

Abstract
Current treatments for inflammation associated with bronchopulmonary dysplasia (BPD) fail to show clinical efficacy. Foxm1, a transcription factor of the Forkhead box family, is a critical mediator of lung development and carcinogenesis, but its role in BPD-associated pulmonary inflammation is unknown. Immunohistochemistry and RNA analysis were used to assess Foxm1 in lung tissue from hyperoxia-treated mice and patients with BPD. LysM-Cre/Foxm1−/− mice, in which Foxm1 was deleted from myeloid-derived inflammatory cells, including macrophages, monocytes, and neutrophils, were exposed to neonatal hyperoxia, causing lung injury and remodeling. Measurements of lung function and flow cytometry were used to evaluate the effects of Foxm1 deletion on pulmonary inflammation and repair. Increased Foxm1 expression was observed in pulmonary macrophages of hyperoxia-exposed mice and in lung tissue from patients with BPD. After hyperoxia, deletion of Foxm1 from the myeloid cell lineage decreased numbers of interstitial macrophages (CD45+CD11b+Ly6C−Ly6G−F4/80+CD68−) and impaired alveologenesis and lung function. The exaggerated BPD-like phenotype observed in hyperoxia-exposed LysM-Cre/Foxm1−/− mice was associated with increased expression of neutrophil-derived myeloperoxidase, proteinase 3, and cathepsin g, all of which are critical for lung remodeling and inflammation. Our data demonstrate that Foxm1 influences pulmonary inflammatory responses to hyperoxia, inhibiting neutrophil-derived enzymes and enhancing monocytic responses that limit alveolar injury and remodeling in neonatal lungs.
Keywords: Foxm1, bronchopulmonary dysplasia, hyperoxia, inflammation, macrophage
Clinical Relevance
Our data demonstrate that Foxm1 influences pulmonary inflammatory responses to hyperoxia, inhibiting neutrophil-derived enzymes and enhancing monocytic responses that limit alveolar injury and remodeling in neonatal lungs. Discovery of pharmacological agents that can modulate Foxm1 function in postnatal lungs could be beneficial to inhibit hyperoxia-induced inflammation and stimulate lung repair in patients with bronchopulmonary dysplasia.
Bronchopulmonary dysplasia (BPD) commonly develops in premature babies born before 36 weeks after conception. With approximately 50% of very-low-birth-weight infants born before 28 weeks gestational age developing BPD, it is the most common chronic lung disease among infants in industrialized nations (1). Every year, 7,500 to 25,000 American infants develop BPD (2). BPD is characterized by lung inflammation and arrested alveolar and vascular lung development (3). Inflammation is a major contributor to the pathogenesis of BPD. Often initiated by a pulmonary fetal inflammatory response, lung inflammation is exacerbated by mechanical ventilation and exposure to supplemental oxygen (4). Persistent lung inflammation has been shown to greatly increase the severity of BPD (2). Lung inflammation is typically characterized by increased pulmonary macrophages and neutrophils (2, 5). Pulmonary inflammation has been characterized to follow a standard timeline, with neutrophils moving from circulation to the lungs and recruiting macrophages, which resolve the inflammatory response (2, 5).
At 28 to 36 weeks gestation, lung development, an alveologenesis in particular, is insufficient to support the respiratory needs of the neonate. Standard clinical practice is mechanical ventilation with higher than atmospheric oxygen to provide an adequate supply of oxygen to the tissues of the body. Oxygen treatment causes undesirable side effects, including extensive inflammatory responses, destruction of the alveolar–capillary barrier, and pulmonary edema (39), all contributing to delayed or permanent disruption of postnatal lung development. Inflammation induced as a result of this insult has been shown to be a major contributor to the development of BPD, yet attempts to limit inflammation have not significantly improved survival, leading to a critical need for new approaches to supplement existing BPD therapy (40).
Exposure of neonatal animals to high concentrations of oxygen causes inflammation and lung remodeling that is similar to pulmonary responses in patients with BPD (6–8). Exposure to hyperoxia causes increased generation of reactive oxygen species, inducing lung injury, which results in cell cycle arrest and/or cell death (9, 10). Hyperoxia causes chemoattractant synthesis and release of inflammatory cytokines to recruit innate immune cells to the lung. Accumulation of neutrophils and macrophages enhances lung injury by the elaboration of proteases, oxidant stress (11), and chemokine release (12), which cause further injury and growth arrest resulting in loss of alveoli. In mouse models, prevention of neutrophil accumulation reduced acute lung injury (13). The beneficial or harmful effects of various inflammatory cells in the pathogenesis of BPD are unclear. Insight into the mechanism of inflammation after prolonged hyperoxic exposure could help identify potential new therapeutic targets in patients with BPD.
Foxm1 (formerly known as HFH-11B, Trident, Win, or MPP2), a member of the Forkhead Box (Fox) family of transcription factors, is mainly expressed in proliferating cells. Foxm1 is widely expressed during embryogenesis, consistent with its role in proliferation and cell differentiation. In adult mice, Foxm1 is more restricted, being observed in intestinal crypts, testes, and thymus (14, 15). During carcinogenesis and after tissue injury, Foxm1 is present in various cell types, including neoplastic, inflammatory, and regenerating cells (16–18). Global deletion of Foxm1 in transgenic mice causes lethal embryonic malformations in multiple organ systems (19). Cell-specific roles for Foxm1 are emerging from studies in transgenic mice with conditional deletion of Foxm1. Embryonic or perinatal lethal phenotypes were observed after Foxm1 deletion from hepatoblasts (20), precursors of cerebellar granule neurons (21), and smooth muscle cells (22). Conditional deletion of Foxm1 in developing respiratory epithelium caused neonatal respiratory failure due to delayed differentiation of type I cells and impaired lung maturation (23). In hepatocytes and in epithelial and endothelial cells, Foxm1 facilitated tissue repair after injury in the lungs and liver (15, 18, 24–26). Mice with conditional deletion of Foxm1 from myeloid inflammatory cells (LysM-Cre/Foxm1−/− mice) were recently generated and exhibited delayed liver repair (27). Foxm1 deletion inhibited carcinogenesis and airway allergic responses (28, 29). Although previous studies indicate that Foxm1 plays a critical role in myeloid-derived inflammatory cells, its role in the pathogenesis of neonatal lung disorders remains unknown.
