

Abstract
The variability in radiosensitivity across the human population is in part governed by genetic factors. The ability to predict therapeutic response, identify individuals at greatest risk for adverse clinical responses after therapeutic radiation doses, or identify individuals at high risk for carcinogenesis from environmental or medical radiation exposures has a medical and economic impact on both the individual and society at-large. As radiotherapy incorporates particles, particularly particles larger than protons, into therapy the need for such discriminators, that is, biomarkers will become ever more important. Cellular assays for survival, DNA repair or chromatid/chromosomal analysis have been used to identify at risk individuals but they are not clinically applicable. Newer approaches such as genome wide analysis of gene expression or single nucleotide polymorphisms, and small copy number variations within chromosomes are examples of technologies being applied to the discovery process. Gene expression analysis of primary or immortalized human cells suggests that there are distinct gene expression patterns associated with radiation exposure to both low and high linear energy transfer radiations and that those most radiosensitive are discernible by their basal gene expression patterns. However, because the genetic alterations that drive radioresponse may be subtle and cumulative, the need for large sample sizes of specific cell or tissue types is required. A systems biology approach will ultimately be necessary. Potential biomarkers from cell lines or animal models will require validation in a human setting where possible, and before being considered as a credible biomarker some understanding of the molecular mechanism is necessary.
Keywords: National Council on Radiation Protection and Measurements, intrinsic radiosensitivity, gene expression, HZE particles, carcinogenesis
INTRODUCTION
It is clear that the radiosensitivity of individuals is quite variable. At least some of this variability is known to be genetic as clinical, epidemiological, and laboratory data have shown that alterations in the structure or expression of specific gene products affect radiosensitivity. This variability is likely not without consequence as clinical and laboratory studies have shown a significant correlation between cellular radiosensitivity and the risk for late normal tissue morbidity following radiotherapy (Burnet et al. 1992, 1994, 1998; Geara et al. 1993; Johansen et al. 1994; Brock et al. 1995). And, because successful radiotherapeutic treatment is a trade-off between tumor control and acceptable risks for normal tissue injury within a given population of patients, modeling studies project that significant gains in tumor control probability would result from individualization of treatment planning based on normal tissue tolerance (Tucker et al. 1996). This is a very important point. Given the percentage of cancer patients treated by radiotherapy with curative intent, the opportunity to raise the total dose for most patients without inflicting severe normal tissue responses could have a profound impact on treatment efficacy for local tumor control. Furthermore, from the standpoint of radiation protection, individuals who are intrinsically more radiosensitive, may be more susceptible to the carcinogenic effects of radiation exposure through the use of diagnostic or therapeutic medical devices, therapy devices that now include heavy particle exposures. Finding alternative procedures for such individuals would likely be medically appropriate.
CLINICAL RADIOSENSITIVITY
Historically, a number of investigators have shown that the in vitro radiosensitivity of normal cells from patients are both highly variable and predictive of normal tissue injury after radiotherapy (Burnet et al. 1992, 1994; Geara et al. 1993; Johansen et al. 1994, 1996; Brock et al. 1995; Dikomey et al. 2000). Typically the normal cells chosen for this analysis have been either skin fibroblasts or peripheral blood lymphocytes. The measurement of choice is typically clonogenic survival as measured by surviving fraction at 2 or 3.5 Gy (SF2, SF3.5, respectively). The SF2 of skin fibroblasts in particular, has been shown to predict late tissue effects such as skin fibrosis, telangiectasia, erythema, bone necrosis, and other complications of the skin, subcutaneous tissue, mucous membrane, and bone (Tucker et al. 1992; Geara et al. 1993; Johansen et al. 1994, 1996). In addition, the probability of a radiation-induced cancer in an individual who is intrinsically radiosensitive is clearly enhanced as there is a direct link between diminished DNA repair capacity and cancer.
