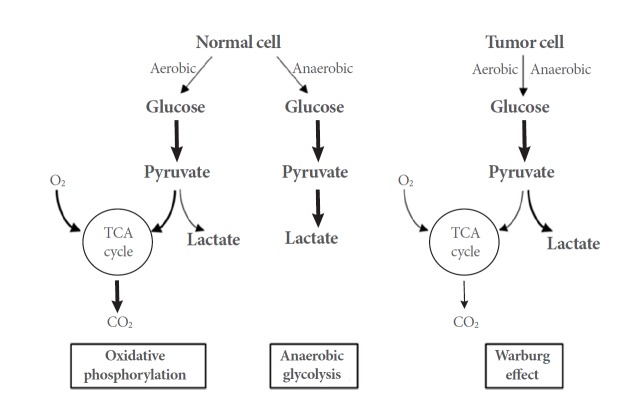
The differences between oxidative phosphorylation, anaerobic glycolysis, and aerobic glycolysis. TCA, tricarboxylic acid.
In several types of cancer, genetic defects in the enzymes of the tricarboxylic acid cycle are involved in tumorigenesis, whereas oncogenic H-Ras transformation damages mitochondrial function [13,14]. Upon initiation of the Warburg effect, cells depend on aerobic glycolysis with lactate production, rather than oxidative phosphorylation, to maintain adenosine triphosphate (ATP) levels. AMPK regulates glycolysis by phosphorylating and activating 6-phosphofructo-2-kinase/fructose-2,6-bisphosphatases (PFKFBs). PFKFB3 is overexpressed and its phosphorylation is elevated in several types of cancer (including colon and breast cancers) as a consequence of AMPK activation [15]. Various stress signals, such as hypoxia and nutrient depletion, can drive PFKFB3 expression, and AMPK contributes to the elevation of PFKFB3 levels and glycolytic flux in response to reduced pH [16]. In addition, expression of PFKFB2, a phosphorylation target of both Akt and AMPK, is elevated in human cancers. Thus, depending on the cellular environment, Akt and AMPK may promote glycolysis.
AMPK can act as a negative regulator of the Warburg effect under nutrient- and growth factor-rich conditions. AMPK depletion further increases mammalian target of rapamycin complex 1 (mTORC1) activation, hypoxia-inducible factor 1 (HIF1) levels, and aerobic glycolysis in Myc over-expressing cells [17]. Depletion of AMPK leads to increased lactate production [17], promoting the use of nonglucose carbon sources for anabolism, maintenance of bioenergetics, and lactate production. The role of lactate production in proliferating tumor cells, the primary characteristic of the Warburg effect, remains poorly understood. The key glycolysis enzyme phosphofructokinase-1 is inhibited by O-GlcNAcylation, which is necessary for optimum tumor growth [18], suggesting that suppression of lactate-producing glycolysis can also confer survival and growth advantages on cancer cells, depending on the genetic background and cellular context.
AMPK: AN ONCOGENIC REGULATOR?
Although the AMPK signaling pathway was originally characterized as a tumor-suppressive pathway, recent studies have demonstrated that many factors in the tumor microenvironment, as well as some oncogenic signals, are AMPK activators. The oncogene Src (proto-oncogene tyrosine-protein kinase), which is overexpressed and activated in many cancers, activates AMPK through the activation of the protein kinase C α-LKB1 pathway in OVCAR3 and A431 cells [19]. Similarly, the expression of MYC and H-RasV12 activates AMPK in some cancers [20,21]. Moreover, androgen receptor (AR) signaling activates AMPK through the transactivation of CaMKK2 in prostate cancer [22-24]. In addition to the activation of oncogenes, the loss of tumor suppressor gene folliculin (FLCN), which is associated with Birt-Hogg-Dubé syndrome, also activates AMPK [25,26].
Initially, two groups reported that LKB1 and AMPK play protumorigenic roles in the context of oncogene-induced transformation of mouse embryonic fibroblasts (MEFs). They showed that both LKB1-null MEFs and AMPKα1/α2-null (AMPKα knockout) MEFs are resistant to oncogenic effects such as H-RasV12/SV40T-induced anchorage-independent growth and solid tumor growth in vivo [27,28]. Remarkably, several studies demonstrated that AMPK is also essential for other types of oncogene-induced tumorigenesis, such as Mycinduced development of hepatocellular carcinoma (HCC), H-RasV12±PTEN deletioninduced astrocytic tumor cell proliferation, and kinase suppressor of Ras 2-induced anoikis resistance in MEFs [20,21,29]. Several recent reports show that AMPK plays a key role in various cancers. Depletion of AMPK activity in breast and pancreatic cancer cells hampers anchorage-independent growth and tumor formation in vivo [30-32]. The LKB1-AMPK signaling pathway is also essential for glioma cell survival under low-glucose conditions [21,33]. Two recent approaches have identified the AMPKα1 and AMPKβ1 subunits as essential protumorigenic genes in melanoma [34] and prostate cancer [35], respectively. Activation of AMPK following loss of the tumor suppressor FLCN increases mitochondrial biogenesis by inducing peroxisome proliferator-activated receptor γ (PPARγ) coactivator 1α (PGC1α) expression, which in turn results in the production of reactive oxygen species (ROS). Yan et al. [26] also demonstrated that high levels of ROS activate HIF1α transcriptional activity, resulting in elevated expression of glycolytic enzymes, which induce ATP production. The AR-CaMKK2-AMPK axis in prostate cancer increases aerobic glycolysis and anabolic metabolism partly through PGC1α-mediated mitochondrial biogenesis, which can provide both the energy and the building blocks required for rapid cell growth [22,23]. Recently, mechanisms associated with the metabolic functions of AMPK, other than those involved in the production of energy or building blocks, have attracted increasing attention. Jeon et al. [36] showed that in the absence of AMPK activity, oxidative stress is the major cause of cancer cell death during glucose deprivation and matrix detachment. Under such stressful metabolic conditions, maintenance of NADPH levels rather than ATP levels is crucial for AMPK-induced solid tumor formation in vivo, tumor cell survival, and anchorage-independent growth. Acetyl-CoA carboxylase (ACC) 1 and ACC2 decrease NADPH consumption by inhibiting fatty acid synthesis and increase NADPH production by activation of fatty acid oxidation, respectively. They both need to be inhibited to maintain the glutathione/oxidized glutathione ratio and to reduce H2O2 levels, thereby protecting against oxidative stress-induced cell death. In osteosarcoma, AMPK protects against H2O2-induced cell death by maintaining NADPH levels [37]. Loss of FLCN results in the constitutive activation of AMPK, which in turn induces autophagy, inhibits apoptosis, improves cellular bioenergetics, and confers resistance to energy-depleting stresses such as oxidative stress, heat, anoxia, and serum deprivation [25].
