

Abstract
Mucociliary clearance (MCC) and submucosal glands are major components of airway innate immunity that have impaired function in cystic fibrosis (CF). Although both of these defense systems develop postnatally in the ferret, the lungs of newborn ferrets remain sterile in the presence of a functioning cystic fibrosis transmembrane conductance regulator gene. We evaluated several components of airway innate immunity and inflammation in the early CF ferret lung. At birth, the rates of MCC did not differ between CF and non-CF animals, but the height of the airway surface liquid was significantly reduced in CF newborn ferrets. CF ferrets had impaired MCC after 7 days of age, despite normal rates of ciliogenesis. Only non-CF ferrets eradicated Pseudomonas directly introduced into the lung after birth, whereas both genotypes could eradicate Staphylococcus. CF bronchoalveolar lavage fluid (BALF) had significantly lower antimicrobial activity selectively against Pseudomonas than non-CF BALF, which was insensitive to changes in pH and bicarbonate. Liquid chromatography–tandem mass spectrometry and cytokine analysis of BALF from sterile Caesarean-sectioned and nonsterile naturally born animals demonstrated CF-associated disturbances in IL-8, TNF-α, and IL-β, and pathways that control immunity and inflammation, including the complement system, macrophage functions, mammalian target of rapamycin signaling, and eukaryotic initiation factor 2 signaling. Interestingly, during the birth transition, IL-8 was selectively induced in CF BALF, despite no genotypic difference in bacterial load shortly after birth. These results suggest that newborn CF ferrets have defects in both innate immunity and inflammatory signaling that may be important in the early onset and progression of lung disease in these animals.
Keywords: cystic fibrosis, cystic fibrosis transmembrane conductance regulator, animal model, innate immunity, inflammation
Clinical Relevance
Animal models are invaluable tools for investigating the pathogenesis of cystic fibrosis (CF) lung disease. In this study, we characterized several components of lung innate immunity and inflammation in cystic fibrosis transmembrane conductance regulator–knockout ferrets at birth. We found that newborn CF ferret airways are dehydrated, are unable to eradicate bacteria due to pH-independent defective antimicrobial activity of lung secretions, and have dysregulated inflammatory signaling pathways in the lung both before birth and after first bacterial exposure during birth.
Lung disease remains the major cause of morbidity and mortality in cystic fibrosis (CF), a disease caused by defects in the cystic fibrosis transmembrane conductance regulator (CFTR) chloride channel (1). One model of airway disease pathogenesis in CF posits that the absence of CFTR leads to hyperactivation of the epithelial sodium channel and dehydration of the periciliary layer and the overlying mucus layers, and that this creates an environment that renders ciliary beating ineffective, thereby compromising mucociliary clearance (MCC) and promoting bacterial colonization (2–4). Other potential models invoke a more direct role for CFTR, with defects in innate immune killing of bacteria caused by altered concentrations of chloride (5) or bicarbonate (6) in the airway surface fluid due to defective CFTR-dependent transport.
In addition to MCC, the airway possesses numerous other innate immune defense mechanisms that have been found, or postulated, to be defective in CF (7, 8). Antimicrobial proteins and small molecules secreted by the airway epithelium or by submucosal glands (SMGs) are necessary for airway defense, and may be functionally affected by the loss of CFTR (9–13). Neutrophils, macrophages, and dendritic cells also play important roles in the cellular immune response to foreign pathogens in the lung, and mounting evidence suggests that antimicrobial functions of these cells are compromised in CF (7, 8, 14–17). Despite these indications that many aspects of the innate immune system are directly compromised in CF, the functional redundancy of innate immune pathways makes it difficult to determine the extent to which each defective component contributes to CF lung disease.
Recent developments have dramatically aided the CF research field in investigating these mechanisms through the creation of the CF pig and ferret models (18). Both of these animal models develop progressive lung disease similar to humans with CF, including thickening of mucus, plugging of airways, and lung colonization by numerous species of bacteria (19–21). However, the CF ferret appears to be much more sensitive to bacterial lung infections in the early neonatal period, requiring multiple antibiotics to survive to weaning (20, 21). Pigs are born with a ciliated airway epithelium and fully developed SMGs. By contrast, ferrets lack ciliated cells and SMGs in their airways at birth, and both of these structures develop within the first month of life (22). Given the significant dependence of the newborn ferret lung on CFTR for preventing bacterial infections, we hypothesized that this model provides an opportunity to dissect new mechanisms of host defense that may be relevant to the distal airway in CF humans (i.e., fewer ciliated cells and lack of SMGs). We further postulated that, due to the postnatal development of ciliated cells and SMGs in the ferret airway, CFTR-dependent innate immune mechanisms in the lung of this species are dependent on the properties of the airway surface liquid (ASL). In the present study, we evaluated multiple components of the innate immune system of the newborn and late-embryonic CF ferret lung. We identified several important defects in innate immunity of the newborn CF ferret lung, including ASL height, the secretion and function of antimicrobial proteins, and bacterial killing. Furthermore, bronchoalveolar lavage fluid (BALF) proteomics and cytokine analysis identified significant perturbation in inflammatory and immunity pathways in the CF lung after natural birth. Interestingly, proteomics and cytokine analysis of sterile BALF from Caesarean-section (C-sectioned) ferrets suggests that disturbances in inflammatory pathways may exist in the CF lung before birth and prime the lung for excessive inflammation after the first bacterial exposure. Overall, our results reveal that the newborn CF ferret lung has several innate immunity defects that are not dependent on airway clearance or SMGs, and mounts a heightened inflammatory response to early bacterial exposure at birth.
Materials and Methods
Animals
All animal experimentation was performed according to protocols approved by the Institutional Animal Care and Use Committees of the University of Iowa. The CFTR knockout (exon-10 disrupted) ferrets used in this report have been described previously (20, 21, 23) and were either used after C-section delivery or at various time points after natural birth. For details, see the supplemental Materials and Methods in the online supplement.
Analyses on BALF
BALF was harvested from ferrets by collecting successive lavages of 5% mannitol. IL-1β, IL-8, and TNF-α levels in the BALF were determined by ELISA. Concentrations of nitric oxide (NO) in newborn CF and non-CF ferret BALF were determined using the Sievers 280i Nitric Oxide Analyzer (GE Analytical Instruments, Boulder, CO). BALF samples were also processed and subjected to liquid chromatography–tandem mass spectrometry (LC-MS/MS). Datasets generated using this technique were analyzed using Scaffold (Proteome Software, Portland OR) and Ingenuity Pathway Analysis (Ingenuity Systems, Redwood City, CA). For details, see the supplemental Materials and Methods in the online supplement.
Immunofluorescence, Fluorescent Microscopy, and Western Blotting
Ferret trachea sections were stained for lysozyme and lactoferrin by immunofluorescence, and analyzed using fluorescence microscopy. BALF samples from newborn or adult ferrets were analyzed for the presence of lysozyme, lactoferrin, lactoperoxidase (LPO), and surfactant protein (SP)-A by Western blot. For details, see the supplemental Materials and Methods in the online supplement.
Tracheal MCC Assay and Microresolution Spectral Domain Optical Coherence Tomography
Rates of MCC were evaluated on ferret tracheas ex vivo by measuring the movement of 1.0-μm fluorescent beads on the lumenal surface. Ferret tracheas were also analyzed ex vivo by microresolution spectral domain optical coherence tomography at the University of Alabama (Birmingham, AL). For a detailed description of these procedures, see the supplemental Materials and Methods in the online supplement.
Bacterial Challenge Assays
Bacterial challenge experiments were performed in vivo after intratracheal injection into newborn ferrets, and in vitro by incubating bacteria with BALF isolated from CF and non-CF ferrets. For a detailed description of these procedures, see the supplemental Materials and Methods in the online supplement.
