

Significance
Mutations in profilin 1 (PFN1) were recently shown to cause amyotrophic lateral sclerosis (ALS); however, little is known about the pathological mechanism of PFN1 in disease. We demonstrate that ALS-linked mutations cause PFN1 to become destabilized in vitro and in cells, likely through a mechanism that involves mutation-induced cavities within the protein core. Changes in protein stability due to disease-causing mutations can play a pivotal role across different disease mechanisms. The destabilized mutant-PFN1 species identified here can serve as an upstream trigger for either loss-of-function or gain-of-toxic-function mechanisms and thus emerges from these studies as a pertinent therapeutic target for the incurable disease ALS.
Keywords: amyotrophic lateral sclerosis, profilin 1, protein stability, X-ray crystallography, protein misfolding
Abstract
Mutations in profilin 1 (PFN1) are associated with amyotrophic lateral sclerosis (ALS); however, the pathological mechanism of PFN1 in this fatal disease is unknown. We demonstrate that ALS-linked mutations severely destabilize the native conformation of PFN1 in vitro and cause accelerated turnover of the PFN1 protein in cells. This mutation-induced destabilization can account for the high propensity of ALS-linked variants to aggregate and also provides rationale for their reported loss-of-function phenotypes in cell-based assays. The source of this destabilization is illuminated by the X-ray crystal structures of several PFN1 proteins, revealing an expanded cavity near the protein core of the destabilized M114T variant. In contrast, the E117G mutation only modestly perturbs the structure and stability of PFN1, an observation that reconciles the occurrence of this mutation in the control population. These findings suggest that a destabilized form of PFN1 underlies PFN1-mediated ALS pathogenesis.
Mutations in the profilin 1 gene (PFN1) were recently associated with both familial and sporadic forms of amyotrophic lateral sclerosis (ALS) (1, 2), an incurable and fatal neurodegenerative disease that primarily targets motor neurons (3). The etiology of sporadic ALS is poorly understood, whereas familial ALS is caused by inheritable genetic defects in defined genes such as PFN1 (3). PFN1 is a 15-kDa protein that is best known for its role in actin dynamics in the context of endocytosis, membrane trafficking, cell motility, and neuronal growth and differentiation (4). In addition to binding monomeric or G-actin, PFN1 also binds to a host of different proteins through their poly-l-proline motifs and to lipids such as phosphatidylinositol 4,5-bisphosphate (4, 5). However, little is known about the mechanism(s) associated with PFN1-mediated ALS pathogenesis. The observation that most ALS-linked PFN1 variants are highly prone to aggregation in mammalian cultured cells suggests that disease-causing mutations induce an altered, or misfolded, conformation within PFN1 (2). Protein misfolding is a hallmark feature of most neurodegenerative diseases, including ALS (3), and can contribute to disease through both gain-of-toxic-function and loss-of-normal-function mechanisms (6). Although mutations in PFN1 cause ALS through a dominant inheritance mode (2), there is some evidence supporting a loss-of-function mechanism for mutant PFN1. For example, ALS-linked mutations were shown to abrogate the binding of PFN1 to actin (2) and to impair the incorporation of PFN1 into cytoplasmic stress granules during arsenite-induced stress (7) in cultured cells. Moreover, ectopic expression of these variants in murine motor neurons led to a reduction in both axon outgrowth and growth cone size, consistent with a loss of function through a dominant-negative mechanism (2).
Although ALS-linked mutations were shown to induce PFN1 aggregation, the effect of these mutations on protein stability and structure has not been studied. Because the impact of disease-causing mutations on protein stability varies from protein to protein (8–10), these parameters must be determined empirically. Here, we demonstrate that certain familial ALS-linked mutations severely destabilize PFN1 in vitro and cause faster turnover of the protein in neuronal cells. To gain insight into the source of this mutation-induced instability, the 3D crystal structures for three PFN1 proteins, including the WT protein, were solved by X-ray crystallography. We discovered that the M114T mutation created a cleft that extended into the interior of PFN1. Further, we predict that the most severely destabilizing C71G mutation also creates a cavity near the core of the PFN1 protein, proximal to the cleft formed by M114T. Experimental mutations that create enlarged pockets or cavities are known to exert a destabilizing effect on the protein’s native conformation (11), and there are several examples of mutation-induced cavity formation occurring in nature and disease (12, 13). Interestingly, the variant predicted to be the least pathogenic according to recent genetics studies, E117G, was relatively stable and closely resembled the WT protein in every assessment performed herein (2, 14). These data implicate a destabilized form of PFN1 in ALS pathogenesis and call for therapeutic strategies that can stabilize mutant PFN1.
Results
ALS-Linked Mutations Destabilize PFN1 in Vitro.
To investigate the effect of ALS-linked mutations on the stability of PFN1, PFN1 proteins were expressed and purified from Escherichia coli and subjected to chemical and thermal denaturation analyses. A novel purification protocol that includes sequential cation-exchange and gel filtration chromatography steps was developed here and applied to all PFN1 variants (Materials and Methods). PFN1 C71G was found to be highly prone to aggregation in E. coli, consistent with observations that this variant exhibited particularly low solubility in mammalian cells (2), and therefore was isolated from inclusion bodies (Materials and Methods). The biochemical properties of PFN1 C71G purified from inclusion bodies are indistinguishable from PFN1 C71G purified from the soluble lysate of E. coli as determined by several assays (Fig. S1), providing confidence that PFN1 proteins purified by these two methods can be directly compared.
Fig. S1.

A comparison of PFN1 C71G purified from the soluble lysate of Escherichia coli vs. from inclusion bodies. (A) Equilibrium unfolding and (B) thermal denaturation curves (described in Fig. 1) for PFN1 C71G purified from the soluble lysate and inclusion bodies. The apparent melting temperature of PFN1 C71G purified from inclusion bodies (34.62 ± 0.05 °C) is the same as that purified from soluble lysate (34.60 ± 0.03 °C). (C) PFN1 C71G has similar affinities to poly-l-proline as determined by the binding assay described in Fig. 6 irrespective of whether this variant was purified from the soluble lysate or inclusion bodies.
To examine the stability of PFN1 proteins, fluorescence from tryptophans (W4 and W32) in PFN1 WT and ALS-linked variants was measured as a function of increasing urea concentration (Fig. 1A). To ensure reversibility, the reciprocal analysis was also performed, where denatured PFN1 proteins in urea were refolded upon dilution with buffer (Fig. S2 A–E). Only one transition was observed between the folded or native (N) and unfolded (U) states for all PFN1 proteins, indicative of a two-state (NΔU) unfolding mechanism. This two-state unfolding model was further substantiated with an unfolding study of two PFN1 proteins (WT and M114T) using CD spectroscopy (Fig. S2F). The following thermodynamic parameters were determined by fitting the fluorescence data to a two-state folding model: apparent ΔG°, the free energy of folding; m, the denaturant dependence of ΔG°; and Cm, the midpoint of the unfolding transition (Table 1). Both ΔG° and Cm were reduced for ALS-linked variants relative to PFN1 WT, particularly for the PFN1 variants C71G, M114T, and G118V, indicating these variants are severely destabilized compared with PFN1 WT (Fig. 1A and Table 1). Differential scanning fluorimetry (DSF) with SYPRO Orange, a fluorescent indicator of hydrophobic regions exposed upon protein unfolding, was used next to determine the apparent melting temperature, Tm, for all PFN1 proteins used in this study (15). Consistent with the chemical denaturation results, all ALS-linked variants except E117G exhibited a Tm that was at least 10 °C lower than WT (Fig. 1B and Table 1). Based on the denaturation studies, C71G emerges as the most destabilizing mutation in the context of PFN1, whereas the E117G mutation has a relatively modest impact on PFN1 stability.
Fig. 1.
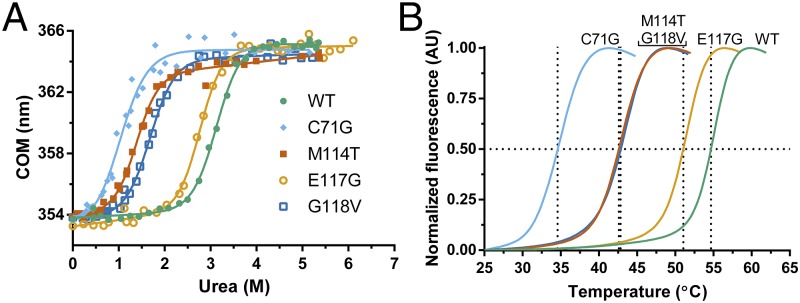
ALS-linked mutations destabilize PFN1. Chemical and thermal denaturation studies reveal that ALS-linked variants C71G, M114T, and G118V, but not E117G, are severely destabilized relative to PFN1 WT. (A) Equilibrium unfolding curves for PFN1 WT and ALS-linked variants generated by measuring the intrinsic tryptophan fluorescence of the indicated protein equilibrated in increasing concentrations of urea. Data were processed to obtain the center of mass (COM) of the emission spectrum and then fit to a two-state model for protein folding. The resulting fits are displayed as solid lines. The corresponding thermodynamic parameters obtained from the fitted data are shown in Table 1. (B) Thermal denaturation profiles of PFN1 proteins measured by SYPRO Orange fluorescence as a function of increasing temperature were used to determine the apparent Tm, which is the temperature corresponding to 0.50 fluorescence signal as denoted by the intersection of the dashed lines for each curve.
Fig. S2.