AMPK: A TUMOR SUPPRESSOR?
Despite accumulating evidence highlighting the oncogenic role of AMPK, this enzyme was originally identified as a tumor suppressor following the discovery of LKB1 and mTORC1 [38-41]. Germline mutations in the gene encoding AMPK were detected in Peutz-Jeghers Syndrome [42]. Additionally, somatic mutations in the tumor suppressor LKB1 were observed in lung and cervical cancers [43]. mTORC1, which is inactivated by the LKB1-AMPK axis, is an important regulator of growth factor and nutrient signaling that controls protein synthesis and cell growth, and is over-activated in most human cancers [43]. Since LKB1 activates AMPK and AMPK inhibits mTORC1, it is reasonable to classify AMPK as a tumor suppressor. Although LKB1 deletion alone is not sufficient to cause lung cancer, it elevates mTORC1 and Src activity, which enhances lung cancer induced by K-Ras activation or PTEN deletion [44-46]. LKB1 deletion accelerates ErbB2-mediated breast cancer associated with elevated mTORC1 activity [47]. Conversely, in the endometrium, LKB1 deletion alone is sufficient for the development of invasive endometrial cancer, which can be effectively treated by rapamycin, an mTORC1 inhibitor, suggesting that mTORC1 activation is the major downstream target of LKB1 inhibition [48-50]. Notably, LKB1 also plays an AMPK/mTORC1-independent role in tumor suppression that depends on microtubule affinity-regulating kinase-mediated regulation of Snail or Hippo signaling [51,52].
Deletion of PRKAA1, the gene that encodes AMPKα1, accelerates development of Myc-induced lymphoma [17]. This observation contradicts the findings of Liu et al. [20], who showed that AMPK plays an important role in the development of Myc-induced HCC. Faubert et al. [17,53] also demonstrated that deletion of either LKB1 or PRKAA1 induces mTORC1/HIF1α-dependent metabolic reprogramming that helps cancer cells meet their energetic and anabolic demands. This observation also conflicts with the findings of Yan et al. [26], who suggested that AMPK activates aerobic glycolysis through the PGC1α-ROS-HIF1α axis in the absence of FLCN. Although this discrepancy could be due to tissue-specific and context-dependent functions of AMPK, further research is needed to obtain an unequivocal interpretation of these seemingly contradictory findings.
Several additional studies have reported conflicting pharmacological evidence regarding the role of AMPK in cancer. The antidiabetic drug metformin, which indirectly activates AMPK by inhibiting mitochondrial metabolism, decreases the incidence and mortality rate of breast cancer [54,55]. Moreover, several lines of evidence support the idea that AMPK activators, such as 5-aminoimidazole-4-carboxamide-1-b-D-ribofuranoside (AICAR), metformin, and phenformin, suppress cancer cell growth and proliferation [56-59]. However, in contradiction to these findings, Liu et al. [60] showed that AICAR and metformin reduce glioblastoma proliferation and viability. Furthermore, the direct AMPK activator A769662 has no effect on proliferation, suggesting that the antitumor effects of AICAR and metformin are AMPK-independent. Shackelford et al. [61] also demonstrated that phenformin selectively kills LKB1-deficient lung tumor cells in vivo, suggesting that the antitumor effect of phenformin is not dependent on activation of AMPK. Notably, Vincent et al. [62] showed that all these nonspecific and indirect AMPK activators strongly inhibit cell proliferation and viability in the absence of AMPK, whereas such cytotoxic effects are reduced in the presence of AMPK, suggesting that the activation of AMPK actually opposes the cytotoxic effect of the indirect AMPK activators. Along these lines, Vincent et al. [62] demonstrated that the direct AMPK agonist A769662 promoted cell proliferation under metabolically stressful conditions, supporting the idea that AMPK plays an oncogenic role in the tumor microenvironment. AMPK regulates diverse aspects of metabolism and cell physiology in both health and disease. Therefore, we must consider the effects of organ, cancer type, and acute vs. long-term AMPK activation and inactivation when investigating the mechanistic contribution of AMPK to cancer [63-65].
REGULATION OF AMPK IN BLADDER CANCER
Non–muscle-invasive bladder cancer (NMIBC) and muscle-invasive bladder cancer (MIBC) are associated with distinct molecular pathways. NMIBC is characterized by activation of the Ras pathway through mutations in H-Ras, FGFR-3, and PI3K, while MIBC is associated with a loss of tumor suppressor genes, including p53, Rb, and PTEN [66-72]. Activation of the Ras pathway occurs in approximately 80% of all NMIBCs [66,68,69], whereas more than 50% of MIBCs exhibit loss of p53 function [70-72]. Importantly, there is crosstalk between these pathways and the mTOR pathway [67,68,73], which means that alterations in either pathway are predicted to influence mTOR activity. Seager et al. [70] reported that intravesical rapamycin instillation exerts striking inhibitory effects on tumor progression in a bladder-specific PTEN and p53 double knockout transgenic mouse model, which develops carcinoma in situ lesions that progress to MIBC.
In addition to the H-Ras, p53, and PTEN pathways, diverse upstream signals such as AMPK and insulin can affect the mTOR pathway. Although no direct evidence about the role of the AMPK pathway in bladder cancer is currently available, several studies support the idea that the AMPK pathway might influence both bladder cancer development and progression. Liu et al. [74] reported that Rhodiola rosea extract and salidroside inhibit the mTOR pathway and translational initiation via activation of AMPKα in UMUC-3 bladder cancer cells. Metformin inhibits the growth of bladder cancer cells via indirect activation of AMPK [75,76], which in turn suppresses the mTOR/p70 S6 kinase-1 (S6K1) pathway in 253J and RT4 bladder cancer cell lines. Yan et al. [77] demonstrated that troglitazone (a synthetic ligand of PPARγ) activates autophagy concurrent with the activation of AMPK and suppression of the mTOR signaling pathway in T24 cells. Activation of mTOR occurs via a multistep process including upstream PI3K and Ras activation, or inactivation of AMPK, leading to phosphorylation and inactivation of the tuberous sclerosis complex 1 and 2 (TSC1/TSC2) heterodimer [78,79]. Inactivation of TSC1/TSC2 results in release of Rheb inhibition and subsequent mTOR activation through Rheb-GTPase activity. Finally, mTOR activity regulates the effects of a number of downstream molecules, including S6K and elongation-initiation factor 4E binding protein-1 [78,79].