Results
MCC Defects Emerge in CF Animals as Tracheal Ciliogenesis Progresses
We first set out to test the hypothesis that CF ferret tracheas had reduced MCC over the time course during which ciliogenesis and SMG morphogenesis occurs. Ciliogenesis during the first 3 weeks of life was similar between genotypes, progressing from roughly 7% to over 50% coverage of the epithelium with cilia between birth and 3 weeks of age, respectively (Figure 1A). MCC rates in newborn animals were extremely low, and no significant differences between genotypes were observed in animals 0–3 days old (Figure 1B). However, between the second and third week of life, there were major increases in the rates of tracheal MCC for non-CF ferrets, but not CF animals (Figure 1B). Rates of MCC were 3.4- and 8.1-fold lower in CF (as compared with non-CF) animals at developmental time points corresponding to approximately 30% (7–12 d) and approximately 60% (13–24 d) cilia coverage of the tracheal epithelium, respectively. Furthermore, linear regression of MCC rates as a function of percent ciliation demonstrated a significant positive correlation in non-CF (P < 0.01; r2 = 0.5872), but not CF (P = 0.06; r2 = 0.3200), animals. These results show that MCC is more or less functionally absent in the ferret trachea at birth, suggesting that MCC defects likely do not contribute to CF lung disease in the neonatal period. However, by 1–3 weeks of life, the impairment of MCC in CF ferrets relative to non-CF animals was significant, and thus, at this developmental stage, could potentially play a role in pathogenesis of lung disease.
Figure 1.

Properties of the developing cystic fibrosis (CF) and non-CF ferret trachea. (A) Ciliogenesis in neonatal ferrets. Sections of non-CF and CF ferret tracheas at different developmental stages were stained for acetylated tubulin to mark cilia, and the percentage of the epithelial surface that was ciliated was determined by fluorescence microscopy. The percentage of the tracheal epithelial surface covered by cilia was calculated for each age group as described. The number of animals in each age group was as follows: Day 0, non-CF = 11, CF = 9; Day 1, non-CF = 9, CF = 14; Days 2–3, non-CF = 7, CF = 4; Days 4–6, non-CF = 3, CF = 3; Days 7–12, non-CF = 5, CF = 5, Days 13–24, non-CF = 5, CF = 4. (B) Tracheal mucociliary clearance (MCC) rates during differentiation. MCC was measured in non-CF and age-matched CF tracheas ex vivo by imaging the movement of fluorescent beads. Animals were divided into groups by age as in (A), and the average MCC rates for animals in each group were calculated. The number of animals in each age group was as follows: Day 0, non-CF = 10, CF = 10; Day 1, non-CF = 10, CF = 10; Days 2–3, non-CF = 3, CF = 4; Days 4–6, non-CF = 3, CF = 3; Days 7–12, non-CF = 10, CF = 10; Days 13–24, non-CF = 5, CF = 7. Error bars represent SEM. *P < 0.05, ****P < 0.0001 by Bonferroni’s multiple comparison post test. (C and D) Airway surface liquid (ASL) height is reduced in newborn CF ferret tracheas. Tracheas from non-CF and CF newborn kits were placed on Dulbecco’s modified Eagle medium–soaked Gelfoam in a humid chamber and allowed to equilibrate for 30 minutes. Microresolution spectral domain optical coherence tomography (μOCT) imaging was then performed in the longitudinal direction, and the height of the ASL in the tracheal lumen was measured. (C) Representative μOCT images from non-CF and CF tracheas, depicting the epithelial layer (ep) and ASL height (white vertical line). Scale bars in the upper right corner, 20 μm. (D) Plot of ASL height in non-CF and CF newborn tracheas. Error bars represent SEM. **P = 0.0014 by Mann-Whitney U test; n = 12 independent tracheas for each genotype.
ASL Levels Are Diminished in Newborn CF Ferrets
Recently, a debate over whether the height of the ASL contributes to pathogenesis in CF lung disease was renewed by the finding that periciliary liquid height does not differ in CF and non-CF pigs (24). Previously, others had suggested that CFTR function is essential for hydration of the luminal airway surface, and therefore for proper mucociliary transport (4, 25). To establish whether ASL defects are present in CF ferrets, we analyzed steady-state ASL height in the tracheas of newborn animals (1–2 d old) by microresolution spectral domain optical coherence tomography, a live imaging technique that has been used successfully to determine ASL height in human and porcine airway samples (26). We found that tracheal explants from non-CF newborn ferrets exhibited a significantly higher (2.5-fold; P = 0.0014) ASL height than their CF counterparts (Figures 1C and 1D). This suggests that either impaired fluid secretion or enhanced fluid absorption occurs in newborn CF ferret airways, and could potentially play a role in the development of lung disease.
Killing of Pseudomonas aeruginosa Is Impaired in the Airway of the Newborn CF Ferret
CF ferrets develop rapid bacterial lung infections within the first few weeks of life (20), and multiple antibiotics are required to rear CF animals to weaning (21). We hypothesized that innate immune mechanisms responsible for bacterial killing are inherently defective in the CF ferret at birth. To directly test this hypothesis, we challenged newborn non-CF and CF kits with antibiotic-resistant strains of either Pseudomonas aeruginosa (PA01) or Staphylococcus pseudintermedius (isolated from an adult CF ferret lung) (21) and directly assayed for killing of these pathogens in vivo (Figure 2A). In this experiment and in following experiments, we interpreted measured bacterial CFUs of lower than input as overall killing, and higher than input as overall bacterial growth or proliferation. Non-CF animals successfully eliminated PA01 relative to input CFU (∼ 100-fold decrease; P < 0.001) and to a significantly greater extent (∼ 100-fold; P < 0.05) than CF animals (Figure 2B). Notably, however, bacterial counts did not increase in the CF group, indicating that significant bacterial proliferation did not occur in vivo. By contrast, we observed that both non-CF and CF newborn ferrets successfully killed S. pseudintermedius to the same degree (∼ 50-fold decrease; P < 0.05) (Figure 2C). These data suggest that the in vivo innate immunity defect in the newborn CF lung has bacterial-specific components.
Figure 2.

CF newborn ferret lungs fail to irradiate Pseudomonas aeruginosa. (A) Schematic of bacterial challenge assays. Newborn non-CF and CF kits (6–24 h old) were challenged via intratracheal injection with 1.5 × 104 CFU of ampicillin-resistant P. aeruginosa (PA01) or erythromycin-resistant Staphylococcus pseudintermedius into the airways for 6 hours. The lung and tracheal tissue was then harvested and homogenized. The total number of CFU was determined by plating serial dilutions of lung homogenate on Luria-Bertani agar plates containing the appropriate antibiotic and counting colonies the next day. (B and C) Plot of results of in vivo bacterial challenge after intratracheal injection. For each experiment, input CFU was determined as well to control for variation on different days, and the total lung CFU was calculated as a percentage of the input CFU. Average input CFU (100%) is denoted with a line on each graph. Error bars represent SEM. *P < 0.05, ***P < 0.001, using the nonparametric Kruskal-Wallis test with Dunn’s multiple comparison post test. (B) n = 8 non-CF, 12 CF animals; (C) n = 9 non-CF, 6 CF animals.
Characterization of Secreted Proteins in BALF of Newborn and C-Section Ferrets
The airway is capable of secreting a host of factors with direct and indirect functions in innate immunity (27, 28). We hypothesized that defects in CFTR result in a reduction in the secretion of antibacterial factors in the CF ferret airway before and after birth. We thus undertook a proteomics approach, analyzing the composition of BALF from natural born (from here on referred to as newborn, 6–24 h old) and C-section–delivered, air-breathing (embryonic day [E]40–41) non-CF and CF ferrets by LC-MS/MS. Selected proteins from newborn and C-sectioned animals of interest to this study are shown in Table 1, and the complete list can be found in Table E1A in the online supplement (newborn) and Table E1B (C-sectioned). Results from this analysis demonstrated a considerably larger percentage of unique peptides and proteins in both the newborn and C-sectioned CF BALF sample than that observed in non-CF BALF samples (Figures 3A–3D), demonstrating that the protein composition of fluid in the CF lung is substantially altered before and after birth. The pulmonary SP-A, SP-B, SP-C, and SP-D were among the most abundant proteins in newborn BALF, and were elevated in CF (Table 1; Table E1A). The abundance of these surfactant proteins was lower in C-sectioned BALF and lacked genotypic differences (Table 1; Table E1B). Lactoferrin and lysozyme were elevated in newborn CF BALF, whereas several complement proteins, α1-antitrypsin, and apolipoprotein A1 were diminished in newborn CF samples. Some complement proteins were similarly diminished in CF C-sectioned BALF, but others, such as C3 and C6, were elevated. Lactoferrin was, overall, diminished in C-sectioned CF BALF, whereas lysozyme was only detectable in one of the three runs in CF. α1-Antitrypsin and apolipoprotein A1 were relatively unchanged in CF C-sectioned BALF. Several proteins associated with neutrophil function, such as myeloperoxidase, neutrophil elastase, and neutrophil collagenase, were either elevated in newborn CF BALF or completely absent in non-CF BALF; none of these proteins was detected in C-sectioned ferret BALF.
Table 1.