In the present study, the role of Foxm1 in regulation of pulmonary inflammation and remodeling in BPD-like lung disease was investigated. Foxm1 expression was increased in lung tissue from patients with BPD and in hyperoxia-exposed neonatal mice. Selective deletion of Foxm1 from myeloid cells exacerbated lung injury and remodeling in hyperoxia-exposed mice, disrupting formation of peripheral alveoli and reducing lung function.
Materials and Methods
Mouse Strains
Generation of Foxm1flox/flox (Foxm1fl/fl) mice was previously described (20). Foxm1fl/fl female mice were bred with LysM-Cretg/− male mice (Jackson Laboratories, Bar Harbor, ME) to generate LysM-Cretg/− Foxm1fl/fl double transgenic mice (mFoxm1−/−). LysM-Cre deletes Foxm1 in all myeloid cells, including macrophages, monocytes, and granulocytes (30). Foxm1fl/fl littermates were used as controls. The following primers were used to genotype mouse genomic DNA: Foxm1 sense, 5′-TGGCTTCCCAGCAGTACAAATC-3′; Foxm1 antisense intron, 5′-TGCTTACAAAAGACACACTTGGACG-3′; Foxm1 antisense 3′-UTR, 5′-TCTCGCTCAATTCCAAGACCAG-3′; LysM-Cre sense, 5′-CTTGGGCTGCCAGAATTTCTC-3′; LysM-Cre antisense, 5′-CCCAGAAATGCCAGATTACG-3′. BPD responses were similar between control Foxm1fl/fl and wild-type mice. The experimental protocol was approved by Cincinnati Children’s Hospital Medical Center Animal Care and Use Committee.
Exposure to Hyperoxia
Pups (≤12 h) were placed in hyperoxic chambers (85% oxygen) or room air for up to 3 weeks. Nursing mothers were rotated between hyperoxia and room air litters daily to avoid maternal oxygen toxicity and maternal effects between groups. Oxygen levels were monitored with a Miniox II monitor (Catalyst Research, Owings Mills, MD). Survival was recorded daily. Baseline lung function was determined by a computer-controlled small animal ventilator (Flexivent, Scireq, VA) as previously described (31).
Morphometric Analysis
Morphometric measurements were performed using Image-1/Metamorph Imaging System (Universal Imaging, West Chester, PA). Radial alveolar counts were performed as previously described (32).
Immunohistochemical Staining and Flow Cytometry
Paraffin lung sections were used for immunohistochemical staining as described previously (31, 33). Human lung samples were collected as part of Cincinnati Children’s Hospital Medical Center Institutional Review Board Study 2008–0844. The study includes autopsy samples from infants with BPD and from infants who did not have BPD. The following antibodies were used for immunohistochemistry: Mac-3 (1:2,000 [IHC]) (BD Pharmingen, San Jose, CA), myeloperoxidase (MPO) (1:1,000) (R&D Systems, Minneapolis, MN), and Foxm1 (1:750 [IHC]) (C-20; Santa Cruz Biotech, Dallas, TX). Immunofluorescent staining was performed as previously described (16, 28, 34). Diff-Quik (Siemens, Malvern, PA) staining was performed according to the manufacturer’s instructions.
Inflammatory cells were isolated from lungs of normoxic and hyperoxic mFoxm1−/− and Foxm1fl/fl mice by flow cytometry as described previously (35). The following antibodies were used to stain inflammatory cells: anti-F4/80 (clone BM8; eBiosciences, San Diego, CA), anti-CD11b (clone M1/70; eBiosciences), anti-Ly-6C (clone HK1.4; BioLegend, San Diego, CA), anti–Ly-6G (clone 1A8; BioLegend), anti-CD68 (clone FA-11; Biolegend), and anti-CD45 (clone 30-F11; BD Pharmingen). Dead cells were excluded using 7-aminoactinomycin stain (eBiosciences). Stained cells were separated using cell sorting (five-laser FACSAria II; BD Biosciences). Specific cell subsets were identified using the indicated surface marker phenotypes: neutrophils, CD45+CD11b+Ly6C+Ly6G+; monocytes, CD45+CD11b+Ly6ChiLy6G−F4/80+; interstitial macrophages, CD45+CD11b+Ly6C−Ly6G−F4/80+CD68−; and alveolar macrophages, CD45+CD11b+Ly6C−Ly6G−F4/80+CD68+. Purified cells were used for RNA preparation followed by quantitative RT-PCR (qRT-PCR).
Alveolar type II epithelial cells were identified using antibodies against CD324 (clone DECMA-1; eBiosciences), CD326 (clone G8.8; eBiosciences), and MHC II (clone M5/114.15.2; eBiosciences).
qRT-PCR and Western Blot
StepOnePlus real-time PCR system (Applied Biosystems, Foster City, CA) and inventoried TaqMan gene expression assays were used as described previously (23, 36). Reactions were analyzed in triplicate, and expression levels were normalized to β-actin mRNA. Western blots were performed as previously described (37) with antibodies against MPO (1:1,000) (R&D Systems) and β-actin (1:3,000) (C-11; Santa Cruz Biotech).
Statistical Analysis
Student’s t test and multivariant ANOVA were used to determine statistical significance. P values less than 0.05 were considered significant. All measurements are expressed as the mean ± SD.
Results
Foxm1 Expression Is Increased in BPD Lungs
Autopsy samples from infants without BPD showed little to no Foxm1 staining, whereas lungs from patients with BPD showed increased Foxm1 staining in inflammatory cells (Figure 1A). Colocalization experiments demonstrated that Mac3-positive macrophages were the primary source of Foxm1 staining in BPD lungs (Figure 1B). Because neonatal hyperoxia exposure in mice recapitulates many features of BPD (6–8), we examined Foxm1 expression in this mouse BPD model. Newborn mice were exposed to hyperoxia for 3 weeks, followed by recovery in room air for an additional 3 weeks. Although hyperoxia itself did not influence Foxm1 mRNA levels, Foxm1 was increased during the recovery period (Figure 1C). Consistent with findings in human BPD lungs, Foxm1 staining was observed in pulmonary macrophages of hyperoxia-exposed mice but not in control mice exposed to room air (Figure 1D). These results indicate increased Foxm1-positive pulmonary macrophages in lungs of patients with BPD and a mouse model of BPD-like disease.
