

Abstract
During T-cell development, Cd8 expression is controlled via dynamic regulation of its cis-regulatory enhancer elements. Insufficiency of enhancer activity causes variegated Cd8 expression in CD4 +CD8 + double-positive (DP) thymocytes. Brd1 is a subunit of the Hbo1 histone acetyltransferase (HAT) complex responsible for acetylation of histone H3 at lysine 14 (H3K14). Here we show that deletion of Brd1 in haematopoietic progenitors causes variegated expression of Cd8, resulting in the appearance of CD4 +CD8 −TCRβ−/low thymocytes indistinguishable from DP thymocytes in their properties. Biochemical analysis confirms that Brd1 forms a HAT complex with Hbo1 in thymocytes. ChIP analysis demonstrates that Brd1 localizes at the known enhancers in the Cd8 genes and is responsible for acetylation at H3K14. These findings indicate that the Brd1-mediated HAT activity is crucial for efficient activation of Cd8 expression via acetylation at H3K14, which serves as an epigenetic mark that promotes the recruitment of transcription machinery to the Cd8 enhancers.
During T-cell development, expression of the Cd4 and Cd8 co-receptor genes, which are required for differentiation of helper- and killer-lineage T cells, respectively, is strictly regulated. Extensive studies have proposed that stage- and lineage-specific expression of Cd8a and Cd8b1 genes is regulated by combined regulation of at least five different Cd8 enhancers (E8I–E8V) in the Cd8 locus1. Manipulation of Cd8 enhancers in mice has unravelled the role of each enhancer in Cd8 gene regulation. Importantly, variegated expression of CD8 in double-positive (DP) thymocytes is observed in mice with combined deletion of E8I and E8II or deletion of E8II and E8III enhancers2–4. Similarly, among transcription factors implicated in the regulation of Cd8 enhancers, combined deletion of genes of Ikaros family (Ikaros +/−Aiolos −/−) was shown to cause variegated CD8 expression in DP thymocytes5. This Cd8 variegation also occurs in mice with attenuated activity of the BAF (Brahma-related gene/Brahma-associated factor) chromatin-remodelling complex. Haplo-insufficiency of Brg1, which is an ATPase required for BAF-mediated chromatin remodelling, leads to the appearance of CD8-negative DP cells, suggesting a link between Cd8 enhancers and the BAF chromatin remodelling complex6. Moreover, variegated CD8 expression could be partially reverted by intercrossing E8I/E8II doubly deficient mice with DNA methyltransferase 1 (Dnmt1)-deficient mice7. These findings suggest a link between transcription factors such as Ikaros and inhibition of DNA methylation in Cd8 gene activation. However, it remains unclear whether histone modifications are crucial for Cd8 gene activation.
The histone acetyltransferases (HATs) of the MYST family include Tip60, Hbo1, Moz/Morf and Mof and function in multisubunit protein complexes. We previously reported that Bromodomain-containing protein 1 (Brd1), also known as Brpf2, forms a novel HAT complex with Hbo1 and is responsible for the global acetylation of histone H3 at lysine 14 (H3K14)8. However, because of embryonic lethality of Brd1 −/− mice, the function of Brd1 in regulating lymphopoiesis remains uncharacterized. In this study, we generated mice in which Brd1 is inactivated in haematopoietic progenitors and found that Brd1 plays a crucial role in the induction of CD8 expression at several stages of T-lymphocyte development. Our findings revealed that H3K14 acetylation by Brd1 is essential for efficient activation of the Cd8 gene.
Results
Deregulated T-cell development in Brd1-deficient thymus
We previously reported that Brd1 −/− mice die from severe anaemia by E13.5 or earlier8. To understand the role of Brd1 in adult haematopoiesis, we generated mice harbouring Brd1fl mutation in which exon 2 containing the first ATG is floxed (Fig. 1a) and then crossed Brd1fl/fl mice with Tie2-Cre mice that specifically express Cre in haematopoietic and endothelial cells (Tie2-Cre;Brd1fl/fl mice)9. Complete deletion of Brd1 in peripheral blood (PB) CD45 + mononuclear haematopoietic cells was confirmed by genomic PCR (Supplementary Fig. 1). In contrast to germline deletion, which causes lethal anaemia in embryos, deletion of Brd1 in haematopoietic and endothelial cells resulted in a mild differentiation block in erythroblasts in the fetal liver, thus allowing Tie2-Cre;Brd1fl/fl mice to be born and grow normally.
Figure 1. Deregulated T-cell development in the Brd1-deficient thymus.

(a) Strategy for making a conditional knockout allele for Brd1 by homologous recombination in ES cells. FRT recombinase was used to remove the TK-Neo cassette. (b) Absolute numbers of total thymocytes from Tie2-Cre (Brd1 +/+) and Tie2-Cre;Brd1fl/fl (Brd1Δ/Δ) mice. Data are presented as mean±s.d. (Brd1 +/+, n =5; Brd1Δ/Δ, n =4). (c) Representative flow cytometric profiles of CD4 and CD8 expression in thymic T cells from 8- to 11-week-old Tie2-Cre (Brd1 +/+) and Tie2-Cre;Brd1fl/fl (Brd1Δ/Δ) mice. Proportion of each gate is indicated. (d) Representative flow cytometric profiles of TCRβ expression in DP, CD4 SP and CD8 SP cells gated in (c). Percentages of TCRβhi and TCRβ−/low cells are shown (Brd1 +/+ in blue and Brd1Δ/Δ in red). (e) Frequencies of indicated cell populations in total thymocytes from 8- to 11-week-old Tie2-Cre (Brd1 +/+) and Tie2-Cre;Brd1fl/fl (Brd1Δ/Δ) mice. Data are presented as mean±s.d. (Brd1 +/+, n =5; Brd1Δ/Δ, n =4). (f) Representative flow cytometric profiles of CD8 expression in thymic T cells in (c). (g) Frequencies of myeloid (Gr-1 + and/or Mac-1+), B (B220+) or T (CD4 + and CD8 +) cells in CD45+ PB mononuclear cells from 8- to 11-week-old Tie2-Cre (Brd1 +/+) and Tie2-Cre;Brd1fl/fl (Brd1Δ/Δ) mice. Data are presented as mean±s.d. (Brd1 +/+, n =10; Brd1Δ/Δ, n =8). *P<0.05, **P<0.01 and ***P<0.001 (Student’s t-test).