Mass Spectrometry–Identified Proteins in Bronchoalveolar Lavage Fluid
| Newborn |
C-Sectioned |
|||||||
|---|---|---|---|---|---|---|---|---|
| Protein Name | ENSEMBL Accession No. | Molecular Weight (kD) | Non-CF | CF | Fold Change | Non-CF | CF | Fold Change |
| α1-antitrypsin* | ENSMPUP00000003207 | 47 | + | + | −1.51 | + | + | 1.04 |
| Apolipoprotein A-I† | ENSMPUP00000004184 | 36 | + | + | −2.30 | + | + | −1.07 |
| Cathepsin B‡ | ENSMPUP00000001812 | 38 | + | + | 1.45 | + | + | −1.05 |
| Cathepsin D‡ | ENSMPUP00000006037 | 41 | + | + | 1.11 | + | + | 1.59 |
| Cathepsin H‡ | ENSMPUP00000015322 | 38 | + | + | −3.52 | + | + | 1.06 |
| Cathepsin L2‡ | ENSMPUP00000006876 | 38 | + | + | 4.59 | + | + | −1.22 |
| C1 inhibitor (serpin G1)† | ENSMPUP00000000263 | 53 | + | + | 2.85 | + | + | 1.15 |
| CD55† | ENSMPUP00000009746 | 60 | + | + | −2.08 | — | — | N/A |
| CD59† | ENSMPUP00000011434 | 13 | — | — | N/A | + | + | −1.68 |
| Complement C3† | ENSMPUP00000006928 | 188 | + | + | −1.68 | + | + | 2.00 |
| Complement component C4B† | ENSMPUP00000010461 | 200 | + | + | 1.54 | + | + | 1.00 |
| Complement component C5† | ENSMPUP00000007560 | 189 | + | + | −2.54 | — | — | N/A |
| Complement component C6† | ENSMPUP00000014449 | 107 | + | — | N/A | + | + | 9.53 |
| Complement component C7† | ENSMPUP00000014635 | 93 | + | + | −1.19 | + | + | −2.33 |
| Complement component C8 beta chain† | ENSMPUP00000007239 | 67 | + | + | −1.26 | — | — | N/A |
| Complement component C9† | ENSMPUP00000014684 | 62 | + | + | −2.94 | + | + | −1.52 |
| Complement factor B† | ENSMPUP00000010553 | 133 | + | + | −1.06 | + | + | 1.46 |
| Complement factor H isoform a† | ENSMPUP00000012049 | 139 | + | + | −1.40 | + | + | −1.01 |
| Complement factor I† | ENSMPUP00000008554 | 60 | + | + | −4.09 | + | + | 1.58 |
| Lactotransferrin† | ENSMPUP00000015079 | 78 | + | + | 4.03 | + | + | −1.63 |
| Leukocyte elastase inhibitor* | ENSMPUP00000000418 | 43 | + | + | 2.73 | — | — | N/A |
| Lysozyme† | ENSMPUP00000017391 | 19 | + | + | 2.83 | — | + | N/A |
| Macrophage migration inhibitory factor* | ENSMPUP00000002945 | 12 | + | + | 1.94 | + | + | −3.21 |
| Mucin 1§ | ENSMPUP00000005582 | 58 | + | + | 20.55 | — | — | N/A |
| Mucin 5AC§ | ENSMPUP00000006373 | 482 | + | + | 1.13 | + | + | −2.90 |
| Neutrophil collagenase (matrix metallopeptidase 8)§ | ENSMPUP00000006829 | 54 | — | + | N/A | — | — | N/A |
| Neutrophil cytosolic factor 2† | ENSMPUP00000011649 | 69 | — | + | N/A | — | — | N/A |
| Neutrophil elastase† | ENSMPUP00000009330 | 28 | — | + | N/A | — | — | N/A |
| Surfactant-associated protein A1† | ENSMPUP00000003546 | 26 | + | + | 1.50 | + | + | 1.03 |
| Surfactant protein B§ | ENSMPUP00000009033 | 40 | + | + | 1.54 | + | + | −1.12 |
| Surfactant protein C§ | ENSMPUP00000003845 | 20 | + | + | 1.39 | — | — | N/A |
| Surfactant protein D† | ENSMPUP00000003557 | 38 | + | + | 2.82 | + | + | 1.12 |
Definition of abbreviations: C-sectioned, Caesarean-sectioned; CF, cystic fibrosis; ENSEMBL, a joint project between the European Bioinformatics Institute, an outstation of the European Molecular Biology Laboratory, and the Wellcome Trust Sanger Institute; N/A, not applicable.
Liquid chromatography–tandem mass spectrometry (MS) was performed three independent times on non-CF and CF bronchoalveolar lavage fluid samples from three pooled animals each (in total, n = 9 non-CF and n = 9 CF animals). Fold change, using the average fold change in MS ionization intensity, is calculated as CF/non-CF. N/A indicates that no protein was found in either or one of the genotypes.
Inhibitor of inflammatory response.
Component of antimicrobial/innate immune defense.
Potential modifier of airway fluid components.
Structural component/modifier of airway surface fluid.
Figure 3.
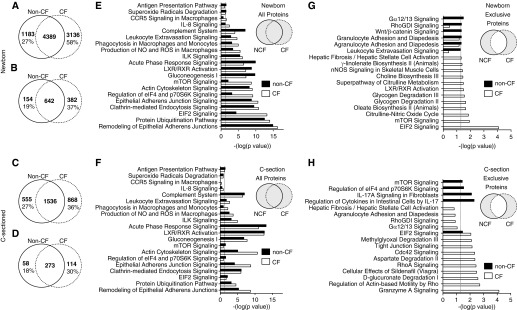
Proteomics and pathway analysis of newborn and Caesarean-sectioned (C-sectioned) ferret bronchoalveolar lavage fluid (BALF). Liquid chromatography–tandem mass spectrometry (LC-MS/MS) was performed on strong cation exchange fractions of newborn (A, B, E, and G) or C-sectioned (C, D, F, and H) non-CF and CF BALF. Peptides and proteins were identified using Mascot, and the results of three separate runs on pooled samples from three animals of each genotype (n = 9 non-CF animals; n = 9 CF animals in total) were compiled in Scaffold software. (A–D) Venn diagrams summarize total number of exclusive and nonexclusive peptides (A and C) and proteins (B and D) in each genotype. The bolded numbers in each Venn diagram are the number of peptides or proteins identified in each category. The percent values given in the Venn diagrams represent the percentage of unique peptides or proteins observed within a given genotype. (E–H) Lists of proteins from each genotype were subjected to Integrated Pathway Analysis (Qiagen) for canonical pathways, using (E and F) all proteins found in each genotype and (G and H) proteins exclusively identified in only one genotype. The schematic insert in each panel denotes the protein groups interrogated. Positive pathway identification was set at a threshold of P < 0.05 (depicted by the dashed line in E–H; Fisher’s exact test). The results from selected pathways graphed against −log(P value) are shown. The data used to generate E and G, and F and H can be found in Tables E2B and E2C and Tables E3B and E3C, respectively. CCR5, chemokine (C-C motif) receptor 5; Cdc42, cell division control protein 42; eIF2, eukaryotic initiation factor 2; ILK, integrin-linked kinase; LXR, liver X receptor; mTOR, mammalian target of rapamycin; NCF, non-CF; nNOS, nitric oxide synthase 1; NO, nitric oxide; ROS, reactive oxygen species; RXR, retinoid X receptor.