All PFN1 variants unfold by a two-state process. (A–E) PFN1 variants denatured in urea were refolded by diluting the urea. The final concentration of PFN1 in each sample was 10 μM and tryptophan fluorescence was used to monitor folding. The equilibrium transition regions overlay closely for the unfolding and refolding curves, indicating that the unfolding reaction is reversible. Filled and open circles represent unfolding and refolding, respectively. (F) The two-state unfolding of PFN1 observed by intrinsic fluorescence (data from Fig. 1A; Fluor) was verified by CD measurements for PFN1 WT and M114T. The concentration of protein used was 2 μM and 10 μM for tryptophan fluorescence and CD measurements, respectively. The y axis on the left is the mean residue ellipticity at 220 nm (MRE220) obtained from CD experiments, whereas the y axis on the right reflects the change in the COM (as shown in Fig. 1). The thermodynamic parameters obtained by fitting the CD data agree well with those obtained from the fluorescence data (Table 1) and are as follows: for WT ΔG° = 7.16 ± 0.11 kcal⋅mol−1, m = 2.36 ± 0.04 kcal⋅mol−1⋅M−1, Cm = 3.03 ± 0.07 M; for M114T ΔG° = 4.35 ± 0.10 kcal⋅mol−1, m = 2.95 ± 0.06 kcal⋅mol−1⋅M−1, Cm = 1.47 ± 0.05 M.
Table 1.
Summary of experimental stability and binding measurements for PFN1 variants
| Variant | Equilibrium unfolding (NΔU)* | Tm,† °C | Binding to poly-l-proline†,‡ Kd, μM | |||
| ΔG°, kcal⋅mol–1 | m, kcal⋅mol–1⋅M–1 | Cm, M | Protein alone | + 4 mM proline | ||
| WT | 7.04 ± 0.49 | 2.25 ± 0.16 | 3.13 ± 0.31 | 54.68 ± 0.04 | 57.25 ± 0.03 | 463 ± 26 |
| C71G | 1.89 ± 0.70 | 1.95 ± 0.40 | 0.97 ± 0.41 | 34.60 ± 0.03 | 39.96 ± 0.03 | 687 ± 77 |
| M114T | 3.51 ± 0.40 | 2.51 ± 0.24 | 1.40 ± 0.21 | 42.62 ± 0.03 | 46.52 ± 0.02 | 572 ± 23 |
| E117G | 6.90 ± 0.74 | 2.49 ± 0.26 | 2.77 ± 0.42 | 51.05 ± 0.04 | 53.78 ± 0.03 | 407 ± 27 |
| G118V | 3.70 ± 0.44 | 2.20 ± 0.23 | 1.68 ± 0.26 | 42.84 ± 0.04 | 46.92 ± 0.04 | 397 ± 40 |
Errors are shown as SD.
Errors are shown as SE.
Kd values are reported in terms of proline residues.
ALS-Linked PFN1 Exhibits Faster Turnover in a Neuronal Cell Line.
The turnover rate for proteins with destabilizing mutations is often faster relative to their WT counterparts, generally because destabilized proteins are misfolded and targeted for degradation by the cellular quality control machinery (16). To determine whether the results of our in vitro denaturation studies extend to a cellular environment, V5-tagged PFN1 variants were transiently transfected into human neuronal SKNAS cells, and PFN1 turnover was assessed by tracking V5-PFN1 protein expression over a 12.5-h time course in the presence of cycloheximide. At the start of the experiment (t = 0 of the cycloheximide time course), all V5-tagged PFN1 variants were expressed at similar levels except that V5-PFN1 C71G, M114T, and G118V partitioned into the insoluble fraction (Fig. 2 A and B) as reported previously (2). The turnover of both PFN1 C71G and M114T occurred significantly faster than that of PFN1 WT. As early as 2.5 h, the majority of PFN1 C71G and M114T within the soluble fraction had already degraded (Fig. 2 A and C). This decrease in soluble PFN1 content was not simply due to further PFN1 aggregation, which could confound our analysis, as evidenced by the concomitant clearance of PFN1 from the insoluble fraction at the early time points of cycloheximide exposure (Fig. 2B). The faster turnover of PFN1 C71G and M114T in cells closely correlates with their reduced stabilities in vitro, confirming the destabilizing effect of the C71G and M114T mutations. We note that the turnover of PFN1 C71G was faster in the soluble fraction compared with the insoluble fraction (Fig. S3), likely because clearance of insoluble cellular aggregates by the quality control machinery is less efficient compared with the turnover of smaller, soluble species (17). Although PFN1 G118V was destabilized to a similar degree as M114T in vitro, the turnover of this variant within the soluble fraction seemed slower in cells (Fig. 2C), which may reflect a stabilizing effect of other proteins and/or factors that interact with PFN1 in the cellular milieu (4), or that this variant is not properly handled by the quality control machinery in the cell. In fact, we detected a low level of insoluble PFN1 G118V that persisted throughout the 12.5-h time course (Fig. 2B and Fig. S3).
Fig. 2.

ALS-linked PFN1 variants exhibit faster turnover in a neuronal cell line. SKNAS cells transiently transfected with V5-PFN1 constructs were treated with cycloheximide (CHX) for up to 12.5 h, during which time lysates were collected and probed by Western analysis with a V5-specific antibody to assess the rate of PFN1 turnover in cells. (A and B) A representative Western blot analysis of soluble and insoluble fractions from cell lysates demonstrates a decrease in V5-PFN1 protein with time. GAPDH serves a loading control for the soluble fraction. (C) Densitometry analysis of A reveals that the turnover of PFN1 C71G and M114T is significantly faster than that of PFN1 WT. Statistical significance was determined using a two-way ANOVA followed by a Tukey’s post hoc analysis (*P < 0.05, **P < 0.01, #P < 0.0001). Error bars represent SEM. WT and E117G, n = 3; G118V, M114T and C71G, n = 4 independent experiments.
Fig. S3.

The turnover of insoluble PFN1 in SKNAS cells. The experiment was carried out as described in Fig. 2, and a representative Western blot analysis of the insoluble fraction is shown in Fig. 2B. The data above reflect the densitometry results from an average of n = 2 (M114T) or n = 3 (C71G and G118V) independent experiments and error bars represent SEM. Each sample was normalized to the PFN1 C71G band corresponding to “time 0.” The turnover of C71G within the insoluble fraction was slower relative to C71G within the soluble fraction (compare this graph to that in Fig. 2C). There was relatively less M114T and G118V in the insoluble fraction compared with C71G, and the small fraction of insoluble G118V persisted throughout the experimental time course.
ALS-Linked Mutations Induce a Misfolded Conformation Within PFN1.
We reasoned that ALS-linked variants must undergo some degree of structural or conformational change to account for their destabilization. However, ALS-causing mutations did not perturb the secondary structural elements of PFN1 as determined by CD spectroscopy (Fig. S4), and the fact that similar m values were determined for all PFN1 variants by the urea denaturation analysis suggested these proteins adopt similar tertiary structures as well (Table 1) (18). To probe further for potential structural differences between PFN1 WT and ALS-linked variants, these proteins were subjected to native gel electrophoresis, a biochemical technique capable of detecting conformational differences between misfolded variants and their WT counterparts (19). PFN1 WT and E117G migrated predominately as single, distinct bands with similar mobility, whereas multiple bands of slower mobility were observed for PFN1 variants C71G, M114T, and G118V (Fig. S5A). The slower mobility bands likely reflect the larger hydrodynamic volume due to partial unfolding of these variants. In addition, PFN1 C71G, M114T, and G118V produced relatively large-molecular-weight species that were retained in the stacking gel and unable to electrophorese through the separating native gel but were resolublized under conditions used for the denaturing gel (Fig. S5A). Analytical size-exclusion chromatography revealed that all PFN1 proteins eluted as expected for soluble, monomeric PFN1 (Fig. S5 B–G). However, despite equal loading of PFN1 proteins onto the analytical size-exclusion column, the peak area corresponding to soluble monomer PFN1 is reduced for ALS-linked variants, particularly for the most aggregation-prone variant, C71G. These data are consistent with a loss of soluble monomer PFN1 in the form of insoluble species that cannot pass through the analytical size-exclusion column filter.
Fig. S4.