In MIBC, PTEN loss appears to correlate with an increased mTOR activity, suggesting that it exerts a direct effect on downstream signaling components in bladder cancer [80,81]. Thirty-nine percent of MIBC cases exhibit either loss-of-heterozygosity (LOH) or homozygous deletion of the PTEN locus, and occasionally, mutations in the PTEN coding region occur [82]. Additionally, loss of p53 function appears to synergize with PTEN loss to promote activation of the mTOR signaling pathway [80]. Finally, LOH of TSC1 has been reported in approximately 50% of bladder cancer cases [83]. Taken together, the deregulation of components upstream of the mTOR pathway may occur in bladder tumors, suggesting that mTOR signaling may be elevated in this type of cancer.
Recently, we performed metabolomic profiling of bladder cancer patients. The results of our analysis revealed that patients with bladder cancer have elevated levels of urinary acetyl-CoA and carnitine. Because carnitine is crucial for the entry of fatty acids into the mitochondria for oxidation, and acetyl-CoA is the final product of this oxidation event, we speculate that β-oxidation of fatty acids might be an important factor in the development of bladder cancer [84]. When we examined the gene expression levels of the enzymes involved in fatty acid oxidation using our published microarray data [85], we found that bladder cancer specimens expressed significantly higher levels of carnitine palmitoyl transferase 1A (CPT1A) than normal bladder mucosae. CPT is a key enzyme that uses carnitine to transfer fatty acids to the mitochondria for oxidation. In particular, the CPT levels were higher in MIBC than in NMIBC. Thus, CPT1A may be associated with the aggressiveness of bladder cancer. Several investigators have attempted to develop inhibitors of CPT as anticancer agents [86,87]. Our microarray data also showed that bladder cancers express carnitine acylcarnitine translocase-like protein (CACL), another enzyme involved in fatty acid transport to mitochondria [88,89], at higher levels than normal bladder mucosae. Both MIBC and NMIBC express significantly higher levels of CACL than normal bladder mucosae. Although CACL has not been studied as extensively as CPT1A, strategies that target it may also have therapeutic potential in bladder cancer. Thus, in line with other studies implicating fatty acid oxidation in various types of tumorigenesis [90,91], our metabolomics study [84] and microarray analysis [85] indicated that β-oxidation of fatty acid plays an important role in bladder tumorigenesis and aggressiveness. The level of acetyl-CoA, another molecule associated with β-oxidation, can be influenced by input from pyruvate through the pyruvate dehydrogenase complex. We also found that the level of a third component of the complex, dihydrolipoyl dehydrogenase, is significantly reduced in bladder cancer. This observation suggests that the higher acetyl-CoA levels in bladder cancer are largely due to elevated β-oxidation, rather than conversion from pyruvate. Our speculation is concordant with the Warburg effect in cancer cells [92], in which pyruvate is converted to lactate rather than acetyl-CoA. Although the AMPK-fatty acid β-oxidation pathway has not been completely characterized in the context of bladder cancer, fatty acid β-oxidation is mainly controlled by the AMPK pathway, suggesting that AMPK acts as an oncogenic regulator of bladder cancer. The pathways related to AMPK-fatty acid β-oxidation that are putatively affected by bladder cancer, based on data from our two studies [84,85], are summarized in Fig. 4.
Fig. 4.
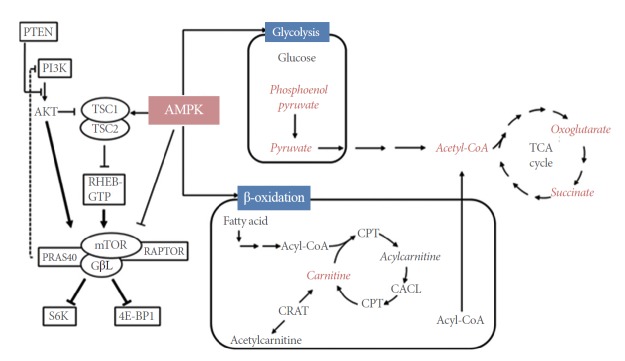
Putative pathways that may be altered in bladder cancer. Metabolites detected in our study [84,85] are indicated in red. AMPK, 5´-AMP-activated protein kinase; CACL, carnitine acylcarnitine translocase-like protein; CPT, carnitine palmitoyltransferase; CRAT, carnitine O-acetyltransferase; PTEN, phsophatase and tensin homolog; PI3K, phosphatidylinositol-4,5-bisphosphate 3-kinase; GβL, G-protein β-subunit-like protein; PRAS40, proline-rich Akt substrate of 40 kDa; RAPTOR, regulatory-associated protein of mTOR; RHEB-GTP, Ras homolog enriched in brain-GTP; 4E-BP1, eukaryotic translation initiation factor 4E binding protein 1; TCA, tricarboxylic acid; TSC1/2, tuberous sclerosis 1/2; S6K, S6 kinase.
Several lines of evidence suggest that activation of the mTOR pathway and stimulation of the fatty acid β-oxidation pathway might be important in the development of bladder cancer. Both pathways are connected with the AMPK pathway. However, no study to date has described the connection between the AMPK, mTOR, and β-oxidation pathways. Additional research into the missing link between mTOR and β-oxidation of fatty acids following AMPK activation may reveal novel mechanisms underlying the roles of AMPK in bladder cancer.
AMPK AS A THERAPEUTIC TARGET FOR CANCER
As discussed above, AMPK activation exerts two seemingly opposing functions in cancer. Although AMPK activation (resulting in TORC1 inhibition) is considered to be a reasonable anticancer tactic [93], AMPK may in fact exert very complex effects in tumor cells. The antidiabetic drug metformin, which inhibits mitochondrial metabolism, inhibits mTORC1 in both an AMPK-dependent and AMPK-independent manner [60,62,94,95]. Compound C, a selective AMPK inhibitor [96], binds to the AMPKα subunit and acts as an ATP-competitive inhibitor. However, several studies have demonstrated that compound C can also inhibit many other kinases as well as the bone morphogenetic protein receptor, and this promiscuity raises doubts about whether its effects are mediated by AMPK inhibition [97]. Sunitinib, a multiple tyrosine kinase inhibitor that is used clinically against advanced clear-cell renal cell carcinoma, can directly inhibit AMPK by binding the AMPKα subunit [98]. Table 1 summarizes how AMPK activity can be affected not only in vitro, but also in growing cells, organs, or whole organisms. These data were collected from several publications describing the molecular mechanisms and tissue-specific effects of AMPK activity [64,99,100].
Table 1.