These proteomic results suggested that innate immunity and inflammatory pathways may be differently regulated in the CF lung during the transition from a sterile (i.e., in utero) to nonsterile environment (i.e., during and after birth). Using Ingenuity Pathway Analysis, we identified several key genotypic differences in canonical pathways involved in the regulation of immunity and inflammatory responses. For example, networks generated using newborn CF BALF proteins were more significantly geared toward inflammatory pathways, including IL-8, mammalian target of rapamycin (mTOR), and leukocyte extravasation signaling, as well as macrophage-related pathways, such as phagocytosis and production of reactive oxygen species and NO, than those generated with newborn non-CF proteins (Figure 3E). Genotypic differences in the significance of these pathways were not as pronounced in C-sectioned BALF (Figure 3F), supporting the concept that first bacterial exposure drives excessive inflammation in the CF lung. In addition, the eukaryotic initiation factor 2 (eIF2) signaling pathway, which is associated with macrophage induction of proinflammatory cytokines and NF-κB after bacterial infection (29), was also significantly enriched in newborn, but not C-section, CF BALF proteins (Figures 3E and 3F). Interestingly, mTOR and eIF2 signaling pathways were only identified in newborn CF BALF when genotype-exclusive proteins were interrogated (Figure 3G). In contrast, genotype-exclusive C-sectioned proteins identified mTOR and IL-17 signaling pathways only in non-CF, granzyme A signaling (a T cell pathway) only in CF, and the eIF2 signaling pathway was more significant in CF (Figure 3H). Pathways that had decreased significance in newborn CF BALF (i.e., enriched in the non-CF BALF protein datasets) included the complement system (an important component of innate immunity), acute-phase response signaling (involving up-regulation of several serpin peptidase inhibitors, such as α1-antitrypsin), and liver X receptor/retinoid X receptor (LXR/RXR) activation (Figure 3E); none of these pathways displayed large genotypic differences in P value using the C-sectioned BALF datasets (Figure 3F). A potential down-regulation of the LXR/RXR pathway in CF BALF is particularly interesting, as excessive NF-κB–mediated inflammation in CF has been suggested to be due to impaired LXR/RXR expression in CF macrophages (30). A full list of newborn and C-sectioned canonical pathways is displayed in Table E2 and E3, respectively. We also directly compared C-sectioned to newborn canonical pathways for each genotype (Figure E1). This analysis again identified CF-associated changes in inflammatory response pathways, including IL-8, mTOR, LXR/RXR, and eIF2 signaling, that were uniquely different than non-CF (Figure E1). For example, using proteins exclusive to one genotype, significance of mTOR and EIF2 signaling pathways declined after natural birth for non-CF animals, whereas the opposite occurred in CF animals (Figures E1E–E1F).
Using data tables comprising protein names and the average fold change in abundance of proteins between newborn CF and non-CF BALF (Table E1A), notable canonical pathways of high significance included: (1) LXR/RXR activation; (2) acute-phase response signaling; and (3) complement system (Figure E2A; Table E2A). Most of the proteins involved in these canonical pathways were down-regulated in CF BALF. Interestingly, the significance all three of these pathways was higher in C-sectioned BALF (Figure E3A and Table E3A). Most proteins identified in the acute-phase response signaling and LXR/RXR activation pathways in C-sectioned animals were down-regulated in CF, suggesting that pre-existing abnormalities in these inflammatory pathways exist before infection of the CF lung. Most proteins identified in newborn BALF as part of the complement system were decreased in CF (Figure E2A); these changes would be predicted to affect several parts of each of the three complement pathways (Figure E2B). Interestingly, there were distinct and opposite alterations to many of these complement proteins in C-sectioned CF BALF (Figure E3 and Table 1), again suggesting that pre-existing alterations in this pathway might precede infection.
We also performed pathway analysis on diseases and biological functions using the genotypic fold change in the abundance of proteins from newborn (Table E4A) or C-sectioned (Table E5A) BALF. Surprisingly, the top disease categories (by P value) altered by genotype were highly similar for both newborn and C-section comparisons, and included immunological disease, inflammatory disease, and immune cell trafficking (Figures E4A and E4B). Interestingly, the most elevated (by z score) disease functions in newborn CF BALF related to lymphocyte and leukocyte chemotaxis/migration (Figure E4C), whereas cell death and apoptosis of lymphocytes were among the most elevated disease functions in C-sectioned CF BALF (Figure E4D). Several of the most down-regulated disease functions in newborn CF BALF involved apoptosis of immune cells and the production of reactive oxygen species (Figure E4E), whereas disease functions related to migration and activation of phagocytes, macrophages, leukocytes, and lymphocytes were among the most significantly decreased (by z score) in CF C-section BALF (Figure E4F). In summary, related biological functions that were increased in CF newborns were decreased in CF C-sectioned animals, and vice versa.
Pathway analysis comparing changes between C-section to naturally-born (i.e., newborn) BALF within a given genotype also demonstrated that many of the disease biofunctions that were down-regulated by natural birth in non-CF were up-regulated in CF, particularly those involved in immune cell trafficking and migration (Table E6 and Figure E5; in particular, see Figure E5E). These comparisons of disease pathways between C-section and newborn BALF strongly suggest that alterations to immune cell function in the CF lung precedes infection, and may prime the lung for an excessive inflammatory response after first bacterial exposure at birth.
We chose several proteins found in our newborn LC-MS/MS screen to further analyze by immunoblot (Figure 4A). These analyses confirmed that the levels of lysozyme and LPO (compared with adult) were extremely low or undetectable in the BALF of newborn ferrets (both genotypes), and likewise confirmed that lactoferrin and SP-A were present. The levels of both lysozyme and lactoferrin were significantly lower in BALF from non-CF and CF newborns than in corresponding samples from adults, whereas no significant developmental difference was seen in the case of SP-A (Figure 4B). Immunofluorescence analysis of lysozyme expression in newborn and adult ferret tracheas confirmed the results of the Western blot analysis, showing extensive staining in the SMGs of the adult, but none in the newborn (Figure 4C). Notably, in the newborn ferret, lactoferrin was present at high levels within the simple columnar surface airway epithelium, but, in the adult, it was localized to the SMGs (Figure 4D). Taken together, these results suggest that lactoferrin, rather than lysozyme or LPO, may be an important determinant of innate immunity in the developing ferret airway.
Figure 4.

Immunoblot and immunofluorescence analysis of BALF antimicrobial proteins. (A) Western blotting demonstrating the expression of BALF components in adult non-CF, newborn non-CF, and newborn CF BALF animals. Samples were collected, and 15 μg of total BALF protein from three animals per group was separated using SDS-PAGE. Antibodies used in Western blotting were against the following: lysozyme (LYZ), lactoferrin (LTF), lactoperoxidase (LPO), and pulmonary surfactant-associated protein (SP)-A. Shown are images of several representative Western blot experiments. (B) Quantification of immunoblotting data. Densitometry was performed on bands for each protein using Metamorph software. Values were normalized to the average value for the adult samples (set at 100%). Owing to lack of normality, these values were rank transformed before analysis by one-way ANOVA, and the Bonferroni correction was applied for pairwise comparisons (SPSS version 18; SPSS, Chicago, IL). Error bars represent SEM. Significant differences between adult and newborn level are marked as: *P < 0.05, †P < 0.01, ^P < 0.001. (C and D) Localization of lysozyme (C) and lactoferrin (D) in the ferret trachea. Cyrosections of non-CF newborn and adult ferret trachea were stained by immunofluorescence (red), and images were captured using a fluorescence microscope at 200× total magnification. Samples were counterstained with 4′,6-diamidino-2-phenylindole to mark nuclei (blue). nb, newborn; SAE, surface airway epithelium; SMG, submucosal gland(s).
Levels of Inflammatory Cytokines Are Altered in the BALF of the Newborn CF Ferret
Excessive and ineffective airway inflammation has been proposed as a component of CF lung disease that contributes to bacterial colonization (31). Our BALF proteomics results suggested that inflammatory abnormalities in the CF ferret lung may initiate in utero and lead to an abnormal proinflammatory phenotype after bacterial exposure at birth. To further examine this, we measured the levels of the cytokines, IL-1β, IL-8, and TNF-α, in BALF from newborn and C-sectioned non-CF and CF ferrets. We also measured levels of NO, which is important for innate immunity in the lung and is diminished in the exhaled breath of patients with CF and airways of CF mice (32, 33). As shown in Figures 5A–5D, the concentrations of IL-1β and NO detected in newborn CF BALF were significantly lower than those in the non-CF BALF, whereas those of IL-8 and TNF-α were significantly elevated. Changes in these BALF cytokines and the abundance of NO were consistent with proteomics analysis involving these pathways. By contrast, BALF from non-CF and CF ferrets delivered sterilely by C-section at E40–41, and allowed to breathe for a short period of time, did not display significant genotypic differences in the level of these cytokines (Figures 5F–5H). However, significant and interesting genotypic differences in the induction of these cytokines by natural birth were observed (Figure 5I). Natural birth led to a significant induction of TNF-α in both non-CF (P < 0.0001) and CF (P = 0.0007) BALF. However, the induction of TNF-α was significantly (P < 0.0001) higher in CF (27.3-fold) than in non-CF (12.7-fold). By contrast, natural birth led to a significant induction of IL-1β in non-CF BALF (5.4-fold; P = 0.0014), but no significant induction was observed in CF BALF. Most interestingly, natural birth significantly induced IL-8 in CF BALF (42.3-fold, P = 0.036), but no significant induction was observed in non-CF BALF. These changes in secreted lung cytokines occurred despite the fact that there were no differences in BALF bacterial load between genotypes, although two CF animals demonstrated nonsterile BALF, and three CF animals had bacterial genomes above background levels of detection (Figures 5E and 5J). These findings suggest that the CF newborn ferret lung mounts an exaggerated inflammatory response to early bacterial exposure after birth and before excessive bacterial proliferation.
