

Significance
Fast–protein-folding experiments and molecular-dynamics simulations can nowadays be compared on the same timescale. The comparison is limited by the lack of time-resolved structural information from experiment, which usually provides only information on global kinetics or stability. Structural data from microsecond folding experiments enables more rigorous testing of the detailed predictions available from simulations, which will in turn make future simulations more reliable. Here, we tackled this long-standing problem by measuring tertiary contact formation between three helices in a fast-folding protein. The folding of this protein is not as simple as it seems but matches up nicely with a long all-atom simulation trajectory that monitors multiple folding and unfolding events.
Keywords: fluorescence, helix bundle, protein folding, thermal denaturation, molecular dynamics
Abstract
Fast protein folding involves complex dynamics in many degrees of freedom, yet microsecond folding experiments provide only low-resolution structural information. We enhance the structural resolution of the five-helix bundle protein λ6–85 by engineering into it three fluorescent tryptophan–tyrosine contact probes. The probes report on distances between three different helix pairs: 1–2, 1–3, and 3–2. Temperature jump relaxation experiments on these three mutants reveal two different kinetic timescales: a slower timescale for 1–3 and a faster one for the two contacts involving helix 2. We hypothesize that these differences arise from a single folding mechanism that forms contacts on different timescales, and not from changes of mechanism due to adding the probes. To test this hypothesis, we analyzed the corresponding three distances in one published single-trajectory all-atom molecular-dynamics simulation of a similar mutant. Autocorrelation analysis of the trajectory reveals the same “slow” and “fast” distance change as does experiment, but on a faster timescale; smoothing the trajectory in time shows that this ordering is robust and persists into the microsecond folding timescale. Structural investigation of the all-atom computational data suggests that helix 2 misfolds to produce a short-lived off-pathway trap, in agreement with the experimental finding that the 1–2 and 3–2 distances involving helix 2 contacts form a kinetic grouping distinct from 1 to 3. Our work demonstrates that comparison between experiment and simulation can be extended to several order parameters, providing a stronger mechanistic test.
The mechanism of protein folding is one of the central questions in biological science (1). The first all-atom simulation to capture substantial protein-refolding dynamics, which lasted 1 μs, was published in 1998 (2). Since then, the timescales of protein folding achieved experimentally and computationally have met (3). Advances in computation now produce distributed (4) and single-trajectory (5) protein-folding simulations on the same nanosecond-to-millisecond timescale as the fastest folding experiments.
With rich computational data becoming available, experimental testing of simulations is now hampered by the difficulty of acquiring experimental structural data with microsecond or faster time resolution. Fast ensemble and single-molecule experiments commonly probe only one order parameter such as the fluorescence lifetime of a single tryptophan residue, a broad infrared (IR) spectral response, or one Förster resonant energy transfer (FRET) efficiency (3). Fortunately, a quantitative comparison of such experimental order parameters with simulations is now possible: for example, solvent-accessible surface area of tryptophan can serve as a proxy for experimentally detected fluorescence (6), or computed 2D-IR spectra can track secondary structure (7). Still, a single-order parameter, even when compared accurately, cannot capture the complexity of folding on a funneled energy landscape (3). It becomes essential to capture experimental information along several coordinates that are amenable to straightforward modeling. One way of achieving this is through the use of multiple probes in a single protein.
The problem of multiple probes reporting on several order parameters of protein folding has been addressed previously. For example, stopped-flow experiments have been combined with multiple fluorescent probes to study slow protein folding or membrane insertion processes (8, 9). Noninvasive equilibrium NMR experiments, including some coupled with molecular dynamics simulations, have evaluated two-state fits and provided high resolution folding landscapes by using multiple probe nuclei (10–12). Notable progress in multiprobe analysis of folding kinetics has been achieved by using nonnatural amino acid IR probes (13) in combination with T-jump relaxation spectroscopy (14, 15). Comparisons between fast protein folding kinetics probed by fluorescence and/or IR spectroscopy identified heterogeneous folding of trpzip2 (16), λ repressor (17), and α3D (18). Tryptophan fluorescence probes local solvent exposure, whereas the amide I IR fingerprint probes global backbone hydrogen bonding. In downhill or incipient downhill folders, a free-energy landscape with a single-order parameter cannot reproduce both fluorescence and IR results because downhill folding is defined by a rapid interconversion between various conformational subpopulations.
Here, we approached the problem of multiple structural coordinates for fluorescence-detected fast kinetics by designing new fluorescence-quenching mutants capable of detecting site-specific tertiary contact formation in the five-helix bundle λ6–85. The required conservative amino acid side-chain mutations fall between extrinsic dye labeling (e.g., FRET detection) and isotope labeling (IR spectroscopy) in their intrusiveness, so simulations will be critical for interpreting them. Our hypothesis, to be tested by comparison with simulation, is that the probes are sufficiently nonintrusive to reveal a common mechanism.
Many folding studies of λ6–85 have been performed by using the fluorescence of a tryptophan (Trp) engineered into position 22 of helix 1 (19), quenched by a tyrosine (Tyr) (20) engineered into position 33 of helix 2. Trp-22 and Tyr-33 form a contact pair in the crystal structure (21). When Tyr-33 is absent, Trp-22 fluorescence increases slightly upon folding; with Tyr-33 present, Trp fluorescence is strongly quenched upon folding.
