

Abstract
Increased glucose metabolism and reprogramming toward aerobic glycolysis are a hallmark of cancer cells, meeting their metabolic needs for sustained cell proliferation. Metabolic reprogramming is usually considered as a downstream consequence of tumor development and oncogene activation; growing evidence indicates, however, that metabolism on its turn can support oncogenic signaling to foster tumor malignancy. Here, we explored how glucose metabolism regulates gene transcription and found an unexpected link with YAP/TAZ, key transcription factors regulating organ growth, tumor cell proliferation and aggressiveness. When cells actively incorporate glucose and route it through glycolysis, YAP/TAZ are fully active; when glucose metabolism is blocked, or glycolysis is reduced, YAP/TAZ transcriptional activity is decreased. Accordingly, glycolysis is required to sustain YAP/TAZ pro-tumorigenic functions, and YAP/TAZ are required for the full deployment of glucose growth-promoting activity. Mechanistically we found that phosphofructokinase (PFK1), the enzyme regulating the first committed step of glycolysis, binds the YAP/TAZ transcriptional cofactors TEADs and promotes their functional and biochemical cooperation with YAP/TAZ. Strikingly, this regulation is conserved in Drosophila, where phosphofructokinase is required for tissue overgrowth promoted by Yki, the fly homologue of YAP. Moreover, gene expression regulated by glucose metabolism in breast cancer cells is strongly associated in a large dataset of primary human mammary tumors with YAP/TAZ activation and with the progression toward more advanced and malignant stages. These findings suggest that aerobic glycolysis endows cancer cells with particular metabolic properties and at the same time sustains transcription factors with potent pro-tumorigenic activities such as YAP/TAZ.
Keywords: aerobic glycolysis, glucose metabolism, Hippo pathway, TEAD, YAP/TAZ
Introduction
YAP and TAZ are important transcriptional coactivators regulating proliferation, survival and self-renewal ability in a number of cellular systems (Pan, 2010; Halder & Johnson, 2011; Tremblay & Camargo, 2012; Piccolo et al, 2014). YAP/TAZ regulate transcription mainly by interacting with the TEAD family of transcription factors, and their activity is regulated by different inputs, including the Hippo kinase cascade, Wnt signaling, RHO GTPases and mechanical cues acting through the F-actin cytoskeleton (Halder et al, 2012; Yu & Guan, 2013). This is fundamental for the growth and homeostasis of tissues and organs, such that YAP/TAZ are recognized as universal regulators of organ size from Drosophila to mammals. Reflecting these key functions, unleashed YAP/TAZ activity is sufficient to promote tumorigenesis, and YAP/TAZ are required for cancer stem cell self-renewal and tumor-seeding ability in different tumor types (Harvey et al, 2013; Johnson & Halder, 2013).
One hallmark of cancer cells is the shift of their glucose metabolism from oxidative respiration to aerobic glycolysis; in these conditions, cells display high glucose metabolism and mainly produce ATP through glycolysis, even if this is far less efficient compared to mitochondrial respiration (Levine & Puzio-Kuter, 2010; Hanahan & Weinberg, 2011; Lunt & Vander Heiden, 2011). The rationale for shifting to such a poorly efficient energy generation process is the chronic and uncontrolled proliferation observed in tumors: cancer cells need not only to produce energy, but also to increase their biomass to sustain production of daughter cells. Aerobic glycolysis would fulfill this duty by allowing the diversion of metabolic intermediates toward various biosynthetic pathways and ultimately favoring the synthesis of macromolecules and new organelles (Lunt & Vander Heiden, 2011; Schulze & Harris, 2012). Further extending the link between aerobic glycolysis and proliferation, a similar glycolytic metabolism is also observed in non-transformed rapidly dividing cells such as embryonic tissues and stem cell compartments (Ochocki & Simon, 2013; Shyh-Chang et al, 2013; Ito & Suda, 2014).
Much of the current literature considers glucose metabolism and aerobic glycolysis as endpoints that occur as a consequence of transformation. Indeed, several oncogenes such as Ras, cMyc and HIF1 (Hypoxia Induced Factor-1) regulate expression of glucose transporters and glycolytic enzymes and ensure the balancing of glycolysis with other metabolic pathways (Gordan et al, 2007; Kroemer & Pouyssegur, 2008). Increasing evidence, however, indicates that metabolic pathways also incorporate signaling mechanisms that inform and coordinate other cellular functions, including nuclear gene transcription and epigenetics. In this manner, metabolic pathways can even play causative roles in regulating cell behavior, in addition to their core biochemical functions (Chaneton & Gottlieb, 2012; Dang, 2012; Hardie et al, 2012; Laplante & Sabatini, 2012; Luo & Semenza, 2012; Wellen & Thompson, 2012; Chang et al, 2013).
Results
Glucose metabolism regulates YAP/TAZ activity
To explore new possible links between glucose metabolism and gene transcription, we asked whether glucose metabolism could regulate known signaling pathways relevant for embryonic development, adult tissue homeostasis and disease. To this end, we performed genome-wide microarray expression profiling to compare cells growing in high glucose with cells treated for 24 h with 2-deoxy-glucose (2DG, 50 mM), a widely used competitive inhibitor of glucose metabolism acting at the level of hexokinase (Tennant et al, 2010), and obtained a list of genes regulated by glucose metabolism. This dose of 2DG is commonly used in cell cultures to block glucose metabolism and was sufficient to inhibit aerobic glycolysis and to increase mitochondrial respiration in our cells, as measured with an extracellular flux analyzer (see below), and to inhibit cell growth.
We then performed a gene set enrichment analysis (GSEA), searching for statistical associations between the genes regulated by 2DG (either up- or down-regulated) and those contained in a collection of gene signatures denoting activation of transcription factors and signaling pathways (see Materials and Methods for details). Since most of these signatures were derived from mammary cell lines, we performed the experiments in MDA-MB-231 breast cancer cells and MCF10A mammary epithelial cells. Several signatures overlapped with genes regulated by 2DG treatment; in both cell lines, the genes induced by YAP/TAZ were significantly enriched among the genes inhibited by 2DG treatment, whereas the genes repressed by YAP were enriched among the genes activated by 2DG (Fig1A and B; Supplementary Fig S1A).
Figure 1.

- Over-representation analysis was performed with gene signatures highlighting activation of specific pathways using gene set enrichment analysis (GSEA) on microarray data obtained from MCF10A or MDA-MB-231 mammary cells untreated or treated with 2-deoxy-glucose (2DG, 50 mM) to inhibit glucose metabolism. The normalized enrichment score (NES) is the primary statistic for examining GSEA results; a positive NES (highlighted in red) indicates signatures expressed more in control cells than upon 2DG treatment (i.e. signatures activated when glucose metabolism is active); a negative NES (highlighted in blue) indicates signatures expressed more upon 2DG treatment. The false discovery rate (FDR) is the estimated probability that a gene set with a given NES represents a false positive; we considered signatures to be significantly enriched at FDR < 0.05. Gene expression data have been obtained from n = 4 biological replicates for each condition. See Supplementary Table S1 for a GSEA analysis including also Biocarta gene sets.
- 2DG treatment downregulates the overall levels of the ‘YAP/TAZ’ gene signature used in (A) as calculated from microarray data of cells untreated (white bars) or treated with 2DG (black bars). See Materials and Methods for details on the statistical methods to quantify average signature expression. Data are shown as mean ± standard error of the mean (SEM). Of note, in this analysis, the basal levels of YAP/TAZ target genes were higher in the cell line displaying higher glycolysis/respiration ratio, that is, in MDA-MB-231 cells (Supplementary Fig S1B).
- Luciferase assay in MDA-MB-231 breast cancer cells transfected with the synthetic YAP/TAZ reporter 8XGTIIC-lux. Starting on the day after DNA transfection, cells were treated for 24 h with the indicated small-molecule inhibitors to block glucose metabolism (50 mM 2DG; 1 mM lonidamine, Loni) or with an inhibitor of the mitochondrial respiratory chain (1 μM oligomycin, Oligo). Activity of the reporter is normalized to cotransfected CMV-lacZ and expressed relative to the cells treated with vehicle only (Co.). See Supplementary Fig S1E–K for controls on the specificity of 2DG treatment and similar results obtained in Hs578T and HepG2 cells. Representative results of a single experiment with n = 2 biological replicates; four independent experiments were consistent.
- Luciferase assay in MDA-MB-231 cells bearing a stably integrated TRE-8XGTIIC-lux reporter, whose transcription can be released following doxycycline treatment to visualize early YAP/TAZ responses (see Supplementary Fig S1N for controls). Control cells (Co.) were left unstimulated (0) or supplemented with doxycycline (4, 6, 8 and 10 h of treatment) to release YAP/TAZ-dependent transcription. 2DG (100 mM) was added together with doxycycline to acutely block glucose metabolism. See Supplementary Fig S1P–R for similar results obtained in MCF10A-MII cells. Representative results of a single experiment with n = 2 biological replicates; three independent experiments were consistent.
- Luciferase assay was carried out as in (D), by removing glucose from the culture medium at the moment of doxycycline supplementation (−Glu). Cells were harvested 24 h after treatment. See Supplementary Fig S1O and R for similar results obtained in HepG2 and MCF10A-MII cells. Representative results of a single experiment with n = 2 biological replicates; three independent experiments were consistent.
- YAP/TAZ are required for transcription of 2DG-regulated genes. qPCR for endogenous target genes in MDA-MB-231 cells treated with water (Co.) or with 2DG or transfected with the indicated siRNAs: control (siCo.), YAP/TAZ mix #1 (siYT1), YAP/TAZ mix #2 (siYT2). Expression levels were calculated relative to GAPDH and are given relative to Co. cells (arbitrarily set to 1). Genes were selected among the probes commonly regulated in microarray profiling (see Supplementary Table S3). Note how both 2DG-induced and 2DG-inhibited genes were coherently regulated by YAP/TAZ knockdown. See Supplementary Fig S1S for other targets and controls, and Supplementary Fig S1T for similar results in Hs578T cells. n = 4 biological replicates from two independent experiments. All differences had P-value < 0.01.
Data information: Unless indicated otherwise, error bars represent mean ± SD. *P-value < 0.01 relative to control.
Our GSEA analysis suggested a link between glucose and YAP/TAZ, but did not inform us about what is upstream and what is downstream. We initially investigated whether YAP/TAZ regulate glucose metabolism by monitoring aerobic glycolysis and mitochondrial respiration levels in mammary epithelial cells expressing activated TAZ. Even if TAZ activation is per se sufficient to endow these cells aggressive traits in vitro and in vivo (Cordenonsi et al, 2011), we did not observe notable changes in glycolysis or respiration (Supplementary Fig S1C and D). Moreover, by surveying microarrays obtained by activation or inhibition of YAP/TAZ in multiple cellular systems (Ota & Sasaki, 2008; Zhao et al, 2008; Zhang et al, 2009; Mohseni et al, 2014 and see below), we failed to observe consistent regulation of glucose transporters or glycolytic genes that are instead typically induced by oncogenes (Gordan et al, 2007; Kroemer & Pouyssegur, 2008). Thus, YAP/TAZ are not obvious inducers of aerobic glycolysis.
We then tested whether glucose metabolism regulates YAP/TAZ. For this, we directly monitored their transcriptional activity with the established YAP/TAZ luciferase reporter 8XGTIIC-lux (Dupont et al, 2011) in MDA-MB-231 cells treated for 24 h with 2DG or with lonidamine [another widely used inhibitor of hexokinase (Tennant et al, 2010)]; as shown in Fig1C, these treatments inhibited YAP/TAZ activity. The effect of 2DG was reversible (Supplementary Fig S1E) and was not caused by aspecific competition for mannose (Kurtoglu et al, 2007) (Supplementary Fig S1F); treatment of cells with oligomycin-A, at doses inhibiting mitochondrial respiration, did not inhibit YAP/TAZ activity (Fig1C), indicating a specific effect of glucose metabolism. Similar results were obtained with the CTGF-lux reporter (Supplementary Fig S1G), in another highly glycolytic breast cancer cell line, Hs578T (Dong et al, 2013) (Supplementary Fig S1H), and in HepG2 cells (Supplementary Fig S1I), characterized by high YAP/TAZ activity (Azzolin et al, 2012; Yimlamai et al, 2014) and high glycolysis (Marroquin et al, 2007). Moreover, a reporter driven by the CMV promoter and a reporter for the Notch pathway were not inhibited, ruling out general effects on transcription (Supplementary Fig S1J and K). Thus, inhibition of glucose metabolism inhibits YAZ/TAZ activity.
Glucose inhibition induces a fast adaptation of cellular metabolism and then secondarily induces other cellular responses, including growth arrest; thus, among the genes regulated by glucose, some could be regulated as a direct consequence of glucose inhibition, while others may be indirect targets of growth arrest. An example of the second class are the E2F-regulated genes (Chen et al, 2009; Dick & Rubin, 2013) that were highly enriched in our GSEA analysis (Fig1A, E2F3 signature); indeed, we found by luciferase assays that E2F activity is inhibited by 2DG and also by inducing growth arrest through expression of established CDK inhibitors (p21/CDKN1A, p16/CDKN2A, p27/CDKN1B) (Supplementary Fig S1L). In contrast, CDK inhibitors do not inhibit YAP/TAZ activity (Supplementary Fig S1M). Thus, growth arrest is not sufficient to explain YAP/TAZ inhibition by 2DG. In line, by using a doxycycline-inducible version of the 8XGTIIC-lux reporter enabling us to visualize early YAP/TAZ transcriptional activity (Supplementary Fig S1N), we found that 2DG treatment inhibited YAP/TAZ-induced transcription at the earliest time points, indicating a rapid response (Fig1D). Importantly, also in cells cultured in absence of glucose, we observed a reduced YAP/TAZ activity (Fig1E). Similar results were obtained in HepG2 cells (Supplementary Fig S1O) and in MCF10A-MII (Supplementary Fig S1P–R). This observation is thus compatible with a direct regulation of YAP/TAZ activity.
To validate further the link between glucose and YAP/TAZ, we checked whether endogenous target genes inhibited by 2DG were coherently regulated also upon knockdown of YAP/TAZ. To select candidate co-regulated genes, we compared microarray profiling of genes regulated by 2DG with microarrays of cells depleted of YAP/TAZ by siRNA transfection (see Materials and Methods). As shown in Supplementary Tables S2 and S3, we could identify several probes that were similarly co-regulated (either up or down) by glucose and by YAP/TAZ in both cell lines. Among the strongest co-regulated microarray probes in MDA-MB-231 cells, we successfully validated a series of transcriptional targets by qPCR, concordantly repressed or activated by 2DG and YAP/TAZ knockdown, including the established YAP/TAZ target HMMR (Wang et al, 2014) (Fig1F; Supplementary Fig S1S). Other YAP/TAZ targets, such as CTGF or ANKRD1, were not regulated (Supplementary Fig S1S), likely due to compensating inputs. The same target genes were also regulated by 2DG treatment and YAP/TAZ knockdown in another breast cancer cell line, Hs578T (Supplementary Fig S1T). Interestingly, Gene Ontology analysis indicates that the genes coregulated by glucose and YAP/TAZ are particularly related to cell cycle control and to DNA synthesis, repair and metabolism (Supplementary Fig S1U), in keeping with the validation of TK1, TYMS, RRM2 and CDC factors shown above. Collectively, these results indicate that YAP/TAZ transcriptional activity is sustained by glucose metabolism.
YAP/TAZ activity is regulated by glycolysis
Glucose fuels multiple metabolic pathways; we then sought to understand which of these was more relevant to regulate YAP/TAZ. Once entrapped in the cell in the form of glucose-6-phosphate (G6P) by hexokinase, glucose can be either converted to fructose-6-phosphate (F6P) by the enzyme glucose-6-phosphate isomerase (GPI), or it is directed into the pentose phosphate pathway (see the simplified scheme in Fig 2A). To test whether GPI was involved in YAP/TAZ regulation, we depleted cells of endogenous GPI with two independent siRNAs and found this was sufficient to recapitulate the effects of 2DG treatment (Fig2B; Supplementary Fig S2A).
Figure 2.