Figure 5.
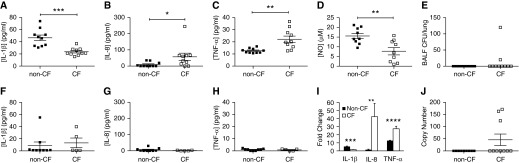
Inflammatory markers dynamically change in CF BALF at birth during the transition from a sterile to nonsterile environment. (A–C) Concentrations of (A) IL-1β, (B) IL-8, and (C) TNF-α in BALF from newborn ferrets were determined by ELISA. (D) Concentration of NO in BALF from newborn ferrets measured using a Siever 280i Nitric Oxide Analyzer. (E) Bacterial CFU counts in non-CF and CF newborn ferret BALF determined by growth on blood agar plates. *P < 0.05, **P < 0.01, ***P < 0.001 by Mann-Whitney U test. (F–H) Concentrations of (F) IL-1β, (G) IL-8, and (H) TNF-α in BALF from breathing, C-sectioned ferrets were determined by ELISA. (I) Fold changes in cytokine levels between C-section and newborn for each genotype. Cytokine concentrations shown (A–C) were divided by the average C-section cytokine concentrations (F–H) to obtain the fold changes for each genotype, and the averages of these values were plotted on the same axis. Mann-Whitney U tests were performed between the data sets in A–C and F–H to determine whether the induction by natural birth was significant for each genotype; the results were as follows: IL-1β, non-CF, P = 0.0014, CF, P = 0.2954; IL-8, non-CF, P = 0.9664, CF, P = 0.036; TNF-α, non-CF, P < 0.0001, CF, P = 0.0007. Genotypic comparisons for the fold change in cytokines between C-section and newborn animals shown in I used the Mann Whitney U test: **P < 0.01, ***P < 0.001, ****P < 0.0001. (J) Bacterial load in newborn ferret BALF was assessed by quantitative PCR for bacterial 16S ribosomal DNA. The limits of sensitivity for this assay were set to zero. Error bars represent SEM throughout.
Species-Specific Proliferation of Bacteria Occurs in BALF from CF Newborn Ferrets in a pH- and Bicarbonate-Independent Manner
Airway surface fluid from patients with CF (5, 34), as well as newborn CF pigs (6), has been found to be defective in bacterial killing. We sought to determine whether BALF isolated from newborn CF ferrets is likewise impaired in its ability to kill bacteria. As shown in Figure 6A, no differences were found between non-CF and CF BALF with respect to the ability to inhibit the proliferation of Escherichia coli, as well as S. pseudintermedius (Figure 6B). However, we observed a nearly 10-fold increase in CFU of P. aeruginosa incubated in BALF from newborn CF ferrets compared with the BALF from non-CF animals (Figure 6C). This finding, of a specific innate immunity defect against P. aeruginosa, is similar to in vivo bacterial challenge experiments (Figure 2), and may have bearings on why this species is such a common lung pathogen in patients with CF (8).
Figure 6.
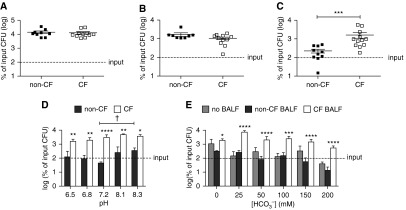
Antimicrobial activity against P. aeruginosa is reduced in BALF from newborn CF ferrets in a pH- and bicarbonate-independent manner. (A–C) Proliferation of various strains of bacteria in BALF from non-CF and CF newborn ferrets. BALF from non-CF and CF newborn ferrets was concentrated to 2–5 μg/μl by centrifugation as described in Materials and Methods. A 15-μg sample of the BALF was inoculated with 5,000 CFU of Escherichia coli (EC838) (A), S. pseudintermedius (clinical isolate from an adult CF ferret lung) (B), or P. aeruginosa (PA01) (C) and incubated at 37°C for 3 hours. The surviving bacteria were then quantified by CFU assay. (D) Proliferation of PA01 bacteria in BALF at various pH levels. Concentrated non-CF or CF BALF (15 μg; prepared as in A–C) was inoculated with 5,000 CFU PA01 in sodium phosphate buffer at the indicated pH, at 37°C for 3 hours, before CFU quantification. (E) Proliferation of PA01 in the presence or absence of BALF at various bicarbonate concentrations. Concentrated non-CF or CF BALF (15 μg) in sodium phosphate buffer (prepared as in A–C) was supplemented with a vehicle or a concentrated bicarbonate solution to the indicated final concentration. Mock samples (no BALF) were also carried along as controls. These samples were then inoculated with 5,000 CFU PA01 at 37°C for 3 hours before CFU quantification. The data in D and E were log transformed before being plotted. Average input CFU (100%) is denoted with a dashed line on each graph. Error bars represent SEM. ***P < 0.001 by Mann-Whitney test (C); significant differences between genotypes at each pH or bicarbonate concentration are marked as: *P < 0.05, **P < 0.01, ***P < 0.001, and ****P < 0.0001 by Sidak’s multiple comparison posttest (D and E); a significant difference (†P < 0.05) between non-CF pH 7.2 and non-CF pH 8.3 was also observed (one-way ANOVA, Sidak’s multiple comparison posttest).
Airway surface pH is significantly lower in newborn CF pigs than in non-CF pigs, and the addition of bicarbonate to the CF pig airway in vivo has been shown to enhance bacterial killing (∼1.8-fold), while elevating the airway pH (6). We tested similar characteristics in the CF ferret. Variations in pH, ranging from 6.5 to 8.3, did not significantly alter P. aeruginosa killing by CF ferret BALF (Figure 6D). However, a significant reduction (∼ 10-fold) in bacterial killing was observed between non-CF BALF when the pH was raised from 7.2 to 8.3 (P < 0.05). At every pH tested, CF BALF facilitated significantly more growth of P. aeruginosa than non-CF BALF (P < 0.0001 by two-way ANOVA). Although we found that bicarbonate did have a significant effect on bacterial growth overall (P < 0.0001; 16.21% total variation by two-way ANOVA), it had this effect even in the absence of BALF and only at 200 mM (P < 0.05 by Sidak’s multiple comparison post test) (Figure 6E). BALF genotype had a greater overall effect on bacterial growth (P < 0.0001; 39.24% total variation by two-way ANOVA), and there was no statistical interaction between the BALF genotype and bicarbonate concentration overall (P = 0.0916). At every concentration of bicarbonate tested, bacterial growth in CF BALF was greater than in non-CF BALF, and the significance of this difference was higher in the presence than in the absence of bicarbonate. Collectively, the results shown in Figure 6 suggest that P. aeruginosa has a unique survival advantage in airway fluid from newborn CF ferret, and that this property is not dependent on pH or bicarbonate.
Discussion
The development of animal models for CF in the mouse, pig, and ferret has been invaluable in deciphering the origins of disease pathogenesis in the lung and other organs (18). In the current study, we examined several major aspects of innate immunity in the newborn ferret lung, and whether these are defective in CF animals. The lack of ciliated epithelial cells and SMGs in the ferret trachea at birth allowed us to specifically interrogate other aspects of CFTR-dependent airway innate immunity in this species. Findings from these studies clearly demonstrate that CFTR-dependent processes in airways without SMGs or MCC do contribute to innate immunity. These processes also appear to be uniquely different than in newborn CF pig airways that contain SMGs and abundant ciliated cells.
