

Abstract
Patients who respond poorly to therapies for hepatitis C virus (HCV) infection display a characteristic phenotype with high basal hepatic interferon-stimulated gene (ISG) expression, but limited induction following interferon (IFN) treatment. The molecular pathways that mediate this refractory state are not known. We examined whether the AMPK activator metformin, the PPARγ agonist pioglitazone, or the PPARα agonist WY-14643 could potentiate IFN responses, reverse IFN refractoriness, and enhance viral eradication in hepatocytes. WY-14643 demonstrated the strongest antiviral synergy with IFN-α and so was tested in the context of chronic IFN activation. Cells rendered refractory to IFN by IFN-α pretreatment were resensitized by WY-14643, as demonstrated by improved STAT1 phosphorylation, promoter activation, and ISG expression. WY-14643 treatment reduced the expression of key negative regulators of IFN signaling: the AXL receptor tyrosine kinase, suppressor of cytokine signaling (SOCS) 1 and 3, which are upregulated in the IFN-refractory state. AXL is a novel regulator of IFN-α signaling that is induced by HCV infection in vitro and which may drive SOCS3 expression. Our data suggests that PPARα agonists could be a useful adjunct treatment for chronic HCV infection by reducing the expression of AXL/SOCS and increasing the sensitivity to IFN.
Introduction
Interferons (IFNs) are key mediators of acute antiviral immunity, but in chronic viral infections, ongoing IFN expression can be counterproductive, paradoxically inhibiting the antiviral immune response (Odorizzi and Wherry 2013). Increased expression of IFN-stimulated genes (ISGs) has been associated with disease progression in human immunodeficiency virus (HIV) infection (Herbeuval and others 2006) and in active versus latent Mycobacterium tuberculosis infection (Berry and others 2010). Similarly, in chronic hepatitis C virus (HCV) infection, IFN responsiveness is a major predictor of treatment outcome. Even for non-IFN containing, all oral, direct acting antiviral (DAA) regimens, IFN sensitivity is strongly linked to treatment response (Poordad and others 2011; Chu and others 2012).
In chronic HCV infection, it is well established that increased expression of ISGs in the liver before treatment is associated with a decreased likelihood of achieving a sustained virological response (SVR), irrespective of whether the treatment is IFN-based or not (Sarasin-Filipowicz and others 2008). In these individuals, overactivation of the type I/III IFN signaling pathways results in elevated baseline ISG expression inducing a state of interferon refractoriness, with minimal further induction following IFN-α treatment, and reduced viral clearance (Sarasin-Filipowicz and others 2008). HCV, therefore, represents a useful model to study the mechanisms of IFN activation during chronic infection, and to explore novel ways to enhance the response.
Interestingly, in chronic hepatitis C, the development of insulin resistance (IR) is also associated with nonresponse to treatment (Romero-Gomez and others 2005; Camma and others 2006); conversely, those responding to therapy demonstrate improved insulin sensitivity (Kawaguchi and others 2007). This relationship may result from HCV inducing suppressor of cytokine signaling (SOCS) 3, a negative regulator of both the insulin and IFN signaling pathways, (Kawaguchi and others 2004; Persico and others 2007). In seeking to determine if there are common mechanisms underlying ISG and SOCS3 regulation that might explain the outcomes of HCV, we examined the role of insulin sensitization agents. We show that the PPARα agonist WY-14643 enhances the effect of IFN-α in cells rendered refractory to IFN stimulation, by increasing STAT phosphorylation, ISG promoter activation, and ISG expression. WY-14643 additionally impaired IFN-mediated expression of the negative regulators AXL, SOCS1, and SOCS3. PPARα agonists reverse hepatic ISG upregulation and, therefore, may improve treatment outcome for both IFN-based and exclusively DAA regimens. Furthermore, treatments that reduce overactive IFN signaling offer potential to treat a wide range of chronic infections, and possibly even nonviral disease (Odorizzi and Wherry 2013).
Materials and Methods
Cell culture, viral RNA production, and infection
All HCV clones were propagated in the Huh-7 hepatoma cell line. Cells were maintained in Dulbecco's minimal essential medium with 10% fetal bovine serum. JFH1 and JFH1-Rluc2A (Gorzin and others 2012) (kindly provided by Prof Eric Gowans, University of Adelaide, South Australia) viral RNA was transcribed in vitro using the T7 RiboMAX express large-scale RNA production system (Promega, Madison, WI) and transfected into Huh-7 cells. A nonclonal stable cell line containing a tricistronic, firefly luciferase expressing the HCV subgenomic replicon (SGR), Tri-JFH1, hereafter referred to as SGR-luc (Jones and others 2010) (kindly provided by Dr. John McLauchlan, University of Glasgow Centre for Virus Research, Glasgow, United Kingdom) was created by selection with 500 μg/mL G418 (Roche, Basel, Switzerland) for 2 weeks before experimentation.
Cell line treatments
JFH1-infected Huh-7 cells were treated with either 100 μM metformin (Sigma Aldrich, St. Louis, MO), 10 μM pioglitazone, or 100 μM WY-14643 (Cayman Chemicals, Ann Arbor, MI). Twenty-four hours after treatment with these insulin sensitizers, IFN-α was added (Roferon®-A; Roche). Cells containing the SGR or luciferase-expressing JFH1 were treated with 10 U/mL IFN-α. Cells infected with wild type JFH1 were treated with 5 and 0.5 U/mL IFN-α. PPARα knockdown was performed by transfecting cells with MISSION PPARα siRNA (Sigma-Aldrich) using Lipofectamine RNAiMax™ (Life Technologies, Carlsbad, CA). Cells were treated with siRNA 24 h before WY-14643 treatment, and remained for the duration of the experiment.
To induce IFN refractoriness in vitro, JFH1-infected Huh-7 cells were treated for 3 weeks with 2 U/mL IFN-α (Roferon-A; Roche) alone, or in combination with 100 μM WY-14643. To ensure continuous drug treatment, media was replaced every 2 days. After 3 weeks of exposure to low-dose IFN-α, cells were treated with 20 U/mL IFN-α then cell protein/RNA was extracted over the following 24 h and analyzed.