Why does a generalized assay of clonogenic survival correlate with late normal tissue responses to radiation, while specific genetic associations are so rare? The answer must be that the ability of a cell to form a colony after exposure to radiation is determined by the correct expression and/or interaction of a large number of gene products, perhaps hundreds, which act to repair damage and recover normal cellular function. Some of the genetic changes associated with altered radiosensitivity may be difficult to measure, but their subtle effects on pathways involved in DNA repair, cell-cycle regulation, or oxidative phosphorylation, for instance, may be readily detectable when a damaged cell is challenged to proliferate and form a colony. Unfortunately, in spite of their value for prediction, high-level global assays such as clonogenic survival, have limited value in providing mechanistic data on the basis of radiosensitivity and will never be sufficiently robust for recommending individual treatment planning. SF2 assays have, however, established the proof-of-principle that the correlation between in vitro radiosensitivity and late normal tissue reactions is highly significant and suggests the possibility that a predictive assay could be developed that a therapist could use to modify radiation dose prescriptions based upon individual risk factors (Tucker et al. 1996; Bentzen and Tucker 1998). The challenge now is to discover other biological parameters that better assess radiosensitivity and other important determinants of the normal tissue response to radiation including risk for adverse normal tissue effects or second cancers.
If the gold standard for radiosensitivity and normal tissue radioresponse, the SF2 assay, is not clinically useful there are other surrogates with strong biological underpinnings that might serve. Typically, these are assays that involve the biochemical repair of either chromosome/chromatid breaks or DNA double-strand breaks (DSBs) (Wurm et al. 1994; Jones et al. 1995; Woudstra et al. 1998; Dikomey et al. 1998, 2000). In particular, measurements of residual lesions by pulsed-field gel electrophoresis have been correlated with fibroblast sensitivity (Wurm et al. 1994; Tucker et al. 1996; Dikomey et al. 1998, 2000). This correlation of DNA repair capacity with fibroblast SF2 implies that fibroblast SF2 is influenced by altered DNA repair pathways or other pathways that interact with or impinge upon DNA repair pathways like the cell-cycle checkpoint pathways.
The list of genes associated with the repair of radiation-induced DNA damage is extensive. So far, only individuals homozygous for mutations in a small number of those genes are phenotypically obvious such as individuals with ataxia telangiectasia (AT) or Nijmegen breakage syndrome (NBS) (Morgan et al. 1968; Taylor et al. 1975). More interesting is the possibility that fibroblasts from normally appearing individuals, who are heterozygous for the ATM, NBS, BRCA1, and BRCA2 genes, are more sensitive than the average individual to radiation. This haplo-insufficiency may partially account for their greater risk for cancer and even make their normal tissues more sensitive to radiation (Swift et al. 1976; Paterson et al. 1979; Kinsella et al. 1982; Pippard et al. 1988; Gowen et al. 1998; Cuneo et al. 2000; Buchholz et al. 2002). Another group of individuals would be those who have polymorphisms of functional significance within specific repair or repair-associated genes. These individuals may also appear phenotypically normal yet have subtle but significant changes in radiosensitivity or cancer risk. Taken together, all of these individuals might be at increased risk for normal tissue injury and, to detect them in a population of patients, it would be necessary to screen for the activity levels of more than just a few key repair or repair-associated genes. Thus, predicting radiosensitivity at the molecular level might require screening of hundreds, if not thousands, of genes or proteins for potential reduced or aberrant expression, some associated with repair, and others associated with growth, stress response, the cell cycle, differentiation, energy metabolism, apoptosis, and other processes. In other words, the successful development of surrogate assays for radiation sensitivity must be designed to assess a wide range of cellular and biochemical processes.
Gene expression analysis through the use of high density microarrays is a technique with such potential. Microarray technology has proven itself by yielding significant new knowledge of the biology of normal development (Colantuoni et al. 2000; de Vries et al. 2000), disease (Whitney et al. 1999; Hwang et al. 2000), particularly cancer (Golub et al. 1999; Sgroi et al. 1999; Walker et al. 1999; Wang et al. 1999; Alizadeh et al. 2000), and in the changes in gene expression that result from exposure to carcinogens, toxins (Bartosiewicz et al. 2000), chemotherapeutic agents (Chen et al. 2000) and radiation (Fornace et al. 1999; Amundson et al. 2000). These RNA-based technologies identify differences in mRNA levels due to altered signal transduction, transcriptional regulation, RNA processing and RNA turnover. They will not detect point mutations, abnormalities or differences in translation, or any type of protein modification or turnover. This is potentially a problem for studies of radiation response where initial protein events such as phosphorylation can occur within seconds.