We expand the use of Trp–Tyr pairs to probe contact formation between three helix pairs during the folding of λ6–85 (Fig. 1). In addition to helices 1–2 probed by Trp-22 and Tyr-33, we generate two new mutants whose fluorescence quenching links helices 1–3 and 2–3. These mutants are thermally stable α-helical proteins with high expression yield and strong Trp quenching by Tyr. Temperature jump measurements on the helix 1–2, 1–3, and 3–2 mutants allowed us to compare experimental kinetics with the corresponding autocorrelation analysis of long molecular-dynamics (MD) trajectories published previously (5). We find agreement between the experimental and computational interhelix contact formation in the helix triad 1–2–3, suggesting that a common mechanism underlies the three different mutants. Both experiments and computational analysis suggest that the free-energy landscape of λ6–85 includes a transiently populated trap at a free energy of about 3.5–4.5 kBT that involves helix 2, altering the kinetics of the 1–2 and 3–2 probes relative to the 1–3 probe. Our results show that the match between fast-folding experiments and MD simulations can advance in its structural resolution into the regime of secondary structural building blocks.
Fig. 1.

Construction of λ6–85 mutants. (A) Crystal structure of the template mutant λ12 (PDB ID code 3KZ3) with residues 22, 33, 51, and 69 highlighted in helices 1, 2, 3, and 4, respectively. Helices 1 through 4 in which mutations were made are shown in orange; the rest of the sequence is shown in gray. Arrows between pairs of residues and mutant names λij follow the coloring and naming scheme used throughout the paper. Mutants λij are named according to the helix pair contact probed. (B) Tryptophan–tyrosine (W-Y) distances calculated for six λ6–85 mutants. Error bars represent the SD from fluctuations of the protein structures in the 100 ns of native-state MD simulations (SI Appendix, Fig. S1 and Table S2). All sequences are in SI Appendix, Fig. S1.
Results
Design of Mutants.
Our goal was to engineer new mutants of λ6–85, such that several helix contact formations could be probed by Trp–Tyr fluorescence quenching. We started with the mutant Y22W, Q33Y, G46A, G48A. Its crystal structure is known [Protein Data Bank (PDB) ID code 3KZ3] (21). We name mutants according to the nomenclature “λij,” where the subscripts stand for the helices in which tryptophan and tyrosine residues are positioned. Thus, we call the above mutant because in this mutant Trp is in helix 1 and Tyr is in helix 2.
Fig. 1A illustrates the crystal structure of , with residues in positions 22, 33, 51, and 69 highlighted to indicate positions that were mutated in helices 1, 2, 3, and 4. We designed five additional mutants to probe other pairs of helices by Trp–Tyr contact (Fig. 1B and SI Appendix, Fig. S1). We mutated large hydrophobic amino acids to accommodate the probes, targeting Trp–Tyr distances <7 Å to obtain quenching upon folding of the protein. In helix 4, positions 62, 66, and 69 were considered for mutations. Position 66 was not pursued because a previous report indicated that such mutations can substantially decrease the thermal stability of the protein (22). Therefore, we tried mutating the remaining two hydrophobic-core–facing residues in positions 62 and 69. Mutations in helix 5 were not attempted because helix 5 is believed to be relatively unstructured in solution based on previously published mutational analysis and MD simulations (22–24). We ran 100-ns MD calculations of the proposed mutants in the native state (SI Appendix, Fig. S1) to ensure that the distances between probes would be short (<7 Å)—a necessary requirement for quenching of tryptophan fluorescence by tyrosine in the native state (Fig. 1B). Expression of mutants that showed larger interprobe distances in MD simulations, and λ42b, was attempted as nonquenching controls.
Expression and Validation of Mutants.
We attempted to express five new mutants of λ6–85 using standard protein overexpression in BL21 Escherichia coli cells (Materials and Methods and SI Appendix, Fig. S1). Images of acrylamide gels containing corresponding chromatography fractions of each mutant are shown in Fig. 2A. Mutants λ12, λ13, and λ32 showed a good expression yield of several milligrams of protein per liter of culture. Mutant λ42 had a 10× lower expression yield. The other two mutants, λ42b and λ14, did not show detectable expression.
Fig. 2.

Expression and thermodynamic validation of engineered mutants. (A) Corresponding fractions of the protein purification gels for each mutant showing the relative expression yield. The midpoint denaturation temperatures in °C as determined by fluorescence intensity analysis (D; SI Appendix, Table S1) and circular dichroism (CD) at 222 nm as a proxy for α-helical content (C; SI Appendix, Table S1) are also shown. (B) CD spectra of four λ6–85 mutants collected at 23 °C. Negative peaks at 209 and 222 nm are characteristic of α-helical content. Data are in SI Appendix, Fig. S2 and Table S1. (C) Equilibrium temperature denaturation of four λ6–85 mutants monitored by CD at 222 nm as a proxy for α-content. Solid lines are two-state thermodynamic fits, which were used to determine the midpoint denaturation temperatures CD-Tm in A. λ42 was not fitted. (D) Equilibrium temperature denaturation of four λ6–85 mutants as monitored by fluorescence intensity integrated from 290 to 450 nm. The traces are normalized to begin at unity and end at zero. Solid lines are two-state fits, which were used to determine the midpoint denaturation temperatures FL-Tm in A. An increase in fluorescence intensity (arrows) is characteristic of the disruption of the Trp–Tyr interaction. Data are in SI Appendix, Fig. S3 and Table S1. Linear baselines are clearly a good approximation over our temperature range.