- A simplified scheme indicating the main metabolic routes followed by glucose, the key intermediates and enzymes involved, and the inhibitors used in this study. Only the pathways and enzymes discussed in the text are shown here for simplicity. G6P: glucose-6-phosphate; F6P: fructose-6-phosphate; F1,6P: fructose-1,6-bisphosphate; F2,6P: fructose-2,6-bisphosphate; GlcNAc: N-acetyl glucosamine; HK: hexokinase; GPI: phosphoglucoisomerase; PFK1: 6-phosphofructo-1-kinase; PFKFB3: 6-phosphofructo-2-kinase/fructose-2,6-bisphosphatase 3. Lonidamine (Loni.) inhibits HK (Tennant et al, 2010); 2DG inhibits both HK and GPI (Wick et al, 1957; Tennant et al, 2010); DON and AZS inhibit the enzyme mediating the first step of the hexosamine pathway (Wellen et al, 2010; Ostrowski & van Aalten, 2013; Onodera et al, 2014). The green arrow indicates the agonistic effect of F2,6P on PFK1. Dashed arrows indicate downstream intermediates or metabolic pathways.
- Phosphoglucoisomerase (GPI) is required for YAP/TAZ activity. Luciferase assay in MDA-MB-231 cells transfected with the indicated siRNAs. See Supplementary Fig S2A for validation of siRNA efficiency. Representative results of a single experiment with n = 2 biological replicates; three independent experiments were consistent.
- The plot indicates basal oxygen consumption rate (OCR) and extracellular acidification rate (ECAR) of TRE-8XGTIIC-lux MDA-MB-231 cells grown in glucose (red, Glu) or 10 mM galactose (blue, Gal). As expected, galactose induces a metabolic shift from aerobic glycolysis to oxidative phosphorylation compared to glucose. See Supplementary Fig S2D–F for detailed OCR and ECAR traces. Representative results of a single experiment with n = 5 biological replicates; two independent experiments were consistent.
- Comparison of YAP/TAZ activity in MDA-MB-231 cells bearing a stably integrated TRE-8XGTIIC-lux reporter and grown in glucose (red, Glu), in galactose to induce a shift toward oxidative respiration (blue, Gal), or shifted back to glucose during doxycycline treatment (blue bars with red stripes, Gal + Glu). Cells were treated with doxycycline to release YAP/TAZ-dependent luciferase transcription for 8 or 24 h. Galactose-fed cells display reduced glycolysis and downregulate YAP/TAZ activity. Glucose rapidly reactivates glycolysis (Supplementary Fig S2F) and YAP/TAZ activity (Gal + Glu). Representative results of a single experiment with n = 2 biological replicates; three independent experiments were consistent.
- Luciferase assay in UOK262 kidney cancer cells, bearing mutation of the fumarate hydratase (FH) enzyme of the tricarboxylic acid cycle (TCA). FH-reconstituted cells (gray bars) display a reduction of aerobic glycolysis and increased respiration (Yang et al, 2013). 2DG treatment (12 mM) of parental cells serves as a positive control for inhibition of the glycolysis–YAP/TAZ axis in parental cells. Representative results of a single experiment with n = 2 biological replicates; two independent experiments were consistent.
Data information: Throughout the figure, error bars represent mean ± SD. *P-value < 0.01.
Downstream of GPI, F6P can be used in glycolysis or in the hexosamine biosynthetic pathway (HBP), the latter providing the metabolic intermediates for protein glycosylation (Wellen & Thompson, 2012). To address a potential involvement of the HBP, we used two strategies: first, we blocked the activity of glucosamine-fructose-6-phosphate transaminase (GFPT), the entry point enzyme of HBP, by treating cells with 6-diazo-5-oxo-L-norleucine (DON) or O-diazoacetyl-L-serine (AZS), at doses commonly used in cancer cells (Wellen et al, 2010; Ostrowski & van Aalten, 2013; Onodera et al, 2014), but these compounds did not affect YAP/TAZ activity (Supplementary Fig S2B). Second, we tested whether protein glycosylation was involved by supplementing N-acetyl glucosamine (GlcNAc) in the culture medium, which can fuel glycosylation in absence of glucose (Wellen et al, 2010; Ostrowski & van Aalten, 2013). Also in this case, GlcNAc was not sufficient to rescue YAP/TAZ inhibition by 2DG (Supplementary Fig S2C). Altogether, this made unlikely that the HBP and protein glycosylation are major regulators of YAP/TAZ; this also suggested glycolysis as the key metabolic pathway regulating YAP/TAZ.
To verify this hypothesis, we modulated the levels of glycolysis by growing cells in the presence of galactose: in these conditions, cells can transform galactose into glucose, but this occurs at a slow rate, aerobic glycolysis is unfavorable, and cells shift their metabolism toward oxidative phosphorylation (Bustamante & Pedersen, 1977; Rossignol, 2004; Marroquin et al, 2007; Chang et al, 2013). In line, we observed a strong reduction of glycolysis and an increase in respiration in MDA-MB-231 cells grown in galactose (Fig2C; Supplementary Fig S2D–F). Strikingly, cells with reduced glycolysis displayed a corresponding reduction of YAP/TAZ activity (Fig2D); moreover, this was restored when we added back glucose during the last part of the experiment only (Fig2D; Supplementary Fig S2G), in line with a rapid response of YAP/TAZ to glucose shown before. A downregulation of YAP/TAZ activity was also observed in UOK262 cells upon reconstitution of the tricarboxylic acid (TCA) cycle enzyme fumarate hydratase (FH), enabling these cells to resume oxidative phosphorylation and causing a parallel reduction of aerobic glycolysis levels (Sudarshan et al, 2009; Yang et al, 2013) (Fig2E). These results collectively support the notion that glycolysis plays a role in regulating YAP/TAZ. This is also indicated by the observation that mitochondrial respiration per se does not regulate YAP/TAZ (Fig1C) and that supplementing pyruvate could not rescue 2DG effects (Supplementary Fig S2H).
Exploring the mechanisms of YAP/TAZ regulation
To understand how glycolysis regulates YAP/TAZ, we initially tested the involvement of known signaling pathways such as the AMPK/mTOR energy-sensing network (Hardie et al, 2012; Laplante & Sabatini, 2012), the Hippo kinase cascade (Pan, 2010; Halder & Johnson, 2011) and the recently identified axislinking mevalonate metabolism to YAP/TAZ regulation (Sorrentino et al, 2014; Wang et al, 2014). Experimental evidence, however, failed to connect any of these pathways to regulation of YAP/TAZ by glucose:
In cells cultured in high glucose, that is, with low AMPK and high mTOR activity, treatment with the dual mTORC1/2 inhibitor AZD2014 (Zhang et al, 2011; Pike et al, 2013) did not affect YAP/TAZ, while it efficiently induced dephosphorylation of the established mTOR downstream target ribosomal protein S6 (Hardie et al, 2012; Laplante & Sabatini, 2012) (Supplementary Fig S3A and B). This finding is in line with DeRan et al (2014) and Fan et al (2013).
Upon 2DG treatment, that is, in conditions where AMPK is activated, blockade of AMPK activity was unable to rescue YAP/TAZ inhibition, while it was sufficient to completely rescue protein S6 phosphorylation (Fig3A; Supplementary Fig S3C–E). Thus, activation of AMPK is not sufficient to account for the effects of glucose metabolism on YAP/TAZ activity (DeRan et al, 2014).
Knockdown of LATS1/2 kinases with two previously validated independent sets of siRNAs (Aragona et al, 2013; Sorrentino et al, 2014) was not sufficient to rescue 2DG treatment, while it was sufficient to completely rescue inhibition caused by NF2/Merlin overexpression (Fig3B). This also indirectly ruled out an involvement of the AMPK-related salt-inducible kinases that regulate Yorkie/YAP through Sav and LATS (Wehr et al, 2012).
Acetyl-CoA, a main derivative of glycolysis, is a precursor for mevalonate metabolism, which in turn is required for RHO GTPase geranylation, hence potentially impacting YAP/TAZ activity (Sorrentino et al, 2014; Wang et al, 2014). We tested whether providing mevalonate to cells could rescue 2DG inhibition, but this was not the case (Fig3C). As a control, mevalonate was instead sufficient to rescue inhibition caused by cerivastatin (Sorrentino et al, 2014; Wang et al, 2014), a small-molecule inhibitor of mevalonate production (Fig3C). In line, 2DG induced a much milder YAP phosphorylation and almost no nuclear exclusion when compared to cerivastatin treatment (Supplementary Fig S3F and G).
Figure 3.
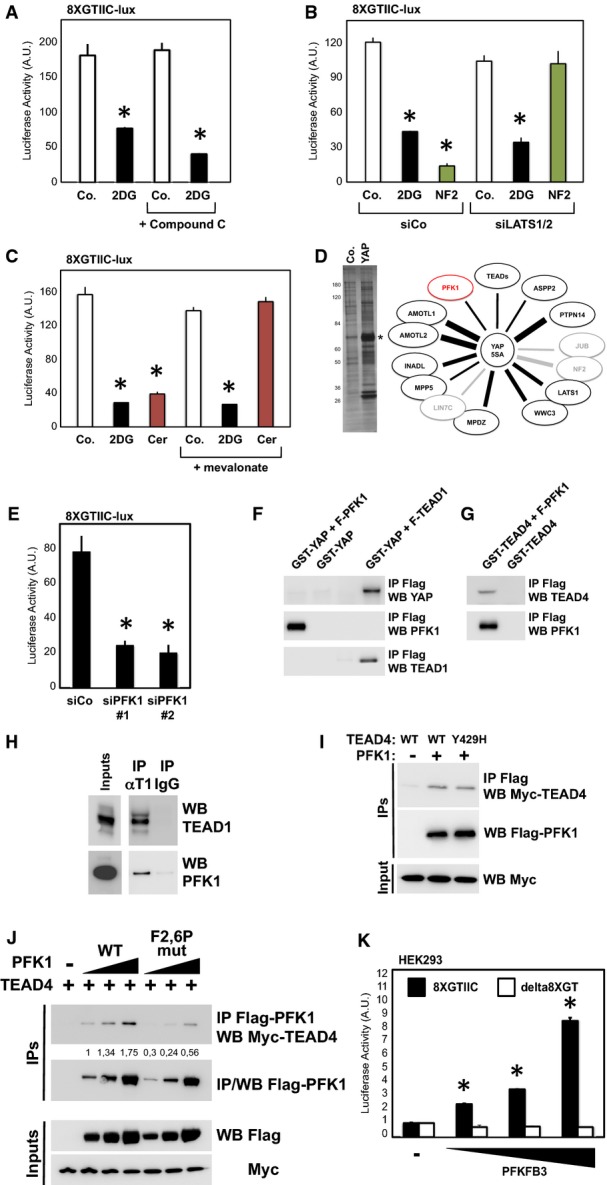
- Luciferase assay in MDA-MB-231 cells treated for 24 h with 2DG (black bars) and/or with compound-C (30 μM), an established inhibitor of AMPK. The same dose of compound-C is sufficient to prevent AMPK activation by 2DG (see Supplementary Fig S3C), but not YAP/TAZ inhibition. Representative results of a single experiment with n = 2 biological replicates; three independent experiments were consistent. See Supplementary Fig S3D and E for similar results with AMPKa1/2 silencing.
- Luciferase assay in cells transfected with control (siCo.) or with an established LATS1/2 siRNA mix (siLATS1/2) (Aragona et al, 2013) and then either treated with 2DG (black bars) or transfected with NF2 expression plasmid to specifically activate the Hippo pathway (green bars). Depletion of LATS1/2 blocked the inhibitory effect of overexpressed NF2, but not of 2DG. Similar results were obtained with an independent mix of LATS1/2 siRNA (data not shown). Representative results of a single experiment with n = 2 biological replicates; two independent experiments were consistent.
- Inhibition of glycolysis could potentially deplete cells of acetyl-CoA, the main precursor for mevalonate, and mevalonate is required for YAP/TAZ activity by regulating RHO GTPases (Sorrentino et al, 2014; Wang et al, 2014). Cells were transfected with the 8XGTIIC-lux YAP/TAZ reporter and treated with 2DG (black bars) or with cerivastatin (3 μM, red bars), an inhibitor of the mevalonate pathway at the level of HMG-CoA reductase. Adding back mevalonate in the culture medium (+ mevalonate, 1 mM) rescues YAP/TAZ inhibition from cerivastatin, but not from 2DG. Representative results of a single experiment with n = 2 biological replicates; two independent experiments were consistent.
- Proteomic analysis of YAP-binding partners reveals interaction with phosphofructokinase (PFK1). Flag-tagged YAP-5SA stably expressed in MCF10A and MDA-MB-231 cells was immunoprecipitated, and associated proteins identified using mass spectrometry. Left panel: silver staining of the purified proteins in representative control (Co.) or YAP immunopurifications. Molecular weight markers are indicated. The asterisk indicates the band corresponding to YAP. Right scheme: The thickness of the lines connecting YAP to its partners is proportional to the number of peptides isolated for each partner. Black proteins (known YAP partners) and PFK1 (in red) were isolated in both cell lines; gray proteins are known regulators of YAP that were only purified from MCF10A cells. See Supplementary Table S4 for a complete list of the identified peptides.
- Luciferase assay in MDA-MB-231 cells transfected with control (siCo.) or two independent PFK1 siRNAs (siPFK1 #1, #2). Representative results of a single experiment with n = 2 biological replicates; four independent experiments were consistent. See Supplementary Fig S3H for validation of PFK1 siRNAs and Supplementary Fig S3I for similar results on CTGF-lux.
- In vitro pull-down assay with purified FLAG-PFK1 and recombinant GST-YAP. GST-YAP was incubated with (first lane) or without (second lane) FLAG-PFK1; as positive control, GST-YAP was incubated with purified FLAG-TEAD1 (right-most lane). Proteins were then subjected to anti-FLAG immunoprecipitation, and purified complexes were probed for coprecipitation of GST-YAP (anti-YAP immunoblot).
- In vitro pull-down assay with purified FLAG-PFK1 and recombinant GST-TEAD4. GST-TEAD4 was incubated with (first lane) or without (second lane) FLAG-PFK1. Proteins were then subjected to anti-FLAG immunoprecipitation, and purified complexes were probed for coprecipitation of GST-TEAD4 (anti-TEAD4 immunoblot).
- MDA-MB-231 cell lysates were immunoprecipitated with anti-TEAD1 antibody, and the precipitating proteins were probed for TEAD1 or PFK1. Immunoprecipitation with an unrelated IgG serves as negative control. Of note, this interaction is in line with the requirement of TEAD1 and TEAD4 for YAP/TAZ activity in our cellular systems (Supplementary Fig S3L and M).
- Lysates from HEK293 cells transfected with the indicated proteins were subjected to anti-FLAG-PFK1 immunoprecipitation, and purified complexes were probed for coprecipitation of MYC-TEAD4. Mutation of a key amino acid required for interaction between TEAD4 and YAP/TAZ (Y429H) did not interfere with PFK1 interaction.
- Mutation of the fructose-2,6-P allosteric site of PFK1 negatively regulates its interaction with TEAD4. HEK293 cells were transfected with MYC-TEAD4 and increasing doses of wild-type (WT) or mutated (F2,6P-mut) FLAG-PFK1 plasmids; cell extracts were immunoprecipitated with anti-FLAG, and the coprecipitating MYC-TEAD4 protein was detected by Western blotting. Immunoprecipitation in the absence of FLAG-PFK1 (lane 1) serves as a negative control. Quantifications of the TEAD4/PFK1 ratio are provided, relative to lane 2.
- Luciferase assay in HEK293 cells transfected with 8XGTIIC-lux reporter (black bars) or with the reporter deleted of the TEAD-binding sites (delta8XGT), and with increasing doses of PFKFB3 expression plasmid. PFKFB3 converts fructose-6-P into fructose-2,6-P, a potent allosteric activator of PFK1 (Sola-Penna et al, 2010). Representative results of a single experiment with n = 2 biological replicates; three independent experiments were consistent. See Supplementary Fig S3P for controls of the delta8XGT reporter.
Data information: Throughout the figure, error bars represent mean ± SD. *P-value < 0.01.
Source data are available online for this figure.