Real-time polymerase chain reaction analysis of RNA
At 0, 24, and 48 h post-IFN addition, RNA was extracted using RNeasy columns (Qiagen, Venlo, Netherlands). cDNA was synthesized from 500 ng of RNA using the Superscript III first-strand synthesis system (Life Technologies) according to the manufacturer's protocol. Quantitative PCR (qPCR) was then performed on the Rotor-Gene 6000 (Qiagen) using either Taqman or SYBR green protocols to amplify ISG15 (CGCAGATCACCCAGAAGATC, GCCCTTGTTATTCCTCACCA), USP18 (CAGACCCTGACAATCCACCT, AGCTCATACTGCCCTCCAGA), Viperin (CTTTTGCTGGGAAGCTCTTG, CAGCTGCTGCTTTCTCCTCT) AXL (AGCGATGTGTGGTCCTTCG, TCCCTGGCGCAGATAGTCAT), SOCS1 (TTCGCCCTTAGCGTGAAGAT, AGCAGCTCGAAGAGGCAGTC), and SOCS3 (CACATGGCACAAGCACAAGA, CCCTCCAACACATTCCAGGT). All mRNA levels were normalized to 18s ribosomal RNA (4319413E; Applied Biosystems, Foster City, CA).
GAS and ISRE activity reporter assays
Firefly luciferase reporter plasmids p(9-27)4tkΔ(−39)lucter and p(IRF-1*GAS)6tkΔ(−39)lucter containing 4 IFN-stimulated response elements and 6 IFN gamma-activated sequences, respectively were kindly provided by Assoc. Prof. Michael Beard (University of Adelaide, South Australia). JFH1-infected Huh-7 cells pretreated for 3 weeks with 2 U/mL IFN-α±WY-14643 or mock-treated cells were seeded into 6-well plates. Cells were transfected with 2 μg of GAS/ISRE reporter plasmids using Fugene HD (Roche), then treated 24 h later with 20 U/mL IFN-α. At 0, 4, and 8 h post IFN, cells were lysed and luminescence was quantified using the VICTOR plate reader (Perkin Elmer, Waltham, MA). Luminescence was normalized to protein concentration and measured by the DC protein assay (Bio-Rad, Hercules, CA).
Western blotting
Protein lysates were resolved using 10% sodium dodecyl sulfate–polyacrylamide gel electrophoresis and transferred onto a nitrocellulose membrane. Specific antibodies against NS5A (Prof. Mark Harris, University of Leeds, United Kingdom), STAT1, and p-STAT1 (SC-345, Santa Cruz Biotechnology; 9167, Cell Signalling, Dallas, TX) or β-actin (A1978; Sigma-Aldrich) were used, followed by peroxidase-conjugated secondary antibodies. Membranes were visualized using the SuperSignal West Pico chemiluminescence kit (Thermo Fisher Scientific, Waltham, MA) and exposed to x-ray film. NS5A protein expression was quantified using Image J densitometry analysis and normalized to β-actin expression.
Chromatin immunoprecipitation
Approximately 2×107 cells at 80% confluency were used for each chromatin immunoprecipitation (ChIP) experiment. Cells were fixed by adding 37% formaldehyde to media for a final concentration of 1%, and incubated at room temperature (RT) for 10 min. Fixation was stopped by adding 1 M glycine to a final concentration of 125 mM, and incubated at RT for 5 min. Cells were washed twice with cold phosphate buffered saline (PBS) and scraped into 50 mL cold PBS containing 0.01× cOmplete protease inhibitor cocktail (Roche). Cells were then pelleted by centrifugation at 3,000 rpm for 10 min at 4°C. To each sample, 2 mL of cold lysis buffer (1% SDS, 10 mM EDTA, 50 mM Tris-HCl, pH 8.1, 1× cOmplete protease inhibitor cocktail) was added and kept on ice for 10 min. Cells were next sonicated on ice at 40% amplitude and 60% duty for 5 cycles of 15 seconds. Lysates were centrifuged at 13,000 rpm for 10 min at 4°C to pellet cell debris and the 300 μL aliquots were removed. ChIP aliquots were diluted in 1.7 mL ChIP dilution buffer (0.01% SDS, 1.1% Triton X-100, 1.2 mM EDTA, 16.7 m Tris-HCl pH 8.1, 167 mM NaCl, 1× cOmplete protease inhibitor cocktail) and precleared with 20 μL of prewashed protein G beads (sc-2002; Santa Cruz Biotechnology). Extracts were rotated at 4°C for 1 h and beads were pelleted by centrifugation at 2,500 rpm for 2 min and supernatant placed into a new tube. To each tube, 2 μL of the STAT1 antibody (sc-345; Santa Cruz Biotechnology) was added and rotated at 4°C overnight. To each tube, 20 μL of prewashed and blocked protein G beads were added and rotated for 1 h at 4°C. Beads were then gently pelleted at 2,500 rpm for 5 min and washed with the following buffers: Low salt (0.1% SDS, 1% Triton X-100, 2 mM EDTA, 20 mM Tris-HCl pH 8.1, 150 mM NaCl), high salt (0.1% SDS, 1% Triton X-100, 2 mM EDTA, 20 mM Tris-HCl pH 8.1, 500 mM NaCl), LiCl wash buffer (1% Triton X-100, 1% deoxycholic acid, 1 mM EDTA, 10 mM Tris-HCl pH 8.1), TE buffer (10 mM Tris-HCl pH 8.1, 1 mM EDTA). To each tube, 250 μL of elution buffer (1% SDS, 100 mM NaHCO3) was added, incubated at RT for 20 min and centrifuged at 2,500 rpm for 5 min. The supernatant was transferred to a new tube, 250 μL of elution buffer was added to the beads, and the step repeated. To reverse the crosslinks between the DNA and protein, 33 μL of 3 M NaCl was added to 500 μL of the elution and heated to 65°C for 6 h. Importantly, input DNA was diluted to 500 μL with ChIP dilution buffer, and crosslinks were reversed using 3 M NaCl. Lastly, 10 μL of 0.5 M EDTA, 20 μL of 1 M Tris-HCl pH 6.5, and 2 μL of 10 mg/mL proteinase K was added to the elution and incubated at 45°C for 1 h, followed by phenol:chloroform extraction to recover DNA. STAT1-bound DNA from individual samples was quantified by qPCR. Two sets of primers aimed as amplifying targeting AXL intron 4 were assayed, as well as a GAPDH control: AXLp1 (CGCTGCGTCTTCTGTGCTAA, GCCGATTCCAGATGTGCTTT), AXLp4 (CTCCCCTACCCTCCCCTTTC, TGCTTGTGCATCTGTGTTTGTG), GAPDH (ATGGTTGCCACTGGGGATCT, TGCCAAAGCCTAGGGGAAGA). All samples were normalized to their respective inputs.
