

Abstract
Background and purpose
4′-O-methylhonokiol (MH) is a natural product showing anti-inflammatory, anti-osteoclastogenic, and neuroprotective effects. MH was reported to modulate cannabinoid CB2 receptors as an inverse agonist for cAMP production and an agonist for intracellular [Ca2+]. It was recently shown that MH inhibits cAMP formation via CB2 receptors. In this study, the exact modulation of MH on CB2 receptor activity was elucidated and its endocannabinoid substrate-specific inhibition (SSI) of cyclooxygenase-2 (COX-2) and CNS bioavailability are described for the first time.
Methods
CB2 receptor modulation ([35S]GTPγS, cAMP, and β-arrestin) by MH was measured in hCB2-transfected CHO-K1 cells and native conditions (HL60 cells and mouse spleen). The COX-2 SSI was investigated in RAW264.7 cells and in Swiss albino mice by targeted metabolomics using LC-MS/MS.
Results
MH is a CB2 receptor agonist and a potent COX-2 SSI. It induced partial agonism in both the [35S]GTPγS binding and β-arrestin recruitment assays while being a full agonist in the cAMP pathway. MH selectively inhibited PGE2 glycerol ester formation (over PGE2) in RAW264.7 cells and significantly increased the levels of 2-AG in mouse brain in a dose-dependent manner (3 to 20 mg kg−1) without affecting other metabolites. After 7 h from intraperitoneal (i.p.) injection, MH was quantified in significant amounts in the brain (corresponding to 200 to 300 nM).
Conclusions
LC-MS/MS quantification shows that MH is bioavailable to the brain and under condition of inflammation exerts significant indirect effects on 2-AG levels. The biphenyl scaffold might serve as valuable source of dual CB2 receptor modulators and COX-2 SSIs as demonstrated by additional MH analogs that show similar effects. The combination of CB2 agonism and COX-2 SSI offers a yet unexplored polypharmacology with expected synergistic effects in neuroinflammatory diseases, thus providing a rationale for the diverse neuroprotective effects reported for MH in animal models.
Electronic supplementary material
The online version of this article (doi:10.1186/s12974-015-0307-7) contains supplementary material, which is available to authorized users.
Keywords: 4′-O-methylhonokiol, Endocannabinoids, COX-2, CB2 receptor, Polypharmacology, Partial agonist, Endocannabinoid system, 2-arachidonoyl glycerol, Magnolia grandiflora
Introduction
4′-O-methylhokiol (MH) is the major bioactive constituent of Magnolia grandiflora L. seed oil and shows pronounced anti-inflammatory, anti-osteoclastogenic, and neuroprotective effects [1-4]. In mouse models, MH prevents LPS-induced memory loss and β-amyloid (1 to42) accumulation [2]. Moreover, MH appears to efficiently attenuate the development of Alzheimer’s disease in Tg2576 transgenic mice [3]. The proposed underlying mechanisms appear to be linked to the inhibition of nuclear factor kappa B, the gene expression of inducible nitric oxide synthase and cyclooxygenase-2 (COX-2), as well as cannabinoid type-2 (CB2) receptors [2,5]. MH and some derivatives potently inhibit COX-2 with poor selectivity towards the constitutive isoform COX-1 [6,7]. Furthermore, MH attenuates different signaling cascades related to oxidative stress and MAP kinases, inhibits moderately acetylcholinesterase activity (see [5] for review), and it also acts as positive allosteric GABAA receptor ligand [8]. Schuehly et al. [1] for the first time described the relatively potent and selective CB2 receptor binding of MH and different analogs. MH was shown to exert an unusual dualistic modulation of CB2 receptor activity, acting as an inverse agonist at the cAMP pathway while behaving as an agonist at increasing the intracellular levels of [Ca2+] [1]. The CB2 receptor inverse agonism on the cAMP pathway was described as the relevant mechanism associated to anti-osteoclastogenesis effects described in mouse RAW264.7 cells and primary human monocytes/macrophages. Other authors recently reported that MH behaves as a CB2 receptor agonist in the cAMP pathway [9], pointing out discrepancies in receptor assays, which gave impetus to the present in-depth study on the CB2 receptor pharmacology of MH. The main degradation route of endocannabinoids (ECs) is their hydrolysis by fatty acid amide hydrolase (FAAH) (for anandamide (AEA)) and monoacylglycerol lipase/α, β-hydrolases (MAGL/ABHDs) (for 2-arachidonoyl glycerol (2-AG)), but ECs might undergo additional enzymatic modifications including oxygenation mediated by COX-2, lipoxygenases (LOXs), and cytochrome P450 (CYP450) [10-12]. Several studies emphasized the relevance of COX-2-mediated oxygenation of ECs in vitro and in vivo under specific conditions like inflammation and in FAAH knock-out animals [13-15]. EC oxygenation might be particularly relevant in inflammation or tissues that constitutively express the inducible form of COX such as the brain, kidney, and spinal cord [16-18]. Upon COX-2 activity, AEA and 2-AG are converted into prostaglandin ethanolamides (prostamides) and prostaglandin glycerol esters (PG-GEs), respectively [10,11]. Prostamides and PG-GEs exert pro-inflammatory actions probably by interacting with specific targets that are different from cannabinoid and classic prostaglandin receptors. A heterodimeric association between the wild-type prostaglandin F (FP) receptor and an alternative splicing variant (Alt4) of such receptors was identified as molecular target for prostamideF2α [19]. The hyperalgesic actions of prostamideF2α were then confirmed by using selective receptor antagonists which showed antinociceptive effects in vitro and in vivo [15,20]. The inhibition of prostamide and PG-GEs formation by COX-2 substrate-specific inhibitors (SSIs) has been reported. Intriguingly, such inhibitors can lead to an increase of AEA and 2-AG levels without affecting the levels of arachidonic acid (AA) and PGE2 [14,21,22]. Duggan et al. [14] recently showed that (R)-flurbiprofen acts as COX-2 SSI by increasing EC levels and by reducing prostamide formation without affecting AA and PGE2 levels. In addition, the morpholino amide derivative of indomethacin LM-4131 showed potent anxiolytic effects in several animal models by selectively inhibiting EC oxygenation, increasing levels of AEA and 2-AG in the brain [21]. COX-2 SSI might represent a promising anti-inflammatory strategy to circumvent the typical gastrointestinal and cardiovascular aversive effects of the non-selective COX-2 inhibitors.
Here, we show that MH and some derivatives exert dual and potentially synergistic actions on the endocannabinoid system (ECS) by acting as central nervous system (CNS) penetrating SSIs of COX-2 and as CB2 receptor agonists. We provide detailed analyses on CB2 receptor signaling and COX-2-mediated oxygenation of ECs by using purified hCOX-2, RAW264.7 cells, and LPS-challenged mouse brain.
Methods
Materials
[Ethanolamine-1-3H]-anandamide and [1,2,3-3H]-2-arachidonyl glycerol were obtained from American Radiolabeled Chemicals (St. Louis, MO, USA), while the [3H] CP-55,940 was obtained from PerkinElmer Life Sciences (Waltham, MA, USA). AM630, WIN55-212, AEA, 2-AG, DuP-697, arachidonic acid-d8 (5Z,8Z,11Z,14Z-eicosatetraenoic-5,6,8,9,11,12,14,15-d8 acid), AEA-d4 (N-(2-hydroxyethyl-1,1,2,2-d4)-5Z,8Z,11Z,14Z-eicosatetraenamide) 2-AG-d5 (5Z,8Z,11Z,14Z-eicosatetraenoic acid, 2-glyceryl-1,1,2,3,3-d5 ester), LEA-d4 (N-(2-hydroxyethyl-1,1,2,2-d4)-9Z,12Z-octadecadienamide); OEA-d4 (N-(2-hydroxyethyl-1,1,2,2-d4)-9Z-octadecenamide), PEA-d5 (N-(2-hydroxyethyl)-hexadecanamide-15,15,16,16,16-d5); PGE2-d4 (9-oxo-11α,15S-dihydroxy-prosta-5Z,13E-dien-1-oic-3,3,4,4-d4 acid); and PGE2-1-glyceryl ester-d5 (9-oxo-11α,15S-dihydroxy-prosta-5Z,13E-dien-1-oic acid,1-glyceryl ester-d5) were obtained from Cayman Chemicals Europe (Ann Arbor, MI, USA). MH-d6 (2-(4-methoxy-3-prop-2-enylphenyl (d))-4-prop-2-enylphenol (d)). The structures of the MH derivatives were previously published in Schühly et al. [7] and Schuehly et al. [1]. The procedure of isolation and purification of MH and the synthesis of its derivatives was previously reported in [1,7]. All tubes used for assays (plastic and glass) were silanized.
Animals
All study involving animals are reported in accordance with the Swiss Federal guidelines. Female RjOrl: SWISS (60) 7 to 8 weeks old, provided by Janvier Labs (St Berthevin, France), were used for these experiments. The mice were housed in groups of five per cage in a selected pathogen-free unit under controlled 12-h light/12-h dark cycle, ambient temperature 21°C ± 1°C humidity 40% to 50% with free access to standard rodent chow and water. The mice were acclimatized to the Animal House for at least 3 days before the experiment.
Radioligand displacement assays on hCB1 and hCB2 receptors
Receptor binding experiments were performed with membrane preparations as previously reported [23]. Briefly, clean membranes expressing hCB1 or hCB2 were resuspended in binding buffer (50 mM Tris-HCl, 2.5 mM EDTA, 5 mM MgCl2, 0.5 mg mL−1 fatty acid-free bovine serum albumin (BSA), pH 7.4) and incubated with vehicle or compounds and 0.5 nM of [3H] CP55,940 for 2 h at 30°C. Non-specific binding was determined in the presence of 10 μM of WIN55,512. After incubation, membranes were filtered through a pre-soaked 96-well microplate bonded with GF/B filters under vacuum and washed twelve times with 150 μL of ice-cold binding buffer. The radioactivity was measured and the results expressed as [3H] CP55,940 binding.