To gain insights into other possible mechanisms by which glucose regulates YAP/TAZ, we then turned our attention to YAP-binding proteins: For this, we performed immunopurification of FLAG-YAP-5SA from MCF10A or MDA-MB-231 cells and identified candidate interacting proteins by mass spectrometry. As shown in Fig3D and Supplementary Table S4, we isolated several known interactors of YAP, including nuclear complexes (Ribeiro et al, 2010; Varelas et al, 2010; Yi et al, 2011; Couzens et al, 2013). We then turned our attention to novel interacting proteins, with particular attention to enzymes involved in glucose metabolism; indeed, it has been previously shown that some metabolic enzymes, such as PKM2, can fulfill non-metabolic functions by interacting with transcription factors (Chaneton & Gottlieb, 2012; Luo & Semenza, 2012). Our attention was immediately retained by the isolation, in both cell lines, of the phosphofructokinase enzyme isoform P (PFK1), key enzyme of glycolysis.
Phosphofructokinase (PFK1) mediates the first committed step of glycolysis, by phosphorylating F6P into fructose-1,6-bisphosphate (F1,6P). PFK1 is a central enzyme for the regulation of glycolysis, as it is subjected to a variety of regulations, including allosteric activation by fructose-2,6-bisphosphate (F2,6P) produced by PFK2 enzymes (Sola-Penna et al, 2010). Recent reports indicate that cancer cells take control over glycolysis by multiple mechanisms acting, directly or indirectly, on PFK1 activity (Lunt & Vander Heiden, 2011; Mor et al, 2011; Yi et al, 2012). Moreover, PFK1 and PFK2 expression is elevated in advanced breast tumors (Atsumi et al, 2002; Onodera et al, 2014), making PFK1 an interesting candidate to link glucose metabolism and YAP/TAZ activity. To explore the functional relevance of this biochemical observation, we designed independent siRNA duplexes to target endogenous PFK1 expression and challenged YAP/TAZ activity (Supplementary Fig S3H). Remarkably, knockdown of PFK1 with two independent siRNAs caused inhibition of YAP/TAZ activity in MDA-MB-231 cells (Fig3E; Supplementary Fig S3I). Also in this case, LATS1/2 siRNAs, mevalonate or pyruvate could not rescue inhibition of YAP/TAZ activity upon PFK1 knockdown (not shown). Thus, PFK1 is required to sustain YAP/TAZ activity and recapitulates the effects of 2DG shown above.
We then sought to validate PFK1 interaction with YAP. In proteomic experiments, several proteins, such as NF2 (Yin et al, 2013), are indirectly binding to YAP. To test for direct interaction, we performed co-immunoprecipitation between immobilized FLAG-PFK1 and bacterially expressed YAP, but we could hardly detect any binding (Fig3F); as a control FLAG-TEAD1 readily coprecipitated YAP (Fig3F). Since glucose metabolism and PFK1 intersect YAP/TAZ activity downstream of Hippo and mevalonate/RHO (see above), possibly acting on YAP/TAZ transcriptional complexes, we asked whether PFK1 might interact with TEADs: Surprisingly, as shown in Fig3G, we could indeed coprecipitate recombinant TEAD4 with FLAG-PFK1. In line, we detected an interaction between PFK1 and TEADs with transfected and endogenous proteins (Fig3H and I). This interaction occurred in the nucleus, as we found a proportion of the PFK1 protein in this compartment by immunofluorescence (Supplementary Fig S3N) and we could detect nuclear PFK1/TEAD1 complexes by proximity ligation assay (PLA) (Supplementary Fig S3O), a technique enabling in situ detection of endogenous protein–protein complexes (Jarvius et al, 2007). To explore whether the binding of TEADs to PFK1 requires the presence of YAP, we compared wild-type and Y429H-mutant TEAD4 [the latter unable to bind YAP/TAZ (Lai et al, 2011)]; as shown in Fig3I, both proteins interacted similarly with PFK1. To explore whether the binding of TEADs to PFK1 is influenced by PFK1 activity, we compared wild-type PFK1 and a PFK1 isoform bearing point mutations of key amino acid residues of the allosteric site for fructose-2,6-bisphosphate (F2,6P), a potent activator of PFK1 (Ferreras et al, 2009; Banaszak et al, 2011; Mor et al, 2011), and found a reduced binding of the mutated PFK1 isoform (Fig3J). F2,6P production is regulated by PFK2 enzymes (see scheme in Fig2A); among these, PFKFB3 is the isoform displaying the highest F6P to F2,6P kinase activity and which potently promotes PFK1 activity (Herrero-Mendez et al, 2009; Yalcin et al, 2009; De Bock et al, 2013); in line with our biochemical findings, PFKFB3 dose-dependently enhanced YAP/TAZ transcriptional activity in HEK293 cells (Fig3K), which typically display low levels of YAP/TAZ activity (Azzolin et al, 2012). Altogether, these results indicate that it is the enzymatically active pool of PFK1 that binds TEADs and fosters YAP/TAZ activity.
Glucose metabolism regulates the interaction between TEADs and YAP/TAZ
Data gathered so far indicates that glucose favors YAP/TAZ activity and suggests that PFK1 mediates these effects by interacting with TEADs. We then asked whether glucose metabolism could regulate binding of YAP/TAZ to TEADs. For this, we immunoprecipitated endogenous YAP from extracts of MDA-MB-231 cells, treated with vehicle or with 2DG, and monitored by immunoblot the amount of coprecipitating TEAD1. Strikingly, as shown in Fig4A, 2DG treatment reduced the interaction between YAP and TEAD1, without leading to quantitative YAP or TEAD1 nuclear exclusion (Supplementary Figs S3G and S4A). We obtained similar results by immunoprecipitating YAP and TAZ with a different antibody (Supplementary Fig S4B), or from extracts of MCF10A, HepG2 and UOK262 cells (Supplementary Fig S4C–E). Moreover, we also observed inhibition of YAP/TEAD1 complex upon glucose withdrawal, which was restored after adding back glucose (Fig4B; Supplementary Fig S4F). Finally, using chromatin immunoprecipitation (ChIP), we found that 2DG treatment reduced the occupancy of YAP at promoters of several genes with known or predicted TEAD-binding sites, including HMMR, TK1, CTGF and ANKRD1 (Zhao et al, 2008; Benhaddou et al, 2012; Wang et al, 2014) (Fig4C; Supplementary Fig S4G–I). Importantly, this indicates that glucose metabolism directly intersects YAP/TAZ activity.
Figure 4.
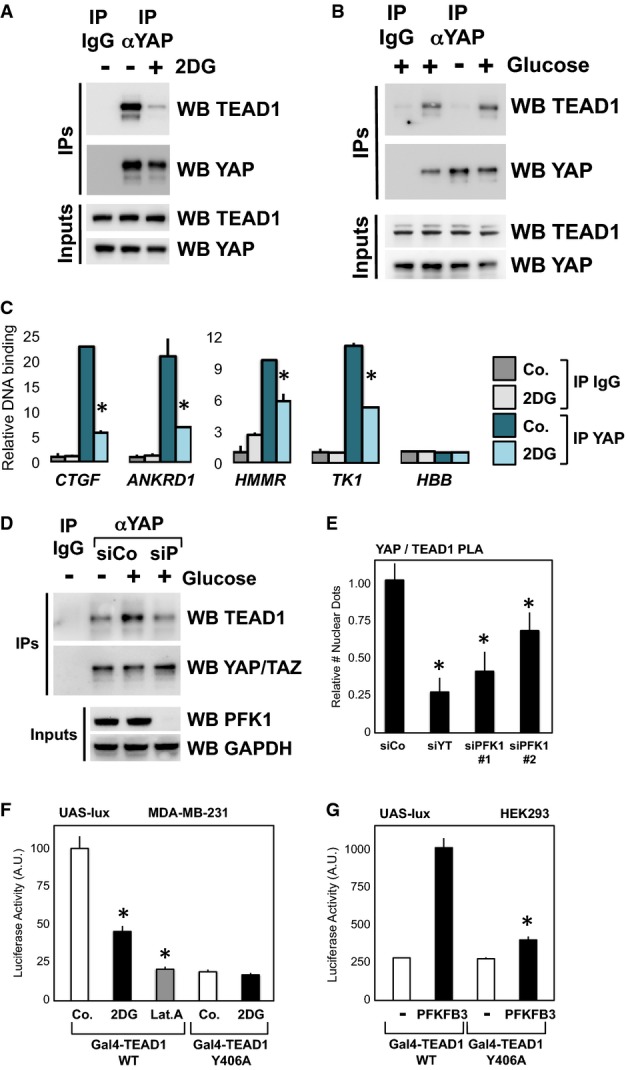
- Extracts of MDA-MB-231 cells treated for 24 h with vehicle (−) or with 2DG (+) were subjected to anti-YAP immunoprecipitation; coprecipitating proteins were then analyzed by Western blotting to detect TEAD1 interaction. Immunoprecipitation with an unrelated IgG serves as a negative control. Similar results were obtained in other cell lines, or in MDA-MB-231 cells by using a different anti-YAP/TAZ antibody (Supplementary Fig S4B–E).
- Coimmunoprecipitations from extracts of MCF10A cells released from contact inhibition by seeding them at low confluence for 36 h with glucose (+, lane 2), without glucose (−, lane 3), or cultured without glucose and then refed of glucose (+, lane 4). Immunoprecipitation with an unrelated IgG (lane 1) serves as a negative control. Similar results were obtained in HepG2 (Supplementary Fig S4F).
- Chromatin immunoprecipitation of MCF10A cells untreated (Co.) or treated with 100 mM 2DG for 24 h. Fragmented chromatin from each experimental condition was immunoprecipitated with control IgG or anti-YAP antibodies and subjected to qPCR to detect the TEAD-binding regions present in the CTGF, ANKRD1, HMMR and TK1 promoters. Amplification of Hemoglobin beta (HBB) serves as a negative control. See Supplementary Fig S4G for similar results in MDA-MB-231 cells. CTGF, ANKRD1 and RHAMM are known targets of YAP/TAZ; TK1 was included in the analysis because it is jointly regulated by glucose and YAP/TAZ (see Fig1F), and its proximal promoter contains two TEAD-binding sites that respond to YAP/TAZ activity (see Supplementary Fig S4H) and to glycolysis coherently (Supplementary Fig S4I). Values in control samples with control IgG were arbitrarily set to 1, and the other values are relative to this (see Materials and Methods). Data are shown as the mean ± SD of two independent experiments.
- Immunoprecipitation of YAP and TEAD1 is reinforced upon glucose supplementation (+), and this requires endogenous PFK1 levels (compare siCo. with siP siRNA transfected extracts).
- In situ interaction of YAP and TEAD1 is regulated by PFK1 by proximity ligation assay (PLA). Depletion of YAP/TAZ (siYT) or PFK1 with two independent siRNAs (siPFK1 #1, #2) reduced the number of nuclear YAP/TEAD1 dots relative to cells transfected with control siRNA (siCo.), suggesting PFK1 is required to stabilize YAP/TEAD1 interaction. See Supplementary Fig S4J for representative pictures of the PLA stainings.
- Luciferase assay in MDA-MB-231 cells transfected with UAS-lux reporter and expression plasmids encoding for in-frame fusions of the GAL4 DNA-binding domain with TEAD1, wild-type (WT) or unable to interact with YAP (Y406A mutant). WT TEAD1 can recruit YAP/TAZ and efficiently promote transcription, while Y406A TEAD1 can only sustain basal transcription (Li et al, 2010). Treatment with 2DG (black bars) inhibited transcription induced by WT TEAD1 but not of the Y406A mutant, in keeping with the observation that 2DG regulates YAP/TEAD1 interaction. Latrunculin A treatment (Lat.A) serves as a positive control for inhibition of YAP/TAZ. Representative results of a single experiment with n = 2 biological replicates; two independent experiments were consistent.
- Luciferase assay in HEK293 cells transfected as in (F). Cotransfection of PFKFB3, an activator of PFK1 activity, fosters the activity of TEAD1 only when it is able to interact with YAP. Representative results of a single experiment with n = 2 biological replicates; two independent experiments were consistent.
Data information: Throughout the figure, error bars represent mean ± SD. *P-value < 0.01.
Source data are available online for this figure.
This glucose-induced YAP/TEAD1 interaction was dependent on endogenous PFK1, as detected by co-immunoprecipitation (Fig4D) or in situ detection of endogenous protein–protein complexes by PLA (Fig4E; Supplementary Fig S4J). Thus, PFK1 stabilizes YAP/TAZ interaction with TEADs. Along this idea, we then surmised that if glucose metabolism regulates the ability of YAP/TAZ to interact with TEADs, then a TEAD isoform unable to bind YAP/TAZ should be insensitive to modulation of glucose metabolism. To test this hypothesis, we used GAL4–TEAD1 fusion proteins and compared wild-type TEAD1 with Y406A-mutant TEAD1, unable to interact with YAP/TAZ (Li et al, 2010): in these conditions, it is possible to uncouple the basal transcriptional effects of TEAD1 (observed in the Y406A mutant) from YAP/TAZ-induced transcription (observed only with WT TEAD1). As shown in Fig4F, 2DG inhibited transcription driven by WT TEAD1, but was unable to inhibit the basal activity of the Y406A mutant in MDA-MB-231 cells. Moreover, overexpressing PFKFB3 in HEK293 selectively enhanced transcription driven by WT TEAD1 (Fig4G). Taken together, these data support the notion that glycolysis regulates the interaction between YAP/TAZ and TEADs.
Interplay of glycolysis, PFK1 and YAP/TAZ for cancer cell growth
Aerobic glycolysis is considered an important hallmark of cancer cells and contributes to cancer cell survival and aggressiveness. Given the known pro-growth and pro-tumorigenic functions of YAP/TAZ, we then sought to determine whether inhibition of glycolysis could oppose these YAP/TAZ-dependent activities. First, we challenged the ability of activated S89A-mutant TAZ to promote self-renewal of MCF10A-MII mammary cells in a mammosphere-forming assay (Cordenonsi et al, 2011; Sorrentino et al, 2014). Inhibition of glycolysis by 2DG treatment, or by transfection of PFK1 and GPI siRNA, potently blocked the effects of TAZ (Fig5A; Supplementary Fig S5A–C). Second, we challenged the ability of YAP to sustain anchorage-independent growth in soft agar, another hallmark of YAP activation (Zhao et al, 2012). Knockdown of PFK1 by siRNA transfection decreased the clonogenic potential of MDA-MB-231 cells, recapitulating inhibition of YAP/TAZ (Fig5B); moreover, blockade of glucose metabolism with 2DG inhibited the growth of colonies experimentally induced by expression of activated YAP-5SA (Fig5C). Third, we challenged cell proliferation induced by cytoskeletal inputs and YAP/TAZ: in dense MCF10A monolayers, cell shape and F-actin remodeling are key for inhibition of YAP/TAZ activity, and thus for contact inhibition of growth; inducing a ‘wound’ in the monolayer causes the cells close to the edge of the wound to stretch, reactivate endogenous YAP/TAZ and proliferate (Zhao et al, 2007; Aragona et al, 2013). As shown in Fig5D, proliferation of the cells abutting the ‘wound’ was inhibited by treating cells with 2DG, indicating that glucose metabolism is required to sustain YAP/TAZ-induced proliferation.
Figure 5.

- A Mammosphere assay with MCF10A-MII cells. Retroviral expression of an activated form of TAZ (S89A mutant) increases the efficiency of primary mammosphere formation compared to parental cells (empty-vector transduced cells). Treatment of TAZ-expressing cells with 2DG (15 mM), or depletion of PFK1 (siP) or GPI (siG), impairs the mammosphere-promoting ability of TAZ. See Supplementary Fig S5A–C for secondary mammospheres and representative pictures. Representative results of a single experiment with n = 4 biological replicates; two independent experiments were consistent.
- B Depletion of PFK1 (siP) impairs the colony-forming ability of MDA-MB-231 cells in soft agar, recapitulating the requirement for YAP/TAZ (siYT). Representative results of a single experiment with n = 2 biological replicates; two independent experiments were consistent.
- C Expression of an activated form of YAP (5SA) strongly promotes the growth of MDA-MB-231 colonies in soft agar, and this is inhibited by 2DG treatment (3 mM). Each box signifies the upper and lower quartiles of data (colony size), while the whiskers extend to the minimum and maximum data points. On the right: representative pictures of colonies growing from 5SA-YAP-expressing cells, treated with vehicle or with 2DG. Representative results of a single experiment with n = 2 biological replicates; four independent experiments were consistent.