Data analysis
Quantitative data was expressed as mean±standard error of the mean. Statistical analysis was performed using the Graphpad Prism. Student's t-tests were performed to compare individual treatments. Correlation was performed and the r2 value determined to identify the relationship between gene expressions.
Results
PPARα agonists enhance the antiviral effect of IFN-α against HCV
Due to the association between IR and treatment nonresponse in patients with HCV infection, it has been suggested that insulin sensitization may increase rates of virological response (D'Souza and others 2005; Romero-Gomez and others 2005). To test this hypothesis, 3 different classes of insulin-sensitizing drugs were tested for their ability to improve the efficacy of IFN-α against HCV: metformin, an AMPK activator; pioglitazone, a thiazolidinedione that acts as a PPARγ agonist; and WY-14643, a PPARα agonist.
To screen for effects of these drugs on HCV replication, luciferase reporter constructs were used. Huh-7 cells were transfected with either a JFH1-based HCV subgenomic replicon SGR-Luc (expressing NS3-NS5A), or the full-length JFH1-Rluc2A. Following the establishment of stable infection, cells were pretreated for 24 h with metformin (100 μM), pioglitazone (10 μM), WY-14643 (100 μM), or mock treated, then treated with IFN-α (10 U/mL).
Pretreatment with the PPARα agonist WY-14643 had the most marked effect, with a significant extra reduction in HCV replication of ∼25% and 40% after 24 and 48 h of IFN-α treatment, respectively, for both the subgenomic (black bars) and full length (white bars) JFH1 constructs (Fig. 1A). The other insulin-sensitizing drugs had a lesser effect, which was only significant after 48 h for cells infected with the JFH1-Rluc2A virus.
FIG. 1.

PPARα agonists and other insulin-sensitizing drugs enhance the antiviral effects of IFN-α against HCV. HCV infected Huh-7 cells were pretreated with different insulin-sensitizing drugs for 24 h, treated with IFN-α (10 U/mL), then HCV replication determined by luciferase reporter output (A). Luminescence in mock-treated cells was normalized to 1 at each time point and compared with treated cells. At 24 and 48 h post IFN-α treatment, WY-14643 alone significantly decreased viral replication compared with control in SGR-luc-containing cells (black) and JFH1-Rluc2A-infected cells (white). To confirm these results, JFH1-infected Huh-7 cells were pretreated with WY-14643 for 24 h, then treated with IFN-α. HCV RNA was measured by qPCR following treatment with 0.5 or 5 U/mL IFN-α (B). Significant reduction in JFH1 replication was observed at both 24 and 48 h using 0.5 U/mL IFN with WY-14643 pretreatment, and at 48 h using 5 U/mL IFN with WY-14643 pretreatment. A maximum ∼40% reduction in HCV protein expression (NS5A) was confirmed by western blot and densitometry analysis of NS5A protein expression (C). Graphs and western blot densitometry demonstrate an average of 3 experimental replicates (*P<0.05, **P<0.01). HCV, hepatitis C virus; IFN, interferon; qPCR, quantitative PCR; SGR, sub-genomic replicon.
Because the PPARα agonist WY-14643 demonstrated the strongest antiviral synergy with IFN-α against HCV, it was chosen for further studies. To exclude any effect of the luciferase reporter used in screening experiments, WY-14643 was tested in the standard JFH1 HCV cell culture model (Wakita and others 2005). JFH1-infected Huh-7 cells were pretreated with WY-14643 (100 μM) for 24 h, then treated with IFN-α (0.5 or 5 U/mL). This low dose of IFN-α was used to enhance the detection of synergy with WY-14643, as higher doses of IFN-α reduced viral replication too rapidly. Figure 1B and C demonstrates that WY-14643 pretreatment significantly reduced both HCV RNA and protein, as quantified by qPCR and western blot, compared with IFN-α treatment alone. There was a significant reduction in HCV RNA after both 24 and 48 h using 0.5 U/mL IFN-α and after 48 h using 5 U/mL IFN-α (Fig. 1B). A ∼50% reduction in NS5A protein after 48 h was observed using both concentrations of IFN-α (Fig. 1C) (P<0.05).
Inhibition of HCV replication by WY-14643 is mediated by PPARα
To confirm that the inhibition of HCV replication by WY-14643 is mediated by PPARα signaling, siRNA knockdown of the PPARα nuclear receptor was performed. Twenty-four hours post PPARα knockdown, JFH1-infected Huh-7 cells were treated with 100 μM WY-14643 followed by IFN-α (0.5 U/mL) for 48 h. An 80%–90% decrease in PPARα mRNA was maintained for the duration of the experiment (Fig. 2A). Knockdown of PPARα abolished the effect of WY-14643 on HCV replication at 24 and 48 h post IFN-α treatment (Fig. 2B), confirming that WY-14643-mediated signaling is dependent on PPARα.
FIG. 2.