Drugs and compounds that modulate AMPK activity
| Drug and compound | Function | Effect on AMPK |
|---|---|---|
| AICAR | AMP analog | Activation |
| Metformin (biguanide) | Reduces mitochondrial ATP production | Activation |
| Phenformin | Inhibition of respiratory chain | Activation |
| Resveratrol | Changes in ATP synthase activity | Activation |
| Prevents acetylation of LKB1 | ||
| Upregulation of adiponectin synthesis | ||
| Multimerization | ||
| TZD derivatives (TZDs, troglitazone; rosiglitazone, pioglitazone) | Stimulate expression of adiponectin | Activation |
| Increase in AMP concentration | ||
| Activation of PPAR | ||
| Antimycin A | Inhibition of respiratory chain | Activation |
| Sodium azide | Inhibition of respiratory chain | Activation |
| NO | Inhibition of respiratory chain | Activation |
| Oligomycin | Inhibition of ATP synthase | Activation |
| Dinitrophenol | Uncoupling of electron transfer/ATP synthesis | Activation |
| 2-Deoxyglucose | Inhibition of glycolysis | Activation |
| Arsenite | Inhibition of TCA cycle | Activation |
| β-guanadinopropionic acid | Increases AMP/ATP ratio | Activation |
| A23187 | Increase in cytosolic calcium ions | Activation |
| A769662 | Direct AMPK activator | Activation |
| Compound C (dorsomorphin) | Reversible, ATP-competitive inhibitor | Inhibition |
| Sunitinib | Binding to AMPK subunit | Inhibition |
| Midostaurin | Binding to AMPK subunit | Inhibition |
AMPK, 5´-AMP-activated protein kinase; AICAR, 5-aminoimidazole-4-carboxamide-1-b-D-ribofuranoside; ATP, adenosine triphosphate; LKB1, liver kinase B1; TZD, thiazolidinedione; PPARγ, peroxisome proliferator-activated receptor γ; NO, nitric oxide; TCA, tricarboxylic acid.
Currently, several lines of evidence suggest that AMPK could act as a tumor promoter rather than tumor suppressor. Notably, secondary activation of AMPK by many metabolic inhibitors actually induces resistance to apoptosis, suggesting that inhibition of AMPK with concurrent induction of metabolic stress represents a more reasonable anticancer therapeutic strategy than activating AMPK. Such an approach might provide an effective way to investigate the synergistic anticancer effects of combination therapy using current treatment modalities in conjunction with the relevant AMPK inhibitors.
CONCLUSIONS
Although AMPK was originally considered a tumor suppressor that acted via mTORC1 inhibition, today the consensus is that AMPK might in fact be a tumor promoter. This suggests that inhibition of AMPK is a more reasonable strategy for treating cancers than activating the enzyme. Although activation of the mTOR pathway and elevated fatty acid β-oxidation are closely associated with the AMPK pathway, no publication has described the connection between the AMPK, mTOR, and β-oxidation pathways. Therefore, further research aimed at identifying the missing link between mTOR and β-oxidation of fatty acid following AMPK activation will provide better insight into the role of AMPK in bladder cancer.
Footnotes
Grant Support
This work was supported by a National Research Foundation of Korea (NRF) grant, funded by the Korea government (MSIP) (No. NRF-2014R1A2A1A09006983).
Conflict of Interest
No potential conflict of interest relevant to this article was reported.
REFERENCES
- 1.Bergeron R, Russell RR, 3rd, Young LH, Ren JM, Marcucci M, Lee A, et al. Effect of AMPK activation on muscle glucose metabolism in conscious rats. Am J Physiol. 1999;276(5 Pt 1):E938–44. doi: 10.1152/ajpendo.1999.276.5.E938. [DOI] [PubMed] [Google Scholar]
- 2.Durante PE, Mustard KJ, Park SH, Winder WW, Hardie DG. Effects of endurance training on activity and expression of AMP-activated protein kinase isoforms in rat muscles. Am J Physiol Endocrinol Metab. 2002;283:E178–86. doi: 10.1152/ajpendo.00404.2001. [DOI] [PubMed] [Google Scholar]
- 3.Ojuka EO. Role of calcium and AMP kinase in the regulation of mitochondrial biogenesis and GLUT4 levels in muscle. Proc Nutr Soc. 2004;63:275–8. doi: 10.1079/PNS2004339. [DOI] [PubMed] [Google Scholar]
- 4.Thomson DM, Porter BB, Tall JH, Kim HJ, Barrow JR, Winder WW. Skeletal muscle and heart LKB1 deficiency causes decreased voluntary running and reduced muscle mitochondrial marker enzyme expression in mice. Am J Physiol Endocrinol Metab. 2007;292:E196–202. doi: 10.1152/ajpendo.00366.2006. [DOI] [PubMed] [Google Scholar]
- 5.Faubert B, Vincent EE, Poffenberger MC, Jones RG. The AMP-activated protein kinase (AMPK) and cancer: many faces of a metabolic regulator. Cancer Lett. 2015;356(2 Pt A):165–70. doi: 10.1016/j.canlet.2014.01.018. [DOI] [PubMed] [Google Scholar]
- 6.Hardie DG, Alessi DR. LKB1 and AMPK and the cancer-metabolism link - ten years after. BMC Biol. 2013;11:36. doi: 10.1186/1741-7007-11-36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Jeon SM, Hay N. The dark face of AMPK as an essential tumor promoter. Cell Logist. 2012;2:197–202. doi: 10.4161/cl.22651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Liang J, Mills GB. AMPK: a contextual oncogene or tumor suppressor? Cancer Res. 2013;73:2929–35. doi: 10.1158/0008-5472.CAN-12-3876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Alfarouk KO, Muddathir AK, Shayoub ME. Tumor acidity as evolutionary spite. Cancers (Basel) 2011;3:408–14. doi: 10.3390/cancers3010408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Gatenby RA, Gillies RJ. Why do cancers have high aerobic glycolysis? Nat Rev Cancer. 2004;4:891–9. doi: 10.1038/nrc1478. [DOI] [PubMed] [Google Scholar]
- 11.Kim JW, Dang CV. Cancer’s molecular sweet tooth and the Warburg effect. Cancer Res. 2006;66:8927–30. doi: 10.1158/0008-5472.CAN-06-1501. [DOI] [PubMed] [Google Scholar]