[35S]GTPyS assay
Assays were performed as previously described [24]. Briefly, 5 μg of clean membranes expressing hCB2 were diluted in binding buffer (50 mM Tris-HCl, 3 mM MgCl2, 0.2 mM EGTA, and 100 mM NaCl at pH 7.4 plus 0.5% fatty acid-free BSA) in the presence of 10 μM of GDP and 0.1 nM of [35S]GTPγS. The mixture was kept on ice until the binding reaction was started by adding the vehicle or compounds. Non-specific binding was measured in the presence of 10 μM of GTPγS. The tubes were incubated at 30°C for 90 min. The reaction was stopped by rapid filtration through a 96-well microplate bonded with GF/B filters previously pre-soaked with washing buffer (50 mM of Tris-HCl pH 7.4 plus 0.1% fatty acid-free BSA). The filters were washed six times with 180 μL of washing buffer under vacuum. Experiments were performed in the presence and after removal of constitutive activity of CB2 receptors. In the latter, membranes were obtained from cells treated overnight with 10 μM of AM630 to shift the vast majority of the receptor population in the inactive state. After the incubation, cells were washed six times with 10 mL of culture medium for 10 min each. Finally, cells were detached and membranes prepared and used to perform [35S]GTPγS binding assays. Membrane obtained from mock-transfected CHO cells were used as negative control. Membranes derived from mouse spleen (20 μg) and human promyelocytic leukemia HL60 cells (40 μg) were used as sources of CB2 receptors expressed in native conditions. In order to reduce the basal [35S]GTPγS binding, the experiments were performed after 30-min pre-treatment with adenosine deaminase (5 units mL−1). The radioactivity was measured, and the results were expressed as [35S]GTPγS binding.
cAMP assay
cAMP assays were performed in CHO-hCB2 stably transfected with the pGloSensorTM 22-F plasmid (Promega, Madison, WI, USA) as previously described [22]. Briefly, cells were seeded in 96-well plate and kept in an equilibration medium (GloSensor™ cAMP reagent diluted in 5% of a CO2-independent medium plus 10% FBS) for 2 h at RT in the dark. Compounds or vehicle were diluted in an assay medium containing 1 μM of forskolin and 250 μM of 3-isobutyl-1-methylxanthine (IBMX). Chemiluminescence was recorded after 20 min. Non-specific signal was determined in cells with only IBMX. Experiments were performed in the presence and after removal of the constitutive activity of the receptors following the same procedure described above. Results were normalized by subtracting the residual cAMP production and expressed amount of cAMP formed.
β-arrestin assay
The PathHunter® β-arrestin cells (CHO-K1-HOMSA-CNR2) were gently donated by DiscoverX (via Hoffman La-Roche Ltd, Basel, Switzerland) and cultured in an F-12 medium containing 10% FBS, 200 μg mL−1 hygromycin and 800 μg mL−1 geneticin. For the experiments, 2 × 104 cells were seeded in 96-well plate and left overnight in the incubator. The day after, vehicle or compounds were diluted in 40 μL of a CO2-independent medium plus 10% FBS. The mixture was added to the cells and incubated for 90 min at 37°C. Chemiluminescence was detected using the PathHunter® detection system (DiscoveRx Corporation, Fremont, CA, USA) according to the instruction protocol. Background levels were subtracted and the results expressed as β-arrestin recruitment.
COX-2 inhibition assay in vitro
The experiments were performed as previously described [22]. Briefly, the inhibition of recombinant hCOX-2 was assessed using a COX fluorescent inhibitor screening assay kit from Cayman Chemicals Europe. Tested compounds or vehicle were pre-incubated with COX-FIS assay buffer (Tris-HCl 100 mM, pH 8), 1 μM of COX-FIS heme hCOX-2 FIS assay agent, and 30 μM of ADHP (10-acetyl-3,7-dihydroxyphenoxazine) fluorometric substrate for 15 min at RT. The reaction was started by adding arachidonic acid, AEA, or 2-AG (10 μM). Fluorescence intensity was measured (535/580 nm) after 5 min of incubation. The results were expressed as hCOX-2 activity.
LC-MS/MS quantification of ECs and PGs in RAW264.7 cells
In case of experiments with homogenates, cells were seeded in T75-cm2 flask and stimulated with LPS (1 μg mL−1) for 12 h at 37°C. After incubation, cells were homogenized in assay buffer (Tris-HCl 10 mM, pH 8) and 900 μg of total protein were used to perform the COX-2 assay. When using intact cells, 0.5 × 106 cells were seeded in a 24-well plate and challenged with LPS (1 μg mL−1) for 12 h at 37°C. The medium was discarded and replaced by a medium containing vehicle or compounds. After 30 min of incubation at 37°C, 2-AG (10 μM) was added and plates were incubated for an additional 30 min at 37°C. Further experiments were carried out without adding 2-AG but stimulating its cellular biosynthesis. In this setting, RAW264.7 cells were treated for 8 h with LPS (1 μg mL−1), ATP (1 mM), and thapsigargin (2 μM) in the presence of vehicle or compounds. In both experimental conditions, after the incubation, cells were detached, transferred into a chloroform:methanol mixture (2:1) containing the internal standards (ISs), and then, phosphate buffered saline (PBS) was added to the final ratio of 6:3:1.5 (chloroform:methanol:PBS). The suspension was vortexed, sonicated, and centrifuged for 5 min at 800 g at 4°C. The organic phase was recovered and dried under nitrogen. The quantification of AA, 2-AG, PGE2, and PGE2GE was performed using LC-MS/MS. The multiple reaction monitoring (MRM) parameters (precursor ion/product ion, declustering potential, collision energy) used in the survey and the respective IS are detailed in Gachet et al. [25] for all analytes except for: MH (Q): 279/264 m/z, −70 eV, −23 eV and (q): 279/248 m/z, −70 eV, −40 eV; MH-d6: 285/279 m/z, −82 eV, −24 eV; PGE2GE (Q): 444/409 m/z, 40 eV, 13 eV and (q): 444/373 m/z, 40 eV, 19 eV; and PGE2GE-d5: 449/396 m/z, 43 eV, 17 eV. The quantification was based on the area ratio of analytical standard/IS.
LC-MS/MS quantification of ECs and PGs in vivo
Swiss albino mice (females, 8 weeks old) were injected intraperitoneally (i.p.) with vehicle or MH at different doses (3, 10, and 20 mg kg−1). Female mice were used for ethical reasons because they were less susceptible for neuroinflammatory damage in this model. After 1 h, the animals (6 to 14 animals per group) were injected i.p. with LPS (2.5 mg kg−1) and kept in the dark for an additional 6 h. After anesthetization, mice were perfused with PBS and the brains were collected. Lipid extraction was performed as recently described (Gachet et al. [25]). In brief, half brain was homogenized on a BeadBeater (Mini-BeadBeater-24, BioSpec, Bartlesville, OK USA) using Chrome-Steel Beats (2.3 mm dia., BioSpec) in the presence of ice-cold chloroform:methanol (2:1) for 1 min (3,450 strokes min−1) at 4°C. Homogenates were added with PBS (containing ISs) to the final ratio of 6:3:1.5 (chloroform:methanol:PBS). The suspension was vortexed, sonicated, and centrifuged for 5 min at 800 g at 4°C. The organic phase was recovered and dried under nitrogen. Subsequently, the samples were reconstituted in ethanol and after the addition of water and pH adjustment (pH = 3), an aliquot was extracted using C-18Sep-Pak cartridge (Waters AG, Zug, Switzerland). Cartridges were washed with 10% ethanol and eluted with acetonitrile (ACN)/ethyl acetate (1:1). The eluates were collected and evaporated to dryness under nitrogen. The samples were reconstituted in ACN, centrifuged for 5 min at 16,100 g at 4°C, and measured by LC-MS/MS in negative and positive modes. The quantification was based on the area ratio of analytical standard/IS.
[3H]AEA and [3H]2-AG hydrolysis assay
The experiments were performed as previously described [23]. Briefly, vehicle or compounds were pre-incubated for 30 min at 37°C with 200 μg of pig brain homogenate for FAAH and MAGL and 100 μg of BV-2 cell homogenate for ABHDs in assay buffer (10 mM Tris-HCl, 1 mM EDTA, pH 7.6 plus 0.1% fatty acid-free BSA). To measure FAAH activity, a mixture of AEA/[3H] AEA was added to the homogenate (final concentration of 1 μM) and incubated for 15 min at 37°C. After incubation, tubes were added with chloroform:methanol (1:1), vortexed, and centrifuged at 10,000 rpm for 10 min at 4°C. To measure MAGL/ABHDs activity, AEA and [3H]AEA were replaced by 2-oleoylglycerol (2-OG) and [3H]2-OG, respectively. The radioactivity associated to the [3H]ethanolamine (or [3H]glycerol) formation was measured in the aqueous phase. The results were expressed as enzymatic activity.
[3H]AEA uptake into U937 cells
The experiments were performed as previously described [23]. Briefly, 106 of U937 cells were suspended in 0.5 mL of PBS and pre-incubated with vehicle or compounds for 30 min at 37°C. Successively, cells were added with a mixture of AEA/[3H]AEA (final concentration of 100 nM) and incubated for 5 min at 37°C. The uptake process was stopped by rapid centrifugation at 800 g for 5 min at 4°C. The supernatant was discarded and the pellet washed with ice-cold PBS plus 1% of fatty acid-free BSA. Cell pellets were resuspended in PBS and extracted with chloroform:methanol (1:1). The organic phase was collected and the radioactivity measured. The results were expressed as [3H]AEA uptake. In case of U937 cell-derived macrophages, U937 cells were treated for 48 h with 2 ng mL−1 of phorbol 12-myristate 13-acetate (PMA) and the uptake experiments were performed as described above.
Data analysis
Data were collected from at least three independent experiments each performed in triplicate. Results are expressed as mean values and standard deviation (SD). The [35S]GTPγS assays performed using CHO-hCB2 membranes were repeated five times, and the error was expressed as standard error of the mean (SEM). The statistical significance difference among groups was determined by Student’s t test (paired, two-tailed t test) or one-way ANOVA followed by Bonferroni’s post-test. Statistical differences between treated and control groups were considered as significant if P ≤ 0.05. GraphPad 5.0 software was used to fit the concentration-dependent curves and for the statistical analysis.