ALS-linked PFN1 variants retain the same secondary structure as PFN1 WT. (A–D) Far UV CD spectra for the indicated PFN1 variant (10 μM) overlaid with CD spectrum for PFN1 WT (10 μM).
Fig. S5.

Analysis of PFN1 proteins by native page and analytical size-exclusion chromatography. (A) PFN1 proteins (10 μg) were subjected to native (Top) or denaturing (Bottom) gel electrophoresis and detected with Coomassie Brilliant Blue stain. The mobility of native PFN1 WT is indicated. PFN1 E117G migrates with a slightly faster mobility than PFN1 WT owing to the addition of a negatively charged amino acid. Misfolded ALS-linked PFN1 variants migrate with slower mobility and form aggregated species that are retained in the stacking gel. This gel is representative of n = 2 experiments using proteins from different purification preparations. (B–F) The indicated PFN1 protein (40 μg) was subjected to analytical size-exclusion chromatography using a Superdex 75 column. A single peak corresponding to the expected elution volume (∼15 mL) for monomeric PFN1 was detected for all PFN1 proteins. The experiments were carried out in duplicate for each variant, indicated by solid (n = 1 experiment) and dashed (n = 2 experiment) lines. The average relative peak area ± the SD is indicated to the right of each curve. Despite equal sample loading, the peak area of PFN1 C71G and M114T is lower than that of WT (within error), consistent with a reduced level of soluble protein for these ALS-linked variants. (G) An overlay of B–F for the n = 1 experiment demonstrates a similar elution profile for all PFN1 proteins.
A Source of Mutation-Induced Destabilization Revealed by X-Ray Crystallography of PFN1.
Crystal structures of PFN1 proteins were determined to identify regions within mutant PFN1 that are conformationally distinct from PFN1 WT at atomic resolution. PFN1 WT, E117G, and M114T produced crystals that diffracted at relatively high resolution (∼2.2 Å; Table S1). The 3D structure of human PFN1 WT agrees well with previously determined structures (20–22). PFN1 WT and E117G crystallized in the same space group, C121, whereas M114T crystallized in the P6 space group, with two molecules (designated as chains A and B) in the asymmetric unit (Table S1).
Table S1.
Crystallographic and refinement statistics of human PFN1 structures
| Statistics | WT | E117G | M114T |
| Resolution, Å | 2.160 | 2.170 | 2.230 |
| Space group | C121 | C121 | P6 |
| a, Å | 74.26 | 73.65 | 81.69 |
| b, Å | 31.84 | 31.71 | 81.69 |
| c, Å | 61.02 | 60.54 | 65.35 |
| Α, ° | 90 | 90 | 90 |
| Β, ° | 122.66 | 122.03 | 90 |
| Γ, ° | 90 | 90 | 120 |
| Z | 1 | 1 | 2 |
| Rmerge, % linear | 0.075 | 0.036 | 0.147 |
| I/sigma | 13.3 | 12.2 | 12.4 |
| Completeness, % | 99.28 | 99.49 | 99.58 |
| Total no. of reflections | 20,783 | 16,453 | 76,801 |
| No. of unique reflections | 6,416 | 6,422 | 12,156 |
| Rfactor, % | 0.2159 | 0.1965 | 0.1952 |
| Rfree, % | 0.2469 | 0.2139 | 0.2383 |
| RMSD in bond lengths, Å | 0.002 | 0.003 | 0.003 |
| RMS angle, ° | 0.62 | 0.67 | 0.61 |
| Temperature, °C | −80 | −80 | −80 |
| Residues missing | 1, 2, 57–62, 92–96 | 1, 2, 59–62, 81,82, 93–95, 140 | |
| Chain A | 1, 93–97 | ||
| Chain B | 1, 13, 91–97 | ||
| PDB ID code | 4X1L | 4X1M | 4X25 |
Residues 22–36, 46–52, 101–105, 112–120, and 125–128 within PFN1 were used for Cα superimposition of the four molecules (PFN1 WT, M114T chains A and B, and E117G). In agreement with the biochemical analyses described above (Table 1 and Fig. S4), the secondary and tertiary structures of all three PFN1 proteins, including chains A and B of M114T, are highly similar (Fig. 3). Although the space groups for PFN1 WT and M114T crystals were different, we calculated the double difference plots between these and the other PFN1 structures to get a sense for structural perturbations potentially induced by the ALS-linked mutations. Double difference plots were constructed by calculating the distances between all of the Cα atoms in PFN1 WT and an ALS-linked variant separately, and then plotting the difference of the difference between PFN1 structures as described previously (23). Virtually no structural deviations were observed between PFN1 WT and E117G, whereas moderate differences were detected between WT and M114T (Fig. S6).
Fig. 3.

Superimposition of the crystal structures for PFN1 WT, E117G, and M114T. (A and B) The secondary and tertiary structures for PFN1 WT (green), E117G (mustard), M114T chain A (pink), and B (red) are highly superimposable. For each structure, sticks and spheres denote the side chains and van der Waals radii, respectively, for residues at position 114 and 117. Residue 117 is located within a solvent-exposed flexible loop that has no discernible secondary structure, whereas Met114 is located within a β-sheet toward the interior of the protein. (B) A zoomed cartoon representation showing residues within 4 Å of residue 114. The side chains of these residues are indicated as sticks with nitrogen, oxygen, and sulfur atoms indicated in blue, red, and yellow, respectively. The van der Waals radii of the atoms comprising residue 114 are reduced upon mutation of methionine (green and mustard structures) to threonine (red and pink structures).
Fig. S6.

Structural changes induced by the M114T mutation revealed in double difference plots. Double different plots (Left) of WT vs. E117G (A), WT vs. M114T chain A (B), WT vs. M114T chain B (C), and M114T chains A vs. B (D). The Avg-Abs-DD values are plotted as a function of residue number for each structural comparison (Middle); these plots provide an indication for residues that undergo a structural change between the proteins that are being compared. Residues with Avg-Abs-DD values of 0.3 Å or greater are plotted onto the structure (Right) of PFN1 WT (A–C) and PFN1 M114T chain A (D) in green. Residues not used in this analysis are colored black.
Next we sought to determine whether these moderate structural changes between PFN1 WT and M114T mapped to regions involved in PFN1 function, namely to residues that make contact with actin (24–31) or poly-l-proline (21, 22, 24, 32, 33). The ternary complex comprised of PFN1 WT, actin, and the poly-l-proline peptide derived from vasodilator-stimulated phosphoprotein (VASP) (21) (PDB ID code 2PAV) is shown in Fig. 4. Residues with the highest (0.3 Å or greater) average of absolute double difference (Avg-Abs-DD) values between PFN1 WT and M114T chain B (Fig. S6C) were mapped onto PFN1 WT (Fig. S7). PFN1 M114T chain B was used for this and all subsequent structural comparisons because chain B had lower B factors compared with chain A (Fig. S8). Indeed, several PFN1 residues that reportedly make contacts with actin (V119, H120, G122, and K126) and poly-l-proline (W4, Y7, H134, and S138) also have relatively high Avg-Abs-DD values (Fig. S7).
Fig. 4.