- 12.Lopez-Lazaro M. The warburg effect: why and how do cancer cells activate glycolysis in the presence of oxygen? Anticancer Agents Med Chem. 2008;8:305–12. doi: 10.2174/187152008783961932. [DOI] [PubMed] [Google Scholar]
- 13.White E. Deconvoluting the context-dependent role for autophagy in cancer. Nat Rev Cancer. 2012;12:401–10. doi: 10.1038/nrc3262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Yang D, Wang MT, Tang Y, Chen Y, Jiang H, Jones TT, et al. Impairment of mitochondrial respiration in mouse fibroblasts by oncogenic H-RAS(Q61L) Cancer Biol Ther. 2010;9:122–33. doi: 10.4161/cbt.9.2.10379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Bando H, Atsumi T, Nishio T, Niwa H, Mishima S, Shimizu C, et al. Phosphorylation of the 6-phosphofructo-2-kinase/fructose 2,6-bisphosphatase/PFKFB3 family of glycolytic regulators in human cancer. Clin Cancer Res. 2005;11:5784–92. doi: 10.1158/1078-0432.CCR-05-0149. [DOI] [PubMed] [Google Scholar]
- 16.Mendoza EE, Pocceschi MG, Kong X, Leeper DB, Caro J, Limesand KH, et al. Control of glycolytic flux by AMP-activated protein kinase in tumor cells adapted to low pH. Transl Oncol. 2012;5:208–16. doi: 10.1593/tlo.11319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Faubert B, Boily G, Izreig S, Griss T, Samborska B, Dong Z, et al. AMPK is a negative regulator of the Warburg effect and suppresses tumor growth in vivo. Cell Metab. 2013;17:113–24. doi: 10.1016/j.cmet.2012.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Yi W, Clark PM, Mason DE, Keenan MC, Hill C, Goddard WA, 3rd, et al. Phosphofructokinase 1 glycosylation regulates cell growth and metabolism. Science. 2012;337:975–80. doi: 10.1126/science.1222278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Mizrachy-Schwartz S, Cohen N, Klein S, Kravchenko-Balasha N, Levitzki A. Up-regulation of AMP-activated protein kinase in cancer cell lines is mediated through c-Src activation. J Biol Chem. 2011;286:15268–77. doi: 10.1074/jbc.M110.211813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Liu L, Ulbrich J, Muller J, Wustefeld T, Aeberhard L, Kress TR, et al. Deregulated MYC expression induces dependence upon AMPK-related kinase 5. Nature. 2012;483:608–12. doi: 10.1038/nature10927. [DOI] [PubMed] [Google Scholar]
- 21.Rios M, Foretz M, Viollet B, Prieto A, Fraga M, Costoya JA, et al. AMPK activation by oncogenesis is required to maintain cancer cell proliferation in astrocytic tumors. Cancer Res. 2013;73:2628–38. doi: 10.1158/0008-5472.CAN-12-0861. [DOI] [PubMed] [Google Scholar]
- 22.Tennakoon JB, Shi Y, Han JJ, Tsouko E, White MA, Burns AR, et al. Androgens regulate prostate cancer cell growth via an AMPK-PGC-1α-mediated metabolic switch. Oncogene. 2014;33:5251–61. doi: 10.1038/onc.2013.463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Massie CE, Lynch A, Ramos-Montoya A, Boren J, Stark R, Fazli L, et al. The androgen receptor fuels prostate cancer by regulating central metabolism and biosynthesis. EMBO J. 2011;30:2719–33. doi: 10.1038/emboj.2011.158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Frigo DE, Howe MK, Wittmann BM, Brunner AM, Cushman I, Wang Q, et al. CaM kinase kinase beta-mediated activation of the growth regulatory kinase AMPK is required for androgen-dependent migration of prostate cancer cells. Cancer Res. 2011;71:528–37. doi: 10.1158/0008-5472.CAN-10-2581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Possik E, Jalali Z, Nouet Y, Yan M, Gingras MC, Schmeisser K, et al. Folliculin regulates ampk-dependent autophagy and metabolic stress survival. PLoS Genet. 2014;10:e1004273. doi: 10.1371/journal.pgen.1004273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Yan M, Gingras MC, Dunlop EA, Nouet Y, Dupuy F, Jalali Z, et al. The tumor suppressor folliculin regulates AMPK-dependent metabolic transformation. J Clin Invest. 2014;124:2640–50. doi: 10.1172/JCI71749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Bardeesy N, Sinha M, Hezel AF, Signoretti S, Hathaway NA, Sharpless NE, et al. Loss of the Lkb1 tumour suppressor provokes intestinal polyposis but resistance to transformation. Nature. 2002;419:162–7. doi: 10.1038/nature01045. [DOI] [PubMed] [Google Scholar]
- 28.Laderoute KR, Amin K, Calaoagan JM, Knapp M, Le T, Orduna J, et al. 5’-AMP-activated protein kinase (AMPK) is induced by low-oxygen and glucose deprivation conditions found in solid-tumor microenvironments. Mol Cell Biol. 2006;26:5336–47. doi: 10.1128/MCB.00166-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Fernandez MR, Henry MD, Lewis RE. Kinase suppressor of Ras 2 (KSR2) regulates tumor cell transformation via AMPK. Mol Cell Biol. 2012;32:3718–31. doi: 10.1128/MCB.06754-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kato K, Ogura T, Kishimoto A, Minegishi Y, Nakajima N, Miyazaki M, et al. Critical roles of AMP-activated protein kinase in constitutive tolerance of cancer cells to nutrient deprivation and tumor formation. Oncogene. 2002;21:6082–90. doi: 10.1038/sj.onc.1205737. [DOI] [PubMed] [Google Scholar]
- 31.Laderoute KR, Calaoagan JM, Chao WR, Dinh D, Denko N, Duellman S, et al. 5’-AMP-activated protein kinase (AMPK) supports the growth of aggressive experimental human breast cancer tumors. J Biol Chem. 2014;289:22850–64. doi: 10.1074/jbc.M114.576371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Hindupur SK, Balaji SA, Saxena M, Pandey S, Sravan GS, Heda N, et al. Identification of a novel AMPK-PEA15 axis in the anoikis-resistant growth of mammary cells. Breast Cancer Res. 2014;16:420. doi: 10.1186/s13058-014-0420-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Godlewski J, Nowicki MO, Bronisz A, Nuovo G, Palatini J, De Lay M, et al. MicroRNA-451 regulates LKB1/AMPK signaling and allows adaptation to metabolic stress in glioma cells. Mol Cell. 2010;37:620–32. doi: 10.1016/j.molcel.2010.02.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Borgdorff V, Rix U, Winter GE, Gridling M, Müller AC, Breitwieser FP, et al. A chemical biology approach identifies AMPK as a modulator of melanoma oncogene MITF. Oncogene. 2014;33:2531–9. doi: 10.1038/onc.2013.185. [DOI] [PubMed] [Google Scholar]