Our finding of a lack of species-specific bacterial killing of P. aeruginosa in newborn CF ferret lungs and BALF is intriguing, given that Staphylococcus is much more frequently observed in the end-stage CF ferret lung (20, 21). Bacteriology of CF ferret and pig lungs both suggest nondiscriminating bacterial defense defects, with a wide range of bacterial pathogens identified (19–21). Numerous mechanisms have been proposed to account for the unique ability of P. aeruginosa to persist in human CF lungs (3, 35), and, although Pseudomonas isolates have been obtained from the adult CF ferret lung, they represent a minor component of the overall bacterial load (21). Our studies also suggest different CFTR-dependent roles for cellular innate immune components in the lung against P. aeruginosa—growth is suppressed in the absence of non-CF lung cellular components (BALF alone), whereas killing (∼ 100-fold) occurs in vivo. Given the immune and inflammatory abnormalities observed in CF ferret BALF before and after birth, one explanation for our findings may involve acquired innate immunity defects in the CF ferret lung later in life that are promoted by the excessive inflammatory responses to minor bacterial species, such as Pseudomonas. Furthermore, the newborn ferret CF airways lack SMGs, and thus may have aspects of innate immunity defects that emerge over time as these structures form. For example, lactoferrin is produced in the newborn CF lung by surface airway epithelial cells and is later replaced by SMGs in older CF ferrets (Figures 4C and 4D). Thus, if lactoferrin is a predominant antimicrobial against Staphylococcus, as CF ferrets mature, one would expect that this pathogen might begin to dominate as less lactoferrin is secreted into the airways due to defective gland secretions.
Defective P. aeruginosa antimicrobial activity in CF ferret BALF was not primarily due to differences in pH or bicarbonate concentration, because altering these in CF BALF did not influence antimicrobial activity. Interestingly, raising the pH of non-CF BALF selectively reduced bacterial killing and/or promoted growth. These observations are in contrast to those observed in pig airway fluid (6), in which raising the pH or adding bicarbonate enhanced antimicrobial activity in airway fluid irrespective of CFTR activity. Thus, it has been proposed that an increase in the acidity of the airway surface fluid, due to the lack of CFTR-dependent bicarbonate secretion, is the cause of impaired bacterial killing in the CF airway (6). Differences in methodology or potential species-specific differences in how airway pH is regulated and relates to bacterial killing may explain why results in the CF ferret model did not support this mechanism. SMGs, which are lacking at birth in the ferrets, have been suggested to be a major source of CFTR-dependent bicarbonate secretion in the airways (36). Thus, the lack of SMG involvement in innate immunity of the newborn ferret airway may in part explain these species-specific differences, and may have relevance to alternative mechanisms of innate immunity in the distal airways where SMGs are absent. Although our studies conclude that neither pH nor bicarbonate influences antimicrobial activity of CF ferret BALF, we cannot conclude that CF-specific changes in bicarbonate or pH in vivo do not have an impact on bacterial growth due to their influences on cellular components.
The innate immunity defect in newborn CF ferrets does not appear to be due to the genotype-specific absence of most major antimicrobial factors in the BALF, suggesting that other mechanisms of CFTR-dependent innate immunity protect newborn ferret lungs. One mechanism may involve disruption of antimicrobial activity due to the altered properties of concentrated ASL or the proinflammatory environment in the absence of CFTR. For example, the antimicrobial activity of lactoferrin has been found to be defective in CF, either due to the increased viscosity of CF airway secretions (3), or possibly due to cleavage by cathepsins B, L, and S (9); cathepsin S has been shown to cleave SP-A in human CF BALF (37). Although we detected several cathepsin proteins in newborn ferret BALF, we did not observe any CF-specific degradation products of either lactoferrin or SP-A in our Western blot analysis, so it remains to be seen whether inflammation-induced proteolytic cleavage of other antimicrobial proteins plays a role in CF lung disease in the ferret.
We found a significant defect in MCC in CF kits older than 1 week of age, and an altered relationship between tracheal MCC and percent ciliation in young CF ferrets. This altered relationship suggests that characteristics of airway fluid may impair MCC in CF kits; the reduced ASL height that we observed in the newborn CF ferret may contribute to the impaired MCC in these animals during the second week of life. However, a recent analysis of the depth of the ASL in CF pigs revealed that it did not differ from that in non-CF pigs, nor was there observed epithelial sodium channel hyperactivation (24) that is typically observed in human CF airway epithelia and thought to promote dehydration of the ASL (4, 25). Despite the absence of increased amiloride-sensitive current in young CF ferrets (21, 38), we found a significant decrease in tracheal ASL height at birth in CF ferrets, suggesting that chloride secretion is dominant in the ferret at this age, and could be compensating for an absence of SMGs, which typically contribute to the ASL depth and content. This discrepancy between pigs and ferrets could reflect a fundamental difference in the regulation of fluid dynamics between species, or simply a difference in the methods used to assess ASL levels.
Proteomics analysis of C-section BALF supports the notion that CF-associated alterations in inflammatory pathways and immune cell function exist in the sterile CF lung (Figures E4B, E4D, and E4F), and that, after pathogen exposure at birth, a hyperinflammatory state exists in the CF ferret lung (Figures E4A, E4C, and E4E). Interestingly, many of the most significant biofunction pathways that were elevated or repressed (by z score) before birth in the CF lung as compared with the non-CF lung were significantly altered in the opposite direction after birth (Figure E4). In terms of specific pathways represented in BALF protein composition before and after natural birth (Figure 3 and Figure E1), we found interesting genotypic differences in significance of the LXR/RXR, eIF2, and mTOR signaling pathways. These pathways are notable, as they regulate inflammatory responses and interact with the NF-κB pathway (29, 39–42), which, in turn, regulates the expression of CF proinflammatory cytokines, such as IL-8, TNF-α, and IL-1β (27, 43, 44). Complementary to these proteomics results, the changes in BALF cytokine during the birth transition (i.e., C-sectioned versus naturally born animals) were uniquely different between genotypes (Figure 5I), implicating dysregulation of the CF lung inflammatory response to first exposure of bacteria. These findings are in stark contrast to what is known about the CF pig, which lacks lung inflammation at birth (19, 45). It has long been debated whether inappropriate inflammatory responses by the CF lung contribute to bacterial colonization. Several signaling pathways, including NF-κB, have been shown to be hyperactivated in CF airway cells, and are thought to contribute to their abnormal cytokine responses (including IL-8 and TNF-α) to bacterial and LPS challenge (31). Some of the inflammatory markers elevated in newborn CF ferret BALF are also elevated in infants with CF (46, 47). Our finding, that NO is diminished in newborn CF ferret BALF, mirrors observations in human patients (32). NO has antimicrobial activity and also plays important roles in regulating inflammation through the NF-κB and signal transducer and activator of transcription 3 pathways (32); thus, this molecule could play a role in the observed phenotype of the newborn CF ferret lung.
Proteomics analysis also demonstrated differences in proteins within the complement system in CF BALF before and after birth. Although complement system factors were generally repressed after natural birth in the CF lung, many of these factors were elevated in the C-sectioned CF lung. This system plays an important role in innate immunity, allowing for the direct killing or opsonization of foreign microbes in the lung, such as P. aeruginosa (44). Similar to CF ferrets, proteomics analysis of human CF BALF showed decreased levels or absence of several complement proteins (48). In addition, CFTR appears to be necessary for the phagocytosis of complement-opsonized P. aeruginosa by monocytes (49), which is interesting, given the functional disturbances observed in CF BALF involving monocytes and macrophages, as well as the specific in vivo defect for P. aeruginosa killing in the CF ferret lung. In mice, the alternative complement pathway and complement component C3 were necessary for an immune response against P. aeruginosa (50), and the alternative complement pathway components were the most reduced in naturally born CF ferret BALF.
Collectively, our results demonstrate that multiple arms of airway innate immune systems and inflammatory pathways are altered before and after birth in CF ferrets. Furthermore, these pathways function in a CFTR-dependent manner in the absence of MCC and SMGs. Altered inflammatory signaling, coupled with decreased function of antimicrobial components in the BALF, likely contribute to the inability of the newborn CF ferret airway to defend against foreign pathogens. Interesting differences in innate immunity and inflammation between CF ferrets and pigs were also uncovered, and likely reflect biologic differences in how CFTR functions to protect airways with and without cilia and SMGs. Defining the mechanisms responsible for these differences may uncover novel targets for therapy for CF lung disease.
Footnotes
This work was supported by National Institutes of Health grants DK047967 and HL108902 (J.F.E.) and HL1116213 (S.M.R. and G.J.T.), University of Iowa Center for Gene Therapy grant DK054759, University of Alabama Cystic Fibrosis Translational Core Center grant DK072482, the Roy J Carver Chair in Molecular Medicine (J.F.E.), and Cystic Fibrosis Foundation grants ROWE10XX0 (S.M.R.) and TEARNE12XX0 (G.J.T.). Mass spectrometry analysis was performed in the Roy J. Carver Charitable Trust–supported CCOM Proteomics Facility at the University of Iowa.