- D MCF10A cells were seeded at high density for 48 h, leading to YAP/TAZ inhibition and growth arrest (High); scratching the monolayer locally enables cell spreading and activates YAP/TAZ, thus inducing cell proliferation (Wound) (Zhao et al, 2007; Aragona et al, 2013). Overnight treatment of cells with 2DG (15 mM) inhibited such YAP/TAZ-induced proliferation. The graph reports the quantification of proliferating cells in the indicated areas, without or with 2DG treatment. To count cells abutting the wound, we arbitrarily set a 100-μm distance from the wound. See Supplementary Fig S5D for representative pictures of a wounded area. Representative results of a single experiment with n = 2 biological replicates (> 700 cells/replicate); three independent experiments were consistent.
- E Clonal expansion induced by overexpression of the YAP homologue Yki in the Drosophila wing imaginal disk is restricted by phosphofructokinase (Pfk) RNAi. Panels on the left show pictures of wing imaginal disks bearing clones of cells (marked by GFP) with mutation of the lethal giant larvae tumor suppressor gene (lgl−) and overexpression of Yki (ykiover), induced by the MARCM technique (Lee & Luo, 2001). The dotted line indicates the outline of the disks. In this genetic setup, the survival of clones within the wing pouch, that is, in the distal region of the wing disk, is strictly dependent on Yki activation (Grzeschik et al, 2010; Menendez et al, 2010; Khan et al, 2013). Upon downregulation of phosphofructokinase, the growth of lgl−; ykiover clones was inhibited, as shown by quantification of clone area (****P < 0.0001, unpaired t-test). n = 34 disks for each genotype. Scale bars 80 μm.
- F Pfk silencing downregulates the Yki target gene DIAP1 in lgl−; ykiover clones. Panels show whole-mount immunostainings for DIAP1 protein levels, a hallmark of Yki transcriptional activity (Huang et al, 2005), on wing imaginal disks of the indicated phenotypes, as in (E). GFP identifies mutant cells, growing within an otherwise wild-type tissue. DIAP1 is autonomously upregulated in lgl−; ykiover clones, while it appears downregulated upon Pfk RNAi. This is consistent with a role for Pfk in regulating Yki transcriptional activity. See Supplementary Fig S5E for lower magnifications of the same wing disks. See Supplementary Fig S5F and G for similar results obtained with dMyc, another target of Yki (Neto-Silva et al, 2010; Ziosi et al, 2010). Scale bars, 20 μm.
- G MDA-MB-231 cells were growth-inhibited by glucose withdrawal for 48 h (−Glu), and then proliferation was induced by supplementing glucose in the medium for 24 h (+Glu). Culture medium was without glutamine to specifically measure glucose-dependent growth. Quantification of proliferation, as measured by BrdU incorporation, indicates that cells depleted of YAP/TAZ (siYT #1) are unable to efficiently restart proliferation in response to glucose compared to cells transfected with control siRNA (siCo.). Similar results were obtained with an independent YAP/TAZ siRNA mix (not shown). Representative results of a single experiment with n = 2 biological replicates (> 1,000 cells/replicate); three independent experiments were consistent.
- H, I Clonogenic assay with UOK262 cells. Parental cells (black bars) are highly glycolytic, while their FH-reconstituted counterpart (gray bars) has reduced glycolysis as they can efficiently perform mitochondrial respiration (Yang et al, 2013). Cells were seeded at clonogenic density and grown in the presence of titrated doses of 2DG (0.25, 0.5, 1 mM) to inhibit glucose metabolism (H) or in the presence of VP (0.3, 1, 3 μM) to inhibit the cooperation between YAP/TAZ and TEADs (Liu-Chittenden et al, 2012) (I). Graphs show the quantification of colonies after 10 days, relative to untreated cells. UOK262 cells are more sensitive than UOK262-FH to 2DG; UOK262 cells are also more sensitive to small-molecule inhibition of YAP/TAZ, in keeping with higher YAP/TAZ activity (shown above). Representative results of a single experiment with n = 3 biological replicates; two independent experiments were consistent.
We then asked whether glycolysis could regulate YAP/TAZ in vivo, by using Drosophila as an established model system in which activation of the YAP/TAZ homologue Yki induces hyperplastic growth (Halder & Johnson, 2011; Harvey et al, 2013). Specifically, we induced clones of cells with mutation of the lethal (2) giant larvae (lgl) tumor suppressor and overexpression of Yki, which cause the Yki-dependent overgrowth of mutant cells in the wing pouch, eventually developing into tumors (Grzeschik et al, 2010; Menendez et al, 2010; Khan et al, 2013). As shown in Fig5E, silencing of phosphofructokinase by RNAi quantitatively counteracted the growth of these clones. Importantly, this was accompanied by inhibited expression of DIAP1 and Myc, two established Yki/TEAD target genes in Drosophila (Harvey et al, 2003; Pantalacci et al, 2003; Udan et al, 2003; Huang et al, 2005; Wu et al, 2008; Neto-Silva et al, 2010; Ziosi et al, 2010) (Fig5F; Supplementary Fig S5E–G). This indicates that phosphofructokinase regulates and is instrumental for Yki activity during tumorigenesis of fly larval tissues.
To further extend the functional connections between glucose metabolism and YAP/TAZ, we tested whether proliferation promoted by glucose would depend, at least to some extent, on YAP/TAZ. In control MDA-MB-231 cells, glucose deprivation for 48 h induces growth arrest, and supplementing back glucose in the medium restarts proliferation; upon YAP/TAZ silencing, however, glucose-induced proliferation is greatly impaired .(Fig5G), although not completely as expected from the general (and YAP/TAZ independent) functions of glucose. We also compared parental UOK262 cells (glycolytic) with FH-reconstituted UOK262 cells [that rely more on mitochondrial respiration (Yang et al, 2013; Sudarshan et al, 2009)] for their sensitivity to inhibition of glucose metabolism and to inhibition of YAP/TAZ. As expected, in clonogenic assays, parental UOK262 cells are more sensitive to 2DG (Fig5H). Strikingly, parental cells were also more sensitive to inhibition of YAP/TAZ activity by verteporfin (VP), a small molecule that specifically impairs the binding of YAP/TAZ to TEADs (Liu-Chittenden et al, 2012) (Fig5I). In other words, this suggests that cells relying on high levels of aerobic glycolysis for growth display a corresponding high requirement for YAP/TAZ activity.
Collectively, our findings indicate that glycolysis is required for the full deployment of YAP/TAZ tumorigenic activities and that YAP/TAZ mediate a segment of the proliferative effects of glucose metabolism, reflecting regulation of YAP/TAZ transcription downstream of glucose in human cells and fly tissues. These results are also in line with the finding that glycolysis regulates YAP/TEAD complex formation, because TEAD binding is required for most YAP/Yki tumorigenic activities, and in particular for the biological assays used above (Ota & Sasaki, 2008; Wu et al, 2008; Zhao et al, 2008; Zhang et al, 2009).
A transcriptional signature associated to aerobic glycolysis correlates with elevated YAP/TAZ activity and is predictive of poor prognosis in primary tumors
YAP/TAZ activation and a shift toward a glycolytic metabolism are commonly observed during tumor progression. This is true in particular for breast cancers, where YAP/TAZ activity is associated with high-grade (G3) tumors and with the cancer stem cell (CSC) content of the tumors, reflecting YAP/TAZ requirement for CSC self-renewal and cancer aggressiveness (Cordenonsi et al, 2011; Chen et al, 2012). Moreover, mammary tumor-initiating cells and undifferentiated basal-like breast cancers display a shift toward aerobic glycolysis, which in turn is required for their self-renewal ability (Dong et al, 2013; Feng et al, 2014). We thus sought to test whether YAP/TAZ activation associates with glucose metabolism in human mammary tumors.
We first derived a gene expression signature experimentally associated with high glucose metabolism in cells of mammary origin (glucose signature) by selecting the genes that were downregulated by 2DG treatment both in MCF10A and in MDA-MB-231 microarrays (see Materials and Methods and Supplementary Table S5). We then analyzed a large metadataset collecting gene expression and associated clinical data of more than 3,600 primary mammary tumors (Cordenonsi et al, 2011; Montagner et al, 2012) and evaluated how the levels of glucose signature were associated with YAP/TAZ activity. Strikingly, we found that the glucose signature is positively and strongly correlated with expression of previously established gene signatures denoting YAP/YAZ activity (Fig6A; Supplementary Fig S6A); moreover, tumors classified according to high (versus low) glucose signature also display higher activity of YAP/TAZ (Fig6B; Supplementary Fig S6B).
Figure 6.

- A Scatter plot (gray dots) and linear regression (red line, slope 0.532) of standardized expression values indicate a positive correlation between a gene signature experimentally associated with active glucose metabolism (glucose signature) and a gene signature denoting YAP/TAZ activity, in a metadataset collecting n = 3,661 primary human breast cancers (see Materials and Methods). The glucose signature is composed of the genes downregulated upon 2DG treatment, that is, requiring active glucose metabolism for their transcription, both in MCF10A and in MDA-MB-231 cells (see Supplementary Table S5). Pearson ρ quantifies the linear dependence between the levels of the two signatures. The coefficient of determination is r2 = 0.731, P-value < 0.0001.
- B Primary human breast cancers of the metadataset were stratified according to high or low glucose signature score, and then, the levels of the YAP/TAZ signature score were determined in the two groups (see Materials and Methods for details on the statistical methods to quantify scores). YAP/TAZ activity is significantly higher in tumors with high levels of the glucose signature, as visualized by box-plot. The bottom and top of the box are the first and third quartiles, and the band inside the box is the median; whiskers represent 1st and 99th percentiles; values lower and greater are shown as circles (P < 0.0001, n = 3,661).
- C, D Primary human breast cancers of the metadataset were classified according to high or low glucose signature score, and then, the levels of the Staminal or stem tumorigenic signature scores, associated to normal and cancer mammary stem cells, were determined in the two groups. Gene expression associated to mammary stem cells is significantly higher in tumors with high levels of the glucose signature, as visualized by box-plot (P < 0.0001, n = 3,661).
- E Genes regulated by glucose metabolism (glucose signature) are elevated in G3 as compared to G1 grade mammary tumors of the metadataset (P < 0.0001; G1 versus G3 unpaired t-test). A similar behavior is observed by using the YAP/TAZ signature (Cordenonsi et al, 2011). See Materials and Methods for details on the statistical methods to quantify average signature expression. Data are shown as mean ± standard error of the mean (SEM).
- F Kaplan–Meier analysis representing the probability of metastasis-free survival in breast cancer patients from the metadataset stratified according to high or low glucose signature score. The log-rank test P-value reflects the significance of the association between high levels of the glucose signature score and shorter survival. A similar behavior is observed by using the YAP/TAZ signature (Cordenonsi et al, 2011).
- G Genes regulated by glucose metabolism but not by YAP/TAZ (glucose NOT YT signature, see Supplementary Table S5) are not expressed at higher levels in G3 grade mammary tumors of the metadataset. See Materials and Methods for details on the statistical methods to quantify average signature expression. Data are shown as mean ± standard error of the mean (SEM).
- H Kaplan–Meier analysis representing the probability of metastasis-free survival in breast cancer patients from the metadataset stratified according to high or low glucose NOT YT signature score, which show no differences.
Prompted by this observation, we tested whether the glucose signature correlates with cancer features previously associated to YAP/TAZ activity, such as tumor grade and the content of CSC (Cordenonsi et al, 2011; Chen et al, 2012). As shown in Fig6C–E, we indeed found that glucose signature expression levels associated to higher expression of mammary stem cell signatures (Liu et al, 2007; Pece et al, 2010), and it was significantly elevated in G3 versus G1 grade tumors (P < 0.0001). Remarkably, by univariate Kaplan–Meier survival analysis, we also found that tumors expressing high levels of the glucose signature (‘High’) displayed a significant higher probability to develop recurrence when compared to the ‘Low’ group (Fig6F). Of note, the higher expression of the glucose signature in G3 tumors, and its ability to stratify patients, was completely lost when we selected from the glucose signature only the genes that were regulated by 2DG but not by YAP/TAZ siRNA (Fig6G and H; Supplementary Table S5). Collectively, the data indicate that during mammary tumor progression, metabolic reprogramming toward aerobic glycolysis is accompanied by elevated YAP/TAZ activity.
Discussion
Cell metabolism and signaling pathways are powerful regulators of cell proliferation and tumorigenesis; how these separate inputs coordinate each other to induce coherent biological responses remains partially understood. Here, we propose that glycolysis, besides its core biochemical role, also contributes to regulate the activity of YAP/TAZ and, in doing so, fuels the proliferative and tumorigenic functions of these powerful oncogenic factors. Blocking glucose metabolism, or shifting cellular metabolism away from glycolysis, impairs YAP/TAZ transcriptional activity and their ability to promote cell proliferation, cancer cell self-renewal and clonogenic abilities in vitro, and tissue overgrowth in Drosophila. In line with this, growth/survival promoted by glucose incorporates YAP/TAZ activity as downstream effectors. Mechanistically, we found that phosphofructokinase (PFK1), mediating the first committed step of glycolysis, interacts with the transcription factors TEADs to stabilize their interaction with YAP/TAZ. In doing so, glucose metabolism regulates the recruitment of YAP at target promoters, providing a rationale for the regulation of YAP/TAZ transcriptional activity.
Recently, DeRan and colleagues reported an effect of energy stress on YAP/TAZ activity: AMPK-activating drugs such as phenformin and AICAR induce Angiomotin (AMOT) phosphorylation, activation of the LATS1/2 kinases and inhibition of YAP/TAZ (DeRan et al, 2014). Although inhibition of glucose metabolism clearly induces activation of AMPK, our data indicate that the AMPK-AMOT-LATS1/2 axis is not sufficient to explain regulation of YAP/TAZ by glucose and glycolysis. Thus, it is likely that multiple parallel mechanisms cooperate to coordinate glucose metabolism with YAP/TAZ activity.
The finding that glucose metabolism intersects YAP/TAZ signaling at the level of TEAD factors underscores the idea that TEADs are central players for the regulation of YAP/TAZ. Indeed, recent findings indicate that disruption of YAP–TEADs interaction may be a promising therapeutic strategy against YAP-driven human cancers (Liu-Chittenden et al, 2012; Jiao et al, 2014). Both in Drosophila and in mammalian systems, TEADs normally interact with transcriptional inhibitors, such as the TGI and VGLL4 Tondu-domain-containing proteins, and YAP/TAZ replace these factors to activate gene transcription (Koontz et al, 2013; Jiao et al, 2014); in future, it will be interesting to understand how these cofactor exchanges are reciprocally regulated and whether these occur while TEAD factors are on or off DNA.
This study also highlights some peculiarities of YAP/TAZ. Classical oncogenes such as Ras, Myc and HIFs actively induce metabolic reprogramming toward aerobic glycolysis (Gordan et al, 2007; Kroemer & Pouyssegur, 2008; Schulze & Harris, 2012), while YAP/TAZ are instead regulated downstream to changes in glucose and mevalonate metabolism. Since YAP/TAZ are also regulated by cues embedded in the cell microenvironment (Hippo kinase cascade, ECM mechanics, cell-cell adhesion structures, GPCR and Wnt signaling), it is tempting to speculate that this mechanism might represent a way by which cell metabolism and tissue-level information are integrated into a common output. YAP/TAZ also appear different from another transcription factor regulated by glucose, ChREBP/MondoA: ChREBP mainly regulates expression of metabolic enzymes and thus serves as a feedback transcriptional mechanism to adjust enzyme levels to nutrient availability and to coordinate lipid and glucose metabolism (Dang, 2012; Havula & Hietakangas, 2012); YAP/TAZ instead incorporate clues from glucose metabolism into cancer cell growth and self-renewal. Supporting this view, by bioinformatics analyses, we found a striking enrichment of YAP/TAZ target genes in primary mammary tumors displaying expression of glucose-regulated genes, and this correlated with a higher grade and aggressiveness of the tumors. Interestingly, this also correlated with gene expression associated to the cancer stem cell (CSC) content of the tumors, fitting well with the CSC-promoting ability of YAP/TAZ (Cordenonsi et al, 2011; Chen et al, 2012) and with the finding that breast tumor-initiating cells display a shift toward glycolysis, which in turn is required for their self-renewal ability (Dong et al, 2013; Feng et al, 2014).