The difficulties encountered in genetic linkage studies such as those used for ATM, or BRCA1/2 have stimulated a renewed interest in genetic association studies, which have been used extensively to examine the relationship between sequence variations in candidate genes and phenotypes. There are hundreds of studies on the ATM gene (Bonnen et al. 2000; Buchholz et al. 2004) that have tried to establish a relationship between altered genotype and phenotypic changes. However, the highly polymorphic nature of the genome makes it extremely unlikely that an arbitrarily selected candidate SNP will prove to be functionally significant. Of the literally thousands of association studies reported, the overwhelming majority considered a single SNP, or at most a handful of SNPs, in a single candidate gene. In a landmark paper, Risch and Merikangas (1996) proposed that loci with modest phenotypic effects could be detected by genome-wide association studies. Analysis of a dense panel of SNPs spanning the genome would obviate the need for a priori selection of candidate genes, the major limitation of traditional association studies.
It is not the phenotypically obvious case that is of interest. Rather it is those seemingly sporadic cases of intrinsic radiosensitivity in individuals who do not have profound and obvious radiosensitivity, that is, those who would present in the clinic as outwardly normal. Assume that their radiosensitivity is due to low penetrance genes, one gene or a combination of many, identifying these genes will be difficult by classical methods as there are likely epistatic events that could confound any interpretation of biological data. However, with large scale SNP genotyping across the entire genome one could expect that numerous SNPs will be identified that associate with radiosensitivity. Evidence for such a relationship can be found in the work of Jones and Mohrenweiser who have over the years identified hundreds of SNPs within genes associated with DNA repair through the National Institutes of Health DNA Polymorphism Discovery Resource. Initially, Mohrenweiser et al. (2003), described over 400 variants within 74 DNA repair genes, and in a subsequent report the authors identified 520 amino acid substitutions in 91 repair genes (Jones et al. 2006). These polymorphic variants were identified through the DNA resequencing of these repair genes in lymphocytes from 90 unrelated individuals that represent the ethnic diversity of the U.S. population. Their focus has been on genes associated with base and nucleotide excision repair although SNPs within genes associated with nonhomologous end joining, homologous recombination and cell-cycle checkpoint genes, genes with a relationship to the response to ionizing radiation exposure, have also been identified (Mohrenweiser et al. 2003). The authors argue in this and subsequent publications that rare SNPs can impede gene function resulting in genomic instability and ultimately cancer (Mohrenweiser et al. 2003; Xi et al. 2004; Jones et al. 2006). Using two software algorithms that predict the impact of amino acid substitutions, they argue that many of these amino acid substitutions impact protein function (Xi et al. 2004). Biological confirmation would be the next logical step and, in a recent publication Jones et al. (2006), tried to link genotype with phenotype by using a functional assay for DNA repair following ionizing radiation in this same lymphocyte sample set described above. The authors then tested several statistical models to try and build SNP predictors of phenotype. One can question the choice of functional assay (alkaline comet), the dose used (5 Gy), the lack of experimental replication, and the genes they chose to use in their candidate approach (genes involved in base excision repair and “anti-oxidation genes”). The authors also had no clear indication of the extent of inter-individual radiosensitivity. Still, the authors advanced several statistical approaches to linking genotype with phenotype that are worth considering.
In addition to the potential for amino acid substitutions to be responsible for some of the diversity of inter-individual radioresponse is the potential for gene copy number variation to provide for the same phenotypic diversity. In genome wide studies of genetic variation two groups have shown that there can be widespread copy number variation between individuals without any evidence of genetic disorders (Iafrate et al. 2004; Sebat et al. 2004; Freeman et al. 2006). Furthermore, Tuzun et al. (2005) identified 241 copy number variations of 8–40 kb when comparing the National Center for Biotechnology Information reference human genome sequence against an anonymous North American female of European descent. These studies have been expanded upon such that copy number variation is now considered to be a significant source of genomic variation (Freeman et al. 2006). This gene copy number variation may impact phenotypic variation through differential levels of gene expression. Array-based comparative genomic hybridization has been valuable in discovering regions of copy number variation and the Copy Number Variation Project is comprehensively characterizing copy number variation in the samples used for the international HapMap Project.
INTRINSIC RADIOSENSITIVITY
One aspect of this program has been to focus on a population of individuals where intrinsic radiosensitivity has clinical consequences. There are three major reasons that such a study is now possible. First, a number of groups are sequencing the genome in an effort to identify SNPs associated with disease, the response to chemicals and drugs, and the response to therapeutic protocols. These data are in large part available through the SNP Consortium and the dbSNP database. Second, technology now allows for the examination of an ever increasing number of previously identified and well characterized SNPs, DNA copy number variation and with the advent of next generation sequencing, novel SNPs, mutations, methylation sites, gene fusions and alternative splicing sites. Third, normal skin fibroblasts from 165 cancer patients treated with curative intent by radiotherapy have been collected To date, the radiosensitivity of 90 of these cultures has been determined and they have been triaged into sensitive, normal, or radioresistant groups. Radiosensitivity changes more than fourfold across this panel. The collection of samples is ongoing, however the focus is now exclusively on those who exhibit adverse normal tissue responses.