Structure of actin–PFN1–VASP peptide ternary complex with the actin and poly-l-proline binding residues mapped on PFN1. The X-ray structure of the PFN1 WT (gray)–actin (blue)–poly-l-proline peptide (gold) complex (PDB ID code 2PAV) is shown. Residues reportedly involved in actin binding (V61, K70, S72, V73, I74, R75, E83, R89, K91, P97, T98, N100, V119, H120, G122, N125, K126, Y129, and E130) and poly-l-proline binding (W4, Y7, N10, A13, S28, S30, W32, H134, S138, and Y140) are highlighted in blue and gold, respectively. The sites of ALS-linked mutations investigated in this study are highlighted and labeled in black with side chains displayed as black sticks. Residues involved in actin or poly-l-proline binding that also exhibit Avg-Abs-DD values of 0.3 Å or greater between PFN1 WT and M114T chain B (W4, K126, and S138) are labeled in black (the remaining residues that fulfill this criteria are shown in Fig. S7).
Fig. S7.

Actin and poly-l-proline binding residues exhibit relatively high double difference values. Residues that have Avg-Abs-DD values of 0.3 Å or greater that are also engaged in actin binding (V119, H120, G122, and K126) or poly-Pro binding (W4, Y7, H134, and S138) are mapped onto the structure of PFN1 WT in magenta. All other residues with Avg-Abs-DD values of 0.3 Å or greater are highlighted in green. Residues with Avg-Abs-DD values between chain A and chain B of M114T 0.3 Å or greater (Fig. S6D) were excluded from this analysis. Residues not used in this analysis are colored black.
Fig. S8.

The calculated α-carbon B factors for all PFN1 structures. Cartoon representations of WT (A), E117G (B), and M114T chains A (C) and B (D). Residues are colored according to the α-carbon B factors using the scale shown at the bottom. The average α-carbon B factor for WT, E117G, and M114T chains A and B structures are 30.52, 22.94, 29.47, and 27.33, respectively. Because the average B factor is higher for M114T chain A, M114T chain B was used for structural analyses unless otherwise noted.
To assess whether these mutation-induced structural changes are sufficient to alter the normal binding interactions of PFN1, we first monitored changes in the intrinsic tryptophan fluorescence of PFN1 as a function of poly-l-proline peptide concentration (Fig. 5A). Our results revealed that the effect of ALS-linked mutations on the PFN1-poly-l-proline interaction was modest, because the apparent dissociation constants (Kd) were within twofold for all PFN1 proteins in this study (Table 1). In fact, excess concentrations of poly-l-proline effectively stabilized all PFN1 proteins as determined by DSF, with the largest increase in Tm observed upon poly-l-proline peptide binding to C71G (Fig. 5B and Table 1). Next, we measured the binding capacity of our PFN1 proteins for G-actin by comparing their concentration-dependent abilities to suppress spontaneous polymerization of pyrenyliodoacetamide‐labeled actin monomers (34). This assay is based on the fact that PFN1 binds G-actin and inhibits actin nucleation in the absence of formins (34). As expected, increasing concentrations of recombinant PFN1 WT reduced the rate of actin polymerization, whereas the H120E variant that exhibits impaired binding to actin failed to suppress actin polymerization to the same extent (Fig. 6). Of the four ALS-linked variants, only G118V was defective in suppressing actin polymerization, which was most apparent at the highest concentration of PFN1 used in this assay, although this effect did not reach statistical significance (Fig. 6). These data argue against a general mechanism for PFN1-mediated ALS pathogenesis that involves impaired direct binding between PFN1 and either poly-l-proline or actin.
Fig. 5.
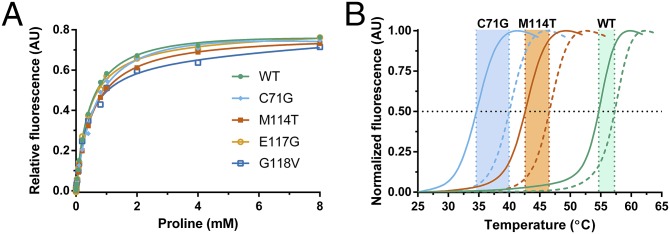
ALS-linked PFN1 variants retain the ability to bind poly-l-proline. (A) Binding of PFN1 to the poly-l-proline peptide was monitored by measuring the intrinsic tryptophan fluorescence of the indicated PFN1 protein as a function of increasing peptide concentration. The data points were fit using a one-site total binding model in GraphPad Prism and the apparent dissociation constants (Kd) obtained from the fit are shown in Table 1. Note that the concentration of the peptide is reported in terms of [proline] because the peptide stock is supplied as a mixture of poly-l-proline species (Materials and Methods). (B) DSF was performed as described in Fig. 1B in the presence (dashed lines) and absence (solid lines) of 4 mM proline. The presence of proline increases the Tm for all PFN1 proteins used in this study (Table 1), as illustrated here for WT, C71G, and M114T.
Fig. 6.

The binding of PFN1 proteins to G-actin. Polymerization of monomeric rabbit muscle actin (3 μM, 5% pyrene-labeled) was monitored in the presence of increasing concentrations of WT or ALS-linked PFN1 variants and used to derive relative rates of polymerization (n = 3). The variant H120E, which is impaired in binding to actin, fails to suppress spontaneous actin polymerization as effectively as WT PFN1. Although G118V is relatively weak in suppressing actin polymerization, the data did not reach statistical significance. Statistical significance was determined using a two-way ANOVA followed by a Tukey’s post hoc analysis. **P ≤ 0.01 for WT vs. H120E at 7 μM concentration. No other significant comparisons with WT were obtained. Other significant comparisons included C71G vs. H120E and E117G vs. H120E (P ≤ 0.05) at 7 μM concentration. Error bars represent SD.
Importantly, the X-ray crystal structures reveal a possible mechanism by which ALS-linked mutations destabilize PFN1. Residues Thr90, Met114, and Gln18 contribute to the formation of a surface exposed pocket that was detected using SiteMap (Fig. 7). Mutation of methionine to threonine at position 114 increased the size of this pocket, thereby forming a cleft, because the residues nearby failed to rearrange and compensate for the loss of van der Waals contacts (Fig. 7B). This cleft is expected to exert a destabilizing effect on the native conformation of PFN1 owing to this loss of van der Waals contacts and the reduced hydrophobicity of the threonine side chain relative to that of methionine (11). Moreover, hydrophobic residues that are otherwise buried in the PFN1 WT structure were exposed by the cleft in the PFN1 M114T structure (Fig. 7 and Fig. S9). To investigate the potential impact of the C71G mutation on PFN1 structure, the cysteine side chain of residue 71 was removed to mimic a glycine amino acid in the PFN1 WT structure using PyMOL. Interestingly, this mutation is predicted to form a void in the core of the protein that partially overlaps with the cleft observed in the PFN1 M114T crystal structure (Fig. 7B). Analysis using PyMOL and SiteMap suggest that, unlike the solvent-accessible WT and the M114T pocket, the proposed C71G void is buried within the core of the protein. Solvent-inaccessible voids have a more destabilizing effect than solvent-exposed cavities (11, 35), providing an explanation for why the C71G mutation is more destabilizing than M114T (Fig. 1).
Fig. 7.

The M114T mutation causes a surface-exposed pocket to expand into the core of the PFN1 protein. (A) Residues are depicted as described in Fig. 3. The van der Waals radii of residues 90, 114, and 18 are in contact in the PFN1 WT structure (Top). These contacts are reduced by the M114T mutation (Bottom) owing to the smaller size of threonine, leading to an enlargement of the surface-exposed pocket. (B) PFN1 WT is shown with a transparent surface and the secondary structure is shown in cartoon representation. The surface pocket volume for PFN1 WT (green) and the cleft volume for PFN1 M114T chain B (red) are depicted as opaque surfaces and were generated using SiteMap. The predicted cavity (blue) for PFN1 C71G (generated using PyMOL) overlays with the M114T void, and unlike the WT and M114T volumes, is not surface-exposed. The insets (Right) show the aforementioned voids for WT (Top), M114T chain B (Middle), and C71G (Bottom).
Fig. S9.