In sum, metabolic reprogramming would fulfill the double duty of providing an optimal metabolism to support cell growth and at the same time sustain, through YAP/TAZ, the genetic program that promotes proliferation and tumor malignancy.
Materials and Methods
Reagents
2-deoxy-D-glucose, lonidamine, cerivastatin, DL-mevalonic acid 5-phosphate, DON, AZS, GlcNAc, D-mannose, D-galactose, D-Luciferin, latrunculin A and verteporfin were from Sigma. CPRG was from Roche. Coelenterazine was from LifeTechnologies.
8XGTIIC-luciferase plasmid is Addgene 34615. The Renilla luciferase version was derived by subcloning of the promoter region into promoterless pRLuc. CTGF-luciferase was created by amplifying the genomic region corresponding to −225 bp from the TSS of the human CTGF promoter, containing three TEAD-binding elements and the TATA box, into pGL3b. TK1-luciferase was created by amplifying the genomic region corresponding to −552 bp from the ATG of the human TK1 locus; the two predicted TEAD-binding elements start at −200 and −453. Doxycycline-inducible reporter systems were obtained by subcloning the tet-responsive element from FudeltaGW plasmid upstream of the promoter-luciferase elements into a puromycin-resistant retroviral backbone. rtTA was subcloned from Addgene 19780 into pBABE-hygro. NF2/Merlin plasmid is Addgene 19699. WT and Y429H MYC-TEAD4 plasmids are Addgene 24638 and 33041. WT and F2,6P mutant FLAG-PFK1 isoform P plasmid was subcloned from Addgene 23869. Mutation of the F2,6P allosteric site of PFK1 was carried out based on Banaszak et al (2011) and Ferreras et al (2009) and entails the following mutations: R481A, R576A, R665A, H671A, R744A (reference sequence is NP_002618). PFKFB3 was subcloned from Addgene 23668. Plasmids encoding GAL4 fusions of WT or Y406A TEAD1 are Addgene 33108 and 33034. GST-YAP and GST-TEAD4 plasmids were obtained by standard subcloning. CMV-lacZ, RBPJ-luciferase, 6XE2F-luciferase and UAS-luciferase have been previously described (Lukas et al, 1997; Dupont et al, 2009; Inui et al, 2011). All plasmids were sequence verified prior to use.
siRNA oligos were standard dsRNA oligos with overhanging dTdT from LifeTechnologies (hereafter, targeted sense sequence of the mRNA): GPI (Qiagen) 1 guuuggaauugacccucaa; GPI 2 gcuugauggcagugcucaa; PFK1 1 ggagaaccgugcccggaaa; PFK1 (Qiagen) 2 cgggcaaccugaacaccua. YAP/TAZ siRNA was as in Dupont et al (2011). LATS1/2 siRNA mixes were as in Aragona et al (2013). AMPKa1/2 siRNA (Qiagen) were as in DeRan et al (2014): A1 1 cacgauauucuguacacaa; A1 2 gggaucaguuagcaacuau; A2 1 gaagucagagcaaaccgua; A2 2 ggaagguagugaaugcaua. TEAD1 ugaaugugcaaugaagcggcg; TEAD2 ccuggugaauuucuugcacaa; TEAD3 uaccuugcucucaaucuggag; TEAD4 uuuccugcacacacgucucuu. Control siRNAs were Qiagen AllStars Negative Control siRNA or a 1:1:1 mix of Scramble, GFP and Luc* siRNAs, which were used back-to-back, with results comparable to non-transfected cells.
Cell culture and transfections
MCF10A (ATCC), MCF10A-MII (a gift from Dr. Santner) and MCF10A-MII-TAZ-S89A cells and their empty-vector controls (Cordenonsi et al, 2011) were cultured in DMEM/F12 supplemented of 5% horse serum, 2 mM glutamine, insulin, cholera toxin, hydrocortisone and EGF (Debnath et al, 2003); MDA-MB-231 (STR verified) in DMEM/F12 with 10% FBS and 2 mM glutamine; Hs578T (ATCC) in DMEM with 10% FBS, 2 mM glutamine and insulin; HEK293 (ATCC) in DMEM with 10% FBS and 2 mM glutamine; HepG2 (ATCC) in MEM with 10% FBS, 2 mM glutamine and NEAA; and UOK262 (a gift from Dr. Linehan and the UOB Tumor Cell Line Repository at NIH Bethesda) in DMEM with 10% FBS, 2 mM glutamine and 1 mM pyruvate. To grow cells in galactose, DMEM/F12 medium without glucose (BioWest) was supplemented with 10 mM galactose, 10% dyalized FBS and 2 mM glutamine. All cells were routinely tested for mycoplasma contaminations with commercial PCR kits (Sigma). siRNA transfections were done with Lipofectamine RNAi MAX (Invitrogen). Plasmid DNA transfections were done with Transit-LT1 (MirusBio). Retroviral infections were carried out following standard procedures and protocols.
Mammosphere assay was carried out by plating 1,000 cells for each 24 well, and by plating 4 wells replicates for each sample; primary mammospheres were counted after 6 days. Differences in cell viability at plating were excluded based on TUNEL assays (not shown). Mammospheres were dissociated in trypsin and an equal number of single cells replated to grow secondary mammospheres. For soft agar assay, we plated 10,000 MDA-MB-231 cells in triplicate in 35-mm dishes in growth medium containing 0.3% low melting agarose, over a 0.6% agarose layer without cells; colonies were grown for 2 weeks (5SA-YAP-expressing cells and their controls) or for 3 weeks (cells transfected with siRNA). Wound assay was performed by plating 1.5 × 106 MCF10A cells in 24 wells containing fibronectin-coated 13-mm glass slides; after 24 h, the cell monolayer was scratched with a sterile P1000 tip, washed and grown overnight. 2DG was added after washing. For clonogenic assay, we plated 800 UOK262 cells in a 35-mm petri dish, in triplicate; after 10 days, colonies were stained with crystal violet, photographed and counted with ImageJ software by quantifying the total stained area.
ECAR and OCR measurements
The XF24 extracellular flux analyzer (Seahorse Bioscience) was used to detect real-time changes in cellular respiration and glycolysis rates. Cells were cultured in standard XF24 plates, by seeding 120,000 (MDA-MB-231) or 90,000 (MCF10A-MII) cells/well 24 h before performing the measures; for experiments with siRNA, cells were transfected, reseeded after 24 h and measured after further 24 h. Analysis of the extracellular acidification rate (ECAR) reflects lactate secretion and serves as indirect measure of glycolysis rate; oxygen consumption (OCR) reflects cellular respiration and is directly determined. All measurements were performed following manufacturer’s instructions and protocols with at least four biological replicates for each condition and normalized to total protein content as determined by Bradford assays.
Real-time PCR
Total RNA was extracted using RNeasy Mini Kit (Qiagen), and contaminant DNA was removed by RNase-Free DNase Set (Qiagen). cDNA synthesis was carried out with dT-primed M-MLV Reverse Transcriptase (LifeTechnologies). Real-time qPCR analyses were carried out with triplicate samplings of each sample cDNA on a Rotor-Gene Q (Qiagen) thermal cycler with FastStart SYBR Green Master Mix (Roche) and analyzed with Rotor-Gene Analysis 6.1 software. Expression levels are calculated relative to GAPDH. See Supplementary Table S6 for the sequences of primers.
Luciferase assays
For transient transfections, cells were typically plated in 24-well format and luciferase reporter plasmids were transfected with CMV-lacZ to normalize for transfection efficiency based on CPRG (Merck) colorimetric assay, together with plasmids encoding for the indicated proteins; DNA content was kept uniform by using pKS Bluescript. Cells were harvested in Luc lysis buffer (25 mM Tris pH 7.8, 2.5 mM EDTA, 10% glycerol, 1% NP-40). Luciferase activity was determined in a Tecan plate luminometer with freshly reconstituted assay reagent (0.5 mM D-Luciferin, 20 mM tricine, 1 mM (MgCO3)4Mg(OH)2, 2.7 mM MgSO4, 0.1 mM EDTA, 33 mM DTT, 0.27 mM CoA, 0.53 mM ATP). For stable cell lines, cells were plated in 12-well format and treated as indicated before harvesting; normalization was based on total protein content, as measured by Bradford assays. Each sample was transfected at least in two biological duplicates in each experiment to determine the experimental variability; each experiment was repeated independently with consistent results.
Microarrays and glucose signatures
For microarrays of genes regulated by glucose uptake, cells were plated in 60-mm plates, treated with 2DG and harvested after 24 h. For YAP/TAZ-regulated genes, cells were plated in 60-mm plates, transfected with siRNA and harvested 48 h after transfection. For each experimental condition, we prepared four biological replicates that were processed in parallel. Total RNA was extracted using RNeasy Mini Kit (Qiagen), and contaminant DNA was removed by RNase-Free DNase Set (Qiagen). RNA quality and purity were assessed on the Agilent Bioanalyzer 2100 (Agilent Technologies); RNA concentration was determined using the NanoDrop ND-1000 Spectrophotometer (NanoDrop Technologies). As control of effective gene modulation and of the whole procedure, we monitored the expression levels of established markers (TXNIP for 2DG, CTGF and ANKRD1 for YAP/TAZ) by qPCR prior to microarray hybridization. Labeling and hybridization were performed according to Affymetrix One Cycle Target Labeling protocol on HG-U133 Plus 2.0 arrays (Affymetrix). Microarray data are available at Gene Expression Omnibus under accession GSE59232.
All data analyses were performed in R (version 3.0.2) using Bioconductor libraries (BioC 2.13) and R statistical packages. Probe level signals were converted to expression values using robust multi-array average procedure RMA (Irizarry et al, 2003) of Bioconductor affy package. Differentially expressed genes were identified using Significance Analysis of Microarray algorithm coded in the samr R package (Tusher et al, 2001). In SAM, we estimated the percentage of false-positive predictions (i.e., false discovery rate, FDR) with 100 permutations.
To identify genes associated with high glucose uptake in cells of mammary origin (glucose signature), we compared the expression levels of MCF10A and MDA-MB-231 cells grown in high glucose or in the presence of 2-deoxy-glucose (2DG) and selected those probe sets with a fold change ≤ −3 in both MCF10A and MDA-MB-231 comparisons. This selection resulted in 298 probe sets downregulated by the presence of 2DG in the culture medium (Supplementary Table S5). We then refined this selection eliminating from the glucose signature the genes that were also regulated upon knockdown of YAP/TAZ, that is removing 2DG-regulated genes with an FDR ≥ 0.1% in the comparisons between YAP/TAZ-depleted MCF10A and MDA-MB-231 cells and their controls (Supplementary Table S5).
To functionally annotate genes coregulated by glucose and YAP/TAZ, we consider the Biological Process Gene Ontology (GO) categories of the Database for Annotation, Visualization and Integrated Discovery (DAVID http://david.abcc.ncifcrf.gov/home.jsp). GO terms were considered significant at a confidence level of 95%.
Drosophila assays
All the strains were obtained from the Bloomington Stock Center and grown under standard conditions. For flippase activation, non-overcrowded cultures of 48 ± 6 h after egg-laying individuals grown at 25°C on standard medium were transferred to a water bath at 37°C for 20 min. After additional 72 h at 25°C, larvae were dissected for analyses. Genotypes analyzed were yw,hs-Flp,tub-Gal4,UAS-GFP/w; tub-Gal80,FRT40A/l(2)gl4,FRT40A; UAS-yki/+ and yw,hs-Flp,tub-Gal4,UAS-GFP/w; tub-Gal80,FRT40A/l(2)gl4,FRT40A; and UAS-yki/UAS-PfkRNAi.
For immunostainings, larvae were dissected in PBS1X and carcasses were fixed, washed and immunostained following standard methods. Primary antibodies were rabbit aPKC (Santa Cruz Biotechnology, 1:200), mouse DIAP1 (B. Hay, 1:200) and mouse dMyc (P. Bellosta, 1:5). Secondary antibodies were anti-mouse Alexa Fluor 555 (Life Technologies) and anti-rabbit DyLight CY5 (Jackson Immunoresearch). Wing disks were isolated and mounted in Vectashield (Vectorlabs), and images were acquired by a Leica TCS SP2 confocal microscope. All figures show single, 1-μm-thick tissue sections.
Clone areas (in pixel2) were measured using ImageJ free software (NIH) on images captured with 90i wide fluorescence microscope (Nikon). Areas of clones grown as multilayers are likely to be underestimated. Disks were scored for clones included in the wing pouch region for a total of 34 disks, and the average clone area was normalized to the wing pouch size.
Antibodies and microscopy
Antibodies were FLAG-M2 HRP or agarose conjugate (Sigma), YAP/TAZ (sc101199), YAP for IP (Abcam 52771), TAZ for IP (Sigma HPA039557), phospho S127 YAP (CST4911), PFK1 (Abcam 119796, sc130227 or CST5412), TEAD1 (BD 610922), TEAD1 for IP (Abcam 133533), TEAD4 (sc101184), MYC (sc789), total S6 (CST2217), phospho S6 (CST5364), AMPK (CST2532) and GAPDH (Millipore MAB374).
For immunofluorescence, cells were fixed for 10 min in 4% PFA, washed in PBS and either stored dried at −80°C or directly permeabilized and processed for immunostaining as described in Dupont et al (2011). Proximity ligation assays were performed as indicated by the provider’s protocol (OLink Bioscience), after an overnight incubation with primary antibodies following our standard protocol. For PLA, antibodies were YAP (CST4912) and TEAD1 BD, and PFK1 sc. pictures were taken at the confocal microscope by selecting the maximal nuclear section. Images were acquired with a Leica SP5 confocal microscope equipped with CCD camera using Volocity software (Perkin Elmer). For BrdU, cells were fixed and processed according to manufacturer’s instructions (BrdU cell proliferation kit, Merck) and images acquired with a Leica DM5000B microscope.
Immunoprecipitations
For immunoprecipitations, cells were lysed in HPO buffer (50 mM Hepes pH 7.5, 100 mM NaCl, 50 mM KCl, 2 mM MgCl2, 1% Triton X-100, 0.5% NP-40, 5% glycerol) with proteases (Merck) and phosphatase (Sigma) inhibitors, and homogenized by sonication (Diagenode Bioruptor). Extracts of equal total protein content and concentration were then subjected to immunoprecipitation with primary antibodies previously bound to protein A-conjugated sepharose (GE Healthcare) in 2% BSA. Control IgG for pull-downs with anti-YAP and anti-TEAD1 rabbit antibodies was anti-HA rabbit polyclonal (sc805). After 2.5 h of incubation on a rotator at 4°C, beads were washed three times in the same buffer and the purified proteins were boiled in Laemmli final sample buffer for Western blotting with species-specific secondary HRP-conjugated antibodies (ExactaCruz).
For studies with recombinant proteins, GST-YAP and GST-TEAD4 were produced and purified from bacteria with standard protocols; FLAG-PFK1 and FLAG-TEAD1 were purified from HEK293-transfected lysates by anti-FLAG-M2 immunoprecipitation, followed by elution with 3xFLAG peptide (Sigma). All proteins were dyalized in BC100. Proteins were mixed in HPO buffer with 5% BSA and incubated for 2 h at 4°C, followed by another 30 min together with anti-FLAG-M2 agarose-conjugated beads. Beads were washed three times in the same buffer, and the purified proteins were boiled in Laemmli final sample buffer for Western blotting.
For mass spectrometry of YAP-interacting proteins, 50% confluent cultures (150-mm dishes) of MCF10A or MDA-MB-231 cells stably expressing FLAG-YAP-5SA were harvested in 1 ml HPO buffer and homogenized by sonication. Controls were parental cells. Freshly prepared extracts from two sibling plates were joined and subjected to immunoprecipitation with agarose-conjugated FLAG M2 antibody for 2.5 h at 4°C on a rotator. After three washes in the same buffer, immunopurified proteins were eluted by adding 3xFLAG peptide (Sigma) for 30 min at 4°C, in order to reduce aspecific purification of proteins on agarose beads. Eluted proteins were run in a 4–12% MOPS-SDS gel (LifeTechnologies) to separate them from the elution peptides, fixed and stained with colloidal Coomassie. The gel was sent to EMBL Proteomic Facility for in-gel tryptic digestion and mass spectrometry. Proteins purified with similar frequency from control and FLAG-YAP-expressing lysates were considered non-specific background; all other proteins were considered for further analysis.