Results
First, we confirmed that MH (Figure 1) selectively binds to CB2 receptors with a Ki value of 188.5 (131.7 to 269.4) nM over 2.4 (1.9 to 2.9) μM for CB1 receptors (Table 1). MH induced a concentration-dependent increase of [35S]GTPγS binding with an EC50 value of 285.7 (125.6 to 647.2) nM and a maximal binding of 185.7% (167.1 to 204.3) (Figure 2A). In the same assay, the positive control 2-AG-induced [35S]GTPγS binding with an EC50 value of 74.0 (28.4 to 192.9) nM, reaching 252.8% (210.2 to 295.4) of maximal response. Statistical analyses confirmed that MH induced a lower maximal G-protein recruitment compared to the endogenous full agonist 2-AG, thus suggesting partial agonism. Partial agonists are versatile ligands which can behave as agonists or antagonists depending on the conditions [26]. In Figure 2B, the incubation of fixed concentrations of MH with increasing concentrations of 2-AG exemplifies these opposite pharmacological behaviors. While MH induced G-protein recruitment at inactive 2-AG concentrations, it reduced the effects induced by high concentrations of the full agonist. We also investigated the behavior of MH using human promyelocytic leukemia HL60 cells [27] and mouse spleen [28] which both endogenously express CB2 receptors. As reported in Figure 2C,D, 2-AG, CP55,940, and MH behaved as agonists by increasing the [35S]GTPγS binding compared to vehicle, while SR144528 and AM630 reduced G-protein recruitment, thus behaving as inverse agonists. In agreement with the results obtained in CHO-hCB2 membranes, MH induced a sub-maximal binding of [35S]GTPγS compared with the full agonists CP55,940 and 2-AG, in both HL60- and spleen-derived membranes, thus confirming its partial agonist behavior. In addition, we also tested 2-AG, CP55,940, and MH effects using membranes derived from mock-transfected CHO cells. All ligands showed no change of the basal level of [35S]GTPγS binding (Additional file 1: Figure S1). Altogether, these data indicate that MH behaves as an agonist at CB2 receptors in both overexpressing and native conditions. We next characterized the effects of MH on the CB2 receptor-mediated inhibition of forskolin-induced cAMP production. As shown in Figure 2E, increasing concentrations of MH inhibited cAMP formation with an IC50 value of 674 (481 to 944) nM. In this assay, in contrast to previous results [1], MH behaved as a full agonist producing the same maximal inhibition as the full agonist 2-AG (data not shown). CB2 receptors are known to possess a high intrinsic constitutive activity. Therefore, compounds that act as ‘protean agonists’ can behave differently depending on the proportion of constitutively activated receptors. ‘Protean agonists’ bind to the receptor and stabilize an active conformation that is capable of initiating signal where there is none (inactive state of the receptor), but they trigger an active state of the receptor that is less efficacious than the naturally occurring, spontaneously formed constitutive active state [29]. Thus, ‘protean agonists’ can behave as an agonist or inverse agonist depending on the active state of the receptor. This phenomenon has been accurately investigated over the past few years, and it was found that by pre-treating CB2 receptors overnight with the inverse agonist AM630, most of the receptors shift towards the inactive state, thus abolishing the constitutive activity [30]. For example, an inverse agonist becomes a silent antagonist [30] and a protean agonist can switch from an inverse agonist to an agonist or antagonist [30,31]. We have validated our assay system by showing that after removing the constitutive activity, AM630 and SR144528 switched from an inverse agonist to a silent antagonist, while CP55,940 did not change its behavior (Additional file 1: Figure S2) in agreement with the literature [30,32]. As shown in Figure 2E, MH acted as a full agonist in both conditions, suggesting that in our assay, the level of constitutive activity of CB2 receptors did not qualitatively affect the pharmacological behavior of MH. We also investigated the effect of MH on β-arrestin recruitment. In Figure 2F, it is clearly shown that similarly to [35S]GTPγS results, the compound induced a sub-maximal effect compared to CP55,940, therefore behaving as a partial agonist. MH has been shown to inhibit the COX-2-mediated AA oxygenation [7]. However, COX-2 is responsible also for the oxygenation of AEA and 2-AG leading to the formation of PG-EAs and PG-GEs, respectively [10,14,21,33]. Therefore, we next studied whether MH selectively inhibits the COX-2-mediated oxygenation of ECs. In Figure 3A, the concentration-dependent inhibition of purified hCOX-2 is shown using 10 μM of AEA, 2-AG, and AA as substrates. Noteworthy, MH inhibited the oxygenation of ECs with IC50 values of 158 (123 to 204) nM and 156 (129 to 189) nM for AEA and 2-AG, respectively, while it blocked AA oxygenation with an IC50 value of 588 (499 to 693) nM (Table 1). We then confirmed the apparent SSI of COX-2 in LPS-stimulated RAW264.7 cells by using cell homogenates and intact cells. We quantified the amount of PGE2 and PGE2-GE formation by LC-MS/MS analysis after incubation for 30 min with 10 μM of 2-AG. As shown in Figure 3B,C, MH inhibited PGE2-GE formation with a higher potency (IC50 value: 301 (257 to 490) nM) compared to PGE2 (IC50 value: 1.21 (0.792 to 1.81) μM) in both systems. The mass chromatograms of PGE2 and PGE2-GE formed upon incubation of LPS-stimulated RAW264.7 cell homogenates as well as from intact cells incubated with 2-AG are shown in Figure 4. The LC-MS/MS method was validated by measuring the non-selective COX-2 inhibitor DuP-697 which showed the same potency in inhibiting the oxygenation of ECs and AA (Figure 3A,B,C,D). RAW264.7 cells can be stimulated to produce significant levels of 2-AG by raising intracellular levels of [Ca2+]. Cells were stimulated for 8 h with LPS, ATP, and thapsigargin in the presence of vehicle or different concentrations of MH. Figure 5A, B shows the concentration-dependent inhibition of PGE2 and PGE2-GE formation by MH. Intriguingly, already at low concentrations (10 to 30 nM), MH led to 20% to 25% inhibition of 2-AG oxygenation while AA oxygenation was not affected. At high concentrations (100 to 300 nM), MH inhibited the formation of both prostaglandin types but with more pronounced effects on PGE2-GE. At 1 μM, MH almost fully inhibited COX-2-mediated oxygenation of AA and 2-AG lacking the substrate specificity (Figure 5C). Since the absolute amount of PGE2 formed was about 100-times higher than PGE2-GE, we wanted to exclude a potential bias in the sensitivity of the system due to different amounts of the initial substrates (AA and 2-AG). Therefore, we tested the effects of DuP-697, and the results confirmed that this non-selective inhibitor potently blocked the formation of PGE2 and PGE2-GE with the same potency (Additional file 1: Figure S3E). To assess the relevance of COX-2 SSI in vivo, we evaluated the effects of MH in LPS-challenged Swiss albino mice. Animals were injected intraperitoneally (i.p.) with vehicle or different concentrations of MH and after 1 h, challenged for 6 h with LPS. Brain levels of AEA, 2-AG, AA, and PGE2, expressed as nmol (or pmol) mg−1 of tissue are shown in Figure 6A,B,C,D. Upon LPS injection, the amount of AA, 2-AG, and AEA did not significantly change, while PGE2 and corticosterone strongly increased (Figure 6E), as expected, and in agreement with the literature [34,35]. MH treatment did not affect the amount of AA and corticosterone at any concentration, while it slightly reduced the levels of PGE2 only at 20 mg kg−1. MH did not significantly affect AEA levels, while significantly increasing the amount of free 2-AG at 10 and 20 mg kg−1, with a tendency of increase also at the lowest dose tested (3 mg kg−1). Since 2-AG levels are about 500 to 1,000 times higher than AEA levels in Swiss albino mice, and assuming that both ECs are equipotent substrates for COX-2, it is not unexpected to see this more pronounced effect (doubling the amount) of a SSI COX-2 inhibitor on 2-AG levels. PGE2-EA and PGE2-GE were below the limit of detection. Moreover, NA-GABA (N-arachidonoyl GABA) and other N-acylethanolamines were not significantly affected by MH (Additional file 1: Figure S4). These findings suggest that MH maintained the ability to specifically inhibit COX-2-mediated EC oxygenation in vivo without affecting the oxygenation of AA. Several publications have recently demonstrated in vitro and in vivo beneficial effects of MH in neurodegenerative disease models [2,4,36]. Nonetheless, the penetration of the compound across the blood-brain barrier has never been investigated. We therefore established an LC-MS/MS method to quantify MH in the brain. Data show that after 7 h after the injection of 3, 10, and 20 mg kg−1, the brain contains 120.1 (93.6 to 147.5), 239.8 (121.1 to 342.3), and 350.2 (248.1 to 426.9) pmol g−1 of MH, respectively (Figure 6F). These results correlate with the significant increase of EC levels induced by 10 and 20 mg kg−1 of MH, while only showing a tendency at 3 mg kg−1. An increase of 2-AG levels can also be achieved by inhibiting its hydrolysis. Therefore, we tested the effects of MH on all the major EC hydrolytic enzymes (FAAH, MAGL, ABHDs) confirming that it does neither affect AEA nor 2-AG hydrolysis (Table 1), while the positive controls URB597, JZL184, and WWL70/tetrahydrolipstatin (for FAAH, MAGL, and ABHDs, respectively) inhibited EC hydrolysis as in agreement with the literature (data not shown) [22]. The trafficking of ECs across plasma membranes is likely regulated by a putative EC membrane transporter (EMT), and several inhibitors prevent intracellular accumulation of AEA and 2-AG [37]. MH only weakly inhibited the cellular accumulation of [3H]AEA in U937 cells with an IC50 value of 6.5 (4.7 to 9) μM (Table 1). A similar potency (EC50 value of 2.7 (1.1 to 6.5) μM) was observed using U937 cells differentiated into macrophages which lack the [3H]ethanolamine incorporation into phospholipids (as shown previously [37]), therefore ruling out potential effects on other targets indirectly related to the process of [3H]AEA uptake (Additional file 1: Figure S5). In the same system, the positive control UCM707 inhibited [3H]AEA uptake with an IC50 value of 1.6 (0.8 to 2.5) μM and 2.4 (1.2 to 3.6) μM in U937 cells and U937 cells differentiated into macrophages, respectively, in agreement with previous reports [23,37]. Thus, with nanomolar brain concentrations of MH, the inhibition of EC trafficking is unlikely to contribute to raising 2-AG levels in vivo.
Figure 1.
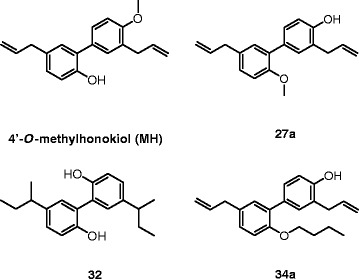
Chemical structures of 4′-O-methylhonokiol (-4′-methoxy-5,3′-di-(2-propenyl)-biphenyl-2-ol), 27a (2-methoxy-5,3′-di-(2-propenyl)- biphenyl-4′-ol), 32 (5,5′-di-sec-butyl- biphenyl-2,2′-diol) and 34a (2-butyloxy-5,3′-di-(2-propenyl)- biphenyl-4′-ol).