Mutants with detectable expression were analyzed for secondary structure content by circular dichroism (CD) (Fig. 2B and SI Appendix, Fig. S2). All mutants showed a characteristic signature of α-helical secondary structure as evidenced by negative peaks at 209 and 222 nm. We used the CD peak at 222 nm to monitor the equilibrium unfolding of λ repressor mutants as a function of temperature (Fig. 2C). All mutants showed an unfolding transition except λ42. Note that λ42 also had the least helical CD signal (Fig. 2B). λ12 was most stable with a midpoint of unfolding at 70 ± 1 °C by CD (Fig. 2 A and C, and SI Appendix, Fig. S2 and Table S1). The other two mutants with high expression yield, λ13 and λ32, were also thermally stable with midpoint temperatures of 58 ± 1 and 60 ± 1 °C.
We also assessed protein thermal stability and quenching upon folding by fluorescence spectroscopy (Fig. 2D and SI Appendix, Fig. S3 and Table S1). λ12 was again the most stable mutant with a midpoint of unfolding at 66 ± 1 °C by fluorescence (Fig. 2 A and D, and SI Appendix, Fig. S3 and Table S1). λ13 and λ32 showed midpoint temperatures of 50 ± 1 and 46 ± 1 °C, respectively. Mutant λ42 showed the lowest midpoint temperature of 39 ± 1 °C. Only the most stable and well-expressed mutants (λ12, λ13, λ32) showed an increase in fluorescence intensity upon denaturation (Fig. 2D). This observation is expected because in the native state, the Trp–Tyr distance for these mutants is short (∼5 ± 1 Å in Fig. 1B) and tryptophan is quenched by tyrosine.
As a control, increased fluorescence intensity upon denaturation is not observed for λ42, a mutant with too large a Trp–Tyr distance for Dexter quenching (20) (∼10 Å; Fig. 1B). As mentioned above, λ42 also showed a lower expression yield, lower secondary structure content, and lower midpoint unfolding temperature compared with the other three mutants. Thus, only the helix 1–2–3 triad of mutants λ12, λ13, and λ32 was retained for further analysis.
Different Relaxation Times for Different Helix Pairs.
We measured the temperature jump relaxation (Materials and Methods) of the three mutants that showed fluorescence quenching upon folding. To compare mutants at similar melting temperatures, we adjusted the melting temperature of λ12 as measured by CD to a value similar to the other two mutants, λ13 and λ32, by using 2.2 M guanidine hydrochloride (GuHCl) (SI Appendix, Fig. S4). We call this sample λ12*. Temperature jumps of 8–9 °C were performed in a range close to the unfolding transition midpoint temperature for each protein [57 ± 1 °C for λ12*, 58 ± 1 °C for λ13, and 60 ± 1 °C for λ32]. Higher temperature data for λ12 without GuHCl extrapolates well to the λ12* data in Fig. 3A (SI Appendix, Figs. S6–S8). The λ12* trace in Fig. 3A does not include a 10% correction for the viscosity of GuHCl, which would make the blue trace even slightly faster than the green trace.
Fig. 3.
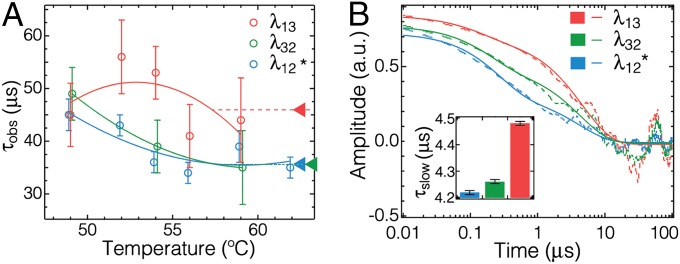
T-jump relaxation experiments and comparison with MD simulations. (A) Relaxation times of three λ6–85 mutants as a function of final T-jump temperature. λ12* was measured in 2.2 M GuHCl to reduce its stability to that of λ13 and λ32. Higher T measurements of λ12 without GuHCl extrapolate well to the data shown here (SI Appendix, Figs. S6–S8 and Tables S2 and S3). Parabolic curves are guides for the eye. Triangular pointers on the Right highlight the observed time constants at the approximate region of equilibrium transition midpoints for each mutant. (B) Autocorrelations from a single MD trajectory (dashed lines) and double-exponential fits (solid lines) of the Dexter-weighted distances δij between positions where Trp and Tyr are located in the experimental mutants. The slow phases are λ13: 4.48 ± 0.01 μs; λ32: 4.26 ± 0.01 μs; and λ12: 4.22 ± 0.01 μs (see SI Appendix, Fig. S10, for details, and analysis of unweighted distances Δij that produce the same ordering). The same trend as in experiment can be seen (Inset), although the simulated kinetics are faster by approximately a factor of 10.
The kinetic traces shown in SI Appendix, Figs. S6 and S7 were fitted to an exponential function FI(t) = A1exp(−t/τobs), where τobs is the observed relaxation time constant. Kinetics faster than 2 μs associated with activated protein population (downhill folding) were not fitted for the present purpose, and for high-concentration measurements (Materials and Methods), a minor slow phase A2exp(−t/τ2) was added to account for a small amount of protein aggregation (SI Appendix, Fig. S5) observed previously (25). All fitting parameters are listed in SI Appendix, Tables S2 and S3. The temperature dependence of the relaxation time τobs for the three mutants from final T-jump temperature ranging from 49 to 59 °C are shown in Fig. 3A. Our results show that λ13 has a slower observed relaxation time, whereas λ32 and λ12* (even without viscosity correction) have faster relaxation times, which are similar within the experimental error in the 50–60 °C range. Measurements over a wider temperature range and without denaturant (SI Appendix, Fig. S8) show that the λ12 relaxation could become the slowest at high temperature.
Comparison with MD Simulations.