Author Contributions: conception and design—N.W.K., S.E.B., I.A.E., S.R.T., A.K.C., X.S., W.Z., J.R.N., E.K.S., K.K.C., G.J.T., M.J.S., J.K.H., S.M.R., and J.F.E.; analysis and interpretation—N.W.K., S.E.B., I.A.E., S.R.T., A.K.C., J.R.N., E.K.S., J.K.H., S.M.R., and J.F.E.; drafting the manuscript for important intellectual content—N.W.K., S.E.B., J.K.H., S.M.R., and J.F.E.
This article has an online supplement, which is accessible from this issue’s table of contents at www.atsjournals.org
Originally Published in Press as DOI: 10.1165/rcmb.2014-0250OC on October 15, 2014
Author disclosures are available with the text of this article at www.atsjournals.org.
References
- 1.Rowe SM, Miller S, Sorscher EJ. Cystic fibrosis. N Engl J Med. 2005;352:1992–2001. doi: 10.1056/NEJMra043184. [DOI] [PubMed] [Google Scholar]
- 2.Boucher RC. Airway surface dehydration in cystic fibrosis: pathogenesis and therapy. Annu Rev Med. 2007;58:157–170. doi: 10.1146/annurev.med.58.071905.105316. [DOI] [PubMed] [Google Scholar]
- 3.Matsui H, Wagner VE, Hill DB, Schwab UE, Rogers TD, Button B, Taylor RM, Superfine R, Rubinstein M, Iglewski BH, et al. A physical linkage between cystic fibrosis airway surface dehydration and Pseudomonas aeruginosa biofilms. Proc Natl Acad Sci USA. 2006;103:18131–18136. doi: 10.1073/pnas.0606428103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Knowles MR, Boucher RC. Mucus clearance as a primary innate defense mechanism for mammalian airways. J Clin Invest. 2002;109:571–577. doi: 10.1172/JCI15217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Smith JJ, Travis SM, Greenberg EP, Welsh MJ. Cystic fibrosis airway epithelia fail to kill bacteria because of abnormal airway surface fluid. Cell. 1996;85:229–236. doi: 10.1016/s0092-8674(00)81099-5. [DOI] [PubMed] [Google Scholar]
- 6.Pezzulo AA, Tang XX, Hoegger MJ, Alaiwa MHA, Ramachandran S, Moninger TO, Karp PH, Wohlford-Lenane CL, Haagsman HP, van Eijk M, et al. Reduced airway surface pH impairs bacterial killing in the porcine cystic fibrosis lung. Nature. 2012;487:109–113. doi: 10.1038/nature11130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hartl D, Gaggar A, Bruscia E, Hector A, Marcos V, Jung A, Greene C, McElvaney G, Mall M, Döring G. Innate immunity in cystic fibrosis lung disease. J Cyst Fibros. 2012;11:363–382. doi: 10.1016/j.jcf.2012.07.003. [DOI] [PubMed] [Google Scholar]
- 8.Ratner D, Mueller C. Immune responses in cystic fibrosis: are they intrinsically defective? Am J Respir Cell Mol Biol. 2012;46:715–722. doi: 10.1165/rcmb.2011-0399RT. [DOI] [PubMed] [Google Scholar]
- 9.Rogan MP, Taggart CC, Greene CM, Murphy PG, O’Neill SJ, McElvaney NG. Loss of microbicidal activity and increased formation of biofilm due to decreased lactoferrin activity in patients with cystic fibrosis. J Infect Dis. 2004;190:1245–1253. doi: 10.1086/423821. [DOI] [PubMed] [Google Scholar]
- 10.Singh PK, Jia HP, Wiles K, Hesselberth J, Liu L, Conway BA, Greenberg EP, Valore EV, Welsh MJ, Ganz T, et al. Production of beta-defensins by human airway epithelia. Proc Natl Acad Sci USA. 1998;95:14961–14966. doi: 10.1073/pnas.95.25.14961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Moskwa P, Lorentzen D, Excoffon KJ, Zabner J, McCray PB, Jr, Nauseef WM, Dupuy C, Banfi B. A novel host defense system of airways is defective in cystic fibrosis. Am J Respir Crit Care Med. 2007;175:174–183. doi: 10.1164/rccm.200607-1029OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Dajani R, Zhang Y, Taft PJ, Travis SM, Starner TD, Olsen A, Zabner J, Welsh MJ, Engelhardt JF. Lysozyme secretion by submucosal glands protects the airway from bacterial infection. Am J Respir Cell Mol Biol. 2005;32:548–552. doi: 10.1165/rcmb.2005-0059OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Sagel SD, Chmiel JF, Konstan MW. Sputum biomarkers of inflammation in cystic fibrosis lung disease. Proc Am Thorac Soc. 2007;4:406–417. doi: 10.1513/pats.200703-044BR. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Bessich JL, Nymon AB, Moulton LA, Dorman D, Ashare A. Low levels of insulin-like growth factor-1 contribute to alveolar macrophage dysfunction in cystic fibrosis. J Immunol. 2013;191:378–385. doi: 10.4049/jimmunol.1300221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Deriy LV, Gomez EA, Zhang G, Beacham DW, Hopson JA, Gallan AJ, Shevchenko PD, Bindokas VP, Nelson DJ. Disease-causing mutations in the cystic fibrosis transmembrane conductance regulator determine the functional responses of alveolar macrophages. J Biol Chem. 2009;284:35926–35938. doi: 10.1074/jbc.M109.057372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Di A, Brown ME, Deriy LV, Li C, Szeto FL, Chen Y, Huang P, Tong J, Naren AP, Bindokas V, et al. Cftr regulates phagosome acidification in macrophages and alters bactericidal activity. Nat Cell Biol. 2006;8:933–944. doi: 10.1038/ncb1456. [DOI] [PubMed] [Google Scholar]
- 17.Painter RG, Valentine VG, Lanson NA, Leidal K, Zhang Q, Lombard G, Thompson C, Viswanathan A, Nauseef WM, Wang G, et al. Cftr expression in human neutrophils and the phagolysosomal chlorination defect in cystic fibrosis. Biochemistry. 2006;45:10260–10269. doi: 10.1021/bi060490t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Keiser NW, Engelhardt JF. New animal models of cystic fibrosis: what are they teaching us? Curr Opin Pulm Med. 2011;17:478–483. doi: 10.1097/MCP.0b013e32834b14c9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Stoltz DA, Meyerholz DK, Pezzulo AA, Ramachandran S, Rogan MP, Davis GJ, Hanfland RA, Wohlford-Lenane C, Dohrn CL, Bartlett JA, et al. Cystic fibrosis pigs develop lung disease and exhibit defective bacterial eradication at birth. Sci Transl Med. 2010;2:29ra31. doi: 10.1126/scitranslmed.3000928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Sun X, Sui H, Fisher JT, Yan Z, Liu X, Cho HJ, Joo NS, Zhang Y, Zhou W, Yi Y, et al. Disease phenotype of a ferret cftr-knockout model of cystic fibrosis. J Clin Invest. 2010;120:3149–3160. doi: 10.1172/JCI43052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Sun X, Olivier AK, Liang B, Yi Y, Sui H, Evans TIA, Zhang Y, Zhou W, Tyler SR, Fisher JT, et al. Lung phenotype of juvenile and adult cftr-knockout ferrets. Am J Respir Cell Mol Biol. 2014;50:510–512. doi: 10.1165/rcmb.2013-0261OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Leigh MW, Gambling TM, Carson JL, Collier AM, Wood RE, Boat TF. Postnatal development of tracheal surface epithelium and submucosal glands in the ferret. Exp Lung Res. 1986;10:153–169. doi: 10.3109/01902148609061490. [DOI] [PubMed] [Google Scholar]
- 23.Sun X, Yan Z, Yi Y, Li Z, Lei D, Rogers CS, Chen J, Zhang Y, Welsh MJ, Leno GH, et al. Adeno-associated virus-targeted disruption of the cftr gene in cloned ferrets. J Clin Invest. 2008;118:1578–1583. doi: 10.1172/JCI34599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Chen JH, Stoltz DA, Karp PH, Ernst SE, Pezzulo AA, Moninger TO, Rector MV, Reznikov LR, Launspach JL, Chaloner K, et al. Loss of anion transport without increased sodium absorption characterizes newborn porcine cystic fibrosis airway epithelia. Cell. 2010;143:911–923. doi: 10.1016/j.cell.2010.11.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Boucher RC. Evidence for airway surface dehydration as the initiating event in cf airway disease. J Intern Med. 2007;261:5–16. doi: 10.1111/j.1365-2796.2006.01744.x. [DOI] [PubMed] [Google Scholar]