- 35.Ros S, Santos CR, Moco S, Baenke F, Kelly G, Howell M, et al. Functional metabolic screen identifies 6-phosphofructo-2-kinase/fructose-2,6-biphosphatase 4 as an important regulator of prostate cancer cell survival. Cancer Discov. 2012;2:328–43. doi: 10.1158/2159-8290.CD-11-0234. [DOI] [PubMed] [Google Scholar]
- 36.Jeon SM, Chandel NS, Hay N. AMPK regulates NADPH homeostasis to promote tumour cell survival during energy stress. Nature. 2012;485:661–5. doi: 10.1038/nature11066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.She C, Zhu LQ, Zhen YF, Wang XD, Dong QR. Activation of AMPK protects against hydrogen peroxide-induced osteoblast apoptosis through autophagy induction and NADPH maintenance: new implications for osteonecrosis treatment? Cell Signal. 2014;26:1–8. doi: 10.1016/j.cellsig.2013.08.046. [DOI] [PubMed] [Google Scholar]
- 38.Hawley SA, Boudeau J, Reid JL, Mustard KJ, Udd L, Mäkela TP, et al. Complexes between the LKB1 tumor suppressor, STRAD alpha/beta and MO25 alpha/beta are upstream kinases in the AMP-activated protein kinase cascade. J Biol. 2003;2:28. doi: 10.1186/1475-4924-2-28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Woods A, Johnstone SR, Dickerson K, Leiper FC, Fryer LG, Neumann D, et al. LKB1 is the upstream kinase in the AMP-activated protein kinase cascade. Curr Biol. 2003;13:2004–8. doi: 10.1016/j.cub.2003.10.031. [DOI] [PubMed] [Google Scholar]
- 40.Corradetti MN, Inoki K, Bardeesy N, DePinho RA, Guan KL. Regulation of the TSC pathway by LKB1: evidence of a molecular link between tuberous sclerosis complex and Peutz-Jeghers syndrome. Genes Dev. 2004;18:1533–8. doi: 10.1101/gad.1199104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Inoki K, Zhu T, Guan KL. TSC2 mediates cellular energy response to control cell growth and survival. Cell. 2003;115:577–90. doi: 10.1016/s0092-8674(03)00929-2. [DOI] [PubMed] [Google Scholar]
- 42.Hemminki A, Markie D, Tomlinson I, Avizienyte E, Roth S, Loukola A, et al. A serine/threonine kinase gene defective in Peutz-Jeghers syndrome. Nature. 1998;391:184–7. doi: 10.1038/34432. [DOI] [PubMed] [Google Scholar]
- 43.Shackelford DB, Shaw RJ. The LKB1-AMPK pathway: metabolism and growth control in tumour suppression. Nat Rev Cancer. 2009;9:563–75. doi: 10.1038/nrc2676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Ji H, Ramsey MR, Hayes DN, Fan C, McNamara K, Kozlowski P, et al. LKB1 modulates lung cancer differentiation and metastasis. Nature. 2007;448:807–10. doi: 10.1038/nature06030. [DOI] [PubMed] [Google Scholar]
- 45.Carretero J, Shimamura T, Rikova K, Jackson AL, Wilkerson MD, Borgman CL, et al. Integrative genomic and proteomic analyses identify targets for Lkb1-deficient metastatic lung tumors. Cancer Cell. 2010;17:547–59. doi: 10.1016/j.ccr.2010.04.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Xu C, Fillmore CM, Koyama S, Wu H, Zhao Y, Chen Z, et al. Loss of Lkb1 and Pten leads to lung squamous cell carcinoma with elevated PD-L1 expression. Cancer Cell. 2014;25:590–604. doi: 10.1016/j.ccr.2014.03.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Dupuy F, Griss T, Blagih J, Bridon G, Avizonis D, Ling C, et al. LKB1 is a central regulator of tumor initiation and pro-growth metabolism in ErbB2-mediated breast cancer. Cancer Metab. 2013;1:18. doi: 10.1186/2049-3002-1-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Cheng H, Liu P, Zhang F, Xu E, Symonds L, Ohlson CE, et al. A genetic mouse model of invasive endometrial cancer driven by concurrent loss of Pten and Lkb1 Is highly responsive to mTOR inhibition. Cancer Res. 2014;74:15–23. doi: 10.1158/0008-5472.CAN-13-0544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Contreras CM, Gurumurthy S, Haynie JM, Shirley LJ, Akbay EA, Wingo SN, et al. Loss of Lkb1 provokes highly invasive endometrial adenocarcinomas. Cancer Res. 2008;68:759–66. doi: 10.1158/0008-5472.CAN-07-5014. [DOI] [PubMed] [Google Scholar]
- 50.Contreras CM, Akbay EA, Gallardo TD, Haynie JM, Sharma S, Tagao O, et al. Lkb1 inactivation is sufficient to drive endometrial cancers that are aggressive yet highly responsive to mTOR inhibitor monotherapy. Dis Model Mech. 2010;3:181–93. doi: 10.1242/dmm.004440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Mohseni M, Sun J, Lau A, Curtis S, Goldsmith J, Fox VL, et al. A genetic screen identifies an LKB1-MARK signalling axis controlling the Hippo-YAP pathway. Nat Cell Biol. 2014;16:108–17. doi: 10.1038/ncb2884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Goodwin JM, Svensson RU, Lou HJ, Winslow MM, Turk BE, Shaw RJ. An AMPK-independent signaling pathway downstream of the LKB1 tumor suppressor controls Snail1 and metastatic potential. Mol Cell. 2014;55:436–50. doi: 10.1016/j.molcel.2014.06.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Faubert B, Vincent EE, Griss T, Samborska B, Izreig S, Svensson RU, et al. Loss of the tumor suppressor LKB1 promotes metabolic reprogramming of cancer cells via HIF-1α. Proc Natl Acad Sci U S A. 2014;111:2554–9. doi: 10.1073/pnas.1312570111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Evans JM, Donnelly LA, Emslie-Smith AM, Alessi DR, Morris AD. Metformin and reduced risk of cancer in diabetic patients. BMJ. 2005;330:1304–5. doi: 10.1136/bmj.38415.708634.F7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Decensi A, Puntoni M, Goodwin P, Cazzaniga M, Gennari A, Bonanni B, et al. Metformin and cancer risk in diabetic patients: a systematic review and meta-analysis. Cancer Prev Res (Phila) 2010;3:1451–61. doi: 10.1158/1940-6207.CAPR-10-0157. [DOI] [PubMed] [Google Scholar]