We compared the results of kinetics experiments with a previously published single-trajectory full-atom MD simulation (λShaw in SI Appendix, Fig. S1, total of 161-μs simulation dataset courtesy of D. E. Shaw Research) (5). This simulation differs from our template protein λ12 by an additional D14A mutation and residues at the N and C terminus (SI Appendix, Fig. S1). Such single-trajectory MD simulations are commonly analyzed in terms of global order parameters such as the contact order (Q) or the root-mean-square deviation of α-carbons from the crystal structure (Cα-RMSD) (SI Appendix, Fig. S9).
Instead, we used the distance between sidechains of amino acids located at the positions of our experimental probes as three order parameters that are directly related to our experimental fluorescence quenching observables. To match computed and experimental observables as accurately as possible, the computed distances were weighted by the Dexter energy transfer efficiency responsible for Tyr–Trp quenching (20). This weighting function is monotonic with distance and does not affect the computed ordering (Fig. 3B and SI Appendix, Fig. S10); neither does varying the screening length from 4 to 7 Å. We calculated autocorrelation functions of the weighted distances as a function of time (Fig. 3B) to compare the ordering of relaxation times between trajectory calculations and relaxation experiments (Fig. 3A). In the linear response regime (where the fluctuation–dissipation theorem is valid) (26), such autocorrelations of trajectories (equilibrium data) correspond directly to the decay signal in a kinetic jump experiment (relaxation data) probing the same variable. We observed that the decay times of computed autocorrelation functions follow the same ordering as the experimentally measured kinetic traces (Fig. 3), grouped into a slower Δ13 distance and faster Δ12 and Δ23 distances near Tm. The slowest phases of the computed decays are about a factor of 10 faster than the experimental kinetics.
Discussion
Comparing fast protein folding experiments and MD simulations at a more detailed structural level is one of the current goals of the protein-folding field. Such comparisons can lead to the detailed mechanistic understanding of protein folding and misfolding, and they are important for further refinement of MD force fields to eventually make them mechanistically accurate. To facilitate such comparisons at higher resolution, microsecond folding experiments with more structural information content are needed. An example directly relevant here is the folding of λ12 monitored by global IR spectroscopy (amide I′ band, reporting on secondary structure) and local tryptophan fluorescence (reporting on the helix 1–2 contact via Trp–Tyr quenching) (17). We found that the two probes relax with different timescales.
To introduce more structural information from fluorescence measurements, we generated three mutants of λ6–85 probing all pairwise combinations of the helix triad 1–2–3 by Trp–Tyr Dexter quenching. In one helix, we substituted a hydrophobic side chain that had little solvent exposure by a tryptophan side chain, to ensure its partial burial and a potentially large change in its fluorescence. Into a neighboring helix, we engineered a contact quencher (Tyr) (20). The 100-ns simulations of all mutants were used to calculate the distance distribution between tryptophan and tyrosine (Fig. 1B). Based on the Trp–Tyr Dexter quenching properties (20), we expected that mutants with average distance ≤7 Å would quench, and longer distance mutants would not. This expectation was borne out by all mutants whose expression yield was sufficient to measure fluorescence quenching (Fig. 2). The fluorescence enhancement upon denaturation is thus an indicator of Trp–Tyr contact formation in the native state.
The variants do not exhibit the exact same thermal stability and identical secondary structure content (Fig. 2). These differences could influence the observed kinetics. However, the ranking of relaxation rates is robust by two different experimental approaches between 50 and 60 °C, a temperature range consistent with the MD simulation when accounting for a global stability shift of the simulated protein due to imperfect solvent modeling (5). In one experimental approach, GuHCl was used to bring the melting temperatures in line for a direct comparison (Fig. 3A), and in the other approach the higher melting mutant data were extrapolated to lower temperature in absence of GuHCl (SI Appendix, Fig. S8). In each case, the helix 1–3 probe has a slower relaxation time than the two helix 1–2 and 3–2 probes involving helix 2.
The MD simulation produces the same ranking of contact formation rate as experiment, albeit 10 times faster (Fig. 3). The lower solvent viscosity of the force field, also reported by others (27, 28), accounts for about a factor of 3. The origin of the remaining factor of 3–4 is not clear at present. Perhaps force field imperfections (e.g., collective behavior of water during the “drying” transition that forms the native state) are responsible; solvation effects are known to increase the computed Tm over the experimental value for λ repressor and other proteins (5).
Comparison of the computed helix 1–3 vs. 1–2 and 3–2 kinetics from the single all-atom simulation with the experiment yields the same order of rates, supporting the hypothesis that the perturbation induced by the probes does not affect the free-energy landscape of the protein under similar free-energy conditions (extrapolated, or achieved by denaturant). We hope that our data encourage long-trajectory simulations of mutants made as site-specific probes, so that such perturbations can be assessed directly. Computational expense is rapidly reaching a point where such direct comparisons are realistic.
The most probable order of events that eventually leads to complete folding/unfolding relaxation, at least as far as helices 1/2/3 are concerned, is as follows: helix 1–3 contact formation occurs more slowly, whereas helix 2 more frequently forms/dissolves contacts with the other two helices. This agreement of experiment and computation suggests that the all-atom trajectories can tell us about the microscopic origin of the difference between the contact probes. Therefore, we further analyzed the single-trajectory MD simulation that we used for autocorrelation analysis (Fig. 4). In particular, we plotted the simulated trajectory in the coordinate space of the three probe distances Δ12, Δ13, and Δ32 with respect to the crystallographic native state, such that Δ13 = Δ12 = Δ32 = 0 for the crystal structure (Fig. 4A and SI Appendix, Fig. S11, for a raw time traces plot).
Fig. 4.