Detailed analysis of the lymphoid organs, however, revealed abnormal thymocyte differentiation. Although thymic cellularity of the Tie2-Cre;Brd1fl/fl mice was comparable to that of the control mice (Fig. 1b), the pattern of CD4/CD8 expression was significantly altered (Fig. 1c). The proportion of CD4 single-positive (SP) thymocytes was significantly increased while that of DP and CD8 SP thymocytes was decreased (Fig. 1c). Analyses of T-cell antigen receptor (TCR) β expression revealed that a proportion of CD4 SP thymocytes of Tie2-Cre;Brd1fl/fl mice expressed no or low levels of TCRβ (TCRβ−/low) like control DP thymocytes (Fig. 1d). DP thymocytes in Tie2-Cre;Brd1fl/fl mice were reduced twofold compared with the control mice. Instead, the CD4+TCRβ−/low thymocytes were found at a frequency comparable to that of DP thymocytes in Tie2-Cre;Brd1fl/fl mice (Fig. 1e). In contrast, the frequency of bona fide CD4 +TCRβhigh SP thymocytes in Tie2-Cre;Brd1fl/fl mice was comparable to that of the control mice (Fig. 1e). Interestingly, the level of CD8 expression in Brd1Δ/Δ thymocytes was moderately but significantly lower than that of the control, with the mean fluorescent intensity (MFI) of DP and CD8 SP thymocytes in flow cytometric analysis being 66.1% and 80.8% of the control, respectively (Fig. 1f). To better distinguish mature from immature thymocytes and reveal their CD4 and CD8 coreceptor expression patterns in Tie2-Cre;Brd1fl/fl mice, we fractionated thymocytes for their expression of TCRβ and CD69 (Supplementary Fig. 2). As expected, WT TCRβ−CD69− thymocytes were mostly CD4+CD8+ DP thymocytes, whereas Brd1Δ/Δ thymocytes contained a large number of CD4 SP cells along with DP cells, suggesting that the large portion of CD4 SP cells are at a similar differentiation stage to DP thymocytes. Of interest, a significant portion of Brd1Δ/Δ mature TCRβ+CD69+ and TCRβ+CD69− thymocytes appeared as CD4−/lowCD8− subset. This result may reflect that some MHC class I-selected thymocytes during maturation towards the CD8 lineage are unable to express CD8 (Supplementary Fig. 2). In addition, CD4−/lowCD8− cells were detected also in the immature TCRβ−CD69− fraction, suggesting that failure of Cd8 gene activation at the transition from DN to DP stage in the absence of Brd1 allows a significant portion of Brd1Δ/Δ DN cells to differentiate into CD4+TCRβ−/low thymocytes directly via CD4lowTCRβ−/low intermediate stage (Supplementary Fig. 2).
We next analysed mature T-cell pools in the periphery, and found that the frequency of mature CD8 + T cells in PB was decreased to 50% of the control (Fig. 1g and Supplementary Fig. 3). All CD4 + T cells showed a high level of TCRβ expression and no CD4 +CD8 −TCR −/low T cells were detected in the PB of Tie2-Cre;Brd1fl/fl mice (Supplementary Fig. 3). Among TCRβ+ cells, however, there was a mild increase in the population of CD4 −CD8 − cells in the PB of Tie2-Cre;Brd1fl/fl mice compared with the controls. It is possible that some class I-restricted cells in the PB are phenotypically CD8 −CD4 − due to the compromised expression of CD8 (Supplementary Fig. 3).
Cell autonomous defect of T cells in the absence of Brd1
The Tie2 promoter drives Cre expression not only in haematopoietic cells, but also in endothelial cells9. To exclude any influence of loss of Brd1 on the thymic microenvironment, we transplanted BM cells from Tie2-Cre control and Tie2-Cre;Brd1fl/fl mice (CD45.2 +) with the same number of BM competitor cells (CD45.1 +) into lethally irradiated wild-type recipient mice (CD45.1 +). Recipient mice reconstituted with Brd1Δ/Δ BM cells showed abnormal thymocyte differentiation similar to that in the Tie2-Cre;Brd1fl/fl mice (Fig. 2a,b). Brd1Δ/Δ T cells were markedly outcompeted by the T cells derived from the competitor cells in the PB (Fig. 2c). We therefore transplanted BM cells from Tie2-Cre;Brd1fl/fl mice without competitor cells and again found that Brd1Δ/Δ T cells, particularly CD8 + T cells, were outcompeted by the residual host T cells (Fig. 2d). These results indicate that the appearance of abnormal CD4 +CD8 − TCRβ−/low thymocytes and the inefficient production of mature CD8 + T cells are intrinsic to Brd1Δ/Δ cells.
Figure 2. Cell autonomous defect of T cells in the absence of Brd1.

(a) Surface expression of CD4 and CD8 on Brd1Δ/Δ cells in recipient mice. BM cells from Tie2-Cre control (Brd1 +/+) and Tie2-Cre;Brd1fl/fl (Brd1Δ/Δ) mice (CD45.2 +) were transplanted into lethally irradiated wild-type recipient mice (CD45.1+) along with the same number of BM competitor cells (CD45.1 +). Representative flow cytometric profiles of CD4 and CD8 expression in CD45.2+ donor-derived T cells in the thymus at 6 months post-transplantation are depicted. The proportion of each gate is indicated. (b) Frequencies of indicated cell populations in CD45.2 + donor-derived T cells in (a). Data are presented as mean±s.d. (Brd1 +/+, n =5; Brd1Δ/Δ, n =5). (c) Contribution of Brd1Δ/Δ haematopoietic cells to the PB T lymphocytes of recipient mice in (a). Chimerism of donor-derived CD45.2+ Brd1Δ/Δ cells in the PB at 12 weeks post-transplantation is shown as mean±s.d. (Brd1 +/+, n =6; Brd1Δ/Δ, n =6). (d) Contribution of Brd1Δ/Δ haematopoietic cells to the PB in recipient mice. BM cells from Tie2-Cre control (Brd1 +/+) and Tie2-Cre;Brd1fl/fl mice (Brd1Δ/Δ) (CD45.2 +) were transplanted into lethally irradiated wild-type recipient mice (CD45.1+) without any BM competitor cells. Chimerism of donor-derived CD45.2 + Brd1Δ/Δ cells in the PB at 12 weeks post-transplantation is shown as mean±s.d. (Brd1 +/+, n =6; Brd1Δ/Δ, n =6). (e) Competitive repopulating capacity of Brd1Δ/Δ peripheral CD4 T cells. CD4 T cells from the Tie2-Cre control and Tie2-Cre;Brd1fl/fl spleen (1 × 106 each, CD45.2) were transferred into sublethally irradiated NOG mice along with the same number of CD45.1 splenic CD4 T cells (competitor cells). Chimerism of CD45.2+ Brd1Δ/Δ CD4 T cells in the PB CD4+TCRβ+ T-cell pool is plotted as dots and mean values are indicated as bars. (f) Expression of S1P receptors in Brd1Δ/Δ CD4 +TCRβhigh thymocytes. Quantitative RT-PCR analysis of the expression of S1P receptor genes in Brd1 +/+ and Brd1Δ/Δ CD4+TCRβhigh thymocytes was performed. Hprt1 was used to normalize the amount of input RNA. ND =not detected. *P<0.05, **P<0.01 and ***P<0.001 (Student’s t-test).