Electrostatic surface potential (ESP) of PFN1 WT and PFN1 M114T. A comparison of the ESP for PFN1 WT (A) and M114T (B) around the surface pocket (for WT) and cleft (for M114T) shown in Fig. 7. Comparison of the ESP was calculated using Maestro (Schrödinger, LLC). The Red_White_Blue color scheme was used to depict the ESP of both surfaces, where red denotes negative, blue denotes positive, and white denotes neutral ESP. The minimum and maximum values are −0.12 and 0.12, respectively. The cleft (boxed region in B) formed by M114T exposes a deeper pocket comprised of hydrophobic residues that would otherwise be buried beneath the surface-exposed pocket (boxed region in A) in PFN1 WT.
Discussion
Here we show that ALS-linked mutations severely destabilize (Fig. 1) and alter the native protein conformation (Fig. 3) of PFN1. Changes in protein stability owing to disease-causing mutations, whether these mutations stabilize or destabilize the protein, are thought to play a pivotal role in various disease mechanisms (13). In the context of ALS, disease-linked mutations destabilize Cu,Zn-superoxide dismutase (SOD1) (9), but instead hyperstabilize TAR DNA-binding protein 43 (TDP-43) (8, 10, 36). These findings underscore the importance of defining the toxic properties of disease-linked proteins, thereby directing the rational design of therapeutic strategies against those offending proteins (3).
Our X-ray crystal structures of PFN1 proteins illuminate a probable source of mutation-induced destabilization. An enlarged surface pocket, or void, forms as a result of the M114T mutation (Fig. 7). The destabilizing effect of similar voids has been demonstrated using a systematic site-directed mutagenesis approach with lysozyme and is thought to arise from a loss of hydrophobic interactions (11, 35). Examples of mutation-induced cavity formation and destabilization have also been observed in nature (13). Interestingly, modeling the removal of the cysteine side chain at position 71 creates an internal cavity that is predicted to partially overlap the cleft formed by M114T, raising the intriguing possibility that both mutations destabilize PFN1 through a common mechanism that involves the loss of hydrophobic and van der Waals contacts within the same region of PFN1 (Fig. 7). Because G118V is located within a solvent-exposed flexible loop, it is difficult to predict whether this mutation propagates structural changes to the same region affected by M114T. We note that the phi and psi angles for Gly118 are in a region of the Ramachandran plot that are generally disallowed for a valine residue, and therefore we speculate that the G118V mutation also induces a conformational change within PFN1 that allows valine to adapt dihedral angles that are energetically more favorable.
Our study also provides insight into the relative pathogenicity of ALS-linked PFN1 variants. The pathogenicity of the E117G variant was called into question after it had been detected in the control population (2, 14, 37, 38). Moreover, this variant exhibited mild phenotypes compared with other ALS-linked PFN1 variants in cell-based functional experiments (2, 7). Here, the E117G mutation had only a modest effect on the stability and structure of PFN1 (Table 1 and Fig. S6), supporting the view that E117G is a risk factor for disease rather than overtly pathogenic (1, 14). Further, the E117G mutation was detected in sporadic ALS and frontotemporal lobar degeneration cases (14, 37–40), consistent with the idea that environmental factors and/or genetic modifiers contribute to PFN1 E117G toxicity. In fact, proteasome inhibition triggered the aggregation of PFN1 E117G (2), suggesting that cellular stress may exacerbate PFN1 misfolding and dysfunction in vivo.
Although the mechanism of PFN1 in ALS has yet to be fully elucidated, the destabilized mutant-PFN1 species identified here can serve as an upstream trigger for either loss-of-function or gain-of-toxic-function mechanisms. Several investigations from cell-based experiments support a loss-of-function mechanism for ALS-linked PFN1 variants with respect to actin binding (2), actin dynamics (2), and stress granule assembly (7). For example, PFN1 variants immunoprecipitated less actin from mammalian cells compared with PFN1 WT (2). Our in vitro results suggest this is unlikely due to a general defect in the inherent ability of mutant PFN1 to directly bind actin (Fig. 6) but may be the consequence of mutant PFN1 being sequestered away from actin and/or engaged in other aberrant interactions within the cell. Moreover, ALS-linked mutations do not simply abrogate the direct-binding interaction between PFN1 and the poly-l-proline motif (Fig. 5A) that is present in many biological PFN1 ligands. These data, however, do not rule out the possibility that mutation-induced misfolding and destabilization culminate in defective actin homeostasis in vivo. PFN1 plays a complex role in actin homeostasis, requiring coordinated interactions between PFN1 and many other cellular factors that ultimately dictate the fate of different actin networks within the cell (41).
The misfolding of PFN1 variants may also induce gain of toxic functions and interactions, the latter via aberrant protein–protein interactions through exposed hydrophobic patches, such as those detected for PFN1 M114T (Fig. S9). Further, the aggregation of PFN1 variants can potentially sequester other vital proteins, including those with poly-l-proline binding motifs (4), culminating in compromised actin and/or cellular homeostasis (6).
Although the downstream effect of ALS-linked PFN1 on actin dynamics and other cellular processes have not been elucidated, our data identify misfolded and destabilized PFN1 as a potential upstream trigger of the adverse events that culminate in ALS, opening new avenues for therapeutic advancement in ALS. One potential direction is the development of pharmacological chaperones (16). For example, small molecules that fill the void formed by the M114T mutation are expected to stabilize the protein (35). Our data with poly-l-proline (Fig. 5B) suggest that small-molecules binding to other regions of PFN1 could also stabilize the protein. We posit that stabilizing mutant PFN1 will restore the normal structure and function of the protein, thereby preventing the pathogenic cascade leading to ALS.
Materials and Methods
A pET vector containing human PFN1 flanked by NdeI and EcoRI restriction sites was kindly provided by Bruce Goode, Brandeis University, Waltham, MA. The mutant PFN1 DNA (2) was amplified using primers 5′- GGACCATATGGCCGGGTGGAAC -3′ and 5′- GCCTGAATTCTCAGTACTGGGAACGC -3′ and ligated into the pET vector using NdeI and EcoRI restriction sites. BL21 (DE3) pLysS cells (200132; Agilent Technologies) transformed with PFN1 constructs were cultured in LB containing 100 μg⋅mL–1 ampicillin and 34 μg⋅mL–1 chloramphenicol at 37 °C until an OD600 of 0.7, at which point PFN1 expression was induced by addition of 1 mM isopropyl β-d-thiogalactopyranoside (0487; Amresco) for either 3 h at 37 °C (for WT and E117G) or 24 h at 18 °C (for C71G, M114T, and G118V). Cells were harvested by centrifugation and stored until purification. Refer to Supporting Information for complete details on methods.
SI Materials and Methods
Purification of Recombinant PFN1.
Cells containing recombinant PFN1 were lysed by sonication in 10 mM citrate and 10 mM NaCl, pH 5.0 (buffer A) containing protease inhibitor (11873580001; Roche). The lysate was cleared by centrifugation and applied to a Nuvia cPrime hydrophobic cation exchange column (35-mL column volume) (156-3402; Bio-Rad) preequilibrated with buffer A using an ÄKTAPurifier FPLC system (GE Healthcare). Bound impurities were eluted with 200 mL linear gradient of 10 mM citrate and 1 M NaCl, pH 5.0 (buffer B). PFN1-containing fractions eluted at 100% buffer B at ∼300 mL from the start of the gradient. SDS/PAGE was used to identify PFN1-containing fractions, which were pooled and dialyzed into buffer A with 6,000–8,000 molecular weight cut-off dialysis tubing (8015-40; Membrane Filtration Products, Inc.) before being applied to an anion (Q-resin) exchange column (17-0510-01; GE Healthcare). PFN1 eluted in the flow-through and was concentrated to 1–2 mL using stirred ultrafiltration cells (5123 and 5121; Millipore) and then applied to a Sephacryl S-100 HR (17-1194-01; GE Healthcare) size-exclusion column preequilibrated with PBS. PFN1 proteins eluted at ∼200 mL and were >95% pure as assessed by SDS/PAGE analysis with Coomassie Brilliant Blue stain. The identity and purity of the PFN1 proteins were verified by intact mass analysis at the Proteomics and Mass Spectrometry Facility (University of Massachusetts Medical School). The concentration of PFN1 was determined spectrophotometrically at an absorbance of 280 nm using a molar extinction coefficient of 18,450 M–1⋅cm–1. Aliquots of PFN1 proteins were stored at –80 °C, typically at concentrations between 60–600 μM.