Figure 1.

Increased expression of Foxm1 in lung tissue of patients with bronchopulmonary dysplasia (BPD) and hyperoxia-exposed mice. (A) Foxm1 expression is increased in lungs of patients with BPD compared with normal lungs. Alveolar macrophages were rare in normal human lung, but Foxm1-positive macrophages (arrows) were prevalent in lung autopsies from patients with BPD (n = 7). Original magnification: ×50 and ×1,000. (B) Colocalization experiments confirmed that Foxm1-positive cells in patients with BPD were macrophages as Foxm1 colocalized with the macrophage marker Mac3 (arrowheads). Original magnification: ×1,000. (C) Foxm1 expression is induced in a mouse model of BPD-like lung injury. Wild-type mice were housed in hyperoxic conditions (HO) for 3 weeks after birth followed by room air recovery (RA). Foxm1 mRNA was increased in mouse lungs after HO and 9-day RA recovery, and this increase was greater at 21-day recovery (n = 5 mice per group). *P < 0.05; **P < 0.01. (D) Foxm1 staining was found in pulmonary macrophages from HO-exposed mice but not RA-exposed mice. Original magnification: ×100 and ×1,000. DAPI, 4′,6-diamidino-2-phenylindole.
Deletion of Foxm1 from Myeloid Cell Lineages Exacerbated Hyperoxia-Induced Alveolar Simplification in Postnatal Lungs
Foxm1 requirements in BPD were examined using mice with myeloid-specific Foxm1 deletion (LysM-Cre/Foxm1fl/fl or mFoxm1−/−). We previously demonstrated that mFoxm1−/− mice had normal lung development (27). Consistent with these results, alveolar structure in mFoxm1−/− mice housed in room air was normal as indicated by measurements of terminal air space (Figures 2A and 2B) and radial alveolar count (Figure 2C). Prolonged neonatal hyperoxia increased terminal air space area (Figure 2B) and decreased radial alveolar counts (Figure 2C) in control Foxm1fl/fl mice, a finding consistent with hyperoxia-induced alveolar simplification. Alveolar simplification was exacerbated in hyperoxia-exposed mFoxm1−/− mice compared with control Foxm1fl/fl mice (Figures 2B and 2C). Consistent with impaired alveologenesis, respiratory system resistance and elasticity were increased, whereas respiratory compliance was decreased in hyperoxia-exposed mFoxm1−/− mice (Figures 3A–3C). Central airway resistance, tissue damping, and tissue elastance were not altered by deletion of Foxm1 (Figures 3D–3F). Thus, deletion of Foxm1 from myeloid cells exacerbated alveolar simplification in a mouse model of BPD-like disease.
Figure 2.

Impaired alveologenesis in mFoxm1−/− mice after prolonged hyperoxia. Newborn mFoxm1−/− and littermate control Foxm1fl/fl mice were exposed to HO or RA from Postnatal Day 1 until 3 weeks of age. Both groups of mice were subsequently housed in RA for an additional 3 weeks. HO disrupted alveologenesis in mFoxm1−/− and control mice, as shown by hematoxylin and eosin staining (A). HO also increased terminal air space area (B) and decreased radial alveolar count (C). Ten random lung sections were measured (n = 5 mice per group). Hyperoxia-induced changes were greater in mFoxm1−/− mice compared with controls. **P < 0.01. There were no differences between mFoxm1−/− and control mice exposed to RA. Original magnification: ×100.
Figure 3.
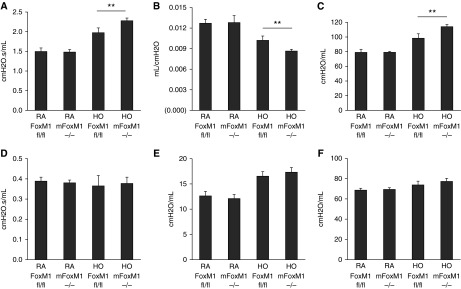
Impaired pulmonary function in mFoxm1−/− mice exposed to prolonged postnatal hyperoxia. Flexivent was used to measure pulmonary function in mice exposed HO or RA for 3 weeks, followed by a 3-week recovery period in RA. Exposure to HO decreased lung function in mFoxm1−/− mice to a greater extent than in control Foxm1fl/fl mice. Respiratory system resistance (A) and respiratory system elasticity (C) were increased in HO mFoxm1−/− mice, whereas respiratory system compliance (B) was decreased. There were no changes in central airway resistance, tissue damping, or tissue elastance (D–F). Five mice were used in each group. **P < 0.01.
Foxm1 Deletion Reduced Accumulation of Interstitial Macrophages in Response to Hyperoxia
Neonatal hyperoxia increases lung inflammation contributing to alveolar simplification in mice and humans (6–8). Therefore, we examined lung inflammation after 3 weeks of hyperoxia exposure. In mFoxm1−/− and control Foxm1fl/fl mice, hyperoxic exposure increased the number of Mac3-positive pulmonary macrophages (Figures 4A and 4B). However, the elevation of Mac3-positive macrophages in mFoxm1−/− lungs was significantly attenuated compared with control mice (Figures 4A and 4B). Because Mac3 antigen is present in multiple subsets of macrophages, FACS analysis was performed to determine the identity of macrophage subsets present in hyperoxia-exposed lungs. Monocytes were identified as CD45+CD11b+Ly6ChiLy6G−F4/80+ cells in total cell suspension from perfused lungs. Alveolar and interstitial macrophages were CD45+CD11b+Ly6C−Ly6G−F4/80+ but were distinguished by the presence of CD68 antigen on their surface (Figure 4C). Consistent with airspace enlargement, total lung cell number was reduced in hyperoxia-exposed mFoxm1−/− mice compared with controls (Figure 4D). The overall percentage of CD45+CD11b+ myeloid cells was increased in both mouse lines, indicating increased inflammation in response to hyperoxia (Figure 4E). Hyperoxic exposure altered the overall balance of macrophage subsets in the lung, decreasing monocytes and alveolar macrophages but elevating the overall percentage of differentiated interstitial macrophages (Figure 4F). Compared with controls, the percentage of interstitial macrophages was decreased in hyperoxia-exposed mFoxm1−/− lungs, whereas monocyte precursors were increased. There was no change in alveolar macrophages in mFoxm1−/− mice compared with controls (Figure 4F). Thus, Foxm1 deletion from myeloid cell lineages decreased accumulation of interstitial macrophages in response to hyperoxia.