PPARα knockdown reverses the IFN enhancing effect of WY-14643. JFH1-infected Huh-7 cells were transfected with PPARα siRNA, or scrambled RNA control. Cells were pretreated with WY-14643 for 24 h, or mock treated, then treated with 0.5 U/mL IFN-α. Treatment of Huh-7 cells with PPARα siRNA significantly reduced PPARα expression at baseline, 24 and 48 h by ∼85%, 80%, and 75%, respectively (A). In cells transfected with scrambled RNA, WY-14643 maintained its IFN-α enhancing activity and reduced JFH1 replication, compared with mock-treated cells (B). PPARα siRNA knockdown prevented the effect of WY-14643, with no significant reduction in HCV RNA. Graphs demonstrate an average of 3 experimental replicates (*P<0.05, **P<0.01).
The addition of a PPARα agonist to cells chronically exposed to IFN-α restores sensitivity to IFN-α and reduces HCV replication
In patients with chronic HCV infection who are refractory to IFN treatment, there is a chronic and sustained induction of hepatic ISGs, most likely due to chronic endogenous IFN stimulation (Sarasin-Filipowicz and others 2008). Huh-7 cells are one of the few cell lines capable of sustaining prolonged HCV (JFH1) infection in vitro, possibly due to deficient RIG-I signaling (Sumpter and others 2005). As a result, Huh-7 cells express little to no type I IFN and, therefore, the HCV cell culture model does not reproduce the chronic IFN exposure seen in the livers of patients with chronic HCV infection. To mimic the persistent exposure to IFN in vivo, JFH1-infected Huh-7 cells were treated with low-dose IFN-α (2 U/mL) for 3 weeks, either alone or in the presence of the PPARα agonist WY-14643 (Fig. 3A). Such treatment has been established to mimic the “interferon refractory” phenotype in vitro (Francois-Newton and others 2011) and in vivo (Sarasin-Filipowicz and others 2009). Cells were then treated with a tenfold increased dose of IFN-α (20 U/mL), and multiple components of the IFN response were measured, including STAT1 phosphorylation, ISRE/GAS promoter activation, ISG expression, and HCV replication.
FIG. 3.

The PPARα agonist WY-14643 enhances the antiviral effect of IFN-α against HCV in cells chronically exposed to low-dose IFN. (A) JFH1-infected Huh-7 cells were pretreated with 2 U/mL IFN-α±WY-14643 for 3 weeks, then treated with 20 U/mL IFN-α. STAT phosphorylation was examined up to 2 h posttreatment with 20 U/mL IFN-α; ISRE/GAS promoter activation, ISG expression, and HCV replication were monitored. (B) In cells pretreated with WY-14643 in addition to low-dose (2 U/mL) IFN-α, there was enhanced response to treatment with high-dose (20 U/mL) IFN-α, with significantly reduced JFH1 replication after 24 h, compared with cells pretreated with IFN alone. Graph demonstrates an average of 3 experimental replicates (*P<0.05). ISG, interferon stimulated gene.
Figure 3B demonstrates the effect of high-dose (20 U/mL) IFN-α treatment on HCV replication in Huh-7 cells, following 3 weeks exposure to low-dose (2 U/mL) IFN-α±the PPARα agonist WY-14643. Cells pretreated with low-dose IFN-α±WY-14643 were normalized to mock treated controls, and as expected, viral replication was significantly reduced (85%–90%), mimicking the partial suppression of HCV replication by endogenous IFNs in vivo (Supplementary Fig. S1; Supplementary Data are available online at www.liebertpub.com/jir). Following treatment with high dose IFN-α (20 U/mL) there was a further steep decline in HCV RNA over 24 h (Fig. 3B), confirming an antiviral response. Significantly, the addition of WY-14643 resulted in a 60% further reduction in JFH1 replication 24 h posttreatment with high-dose IFN-α (Fig. 3B).
The PPARα nuclear receptor is widely known as a modulator of lipid metabolism, but has also been shown to inhibit IFN signaling through effects on STAT1 activation and complex formation (Lee and others 2005; Yoo and others 2011). To determine whether PPARα agonists affected IFN-α-induced signaling, IFN-induced phosphorylation of STAT1 (pSTAT1) was examined following chronic IFN-α pretreatment, alone or in combination with WY-14643. pSTAT1 was detected by western blot, using phospho-specific antibodies, and the relative increase in pSTAT1/STAT1 from baseline was calculated using densitometry (Fig. 4A). There was strong pSTAT1 30 min after treatment with IFN-α (20 U/mL), which persisted for at least 2 h. Consistent with previous reports (Sarasin-Filipowicz and others 2009), pre-exposure to low-dose IFN-α (2 U/mL) induced an IFN refractory state, characterized by increased baseline STAT1 phosphorylation, but reduced STAT1 phosphorylation following treatment with high-dose IFN-α (20 U/mL). The PPARα agonist WY-14643 partially reversed this IFN refractoriness, restoring sensitivity to IFN-α (Fig. 4A). In cells pretreated with WY-14643 as well as IFN-α, baseline STAT1 phosphorylation was reduced to levels similar to untreated cells. More importantly, IFN-α-induced STAT1 phosphorylation was significantly increased, consistent with restored IFN sensitivity.
FIG. 4.

STAT1 phosphorylation and ISG promoter activity is desensitized as a result of IFN-α pretreatment, and resensitized with the addition of PPARα agonist WY-14643. JFH1-infected cells were treated with 2 U/mL IFN-α for 3 weeks, alone or in combination with WY-14643, or mock treated, then treated with 20 U/mL IFN-α. Following the addition of 20 U/mL IFN-α, STAT1 and pSTAT1 western blots were performed (A), and ISRE/GAS promoter activation was examined using luciferase reporter plasmids (B). Pretreatment with IFN-α significantly reduced STAT1 phosphorylation following subsequent IFN-α treatment, which was partially restored with concurrent WY-14643 pretreatment (3 experimental replicates performed in duplicate). IFN-α pretreatment also significantly decreased sensitivity of ISRE activation at 8 and 24 h post-IFN-α treatment. Pretreatment with WY-14643 rescued ISRE sensitivity to levels similar to untreated cells. There was a trend toward reduced GAS activation in cells treated with both WY-14643 and IFN-α, compared with IFN-α alone. *Significant difference between treatment and control, and 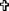 significant difference between IFN and IFN plus WY-14643 treatment. Graphs demonstrate an average of 3 experimental replicates (
significant difference between IFN and IFN plus WY-14643 treatment. Graphs demonstrate an average of 3 experimental replicates (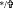 P<0.05,
P<0.05, 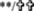 P<0.01). pSTAT1, phosphorylation of STAT1.