- 26.Liu L, Chu KK, Houser GH, Diephuis BJ, Li Y, Wilsterman EJ, Shastry S, Dierksen G, Birket SE, Mazur M, et al. Method for quantitative study of airway functional microanatomy using micro-optical coherence tomography. PLoS One. 2013;8:e54473. doi: 10.1371/journal.pone.0054473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Parker D, Prince A. Innate immunity in the respiratory epithelium. Am J Respir Cell Mol Biol. 2011;45:189–201. doi: 10.1165/rcmb.2011-0011RT. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Waterer GW. Airway defense mechanisms. Clin Chest Med. 2012;33:199–209. doi: 10.1016/j.ccm.2012.03.003. [DOI] [PubMed] [Google Scholar]
- 29.Shrestha N, Bahnan W, Wiley DJ, Barber G, Fields KA, Schesser K. Eukaryotic initiation factor 2 (eIF2) signaling regulates proinflammatory cytokine expression and bacterial invasion. J Biol Chem. 2012;287:28738–28744. doi: 10.1074/jbc.M112.375915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Andersson C, Zaman MM, Jones AB, Freedman SD. Alterations in immune response and PPAR/LXR regulation in cystic fibrosis macrophages. J Cyst Fibros. 2008;7:68–78. doi: 10.1016/j.jcf.2007.05.004. [DOI] [PubMed] [Google Scholar]
- 31.Cohen TS, Prince A. Cystic fibrosis: a mucosal immunodeficiency syndrome. Nat Med. 2012;18:509–519. doi: 10.1038/nm.2715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Grasemann H, Ratjen F. Cystic fibrosis lung disease: the role of nitric oxide. Pediatr Pulmonol. 1999;28:442–448. doi: 10.1002/(sici)1099-0496(199912)28:6<442::aid-ppul10>3.0.co;2-4. [DOI] [PubMed] [Google Scholar]
- 33.Steagall WK, Elmer HL, Brady KG, Kelley TJ. Cystic fibrosis transmembrane conductance regulator–dependent regulation of epithelial inducible nitric oxide synthase expression. Am J Respir Cell Mol Biol. 2000;22:45–50. doi: 10.1165/ajrcmb.22.1.3789. [DOI] [PubMed] [Google Scholar]
- 34.Bals R, Weiner DJ, Meegalla RL, Accurso F, Wilson JM. Salt-independent abnormality of antimicrobial activity in cystic fibrosis airway surface fluid. Am J Respir Cell Mol Biol. 2001;25:21–25. doi: 10.1165/ajrcmb.25.1.4436. [DOI] [PubMed] [Google Scholar]
- 35.Pier GB. Role of the cystic fibrosis transmembrane conductance regulator in innate immunity to Pseudomonas aeruginosa infections. Proc Natl Acad Sci USA. 2000;97:8822–8828. doi: 10.1073/pnas.97.16.8822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ballard ST, Trout L, Bebok Z, Sorscher EJ, Crews A. Cftr involvement in chloride, bicarbonate, and liquid secretion by airway submucosal glands. Am J Physiol. 1999;277:L694–L699. doi: 10.1152/ajplung.1999.277.4.L694. [DOI] [PubMed] [Google Scholar]
- 37.Lecaille F, Naudin C, Sage J, Joulin-Giet A, Courty A, Andrault PM, Veldhuizen RA, Possmayer F, Lalmanach G. Specific cleavage of the lung surfactant protein A by human cathepsin S may impair its antibacterial properties. Int J Biochem Cell Biol. 2013;45:1701–1709. doi: 10.1016/j.biocel.2013.05.018. [DOI] [PubMed] [Google Scholar]
- 38.Fisher JT, Tyler SR, Zhang Y, Lee BJ, Liu X, Sun X, Sui H, Liang B, Luo M, Xie W, et al. Bioelectric characterization of epithelia from neonatal cftr knockout ferrets. Am J Respir Cell Mol Biol. 2013;49:837–844. doi: 10.1165/rcmb.2012-0433OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Smoak K, Madenspacher J, Jeyaseelan S, Williams B, Dixon D, Poch KR, Nick JA, Worthen GS, Fessler MB. Effects of liver X receptor agonist treatment on pulmonary inflammation and host defense. J Immunol. 2008;180:3305–3312. doi: 10.4049/jimmunol.180.5.3305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Birrell MA, Catley MC, Hardaker E, Wong S, Willson TM, McCluskie K, Leonard T, Farrow SN, Collins JL, Haj-Yahia S, et al. Novel role for the liver X nuclear receptor in the suppression of lung inflammatory responses. J Biol Chem. 2007;282:31882–31890. doi: 10.1074/jbc.M703278200. [DOI] [PubMed] [Google Scholar]
- 41.Fielhaber JA, Carroll SF, Dydensborg AB, Shourian M, Triantafillopoulos A, Harel S, Hussain SN, Bouchard M, Qureshi ST, Kristof AS. Inhibition of mammalian target of rapamycin augments lipopolysaccharide-induced lung injury and apoptosis. J Immunol. 2012;188:4535–4542. doi: 10.4049/jimmunol.1003655. [DOI] [PubMed] [Google Scholar]
- 42.Jiang HY, Wek SA, McGrath BC, Scheuner D, Kaufman RJ, Cavener DR, Wek RC. Phosphorylation of the alpha subunit of eukaryotic initiation factor 2 is required for activation of NF-kappab in response to diverse cellular stresses. Mol Cell Biol. 2003;23:5651–5663. doi: 10.1128/MCB.23.16.5651-5663.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Muir A, Soong G, Sokol S, Reddy B, Gomez MI, Van Heeckeren A, Prince A. Toll-like receptors in normal and cystic fibrosis airway epithelial cells. Am J Respir Cell Mol Biol. 2004;30:777–783. doi: 10.1165/rcmb.2003-0329OC. [DOI] [PubMed] [Google Scholar]
- 44.Lavoie EG, Wangdi T, Kazmierczak BI. Innate immune responses to Pseudomonas aeruginosa infection. Microbes Infect. 2011;13:1133–1145. doi: 10.1016/j.micinf.2011.07.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Rogers CS, Hao Y, Rokhlina T, Samuel M, Stoltz DA, Li Y, Petroff E, Vermeer DW, Kabel AC, Yan Z, et al. Production of CFTR-null and CFTR-DeltaF508 heterozygous pigs by adeno-associated virus–mediated gene targeting and somatic cell nuclear transfer. J Clin Invest. 2008;118:1571–1577. doi: 10.1172/JCI34773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Peterson-Carmichael SL, Harris WT, Goel R, Noah TL, Johnson R, Leigh MW, Davis SD. Association of lower airway inflammation with physiologic findings in young children with cystic fibrosis. Pediatr Pulmonol. 2009;44:503–511. doi: 10.1002/ppul.21044. [DOI] [PubMed] [Google Scholar]
- 47.Ranganathan SC, Parsons F, Gangell C, Brennan S, Stick SM, Sly PD Australian Respiratory Early Surveillance Team for Cystic Fibrosis. Evolution of pulmonary inflammation and nutritional status in infants and young children with cystic fibrosis. Thorax. 2011;66:408–413. doi: 10.1136/thx.2010.139493. [DOI] [PubMed] [Google Scholar]
- 48.Gharib SA, Vaisar T, Aitken ML, Park DR, Heinecke JW, Fu X. Mapping the lung proteome in cystic fibrosis. J Proteome Res. 2009;8:3020–3028. doi: 10.1021/pr900093j. [DOI] [PubMed] [Google Scholar]
- 49.Van de Weert-van Leeuwen PB, Van Meegen MA, Speirs JJ, Pals DJ, Rooijakkers SH, Van der Ent CK, Terheggen-Lagro SW, Arets HG, Beekman JM. Optimal complement-mediated phagocytosis of Pseudomonas aeruginosa by monocytes is cystic fibrosis transmembrane conductance regulator-dependent. Am J Respir Cell Mol Biol. 2013;49:463–470. doi: 10.1165/rcmb.2012-0502OC. [DOI] [PubMed] [Google Scholar]
- 50.Mueller-Ortiz SL, Drouin SM, Wetsel RA. The alternative activation pathway and complement component C3 are critical for a protective immune response against Pseudomonas aeruginosa in a murine model of pneumonia. Infect Immun. 2004;72:2899–2906. doi: 10.1128/IAI.72.5.2899-2906.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]