Given the essential requirement for CD8 as a co-receptor to support the differentiation of CD8 T cells, it is likely that the low CD8 expression in the thymus is causal for the reduction of CD8 T cells. However, Brd1Δ/Δ CD4 T cells also showed significantly lower chimerism in PB in competitive repopulation experiments (Fig. 2d). We therefore tested the ability of Brd1Δ/Δ peripheral CD4 T cells to repopulate in immuno-compromised host mice. We transferred 1 × 106 CD45.2 CD4 T cells from the Tie2-Cre control and the Tie2-Cre;Brd1fl/fl spleen into sublethally irradiated NOG mice along with the same number of CD45.1 splenic CD4 T cells (competitor cells). Brd1Δ/Δ CD4 T cells established chimerism that is comparable to or rather higher than that of the control cells (Fig. 2e). Considering that the number of Brd1Δ/Δ CD4 +TCRβhigh SP thymocytes is comparable to that of the WT control, these results suggest that the reduced chimerism of Brd1Δ/Δ CD4 T cells in the periphery could be attributed to the impaired egress from the thymus to the periphery. To test this hypothesis, we checked the expression of S1P receptors (S1P1–5), which regulate the egress of T cells from the thymus10. Although expression of S1P1, which is critical for CD4 SP thymocytes to egress from the thymus10, showed a trend of reduction in Brd1Δ/Δ CD4 +TCRβhigh thymocytes, it did not give a statistical significance (Fig. 2f).
These findings suggest that Brd1 is necessary to endow appropriate integrity to CD4 T cells in the periphery. We then examined whether Brd1 was required for CD4 T-cell function. We purified splenic naive CD4 T cells from WT and Tie2-Cre;Brd1fl/fl mice and cultured them under Th1 or Th2 conditions in vitro. Intriguingly, the numbers of both IFNγ-producing cells under Th1 conditions and IL-4-producing cells under Th2 conditions were decreased substantially by loss of Brd1 (Supplementary Fig. 4). These findings indicate that Brd1 is also required for efficient cytokine production by peripheral CD4 T cells.
Variegated Cd8 expression in the absence of Brd1
We next characterized the cell cycle status of Brd1Δ/Δ CD4 +CD8 − TCRβ−/low thymocytes reconstituted in wild-type thymic environment using a BrdU incorporation assay. Both control and Brd1Δ/Δ DP thymocytes contained a significant proportion of cycling cells, whereas CD4 +CD8 −TCRβhigh and CD8 SP thy-mocytes did not (Fig. 3a). Of note, Brd1Δ/Δ CD4 +CD8 − TCRβ−/low thymocytes contained cycling cells at the same frequency as DP thymocytes.
Figure 3. Inefficient activation of Cd8 genes in Brd1-deficient DP thymocytes.

(a) Cell cycle status of repopulated thymocytes. Percentages of cells in S phase in the indicated populations of thymocytes from the recipient mice in Fig. 2d were measured by detecting incorporated BrdU by flow cytometry at 2 h post-intraperitoneal injection of BrdU. Data are presented as mean±s.d. (Brd1 +/+, n =4; Brd1Δ/Δ, n =3). (b) Hierarchical clustering of gene expression patterns of the indicated cell fractions obtained in microarray analyses. For clustering analysis, the Ward’s linkage rule was applied to all expressed genes in each of the sorted cells. The Ward’s linkage scores are indicated on the top. (c) A scatter diagram of microarray analysis. The average signal levels of indicated cell fractions compared with those of Brd1Δ/Δ CD4 +CD8 −TCRβ−/low cells are plotted. The upper and lower lines represent the borderline for a twofold increase and a twofold decrease, respectively. (d) Quantitative RT-PCR analysis of the expression of the genes indicated in Brd1 +/+ and Brd1Δ/Δ thymocytes. Hprt1 was used to normalize the amount of input RNA. (e) Representative flow cytometric profiles of CD4 and CD8 expression in thymic T cells from 8- to 11-week-old Tie2-Cre (Brd1 +/+) and Tie2-Cre;Brd1fl/fl (Brd1Δ/Δ) mice expressing OT-I TCR transgene. Proportion of each gate is indicated. Data are representative of three independent experiments. (f) Representative flow cytometric profiles of CD4 and CD8 expression in PB of Brd1 +/+ OT-I and Brd1Δ/Δ OT-I mice (left panels). Proportion of each gate is indicated. CD8 expression in PB T cells is also depicted in histograms (right panel).
Furthermore, hierarchical clustering of gene expression profile in several thymocyte fractions from Brd1 +/+ and Brd1Δ/Δ mice revealed that Brd1Δ/Δ CD4 +CD8 −TCRβ−/low thymocytes have an expression profile highly similar to DP thymocytes, particularly to Brd1Δ/Δ DP thymocytes, but quite different from mature (TCRβhi) CD4 SP thymocytes of both Brd1 +/+ and Brd1Δ/Δ genotypes (Fig. 3b,c). Quantitative RT-PCR confirmed that Brd1Δ/Δ CD4 +CD8 −TCRβ−/low thymocytes express genes specific to DP thymocytes, such as Myb and Rag1 (refs 11,12), but not Cd8a and Cd8b1 (Fig. 3d and Supplementary Table 1). Expression of key regulator genes of Cd8 transcription, such as Ikaros, Runx1 and Runx3, was not altered in Brd1Δ/Δ DP and CD4 +CD8 −TCRβ−/low thymocytes compared with that in WT DP thymocytes (Supplementary Table 1). These results indicate that Brd1Δ/Δ CD4 +CD8 −TCRβ−/low thymocytes correspond to immature thymocytes that would normally appear as DP thymocytes. Correspondingly, expression of Zbtb7b that encodes Th-POK, a master regulator of CD4 helper T cells, was not detected in Brd1Δ/Δ CD4 +CD8 −TCRβ−/low thymocytes as was not detected in WT DP thymocytes. However, its expression in Brd1Δ/Δ CD4 +CD8 −TCRβhigh thymocytes was comparable to that in WT CD4 SP thymocytes (Fig. 3d).
To investigate whether the lack of Brd1 affects the direction of class I-restricted thymocytes to CD8 SP cells, we crossed Tie2-Cre;Brd1fl/fl mice with mice expressing OT-I MHC class I-restricted transgenic TCR13. As expected, the vast majority of Brd1Δ/Δ DP thymocytes appeared as CD8 − DP (CD4 +CD8 −TCRβ−/low) thymocytes (Fig. 3e). However, some of the canonical Brd1Δ/Δ DP thymocytes were positively selected and differentiated into mature CD8 SP thymocytes. In the PB, CD8 + T cells dominated even in the absence of Brd1, although they exhibited a reduced level of CD8 expression (Fig. 3f). On the basis of these findings, we conclude that Brd1 deficiency causes variegated expression of CD8 due to inefficient activation of the Cd8 gene.