When PFN1 C71G was purified from inclusion bodies, BL21 (DE3) pLysS cells expressing C71G were cultured as described for PFN1 WT and C71G-containing inclusion bodies were extracted as previously described (42). Inclusion bodies were solubilized in 50 mM Tris⋅HCl, pH 7.0, containing 5 mM EDTA, 5 mM DTT, and 3 M guanidinium hydrochloride (buffer C) at ambient temperature. The solubilized inclusion bodies were diluted in buffer C to a PFN1 C71G concentration of ∼5 mg⋅mL–1. PFN1 C71G was refolded in buffer A containing 0.5 M l-arginine at ambient temperature under conditions where the final concentrations of guanidinium hydrochloride and C71G were below 0.1 M and 0.2 mg⋅mL–1, respectively. The refolded protein was dialyzed in buffer A at 4 °C and purified using a Sephacryl S-100 HR column as described for PFN1 WT.
Equilibrium Unfolding Experiments.
For equilibrium unfolding experiments using tryptophan fluorescence, solutions of increasing urea concentration were prepared from a concentrated stock solution of 10.546 M urea in PBS using a Hamilton Microlab 500 titrator. PFN1 was mixed into the urea solutions to a final concentration of 2 µM with 1 mM Tris(2-carboxyethyl)phosphine) (TCEP) and the samples were equilibrated for 15–30 min. The intrinsic tryptophan fluorescence of PFN1 was measured at 25 °C with a T-format Horiba Fluorolog fluorimeter using an excitation wavelength of 295 nm. Three emission spectra (310 nm to 450 nm) were collected for each sample and averaged. The concentration of the urea in each sample was measured using an Abbe refractometer after data acquisition. Data were processed to obtain the center of mass (COM) of the emission spectrum. The COM was fit to a two-state transition model as previously described and the thermodynamic parameters, apparent ΔG° (the free energy of folding), m (the denaturant dependence of ΔG°), and Cm (the midpoint of the unfolding transition) were determined with the program Savuka (43, 44). Because the quantum yield of the native and unfolded states was within a factor of 2, the use of COM analysis is justified. We explicitly checked this by a rigorous global analysis using singular value decomposition and showed that the fit of the urea dependence basis vector gave thermodynamic parameters that were within the error of the COM and CD spectroscopy analyses, and no indications of non-two-state behavior. For equilibrium unfolding experiments using CD spectroscopy, PFN1 (10 μM) was equilibrated in various concentrations of urea as described above and CD spectra were acquired from 215 nm to 260 nm using a Jasco J-810 spectropolarimeter. Three spectra were averaged and the mean residual ellipticity (MRE) at 220 nm was plotted as a function of urea concentration and fit to a two-state equilibrium unfolding model.
For protein refolding experiments, a concentrated stock of PFN1 (100–250 μM) denatured in urea (4–8.5 M) was diluted in urea/PBS to obtain a series of samples with decreasing concentrations of urea, 10 μM PFN1 and 1 mM TCEP. Samples were equilibrated for 30 min before acquisition of fluorescence emission spectra as described above.
DSF.
Samples containing WT or mutant PFN1 (20 μM) in PBS with 20× SYPRO Orange (S6651; Invitrogen) were pipetted in quadruplicate into a 384-well plate and subjected to heat denaturation using a Bio-RadCFX384 Touch Real-Time PCR Detection System. The temperature was increased from 25 °C to 100 °C in 0.3 °C increments and at each increment fluorescent intensities were acquired using HEX detector (excitation 515–535 nm, emission 560–580 nm). PFN1 proteins were analyzed alone and in the presence of the poly-l-proline peptide (molecular weight 1,000–10,000, P2254; Sigma). Because this peptide was supplied from the manufacturer as a mixture of poly-l-proline species, the concentration is reported here in units of proline (molecular weight 115.13 g⋅mol–1). For experiments with the poly-l-proline peptide, PFN1 was prepared with 4 mM proline. The fluorescence intensities for the four replicates were averaged, normalized to the maximum fluorescence intensity, and plotted as a function of temperature to obtain melting curves, which were fit with a sigmoidal function in GraphPad Prism to determine the midpoint of transition or the apparent Tm.
Measuring PFN1 Turnover in Cells.
Human SKNAS cells were cultured in DMEM (11965; Gibco) containing 10% (vol/vol) FBS (F4135; Sigma-Aldrich) and 1% (wt/vol) penicillin and streptomycin (10378; Gibco) under standard culture conditions (37 °C, 5% CO2/95% air). SKNAS cells were transiently transfected with 0.5 µg of V5-PFN1 plasmids (2) in 24-well plates using 1.75 µL NeuroMag (NM50500; OZ Biosciences) diluted in Opti-MEM (38915; Invitrogen). After 12 h of V5-PFN1 expression, translation was inhibited with 30 µg⋅mL–1 cycloheximide (C7698; Sigma-Aldrich). Cells were lysed at specific time points during a 12.5-h time course following cycloheximide addition using RIPA buffer (BP-115-500; Boston BioProducts) supplemented with protease inhibitors (11836170001; Roche) and centrifuged at 19,357 × g for 15 min, after which the supernatant (containing soluble PFN1) was collected. The remaining pellet (containing insoluble PFN1) was washed once with RIPA lysis buffer, centrifuged again, and resolubilized with 8 M urea in volumes equal to their soluble counterparts. The protein concentration of the soluble fractions was determined using a bicinchoninic acid assay (23227; Thermo Scientific Pierce). Samples were processed and subjected to Western blot and densitometry analyses essentially as described (19). Western blots were probed using V5-specific (1:1,000, R96025; Invitrogen) and GAPDH-specific (1:20,000, G9545; Sigma) antibodies. Bands corresponding to soluble V5-PFN1 were normalized to the loading control, GAPDH, and then to the band corresponding to cycloheximide treatment for “0 h” for each protein. For each biological replicate, visible bands corresponding to insoluble V5-PFN1 were normalized to their respective 0 h PFN1 C71G band. Statistical significance was determined using a two-way ANOVA followed by Tukey’s post hoc analysis.
CD Spectroscopy.
CD spectra of WT PFN1 or mutants (10 μM in PBS) were acquired from 190 nm to 260 nm at a scan speed of 2 s per wavelength with a 1-mm cuvette at 25 °C using a AVIV Biomedical CD spectrometer model 400. Data reflect an average of five scans that were blank subtracted. The resulting ellipticity curves were transformed to mean residue ellipticity as described (45).
Acidic Native PAGE.
The method for acidic native PAGE analysis of basic proteins described by the Mario Lebendiker laboratory (wolfson.huji.ac.il/purification/) was used. Briefly, 29:1 acrylamide-bisacrylamide (BP1408-1; Fisher Scientific) native gels were cast with 7.5% (wt/vol) polyacrylamide in the resolving gel, pH 4.3, and 3% (wt/vol) polyacrylamide in the stacking gel, pH 6.8. The gel sample containing WT or mutant PFN1 (0.8 μg⋅μL–1) was prepared under native conditions using ice-cold acetate–KOH, pH 6.8 and 10% (vol/vol) glycerol with 0.025% (wt/vol) of methylene blue. PFN1 proteins (10 μg) were loaded onto the gel and subjected to reversed polarity electrophoresis under ice-cold conditions for 2 h at 100 V. The protein bands were visualized with Coomassie Brilliant Blue as described above for denaturing gels.
Analytical Size-Exclusion Chromatography.