Chromatin immunoprecipitation
Cells were plated in duplicate, using two 150-mm dishes for each replicate. After the indicated treatments, cells were crosslinked by adding to the culture medium 1/10 of fresh formaldehyde solution (11% formaldehyde 0.1 M NaCl, 1 mM EDTA, 0.5 mM EGTA, 50 mM HEPES pH 7.5) for 10 min at RT, followed by quenching with 125 mM glycine for 5 min. After washing in PBS, cells were harvested in PBS with protease inhibitors and pelleted for 5 min at 1,500 rcf at 4°C. Cells were resuspended in cold LB1 (10 mM NaCl; 1 mM EDTA; 50 mM; HEPES pH 7.5; 10% glycerol; 0.5% NP-40; 0.25% Triton X-100) and incubated for 20 min, pelleted and resuspended in LB2 (10 mM Tris pH 8.0; 200 mM NaCl; 1 mM EDTA; 0.5 mM EGTA) and incubated for 10 min; and pelleted and resuspended in sonication buffer (10 mM Tris pH 8.0; 1 mM EDTA; 100 mM NaCl; 0.5 mM EGTA; 0.1% sodium deoxycholate; 0.5% N-lauroylsarcosine). Sonication was carried out with a Branson Sonfier 450 to obtain sheared chromatin of 200–600 bp fragments. Effective sonication and quantification of the DNA to equalize samples was carried out on 1% of each sample after decrosslinking; this sample was also used as input control for qPCR. The same total amount of sheared chromatin per sample (in the range of 120 μg) was supplemented with 1% Triton X-100 and subjected to immunoprecipitation o.n. at 4°C with anti-YAP (Abcam) or with control rabbit IgG. Protein A dynabeads (LifeTechnologies) were added for 2 h after extensive blocking in 0.5% BSA. We performed the following washes (5 min each): low salt (0.1% SDS, 2 mM EDTA, 1% Triton X-100, 20 mM Tris pH 8, 150 mM NaCl), high salt (500 mM NaCl), low salt, high salt, TE + 50 mM NaCl. Immunoprecipitated material was eluted from the beads by incubating 20 min at 65°C in TE + 1% SDS. Supernatant was decrosslinked o.n. at 65°C, diluted 1:2 in TE, treated 1 h at 37°C with RNase A (0.2 mg/ml) and followed by 1 h at 55°C with Proteinase K. After phenol–chloroform extraction and ethanol precipitation, DNA was resuspended in water for qPCR analysis. The amount of DNA present in each immunoprecipitate was quantified as the fraction of its input. See Supplementary Table S6 for the sequences of primers.
Over-representation GSEA analysis
Over-representation analysis was performed using Gene Set Enrichment Analysis and gene sets derived from previously published gene signatures. In particular, we investigated whether the expression levels of MCF10A and MDA-MB-231 cells grown in high glucose or in the presence of 2-deoxy-glucose (2DG) were associated with elevated expression of Staminal (Pece et al, 2010), stem_tumorigenic (Liu et al, 2007), Myc (Bild et al, 2006), YAP/TAZ (Zhang et al, 2009) and YAP (Dupont et al, 2011), induced by YAP (Zhao et al, 2008) and repressed by YAP (Zhao et al, 2008), Notch A [Notch signature in (Mazzone et al, 2010)], Notch B [NICD signature in (Mazzone et al, 2010)], RAS (Bild et al, 2006), ERBB2 (Mackay et al, 2003), beta-catenin (Bild et al, 2006), Wnt (DiMeo et al, 2009), TGF-beta A (Padua et al, 2008), TGF-beta B (Adorno et al, 2009), TGF-beta C (Montagner et al, 2012), NF-kB (Park et al, 2007), STAT3 (Alvarez et al, 2005), Src (Bild et al, 2006), E2F3 (Bild et al, 2006), mutant-p53 (Miller et al, 2005), wt-p53 (Miller et al, 2005), TCF4 (van de Wetering et al, 2002), HIF (Montagner et al, 2012) and Sharp1 (Montagner et al, 2012). The complete gene signatures are provided in Supplementary Table S7. GSEA software (http://www.broadinstitute.org/gsea/index.jsp) was applied on log2 expression data of MCF10A and MDA-MB-231 cells grown in high glucose or in the presence of 2DG. Gene sets were considered significantly enriched at FDR < 5% when using Signal2Noise as metric and 1,000 permutations of gene sets.
Average signature expression and signature scores
Average signature expression has been calculated as the standardized average expression of all signature genes in sample subgroups (e.g. 2DG treated/controls; histological grade). Signature scores have been obtained summarizing the standardized expression levels of signature genes into a combined score with zero mean (Adorno et al, 2009). The values shown in graphs are thus adimensional.
Collection and processing of breast cancer gene expression data
We started from a collection of 4,640 samples from 27 major datasets comprising microarray data of breast cancer samples annotated with histological tumor grade and clinical outcome (Supplementary Table S8). All data were measured on Affymetrix arrays and have been downloaded from NCBI Gene Expression Omnibus (GEO, http://www.ncbi.nlm.nih.gov/geo/) and EMBL-EBI ArrayExpress (http://www.ebi.ac.uk/arrayexpress/).
Prior to analysis, we re-organized all datasets eliminating duplicate samples and re-naming any original set after the medical center where patients were recruited. Briefly, the datasets have been modified as follows:
Stockholm dataset has been used as is and re-named as KI_Stockholm (Karolinska Institutet, Stockholm);
EMC-286 and EMC-58 were merged to create EMC-344 (Erasmus Medical Center);
MSK has been used as is and re-named as MSKCC (Memorial Sloan-Kettering Cancer Center);
Uppsala-Miller, Ivshina-Miller and Loi datasets (GSE3494, GSE4922 and GSE6532) have been split into KI_Uppsala comprising all 258 unique patients of the Uppsala University Hospital, OXF comprising 178 samples collected at the John Radcliffe Hospital in Oxford and formerly part of GSE6532, and GUY composed of the 87 samples (from the Guys Hospital in London and formerly part of GSE6532) and of 77 samples from the former Tamoxifen study;
Sotiriou dataset has been eliminated since samples of this series are all included in GSE6532;
Tamoxifen dataset has been added to GUY cohort since all patients were recruited at the Guys Hospital in London;
Desmedt dataset has been used as is and re-named as TRANSBIG (after the consortium of cancer centers where samples have been collected);
Schmidt datasets have been used as is and re-named as Mainz (Johannes Gutenberg University in Mainz);
Veridex has been used as is;
Chin (E-TABM-158) and Zhou (GSE7378) were merged to create UCSF since all patients were recruited at the University of California, San Francisco (173 samples). Moreover, a comparison of the hybridization dates on the CEL files of E-TABM-154 and GSE7378 and of the patients’ clinical information revealed that 17 samples were deposited twice for a total of 166 unique samples out of 173 samples;
Top trial (GSE16446) was re-named as IJB_TOP (Institut Jules Bordet/Trial of Principle);
GSE19615 was re-named as US_NCI since all patients were recruited at US National Cancer Institute;
IPC (GSE21653) was re-named as CRCM since all patients were recruited at Centre de Cancérologie de Marseille;
KFSYSCC (GSE20685) was re-named as KOOF since all patients were recruited at Koo Foundation SYS Cancer Center;
GSE31519 was re-named as Goethe since all patients were recruited at Goethe University, Frankfurt;
GSE22093 was re-named as MDACC_IGR (M.D. Anderson Cancer Center/Institut Goustave Russy) and comprises 103 samples, 36 of which included in GSE20271;
Hatzis (GSE25066) includes samples derived from 4 cancer centers, that is, I-SPY-1 (Investigation of Serial Studies to Predict Your Therapeutic Response With Imaging and Molecular Analysis), LBJ_INEN_GEICAM (Lyndon B. Johnson Hospital, Instituto Nacional de Enfermedades Neoplásicas and Grupo Español de Investigación en Cáncer de Mama), USO-02103 (US Oncology) and MDACC (M. D. Anderson Cancer Center, Houston). Moreover, a comparison of the hybridization dates on the CEL files of GSE25066 and GSE20271 and GSE20194 and also of the patients’ clinical information revealed that, despite being deposited twice, some samples are identical. As such, these four datasets have been split into the following:
I-SPY-1 comprising 83 samples;
LBJ_INEN_GEICAM comprising 58 samples; and
MDACC_GSE25066 comprising 313 samples;
GSE23988 was re-named USO-02103, and it is composed of 54 samples included in USO-02103 cohort of GSE25066 and 61 from GSE23988. Twenty samples were removed, because they were deposited twice;
GSE20271 was re-named MDACC_GSE20271 and comprises 100 samples, since 78 were already included in MDACC_GSE25066;
GSE20194 is largely included in MDACC_GSE25066 (187 out of 230 samples; other four samples are included in GSE20271); the remaining 39 samples were included in the cohort named MDACC MAQC-II;
GSE32646, GSE18728 and GSE19697 were re-named as Osaka, UW and St. Louis, respectively, since all patients were recruited at the Osaka University, University of Washington (Seattle) and Washington University School of Medicine (St. Louis), respectively.
This re-organization resulted in a compendium (metadataset) comprising 3,661 unique samples from 25 independent cohorts (Supplementary Table S9). The type and content of clinical and pathological annotations of the metadataset samples have been derived from the original cohorts.
Since raw data (.CEL files) were available for all samples, the integration, normalization and summarization of gene expression signals has been obtained applying the procedure described in Rustighi et al (2014). Briefly, expression values were generated from intensity signals using a custom CDF obtained merging HG-U133A, HG-U133AAofAV2 and HG-U133 Plus2 original CDFs and transforming the original CEL files accordingly. Intensity values for a total of 21,986 probe sets have been background-adjusted, normalized using quantile normalization, and gene expression levels calculated using median polish summarization (multi-array average procedure, RMA). Clinical information among the various datasets has been standardized as described in Cordenonsi et al (2011).
Kaplan–Meier survival analysis
To identify two groups of tumors with either high or low glucose signature, we used the classifier described in Adorno et al (2009), that is a classification rule based on the glucose signature score. Tumors were classified as glucose signature ‘Low’ if the combined score was negative and as glucose signature ‘High’ if the combined score was positive. This classification was applied to expression values of the metadataset. To evaluate the prognostic value of the glucose signature, we estimated, using the Kaplan–Meier method, the probabilities that patients would remain free of metastatic. To confirm these findings, the Kaplan–Meier curves were compared using the log-rank (Mantel–Cox) test. P-values were calculated according to the standard normal asymptotic distribution. Survival analysis was performed in GraphPad Prism.
Statistical analysis
Statistical analyses were performed using Prism software (GraphPad software). Mean values and standard deviations (SD) are shown in graphs that were generated from several repeats of biological experiments, unless otherwise indicated. Immunoprecipitation experiments were performed with n = 2 biological replicates for each sample and repeated at least two times independently with comparable results.
Acknowledgments
We thank Stefano Piccolo and Michelangelo Cordenonsi for support, Jeroen Krijgsveld at EMBL proteomic core facility for assistance with mass spectrometry, Christian Frezza for advice with UOK262 cells, and Graziano Martello and Marco Montagner for critically reading the manuscript. This work was supported by grants from AIRC IG15307, MIUR/PRIN and PRAT University of Padova to SD, and from FIRB Accordi di Programma 2011 RBAP11T3WB to MF.
Author contributions
GS, EE, APo and SD performed and analyzed experiments; SD performed protein–protein interaction studies; MA and SBr performed microarray experiments; DG and APe performed Drosophila assays; MF and SBi performed bioinformatics analyses; FZ performed ChIP; GG performed Seahorse measurements; and SD, EE and GS wrote the paper.
Conflict of interest
The authors declare that they have no conflict of interest.