- 56.Rosilio C, Lounnas N, Nebout M, Imbert V, Hagenbeek T, Spits H, et al. The metabolic perturbators metformin, phenformin and AICAR interfere with the growth and survival of murine PTEN-deficient T cell lymphomas and human T-ALL/T-LL cancer cells. Cancer Lett. 2013;336:114–26. doi: 10.1016/j.canlet.2013.04.015. [DOI] [PubMed] [Google Scholar]
- 57.El-Masry OS, Brown BL, Dobson PR. Effects of activation of AMPK on human breast cancer cell lines with different genetic backgrounds. Oncol Lett. 2012;3:224–8. doi: 10.3892/ol.2011.458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Buzzai M, Jones RG, Amaravadi RK, Lum JJ, DeBerardinis RJ, Zhao F, et al. Systemic treatment with the antidiabetic drug metformin selectively impairs p53-deficient tumor cell growth. Cancer Res. 2007;67:6745–52. doi: 10.1158/0008-5472.CAN-06-4447. [DOI] [PubMed] [Google Scholar]
- 59.Appleyard MV, Murray KE, Coates PJ, Wullschleger S, Bray SE, Kernohan NM, et al. Phenformin as prophylaxis and therapy in breast cancer xenografts. Br J Cancer. 2012;106:1117–22. doi: 10.1038/bjc.2012.56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Liu X, Chhipa RR, Pooya S, Wortman M, Yachyshin S, Chow LM, et al. Discrete mechanisms of mTOR and cell cycle regulation by AMPK agonists independent of AMPK. Proc Natl Acad Sci U S A. 2014;111:E435–44. doi: 10.1073/pnas.1311121111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Shackelford DB, Abt E, Gerken L, Vasquez DS, Seki A, Leblanc M, et al. LKB1 inactivation dictates therapeutic response of non-small cell lung cancer to the metabolism drug phenformin. Cancer Cell. 2013;23:143–58. doi: 10.1016/j.ccr.2012.12.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Vincent EE, Coelho PP, Blagih J, Griss T, Viollet B, Jones RG, et al. Differential effects of AMPK agonists on cell growth and metabolism. Oncogene. 2014 Sep 22; doi: 10.1038/onc.2014.301. [Epub]. http://dx.doi.org/10.1038/onc.2014.301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Steinberg GR, Kemp BE. AMPK in health and disease. Physiol Rev. 2009;89:1025–78. doi: 10.1152/physrev.00011.2008. [DOI] [PubMed] [Google Scholar]
- 64.Viollet B, Horman S, Leclerc J, Lantier L, Foretz M, Billaud M, et al. AMPK inhibition in health and disease. Crit Rev Biochem Mol Biol. 2010;45:276–95. doi: 10.3109/10409238.2010.488215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Mantovani J, Roy R. Re-evaluating the general(ized) roles of AMPK in cellular metabolism. FEBS Lett. 2011;585:967–72. doi: 10.1016/j.febslet.2010.12.015. [DOI] [PubMed] [Google Scholar]
- 66.Platt FM, Hurst CD, Taylor CF, Gregory WM, Harnden P, Knowles MA. Spectrum of phosphatidylinositol 3-kinase pathway gene alterations in bladder cancer. Clin Cancer Res. 2009;15:6008–17. doi: 10.1158/1078-0432.CCR-09-0898. [DOI] [PubMed] [Google Scholar]
- 67.Dinney CP, McConkey DJ, Millikan RE, Wu X, Bar-Eli M, Adam L, et al. Focus on bladder cancer. Cancer Cell. 2004;6:111–6. doi: 10.1016/j.ccr.2004.08.002. [DOI] [PubMed] [Google Scholar]
- 68.Mo L, Zheng X, Huang HY, Shapiro E, Lepor H, Cordon-Cardo C, et al. Hyperactivation of Ha-ras oncogene, but not Ink4a/Arf deficiency, triggers bladder tumorigenesis. J Clin Invest. 2007;117:314–25. doi: 10.1172/JCI30062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Sibley K, Cuthbert-Heavens D, Knowles MA. Loss of heterozygosity at 4p16.3 and mutation of FGFR3 in transitional cell carcinoma. Oncogene. 2001;20:686–91. doi: 10.1038/sj.onc.1204110. [DOI] [PubMed] [Google Scholar]
- 70.Seager CM, Puzio-Kuter AM, Patel T, Jain S, Cordon-Cardo C, Mc Kiernan J, et al. Intravesical delivery of rapamycin suppresses tumorigenesis in a mouse model of progressive bladder cancer. Cancer Prev Res (Phila) 2009;2:1008–14. doi: 10.1158/1940-6207.CAPR-09-0169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.He F, Mo L, Zheng XY, Hu C, Lepor H, Lee EY, et al. Deficiency of pRb family proteins and p53 in invasive urothelial tumorigenesis. Cancer Res. 2009;69:9413–21. doi: 10.1158/0008-5472.CAN-09-2158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Chatterjee SJ, Datar R, Youssefzadeh D, George B, Goebell PJ, Stein JP, et al. Combined effects of p53, p21, and pRb expression in the progression of bladder transitional cell carcinoma. J Clin Oncol. 2004;22:1007–13. doi: 10.1200/JCO.2004.05.174. [DOI] [PubMed] [Google Scholar]
- 73.Feng Z, Hu W, de Stanchina E, Teresky AK, Jin S, Lowe S, et al. The regulation of AMPK beta1, TSC2, and PTEN expression by p53: stress, cell and tissue specificity, and the role of these gene products in modulating the IGF-1-AKT-mTOR pathways. Cancer Res. 2007;67:3043–53. doi: 10.1158/0008-5472.CAN-06-4149. [DOI] [PubMed] [Google Scholar]
- 74.Liu Z, Li X, Simoneau AR, Jafari M, Zi X. Rhodiola rosea extracts and salidroside decrease the growth of bladder cancer cell lines via inhibition of the mTOR pathway and induction of autophagy. Mol Carcinog. 2012;51:257–67. doi: 10.1002/mc.20780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Papanas N, Maltezos E, Mikhailidis DP. Metformin and cancer: licence to heal? Expert Opin Investig Drugs. 2010;19:913–7. doi: 10.1517/13543784.2010.499122. [DOI] [PubMed] [Google Scholar]
- 76.Zhang T, Wang X, He D, Jin X, Guo P. Metformin sensitizes human bladder cancer cells to TRAIL-induced apoptosis through mTOR/S6K1-mediated downregulation of c-FLIP. Anticancer Drugs. 2014;25:887–97. doi: 10.1097/CAD.0000000000000116. [DOI] [PubMed] [Google Scholar]
- 77.Yan S, Yang X, Chen T, Xi Z, Jiang X. The PPARγ agonist Troglitazone induces autophagy, apoptosis and necroptosis in bladder cancer cells. Cancer Gene Ther. 2014;21:188–93. doi: 10.1038/cgt.2014.16. [DOI] [PubMed] [Google Scholar]