Of interest, expression of Brd1 is upregulated during transition of DN to DP stage and is downregulated afterwards (Fig. 3d). Thus, it is possible that an increase in the amount of Brd1 is necessary to initiate Cd8 activation during the DN to DP transition. To test this notion, we cultured DN3 cells on TSt-4/DLL stromal cells and monitored their differentiation. As clearly demonstrated in Fig. 4, the majority of Brd1Δ/Δ DN3 cells differentiated into CD4 +CD8 −TCRβ−/low thymocytes without going through the CD4 +CD8 + DP stage. These findings well correspond to the in vivo finding that a small but significant subpopulation of TCRβ−CD69 − immature thymocytes exhibits a CD4 −/low CD8 − phenotype in the Tie2-Cre;Brd1fl/fl thymus (Fig. 1c and Supplementary Fig. 2).
Figure 4. In vitro differentiation of Brd1Δ/Δ DN3 cells.
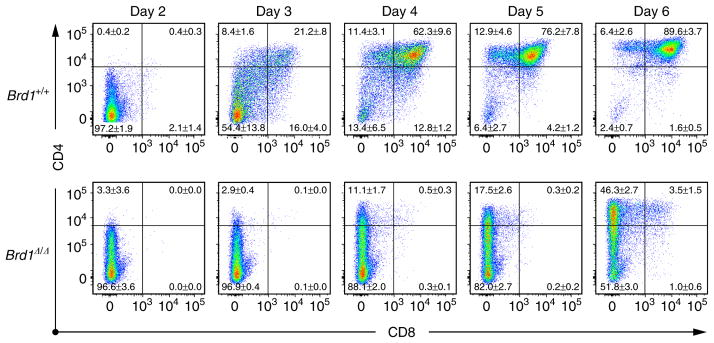
DN3 cells from Tie2-Cre control (Brd1 +/+) and Tie2-Cre;Brd1fl/fl (Brd1Δ/Δ) mice were seeded on TSt-4/DLL stromal cells in triplicate and their differentiation was monitored. Cytokines (10 ng ml −1 SCF, IL-7 and Flt3L) were supplemented only for the initial 24 h. Representative CD4 and CD8 expression profiles from the three repeated experiments are depicted. The proportion of each gate is indicated.
We next examined the apoptotic status of Brd1Δ/Δ CD8 SP thymocytes with lower CD8 expression and found that they do not show any enhancement in apoptotic cell death compared with the control cells (Annexin V + cells; Brd1 +/+ 8.4%, n =2 versus Brd1Δ/Δ 6.1%, n =2).
Impaired reactivation of Cd8 in Brd1Δ/Δ peripheral T cells
In the gut intraepithelial lymphocyte (IEL) compartment, there are three cell subsets expressing the Cd8a gene other than conventional CD8αβ+ cytotoxic T cells; γδT cells, CD8αα+ and CD4 +CD8αα+ αβT cells14. We therefore examined CD8 expression in those cells. Of interest, the population of CD8αα+ and CD4 +CD8αα+ αβT cells were significantly decreased in Tie2-Cre;Brd1fl/fl mice compared with the Tie2-Cre control mice (Fig. 5). In addition, the percentage of CD8αα expressing γδT cells was significantly reduced (Fig. 5). We interpreted the reduction in CD8αα+ γδT cells as a reflection of variegated CD8 expression in γδT cells in the gut. It has also been reported that CD4 +CD8αα+ IELs are differentiated from CD4 + T cells by reactivating the Cd8a gene after the downregulation of Th-POK, a master regulator of CD4 helper T cells15. Together, these findings suggest that Brd1 is required to initiate Cd8a activation in γδT cells as well as Cd8 reactivation in CD4 + T cells in the gut.
Figure 5. Impaired CD8 expression in IEL subsets in the absence of Brd1.

Expression of CD8αα in TCRβ+, CD4 +TCRβ+ and γδTCR + IELs was analysed in Tie2-Cre control (Brd1 +/+) and Tie2-Cre;Brd1fl/fl (Brd1Δ/Δ) mice. Representative flow cytometric profiles from the two repeated experiments with a proportion of each gate (left panels) and the percentages of CD8αα+ cells in the indicated cell populations with mean values (right panels) are shown. **P<0.01 and ***P<0.001 (Student’s t-test).
Brd1-Hbo1 complex is responsible for H3K14 acetylation at Cd8 gene loci
To confirm that Brd1 acts as a histone modifier, we compared levels of histone acetylation between Tie2-Cre and Tie2-Cre;Brd1fl/fl thymi. As we reported previously in Brd1 −/− erythroblasts8, the global level of H3K14 acetylation was profoundly decreased in Brd1Δ/Δ thymocytes, whereas the levels of H3K9, H4K5, K8, K12 and K16 were not significantly changed (Fig. 6a). Biochemical analysis also confirmed that Brd1 forms a HAT complex with Hbo1 and Ing4/5 in the AKR1 DP T cell line expressing 3xFlag-Brd1 (AKR1/Brd1 cells) (Supplementary Fig. 5). Chromatin immunoprecipitation (ChIP) assays of AKR1/Brd1 and AKR1/Hbo1 cells confirmed that Brd1 and Hbo1 bind to the enhancers and promoters of the Cd8 genes, respectively (Fig. 6b). ChIP assays also revealed drastic reduction in levels of H3K14 acetylation at the enhancers and promoters of the Cd8a and Cd8b1 loci in Brd1Δ/Δ DP thymocytes and even more reduction in Brd1Δ/Δ CD8 − DP thymocytes. In contrast, the reduction was only moderate at the Cd4 silencer in both Brd1Δ/Δ DP and CD8 − DP thymocytes, which express CD4 (Fig. 6b). These findings suggest that the Hbo1-Brd1 HAT complex acetylates H3K14 at the transcriptional regulatory elements of the Cd8 gene, and that this step is necessary for efficient activation of the Cd8 gene.
Figure 6. Brd1-Hbo1 complex acetylates H3K14 at the Cd8 gene loci.