P<0.01). pSTAT1, phosphorylation of STAT1.
When IFN-α binds the type 1 IFN receptor, phosphorylated STAT1 forms heterodimers with STAT2 and binds the ISRE in the promoter of hundreds of ISGs. To a lesser extent, STAT1 can homodimerize and bind GAS elements in the promoter of an overlapping subset of ISGs (Reich 2007). Interestingly, STAT1 homodimers have been specifically implicated in the induction of SOCS1 and SOCS3, both of which negatively regulate type I IFN signaling (Rothlin and others 2007).
To examine whether the effects of WY-14643 on STAT phosphorylation results in differential activation of ISRE and GAS promoters, ISRE and GAS luciferase reporter constructs were transfected into HCV (JFH1)-infected Huh-7 cells that had been pretreated with IFN-α (2 U/mL) for 3 weeks±WY-14643, or mock treated. Cells were then treated with IFN-α (20 U/mL) and luminescence was measured over 24 h (Fig. 4B). Consistent with chronic IFN-α exposure inducing IFN refractoriness, in cells pretreated with low-dose IFN-α there was significantly reduced ISRE activation following high-dose IFN-α treatment, compared with mock-treated cells. Interestingly, the addition of WY-14643 to IFN-α pretreatment restored the sensitivity to IFN-α, with significantly increased ISRE activation after 24 h.
GAS activation showed no significant differences among treatments due to its minimal activation by IFN-α. Nonetheless, pretreatment with IFN-α modestly enhanced baseline GAS activation at all time points (Fig. 4B).
Pretreatment with PPARα agonist increases IFN-α-mediated ISG induction and decreases the expression of the negative regulators AXL and SOCS3
A number of antiviral and regulatory ISGs were next examined to determine whether WY-14643 pretreatment significantly increased ISG induction at the transcriptional level. Figure 5 demonstrates the expression of the well-characterized ISGs viperin (RSAD2), ISG15, and USP18. In agreement with the STAT1 phosphorylation results, IFN-α pretreatment increased the baseline ISG expression, whereas the addition of WY-14643 reduced their baseline (0 h) expression (Fig. 5A). Induction of ISGs by high-dose IFN-α (20 U/mL) was blunted following pretreatment with low-dose IFN-α, but was partially restored in the presence of WY-14643 (Fig. 5B).
FIG. 5.

IFN-α and WY-14643 pretreatment modulates ISG expression at baseline and in response to IFN-α. Pretreatment with IFN-α (2 U/mL) significantly upregulated the baseline expression of ISG15, USP18, and viperin, all of which were significantly reduced with the addition of WY-14643 (A). When cells were treated with high-dose IFN-α (20 U/mL), induction of all 3 ISGs was highest in mock-treated cells and significantly reduced following IFN-α pretreatment (B). The addition of WY-14643 to IFN-α pretreatment significantly increased induction of USP18 at 4 h, ISG15 and viperin at 8 h, and viperin at 24 h. Significant difference between treatment and control, and *significant difference between IFN and IFN plus WY-14643 treatment. Graphs demonstrate an average of 3 experimental replicates (P<0.05, P<0.01).
Expression of ISGs responsible for the negative regulation of the IFN-α signaling pathway, SOCS1, SOCS3, and AXL, were also examined. In our HCV model, pretreatment with low-dose IFN-α alone increased the expression of both AXL and SOCS3, both at baseline and following further treatment with IFN-α (20 U/mL), compared with mock-treated cells (Fig. 6). In contrast, pretreatment with both WY-14643 and IFN-α significantly reduced IFN-α-mediated induction of these negative regulators. An early reduction in AXL expression was observed at baseline and 4 h, with reduced induction of SOCS1 at 8 h and SOCS3 at 4 and 8 h (P<0.05). The early downregulation of AXL, with subsequent blunting of SOCS3 expression, supports the hypothesis that AXL induces SOCS genes in hepatocytes, as has been previously demonstrated in dendritic cells (Rothlin and others 2007).
FIG. 6.

Pretreatment with IFN-α±WY-14643 modulates IFN-α-induced AXL, SOCS1, and SOCS3 expression. IFN-α pretreatment upregulated the negative regulators of the IFN-α signaling pathway. When WY-14643 was added, AXL expression was reduced at baseline and 4 h post-IFN-α treatment. SOCS1 expression was reduced at 8 and 24 h post-IFN-α treatment, whereas SOCS3 expression was reduced at 4 and 8 h. *Significant difference between treatment and control, and significant difference between IFN and IFN plus WY-14643 treatment. Graphs demonstrate an average of 3 experimental replicates (P<0.05). SOCS, suppressor of cytokine signaling.
AXL STAT1 promoter occupancy and expression is increased following HCV infection and decreased following PPARα agonist treatment
Next, we wanted to determine whether WY-14643 treatment alters STAT1 binding to the AXL promoter/enhancer. The UCSC genome browser (http://genome.ucsc.edu) was interrogated to identify putative STAT binding sites within the AXL gene (Kent and others 2002). Although no STAT binding sites were identified within the AXL promoter, putative binding sites (p1 and p4) were found in the fourth intron of AXL; a region of strong histone acetylation indicative of histone unwinding (Fig. 7A). To confirm binding to these potential gene enhancer sites, STAT1 ChIP was performed.
FIG. 7.