Figure 4.

Foxm1 deletion from myeloid cell lineages decreases accumulation of pulmonary interstitial macrophages after prolonged postnatal hyperoxia. Newborn mFoxm1−/− and control Foxm1fl/fl mice were exposed to HO or RA and harvested at 3 weeks of age. (A and B) Immunostaining for Mac-3 showed reduced numbers of macrophages in HO-exposed mFoxm1−/− lungs. The number of Mac-3–positive cells was counted in 10 random sections using five mice in each group. Original magnification: ×400. **P < 0.05. (C) Inflammatory cells were isolated from lung tissue of mFoxm1−/− or Foxm1fl/fl mice after 1 week of HO-exposure. Dead cells were excluded using 7-aminoactinomycin stain. The following cell surface markers were used to identify cell types: neutrophils, CD45+CD11b+Ly6C+ Ly6G+; monocytes, CD45+CD11b+ Ly6ChiLy6G−F4/80+; interstitial macrophages, CD45+CD11b+ Ly6C−Ly6G− F4/80+CD68−; alveolar macrophages, CD45+CD11b+ Ly6C−Ly6G− F4/80+CD68+. (D) HO-exposed mFoxm1−/− mice had a lower total cell count than Foxm1fl/fl mice. (E) The percentage of myeloid cells was calculated by flow cytometry using CD45+CD11b+ cell surface markers. Although the percentage of myeloid cells increased significantly after HO exposure, there were no significant differences between mFoxm1−/− and Foxm1fl/fl mice. (F) Decreased numbers of interstitial macrophages were observed in HO-exposed mFoxm1−/− lungs. Cell numbers were counted after enzymatic digestion of lung tissue (n = 5 mice per group).The percentage of interstitial macrophages was decreased in HO-exposed mFoxm1−/− lungs, whereas the number of alveolar macrophages was unaltered. The number of monocytes was increased in HO-exposed mFoxm1−/− lungs compared with controls. *P < 0.05; **P < 0.01. FSC-A, forward scatter defined by area; PE-A, intensity of phycoerythrin-stained cells defined by area; SSC-A, side scatter defined by area.
qRT-PCR analysis of FACS-sorted myeloid cell subsets indicated robust Cre mRNA expression in all myeloid cell lineages investigated, including monocytes, interstitial macrophages, alveolar macrophages, and neutrophils (Figure 5B). Foxm1 mRNA was decreased 40 to 60% in all myeloid cell lineages (Figure 5A). PCR analysis of genomic DNA confirmed the presence of the Foxm1-null allele in mFoxm1−/− mice (Figure 5F). Neutrophils contained very little Foxm1 mRNA, and its levels were unchanged after hyperoxia. Despite significant Foxm1 deletion in all monocytic cell lineages, only interstitial macrophages showed a significant decrease in cyclin B1 and cdc 25B (Figures 5C–5E), both critical cell cycle regulators and known Foxm1 target genes (37). There was no difference in expression of the Foxm1-independent cell cycle regulators cMyc or cyclin D1. These results demonstrate that Foxm1 downstream signaling events were inhibited in interstitial macrophages of mFoxm1−/− mice after hyperoxia.
Figure 5.

Decreased expression of cyclin B1 and cdc25B in interstitial macrophages from hyperoxia-exposed mFoxm1−/− lungs. Cells were purified from lungs of mFoxm1−/− and control mice by flow cytometry (see Figure 4 for cell surface markers) and used for quantitative RT-PCR (qRT-PCR). (A) Foxm1 mRNA expression was decreased in monocytes (Mon), interstitial macrophages (IM), and alveolar macrophages (AM) from mFoxm1−/− mice. Foxm1 mRNA levels were low in neutrophils (Neu). Six mice were used in each group. (B) Robust Cre mRNA expression was detected in all subsets of myeloid cells from mFoxm1−/− mice. (C) qRT-PCR showed decreased mRNAs of cyclin B1 and cdc25B in interstitial macrophages from hyperoxia-exposed mFoxm1−/− lungs compared with controls. (D and E) There were no statistical differences in mRNA levels of cell cycle regulatory genes in mFoxm1−/− alveolar macrophages or monocytes. *P < 0.05; **P < 0.01. (F) PCR analysis of genomic DNA was used to identify the presence of the Foxm1-null allele in mFoxm1−/− mice.
Unaltered Development of Alveolar Type II Cells in mFoxm1−/− Mice
Because LysM-Cre targets a subset of type II cells (38), FACS sorting was used to isolate alveolar type II cells (ATII) for analysis of Foxm1 expression (Figure 6A). Type II cells were identified as cells expressing EpCam+E-cadherin+MHC II+ but were negative for CD45 and PECAM-1 (Figure 6A). qRT-PCR confirmed the purity of ATII cell isolation, showing high expression of ATII-specific surfactant protein (SP)-A, SP-B, and SP-C but little to no expression of aquaporin 5, Sox17, Foxj1, and smooth muscle myosin heavy chain, which are expressed in various non-ATII cell types (Figure 6B). Compared with monocytes, ATII cells from mFoxm1−/− mice contained relatively high levels of Foxm1 mRNA (Figure 6C) and low levels of Cre mRNA (Figure 6D). However, there were no significant changes in Foxm1 mRNA in normal or hyperoxia-exposed ATII cells from either group of mice. Thus, ATII cells are not targeted in mFoxm1−/− mice. These results also suggest that alveolar simplification in hyperoxia-exposed mFoxm1−/− mice occurs independently of Foxm1 deficiency in ATII cells.
Figure 6.