(a) Levels of acetylation at histone H3 and H4 in total thymocytes from Tie2-Cre (Brd1 +/+) and Tie2-Cre;Brd1fl/fl (Brd1Δ/Δ) thymus. Histones purified from thymocytes were analysed by western blotting using the indicated antibodies. (b) Levels of H3K14 acetylation at the enhancers and promoters of the Cd8a and Cd8b1 genes. Schematic representation of the Cd8a and Cd8b1 gene loci and the location of the primer sets are depicted (upper panel). The binding of Brd1 and Hbo1 to the Cd8a and Cd8b1 gene loci was determined by ChIP using an anti-Flag antibody in AKR1/3 × Flag-Brd1 cells and anti-HA antibody in AKR1/HA-Hbo1 cells, respectively. The relative amount of immunoprecipitated DNA is depicted as a percentage of input DNA. An isotype control IgG was used as a negative control. The Cd4 silencer element and p16 promoter served as controls. The data are shown as the mean±s.d. for triplicate PCRs (middle panel). ChIP analyses were performed using Brd1 +/+ and Brd1Δ/Δ DP cells and Brd1Δ/Δ CD4 +CD8 −TCRβ−/low cells purified by cell sorting and an anti-acetylated H3K14 antibody. The relative amount of immunoprecipitated DNA is depicted as a percentage of input DNA. The Cd4 silencer element served as a control. The data are shown as the mean±s.d. for triplicate PCRs (lower panel). (c) Brd1 and Hbo1 directly interact with Runx proteins. 293T cells were transfected with Flag-tagged Runx1 or Runx3 together with either HA-tagged Brd1 or Hbo1. Runx proteins were immunoprecipitated using an anti-Flag antibody, and then immunoprecipitates were detected by immunoblotting using anti-Flag and anti-HA antibodies (upper and middle panels). The direct interactions between Runx proteins with Brd1 and Hbo1 were confirmed by GST-pull down assays using GST-Runx1 and GST-Runx3 fusion proteins and in vitro translated HA-tagged Brd1 and Hbo1 following immunoblotting using an anti-HA antibody (lower panels). (d) Brd1 and Hbo1 directly interact with Ikaros proteins. 293T cells were transfected with Flag-tagged Ikaros together with either HA-tagged Brd1 or Hbo1. Ikaros protein was immunoprecipitated using an anti-Flag antibody, and then immunoprecipitates were detected by immunoblotting using anti-Flag and anti-HA antibodies.
HATs are known to collaborate with the transcription factors critical for the transcriptional activation of the given genes. We therefore searched for physical interactions between the Hbo1-Brd1 HAT with Runx family transcription factors and Ikaros, which are known critical regulators that activate the Cd8 genes5,16. To identify physical interactions, we co-transfected 293T cells with Runx1 or Runx3 together with either Brd1 or Hbo1 and performed immunoprecipitation. Both Brd1 and Hbo1 were readily co-immunoprecipitated with Runx1 and Runx3 (Fig. 6c). These direct interactions were confirmed by GST-pull down assays using GST-Runx1 and GST-Runx3 and in vitro translated Brd1 and Hbo1 (Fig. 6c). We also detected co-immunoprecipitation of Ikaros with Brd1 or Hbo1 in 293T cells in a similar transfection assay (Fig. 6d). These results support the idea of crosstalk between transcription factors and the Hbo1-Brd1 HAT complex in the activation of Cd8 genes.
Finally, to confirm the role of Brd1 in the initial activation of Cd8 gene, we transduced Brd1Δ/Δ DN cells with a Brd1 retrovirus and cultured them on TSt-4/DLL stromal cells. Defective CD8 expression on differentiating Brd1Δ/Δ DN cells was completely rescued by exogenous Brd1, allowing Brd1Δ/Δ DN cells to become DP cells in vitro (Fig. 7a). Corresponding to this finding, the global level of acetylation at H3K14 in DP cells was largely restored by retroviral complementation of Brd1 (Fig. 7b). These results confirm the critical role of the acetylation at H3K14 by the Hbo1-Brd1 HAT complex in the transcriptional activation of Cd8 gene.
Figure 7. In vitro differentiation of Brd1Δ/Δ DN3 cells.

(a) DN3 thymocytes from Tie2-Cre;Brd1fl/fl (Brd1Δ/Δ) mice were seeded on TSt-4/DLL stromal cells in the presence of 10 ng/ml−1 SCF, IL-7 and Flt3L and transduced with a Brd1 retrovirus. At day 3 post-transduction, transduced GFP + cells were purified by cell sorting and further cultured on TSt-4/DLL stromal cells in the presence of 2 ng ml −1 SCF, IL-7 and Flt3L for 7 to 10 days. DN3 thymocytes from Tie2-Cre (Brd1 +/+) and Tie2-Cre;Brd1fl/fl (Brd1Δ/Δ) mice without transduction served as controls. Representative CD4 and CD8 expression profiles from three repeated experiments are depicted. The proportion of each gate is indicated. (b) Levels of acetylation at H3K14 in rescued DP cells. DP cells were derived from Brd1-transduced Brd1Δ/Δ DN1/2 thymocytes (Brd1Δ/Δ DP/Brd1, lane 4) and from WT DN1/2 thymocytes (Brd1 +/+ DP/mock, lane 3) as in (a) and were subjected to western blotting using an anti-H3K14ac antibody (left panel). DP thymocytes from Tie2-Cre (Brd1 +/+ DP, lane 1) and Tie2-Cre;Brd1fl/fl (Brd1Δ/Δ DP, lane 2) thymus served as controls.
Discussion
In this study, variegated expression of CD8 was observed in Brd1Δ/Δ DP thymocytes in a manner similar to DP thymocytes with insufficient activity of Cd8 enhancers2–4. Several molecules have been implicated in the regulation of Cd8 enhancers, including Ikaros and Runx family transcription factors5,16, BAF chromatin-remodelling complex6 and Dnmt17. However, the role of histone modifications has never been characterized before.
N-terminal tail domains of histones are acetylated in the promoter region of actively transcribed genes17. Acetyl-lysine residues are recognized by bromodomain-containing factors. Although the role of acetylation at H3K14 is not well understood, it has been reported that several bromodomain-containing proteins such as BAF57, a subunit of the BAF chromatin remodelling complex, and TAFII250, which recruits TFIID, bind to acetylated H3K14 (refs 18,19). Indeed, compromised BAF activity causes variegated CD8 expression in a manner similar to that caused by the loss of Brd1, suggesting a functional crosstalk between the Hbo1-Brd1 HAT complex and the BAF complex6. It is therefore possible that the Hbo1-Brd1 complex is involved in the chromatin relaxation via H3K14 acetylation, which is followed by subsequent recruitment of transcriptional complexes at the Cd8 enhancers for full activation of the Cd8 locus.
Although the expression of CD8 on the cell surface was moderately decreased in Brd1Δ/Δ DP and CD8 SP thymocytes and Brd1Δ/Δ CD8 + T cells in the PB, mRNA expression of Cd8a and Cd8b1 genes in these CD8 expressing Brd1Δ/Δ cells was comparable to that of the control cells. These results suggest that the Hbo1-Brd1 HAT complex and presumably acetylation at H3K14 are required in the initiation of Cd8 transcription at the transition of DN to DP stage, but once the Cd8 transcription is activated by the compensatory action of other HATs, it can be maintained even in the absence of Brd1. Nevertheless, failure of CD8αα induction in CD4 T cells in the gut indicates that Brd1 is required to reactivate Cd8 transcription even in the peripheral T cells. These findings highlight the role of the Hbo1-Brd1 HAT complex in the activation but not the maintenance of Cd8 expression.