Analysis of the folding MD simulations (161-μs simulation data sampled at 0.2 ns; courtesy of D. E. Shaw Research) (5). (A) Smoothed time traces of distances (with respect to the native-state values) between side chains of the simulated mutant where Trp and Tyr are located in λ12, λ13, and λ32. (Raw distance time traces are in SI Appendix, Fig. S11.) Pink background indicates high Q states (folded). (B) Histogram of populations (Left) and corresponding estimates of the free-energy profiles (Right) with respect to interhelical distances for the time trace in A; the three order parameters differ slightly. (C–E) Close-up of interhelical distances within the time trace in A. (F–H) Sample structures of (F) a trapped state (T) with high Q, short distance between helices 1 and 3 (Δ13), but long distances between helices 1 and 2 (Δ12) and helices 3 and 2 (Δ32); of (G) a native state (N) with high Q and short distances between all three probe pairs; and of (H) denatured structure (D) with low Q and long interhelical distances. (I) Time trace plotted in the coordinates of interhelical distances and color-coded by the Q values (color scale is on the Right). Three states are circled: the native state (N), where Q is high and all interhelical distances are native-like (i.e., close to the origin in this coordinate system); the denatured state (D), where Q is low and all interhelical distances are long; and the trap (T), where Q is high (Q > 0.6), Δ13 is short (<3 Å), but Δ12 and Δ32 are long (>5 Å). (J) Three-state Markov model based on the complete simulation data for the system in I (data are in SI Appendix, Tables S5–S7).
Fig. 4B shows the corresponding histograms of populations and also estimated potentials of mean force (PMFs) for the three order parameters. The mean first-passage times of folding on the three PMFs follow the trend captured by the autocorrelation functions (SI Appendix, Table S8). This result reinforces the notion that our chosen 1D order parameters self-consistently describe the folding process as captured by the MD simulations and experiments. Fig. 4 C–E shows highlights from the trajectory; in particular, Fig. 4C shows one of the many excursions that make Δ12 and Δ32 “faster” than Δ13 in the autocorrelation of Fig. 3B.
Fig. 4I shows the trajectory in the space of Δ12, Δ13, and Δ32 (color represents contact order Q; see SI Appendix for details.) The visualization in Fig. 4I allowed us to identify three distinct states: a native state (denoted N; structure shown in Fig. 4G), a denatured state (denoted D; sample structure shown in Fig. 4H), and a transiently populated trap (denoted T; structure shown in Fig. 4F). The trapped state showed a high, folded-like Q and short distance between helices 1 and 3 (Δ13), but a long distance between helix 2 and the other two helices (Δ32 and Δ12 > Δ13). Helix 2 is not properly docked in this otherwise native-like but short-lived trap (about 0.3-μs residence time in the example in Fig. 4C). The existence of a short-lived trap is consistent with a previous two-probe experimental study (17) of λ6–85, and with more recent computational results suggesting that λ6–85 is not a perfect two-state folder (23, 29). Recent pressure-jump experiments and simulations also show that helix 2 lags by ∼0.5 μs during the transition from U to N, and that traps exist during pressure-jump refolding (24). The short-lived trap is also consistent with previous single-probe downhill folding experiments on λ12 and its variants that show (km)−1 ∼ 1- to 2-μs transit times between N and D attributed to landscape roughness (=short-lived traps) in the folding free-energy surface (25). However, these single-probe analyses are intrinsically limited. Identification of transient nonnative states requires a significant separation of timescales with a single probe, which is not necessary when independent probes are used.
We used the MD simulation from D. E. Shaw Research to generate a three-state Markov model (Fig. 4J and SI Appendix, Tables S5–S7) based on the N, T, and D states observed in the simulation. A single major trap state is consistent with our experimental data (Fig. 3A). If the rates of λ12 and λ32 were also significantly different near Tm (three interconversion timescales), one would expect to observe at least one additional trap or intermediate state (four states). The transition matrix revealed that the native protein N is almost 10 times more likely to convert to the denatured state D directly without visiting trap T, and the denatured state D is 14 times more likely to convert back to the native state N before being trapped in T. The trap is thus a high free-energy state rarely visited from D and N. T lies at about kBTln(4.42/0.05) ∼ 4.5 kBT above the average free energy of D, and 3.4 kBT above the average free energy of N at the temperature sampled by the trajectory in Fig. 4. T is best characterized as a short-lived high-energy intermediate only occasionally populated in transit between the native and denatured states, and thus requiring two order parameters, as proposed by Bieri and Kiefhaber (30) for the much slower lysozyme folding process. SI Appendix, Tables S6 and S7 show that trapping is also very rarely observed when helices 1 or 3 are not properly docked, indicating further traps at free energies at least 9 kBT above the state N. When a protein like λ6–85 is biased toward downhill folding, such excited states are the source of free-energy landscape roughness that limits the molecular timescale (or transition state transit time) to ≥1 μs (21).
According to our Markov model, T lies neither on a highly populated parallel path, nor is it completely off the N–D path. Its accessibility from N is four times greater than its accessibility from D, but not much larger (as required for a strict off-pathway trap accessible only from the native state). This observation illustrates that “the glass is half empty,” when it comes to classifying nonnative states exclusively as traps, parallel pathways, or productive intermediates.
In conclusion, we have investigated more deeply the folding mechanism of λ6–85 with conservative mutants that probe contacts between three different helix pairs using Dexter fluorescence quenching. A nonquenching mutant experimentally confirmed the design. We analyzed single-trajectory full-atom MD simulations of λ6–85 to gain a microscopic understanding of the experimentally observed kinetics and discovered a native-like trap with helix 2 unraveled a few kBT above the native and denatured states. The trap is populated transiently from both the native and the denatured states, but to different extent. The T-jump experiments show that helix 2 undocks from 1 and 3 faster than 1 does from 3, consistent with a short-lived state without helix 2 properly folded into place. This result shows that multiprobe mapping of protein folding can be combined successfully with state-of-the-art MD simulations to gain a higher resolution picture of apparent two-state folding.