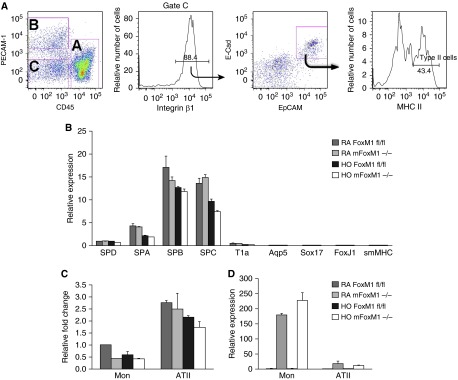
Foxm1 is not deleted from alveolar type II cells of mFoxm1−/− mice. Alveolar type II epithelial cells (ATII) were isolated from either mFoxm1−/− or Foxm1fl/fl lungs by FACS. (A) ATII cells were identified using the following cell surface markers: EpCAM+E-cadherin+MHC II+CD45−PECAM-1−. (B) Gene expression profile of ATII cells by qRT-PCR showed the presence of surfactant protein (SP)-A, SP-B, SP-C, and SP-D mRNAs and the absence of T1A, aqp5, Sox17, FoxJ1, and smooth muscle myosin heavy chain (smMHC) mRNAs. smMHC, smooth muscle myosin heavy chain. (C) There were no significant differences in Foxm1 mRNA in ATII cells from mFoxm1−/− and control Foxm1fl/fl mice. (D) qRT-PCR showed low levels of Cre mRNA in ATII cells from mFoxm1−/− mice. Five mice were used in each group.
Increased Expression of MPO, Proteinase 3, and Cathepsin g in mFoxm1−/− Lungs after Hyperoxia
Neutrophils produce various enzymes, such as MPO, proteinase 3, and cathepsin g, which degrade extracellular matrix and other components of the alveolar wall, directly contributing to alveolar simplification (2). To determine whether neutrophilic infiltration is associated with the mFoxm1−/− lung phenotype, peripheral blood was collected from mFoxm1−/− and control Foxm1fl/fl mice, and Diff-Quik staining was used to analyze circulating neutrophils. Hyperoxia increased the percentage of circulating neutrophils in both groups of mice, but these changes were exacerbated in mFoxm1−/− mice compared with controls (Figures 7A and 7B). Although FACS analysis of whole lung tissue showed an increased percentage of neutrophils (CD45+CD22G+Ly6C+Ly6G+) in hyperoxia-exposed mFoxm1−/− lungs versus hyperoxia-exposed Foxm1fl/fl lungs (Figure 7E), these changes were not induced by hyperoxia because there was no difference between hyperoxia-exposed and room air–housed mice. Immunostaining of lung tissue for MPO, a neutrophil-specific enzyme (39), demonstrated increased abundance of MPO-positive neutrophils in hyperoxia-exposed mFoxm1−/− mice (Figures 7C and 7D). Elevated MPO protein levels were confirmed by Western blot (Figure 7G). qRT-PCR analysis of whole lung RNA showed increased mRNA levels of neutrophil-derived enzymes MPO, proteinase 3, and cathepsin g in hyperoxia-exposed mFoxm1−/− mice compared with controls (Figure 7F). Altogether, our results show that Foxm1 deletion from myeloid cell lineages increased the percentage of circulating neutrophils and increased expression of neutrophil-derived enzymes in lung tissue.
Figure 7.

Foxm1 deletion in myeloid cell lineages increases circulating neutrophils and stimulates expression of neutrophil-derived enzymes in hyperoxia-exposed lungs. (A) The percentage of neutrophils in peripheral blood was increased in HO mFoxm1−/− mice as shown by Diff-quik staining of blood smears. Original magnification: ×400. (B) The number of neutrophils was counted in 10 random sections using five mice in each group. (C) Lung sections from mFoxm1−/− and Foxm1fl/fl mice were stained with myeloperoxidase (MPO) antibodies (dark brown) and counterstained with nuclear fast red (red nuclei). In the HO group, the number of MPO-positive neutrophils was increased compared with controls. Original magnification: ×400. (D) The number of MPO-positive cells was counted in 10 random sections using five mice in each group. (E) Myeloid cells were isolated from either mFoxm1−/− or Foxm1fl/fl lungs by FACS. The percentage of lung myeloid cells that were neutrophils (CD45+CD11b+ Ly6C+Ly6G+) was increased in HO-exposed mFoxm1−/− mice compared HO-exposed Foxm1fl/fl mice. (F) qRT-PCR showed increased mRNAs of MPO, proteinase 3, and cathepsin g in HO-exposed mFoxm1−/− lungs. mRNA levels were normalized to β-actin mRNA. (G) Western blot showed increased MPO protein in HO-exposed mFoxm1−/− lungs. *P < 0.05; **P < 0.01.
Discussion
The results of this study show that Foxm1-positive macrophages are abundant in lungs of patients with BPD and in a mouse model of BPD-like disease. Myeloid cells are important mediators of pulmonary remodeling after hyperoxia exposure, with perturbations in the types of inflammatory cells present in the lung being critical to disease progression. Our data indicate that expression of Foxm1 transcription factor in myeloid inflammatory cells is critical for the recruitment and accumulation of interstitial macrophages in response to hyperoxia. Deletion of Foxm1 from the myeloid cell lineage decreased interstitial macrophage accumulation in the lung after prolonged hyperoxic exposure but increased circulating neutrophils and neutrophil-derived enzymes in lung tissue, ultimately exacerbating lung remodeling and decreasing lung function. Hyperoxia-exposed mFoxm1−/− lungs had increased expression of the neutrophilic enzymes MPO, proteinase 3, and cathepsin g, all of which are capable of disrupting alveologenesis by degrading extracellular matrix proteins critical for maintaining integrity of the alveolar wall. Thus, Foxm1 deletion from myeloid cell lineages changes the inflammatory response to hyperoxia, decreasing the accumulation of macrophages and increasing the abundance of neutrophil-derived enzymes, a phenotype that is detrimental to lung function and repair after neonatal hyperoxic exposure. Our results are consistent with clinical data showing a direct correlation between neutrophil-derived enzymes and severity of BPD in preterm infants.