Of note, Brd1Δ/Δ CD4 T cells also showed significantly lower chimerism in PB, although the capacity of Brd1Δ/Δ peripheral CD4 T cells to repopulate in immunocompromised host mice was almost intact. We therefore suspected that the reduced chimerism of Brd1Δ/Δ CD4 T cells in the periphery could be attributed to the impaired egress from the thymus to the periphery. Indeed, the expression of S1P1, which is critical for CD4 SP thymocytes to egress from the thymus, showed a mild, albeit not statistically significant, reduction in Brd1Δ/Δ CD4 +TCRβhigh thymocytes, suggesting a moderate defect of Brd1Δ/Δ CD4 SP thymocytes in egression into the periphery. In addition, given the substantial difference in gene expression profiles between WT and Brd1Δ/Δ peripheral CD4 T cells (Fig. 3b), Brd1 could also be necessary to endow appropriate integrity to CD4 SP thymocytes during terminal differentiation process. Furthermore, Brd1Δ/Δ peripheral CD4 T cells were demonstrated to be defective in producing cytokines. Together, all these findings implicate Brd1 in various aspects of the immune cell development and functions.
Advances in epigenetic research are revealing the fine epigenetic regulations that control developmentally regulated gene expression, including those involved in haemato-lymphopoiesis20,21. Our findings in this study provide novel insights into epigenetic control of T-cell development. Characterization of the crosstalk between the Hbo1-Brd1 HAT complex and other regulators would promote the understanding of the complex regulation of the co-receptor expression, an essential step in the development of thymocytes.
Methods
Mice and gene targeting of Brd1
For conditional deletion of Brd1, the targeting vector was designed as in Fig. 1, and Brd1fl/fl mice were generated using R1 embryonic stem (ES) cells according to the conventional protocol. Brd1fl/fl mice were backcrossed to the C57BL/6 background more than seven times and crossed with Tie2-Cre mice9. Eight-week-old female C57BL/6 (CD45.2) mice were purchased from Japan SLC. Eight-week-old female C57BL/6 mice congenic for the Ly5 locus (CD45.1) were purchased from Sankyo-Lab Service Corporation. OT-I mice (C57BL/6) were purchased from the Jackson Laboratory. Mice were bred and maintained in the Animal Research Facility of the Graduate School of Medicine, Chiba University in accordance with institutional guidelines.
Flow cytometry and antibodies
The mAbs were purchased from BD Biosciences, eBioscience or BioLegend. For analysis of co-receptor expression in adult thymocytes, cells were stained with CD45.2-PacificBlue (1:200, 104), CD4-APC (1:50, GK1.5), CD8α-PE (1:25, 53–6.7) and TCRβ chain-APC/Cy7 (1:50, H57–597). For fetal DN thymocytes, cells were stained with CD3ε-Brilliant Violet (1:50, 145–2C11), CD4-Biotin (1:50, GK1.5), CD8-APC/Cy7 (1:50, 53–6.7), CD25-APC (1:50, PC61), CD44-PE (1:50, IM7) and CD69-PE/Cy7 (1:50, H1.2F3). A biotin-labelled CD4 antibody was visualized with Streptavidin (1:50, PE/Cy7). For cell cycle analysis, incorporated BrdU was measured by using APC BrdU Flow Kit (BD Biosciences) according to the manufacturer’s protocol on gated DP, CD4 SP, CD4 +CD8 −TCRβ−/low and CD8 SP cells. For PB cells, CD45.1-PE/Cy7 (1:50, A20), CD45.2-FITC (1:50, 104), B220-APC (1:50, RA3–6B2), Gr-1-PE (1:200, RB6–8C5), Mac-1-PE (1:200, M1/70), CD4-APC/Cy7 or -APC (1:50, RM4–5 or 1:50, GK1.5) and CD8α-APC/Cy7 or -PE (1:50, 53-6.7) were used. Other antibodies used are γδTCR-biotin (1:50, GL3), NK1.1 (PK136), CD11c (N418), CD25-APC (1:50, PC61) and CD44-PE (1:50, IM7). Dead cells were eliminated by staining with propidium iodide (1 μg ml −1; Sigma-Aldrich). For Annexin V staining, cells were suspended with 1xAnnexin binding buffer and stained with FITC-Annexin V following the manufacturer’s protocol (BD Biosciences). All flow cytometric analyses and cell sorting were performed on FACSAria II or FACS-Canto II (BD Biosciences). IELs were isolated as previously described22.
DN3 culture
Using a biotinylated lineage mixture [Gr-1, Mac-1, Ter-119, B220 (1:40), CD4, CD8α (1:10), γΔTCR, CD3ε, CD11c and NK1.1 (1:20)], CD25-APC(1:50, PC61) and CD44-PE(1:50, IM7), Lin −CD25 +CD44 + DN3 thymocytes were sorted on a FACSAria II. DN3 cells were cultured on TSt-4/DLL stromal cells with 10 ng ml −1 SCF, IL-7 and Flt3L (Peprotech) supplemented only for the initial 24 h. For rescue experiments, Brd1Δ/Δ DN1/2 or DN3 cells were transduced with a Brd1 retrovirus (pMC-3xFlag-Brd1-IRES-EGFP8) on TSt-4/DLL stromal cells. Transduced GFP + cells were purified by cell sorting and then cultured as described above.
Competitive repopulating assay of peripheral CD4 T cells
We transferred 1 × 106 CD45.2 CD4 T cells purified from the Tie2-Cre control and Tie2-Cre;Brd1fl/fl spleen into sublethally irradiated NOG mice (2.0 Gy) along with the same number of CD45.1 splenic CD4 T cells (competitor cells). Chimerism of CD45.1 competitor CD4 T cells and CD45.2 test CD4 T cells in the PB was monitored weekly.
Induction of Th1 and Th2 cytokine expression
CD4 + T cells with a naive phenotype (CD4 +CD62Lhi) were purified with a FACSAria cell sorter (Becton Dickinson), which yielded a purity of >98%, and were used as naive CD4 + T cells. Naive CD4 + T cells (1.5 × 106) were stimulated for 2 days with immobilized mAb to TCRβ (10 μg ml −1; H57-597) plus soluble mAb to CD28 (1 μg ml −1; 37.51; BioLegend) in the presence of IL-2 (15 ng ml −1), IL-12 (10 ng ml −1; Wako) and mAb to IL-4 (5 μg ml −1; 11B11) for Th1 conditions, or in the presence of IL-2 (15 ng ml −1), IL-4 (100 ng ml −1; PeproTech) and mAb to IFN-γ (5 μg ml −1; R4-6A2) for Th2 conditions. Cells were cultured for an additional 4 days without stimulation of the TCR in the presence of the original cytokines. Cultured cells were restimulated for 6 h with immobilized mAb to TCRβ (10 μg ml −1), and intracellular staining was done as described23.