Table 1.
Summary of the pharmacological effects exerted by MH, 27a, 32, and 34a
| IC 50 or K i values (mean value ± SD, μM) | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| h COX-2 | RAW264.7 Intact cells | CHO membranes | CHO cells | Pig brain homog. | BV-2 homog. | U937 cells | ||||||
| AA | 2-AG | AA | 2-AG | h CB 1 | h CB 2 | h CB2 [ 35 S] GTPγS | h CB2 cAMP | FAAH | MAGL | ABHDs | AEA uptake | |
| MH | 0.58 ± 0.08 | **0.15 ± 0.09 | 1.56 ± 0.12 | **0.28 ± 0.09 | 2.4 ± 0.6 | 0.19 ± 0.12 | 0.20 ± 0.10 | 0.67 ± 0.07 | >10 | >10 | >10 | 6.5 ± 1.2 |
| 27a | 1.21 ± 0.08 | **0.42 ± 0.09 | 0.98 ± 0.08 | **0.28 ± 0.11 | 0.79 ± 0.10 | 0.11 ± 0.09 | 0.18 ± 0.09 | 0.71 ± 0.09 | >10 | >10 | >10 | >10 |
| 32 | 10.2 ± 0.12 | **0.63 ± 0.05 | 2.8 ± 0.22 | **0.21 ± 0.14 | >10 | >10 | n.d. | n.d. | >10 | >10 | >10 | 6.3 ± 1.6 |
| 34a | 4.31 ± 0.10 | *1.29 ± 0.07 | >30 | **1.57 ± 0.13 | 1.12 ± 0.15 | 0.35 ± 0.11 | 1.82 ± 0.14 | 1.52 ± 0.13 | >10 | >10 | >10 | >10 |
| DuP-697 | 0.061 ± 0.005 | 0.07 ± 0.01 | 0.004 ± 0.004 | 0.005 ± 0.006 | n.d. | n.d. | n.d. | n.d. | n.d. | n.d. | n.d. | n.d. |
The data are expressed as mean ± SD of at least three independent experiments each performed in triplicates. Binding affinity to CB1 and CB2 receptors is expressed as K i value while modulation of CB2 receptor activation (cAMP and [35S] GTPγS) and inhibition of enzyme activity and AEA uptake are expressed as IC50 value. *P < 0.05; **P < 0.01 PGE2-GE vs. PGE2. n.d.: not determined P < 0.05; **P < 0.01 PGE2-GE vs. PGE2. Items in bold are compound abbreviations.
Figure 2.
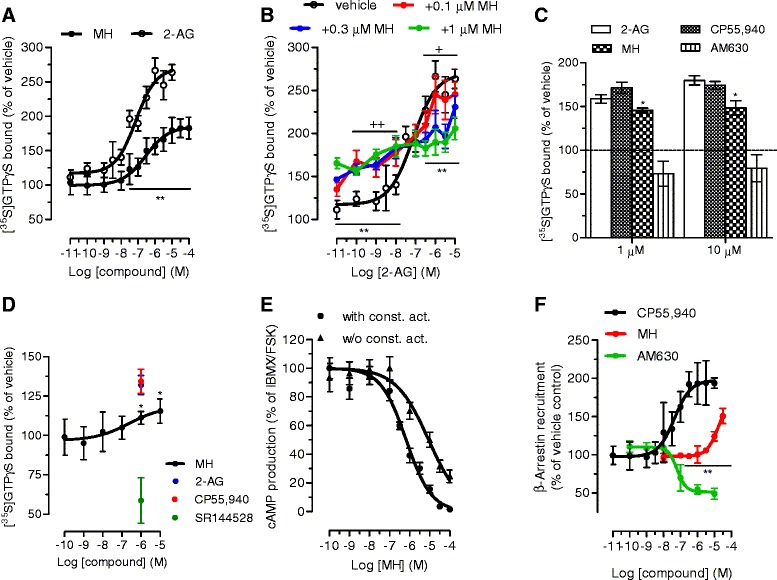
Modulation of CB2 receptor activity by MH. (A) [35S]GTPγS binding assay was performed in stably hCB2 receptor overexpressing CHO-K1 membranes, in the presence of different concentrations of MH and 2-AG alone or (B) in combination. The same experiments were performed in endogenously CB2 expressing (C) HL60 cells and (D) mouse spleen membranes. (E) Inhibition of forskolin-induced cAMP formation by MH in CHO-hCB2 cells transfected with pGloSensorTM 22-F plasmid. The experiments were performed in the presence (circles) and in the absence (triangles) of constitutive activity. (F) β-arrestin recruitment induced by increasing concentrations of MH, CP55,940, and AM630, measured in PathHunter® β-arrestin cells (CHO-K1-HOMSA-CNR2). ** P < 0.01 MH vs. 2-AG (Figure 2A); ** P < 0.01 MH (1 μM) plus 2-AG vs. 2-AG, ++ P < 0.01, + P < 0.05 MH (0.3 μM) plus 2-AG vs. 2-AG (Figure 2B). * P < 0.05 MH vs. 2-AG and CP55,940 (Figures 2C, D).** P < 0.01 MH vs. CP55,940 (Figure 2F).
Figure 3.
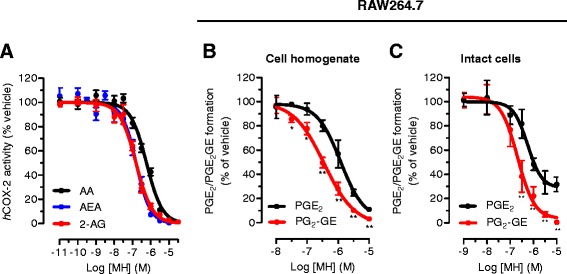
Dose-dependent COX-2 inhibition induced by MH in different biological matrices. (A) MH-mediated inhibition of hCOX-2 using 10 μM of AA (black), AEA (blue), and 2-AG (red) as substrates. MH-mediated inhibition of PGE2 and PGE2-GE formation in (B) RAW264.7 cell homogenate and (C) intact cells after 12 h of incubation with LPS (1 mg mL−1). Homogenates and intact cells were incubated for 30 min with 10 μM of 2-AG after the pre-treatment (30 min) with different concentrations of MH or vehicle. * P < 0.05, ** P < 0.01 PGE2 vs. PGE2-GE.
Figure 4.
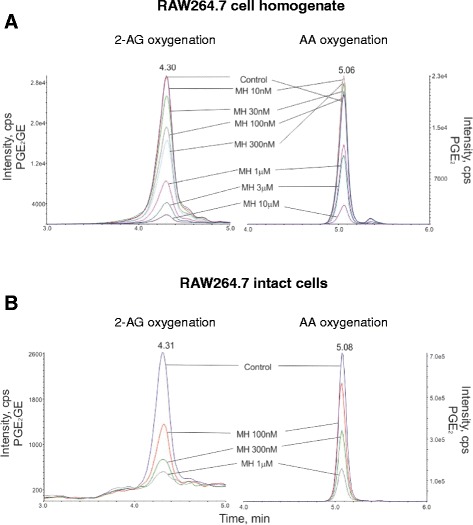
MS chromatograms of PGE2 and PGE2-GE measured in different biological matrices. Quantification of PGE2 and PGE2-GE formation measured after incubation of (A) RAW264.7 cell homogenate and (B) intact cells with 10 μM of AA or 2-AG in the presence of different concentrations of MH or vehicle. Homogenates and cells were treated for 12 h with LPS (1 mg mL−1) followed by 30-min incubation with 10 μM of 2-AG after the pre-treatment (30 min) with different concentrations of MH or vehicle.
Figure 5.
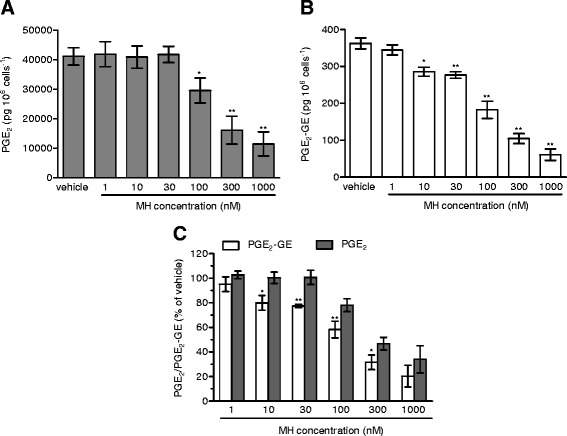
MH-mediated inhibition of PGE2 and PGE2-GE formation in RAW264.7 cells without external addition of 2-AG. Concentration-dependent inhibition of (A) PGE2 and (B) PGE2-GE formation (picogram per million of cell) in RAW264.7 intact cells previously stimulated with LPS (1 mg mL−1) ATP (1 mM) and thapsigargin (2 μM) for 8 h. (C) PGE2 and PGE2-GE formation inhibition upon different concentrations of MH (expressed as % of vehicle-treated cells). * P < 0.05, ** P < 0.01 MH treated samples vs. vehicle for panels (A) and (B); * P < 0.05, ** P < 0.01 PGE2-GE vs. PGE2 for panel (C).
Figure 6.
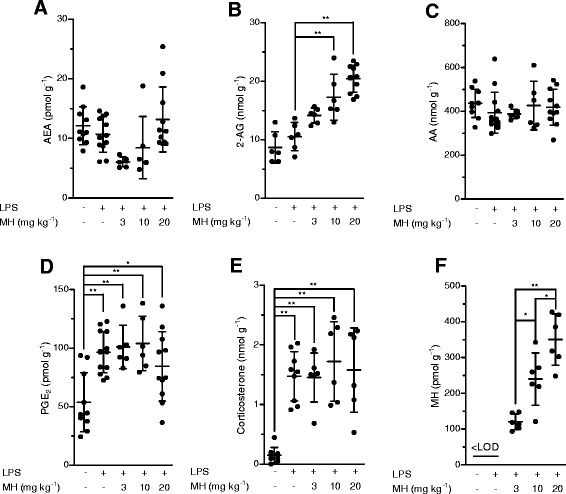
LC-MS/MS quantification of different analytes in mouse brain. (A) AEA, (B) 2-AG, (C) AA, (D) PGE2, and (E) corticosterone were quantified in the brains from mice (6 to 14 animals per group) challenged for 6 h with LPS (i.p., 2.5 mg kg−1, or saline), after 1 h of pre-treatment with MH (i.p., 3, 10, and 20 mg kg−1) or vehicle. (F) MH quantification in the brain of LPS-challenged mice. * P < 0.05, ** P < 0.01 treated vs. not-treated animals (Figure 6A-E) and * P < 0.05 MH 10 mg kg−1 vs. 20 mg kg−1 (Figure 6F).