Hyperoxia-induced lung injury is initiated by reactive oxygen species, followed by extensive infiltration of inflammatory leukocytes into the lung (40). After an insult, resident macrophages and epithelial cells secrete proinflammatory chemokines and cytokines, inducing migration of neutrophils out of pulmonary capillaries into the air space (41). Neutrophils migrate to the inflamed tissue, promote recruitment of inflammatory monocytes, and potentiate the proinflammatory environment, allowing appropriate resolution of the insult (42). Neutrophils undergo apoptosis after performing their action at the inflamed site, and macrophages ingest apoptotic neutrophils (43). Clearance of apoptotic neutrophils prompts a switch from a pro- to an antiinflammatory macrophage phenotype (44), which is a prerequisite for macrophage egress via the lymphatic vessels favoring return to tissue homeostasis. The increase in circulating neutrophils and neutrophil-derived enzymes in the lung observed in mFoxm1−/− mice likely exacerbates the proinflammatory response within the lung after hyperoxic insult. This in turn may prevent resolution of the inflammatory response and delay the ability of the lung to sustain proper alveologenesis, likely contributing to increased terminal air space observed in mFoxm1−/− mice after prolonged hyperoxia. Furthermore, there is accumulating evidence that macrophages reduce inflammatory responses through ingestion of dying cells or dead cell debris, thereby mitigating tissue damage (45–47). Chronic inflammation in the lung appears to be associated with delayed removal of dying cells, which may directly affect the natural ability of the injured organism to shut down inflammation and initiate tissue repair (48). Resolving lung inflammation depends on timely and adequate removal of acute inflammatory cells by macrophages. In this study, deletion of Foxm1 from myeloid cells reduced the number of interstitial macrophages after hyperoxia. This likely produces delayed resolution of the proinflammatory microenvironment, which, together with exacerbated proteolytic activities of neutrophilic enzymes, alters pulmonary remodeling after hyperoxia exposure.
In summary, the results of this study show that patients with BPD and hyperoxia-exposed mice have increased Foxm1-positive macrophages in lung tissue. Using mice with myeloid-specific Foxm1 deletion, we demonstrated that Foxm1 expression in myeloid cell lineages is required to maintain proper balance between neutrophils and interstitial macrophages in response to prolonged neonatal hyperoxia. Loss of Foxm1 increases neutrophil-derived enzymes in lung tissue, disrupting alveologenesis and exacerbating BPD-like disease. Thus, discovery of pharmacological agents that can modulate Foxm1 function in postnatal lungs could be beneficial to inhibiting hyperoxia-induced inflammation and stimulate lung repair in patients with BPD.
Footnotes
This work was supported by National Institutes of Health grants HL84151 (V.V.K.), HL123490 (V.V.K.), and CA142724 (T.V.K.).
Author Contributions: Conception and design: H.X., X.R., and V.V.K. Analysis and interpretation: H.X., X.R., C.S.B., V.U., Y.Z., T.A.S., and V.V.K. Drafting the manuscript for important intellectual content: H.X., C.S.B., T.V.K., J.A.W., and V.V.K.
Originally Published in Press as DOI: 10.1165/rcmb.2014-0091OC on October 2, 2014
Author disclosures are available with the text of this article at www.atsjournals.org.
References
- 1.Stoll BJ, Hansen NI, Bell EF, Shankaran S, Laptook AR, Walsh MC, Hale EC, Newman NS, Schibler K, Carlo WA, et al. Neonatal outcomes of extremely preterm infants from the NICHD Neonatal Research Network. Pediatrics. 2010;126:443–456. doi: 10.1542/peds.2009-2959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ryan RM, Ahmed Q, Lakshminrusimha S. Inflammatory mediators in the immunobiology of bronchopulmonary dysplasia. Clin Rev Allergy Immunol. 2008;34:174–190. doi: 10.1007/s12016-007-8031-4. [DOI] [PubMed] [Google Scholar]
- 3.Jobe AH, Ikegami M. Mechanisms initiating lung injury in the preterm. Early Hum Dev. 1998;53:81–94. doi: 10.1016/s0378-3782(98)00045-0. [DOI] [PubMed] [Google Scholar]
- 4.Bose CL, Dammann CE, Laughon MM. Bronchopulmonary dysplasia and inflammatory biomarkers in the premature neonate. Arch Dis Child Fetal Neonatal Ed. 2008;93:F455–F461. doi: 10.1136/adc.2007.121327. [DOI] [PubMed] [Google Scholar]
- 5.Speer CP. Inflammation and bronchopulmonary dysplasia: a continuing story. Semin Fetal Neonatal Med. 2006;11:354–362. doi: 10.1016/j.siny.2006.03.004. [DOI] [PubMed] [Google Scholar]
- 6.Han RN, Buch S, Tseu I, Young J, Christie NA, Frndova H, Lye SJ, Post M, Tanswell AK. Changes in structure, mechanics, and insulin-like growth factor-related gene expression in the lungs of newborn rats exposed to air or 60% oxygen. Pediatr Res. 1996;39:921–929. doi: 10.1203/00006450-199606000-00001. [DOI] [PubMed] [Google Scholar]
- 7.Wagenaar GT, ter Horst SA, van Gastelen MA, Leijser LM, Mauad T, van der Velden PA, de Heer E, Hiemstra PS, Poorthuis BJ, Walther FJ. Gene expression profile and histopathology of experimental bronchopulmonary dysplasia induced by prolonged oxidative stress. Free Radic Biol Med. 2004;36:782–801. doi: 10.1016/j.freeradbiomed.2003.12.007. [DOI] [PubMed] [Google Scholar]
- 8.Warner BB, Stuart LA, Papes RA, Wispe JR. Functional and pathological effects of prolonged hyperoxia in neonatal mice. Am J Physiol. 1998;275:L110–L117. doi: 10.1152/ajplung.1998.275.1.L110. [DOI] [PubMed] [Google Scholar]