Materials and Methods
Protein Expression and Purification.
The gene of interest subcloned into a pET-15b vector was obtained from GenScript. BL21(DE3)-RipL cells (Agilent) were transformed with the plasmid and grown on selective media. The cells were then grown in liquid culture at 37 °C to OD600 of 0.6, and the cultures were induced with 1 mM isopropyl β-d-1-thiogalactopyranoside overnight at 23 °C. The cells were pelleted and lysed using sonication in a solution containing 1 mM phenylmethylsulfonyl fluoride for protease inhibition. The supernatant was filtered with 0.45-μm syringe filters and applied to a Ni:NTA column (Qiagen). The column was eluted using an imidazole gradient. Fractions containing the protein of interest were dialyzed against 50 mM K3PO4, pH 7.0, at 4 °C. Mutations were done using QuikChange site-directed mutagenesis (Stratagene). All chemicals were purchased from Sigma-Aldrich and used without further purification.
Equilibrium Experiments and Data Analysis.
CD experiments were done using a Jasco-715 spectropolarimeter (Jasco). Each measurement consisted of three accumulations of spectra, which were collected between 200 and 250 nm. The temperature was controlled using a Peltier element. The temperature was incremented by 3 °C starting with 5 °C and increasing to 95 °C. Protein concentration was 2.5 μM. Fluorescence experiments were done using a Cary Eclipse (Varian) spectrophotometer equipped with a Peltier heating stage. Tryptophan was excited at 280 nm, and fluorescence was collected between 290 and 450 nm. Temperature was incremented in the same way as in CD measurements. Protein concentration was 10 μM. Starna cuvettes (Starna Cells) were used for both measurements. Igor Pro (Wavemetrics) and Matlab (MathWorks) were used for data analysis. Fluorescence intensity was calculated by integrating the entire spectrum at a specific temperature: Itotal(T) = ∫I(λ,T)dλ. The mean wavelength was calculated according to the formula: 〈I(T)〉 = ∫ λ I(λ,T) dλ/∫I(λ,T) dλ. Two-state thermodynamic fits had the form: F(T) = [FD/(1 + Keq)] + [FNKeq/(1 + Keq)], where Keq = exp[−ΔG(T)/RT]; Fi = bi + mi(T − Tm); and ΔG(T) ≅ ΔG(Tm) + ΔS(Tm)Tm – ΔS(Tm)T. We note that a linear temperature dependence of Trp fluorescence intensity is an approximation used here for simplicity (10).
Kinetics Experiments and Data Analysis.
Temperature jumps were performed using an in-house built instrument, which was described in detail previously (31). Briefly, a sample consisting of the tested protein in 50 mM K3PO4 buffer at pH 7 was placed in a cuvette made out of a rectangular capillary (Vitrocom) welded on one side. The sample concentrations were 309, 373, and 50 μM, for , λ13, and λ32, respectively. Low concentration was used in λ32 because a significant amount of sample aggregation was visually observed during the measurements in high concentration. The equilibrium temperature of the sample was maintained using a water flow system. The temperature of the solution was suddenly jumped using a nanosecond pulse of a Q-switched Nd:YAG laser (Continuum). The fundamental wavelength of 1,064 nm of the laser was Raman-shifted to 1.9 μm using a 1-m-long chamber with hydrogen compressed to 300 psi. The beam was split using a 50% reflective mirror, and the sample was illuminated from both sides to achieve uniform heating. Tryptophan was excited every 12.5 ns using light centered at 280 nm from a frequency-tripled mode-locked Ti:sapphire laser (KMLabs). The repetition period of the Ti:sapphire laser (12.5 ns) puts a limit on the time resolution of our kinetics data even in the absence of noise. Fluorescence was collected at 90° angle using an optical waveguide (Oriel), filtered from incident radiation with a bandpass filter (B370; Hoya), and detected by a photomultiplier tube (R7400U-03; Hamamatsu). The signal was digitized with a period of 100 ps (10 GHz; 125 points per fluorescence decay) and a bandwidth of 2.5 GHz using an oscilloscope (DPO7254; Tektronix). The data were analyzed using Igor Pro (Wavemetrics) and Matlab (MathWorks). One-hundred kinetic traces were averaged at each temperature unless stated otherwise. The decays were normalized to yield a parameter χ(t) such that χ(t) = a1/(a1 + a2), where each fluorescence decay is represented as a linear combination of a decay before the temperature jump (f1) and a decay at the very end of the kinetic trace (f2), f = a1f1 + a2f2. Thus, every trace begins with χ(t = 0) = 1 and ends with χ(t = 1 ms) = 0. The resulting kinetic traces were then fit to a single- or a double-exponential function of the forms FI(t) = A exp(−t/τ) and FI(t) = A1 exp(−t/τ1) + A2 exp(−t/τ2), respectively.
MD Simulations.