WT or mutant PFN1 (50 μL of PFN1 at 0.8 μg⋅μL–1) were subjected to analytical size-exclusion chromatography at 4 °C using a Superdex 75 column (17-5174-01; GE Healthcare) equilibrated with PBS and a flow rate of 0.5 mL⋅min−1. For each trial (n = 2), elution profiles were acquired using absorbance at 280 nm and normalized to the peak value of WT PFN1. The area under peak was calculated using GraphPad Prism.
Protein Crystallization and X-Ray Structural Determination.
PFN1 crystals were grown by hanging drop vapor diffusion after mixing the PFN1 protein with a 1:1 ratio of reservoir solution at 25 °C for WT and E117G and at 18 °C for M114T. Reservoir solution for WT contained 50 mM KH2PO4, 36% (wt/vol) PEG 8,000 and 100 mM MES, pH 6.0. Reservoir solution for E117G contained 50 mM KH2PO4, 41% (wt/vol) PEG 8,000 and 100 mM MES, pH 6.0. Reservoir solution for M114T contained 750 mM sodium citrate, 200 mM NaCl, and 100 mM Tris, pH 7.5.
E117G crystals where soaked in cryoprotectant composed of 25% (vol/vol) ethylene glycol and 75% (vol/vol) reservoir solution and M114T crystals were passed through mineral oil before mounting for data collection. Diffraction data were collected using a Rigaku 007 MicroMax HF rotating anode X-ray generator, under a nitrogen cryostream at 100 K (Oxford Cryosystems), on a Saturn944+ CCD detector.
The data were processed using Xia2 (46) [running XDS (47)] for WT and M114T and HKL2000 (HKL Research) for E117G. All three structures were solved via molecular replacement with Phaser (48) using the profilin structure PDB ID code 1FIK (20) as the starting model followed by multiple rounds of manual model building performed with Coot (49). WT was refined with PHENIX (50) and E117G with REFMAC5 (51) using standard refinement protocols. M114T was refined with PHENIX using twin refinement with the twin law {h,-h-k,l} applied through refinement, because the data were highly twinned with a twin fraction estimated to be 0.48.
Structural Analysis.
SiteMap (Schrödinger, LLC) was used to identify and evaluate the mutation-site cavity volumes. Figures were generated using PyMOL (Schrödinger, LLC).
Poly-l-Proline Peptide Binding Experiments.
The intrinsic tryptophan fluorescence of WT or ALS-PFN1 (2 μM) as a function of increasing concentrations of the poly-l-proline peptide described above at 25 °C was used to measure binding of PFN1 to poly-l-proline as previously described (52). The samples were excited at 295 nm and three emission spectra between 310 nm and 450 nm were collected for each sample and averaged. The fluorescence emission intensity at 323 nm was baseline-corrected, normalized, plotted as a function of poly-l-proline and fit to a one-site total binding model in GraphPad Prism to yield apparent Kd values.
Inhibition of Spontaneous Actin Assembly.
Gel-filtered monomeric rabbit muscle actin (3 μM, 5% pyrene-labeled) was converted to Mg–ATP–actin immediately before use in each reaction and mixed with 7 μL of different concentrations of PFN1 WT, and PFN1 mutants or control buffer and 3 μL of 20× initiation mix (40 mM MgCl2, 10 mM ATP, and 1 M KCl) in 60-μL reactions. Actin polymerization was monitored over time at 365 nm excitation and 407 nm emission in a PTI fluorometer at 25 °C. Average relative rates of actin polymerization (n = 3) were determined based on the slopes of the assembly curves during the first 500 s of each reaction and plotted against increasing concentrations of PFN1 (mutants). Statistical significance was determined using a two-way ANOVA followed by Tukey’s post hoc analysis.
Supplementary Material
Acknowledgments
We thank K. Boggio and the Proteomics and Mass Spectrometry Facility [University of Massachusetts Medical School (UMMS)] for analysis of recombinant PFN1 proteins; C. R. Matthews (UMMS) for the use of his circular dichroism spectrophotometers and fluorimeter; and O. Bilsel, C.-H. Wu, C. Fallini, N. Cohen, and B. Mackness for helpful advice with experiments and the manuscript. We acknowledge funding from National Institutes of Health Grants R01 NS090352, R01 NS078145, and R01 NS067206 (to D.A.B.); R01 NS073873 (to J.E.L.); P01 GM091743 (to C.A.S. and S.M.S.); P01 GM091743-03S1 (to T.V.S.); R01 GM063691 (to B.L.G.); and R01 GM54836 (to J.A.Z.); and from Target ALS (to J.E.L., D.A.B., and B.L.G.).
Footnotes
The authors declare no conflict of interest.
This article is a PNAS Direct Submission.
Data deposition: The crystallography, atomic coordinates, and structure factors have been deposited in the Protein Data Bank, www.pdb.org (PDB ID codes 4X1L, 4X1M, and 4X25).
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1424108112/-/DCSupplemental.
References
- 1.Smith BN, et al. 2015. Novel mutations support a role for Profilin 1 in the pathogenesis of ALS. Neurobiol Aging 36(3):1602.e17–1602.e27.
- 2.Wu CH, et al. Mutations in the profilin 1 gene cause familial amyotrophic lateral sclerosis. Nature. 2012;488(7412):499–503. doi: 10.1038/nature11280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bosco DA, LaVoie MJ, Petsko GA, Ringe D. Proteostasis and movement disorders: Parkinson’s disease and amyotrophic lateral sclerosis. Cold Spring Harb Perspect Biol. 2011;3(10):a007500. doi: 10.1101/cshperspect.a007500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Witke W. The role of profilin complexes in cell motility and other cellular processes. Trends Cell Biol. 2004;14(8):461–469. doi: 10.1016/j.tcb.2004.07.003. [DOI] [PubMed] [Google Scholar]
- 5.Lambrechts A, et al. The mammalian profilin isoforms display complementary affinities for PIP2 and proline-rich sequences. EMBO J. 1997;16(3):484–494. doi: 10.1093/emboj/16.3.484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Winklhofer KF, Tatzelt J, Haass C. The two faces of protein misfolding: Gain- and loss-of-function in neurodegenerative diseases. EMBO J. 2008;27(2):336–349. doi: 10.1038/sj.emboj.7601930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Figley MD, Bieri G, Kolaitis RM, Taylor JP, Gitler AD. Profilin 1 associates with stress granules and ALS-linked mutations alter stress granule dynamics. J Neurosci. 2014;34(24):8083–8097. doi: 10.1523/JNEUROSCI.0543-14.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Austin JA, et al. Disease causing mutants of TDP-43 nucleic acid binding domains are resistant to aggregation and have increased stability and half-life. Proc Natl Acad Sci USA. 2014;111(11):4309–4314. doi: 10.1073/pnas.1317317111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Rotunno MS, Bosco DA. An emerging role for misfolded wild-type SOD1 in sporadic ALS pathogenesis. Front Cell Neurosci. 2013;7:253. doi: 10.3389/fncel.2013.00253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Watanabe S, Kaneko K, Yamanaka K. Accelerated disease onset with stabilized familial amyotrophic lateral sclerosis (ALS)-linked mutant TDP-43 proteins. J Biol Chem. 2013;288(5):3641–3654. doi: 10.1074/jbc.M112.433615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Eriksson AE, et al. Response of a protein structure to cavity-creating mutations and its relation to the hydrophobic effect. Science. 1992;255(5041):178–183. doi: 10.1126/science.1553543. [DOI] [PubMed] [Google Scholar]
- 12.Joerger AC, Ang HC, Fersht AR. Structural basis for understanding oncogenic p53 mutations and designing rescue drugs. Proc Natl Acad Sci USA. 2006;103(41):15056–15061. doi: 10.1073/pnas.0607286103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Yue P, Li Z, Moult J. Loss of protein structure stability as a major causative factor in monogenic disease. J Mol Biol. 2005;353(2):459–473. doi: 10.1016/j.jmb.2005.08.020. [DOI] [PubMed] [Google Scholar]
- 14.Fratta P, et al. Profilin1 E117G is a moderate risk factor for amyotrophic lateral sclerosis. J Neurol Neurosurg Psychiatry. 2014;85(5):506–508. doi: 10.1136/jnnp-2013-306761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Vedadi M, et al. Chemical screening methods to identify ligands that promote protein stability, protein crystallization, and structure determination. Proc Natl Acad Sci USA. 2006;103(43):15835–15840. doi: 10.1073/pnas.0605224103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ringe D, Petsko GA. What are pharmacological chaperones and why are they interesting? J Biol. 2009;8(9):80. doi: 10.1186/jbiol186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Verhoef LG, Lindsten K, Masucci MG, Dantuma NP. Aggregate formation inhibits proteasomal degradation of polyglutamine proteins. Hum Mol Genet. 2002;11(22):2689–2700. doi: 10.1093/hmg/11.22.2689. [DOI] [PubMed] [Google Scholar]