HCV-induced AXL expression is reduced by WY-14643. (A) The UCSC genome browser was used to identify 2 potential STAT1 binding sites (p1 and p4) in the fourth intron of AXL. (B) Binding of STAT1 to AXL was analyzed by ChIP followed by qPCR. JFH1-infected and mock-infected Huh-7 cells were treated with IFN-α (50 U/L), with or without pretreatment with the PPARα agonist WY-14643. In response to IFN-α treatment, STAT1 binding was reduced by ∼80% in cells pretreated with WY-14643 (P<0.01). STAT1 binding to AXL p1 (P<0.05) and p4 (N.S.) was increased by ∼2-fold in JFH1-infected cells compared with mock-infected Huh-7 cells (2 experimental ChIP replicates, performed in duplicate). (C) AXL expression in JFH1-infected cells was increased by a similar 2-fold over Huh-7 cells. SOCS3 expression was also significantly upregulated in JFH1-infected cells, and correlated with AXL expression (D), suggesting that AXL may induce SOCS3 expression in hepatocytes, as previously demonstrated in dendritic cells (Rothlin and others 2007). Graphs demonstrate an average of 3 experimental replicates (*P<0.05; **P<0.01). ChIP, chromatin immunoprecipitation; N.S., not significant.
Huh-7 and JFH1-infected Huh-7 cells were treated with the PPARα agonist WY-14643 (100 μM) for 48 h, or mock treated, then treated with IFN-α (50 U/mL) for 2 h. As shown in Fig. 7B, the PPARα agonist significantly reduced STAT1 binding to the AXL enhancer p1 by >80% in both Huh-7 and JFH1-infected Huh-7 cells. Interestingly, STAT1 binding was significantly increased in JFH-1-infected cells, compared with mock-infected cells, suggesting that HCV infection may upregulate the AXL expression. To confirm this, qPCR was performed to measure the expression of AXL and its downstream targets SOCS1 and SOCS3. Interestingly, both AXL and SOCS3 were significantly upregulated in JFH1-infected Huh-7 cells by ∼2-fold, with no significant change in SOCS1 expression (Fig. 7C). A strong correlation between AXL and SOCS3, but not SOCS1 expression was also observed, supporting a role for AXL in SOCS3 induction (Fig. 7D).
Discussion
To explore the mechanisms underlying IFN refractoriness in chronic viral infections, we examined different classes of insulin sensitizers for synergy with IFN-α. While metformin and pioglitazone demonstrated modest synergy, the PPARα agonist WY-14643 most potently enhanced the cellular antiviral response. This was accomplished by inhibiting the binding of activated STAT1 to enhancer regions of the AXL gene, thereby reducing AXL induction and subsequent SOC3 upregulation; the net effect is restoration of IFN sensitivity and enhanced viral clearance. This data suggests that PPARα agonists could be a useful adjunct treatment for chronic HCV infection, as well as other chronic infections associated with IFN refractoriness.
Huh-7 cells secrete very low levels of Type I IFN, so we adopted an established in vitro model of IFN refractoriness. Pretreating JFH1-infected Huh-7 cells with low-dose IFN-α mimics the interferon-refractory phenotype that is common among patients with poor treatment response (Lanford and others 2007; Sarasin-Filipowicz and others 2008). This phenotype, characterized by high baseline ISG expression but decreased STAT1 and ISRE activation in response to treatment, with blunted ISG induction, was confirmed in our model. Most importantly, the refractoriness to subsequent IFN exposure was partially reversed by treatment with the PPARα agonist WY-14643. In contrast to antiviral ISGs, in this setting we observed decreased baseline expression and IFN-α-mediated induction of the negative regulators of the IFN-α signaling pathway, namely AXL, SOCS1, and SOCS3.
In dendritic cells, the tyrosine kinase AXL provides a negative feedback loop for IFN signaling: phosphorylated STAT1 induces AXL, which (1) directly inhibits signaling at the type I IFNα receptor; and (2) induces expression of SOCS1 and SOCS3, which in turn suppresses the expression of ISGs (Rothlin and others 2007). The role of AXL in the IFN response in hepatocytes has not been studied. We have now clearly demonstrated the upregulation of AXL following HCV infection in vitro. We further identified a strong correlation between AXL and SOCS3 expression, indicating that AXL may drive SOCS3 expression in the HCV-infected liver, thereby contributing to treatment failure. Consistent with these data, previous studies have demonstrated that hepatic SOCS3 expression is elevated in nonresponders to IFN-based therapy (Huang and others 2007; Persico and others 2007).
In addition to their effects on lipids, PPARα agonists have anti-inflammatory effects, modulating STAT1 and improving a variety of inflammatory disorders in mice (Michalik and Wahli 2008). We, therefore, hypothesized that PPARα agonists may enhance the antiviral effects of IFN by dampening ISG overactivation in infected cells, thereby restoring sensitivity to IFN. As expected, chronic exposure to low-dose IFN-α resulted in overactive ISG expression and a blunted antiviral response to IFN treatment, consistent with previous reports (Sarasin-Filipowicz and others 2009; Francois-Newton and others 2011). Significantly, treatment with the PPARα agonist partially restored baseline STAT1 phosphorylation to control levels (Fig. 4), and reduced baseline ISG expression (Fig. 5). This resulted in a significant resensitization to IFN-α, with increased ISG induction and ISRE promoter activity.
Our ChIP analysis of putative AXL promoters (Fig. 7) provides a plausible and novel mechanism for this effect. Binding of STAT1 to the AXL enhancer region in the fourth intron was significantly increased in HCV-infected cells, consistent with chronic IFN activation. However, STAT1 binding to this region was dramatically reduced (over 80%) following treatment with the PPARα agonist. This supports our hypotheses that reduced IFN responsiveness in the context of chronic HCV infection is due to increased AXL expression, and that PPARα agonists can restore the antiviral response by inhibiting AXL.
Enrichment of PPARα binding sites in close proximity to STAT1/3 promoter regions has previously been demonstrated in HepG2 cells by ChIP and transcriptional profiling (ChIP-chip) (van der Meer and others 2010). This suggests that in addition to inducing global changes in STAT phosphorylation, PPARα may also physically interfere with STAT1 promoter binding, resulting in gene-specific downstream inhibition of inflammatory genes.