- 9.Li Y, Zhang W, Mantell LL, Kazzaz JA, Fein AM, Horowitz S. Nuclear factor-kappaB is activated by hyperoxia but does not protect from cell death. J Biol Chem. 1997;272:20646–20649. doi: 10.1074/jbc.272.33.20646. [DOI] [PubMed] [Google Scholar]
- 10.Zaher TE, Miller EJ, Morrow DM, Javdan M, Mantell LL. Hyperoxia-induced signal transduction pathways in pulmonary epithelial cells. Free Radic Biol Med. 2007;42:897–908. doi: 10.1016/j.freeradbiomed.2007.01.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Nader-Djalal N, Knight PR, III, Thusu K, Davidson BA, Holm BA, Johnson KJ, Dandona P. Reactive oxygen species contribute to oxygen-related lung injury after acid aspiration. Anesth Analg. 1998;87:127–133. doi: 10.1097/00000539-199807000-00028. [DOI] [PubMed] [Google Scholar]
- 12.Speer CP. Inflammatory mechanisms in neonatal chronic lung disease. Eur J Pediatr. 1999;158:S18–S22. doi: 10.1007/pl00014314. [DOI] [PubMed] [Google Scholar]
- 13.Bhatia M, Saluja AK, Hofbauer B, Lee HS, Frossard JL, Steer ML. The effects of neutrophil depletion on a completely noninvasive model of acute pancreatitis-associated lung injury. Int J Pancreatol. 1998;24:77–83. doi: 10.1007/BF02788564. [DOI] [PubMed] [Google Scholar]
- 14.Korver W, Roose J, Clevers H. The winged-helix transcription factor Trident is expressed in cycling cells. Nucleic Acids Res. 1997;25:1715–1719. doi: 10.1093/nar/25.9.1715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ye H, Holterman AX, Yoo KW, Franks RR, Costa RH. Premature expression of the winged helix transcription factor HFH-11B in regenerating mouse liver accelerates hepatocyte entry into S phase. Mol Cell Biol. 1999;19:8570–8580. doi: 10.1128/mcb.19.12.8570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Balli D, Ustiyan V, Zhang Y, Wang IC, Masino AJ, Ren X, Whitsett JA, Kalinichenko VV, Kalin TV. Foxm1 transcription factor is required for lung fibrosis and epithelial-to-mesenchymal transition. EMBO J. 2013;32:231–244. doi: 10.1038/emboj.2012.336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Cai Y, Balli D, Ustiyan V, Fulford L, Hiller A, Misetic V, Zhang Y, Paluch AM, Waltz SE, Kasper S, et al. Foxm1 expression in prostate epithelial cells is essential for prostate carcinogenesis. J Biol Chem. 2013;288:22527–22541. doi: 10.1074/jbc.M113.455089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Liu Y, Sadikot RT, Adami GR, Kalinichenko VV, Pendyala S, Natarajan V, Zhao YY, Malik AB. FoxM1 mediates the progenitor function of type II epithelial cells in repairing alveolar injury induced by Pseudomonas aeruginosa. J Exp Med. 2011;208:1473–1484. doi: 10.1084/jem.20102041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ramakrishna S, Kim IM, Petrovic V, Malin D, Wang IC, Kalin TV, Meliton L, Zhao YY, Ackerson T, Qin Y, et al. Myocardium defects and ventricular hypoplasia in mice homozygous null for the Forkhead Box M1 transcription factor. Dev Dyn. 2007;236:1000–1013. doi: 10.1002/dvdy.21113. [DOI] [PubMed] [Google Scholar]
- 20.Krupczak-Hollis K, Wang X, Kalinichenko VV, Gusarova GA, Wang IC, Dennewitz MB, Yoder HM, Kiyokawa H, Kaestner KH, Costa RH. The mouse Forkhead Box m1 transcription factor is essential for hepatoblast mitosis and development of intrahepatic bile ducts and vessels during liver morphogenesis. Dev Biol. 2004;276:74–88. doi: 10.1016/j.ydbio.2004.08.022. [DOI] [PubMed] [Google Scholar]
- 21.Schuller U, Zhao Q, Godinho SA, Heine VM, Medema RH, Pellman D, Rowitch DH. Forkhead transcription factor FoxM1 regulates mitotic entry and prevents spindle defects in cerebellar granule neuron precursors. Mol Cell Biol. 2007;27:8259–8270. doi: 10.1128/MCB.00707-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ustiyan V, Wang IC, Ren X, Zhang Y, Snyder J, Xu Y, Wert SE, Lessard JL, Kalin TV, Kalinichenko VV. Forkhead box M1 transcriptional factor is required for smooth muscle cells during embryonic development of blood vessels and esophagus. Dev Biol. 2009;336:266–279. doi: 10.1016/j.ydbio.2009.10.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kalin TV, Wang IC, Meliton L, Zhang Y, Wert SE, Ren X, Snyder J, Bell SM, Graf L, Jr, Whitsett JA, et al. Forkhead Box m1 transcription factor is required for perinatal lung function. Proc Natl Acad Sci USA. 2008;105:19330–19335. doi: 10.1073/pnas.0806748105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kalinichenko VV, Lim L, Shin B, Costa RH. Differential expression of forkhead box transcription factors following butylated hydroxytoluene lung injury. Am J Physiol Lung Cell Mol Physiol. 2001;280:L695–L704. doi: 10.1152/ajplung.2001.280.4.L695. [DOI] [PubMed] [Google Scholar]
- 25.Kalinichenko VV, Gusarova GA, Tan Y, Wang IC, Major ML, Wang X, Yoder HM, Costa RH. Ubiquitous expression of the forkhead box M1B transgene accelerates proliferation of distinct pulmonary cell types following lung injury. J Biol Chem. 2003;278:37888–37894. doi: 10.1074/jbc.M305555200. [DOI] [PubMed] [Google Scholar]