We tested a library of 40 MH, magnolol, and honokiol analogs for COX-2 SSI previously described [1,7]. Compounds which showed >50% inhibition of 2-AG oxygenation at 2 μM were selected for further investigations. As shown in Additional file 1: Figure S6A, B, from the whole library, only three molecules (27a, 32, and 34a, see Figure 1 for chemical structures) showed a stronger, selective inhibition of 2-AG oxygenation over AA. We characterized the effects of these three compounds in purified hCOX-2, RAW264.7 cell homogenates, and intact cells similarly to MH. As shown in Figure 7, all compounds selectively inhibited 2-AG oxygenation with potencies ranging from 210 nM (32) to 1.57 μM in living cells (34a). Compounds 32 and 34a showed the greatest selectivity for 2-AG oxygenation (10/15-fold more potent than AA oxygenation (Table 1)). At 10 μM, 32 did not show any relevant binding at both CB receptor subtypes, while 27a and 34a showed selective CB2 binding (Table 1). Next, we characterized the functional effects of both ligands by assessing G-protein and β-arrestin recruitment and cAMP formation. Compound 27a, which was previously reported to be an inverse agonist [1], in our settings like MH behaved as a partial agonist in the [35S]GTPγS assay (Figure 8A), exhibiting full agonism at inhibiting the forskolin-induced cAMP production (Figure 8B) and sub-maximal recruitment of β-arrestin (Figure 8C). However, compound 34a reduced [35S]GTPγS binding and increased cAMP formation, thus behaving as an inverse agonist (Figure 8A, B). Interestingly, when the constitutive activity was removed, 34a behaved as an agonist by increasing the [35S]GTPγS binding and inhibiting the forskolin-induced cAMP formation (Figure 8A, B). This dual effect is a typical feature of ‘protean agonists’ [29]. In the β-arrestin assay, 34a did not show any significant effect despite a tendency to weakly reduce β-arrestin recruitment by 10% (Figure 8C). The IC50 values of 27a and 34a for the [35S]GTPγS assay and cAMP assay are reported in Table 1. All three compounds (27a, 32, and 34a) did not show any significant interaction with other components of the ECS apart from the magnolol derivative (32) which weakly inhibited the [3H]AEA uptake (IC50 value of 6.3 (3.2 to 9.2) μM) similarly to MH. Similarly to MH, also compound 32 showed the same potency of inhibiting [3H]AEA uptake in U937 cells and U937-derived macrophages (EC50 value of 2.4 (0.9 to 6.1 μM) (Additional file 1: Figure S5).
Figure 7.
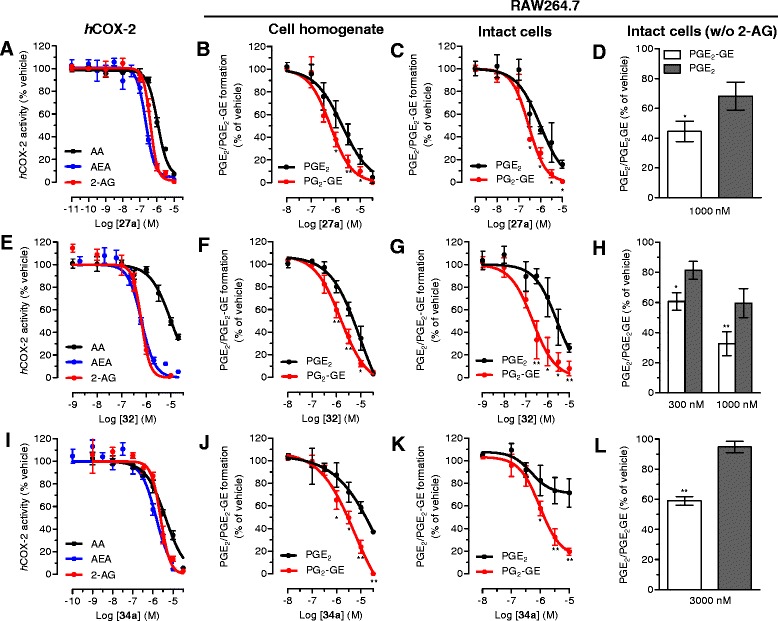
COX-2 inhibition by biphenyl neolignans in different biological matrices. (A-D) 27a, (E-H) 32, and (I-L) 34a inhibition of 2-AG, AEA, and AA oxygenation in hCOX-2 (A, E, I), RAW264.7 cell homogenates (B, F, J), and intact cells (C, G, K) incubated with 10 μM of substrate (2-AG, AEA, or AA depending on the assay). (D, H, L) Inhibition of PGE2 and PGE2-GE formation in RAW264.7 cells upon LPS (1 mg mL−1) ATP (1 mM) and thapsigargin (2 μM) treatment without externally adding 2-AG. * P < 0.05, ** P < 0.01 PGE2-GE vs. PGE2.
Figure 8.
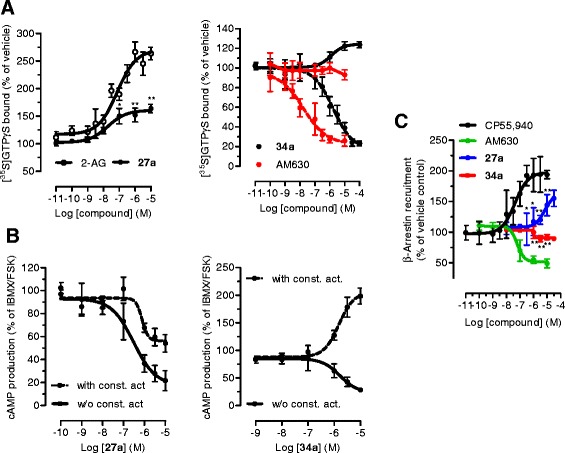
Modulation of CB2 receptor activity by 27a and 34a. (A) [35S]GTPγS binding assay performed in CHO-K1 membranes containing hCB2 receptors, in the presence of different concentrations of 27a and 34a. The CB2 inverse agonist AM630 is reported as positive control (red line). Experiments were performed in the presence (dotted line) and in the absence (solid line) of constitutive activity of the receptors. (B) Inhibition of forskolin-induced cAMP formation by 27a and 34a in CHO-hCB2 cells transfected with pGloSensorTM 22-F plasmid. The experiments were performed in the presence (dotted line) and in the absence (solid line) of constitutive activity. (C) β-arrestin recruitment induced by increasing concentrations of 27a and 34a, measured in PathHunter® β-arrestin cells (CHO-K1-HOMSA-CNR2). * P < 0.05, ** P < 0.01 27a vs. 2-AG (Figure 8A); * P < 0.05, ** P < 0.01 27a and 34a vs. CP55, 940 (Figure 8C).
Discussion
MH is a selective and potent CB2 ligand which was originally described as a full agonist at intracellular [Ca2+] and an inverse agonist at Gi/o recruitment by Schuehly et al. [1]. Recently, another group reported the agonist behavior of MH also on the cAMP pathway [9]. In the present study, using different standardized receptor signaling assays, we found that MH behaves as a partial agonist in the [35S]GTPγS binding in overexpressing systems and native conditions and in β-arrestin recruitment assays, while acting as a full agonist at the cAMP pathway. Similar differential actions between the G-protein recruitment and adenylate cyclase modulation have been reported for other GPCRs like. For example, morphine and fentanyl induced total inhibition of cAMP formation upon binding to μ-opioid receptors, while only showing 75% to 80% of maximal G-protein activation compared to the full agonist etorphine [38]. The [35S]GTPγS binding assay monitors the first step of G-protein activation but does not provide further information about the downstream signaling pathways which can be differently affected by different ligands. Thus, GPCRs may be coupled prevalently with one type of G-protein, but depending on the system (native vs. overexpressing) and the type of tissue, the same receptor might activate different types of G-protein [28,30]. For example, CB1 receptors are usually coupled to Gi/o but under certain conditions can also recruit stimulatory G-protein [39,40], or can induce pertussis toxin-independent increase of intracellular [Ca2+] via Gq/11 recruitment [41]. It has been reported that CB1 receptors expressed in HEK-293 cells can transduce the signal via Gi/o when activated by CP55,940, 2-AG, or ∆9-THC, while WIN55,212-2 induced a CB1-mediated Gq/11 signaling [41]. This suggests that different ligands can trigger distinct active conformations of the receptor which, in turn, can recruit different types of G-proteins. In addition, ligands can also alternatively transduce the signal either via Gα- or Gβγ-subunit as it was recently proposed for ∆9-THC and 2-AG which modulate COX-2 expression in opposite ways by activating CB1 receptors in neurons [42]. The level of complexity is also raised by G-protein-independent signaling pathways such as β-arrestin, whose receptor-mediated recruitment can modulate several downstream effectors (e.g., MAPK, ERK1/2, PI3K) besides its role in the desensitization and internalization process (see [43] for review). The introduction of the ‘biased agonism’ (or functional selectivity) concept triggered a shift from the classic definition of agonism, antagonism, and inverse agonism towards the characterization of ligands which can preferentially activate only one (or few) signaling pathways [44]. It is well established that CB2 receptors possess high levels of constitutive activity and a high degree of trafficking from intracellular pools to the membrane surface and vice versa [45]. As reported by Schuehly et al. [1], MH and 27a inhibit osteoclastogenesis by acting as inverse agonists on the CB2-mediated cAMP pathway. We now show that both ligands behave as full agonists by inhibiting cAMP formation in the presence or in the absence of constitutive CB2 activation. On the other hand, MH and 27a show partial agonism for G-protein activation and β-arrestin recruitment. Partial agonists are very ductile pharmacological tools which can modulate the activation state of a certain receptor in both ways. Indeed, MH could either potentiate or attenuate the CB2-mediated G-protein recruitment triggered by residual 2-AG, depending on the concentration ratio between the two ligands. This pharmacological feature might at least in part explain the discrepancy between the different effects reported for MH at CB2 receptors (agonist vs. inverse agonist), due to the presence of high or low amounts of 2-AG in the culture medium. We recently reported that fetal bovine serum (FBS) contains ECs, and, depending on the batch, the levels of 2-AG can even reach sub-micromolar concentrations [46]. Culturing RAW264.7 cells with 2-AG high-content sera led to significant formation of osteoclasts [46]. In this scenario, the partial agonists MH and 27a might exert their anti-osteoclastogenesis effects indirectly by blocking 2-AG-induced CB2-mediated osteoclastogenesis. Our current results also suggest that alternative signaling pathways than cAMP might be involved. In line with this hypothesis, Schuehly et al. [1] reported the agonistic behavior of MH on CB2-mediated increase of intracellular [Ca2+], while here, we report that MH triggers the recruitment of β-arrestin, which might contribute to the modulation of different downstream effectors. In addition, we described that the 2-O-butyl honokiol derivative (34a) acts as a ‘protean agonist’ as it could behave as an agonist or inverse agonist, depending on the proportion of constitutively activated CB2 receptors. On the contrary the CB2 inverse agonist AM630 switched from an inverse agonist to an inactive ligand upon the removal of the constitutive activity of the receptors in both [35S]GTPγS and cAMP assays (see Figure 8A and Additional file 1: Figure S1), thus confirming that 34a is not simply behaving as an inverse agonist. Noteworthy, upon i.p. application, MH treatment significantly increased the brain levels of 2-AG in a concentration-dependent manner without significantly affecting AEA, AA, PGE2, and corticosterone levels. These findings clearly suggest that the inhibition of COX-2-mediated oxygenation as metabolic pathway for EC degradation in conditions of inflammation is a relevant target for MH. Unfortunately, in vivo we and others could not detect PGE2-GE, probably due to an extensive hydrolysis to the related arachidonoyl derivative (PGE2), as previously shown [47,48]. Also, the effect of SSIs may turn out to be different, dependent on mouse strains. Recently, Hermanson et al. [21] reported using a morpholino amide derivative of indomethacin, LM-4131, a 150% increase of AEA levels and only minor effects on 2-AG levels (~110%) in imprinting control region (ICR) mouse brain. In normal conditions, 2-AG predominantly undergoes hydrolytic degradation [49,50], but upon LPS-challenge, COX-2 levels significantly rise in neurons [51] and astrocytes/microglial cells [52], thus increasing the relevance of the oxygenation pathway. In our setting we detected a significant increase of 2-AG levels by 170% and 200% upon treatment with 10 and 20 mg kg−1 of MH, respectively. AEA levels were not significantly changed by MH treatment despite a tendency to increase (120%) at the dose of 20 mg kg−1. In addition, MH treatment selectively modulated the levels of 2-AG without affecting the levels of AA, PGE2, the FAAH-substrates N-acetylethanolamines, and corticosterone. Therefore, SSI of COX-2 activity appears to be a promising strategy to modulate EC levels in the brain, especially under inflammatory conditions. Prostamides and PG-GEs play an active role in the inflammatory process both in vitro and in vivo by acting through CB1/2 receptor-independent targets [15,19,53]. In hippocampal neurons, COX-2 inhibition rather than FAAH blockage led to a prolonged depolarization-induced suppression of inhibition suggesting a prominent role of 2-AG in the retrograde signal and COX-2-mediated oxygenation as relevant mechanism of reducing 2-AG levels at the synaptic level [54]. Straiker et al. [55] also reported the relevant role of COX-2 activity in reducing EC-mediated retrograde signaling. Recently it was shown that MH can weakly potentiate GABA-induced chloride currents in Xenopus oocytes by acting at the most common α1β2γ2 GABAA receptor subtype [8]. In an animal model, repeated low doses (0.5 mg kg−1per os) of MH produced weak anxiolytic effects [56] similar to the SSI LM-4131 [21]. Thus, MH could represent a prototype of multi-target compound which can directly and indirectly potentiate the ECS. Nonetheless, the pharmacokinetic profile of MH indicates a poor oral bioavailability and extensive hepatic metabolism in rats [57]. Our data show that upon i.p. injection, MH penetrates the brain at concentrations which are in line with the effects on CB2 receptors and the SSI of COX-2 activity (approximately 200 and 300 nM for 10 and 20 mg kg−1, respectively). Mice were sacrificed 7 h after the injection, thus indicating that MH could reach and accumulate in the brain without being significantly metabolized.
Despite the reported poor bioavailability after single oral administration [57], upon repeated doses, MH might accumulate and exert pharmacological effects in the brain. This could also explain the beneficial effects reported for long-term treatment with low doses of MH administered per os (0.1 to 1 mg kg−1) in neurodegenerative animal models [2,4,36]. CB2 receptors are almost absent in the CNS under normal conditions, but upon inflammation, they are significantly upregulated in microglial cells. Neuroinflammation is a hallmark of neurodegenerative diseases which contributes to the onset and progression of the disease [58,59] and several results support the positive role of CB2 activation in neurodegenerative diseases including the stimulation of β-amyloid removal by macrophages and down-regulation of TNF-α production [60-62]. MH showed beneficial effects in Alzheimer’s disease (AD) models by reducing LPS-induced β-amyloid accumulation, pro-inflammatory cytokine production, and memory impairment [2,4]. Since in addition to the direct activation of CB2 receptors, MH also elevates 2-AG levels, which is the main endogenous agonist for CB2, and it further inhibits the formation of pro-inflammatory PG-EAs and PG-GEs; synergistic effects for attenuating the inflammatory process are expected to represent one of the main underlying mechanisms of the neuroprotective effects exerted by MH. Interestingly, (R)-flurbiprofen is a COX-2 SSI with negligible effects on AA metabolism [14] which showed COX-2-independent anti-inflammatory effects in AD and experimental autoimmune encephalomyelitis multiple sclerosis models [63-65]. Remarkably, in this model, (R)-flurbiprofen induced a significant increase of EC levels in the spinal cord and somatosensory cortex while only mildly affecting PGs levels [65]. Therefore, based on our data we suggest that the major mode of action related to the pronounced neuroprotective effects of MH may be related to its outstanding polypharmacology in the ECS.
Conclusions
We show for the first time that MH penetrates the blood-brain barrier and accumulates in the brain in amounts that are consistent with the concentrations that modulate the ECS in vitro. MH might serve as prototype scaffold to develop compounds exhibiting a novel combination of polypharmacology to modulate the ECS through a concurrent activation of CB2 receptors and the SSI of COX-2-mediated EC oxygenation. For MH, the latter mechanism was shown here for the first time both in vitro and in vivo. This polypharmacology might achieve synergistic anti-inflammatory and protective actions in certain tissues, including the brain, by directly and indirectly ( increasing 2-AG levels) activating CB2 receptors, avoiding the typical gastrointestinal and cardiovascular side effects of NSAIDs. Moreover, these two mechanisms of action might be more relevant in neuroinflammatory diseases where both COX-2 and CB2 receptors are overexpressed in the brain, thus providing a rationale for the diverse neuroprotective effects reported for MH in animal models.
Acknowledgements
We would like to acknowledge Christoph Ullmer and Uwe Grether from F. Hoffmann-La-Roche Ltd. for independently confirming our data with MH in their cAMP assays. We thank Antje Hüfner (University of Graz) for synthesizing the MH-d6 standard. Finally, we would like to thank Stefano Chicca for performing [35S]GTPγS assays in spleen membranes.
Abbreviations
- AA
arachidonic acid
- AD
Alzheimer’s disease
- ABHDs
α, β-hydrolases
- AEA
anandamide
- 2-AG
2-arachidonoyl glycerol
- AM630
[6-iodo-2-methyl-1-[2-(4-morpholinyl) ethyl]-1H-indol-3-yl] (4-methoxyphenyl)-methanone
- cAMP
cyclic AMP
- BSA
bovine serum albumin
- CB1
type-1 cannabinoid receptor
- CB2
type-2 cannabinoid receptor
- CP55
940, (−)-cis-3-[2-hydroxy-4-(1,1 dimethylheptyl) phenyl]-trans-4-(3-hydroxypropyl) cyclohexanol
- COX-2
cyclooxygenase-2
- CNS
central nervous system
- EC
endocannabinoid
- ECS
endocannabinoid system
- FAAH
fatty acid amide hydrolase
- FSK
forskolin
- ICR
imprinting control region
- IS
internal standard
- JZL184
4-nitrophenyl-4-[bis (1,3-benzodioxol-5-yl) (hydroxy) methyl] piperidine-1-carboxylate
- MAGL
monoacylglycerol lipase
- MH
4′-O-methylhonokiol
- MRM
multiple reaction monitoring
- NA-GABA
N-arachidonoyl GABA
- NSAIDs
non-steroidal anti-inflammatory drugs
- PGs
prostaglandins
- PG-EAs
prostaglandin-ethanolamides (prostamides)
- PG-GEs
prostaglandin glycerol esters
- SSI
substrate selective inhibition/inhibitor
- UCM707
(5Z, 8Z, 11Z, 14Z)-N-(3-furanylmethyl)-5,8,11,14-eicosatetraenamide
- URB597
(3′-(aminocarbonyl) [1, 1′-biphenyl]-3-yl)-cyclohexylcarbamate
- WIN55212-2
(R)-(+)-[2, 3-dihydro-5-methyl-3-(4-morpholinylmethyl) pyrrolo-[1,2,3-de]-1,4-benzoxazin-6-yl]-1-naphtaleneylmethanone
- WWL70
N-methyl-N-[[3-(4-pyridinyl) phenyl] methyl]-carbamic acid 4′ - (aminocarbonyl) [1, 1′-biphenyl]-4-yl ester
Additional file
[35S] GTPS binding assays performed in mock-transfected CHO cells. Figure S2: Pharmacological behavior of AM630 and CP55,940 in the presence or absence of constitutive activity of CB2 receptors. Figure S3: The effects of DuP-697 in different cellular systems. Figure S4: LC-MS/MS quantification of N-acetylethanolamines in mouse brain. Figure S5: [3H] AEA uptake into U937 cell-derived macrophages. Figure S6: Screening of several MHK, honokiol, and magnolol derivatives for SSI of COX-2 activity.
Footnotes
Competing interests
The authors declare that they have no competing interests.
Authors’ contributions
AC, MSG, VP, and RPC performed the experiments; JG and AC designed the research study; WS contributed the essential tools; AC, MSG, and JG analyzed the data; AC and JG wrote the paper. All authors read and approved the final manuscript.
Contributor Information
Andrea Chicca, Email: chicca@ibmm.unibe.ch.