The coordinates of wild-type protein were obtained from a crystal structure (PDB ID code 3KZ3; resolution of 1.64 Å). Mutants were prepared using the Mutator plugin of Visual Molecular Dynamics (VMD) software (32). Each system was minimized for 1,000 time steps, and equilibrium MD was performed for 100 ns using NAMD2 software (33) with the CHARMM27 force field (34), the TIP3P model of water (35), and the CMAP corrections (36) for proteins in the NPT ensemble. Langevin dynamics with a 0.5 ps−1 damping coefficient and the Langevin piston Nosé–Hoover procedure was used to maintain temperature and pressure to constant values (300 K and 1 atm, respectively) (37, 38). The particle mesh Ewald (PME) method (39) was used to calculate long-range electrostatic forces without truncation, using a grid density of 1 Å−3 . The van der Waals interaction cutoff value was 12 Å, whereas the integration time steps for bonded, nonbonded, and PME calculations were 2, 1, and 2 fs, respectively. Trajectory geometry analysis was performed using Tcl scripts within VMD (32).
D. E. Shaw Research MD Simulation Data Analyses.
We obtained the all-atom λ-repressor simulation dataset from D. E. Shaw Research. Simulations were performed by D. E. Shaw Research in the CHARMM22* force field, and the frames were saved every 200 ps (5). The simulations data were analyzed using scripts for VMD (32) and Matlab.
Supplementary Material
Acknowledgments
We thank D. E. Shaw Research for providing access to the simulation data. T.V.P. thanks Michael Hallock and Mahmoud Moradi for helpful and stimulating discussions. The 100-ns MD simulations were performed using the Lipid cluster at the School of Chemical Sciences Computer Center at the University of Illinois at Urbana–Champaign. T.V.P. is grateful for support from the Illinois Campus Research Board and the Extreme Science and Engineering Discovery Environment (Grant TG-MCB130112), which is supported by National Science Foundation Grant ACI-1053575. T.V.P. was a Faculty Fellow of the National Center for Supercomputing Applications when this work was completed. M.B.P. was a Howard Hughes Medical Institute International Student Research Fellow. This work was supported by Grant 2R01 GM093318-04 from the National Institutes of Health.
Footnotes
The authors declare no conflict of interest.
This article is a PNAS Direct Submission.
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1422683112/-/DCSupplemental.
References
- 1.Wolynes PG. Recent successes of the energy landscape theory of protein folding and function. Q Rev Biophys. 2005;38(4):405–410. doi: 10.1017/S0033583505004075. [DOI] [PubMed] [Google Scholar]
- 2.Duan Y, Kollman PA. Pathways to a protein folding intermediate observed in a 1-microsecond simulation in aqueous solution. Science. 1998;282(5389):740–744. doi: 10.1126/science.282.5389.740. [DOI] [PubMed] [Google Scholar]
- 3.Prigozhin MB, Gruebele M. Microsecond folding experiments and simulations: A match is made. Phys Chem Chem Phys. 2013;15(10):3372–3388. doi: 10.1039/c3cp43992e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Snow CD, Nguyen H, Pande VS, Gruebele M. Absolute comparison of simulated and experimental protein-folding dynamics. Nature. 2002;420(6911):102–106. doi: 10.1038/nature01160. [DOI] [PubMed] [Google Scholar]
- 5.Lindorff-Larsen K, Piana S, Dror RO, Shaw DE. How fast-folding proteins fold. Science. 2011;334(6055):517–520. doi: 10.1126/science.1208351. [DOI] [PubMed] [Google Scholar]
- 6.Samanta U, Bahadur RP, Chakrabarti P. Quantifying the accessible surface area of protein residues in their local environment. Protein Eng. 2002;15(8):659–667. doi: 10.1093/protein/15.8.659. [DOI] [PubMed] [Google Scholar]
- 7.Baiz CR, et al. A molecular interpretation of 2D IR protein folding experiments with Markov state models. Biophys J. 2014;106(6):1359–1370. doi: 10.1016/j.bpj.2014.02.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Tang J, et al. Using two fluorescent probes to dissect the binding, insertion, and dimerization kinetics of a model membrane peptide. J Am Chem Soc. 2009;131(11):3816–3817. doi: 10.1021/ja809007f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Sherman MA, Beechem JM, Mas MT. Probing intradomain and interdomain conformational changes during equilibrium unfolding of phosphoglycerate kinase: Fluorescence and circular dichroism study of tryptophan mutants. Biochemistry. 1995;34(42):13934–13942. doi: 10.1021/bi00042a027. [DOI] [PubMed] [Google Scholar]
- 10.Naganathan AN, Muñoz V. Determining denaturation midpoints in multiprobe equilibrium protein folding experiments. Biochemistry. 2008;47(26):6752–6761. doi: 10.1021/bi800336x. [DOI] [PubMed] [Google Scholar]
- 11.Naganathan AN, Muñoz V. Thermodynamics of downhill folding: Multi-probe analysis of PDD, a protein that folds over a marginal free energy barrier. J Phys Chem B. 2014;118(30):8982–8994. doi: 10.1021/jp504261g. [DOI] [PubMed] [Google Scholar]