Lastly, we suggest that supplementing current HCV treatments in resource-poor countries may be a cost-effective method to improve rates of SVR. DAAs such as simeprevir and sofosbuvir in combination with ribavirin are equally/more effective than IFN-based therapy, but remain expensive even in resource-rich countries (Hill and others 2014; Lawitz and others 2014). Resource-poor countries possess the highest rates of HCV infection and will require IFN-based treatments for years to come whereas the price of current DAAs drops (Hill and others 2014). Supplementing pegylated IFN and ribavirin treatment with PPARα agonists such as fenofibrate or natural PPARα ligands such as fatty acids may be sufficient to improve SVR, while simultaneously improving insulin sensitivity (Fujita and others 2006).
In conclusion, we have demonstrated a novel mechanism for reduced cellular responses to IFN during chronic HCV infection, and a potential treatment to restore this response. IFN pathway activation during chronic infection induces AXL in infected hepatocytes, directly blocking signaling of the type I IFN receptor, and inducing SOCS3, which in turn impairs subsequent induction of ISGs. Treatment with a PPARα agonist reverses this effect by reducing the binding of activated STAT1 to enhancer regions of the AXL gene. This suggests that PPARα agonists, which are widely available and safe, could be useful adjunct treatments for chronic HCV infection, particularly in resource-poor countries. Furthermore, PPARα agonists could potentially be used for other chronic infections associated with IFN pathway overactivation, such as HIV and tuberculosis.
Supplementary Material
Acknowledgments
Work on this article was, in part, supported by program and project grants from the National Health and Medical Research Council of Australia (358772, 1003767, 1047417, and 1053206), the Natural Sciences and Engineering Research Council of Canada PhD scholarship PGSD3-346640-2008, and by the Robert W. Storr bequest to the Sydney Medical Foundation, University of Sydney.
Author Disclosure Statement
The authors declare that no competing financial interests exist.
References
- Berry MP, Graham CM, McNab FW, Xu Z, Bloch SA, Oni T, Wilkinson KA, Banchereau R, Skinner J, Wilkinson RJ, Quinn C, Blankenship D, Dhawan R, Cush JJ, Mejias A, Ramilo O, Kon OM, Pascual V, Banchereau J, Chaussabel D, O'Garra A. 2010. An interferon-inducible neutrophil-driven blood transcriptional signature in human tuberculosis. Nature 466(7309):973–977 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Camma C, Bruno S, Di Marco V, Di Bona D, Rumi M, Vinci M, Rebucci C, Cividini A, Pizzolanti G, Minola E, Mondelli MU, Colombo M, Pinzello G, Craxi A. 2006. Insulin resistance is associated with steatosis in nondiabetic patients with genotype 1 chronic hepatitis C. Hepatology 43(1):64–71 [DOI] [PubMed] [Google Scholar]
- Chu TW, Kulkarni R, Gane EJ, Roberts SK, Stedman C, Angus PW, Ritchie B, Lu XY, Ipe D, Lopatin U, Germer S, Iglesias VA, Elston R, Smith PF, Shulman NS. 2012. Effect of IL28B genotype on early viral kinetics during interferon-free treatment of patients with chronic hepatitis C. Gastroenterology 142(4):790–795 [DOI] [PubMed] [Google Scholar]
- D'Souza R, Sabin CA, Foster GR. 2005. Insulin resistance plays a significant role in liver fibrosis in chronic hepatitis C and in the response to antiviral therapy. Am J Gastroenterol 100(7):1509–1515 [DOI] [PubMed] [Google Scholar]
- Francois-Newton V, Magno de Freitas Almeida G, Payelle-Brogard B, Monneron D, Pichard-Garcia L, Piehler J, Pellegrini S, Uze G. 2011. USP18-based negative feedback control is induced by type I and type III interferons and specifically inactivates interferon alpha response. PLoS One 6(7):e22200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fujita N, Kaito M, Kai M, Sugimoto R, Tanaka H, Horiike S, Konishi M, Iwasa M, Watanabe S, Adachi Y. 2006. Effects of bezafibrate in patients with chronic hepatitis C virus infection: combination with interferon and ribavirin. J Viral Hepat 13(7):441–448 [DOI] [PubMed] [Google Scholar]
- Gorzin AA, Ramsland PA, Tachedjian G, Gowans EJ. 2012. Identification of residues involved in NS2 homodimerization and elucidation of their impact on the HCV life cycle. J Viral Hepat 19(3):189–198 [DOI] [PubMed] [Google Scholar]
- Herbeuval JP, Nilsson J, Boasso A, Hardy AW, Kruhlak MJ, Anderson SA, Dolan MJ, Dy M, Andersson J, Shearer GM. 2006. Differential expression of IFN-alpha and TRAIL/DR5 in lymphoid tissue of progressor versus nonprogressor HIV-1-infected patients. Proc Natl Acad Sci U S A 103(18):7000–7005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hill A, Khoo S, Fortunak J, Simmons B, Ford N. 2014. Minimum costs for producing hepatitis C direct-acting antivirals for use in large-scale treatment access programs in developing countries. Clin Infect Dis 58(7):928–936 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang Y, Feld JJ, Sapp RK, Nanda S, Lin JH, Blatt LM, Fried MW, Murthy K, Liang TJ. 2007. Defective hepatic response to interferon and activation of suppressor of cytokine signaling 3 in chronic hepatitis C. Gastroenterology 132(2):733–744 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones DM, Domingues P, Targett-Adams P, McLauchlan J. 2010. Comparison of U2OS and Huh-7 cells for identifying host factors that affect hepatitis C virus RNA replication. J Gen Virol 91(Pt 9):2238–2248 [DOI] [PubMed] [Google Scholar]