Maria Salomé Gachet, Email: gachet@ibmm.unibe.ch.
Vanessa Petrucci, Email: petrucci@ibmm.unibe.ch.
Wolfgang Schuehly, Email: wolfgang.schuehly@uni-graz.at.
Roch-Philippe Charles, Email: charles@ibmm.unibe.ch.
Jürg Gertsch, Email: gertsch@ibmm.unibe.ch.
References
- 1.Schuehly W, Paredes JM, Kleyer J, Huefner A, Anavi-Goffer S, Raduner S, et al. Mechanisms of osteoclastogenesis inhibition by a novel class of biphenyl-type cannabinoid CB (2) receptor inverse agonists. Chem Biol. 2011;18:1053–64. doi: 10.1016/j.chembiol.2011.05.012. [DOI] [PubMed] [Google Scholar]
- 2.Lee YJ, Choi DY, Choi IS, Kim KH, Kim YH, Kim HM, et al. Inhibitory effect of 4-O-methylhonokiol on lipopolysaccharide-induced neuroinflammation, amyloidogenesis and memory impairment via inhibition of nuclear factor-kappaB in vitro and in vivo models. J Neuroinflammation. 2012;9:35. doi: 10.1186/1742-2094-9-35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Lee YJ, Choi DY, Lee YK, Lee YM, Han SB, Kim YH, et al. 4-O-methylhonokiol prevents memory impairment in the Tg2576 transgenic mice model of Alzheimer’s disease via regulation of β-secretase activity. J Alzheimers Dis. 2012;29:677–90. doi: 10.3233/JAD-2012-111835. [DOI] [PubMed] [Google Scholar]
- 4.Jung YY, Lee YJ, Choi DY, Hong JT. Amelioration of cognitive dysfunction in APP/ps1 double transgenic mice by long-term treatment of 4-O-Methylhonokiol. Biomol Ther (Seoul). 2014;22:232–8. doi: 10.4062/biomolther.2014.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Gertsch J, Anavi-Goffer S. Methylhonokiol attenuates neuroinflammation: a role for cannabinoid receptors? J Neuroinflammation. 2012;9:135. doi: 10.1186/1742-2094-9-135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kim HS, Ryu HS, Kim JS, Kim YG, Lee HK, Jung JK, et al. Validation of cyclooxygenase-2 as a direct anti-inflammatory target of 4-O-methylhonokiol in zymosan-induced animal models. Arch Pharm Res. 2014. In press. [DOI] [PubMed]
- 7.Schühly W, Hüfner A, Pferschy-Wenzig EM, Prettner E, Adams M, Bodensieck A, et al. Design and synthesis of ten biphenyl-neolignan derivatives and their in vitro inhibitory potency against cyclooxygenase-1/2 activity and 5-lipoxygenase-mediated LTB4-formation. Bioorg Med Chem. 2009;17:4459–65. doi: 10.1016/j.bmc.2009.05.018. [DOI] [PubMed] [Google Scholar]
- 8.Baur R, Schuehly W, Sigel E. Moderate concentrations of 4-O-methylhonokiol potentiate GABAA receptor currents stronger than honokiol. Biochim Biophys Acta. 2014;1840:3017–21. doi: 10.1016/j.bbagen.2014.06.016. [DOI] [PubMed] [Google Scholar]
- 9.Fuchs A, Rempel V, Müller CE. The natural product magnolol as a lead structure for the development of potent cannabinoid receptor agonists. PLoS One. 2013;8:e77739. doi: 10.1371/journal.pone.0077739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kozak KR, Crews BC, Morrow JD, Wang LH, Ma YH, Weinander R, et al. Metabolism of the endocannabinoids, 2-arachidonylglycerol and anandamide, into prostaglandin, thromboxane, and prostacyclin glycerol esters and ethanolamides. J Biol Chem. 2002;277:44877–85. doi: 10.1074/jbc.M206788200. [DOI] [PubMed] [Google Scholar]
- 11.Kozak KR, Gupta RA, Moody JS, Ji C, Boeglin WE, DuBois RN, et al. 15-Lipoxygenase metabolism of 2-arachidonylglycerol. Generation of a peroxisome proliferator-activated receptor alpha agonist. J Biol Chem. 2002;277:23278–86. doi: 10.1074/jbc.M201084200. [DOI] [PubMed] [Google Scholar]
- 12.Snider NT, Walker VJ, Hollenberg PF. Oxidation of the endogenous cannabinoid arachidonoyl ethanolamide by the cytochrome P450 monooxygenases: physiological and pharmacological implications. Pharmacol Rev. 2010;62:136–54. doi: 10.1124/pr.109.001081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Weber A, Ni J, Ling KH, Acheampong A, Tang-Liu DD, Burk R, et al. Formation of prostamides from anandamide in FAAH knockout mice analyzed by HPLC with tandem mass spectrometry. J Lipid Res. 2004;45:757–63. doi: 10.1194/jlr.M300475-JLR200. [DOI] [PubMed] [Google Scholar]
- 14.Duggan KC, Hermanson DJ, Musee J, Prusakiewicz JJ, Scheib JL, Carter BD, et al. (R)-Profens are substrate-selective inhibitors of endocannabinoid oxygenation by COX-2. Nat Chem Biol. 2011;7:803–9. doi: 10.1038/nchembio.663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Gatta L, Piscitelli F, Giordano C, Boccella S, Lichtman A, Maione S, et al. Discovery of prostamide F2α and its role in inflammatory pain and dorsal horn nociceptive neuron hyperexcitability. PLoS One. 2012;7:e31111. doi: 10.1371/journal.pone.0031111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kaufmann WE, Worley PF, Pegg J, Bremer M, Isakson P. COX-2, a synaptically induced enzyme, is expressed by excitatory neurons at postsynaptic sites in rat cerebral cortex. Proc Natl Acad Sci U S A. 1996;93:2317–21. doi: 10.1073/pnas.93.6.2317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Yoshikawa K, Takei S, Hasegawa-Ishii S, Chiba Y, Furukawa A, Kawamura N, et al. Preferential localization of prostamide/prostaglandin F synthase in myelin sheaths of the central nervous system. Brain Res. 2011;1367:22–32. doi: 10.1016/j.brainres.2010.10.019. [DOI] [PubMed] [Google Scholar]
- 18.Ritter JK, Li C, Xia M, Poklis JL, Lichtman AH, Abdullah RA, et al. Production and actions of the anandamide metabolite prostamide E2 in the renal medulla. J Pharmacol Exp Ther. 2012;342:770–9. doi: 10.1124/jpet.112.196451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Liang Y, Woodward DF, Guzman VM, Li C, Scott DF, Wang JW, et al. Identification and pharmacological characterization of the prostaglandin FP receptor and FP receptor variant complexes. Br J Pharmacol. 2008;154:1079–93. doi: 10.1038/bjp.2008.142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ligresti A, Martos J, Wang J, Guida F, Allarà M, Palmieri V, et al. Prostamide F (2) α receptor antagonism combined with inhibition of FAAH may block the pro-inflammatory mediators formed following selective FAAH inhibition. Br J Pharmacol. 2014;171:1408–19. doi: 10.1111/bph.12410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hermanson DJ, Hartley ND, Gamble-George J, Brown N, Shonesy BC, Kingsley PJ, et al. Substrate-selective COX-2 inhibition decreases anxiety via endocannabinoid activation. Nat Neurosci. 2013;16:1291–8. doi: 10.1038/nn.3480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Chicca A, Caprioglio D, Minassi A, Petrucci V, Appendino G, Taglialatela-Scafati O, et al. Functionalization of β-caryophyllene generates novel polypharmacology in the endocannabinoid system. ACS Chem Biol. 2014;9:1499–507. doi: 10.1021/cb500177c. [DOI] [PubMed] [Google Scholar]
- 23.Chicca A, Marazzi J, Gertsch J. The antinociceptive triterpene β-amyrin inhibits 2-arachidonoylglycerol (2-AG) hydrolysis without directly targeting cannabinoid receptors. Br J Pharmacol. 2002;167:1596–608. doi: 10.1111/j.1476-5381.2012.02059.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Rosati O, Messina F, Pelosi A, Curini M, Petrucci V, Gertsch J, et al. One-pot heterogeneous synthesis of Δ (3)-tetrahydrocannabinol analogues and xanthenes showing differential binding to CB (1) and CB (2) receptors. Eur J Med Chem. 2014;85:77–86. doi: 10.1016/j.ejmech.2014.07.062. [DOI] [PubMed] [Google Scholar]
- 25.Gachet MS, Rhyn P, Bosch OG, Quednow BB, Gertsch J. A quantitiative LC-MS/MS method for the measurement of arachidonic acid, prostanoids, endocannabinoids, N-acylethanolamines and steroids in human plasma. J Chromatogr, B. 2015;976–977:6–18. doi: 10.1016/j.jchromb.2014.11.001. [DOI] [PubMed] [Google Scholar]
- 26.Seifert R, Wenzel-Seifert K, Gether U, Kobilka BK. Functional differences between full and partial agonists: evidence for ligand-specific receptor conformations. J Pharmacol Exp Ther. 2001;297:1218–26. [PubMed] [Google Scholar]
- 27.Munro S, Thomas KL, Abu-Shaar M. Molecular characterization of a peripheral receptor for cannabinoids. Nature. 1993;365:61–5. doi: 10.1038/365061a0. [DOI] [PubMed] [Google Scholar]
- 28.Marini P, Cascio MG, King A, Pertwee RG, Ross RA. Characterization of cannabinoid receptor ligands in tissues natively expressing cannabinoid CB2 receptors. Br J Pharmacol. 2013;169:887–99. doi: 10.1111/bph.12191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kenakin T. Functional selectivity through protean and biased agonism: who steers the ship? Mol Pharmacol. 2007;72:1393–13401. doi: 10.1124/mol.107.040352. [DOI] [PubMed] [Google Scholar]