- 26.Zhao YY, Gao XP, Zhao YD, Mirza MK, Frey RS, Kalinichenko VV, Wang IC, Costa RH, Malik AB. Endothelial cell-restricted disruption of FoxM1 impairs endothelial repair following LPS-induced vascular injury. J Clin Invest. 2006;116:2333–2343. doi: 10.1172/JCI27154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ren X, Zhang Y, Snyder J, Cross ER, Shah TA, Kalin TV, Kalinichenko VV. Forkhead box M1 transcription factor is required for macrophage recruitment during liver repair. Mol Cell Biol. 2010;30:5381–5393. doi: 10.1128/MCB.00876-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ren X, Shah TA, Ustiyan V, Zhang Y, Shinn J, Chen G, Whitsett JA, Kalin TV, Kalinichenko VV. FOXM1 promotes allergen-induced goblet cell metaplasia and pulmonary inflammation. Mol Cell Biol. 2013;33:371–386. doi: 10.1128/MCB.00934-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kalin TV, Ustiyan V, Kalinichenko VV. Multiple faces of FoxM1 transcription factor: lessons from transgenic mouse models. Cell Cycle. 2011;10:396–405. doi: 10.4161/cc.10.3.14709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Clausen BE, Burkhardt C, Reith W, Renkawitz R, Forster I. Conditional gene targeting in macrophages and granulocytes using LysMcre mice. Transgenic Res. 1999;8:265–277. doi: 10.1023/a:1008942828960. [DOI] [PubMed] [Google Scholar]
- 31.Kalin TV, Meliton L, Meliton AY, Zhu X, Whitsett JA, Kalinichenko VV. Pulmonary mastocytosis and enhanced lung inflammation in mice heterozygous null for the Foxf1 gene. Am J Respir Cell Mol Biol. 2008;39:390–399. doi: 10.1165/rcmb.2008-0044OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Betz P, Nerlich A, Bussler J, Hausmann R, Eisenmenger W. Radial alveolar count as a tool for the estimation of fetal age. Int J Legal Med. 1997;110:52–54. doi: 10.1007/s004140050030. [DOI] [PubMed] [Google Scholar]
- 33.Kalinichenko VV, Bhattacharyya D, Zhou Y, Gusarova GA, Kim W, Shin B, Costa RH. Foxf1 +/− mice exhibit defective stellate cell activation and abnormal liver regeneration following CCl4 injury. Hepatology. 2003;37:107–117. doi: 10.1053/jhep.2003.50005. [DOI] [PubMed] [Google Scholar]
- 34.Wang IC, Ustiyan V, Zhang Y, Cai Y, Kalin TV, Kalinichenko VV. Foxm1 transcription factor is required for the initiation of lung tumorigenesis by oncogenic Kras. Oncogene. doi: 10.1038/onc.2013.475. (In press) [DOI] [PubMed] [Google Scholar]
- 35.Lewkowich IP, Herman NS, Schleifer KW, Dance MP, Chen BL, Dienger KM, Sproles AA, Shah JS, Kohl J, Belkaid Y, et al. CD4+CD25+ T cells protect against experimentally induced asthma and alter pulmonary dendritic cell phenotype and function. J Exp Med. 2005;202:1549–1561. doi: 10.1084/jem.20051506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Wang IC, Meliton L, Tretiakova M, Costa RH, Kalinichenko VV, Kalin TV. Transgenic expression of the forkhead box M1 transcription factor induces formation of lung tumors. Oncogene. 2008;27:4137–4149. doi: 10.1038/onc.2008.60. [DOI] [PubMed] [Google Scholar]
- 37.Bolte C, Zhang Y, Wang IC, Kalin TV, Molkentin JD, Kalinichenko VV. Expression of Foxm1 transcription factor in cardiomyocytes is required for myocardial development. PLoS One. 2011;6:e22217. doi: 10.1371/journal.pone.0022217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Markart P, Faust N, Graf T, Na CL, Weaver TE, Akinbi HT. Comparison of the microbicidal and muramidase activities of mouse lysozyme M and P. Biochem J. 2004;380:385–392. doi: 10.1042/BJ20031810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Klebanoff SJ. Myeloperoxidase: friend and foe. J Leukoc Biol. 2005;77:598–625. doi: 10.1189/jlb.1204697. [DOI] [PubMed] [Google Scholar]
- 40.Auten RL, Whorton MH, Nicholas Mason S. Blocking neutrophil influx reduces DNA damage in hyperoxia-exposed newborn rat lung. Am J Respir Cell Mol Biol. 2002;26:391–397. doi: 10.1165/ajrcmb.26.4.4708. [DOI] [PubMed] [Google Scholar]
- 41.Bender AT, Ostenson CL, Wang EH, Beavo JA. Selective up-regulation of PDE1B2 upon monocyte-to-macrophage differentiation. Proc Natl Acad Sci USA. 2005;102:497–502. doi: 10.1073/pnas.0408535102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Mantovani A, Bonecchi R, Locati M. Tuning inflammation and immunity by chemokine sequestration: decoys and more. Nat Rev Immunol. 2006;6:907–918. doi: 10.1038/nri1964. [DOI] [PubMed] [Google Scholar]
- 43.Fox S, Leitch AE, Duffin R, Haslett C, Rossi AG. Neutrophil apoptosis: relevance to the innate immune response and inflammatory disease. J Innate Immun. 2010;2:216–227. doi: 10.1159/000284367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Michlewska S, Dransfield I, Megson IL, Rossi AG. Macrophage phagocytosis of apoptotic neutrophils is critically regulated by the opposing actions of pro-inflammatory and anti-inflammatory agents: key role for TNF-alpha. FASEB J. 2009;23:844–854. doi: 10.1096/fj.08-121228. [DOI] [PubMed] [Google Scholar]
- 45.Jang HS, Kim J, Park YK, Park KM. Infiltrated macrophages contribute to recovery after ischemic injury but not to ischemic preconditioning in kidneys. Transplantation. 2008;85:447–455. doi: 10.1097/TP.0b013e318160f0d1. [DOI] [PubMed] [Google Scholar]
- 46.Leor J, Rozen L, Zuloff-Shani A, Feinberg MS, Amsalem Y, Barbash IM, Kachel E, Holbova R, Mardor Y, Daniels D, et al. Ex vivo activated human macrophages improve healing, remodeling, and function of the infarcted heart. Circulation. 2006;114:I94–I100. doi: 10.1161/CIRCULATIONAHA.105.000331. [DOI] [PubMed] [Google Scholar]
- 47.van Amerongen MJ, Harmsen MC, van Rooijen N, Petersen AH, van Luyn MJ. Macrophage depletion impairs wound healing and increases left ventricular remodeling after myocardial injury in mice. Am J Pathol. 2007;170:818–829. doi: 10.2353/ajpath.2007.060547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Vandivier RW, Henson PM, Douglas IS. Burying the dead: the impact of failed apoptotic cell removal (efferocytosis) on chronic inflammatory lung disease. Chest. 2006;129:1673–1682. doi: 10.1378/chest.129.6.1673. [DOI] [PubMed] [Google Scholar]