- 12.Sborgi L, et al. Interaction networks in protein folding via atomic-resolution experiments and long-time-scale molecular dynamics simulations. J Am Chem Soc. 2015;137(20):6506–6516. doi: 10.1021/jacs.5b02324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ma J, Pazos IM, Zhang W, Culik RM, Gai F. Site-specific infrared probes of proteins. Annu Rev Phys Chem. 2015;66:357–377. doi: 10.1146/annurev-physchem-040214-121802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Davis CM, Cooper AK, Dyer RB. Fast helix formation in the B domain of protein A revealed by site-specific infrared probes. Biochemistry. 2015;54(9):1758–1766. doi: 10.1021/acs.biochem.5b00037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Nagarajan S, et al. Differential ordering of the protein backbone and side chains during protein folding revealed by site-specific recombinant infrared probes. J Am Chem Soc. 2011;133(50):20335–20340. doi: 10.1021/ja2071362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Yang WY, Gruebele M. Detection-dependent kinetics as a probe of folding landscape microstructure. J Am Chem Soc. 2004;126(25):7758–7759. doi: 10.1021/ja0493751. [DOI] [PubMed] [Google Scholar]
- 17.Ma H, Gruebele M. Kinetics are probe-dependent during downhill folding of an engineered lambda6-85 protein. Proc Natl Acad Sci USA. 2005;102(7):2283–2287. doi: 10.1073/pnas.0409270102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Liu F, et al. A one-dimensional free energy surface does not account for two-probe folding kinetics of protein alpha(3)D. J Chem Phys. 2009;130(6):061101. doi: 10.1063/1.3077008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ghaemmaghami S, Word JM, Burton RE, Richardson JS, Oas TG. Folding kinetics of a fluorescent variant of monomeric lambda repressor. Biochemistry. 1998;37(25):9179–9185. doi: 10.1021/bi980356b. [DOI] [PubMed] [Google Scholar]
- 20.Chen Y, Barkley MD. Toward understanding tryptophan fluorescence in proteins. Biochemistry. 1998;37(28):9976–9982. doi: 10.1021/bi980274n. [DOI] [PubMed] [Google Scholar]
- 21.Liu F, Gao Y-G, Gruebele M. A survey of lambda repressor fragments from two-state to downhill folding. J Mol Biol. 2010;397(3):789–798. doi: 10.1016/j.jmb.2010.01.071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Larios E, Pitera JW, Swope WC, Gruebele M. Correlation of early orientational ordering of engineered λ6–85 structure with kinetics and thermodynamics. J Chem Phys. 2006;323(1):45–53. [Google Scholar]
- 23.Bowman GR, Voelz VA, Pande VS. Atomistic folding simulations of the five-helix bundle protein λ(6−85) J Am Chem Soc. 2011;133(4):664–667. doi: 10.1021/ja106936n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Liu Y, Prigozhin MB, Schulten K, Gruebele M. Observation of complete pressure-jump protein refolding in molecular dynamics simulation and experiment. J Am Chem Soc. 2014;136(11):4265–4272. doi: 10.1021/ja412639u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Yang WY, Gruebele M. Folding at the speed limit. Nature. 2003;423(6936):193–197. doi: 10.1038/nature01609. [DOI] [PubMed] [Google Scholar]
- 26.Callen HB, Welton TA. Irreversibility and generalized noise. Phys Rev. 1951;83(1):34–40. [Google Scholar]
- 27.Shen My MY, Freed KF. Long time dynamics of Met-enkephalin: Comparison of explicit and implicit solvent models. Biophys J. 2002;82(4):1791–1808. doi: 10.1016/s0006-3495(02)75530-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Feller SE, Pastor RW, Rojnuckarin A, Bogusz S, Brooks BR. Effect of electrostatic force truncation on interfacial and transport properties of water. J Phys Chem B. 1996;100(42):17011–17020. [Google Scholar]
- 29.Liu Y, Strümpfer J, Freddolino PL, Gruebele M, Schulten K. Structural characterization of λ-repressor folding from all-atom molecular dynamics simulations. J Phys Chem Lett. 2012;3(9):1117–1123. doi: 10.1021/jz300017c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Bieri O, Kiefhaber T. Origin of apparent fast and non-exponential kinetics of lysozyme folding measured in pulsed hydrogen exchange experiments. J Mol Biol. 2001;310(4):919–935. doi: 10.1006/jmbi.2001.4804. [DOI] [PubMed] [Google Scholar]
- 31.Prigozhin MB, Gruebele M. The fast and the slow: Folding and trapping of λ6-85. J Am Chem Soc. 2011;133(48):19338–19341. doi: 10.1021/ja209073z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Humphrey W, Dalke A, Schulten K. VMD: Visual molecular dynamics. J Mol Graph. 1996;14(1):33–38, 27–28. doi: 10.1016/0263-7855(96)00018-5. [DOI] [PubMed] [Google Scholar]
- 33.Phillips JC, et al. Scalable molecular dynamics with NAMD. J Comput Chem. 2005;26(16):1781–1802. doi: 10.1002/jcc.20289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Vanommeslaeghe K, et al. CHARMM general force field: A force field for drug-like molecules compatible with the CHARMM all-atom additive biological force fields. J Comput Chem. 2010;31(4):671–690. doi: 10.1002/jcc.21367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Jorgensen WL, Chandrasekhar J, Madura JD, Impey RW, Klein ML. Comparison of simple potential functions for simulating liquid water. J Chem Phys. 1983;79(2):926–935. [Google Scholar]
- 36.Mackerell AD, Jr, Feig M, Brooks CL., 3rd Extending the treatment of backbone energetics in protein force fields: Limitations of gas-phase quantum mechanics in reproducing protein conformational distributions in molecular dynamics simulations. J Comput Chem. 2004;25(11):1400–1415. doi: 10.1002/jcc.20065. [DOI] [PubMed] [Google Scholar]
- 37.Martyna GJ, Tobias DJ, Klein ML. Constant pressure molecular dynamics algorithms. J Chem Phys. 1994;101(5):4177–4189. [Google Scholar]
- 38.Feller SE, Zhang Y, Pastor RW, Brooks BR. Constant pressure molecular dynamics simulation: The Langevin piston method. J Chem Phys. 1995;103(11):4613–4621. [Google Scholar]
- 39.Darden T, York D, Pedersen L. Particle mesh Ewald: An N⋅log(N) method for Ewald sums in large systems. J Chem Phys. 1993;98(12):10089–10092. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.