Supporting Information
Supplementary Figure S1
{kind=link}
Supplementary Figure S2
{kind=link}
Supplementary Figure S3
{kind=link}
Supplementary Figure S4
{kind=link}
Supplementary Figure S5
{kind=link}
Supplementary Figure S6
{kind=link}
Supplementary Table S1
Supplementary Table S2
Supplementary Table S3
Supplementary Table S4
Supplementary Table S5
Supplementary Table S6
Supplementary Table S7
Supplementary Tables S8 and S9
Supplementary Figure Legends
Source Data for Supplementary Figure S4
Review Process File
Source Data for Figure 3
Source Data for Figure 4
References
- Adorno M, Cordenonsi M, Montagner M, Dupont S, Wong C, Hann B, Solari A, Bobisse S, Rondina MB, Guzzardo V, Parenti AR, Rosato A, Bicciato S, Balmain A, Piccolo S. A Mutant-p53/Smad complex opposes p63 to empower TGFbeta-induced metastasis. Cell. 2009;137:87–98. doi: 10.1016/j.cell.2009.01.039. [DOI] [PubMed] [Google Scholar]
- Alvarez JV, Febbo PG, Ramaswamy S, Loda M, Richardson A, Frank DA. Identification of a genetic signature of activated signal transducer and activator of transcription 3 in human tumors. Cancer Res. 2005;65:5054–5062. doi: 10.1158/0008-5472.CAN-04-4281. [DOI] [PubMed] [Google Scholar]
- Aragona M, Panciera T, Manfrin A, Giulitti S, Michielin F, Elvassore N, Dupont S, Piccolo S. A mechanical checkpoint controls multicellular growth through YAP/TAZ regulation by actin-processing factors. Cell. 2013;154:1047–1059. doi: 10.1016/j.cell.2013.07.042. [DOI] [PubMed] [Google Scholar]
- Atsumi T, Chesney J, Metz C, Leng L, Donnelly S, Makita Z, Mitchell R, Bucala R. High expression of inducible 6-phosphofructo-2-kinase/fructose-2,6-bisphosphatase (iPFK-2; PFKFB3) in human cancers. Cancer Res. 2002;62:5881–5887. [PubMed] [Google Scholar]
- Azzolin L, Zanconato F, Bresolin S, Forcato M, Basso G, Bicciato S, Cordenonsi M, Piccolo S. Role of TAZ as mediator of Wnt signaling. Cell. 2012;151:1443–1456. doi: 10.1016/j.cell.2012.11.027. [DOI] [PubMed] [Google Scholar]
- Banaszak K, Mechin I, Obmolova G, Oldham M, Chang SH, Ruiz T, Radermacher M, Kopperschläger G, Rypniewski W. The crystal structures of eukaryotic phosphofructokinases from Baker’s yeast and rabbit skeletal muscle. J Mol Biol. 2011;407:284–297. doi: 10.1016/j.jmb.2011.01.019. [DOI] [PubMed] [Google Scholar]
- Benhaddou A, Keime C, Ye T, Morlon A, Michel I, Jost B, Mengus G, Davidson I. Transcription factor TEAD4 regulates expression of myogenin and the unfolded protein response genes during C2C12 cell differentiation. Cell Death Differ. 2012;19:220–231. doi: 10.1038/cdd.2011.87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bild AH, Yao G, Chang JT, Wang Q, Potti A, Chasse D, Joshi M-B, Harpole D, Lancaster JM, Berchuck A, Olson JA, Marks JR, Dressman HK, West M, Nevins JR. Oncogenic pathway signatures in human cancers as a guide to targeted therapies. Nature. 2006;439:353–357. doi: 10.1038/nature04296. [DOI] [PubMed] [Google Scholar]
- Bustamante E, Pedersen PL. High aerobic glycolysis of rat hepatoma cells in culture: role of mitochondrial hexokinase. Proc Natl Acad Sci USA. 1977;74:3735–3739. doi: 10.1073/pnas.74.9.3735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chaneton B, Gottlieb E. Rocking cell metabolism: revised functions of the key glycolytic regulator PKM2 in cancer. Trends Biochem Sci. 2012;37:309–316. doi: 10.1016/j.tibs.2012.04.003. [DOI] [PubMed] [Google Scholar]
- Chang C-H, Curtis JD, Maggi LB, Faubert B, Villarino AV, O’Sullivan D, Huang SC-C, van der Windt GJW, Blagih J, Qiu J, Weber JD, Pearce EJ, Jones RG, Pearce EL. Posttranscriptional control of T cell effector function by aerobic glycolysis. Cell. 2013;153:1239–1251. doi: 10.1016/j.cell.2013.05.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen H-Z, Tsai S-Y, Leone G. Emerging roles of E2Fs in cancer: an exit from cell cycle control. Nat Rev Cancer. 2009;9:785–797. doi: 10.1038/nrc2696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen D, Sun Y, Wei Y, Zhang P, Rezaeian AH, Teruya-Feldstein J, Gupta S, Liang H, Lin H-K, Hung M-C, Ma L. LIFR is a breast cancer metastasis suppressor upstream of the Hippo-YAP pathway and a prognostic marker. Nat Med. 2012;18:1511–1517. doi: 10.1038/nm.2940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cordenonsi M, Zanconato F, Azzolin L, Forcato M, Rosato A, Frasson C, Inui M, Montagner M, Parenti AR, Poletti A, Daidone MG, Dupont S, Basso G, Bicciato S, Piccolo S. The hippo transducer TAZ confers cancer stem cell-related traits on breast cancer cells. Cell. 2011;147:759–772. doi: 10.1016/j.cell.2011.09.048. [DOI] [PubMed] [Google Scholar]
- Couzens AL, Knight JDR, Kean MJ, Teo G, Weiss A, Dunham WH, Lin Z-Y, Bagshaw RD, Sicheri F, Pawson T, Wrana JL, Choi H, Gingras A-C. Protein interaction network of the mammalian hippo pathway reveals mechanisms of kinase-phosphatase interactions. Sci Signal. 2013;6:rs15. doi: 10.1126/scisignal.2004712. [DOI] [PubMed] [Google Scholar]
- Dang CV. Links between metabolism and cancer. Genes Dev. 2012;26:877–890. doi: 10.1101/gad.189365.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Bock K, Georgiadou M, Schoors S, Kuchnio A, Wong BW, Cantelmo AR, Quaegebeur A, Ghesquière B, Cauwenberghs S, Eelen G, Phng L-K, Betz I, Tembuyser B, Brepoels K, Welti J, Geudens I, Segura I, Cruys B, Bifari F, Decimo I, et al. Role of PFKFB3-driven glycolysis in vessel sprouting. Cell. 2013;154:651–663. doi: 10.1016/j.cell.2013.06.037. [DOI] [PubMed] [Google Scholar]
- Debnath J, Muthuswamy SK, Brugge JS. Morphogenesis and oncogenesis of MCF-10A mammary epithelial acini grown in three-dimensional basement membrane cultures. Methods. 2003;30:256–268. doi: 10.1016/s1046-2023(03)00032-x. [DOI] [PubMed] [Google Scholar]
- DeRan M, Yang J, Shen C-H, Peters EC, Fitamant J, Chan P, Hsieh M, Zhu S, Asara JM, Zheng B, Bardeesy N, Liu J, Wu X. Energy stress regulates hippo-YAP signaling involving AMPK-mediated regulation of angiomotin-like 1 protein. Cell Rep. 2014;9:495–503. doi: 10.1016/j.celrep.2014.09.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dick FA, Rubin SM. Molecular mechanisms underlying RB protein function. Nat Rev Mol Cell Biol. 2013;14:297–306. doi: 10.1038/nrm3567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DiMeo TA, Anderson K, Phadke P, Fan C, Feng C, Perou CM, Naber S, Kuperwasser C. A novel lung metastasis signature links Wnt signaling with cancer cell self-renewal and epithelial-mesenchymal transition in basal-like breast cancer. Cancer Res. 2009;69:5364–5373. doi: 10.1158/0008-5472.CAN-08-4135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dong C, Yuan T, Wu Y, Wang Y, Fan TWM, Miriyala S, Lin Y, Yao J, Shi J, Kang T, Lorkiewicz P, St Clair D, Hung M-C, Evers BM, Zhou BP. Loss of FBP1 by snail-mediated repression provides metabolic advantages in basal-like breast cancer. Cancer Cell. 2013;23:316–331. doi: 10.1016/j.ccr.2013.01.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dupont S, Mamidi A, Cordenonsi M, Montagner M, Zacchigna L, Adorno M, Martello G, Stinchfield MJ, Soligo S, Morsut L, Inui M, Moro S, Modena N, Argenton F, Newfeld SJ, Piccolo S. FAM/USP9x, a deubiquitinating enzyme essential for TGFbeta signaling, controls Smad4 monoubiquitination. Cell. 2009;136:123–135. doi: 10.1016/j.cell.2008.10.051. [DOI] [PubMed] [Google Scholar]
- Dupont S, Morsut L, Aragona M, Enzo E, Giulitti S, Cordenonsi M, Zanconato F, Le Digabel J, Forcato M, Bicciato S, Elvassore N, Piccolo S. Role of YAP/TAZ in mechanotransduction. Nature. 2011;474:179–183. doi: 10.1038/nature10137. [DOI] [PubMed] [Google Scholar]
- Fan R, Kim N-G, Gumbiner BM. Regulation of Hippo pathway by mitogenic growth factors via phosphoinositide 3-kinase and phosphoinositide-dependent kinase-1. Proc Natl Acad Sci. 2013;110:2569–2574. doi: 10.1073/pnas.1216462110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feng W, Gentles A, Nair RV, Huang M, Lin Y, Lee CY, Cai S, Scheeren FA, Kuo AH, Diehn M. Targeting unique metabolic properties of breast tumor initiating cells. Stem Cells. 2014;32:1734–1745. doi: 10.1002/stem.1662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferreras C, Hernandez ED, Martinez-Costa OH, Aragon JJ. Subunit interactions and composition of the fructose 6-phosphate catalytic site and the fructose 2,6-bisphosphate allosteric site of mammalian phosphofructokinase. J Biol Chem. 2009;284:9124–9131. doi: 10.1074/jbc.M807737200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gordan JD, Thompson CB, Simon MC. HIF and c-Myc: sibling rivals for control of cancer cell metabolism and proliferation. Cancer Cell. 2007;12:108–113. doi: 10.1016/j.ccr.2007.07.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grzeschik NA, Parsons LM, Allott ML, Harvey KF, Richardson HE. Lgl, aPKC, and Crumbs regulate the Salvador/Warts/Hippo pathway through two distinct mechanisms. Curr Biol. 2010;20:573–581. doi: 10.1016/j.cub.2010.01.055. [DOI] [PubMed] [Google Scholar]
- Halder G, Johnson RL. Hippo signaling: growth control and beyond. Development. 2011;138:9–22. doi: 10.1242/dev.045500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Halder G, Dupont S, Piccolo S. Transduction of mechanical and cytoskeletal cues by YAP and TAZ. Nat Rev Mol Cell Biol. 2012;13:591–600. doi: 10.1038/nrm3416. [DOI] [PubMed] [Google Scholar]
- Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011;144:646–674. doi: 10.1016/j.cell.2011.02.013. [DOI] [PubMed] [Google Scholar]
- Hardie DG, Ross FA, Hawley SA. AMPK: a nutrient and energy sensor that maintains energy homeostasis. Nat Rev Mol Cell Biol. 2012;13:251–262. doi: 10.1038/nrm3311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harvey KF, Pfleger CM, Hariharan IK. The Drosophila Mst ortholog, hippo, restricts growth and cell proliferation and promotes apoptosis. Cell. 2003;114:457–467. doi: 10.1016/s0092-8674(03)00557-9. [DOI] [PubMed] [Google Scholar]
- Harvey KF, Zhang X, Thomas DM. The Hippo pathway and human cancer. Nat Rev Cancer. 2013;13:246–257. doi: 10.1038/nrc3458. [DOI] [PubMed] [Google Scholar]
- Havula E, Hietakangas V. Glucose sensing by ChREBP/MondoA-Mlx transcription factors. Semin Cell Dev Biol. 2012;23:640–647. doi: 10.1016/j.semcdb.2012.02.007. [DOI] [PubMed] [Google Scholar]
- Herrero-Mendez A, Almeida A, Fernández E, Maestre C, Moncada S, Bolaños JP. The bioenergetic and antioxidant status of neurons is controlled by continuous degradation of a key glycolytic enzyme by APC/C-Cdh1. Nat Cell Biol. 2009;11:747–752. doi: 10.1038/ncb1881. [DOI] [PubMed] [Google Scholar]
- Huang J, Wu S, Barrera J, Matthews K, Pan D. The Hippo signaling pathway coordinately regulates cell proliferation and apoptosis by inactivating Yorkie, the Drosophila Homolog of YAP. Cell. 2005;122:421–434. doi: 10.1016/j.cell.2005.06.007. [DOI] [PubMed] [Google Scholar]
- Inui M, Manfrin A, Mamidi A, Martello G, Morsut L, Soligo S, Enzo E, Moro S, Polo S, Dupont S, Cordenonsi M, Piccolo S. USP15 is a deubiquitylating enzyme for receptor-activated SMADs. Nat Cell Biol. 2011;13:1368–1375. doi: 10.1038/ncb2346. [DOI] [PubMed] [Google Scholar]
- Irizarry RA, Hobbs B, Collin F, Beazer-Barclay YD, Antonellis KJ, Scherf U, Speed TP. Exploration, normalization, and summaries of high density oligonucleotide array probe level data. Biostatistics. 2003;4:249–264. doi: 10.1093/biostatistics/4.2.249. [DOI] [PubMed] [Google Scholar]
- Ito K, Suda T. Metabolic requirements for the maintenance of self-renewing stem cells. Nat Rev Mol Cell Biol. 2014;15:243–256. doi: 10.1038/nrm3772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jarvius M, Paulsson J, Weibrecht I, Leuchowius K-J, Andersson A-C, Wählby C, Gullberg M, Botling J, Sjöblom T, Markova B, Östman A, Landegren U, Söderberg O. In situ detection of phosphorylated platelet-derived growth factor receptor beta using a generalized proximity ligation method. Mol Cell Proteomics. 2007;6:1500–1509. doi: 10.1074/mcp.M700166-MCP200. [DOI] [PubMed] [Google Scholar]
- Jiao S, Wang H, Shi Z, Dong A, Zhang W, Song X, He F, Wang Y, Zhang Z, Wang W, Wang X, Guo T, Li P, Zhao Y, Ji H, Zhang L, Zhou Z. A peptide mimicking VGLL4 function acts as a YAP antagonist therapy against gastric cancer. Cancer Cell. 2014;25:166–180. doi: 10.1016/j.ccr.2014.01.010. [DOI] [PubMed] [Google Scholar]
- Johnson R, Halder G. The two faces of Hippo: targeting the Hippo pathway for regenerative medicine and cancer treatment. Nat Rev Drug Discov. 2013;13:63–79. doi: 10.1038/nrd4161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khan SJ, Bajpai A, Alam MA, Gupta RP, Harsh S, Pandey RK, Goel-Bhattacharya S, Nigam A, Mishra A, Sinha P. Epithelial neoplasia in Drosophila entails switch to primitive cell states. Proc Natl Acad Sci USA. 2013;110:E2163–E2172. doi: 10.1073/pnas.1212513110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koontz LM, Liu-Chittenden Y, Yin F, Zheng Y, Yu J, Huang B, Chen Q, Wu S, Pan D. The hippo effector yorkie controls normal tissue growth by antagonizing scalloped-mediated default repression. Dev Cell. 2013;25:388–401. doi: 10.1016/j.devcel.2013.04.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kroemer G, Pouyssegur J. Tumor cell metabolism: cancer’s Achilles’ heel. Cancer Cell. 2008;13:472–482. doi: 10.1016/j.ccr.2008.05.005. [DOI] [PubMed] [Google Scholar]
- Kurtoglu M, Gao N, Shang J, Maher JC, Lehrman MA, Wangpaichitr M, Savaraj N, Lane AN, Lampidis TJ. Under normoxia, 2-deoxy-D-glucose elicits cell death in select tumor types not by inhibition of glycolysis but by interfering with N-linked glycosylation. Mol Cancer Ther. 2007;6:3049–3058. doi: 10.1158/1535-7163.MCT-07-0310. [DOI] [PubMed] [Google Scholar]
- Lai D, Ho KC, Hao Y, Yang X. Taxol resistance in breast cancer cells is mediated by the hippo pathway component TAZ and its downstream transcriptional targets Cyr61 and CTGF. Cancer Res. 2011;71:2728–2738. doi: 10.1158/0008-5472.CAN-10-2711. [DOI] [PubMed] [Google Scholar]
- Laplante M, Sabatini DM. mTOR signaling in growth control and disease. Cell. 2012;149:274–293. doi: 10.1016/j.cell.2012.03.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee T, Luo L. Mosaic analysis with a repressible cell marker (MARCM) for Drosophila neural development. Trends Neurosci. 2001;24:251–254. doi: 10.1016/s0166-2236(00)01791-4. [DOI] [PubMed] [Google Scholar]
- Levine AJ, Puzio-Kuter AM. The control of the metabolic switch in cancers by oncogenes and tumor suppressor genes. Science (New York, NY) 2010;330:1340–1344. doi: 10.1126/science.1193494. [DOI] [PubMed] [Google Scholar]
- Li Z, Zhao B, Wang P, Chen F, Dong Z, Yang H, Guan K-L, Xu Y. Structural insights into the YAP and TEAD complex. Genes Dev. 2010;24:235–240. doi: 10.1101/gad.1865810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu R, Wang X, Chen GY, Dalerba P, Gurney A, Hoey T, Sherlock G, Lewicki J, Shedden K, Clarke MF. The prognostic role of a gene signature from tumorigenic breast-cancer cells. N Engl J Med. 2007;356:217–226. doi: 10.1056/NEJMoa063994. [DOI] [PubMed] [Google Scholar]
- Liu-Chittenden Y, Huang B, Shim JS, Chen Q, Lee S-J, Anders RA, Liu JO, Pan D. Genetic and pharmacological disruption of the TEAD-YAP complex suppresses the oncogenic activity of YAP. Genes Dev. 2012;26:1300–1305. doi: 10.1101/gad.192856.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lukas J, Herzinger T, Hansen K, Moroni MC, Resnitzky D, Helin K, Reed SI, Bartek J. Cyclin E-induced S phase without activation of the pRb/E2F pathway. Genes Dev. 1997;11:1479–1492. doi: 10.1101/gad.11.11.1479. [DOI] [PubMed] [Google Scholar]