BM transplantation
BM cells from B6-CD45.2 mutant mice (test cells) were transplanted intravenously into 8-week-old B6-CD45.1 recipients irradiated at a dose of 9.5 Gy with or without BM cells from 8-week-old B6-CD45.1 congenic mice (competitor cells).
Immunoprecipitation and GST pull-down assay
293T cells were transfected with pcDNA3-Flag-human Runx1, human Runx3 or mouse Ikaros together with either pcDNA3-HA-mouse Brd1 or mouse Hbo1. The cells were lysed in lysis buffer containing 250 mM NaCl and then immunoprecipitation was performed using anti-FLAG M2 affinity gel. Immunocomplexes were eluted with FLAG peptide. Histone proteins were extracted following the method described previously24. For GST pull-down assays, DNA fragments encoding the full-length human Runx1 and Runx3 were subcloned into the pGEX6p vector. GST fusion proteins were expressed in BL21 (DE3) cells. For the GST pull-down assay, HA-tagged Brd1 or Hbo1 was translated in vitro using a TNT reticulocyte lysate transcription/translation system (Promega) and mixed with GST-fusion proteins. The reaction was carried out in lysis buffer containing 150 mM NaCl at 4 °C for 2 h and the beads were washed four times with the same buffer. Proteins were separated by sodium dodecyl sulfate–PAGE (SDS–PAGE), transferred to a PVDF membrane and detected by western blotting using the following antibodies at a 1:1,000 dilution except for anti-histone H3 (1:2,000): anti-FLAG (clone M2; Sigma), anti-HA (clone 3F10; Roche), anti-HA (rabbit IgG, Santa Cruz), anti-acetyl-histone H3 (lys9), anti-acetyl-histone H3 (lys14), anti-histone H3 (rabbit IgG; Millipore), anti-acetyl-histone H4 (lys5) (clone EP1000Y), anti-acetyl-histone H4 (lys8), anti-acetyl-histone H4 (lys12), anti-acetyl-histone H4 (lys16) and anti-histone H4 (rabbit IgG; Millipore). Uncropped raw scans of blots used in the figures are presented in Supplementary Figs 6–8.
Microarray analysis
Gene expression profiles were evaluated using the Agilent one-colour microarray-based gene expression platform according to the manufacturer’s instructions (Low Input Quick Amp Labeling). Total RNA (10 ng) was amplified and labelled using the one-colour Quick Amp labeling kit (51900612; Agilent Technologies). Complementary RNA (cRNA) samples were hybridized onto SurePrint G3 Mouse GE 8 × 60 K Microarray (G4852A, Agilent Technologies). The microarray numerical values were analysed using the GeneSpring GX 11.5.1 software (Agilent Technologies). For clustering analysis, the Ward’s linkage rule was applied to all expressed genes in each sorted cells. Microarray and ChIP-sequence data were deposited in Gene Expression Omnibus (accession number GSE48797).
qRT-PCR
Total RNA was isolated using TRIZOL LS solution (Invitrogen) and reverse-transcribed by the ThermoScript RT-PCR system (Invitrogen) with an oligo-dT primer or random hexamers. Real-time quantitative RT-PCR (qRT-PCR) was performed with an ABI Prism 7300 Thermal Cycler (Applied Biosystems) using SYBR Premix Ex TaqII (TaKaRa) and primers listed in Supplementary Table 2.
Chromatin immunoprecipitation assay
AKR1 DP T cells expressing 3 × Flag-Brd1 (AKR1/Brd1 cells) or HA-Hbo1 (AKR1/Hbo1 cells) were prepared. Primary thymocytes and AKR1 cells were crosslinked with 1% formaldehyde for 5–10 min at 37 °C, washed two times with W&B buffer (100 mM Na Phosphate (pH7.0) and 0.1% Tween 20), and lysed with RIPA buffer (10 mM Tris (pH 8.0), 140 mM NaCl, 1 mM EDTA, 1% TritonX-100, 0.1% SDS, 0.1% Sodium deoxycholate (DOC) and protease inhibitor cocktail) and sonicated 15 times for 20 s using a Bioruptor (Cosmo Bio). The average chromatin fragment size after sonication was ~200–300 bp. After centrifugation, the soluble chromatin fraction was recovered, precleared for 1 h at 4 °C with a mixture of Protein A and G-conjugated Dynabeads (Invitrogen) blocked with BSA and salmon sperm DNA (Invitrogen), and then incubated with an anti-Flag, an anti-HA or an anti-acetyl H3K14 antibody (07–449; Millipore) for 2 h at 4 °C. Chromatin was immunoprecipitated overnight at 4 °C with antibody-conjugated Dynabeads. The immunoprecipitates were extensively washed with the following combination of wash buffers; RIPA buffer (10 mM Tris (pH 8.0), 150 mM NaCl, 1 mM EDTA, 1% TritonX-100, 0.1% SDS, 0.1% DOC and protease inhibitor cocktail), high salt (500 mM NaCl) RIPA buffer, LiCl wash buffer (10 mM Tris–HCl (pH 8.0), 250 mM LiCl, 1 mM EDTA, 0.5% NP40 and 0.5% DOC) and TE buffer (10 mM Tris–HCl (pH 8.0) and 1 mM EDTA). Bound chromatin and input DNA were placed in elution buffer (10 mM Tris–HCl (pH 8.0), 5 mM EDTA, 300 mM NaCl and 0.5% SDS) and reverse crosslinked. Immunoprecipitated DNA and input DNA were treated with RNase A (Sigma-Aldrich) and proteinase K (Roche). To separate DNA from protein fragments, phenol-chloroform is used in conjunction with a standard DNA purification method. For ChIP assay, quantitative PCR was performed with an ABI prism Step One Plus real time PCR system using SYBR Premix Ex Taq II (Takara Bio). The primer sequences are listed in Supplementary Table 2.
Statistical analysis
Statistical tests were carried out using Graph Pad Prism version 6.
Supplementary Material
Acknowledgments
We thank Dr Motomi Osato for providing the human RUNX1 and RUNX3 cDNAs and George R Wendt and Ola Mohammed Kamel Rizq for critical reading of the manuscript. This work was supported in part by Grants-in-aid for Scientific Research (#24249054) and Scientific Research on Innovative Areas ‘Stem Cell Aging and Disease’ (#25115002), ‘Cancer Stem Cell’ (#25130702) and ‘Genome Science’ (#221S0002) from MEXT, Japan, a Grant-in-aid for Core Research for Evolutional Science and Technology (CREST) from the Japan Science and Technology Corporation (JST), and grants from the Takeda Science Foundation and the Uehara Memorial Foundation.