- 30.Mancini I, Brusa R, Quadrato G, Foglia C, Scandroglio P, Silverman LS, et al. Constitutive activity of cannabinoid-2 (CB2) receptors plays an essential role in the protean agonism of (+) AM1241 and L768242. Br J Pharmacol. 2009;158:382–91. doi: 10.1111/j.1476-5381.2009.00154.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Yao BB, Mukherjee S, Fan Y, Garrison TR, Daza AV, Grayson GK, et al. In vitro pharmacological characterization of AM1241: a protean agonist at the cannabinoid CB2 receptor? Br J Pharmacol. 2006;149:145–54. doi: 10.1038/sj.bjp.0706838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Bolognini D, Cascio MG, Parolaro D, Pertwee RG. AM630 behaves as a protean ligand at the human cannabinoid CB2 receptor. Br J Pharmacol. 2012;165:2561–74. doi: 10.1111/j.1476-5381.2011.01503.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Silvestri C, Martella A, Poloso NJ, Piscitelli F, Capasso R, Izzo A, et al. Anandamide-derived prostamide F2α negatively regulates adipogenesis. J Biol Chem. 2014;288:23307–21. doi: 10.1074/jbc.M113.489906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Nomura DK, Morrison BE, Blankman JL, Long JZ, Kinsey SG, Marcondes MC, et al. Endocannabinoid hydrolysis generates brain prostaglandins that promote neuroinflammation. Science. 2011;334:809–13. doi: 10.1126/science.1209200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Cabrera R, Korte SM, Lentjes EG, Romijn F, Schönbaum E, De Nicola A, et al. The amount of free corticosterone is increased during lipopolysaccharide-induced fever. Life Sci. 2002;66:553–62. doi: 10.1016/S0024-3205(99)00628-1. [DOI] [PubMed] [Google Scholar]
- 36.Lee YK, Choi IS, Ban JO, Lee HJ, Lee US, Han SB, et al. 4-O-methylhonokiol attenuated β-amyloid-induced memory impairment through reduction of oxidative damages via inactivation of p38 MAP kinase. J Nutr Biochem. 2011;22:476–86. doi: 10.1016/j.jnutbio.2010.04.002. [DOI] [PubMed] [Google Scholar]
- 37.Chicca A, Marazzi J, Nicolussi S, Gertsch J. Evidence for bidirectional endocannabinoid transport across cell membranes. J Biol Chem. 2012;287:34660–82. doi: 10.1074/jbc.M112.373241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Zaki PA, Keith DE, Jr, Brine GA, Carroll FI, Evans CJ. Ligand-induced changes in surface mu-opioid receptor number: relationship to G protein activation? J Pharmacol Exp Ther. 2000;292:1127–34. [PubMed] [Google Scholar]
- 39.Felder CC, Joyce KE, Briley EM, Glass M, Mackie KP, Fahey KJ, et al. LY320135, a novel cannabinoid CB1 receptor antagonist, unmasks coupling of the CB1 receptor to stimulation of cAMP accumulation. J Pharmacol Exp Ther. 1998;284:291–7. [PubMed] [Google Scholar]
- 40.Bauer M, Chicca A, Tamborrini M, Eisen D, Lerner R, Lutz B, et al. Identification and quantification of a new family of peptide endocannabinoids (Pepcans) showing negative allosteric modulation at CB1 receptors. J Biol Chem. 2012;287:36944–9667. doi: 10.1074/jbc.M112.382481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Lauckner JE, Hille B, Mackie K. The cannabinoid agonist WIN55,212-2 increases intracellular calcium via CB1 receptor coupling to Gq/11 G proteins. Proc Natl Acad Sci U S A. 2005;102:19144–9. doi: 10.1073/pnas.0509588102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Chen R, Zhang J, Fan N, Teng ZQ, Wu Y, Yang H, et al. Δ9-THC-caused synaptic and memory impairments are mediated through COX-2 signaling. Cell. 2014;155:1154–65. doi: 10.1016/j.cell.2013.10.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Reiter E, Ahn S, Shukla AK, Lefkowitz RJ. Molecular mechanism of β-arrestin-biased agonism at seven-transmembrane receptors. Annu Rev Pharmacol Toxicol. 2012;52:179–97. doi: 10.1146/annurev.pharmtox.010909.105800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Kenakin T, Christopoulos A. Signalling bias in new drug discovery: detection, quantification and therapeutic impact. Nat Rev Drug Discov. 2013;12:205–16. doi: 10.1038/nrd3954. [DOI] [PubMed] [Google Scholar]
- 45.Kleyer J, Nicolussi S, Taylor P, Simonelli D, Furger E, Anderle P, et al. Cannabinoid receptor trafficking in peripheral cells is dynamically regulated by a binary biochemical switch. Biochem Pharmacol. 2012;83:1393–412. doi: 10.1016/j.bcp.2012.02.014. [DOI] [PubMed] [Google Scholar]
- 46.Marazzi J, Kleyer J, Paredes JM, Gertsch J. Endocannabinoid content in fetal bovine sera - unexpected effects on mononuclear cells and osteoclastogenesis. J Immunol Methods. 2011;373:219–28. doi: 10.1016/j.jim.2011.08.021. [DOI] [PubMed] [Google Scholar]
- 47.Kozak KR, Crews BC, Ray JL, Tai HH, Morrow JD, Marnett LJ. Metabolism of prostaglandin glycerol esters and prostaglandin ethanolamides in vitro and in vivo. J Biol Chem. 2001;276:36993–8. doi: 10.1074/jbc.M105854200. [DOI] [PubMed] [Google Scholar]
- 48.Savinainen JR, Kansanen E, Pantsar T, Navia-Paldanius D, Parkkari T, Lehtonen M, et al. Robust hydrolysis of prostaglandin glycerol esters by human monoacylglycerol lipase (MAGL) Mol Pharmacol. 2014;86:522–35. doi: 10.1124/mol.114.094284. [DOI] [PubMed] [Google Scholar]
- 49.Long JZ, Li W, Booker L, Burston JJ, Kinsey SG, Schlosburg JE, et al. Selective blockade of 2-arachidonoylglycerol hydrolysis produces cannabinoid behavioral effects. Nat Chem Biol. 2009;5:37–44. doi: 10.1038/nchembio.129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Marrs WR, Blankman JL, Horne EA, Thomazeau A, Lin YH, Coy J, et al. The serine hydrolase ABHD6 controls the accumulation and efficacy of 2-AG at cannabinoid receptors. Nat Neurosci. 2010;13:951–7. doi: 10.1038/nn.2601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Du H, Chen X, Zhang J, Chen C. Inhibition of COX-2 expression by endocannabinoid 2-arachidonoylglycerol is mediated via PPAR-γ. Br J Pharmacol. 2011;163:1533–49. doi: 10.1111/j.1476-5381.2011.01444.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Font-Nieves M, Sans-Fons MG, Gorina R, Bonfill-Teixidor E, Salas-Pérdomo A, Márquez-Kisinousky L, et al. Induction of COX-2 enzyme and down-regulation of COX-1 expression by lipopolysaccharide (LPS) control prostaglandin E2 production in astrocytes. J Biol Chem. 2012;287:6454–68. doi: 10.1074/jbc.M111.327874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Woodward DF, Carling RW, Cornell CL, Fliri HG, Martos JL, Pettit SN, et al. The pharmacology and therapeutic relevance of endocannabinoid derived cyclo-oxygenase (COX)-2 products. Pharmacol Ther. 2008;120:71–80. doi: 10.1016/j.pharmthera.2008.08.001. [DOI] [PubMed] [Google Scholar]
- 54.Kim J, Alger BE. Inhibition of cyclooxygenase-2 potentiates retrograde endocannabinoid effects in hippocampus. Nat Neurosci. 2004;7:697–8. doi: 10.1038/nn1262. [DOI] [PubMed] [Google Scholar]
- 55.Straiker A, Wager-Miller J, Hu SS, Blankman JL, Cravatt BF, Mackie K. COX-2 and fatty acid amide hydrolase can regulate the time course of depolarization-induced suppression of excitation. Br J Pharmacol. 2011;164:1672–83. doi: 10.1111/j.1476-5381.2011.01486.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Han H, Jung JK, Han SB, Nam SY, Oh KW, Hong JT. Anxiolytic-like effects of 4-O-methylhonokiol isolated from Magnolia officinalis through enhancement of GABAergic transmission and chloride influx. J Med Food. 2011;14:724–31. doi: 10.1089/jmf.2010.1111. [DOI] [PubMed] [Google Scholar]
- 57.Yu HE, Oh SJ, Ryu JK, Kang JS, Hong JT, Jung JK, et al. Pharmacokinetics and metabolism of 4-O-methylhonokiol in rats. Phytother Res. 2014;28:568–78. doi: 10.1002/ptr.5033. [DOI] [PubMed] [Google Scholar]
- 58.Ashton JC, Glass M. The cannabinoid CB2 receptor as a target for inflammation-dependent neurodegeneration. Curr Neuropharmacol. 2007;5(2):73–80. doi: 10.2174/157015907780866884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Zotova E, Nicoll JA, Kalaria R, Holmes C, Boche D. Inflammation in Alzheimer’s disease: relevance to pathogenesis and therapy. Alzheimers Res Ther. 2010;2(1):1. doi: 10.1186/alzrt24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Rom S, Persidsky Y. Cannabinoid receptor 2: potential role in immunomodulation and neuroinflammation. J Neuroimmune Pharmacol. 2013;8:608–20. doi: 10.1007/s11481-013-9445-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Gowran A, Noonan J, Campbell VA. The multiplicity of action of cannabinoids: implications for treating neurodegeneration. CNS Neurosci Ther. 2011;17:637–44. doi: 10.1111/j.1755-5949.2010.00195.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Aso E, Juvés S, Maldonado R, Ferrer I. CB2 cannabinoid receptor agonist ameliorates Alzheimer-like phenotype in AβPP/PS1 mice. J Alzheimers Dis. 2013;35(4):847–58. doi: 10.3233/JAD-130137. [DOI] [PubMed] [Google Scholar]
- 63.Carreras I, McKee AC, Choi JK, Aytan N, Kowall NW, Jenkins BG, et al. R-flurbiprofen improves tau, but not Aß pathology in a triple transgenic model of Alzheimer’s disease. Brain Res. 2013;1541:115–27. doi: 10.1016/j.brainres.2013.10.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Kukar T, Prescott S, Eriksen JL, Holloway V, Murphy MP, Koo EH, et al. Chronic administration of R-flurbiprofen attenuates learning impairments in transgenic amyloid precursor protein mice. BMC Neurosci. 2007;8:54. doi: 10.1186/1471-2202-8-54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Schmitz K, de Bruin N, Bishay P, Männich J, Häussler A, Altmann C, et al. R-flurbiprofen attenuates experimental autoimmune encephalomyelitis in mice. EMBO Mol Med. 2014;6:1398–422. doi: 10.15252/emmm.201404168. [DOI] [PMC free article] [PubMed] [Google Scholar]