- 18.Myers JK, Pace CN, Scholtz JM. 1995. Denaturant m values and heat capacity changes: relation to changes in accessible surface areas of protein unfolding. Protein Sci 4(10):2138–2148.
- 19.Rotunno MS, et al. Identification of a misfolded region in superoxide dismutase 1 that is exposed in amyotrophic lateral sclerosis. J Biol Chem. 2014;289(41):28527–28538. doi: 10.1074/jbc.M114.581801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Fedorov AA, Pollard TD, Almo SC. Purification, characterization and crystallization of human platelet profilin expressed in Escherichia coli. J Mol Biol. 1994;241(3):480–482. doi: 10.1006/jmbi.1994.1522. [DOI] [PubMed] [Google Scholar]
- 21.Ferron F, Rebowski G, Lee SH, Dominguez R. Structural basis for the recruitment of profilin-actin complexes during filament elongation by Ena/VASP. EMBO J. 2007;26(21):4597–4606. doi: 10.1038/sj.emboj.7601874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Mahoney NM, Rozwarski DA, Fedorov E, Fedorov AA, Almo SC. Profilin binds proline-rich ligands in two distinct amide backbone orientations. Nat Struct Biol. 1999;6(7):666–671. doi: 10.1038/10722. [DOI] [PubMed] [Google Scholar]
- 23.Prabu-Jeyabalan M, Nalivaika EA, Romano K, Schiffer CA. Mechanism of substrate recognition by drug-resistant human immunodeficiency virus type 1 protease variants revealed by a novel structural intermediate. J Virol. 2006;80(7):3607–3616. doi: 10.1128/JVI.80.7.3607-3616.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Cedergren-Zeppezauer ES, et al. Crystallization and structure determination of bovine profilin at 2.0 A resolution. J Mol Biol. 1994;240(5):459–475. doi: 10.1006/jmbi.1994.1461. [DOI] [PubMed] [Google Scholar]
- 25.Chik JK, Lindberg U, Schutt CE. The structure of an open state of beta-actin at 2.65 A resolution. J Mol Biol. 1996;263(4):607–623. doi: 10.1006/jmbi.1996.0602. [DOI] [PubMed] [Google Scholar]
- 26.Hájková L, Björkegren Sjögren C, Korenbaum E, Nordberg P, Karlsson R. Characterization of a mutant profilin with reduced actin-binding capacity: Effects in vitro and in vivo. Exp Cell Res. 1997;234(1):66–77. doi: 10.1006/excr.1997.3607. [DOI] [PubMed] [Google Scholar]
- 27.Korenbaum E, et al. The role of profilin in actin polymerization and nucleotide exchange. Biochemistry. 1998;37(26):9274–9283. doi: 10.1021/bi9803675. [DOI] [PubMed] [Google Scholar]
- 28.Porta JC, Borgstahl GE. Structural basis for profilin-mediated actin nucleotide exchange. J Mol Biol. 2012;418(1-2):103–116. doi: 10.1016/j.jmb.2012.02.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Schutt CE, Myslik JC, Rozycki MD, Goonesekere NC, Lindberg U. The structure of crystalline profilin-beta-actin. Nature. 1993;365(6449):810–816. doi: 10.1038/365810a0. [DOI] [PubMed] [Google Scholar]
- 30.Sohn RH, Chen J, Koblan KS, Bray PF, Goldschmidt-Clermont PJ. Localization of a binding site for phosphatidylinositol 4,5-bisphosphate on human profilin. J Biol Chem. 1995;270(36):21114–21120. doi: 10.1074/jbc.270.36.21114. [DOI] [PubMed] [Google Scholar]
- 31.Suetsugu S, Miki H, Takenawa T. The essential role of profilin in the assembly of actin for microspike formation. EMBO J. 1998;17(22):6516–6526. doi: 10.1093/emboj/17.22.6516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Björkegren C, Rozycki M, Schutt CE, Lindberg U, Karlsson R. Mutagenesis of human profilin locates its poly(L-proline)-binding site to a hydrophobic patch of aromatic amino acids. FEBS Lett. 1993;333(1-2):123–126. doi: 10.1016/0014-5793(93)80388-b. [DOI] [PubMed] [Google Scholar]
- 33.Ostrander DB, Ernst EG, Lavoie TB, Gorman JA. Polyproline binding is an essential function of human profilin in yeast. Eur J Biochem. 1999;262(1):26–35. doi: 10.1046/j.1432-1327.1999.00354.x. [DOI] [PubMed] [Google Scholar]
- 34.Pollard TD, Cooper JA. Quantitative analysis of the effect of Acanthamoeba profilin on actin filament nucleation and elongation. Biochemistry. 1984;23(26):6631–6641. doi: 10.1021/bi00321a054. [DOI] [PubMed] [Google Scholar]
- 35.Eriksson AE, Baase WA, Wozniak JA, Matthews BW. A cavity-containing mutant of T4 lysozyme is stabilized by buried benzene. Nature. 1992;355(6358):371–373. doi: 10.1038/355371a0. [DOI] [PubMed] [Google Scholar]
- 36.Ling SC, et al. ALS-associated mutations in TDP-43 increase its stability and promote TDP-43 complexes with FUS/TLS. Proc Natl Acad Sci USA. 2010;107(30):13318–13323. doi: 10.1073/pnas.1008227107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Dillen L, et al. 2013. Explorative genetic study of UBQLN2 and PFN1 in an extended Flanders-Belgian cohort of frontotemporal lobar degeneration patients. Neurobiol Aging 34(6):1711.e1–1711.e5.
- 38.van Blitterswijk M, et al. Profilin-1 mutations are rare in patients with amyotrophic lateral sclerosis and frontotemporal dementia. Amyotroph Lateral Scler Frontotemporal Degener. 2013;14(5-6):463–469. doi: 10.3109/21678421.2013.787630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Tiloca C, et al. 2013. Screening of the PFN1 gene in sporadic amyotrophic lateral sclerosis and in frontotemporal dementia. Neurobiol Aging 34(5):1517.e9–1517.e10.
- 40.Yang S, et al. 2013. Mutation analysis and immunopathological studies of PFN1 in familial and sporadic amyotrophic lateral sclerosis. Neurobiol Aging 34(9):2235.e7–2235.e10.
- 41.Rotty JD, et al. Profilin-1 serves as a gatekeeper for actin assembly by Arp2/3-dependent and -independent pathways. Dev Cell. 2015;32(1):54–67. doi: 10.1016/j.devcel.2014.10.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Palmer I, Wingfield PT. 2012. Preparation and extraction of insoluble (inclusion-body) proteins from Escherichia coli. Curr Protoc Protein Sci Chap 6, Unit 6.3.
- 43.Bilsel O, Yang L, Zitzewitz JA, Beechem JM, Matthews CR. Time-resolved fluorescence anisotropy study of the refolding reaction of the alpha-subunit of tryptophan synthase reveals nonmonotonic behavior of the rotational correlation time. Biochemistry. 1999;38(13):4177–4187. doi: 10.1021/bi9829433. [DOI] [PubMed] [Google Scholar]
- 44.Greene RF, Jr, Pace CN. Urea and guanidine hydrochloride denaturation of ribonuclease, lysozyme, alpha-chymotrypsin, and beta-lactoglobulin. J Biol Chem. 1974;249(17):5388–5393. [PubMed] [Google Scholar]
- 45.Mackness BC, Tran MT, McClain SP, Matthews CR, Zitzewitz JA. Folding of the RNA recognition motif (RRM) domains of the amyotrophic lateral sclerosis (ALS)-linked protein TDP-43 reveals an intermediate state. J Biol Chem. 2014;289(12):8264–8276. doi: 10.1074/jbc.M113.542779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Winter G. xia2: An expert system for macromolecular crystallography data reduction. J Appl Cryst. 2010;43:186–190. [Google Scholar]
- 47.Kabsch W. Xds. Acta Crystallogr D Biol Crystallogr. 2010;66(Pt 2):125–132. doi: 10.1107/S0907444909047337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.McCoy AJ, et al. Phaser crystallographic software. J Appl Cryst. 2007;40(Pt 4):658–674. doi: 10.1107/S0021889807021206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Emsley P, Cowtan K. Coot: Model-building tools for molecular graphics. Acta Crystallogr D Biol Crystallogr. 2004;60(Pt 12 Pt 1):2126–2132. doi: 10.1107/S0907444904019158. [DOI] [PubMed] [Google Scholar]
- 50.Adams PD, et al. PHENIX: A comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr D Biol Crystallogr. 2010;66(Pt 2):213–221. doi: 10.1107/S0907444909052925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Murshudov GN, Vagin AA, Dodson EJ. Refinement of macromolecular structures by the maximum-likelihood method. Acta Crystallogr D Biol Crystallogr. 1997;53(Pt 3):240–255. doi: 10.1107/S0907444996012255. [DOI] [PubMed] [Google Scholar]
- 52.Lu J, Pollard TD. Profilin binding to poly-L-proline and actin monomers along with ability to catalyze actin nucleotide exchange is required for viability of fission yeast. Mol Biol Cell. 2001;12(4):1161–1175. doi: 10.1091/mbc.12.4.1161. [DOI] [PMC free article] [PubMed] [Google Scholar]