- Kawaguchi T, Ide T, Taniguchi E, Hirano E, Itou M, Sumie S, Nagao Y, Yanagimoto C, Hanada S, Koga H, Sata M. 2007. Clearance of HCV improves insulin resistance, beta-cell function, and hepatic expression of insulin receptor substrate 1 and 2. Am J Gastroenterol 102(3):570–576 [DOI] [PubMed] [Google Scholar]
- Kawaguchi T, Yoshida T, Harada M, Hisamoto T, Nagao Y, Ide T, Taniguchi E, Kumemura H, Hanada S, Maeyama M, Baba S, Koga H, Kumashiro R, Ueno T, Ogata H, Yoshimura A, Sata M. 2004. Hepatitis C virus down-regulates insulin receptor substrates 1 and 2 through up-regulation of suppressor of cytokine signaling 3. Am J Pathol 165(5):1499–1508 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kent WJ, Sugnet CW, Furey TS, Roskin KM, Pringle TH, Zahler AM, Haussler D. 2002. The human genome browser at UCSC. Genome Res 12(6):996–1006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lanford RE, Guerra B, Bigger CB, Lee H, Chavez D, Brasky KM. 2007. Lack of response to exogenous interferon-alpha in the liver of chimpanzees chronically infected with hepatitis C virus. Hepatology 46(4):999–1008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lawitz E, Sulkowski MS, Ghalib R, Rodriguez-Torres M, Younossi ZM, Corregidor A, DeJesus E, Pearlman B, Rabinovitz M, Gitlin N, Lim JK, Pockros PJ, Scott JD, Fevery B, Lambrecht T, Ouwerkerk-Mahadevan S, Callewaert K, Symonds WT, Picchio G, Lindsay KL, Beumont M, Jacobson IM. 2014. Simeprevir plus sofosbuvir, with or without ribavirin, to treat chronic infection with hepatitis C virus genotype 1 in non-responders to pegylated interferon and ribavirin and treatment-naive patients: the COSMOS randomised study. Lancet 384(9956):1756–1765 [DOI] [PubMed] [Google Scholar]
- Lee JH, Joe EH, Jou I. 2005. PPAR-alpha activators suppress STAT1 inflammatory signaling in lipopolysaccharide-activated rat glia. Neuroreport 16(8):829–833 [DOI] [PubMed] [Google Scholar]
- Michalik L, Wahli W. 2008. PPARs mediate lipid signaling in inflammation and cancer. PPAR Res 2008:134059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Odorizzi PM, Wherry EJ. 2013. Immunology. An interferon paradox. Science 340(6129):155–156 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Persico M, Capasso M, Persico E, Svelto M, Russo R, Spano D, Croce L, La Mura V, Moschella F, Masutti F, Torella R, Tiribelli C, Iolascon A. 2007. Suppressor of cytokine signaling 3 (SOCS3) expression and hepatitis C virus-related chronic hepatitis: Insulin resistance and response to antiviral therapy. Hepatology 46(4):1009–1015 [DOI] [PubMed] [Google Scholar]
- Poordad F, McCone J, Jr., Bacon BR, Bruno S, Manns MP, Sulkowski MS, Jacobson IM, Reddy KR, Goodman ZD, Boparai N, DiNubile MJ, Sniukiene V, Brass CA, Albrecht JK, Bronowicki JP. 2011. Boceprevir for untreated chronic HCV genotype 1 infection. N Engl J Med 364(13):1195–1206 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reich NC. 2007. STAT dynamics. Cytokine Growth Factor Rev 18(5–6):511–518 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Romero-Gomez M, Del Mar Viloria M, Andrade RJ, Salmeron J, Diago M, Fernandez-Rodriguez CM, Corpas R, Cruz M, Grande L, Vazquez L, Munoz-De-Rueda P, Lopez-Serrano P, Gila A, Gutierrez ML, Perez C, Ruiz-Extremera A, Suarez E, Castillo J. 2005. Insulin resistance impairs sustained response rate to peginterferon plus ribavirin in chronic hepatitis C patients. Gastroenterology 128(3):636–641 [DOI] [PubMed] [Google Scholar]
- Rothlin CV, Ghosh S, Zuniga EI, Oldstone MB, Lemke G. 2007. TAM receptors are pleiotropic inhibitors of the innate immune response. Cell 131(6):1124–1136 [DOI] [PubMed] [Google Scholar]
- Sarasin-Filipowicz M, Oakeley EJ, Duong FH, Christen V, Terracciano L, Filipowicz W, Heim MH. 2008. Interferon signaling and treatment outcome in chronic hepatitis C. Proc Natl Acad Sci U S A 105(19):7034–7039 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sarasin-Filipowicz M, Wang X, Yan M, Duong FH, Poli V, Hilton DJ, Zhang DE, Heim MH. 2009. Alpha interferon induces long-lasting refractoriness of JAK-STAT signaling in the mouse liver through induction of USP18/UBP43. Mol Cell Biol 29(17):4841–4851 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sumpter R, Jr., Loo YM, Foy E, Li K, Yoneyama M, Fujita T, Lemon SM, Gale M., Jr. 2005. Regulating intracellular antiviral defense and permissiveness to hepatitis C virus RNA replication through a cellular RNA helicase, RIG-I. J Virol 79(5):2689–2699 [DOI] [PMC free article] [PubMed] [Google Scholar]
- van der Meer DL, Degenhardt T, Vaisanen S, de Groot PJ, Heinaniemi M, de Vries SC, Muller M, Carlberg C, Kersten S. 2010. Profiling of promoter occupancy by PPARalpha in human hepatoma cells via ChIP-chip analysis. Nucleic Acids Res 38(9):2839–2850 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wakita T, Pietschmann T, Kato T, Date T, Miyamoto M, Zhao Z, Murthy K, Habermann A, Krausslich HG, Mizokami M, Bartenschlager R, Liang TJ. 2005. Production of infectious hepatitis C virus in tissue culture from a cloned viral genome. Nat Med 11(7):791–796 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoo SH, Park O, Henderson LE, Abdelmegeed MA, Moon KH, Song BJ. 2011. Lack of PPARalpha exacerbates lipopolysaccharide-induced liver toxicity through STAT1 inflammatory signaling and increased oxidative/nitrosative stress. Toxicol Lett 202(1):23–29 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.