- 78.Hay N. The Akt-mTOR tango and its relevance to cancer. Cancer Cell. 2005;8:179–83. doi: 10.1016/j.ccr.2005.08.008. [DOI] [PubMed] [Google Scholar]
- 79.Sabatini DM. mTOR and cancer: insights into a complex relationship. Nat Rev Cancer. 2006;6:729–34. doi: 10.1038/nrc1974. [DOI] [PubMed] [Google Scholar]
- 80.Puzio-Kuter AM, Castillo-Martin M, Kinkade CW, Wang X, Shen TH, Matos T, et al. Inactivation of p53 and Pten promotes invasive bladder cancer. Genes Dev. 2009;23:675–80. doi: 10.1101/gad.1772909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Qian CN, Furge KA, Knol J, Huang D, Chen J, Dykema KJ, et al. Activation of the PI3K/AKT pathway induces urothelial carcinoma of the renal pelvis: identification in human tumors and confirmation in animal models. Cancer Res. 2009;69:8256–64. doi: 10.1158/0008-5472.CAN-09-1689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Cappellen D, Gil Diez de Medina S, Chopin D, Thiery JP, Radvanyi F. Frequent loss of heterozygosity on chromosome 10q in muscle-invasive transitional cell carcinomas of the bladder. Oncogene. 1997;14:3059–66. doi: 10.1038/sj.onc.1201154. [DOI] [PubMed] [Google Scholar]
- 83.Pymar LS, Platt FM, Askham JM, Morrison EE, Knowles MA. Bladder tumour-derived somatic TSC1 missense mutations cause loss of function via distinct mechanisms. Hum Mol Genet. 2008;17:2006–17. doi: 10.1093/hmg/ddn098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Jin X, Yun SJ, Jeong P, Kim IY, Kim WJ, Park S. Diagnosis of bladder cancer and prediction of survival by urinary metabolomics. Oncotarget. 2014;5:1635–45. doi: 10.18632/oncotarget.1744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Kim WJ, Kim EJ, Kim SK, Kim YJ, Ha YS, Jeong P, et al. Predictive value of progression-related gene classifier in primary non-muscle invasive bladder cancer. Mol Cancer. 2010;9:3. doi: 10.1186/1476-4598-9-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Samudio I, Harmancey R, Fiegl M, Kantarjian H, Konopleva M, Korchin B, et al. Pharmacologic inhibition of fatty acid oxidation sensitizes human leukemia cells to apoptosis induction. J Clin Invest. 2010;120:142–56. doi: 10.1172/JCI38942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Bebernitz GR, Schuster HF. The impact of fatty acid oxidation on energy utilization: targets and therapy. Curr Pharm Des. 2002;8:1199–227. doi: 10.2174/1381612023394692. [DOI] [PubMed] [Google Scholar]
- 88.Sekoguchi E, Sato N, Yasui A, Fukada S, Nimura Y, Aburatani H, et al. A novel mitochondrial carnitine-acylcarnitine translocase induced by partial hepatectomy and fasting. J Biol Chem. 2003;278:38796–802. doi: 10.1074/jbc.M306372200. [DOI] [PubMed] [Google Scholar]
- 89.Camacho JA, Rioseco-Camacho N. The human and mouse SLC25A29 mitochondrial transporters rescue the deficient ornithine metabolism in fibroblasts of patients with the hyperornithinemiahyperammonemia-homocitrullinuria (HHH) syndrome. Pediatr Res. 2009;66:35–41. doi: 10.1203/PDR.0b013e3181a283c1. [DOI] [PubMed] [Google Scholar]
- 90.Hirsch HA, Iliopoulos D, Joshi A, Zhang Y, Jaeger SA, Bulyk M, et al. A transcriptional signature and common gene networks link cancer with lipid metabolism and diverse human diseases. Cancer Cell. 2010;17:348–61. doi: 10.1016/j.ccr.2010.01.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Menendez JA, Lupu R. Fatty acid synthase and the lipogenic phenotype in cancer pathogenesis. Nat Rev Cancer. 2007;7:763–77. doi: 10.1038/nrc2222. [DOI] [PubMed] [Google Scholar]
- 92.Warburg O. On the origin of cancer cells. Science. 1956;123:309–14. doi: 10.1126/science.123.3191.309. [DOI] [PubMed] [Google Scholar]
- 93.Kim I, He YY. Targeting the AMP-activated protein kinase for cancer prevention and therapy. Front Oncol. 2013;3:175. doi: 10.3389/fonc.2013.00175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Ben Sahra I, Regazzetti C, Robert G, Laurent K, Le Marchand-Brustel Y, Auberger P, et al. Metformin, independent of AMPK, induces mTOR inhibition and cell-cycle arrest through REDD1. Cancer Res. 2011;71:4366–72. doi: 10.1158/0008-5472.CAN-10-1769. [DOI] [PubMed] [Google Scholar]
- 95.Nair V, Sreevalsan S, Basha R, Abdelrahim M, Abudayyeh A, Rodrigues Hoffman A, et al. Mechanism of metformin-dependent inhibition of mammalian target of rapamycin (mTOR) and Ras activity in pancreatic cancer: role of specificity protein (Sp) transcription factors. J Biol Chem. 2014;289:27692–701. doi: 10.1074/jbc.M114.592576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Zhou G, Myers R, Li Y, Chen Y, Shen X, Fenyk-Melody J, et al. Role of AMP-activated protein kinase in mechanism of metformin action. J Clin Invest. 2001;108:1167–74. doi: 10.1172/JCI13505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Sinnett SE, Brenman JE. Past strategies and future directions for identifying AMP-activated protein kinase (AMPK) modulators. Pharmacol Ther. 2014;143:111–8. doi: 10.1016/j.pharmthera.2014.02.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Laderoute KR, Calaoagan JM, Madrid PB, Klon AE, Ehrlich PJ. SU11248 (sunitinib) directly inhibits the activity of mammalian 5’-AMP-activated protein kinase (AMPK) Cancer Biol Ther. 2010;10:68–76. doi: 10.4161/cbt.10.1.12162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Steinberg GR, Watt MJ, Febbraio MA. Cytokine Regulation of AMPK signalling. Front Biosci (Landmark Ed) 2009;14:1902–16. doi: 10.2741/3350. [DOI] [PubMed] [Google Scholar]
- 100.Jeon SM, Hay N. The double-edged sword of AMPK signaling in cancer and its therapeutic implications. Arch Pharm Res. 2015;38:346–57. doi: 10.1007/s12272-015-0549-z. [DOI] [PMC free article] [PubMed] [Google Scholar]