- Lunt SY, Vander Heiden MG. Aerobic glycolysis: meeting the metabolic requirements of cell proliferation. Annu Rev Cell Dev Biol. 2011;27:441–464. doi: 10.1146/annurev-cellbio-092910-154237. [DOI] [PubMed] [Google Scholar]
- Luo W, Semenza GL. Emerging roles of PKM2 in cell metabolism and cancer progression. Trends Endocrinol Metab. 2012;23:560–566. doi: 10.1016/j.tem.2012.06.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mackay A, Jones C, Dexter T, Silva RLA, Bulmer K, Jones A, Simpson P, Harris RA, Jat PS, Neville AM, Reis LFL, Lakhani SR, O’Hare MJ. cDNA microarray analysis of genes associated with ERBB2 (HER2/neu) overexpression in human mammary luminal epithelial cells. Oncogene. 2003;22:2680–2688. doi: 10.1038/sj.onc.1206349. [DOI] [PubMed] [Google Scholar]
- Marroquin LD, Hynes J, Dykens JA, Jamieson JD, Will Y. Circumventing the Crabtree effect: replacing media glucose with galactose increases susceptibility of HepG2 cells to mitochondrial toxicants. Toxicol Sci. 2007;97:539–547. doi: 10.1093/toxsci/kfm052. [DOI] [PubMed] [Google Scholar]
- Mazzone M, Selfors LM, Albeck J, Overholtzer M, Sale S, Carroll DL, Pandya D, Lu Y, Mills GB, Aster JC, Artavanis-Tsakonas S, Brugge JS. Dose-dependent induction of distinct phenotypic responses to Notch pathway activation in mammary epithelial cells. Proc Natl Acad Sci USA. 2010;107:5012–5017. doi: 10.1073/pnas.1000896107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Menendez J, Perez-Garijo A, Calleja M, Morata G. A tumor-suppressing mechanism in Drosophila involving cell competition and the Hippo pathway. Proc Natl Acad Sci USA. 2010;107:14651–14656. doi: 10.1073/pnas.1009376107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller LD, Smeds J, George J, Vega VB, Vergara L, Ploner A, Pawitan Y, Hall P, Klaar S, Liu ET, Bergh J. An expression signature for p53 status in human breast cancer predicts mutation status, transcriptional effects, and patient survival. Proc Natl Acad Sci USA. 2005;102:13550–13555. doi: 10.1073/pnas.0506230102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mohseni M, Sun J, Lau A, Curtis S, Goldsmith J, Fox VL, Wei C, Frazier M, Samson O, Wong K-K, Kim C, Camargo FD. A genetic screen identifies an LKB1-MARK signalling axis controlling the Hippo-YAP pathway. Nat Cell Biol. 2014;16:108–117. doi: 10.1038/ncb2884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Montagner M, Enzo E, Forcato M, Zanconato F, Parenti A, Rampazzo E, Basso G, Leo G, Rosato A, Bicciato S, Cordenonsi M, Piccolo S. SHARP1 suppresses breast cancer metastasis by promoting degradation of hypoxia-inducible factors. Nature. 2012;487:380–384. doi: 10.1038/nature11207. [DOI] [PubMed] [Google Scholar]
- Mor I, Cheung EC, Vousden KH. Control of glycolysis through regulation of PFK1: old friends and recent additions. Cold Spring Harb Symp Quant Biol. 2011;76:211–216. doi: 10.1101/sqb.2011.76.010868. [DOI] [PubMed] [Google Scholar]
- Neto-Silva RM, de Beco S, Johnston LA. Evidence for a growth-stabilizing regulatory feedback mechanism between Myc and Yorkie, the Drosophila homolog of Yap. Dev Cell. 2010;19:507–520. doi: 10.1016/j.devcel.2010.09.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ochocki JD, Simon MC. Nutrient-sensing pathways and metabolic regulation in stem cells. J Cell Biol. 2013;203:23–33. doi: 10.1083/jcb.201303110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Onodera Y, Nam J-M, Bissell MJ. Increased sugar uptake promotes oncogenesis via EPAC/RAP1 and O-GlcNAc pathways. J Clin Invest. 2014;124:367–384. doi: 10.1172/JCI63146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ostrowski A, van Aalten DMF. Chemical tools to probe cellular O-GlcNAc signalling. Biochem J. 2013;456:1–12. doi: 10.1042/BJ20131081. [DOI] [PubMed] [Google Scholar]
- Ota M, Sasaki H. Mammalian Tead proteins regulate cell proliferation and contact inhibition as transcriptional mediators of Hippo signaling. Development. 2008;135:4059–4069. doi: 10.1242/dev.027151. [DOI] [PubMed] [Google Scholar]
- Padua D, Zhang XH-F, Wang Q, Nadal C, Gerald WL, Gomis RR, Massagué J. TGFbeta primes breast tumors for lung metastasis seeding through angiopoietin-like 4. Cell. 2008;133:66–77. doi: 10.1016/j.cell.2008.01.046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pan D. The hippo signaling pathway in development and cancer. Dev Cell. 2010;19:491–505. doi: 10.1016/j.devcel.2010.09.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pantalacci S, Tapon N, Léopold P. The Salvador partner Hippo promotes apoptosis and cell-cycle exit in Drosophila. Nat Cell Biol. 2003;5:921–927. doi: 10.1038/ncb1051. [DOI] [PubMed] [Google Scholar]
- Park BK, Zhang H, Zeng Q, Dai J, Keller ET, Giordano T, Gu K, Shah V, Pei L, Zarbo RJ, McCauley L, Shi S, Chen S, Wang C-Y. NF-kappaB in breast cancer cells promotes osteolytic bone metastasis by inducing osteoclastogenesis via GM-CSF. Nat Med. 2007;13:62–69. doi: 10.1038/nm1519. [DOI] [PubMed] [Google Scholar]
- Pece S, Tosoni D, Confalonieri S, Mazzarol G, Vecchi M, Ronzoni S, Bernard L, Viale G, Pelicci PG, Di Fiore PP. Biological and molecular heterogeneity of breast cancers correlates with their cancer stem cell content. Cell. 2010;140:62–73. doi: 10.1016/j.cell.2009.12.007. [DOI] [PubMed] [Google Scholar]
- Piccolo S, Dupont S, Cordenonsi M. The biology of YAP/TAZ: hippo signaling and beyond. Physiol Rev. 2014;94:1287–1312. doi: 10.1152/physrev.00005.2014. [DOI] [PubMed] [Google Scholar]
- Pike KG, Malagu K, Hummersone MG, Menear KA, Duggan HME, Gomez S, Martin NMB, Ruston L, Pass SL, Pass M. Optimization of potent and selective dual mTORC1 and mTORC2 inhibitors: the discovery of AZD8055 and AZD2014. Bioorg Med Chem Lett. 2013;23:1212–1216. doi: 10.1016/j.bmcl.2013.01.019. [DOI] [PubMed] [Google Scholar]
- Ribeiro PS, Josué F, Wepf A, Wehr MC, Rinner O, Kelly G, Tapon N, Gstaiger M. Combined functional genomic and proteomic approaches identify a PP2A complex as a negative regulator of Hippo signaling. Mol Cell. 2010;39:521–534. doi: 10.1016/j.molcel.2010.08.002. [DOI] [PubMed] [Google Scholar]
- Rossignol R. Energy substrate modulates mitochondrial structure and oxidative capacity in cancer cells. Cancer Res. 2004;64:985–993. doi: 10.1158/0008-5472.can-03-1101. [DOI] [PubMed] [Google Scholar]
- Rustighi A, Zannini A, Tiberi L, Sommaggio R, Piazza S, Sorrentino G, Nuzzo S, Tuscano A, Eterno V, Benvenuti F, Santarpia L, Aifantis I, Rosato A, Bicciato S, Zambelli A, Del Sal G. Prolyl-isomerase Pin1 controls normal and cancer stem cells of the breast. EMBO Mol Med. 2014;6:99–119. doi: 10.1002/emmm.201302909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schulze A, Harris AL. How cancer metabolism is tuned for proliferation and vulnerable to disruption. Nature. 2012;491:364–373. doi: 10.1038/nature11706. [DOI] [PubMed] [Google Scholar]
- Shyh-Chang N, Daley GQ, Cantley LC. Stem cell metabolism in tissue development and aging. Development. 2013;140:2535–2547. doi: 10.1242/dev.091777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sola-Penna M, Da Silva D, Coelho WS, Marinho-Carvalho MM, Zancan P. Regulation of mammalian muscle type 6-phosphofructo-1-kinase and its implication for the control of the metabolism. IUBMB Life. 2010;62:791–796. doi: 10.1002/iub.393. [DOI] [PubMed] [Google Scholar]
- Sorrentino G, Ruggeri N, Specchia V, Cordenonsi M, Mano M, Dupont S, Manfrin A, Ingallina E, Sommaggio R, Piazza S, Rosato A, Piccolo S, Del Sal G. Metabolic control of YAP and TAZ by the mevalonate pathway. Nat Cell Biol. 2014;16:357–366. doi: 10.1038/ncb2936. [DOI] [PubMed] [Google Scholar]
- Sudarshan S, Sourbier C, Kong HS, Block K, Romero VAV, Yang Y, Galindo C, Mollapour M, Scroggins B, Goode N, Lee MJ, Gourlay CW, Trepel J, Linehan WM, Neckers L. Fumarate hydratase deficiency in renal cancer induces glycolytic addiction and hypoxia-inducible transcription factor 1 stabilization by glucose-dependent generation of reactive oxygen species. Mol Cell Biol. 2009;29:4080–4090. doi: 10.1128/MCB.00483-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tennant DA, Durán RV, Gottlieb E. Targeting metabolic transformation for cancer therapy. Nat Rev Cancer. 2010;10:267–277. doi: 10.1038/nrc2817. [DOI] [PubMed] [Google Scholar]
- Tremblay AM, Camargo FD. Hippo signaling in mammalian stem cells. Semin Cell Dev Biol. 2012;23:818–826. doi: 10.1016/j.semcdb.2012.08.001. [DOI] [PubMed] [Google Scholar]
- Tusher VG, Tibshirani R, Chu G. Significance analysis of microarrays applied to the ionizing radiation response. Proc Natl Acad Sci USA. 2001;98:5116–5121. doi: 10.1073/pnas.091062498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Udan RS, Kango-Singh M, Nolo R, Tao C, Halder G. Hippo promotes proliferation arrest and apoptosis in the Salvador/Warts pathway. Nat Cell Biol. 2003;5:914–920. doi: 10.1038/ncb1050. [DOI] [PubMed] [Google Scholar]
- Varelas X, Samavarchi-Tehrani P, Narimatsu M, Weiss A, Cockburn K, Larsen BG, Rossant J, Wrana JL. The Crumbs complex couples cell density sensing to Hippo-dependent control of the TGF-β-SMAD pathway. Dev Cell. 2010;19:831–844. doi: 10.1016/j.devcel.2010.11.012. [DOI] [PubMed] [Google Scholar]
- Wang Z, Wu Y, Wang H, Zhang Y, Mei L, Fang X, Zhang X, Zhang F, Chen H, Liu Y, Jiang Y, Sun S, Zheng Y, Li N, Huang L. Interplay of mevalonate and Hippo pathways regulates RHAMM transcription via YAP to modulate breast cancer cell motility. Proc Natl Acad Sci USA. 2014;111:E89–E98. doi: 10.1073/pnas.1319190110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wehr MC, Holder MV, Gailite I, Saunders RE, Maile TM, Ciirdaeva E, Instrell R, Jiang M, Howell M, Rossner MJ, Tapon N. Salt-inducible kinases regulate growth through the Hippo signalling pathway in Drosophila. Nat Cell Biol. 2012;15:61–71. doi: 10.1038/ncb2658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wellen KE, Lu C, Mancuso A, Lemons JMS, Ryczko M, Dennis JW, Rabinowitz JD, Coller HA, Thompson CB. The hexosamine biosynthetic pathway couples growth factor-induced glutamine uptake to glucose metabolism. Genes Dev. 2010;24:2784–2799. doi: 10.1101/gad.1985910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wellen KE, Thompson CB. A two-way street: reciprocal regulation of metabolism and signalling. Nat Rev Mol Cell Biol. 2012;13:270–276. doi: 10.1038/nrm3305. [DOI] [PubMed] [Google Scholar]
- van de Wetering M, Sancho E, Verweij C, de Lau W, Oving I, Hurlstone A, van der Horn K, Batlle E, Coudreuse D, Haramis AP, Tjon-Pon-Fong M, Moerer P, van den Born M, Soete G, Pals S, Eilers M, Medema R, Clevers H. The beta-catenin/TCF-4 complex imposes a crypt progenitor phenotype on colorectal cancer cells. Cell. 2002;111:241–250. doi: 10.1016/s0092-8674(02)01014-0. [DOI] [PubMed] [Google Scholar]
- Wick A, Drury D, Nakada H, Wolfe J. Localization of the primary metabolic block produced by 2-deoxyglucose. J Biol Chem. 1957;224:963–969. [PubMed] [Google Scholar]
- Wu S, Liu Y, Zheng Y, Dong J, Pan D. The TEAD/TEF family protein Scalloped mediates transcriptional output of the Hippo growth-regulatory pathway. Dev Cell. 2008;14:388–398. doi: 10.1016/j.devcel.2008.01.007. [DOI] [PubMed] [Google Scholar]
- Yalcin A, Telang S, Clem B, Chesney J. Regulation of glucose metabolism by 6-phosphofructo-2-kinase/fructose-2,6-bisphosphatases in cancer. Exp Mol Pathol. 2009;86:174–179. doi: 10.1016/j.yexmp.2009.01.003. [DOI] [PubMed] [Google Scholar]
- Yang Y, Lane AN, Ricketts CJ, Sourbier C, Wei M-H, Shuch B, Pike L, Wu M, Rouault TA, Boros LG, Fan TWM, Linehan WM. Metabolic reprogramming for producing energy and reducing power in fumarate hydratase null cells from hereditary leiomyomatosis renal cell carcinoma. PLoS ONE. 2013;8:e72179. doi: 10.1371/journal.pone.0072179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yi C, Troutman S, Fera D, Stemmer-Rachamimov A, Avila JL, Christian N, Persson NL, Shimono A, Speicher DW, Marmorstein R, Holmgren L, Kissil JL. A tight junction-associated merlin-angiomotin complex mediates merlin’s regulation of mitogenic signaling and tumor suppressive functions. Cancer Cell. 2011;19:527–540. doi: 10.1016/j.ccr.2011.02.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yi W, Clark PM, Mason DE, Keenan MC, Hill C, Goddard WA, Peters EC, Driggers EM, Hsieh-Wilson LC. Phosphofructokinase 1 glycosylation regulates cell growth and metabolism. Science (New York, NY) 2012;337:975–980. doi: 10.1126/science.1222278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yimlamai D, Christodoulou C, Galli GG, Yanger K, Pepe-Mooney B, Gurung B, Shrestha K, Cahan P, Stanger BZ, Camargo FD. Hippo pathway activity influences liver cell fate. Cell. 2014;157:1324–1338. doi: 10.1016/j.cell.2014.03.060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yin F, Yu J, Zheng Y, Chen Q, Zhang N, Pan D. Spatial organization of Hippo signaling at the plasma membrane mediated by the tumor suppressor Merlin/NF2. Cell. 2013;154:1342–1355. doi: 10.1016/j.cell.2013.08.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu F-X, Guan K-L. The Hippo pathway: regulators and regulations. Genes Dev. 2013;27:355–371. doi: 10.1101/gad.210773.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang H, Liu C-Y, Zha Z-Y, Zhao B, Yao J, Zhao S, Xiong Y, Lei Q-Y, Guan K-L. TEAD transcription factors mediate the function of TAZ in cell growth and epithelial-mesenchymal transition. J Biol Chem. 2009;284:13355–13362. doi: 10.1074/jbc.M900843200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y-J, Duan Y, Zheng XFS. Targeting the mTOR kinase domain: the second generation of mTOR inhibitors. Drug Discov Today. 2011;16:325–331. doi: 10.1016/j.drudis.2011.02.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao B, Wei X, Li W, Udan RS, Yang Q, Kim J, Xie J, Ikenoue T, Yu J, Li L, Zheng P, Ye K, Chinnaiyan A, Halder G, Lai Z-C, Guan K-L. Inactivation of YAP oncoprotein by the Hippo pathway is involved in cell contact inhibition and tissue growth control. Genes Dev. 2007;21:2747–2761. doi: 10.1101/gad.1602907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao B, Ye X, Yu J, Li L, Li W, Li S, Yu J, Lin JD, Wang C-Y, Chinnaiyan AM, Lai Z-C, Guan K-L. TEAD mediates YAP-dependent gene induction and growth control. Genes Dev. 2008;22:1962–1971. doi: 10.1101/gad.1664408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao B, Li L, Wang L, Wang C-Y, Yu J, Guan K-L. Cell detachment activates the Hippo pathway via cytoskeleton reorganization to induce anoikis. Genes Dev. 2012;26:54–68. doi: 10.1101/gad.173435.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ziosi M, Baena-López LA, Grifoni D, Froldi F, Pession A, Garoia F, Trotta V, Bellosta P, Cavicchi S, Pession A. dMyc functions downstream of yorkie to promote the supercompetitive behavior of hippo pathway mutant cells. PLoS Genet. 2010;6:e1001140. doi: 10.1371/journal.pgen.1001140. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Figure S1
Supplementary Figure S2
Supplementary Figure S3
Supplementary Figure S4
Supplementary Figure S5
Supplementary Figure S6
Supplementary Table S1
Supplementary Table S2
Supplementary Table S3
Supplementary Table S4
Supplementary Table S5
Supplementary Table S6
Supplementary Table S7
Supplementary Tables S8 and S9
Supplementary Figure Legends
Source Data for Supplementary Figure S4
Review Process File
Source Data for Figure 3
Source Data for Figure 4