Footnotes
Author contributions
Y.M., C.W., S.M., A.S. and I.T. designed and performed experiments, analysed data and wrote the manuscript; H.H., M.M.-K., Y.N.-T, G.S. and S.K. designed and performed experiments and provided intellectual input; M.N., A.O., T. Nakayama, D.G.T. and N.Y. provided intellectual input on experimental design and interpretation of results; T. Naito, T.I. and H.K. provided key reagents and assistance with experimental design and interpretation; A.I. led the investigation, devised the project and wrote the manuscript. All authors reviewed and edited the manuscript.
Supplementary Information accompanies this paper at http://www.nature.com/naturecommunications
Competing financial interests: The authors declare no competing financial interests.
References
- 1.Taniuchi I, Ellmeier W. Transcriptional and epigenetic regulation of CD4/CD8 lineage choice. Adv Immunol. 2011;110:71–110. doi: 10.1016/B978-0-12-387663-8.00003-X. [DOI] [PubMed] [Google Scholar]
- 2.Ellmeier W, Sunshine MJ, Maschek R, Littman DR. Combined deletion of CD8 locus cis-regulatory elements affects initiation but not maintenance of CD8 expression. Immunity. 2002;16:623–634. doi: 10.1016/s1074-7613(02)00309-6. [DOI] [PubMed] [Google Scholar]
- 3.Garefalaki A, et al. Variegated expression of CD8 alpha resulting from in situ deletion of regulatory sequences. Immunity. 2002;16:635–647. doi: 10.1016/s1074-7613(02)00308-4. [DOI] [PubMed] [Google Scholar]
- 4.Feik N, et al. Functional and molecular analysis of the double-positive stage-specific CD8 enhancer E8III during thymocyte development. J Immunol. 2005;174:1513–1524. doi: 10.4049/jimmunol.174.3.1513. [DOI] [PubMed] [Google Scholar]
- 5.Harker N, et al. The CD8alpha gene locus is regulated by the Ikaros family of proteins. Mol Cell. 2002;10:1403–1415. doi: 10.1016/s1097-2765(02)00711-6. [DOI] [PubMed] [Google Scholar]
- 6.Chi TH, et al. Reciprocal regulation of CD4/CD8 expression by SWI/SNF-like BAF complexes. Nature. 2002;418:195–199. doi: 10.1038/nature00876. [DOI] [PubMed] [Google Scholar]
- 7.Lee PP, et al. A critical role for Dnmt1 and DNA methylation in T cell development, function, and survival. Immunity. 2001;15:763–774. doi: 10.1016/s1074-7613(01)00227-8. [DOI] [PubMed] [Google Scholar]
- 8.Mishima Y, et al. The Hbo1-Brd1/Brpf2 complex is responsible for global acetylation of H3K14 and required for fetal liver erythropoiesis. Blood. 2011;118:2443–2453. doi: 10.1182/blood-2011-01-331892. [DOI] [PubMed] [Google Scholar]
- 9.Kisanuki YY, et al. Tie2-Cre transgenic mice: a new model for endothelial cell-lineage analysis in vivo. Dev Biol. 2001;230:230–242. doi: 10.1006/dbio.2000.0106. [DOI] [PubMed] [Google Scholar]
- 10.Matloubian M, et al. Lymphocyte egress from thymus and peripheral lymphoid organs is dependent on S1P receptor 1. Nature. 2004;427:355–360. doi: 10.1038/nature02284. [DOI] [PubMed] [Google Scholar]
- 11.Yuan J, Crittenden RB, Bender T. P c-Myb promotes the survival of CD4 +CD8 + double-positive thymocytes through upregulation of Bcl-xL. J Immunol. 2010;184:2793–2804. doi: 10.4049/jimmunol.0902846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Rothenberg EV, Moore JE, Yui MA. Launching the T-cell-lineage developmental programme. Nat Rev Immunol. 2008;8:9–21. doi: 10.1038/nri2232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hogquist KA, et al. T cell receptor antagonist peptides induce positive selection. Cell. 1994;76:17–27. doi: 10.1016/0092-8674(94)90169-4. [DOI] [PubMed] [Google Scholar]
- 14.Hayday A, Theodoridis E, Ramsburg E, Shires J. Intraepithelial lymphocytes: exploring the Third Way in immunology. Nat Immunol. 2001;2:997–1003. doi: 10.1038/ni1101-997. [DOI] [PubMed] [Google Scholar]
- 15.Mucida D, et al. Transcriptional reprogramming of mature CD4 + helper T cells generates distinct MHC class II-restricted cytotoxic T lymphocytes. Nat Immunol. 2013;14:281–289. doi: 10.1038/ni.2523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Collins A, Littman DR, Taniuchi I. RUNX proteins in transcription factor networks that regulate T-cell lineage choice. Nat Rev Immunol. 2009;9:106–115. doi: 10.1038/nri2489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Rothbart SB, Strahl BD. Interpreting the language of histone and DNA modifications. Biochim Biophys Acta. 2014;1839:627–643. doi: 10.1016/j.bbagrm.2014.03.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Agalioti T, Chen G, Thanos D. Deciphering the transcriptional histone acetylation code for a human gene. Cell. 2002;111:381–392. doi: 10.1016/s0092-8674(02)01077-2. [DOI] [PubMed] [Google Scholar]
- 19.Vicent GP, et al. Two chromatin remodeling activities cooperate during activation of hormone responsive promoters. PLoS Genet. 2009;5:e1000567. doi: 10.1371/journal.pgen.1000567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Cedar H, Bergman Y. Epigenetics of haematopoietic cell development. Nat Rev Immunol. 2011;11:478–488. doi: 10.1038/nri2991. [DOI] [PubMed] [Google Scholar]
- 21.Oshima M, Iwama A. Epigenetics of hematopoietic stem cell aging and disease. Int J Hematol. 2014;100:326–334. doi: 10.1007/s12185-014-1647-2. [DOI] [PubMed] [Google Scholar]
- 22.Mucida D, et al. Reciprocal TH17 and regulatory T cell differentiation mediated by retinoic acid. Science. 2007;317:256–260. doi: 10.1126/science.1145697. [DOI] [PubMed] [Google Scholar]
- 23.Yamashita M, et al. Crucial role of MLL for the maintenance of memory T helper type 2 cell responses. Immunity. 2006;24:611–622. doi: 10.1016/j.immuni.2006.03.017. [DOI] [PubMed] [Google Scholar]
- 24.Kimura H, Hayashi-Takanaka Y, Goto Y, Takizawa N, Nozaki N. The organization of histone H3 modifications as revealed by a panel of specific monoclonal antibodies. Cell Struct Funct. 2008;33:61–73. doi: 10.1247/csf.07035. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.