The intrinsic cellular radiosensitivity of normal skin fibroblasts was determined to be a surrogate for tissue radiosensitivity and the basic principle that cellular radiosensitivity is an important determinant of normal tissue response to radiation was established (Geara et al. 1992a, 1993). In Fig. 1, that relationship is described in a series of fibroblast samples from 23 head and neck patients. However, the precision of the prediction was never sufficient for clinical (e.g., individual patient) use. This is apparent for samples where a late grade reaction of three was determined. Here, five of eight patients would, by current criteria, be considered as having normal radiosensitivity, while three would be considered radiosensitive. However, if SF2 is considered as the independent variable, any patient with a skin fibroblast SF2 of 0.26 or less had a late grade reaction of three or worse. Because the sample size is not sufficient to be predictive of individual response, and the assay is not clinically useful, the collection of skin fibroblast cell lines has been increased to 165 and other biological endpoints will be examined.
Fig. 1.
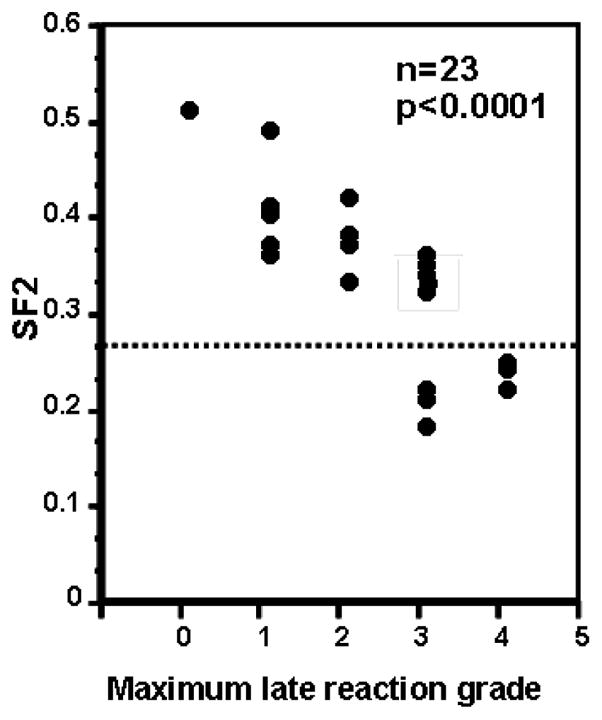
Skin fibroblast SF2 plotted against the maximum late normal tissue complication measured in the donating patient (adapted from Geara et al. 1996).
Most of these patient samples were procured through the head and neck and breast cancer clinics although other clinics identified individuals. Over 20 of these patients have had adverse reactions to radiotherapy with documented late normal tissue reactions (Grade 3 or greater) scored according to the Radiation Therapy Oncology Group/European Organizationfor Research and Treatment of Cancer scoring system. Late effects documented include skin fibrosis, subcutaneous fibrosis, skin retraction, and soft tissue and bone necrosis. For all of these primary cultures, fibroblast SF2 values were determined based upon the generation of a minimum of three complete survival curves. The actual SF2 value is from all data for each cell line fitted by an α/β equation (coefficient of variation: <0.25). Fig. 2 is a rank order plot showing the measured surviving fraction at 2 Gy (SF2) values for 90 of these fibroblast lines. Interestingly, SF2 ranking appears as a near-continuous variable which is best approximated by a normal distribution of phenotype, which could but is not necessarily explained by multiple interacting Mendelian factors, that is, a genetic basis for differences in SF2. This continuous variable may be fortuitous—calculations of association between genotype and phenotype would be easier (Analysis of Variance) —or it may be deceiving as the samples were not collected completely at random as there was a concerted effort to collect samples from patients with adverse late normal tissue effects. From a randomly chosen sampling of patients from a radiotherapy population one would expect roughly 5% of patients to have adverse late normal tissue responses, which is likely not associated 1:1 with radiosensitivity (see Grade 3 reactions, Fig. 1).
Fig. 2.
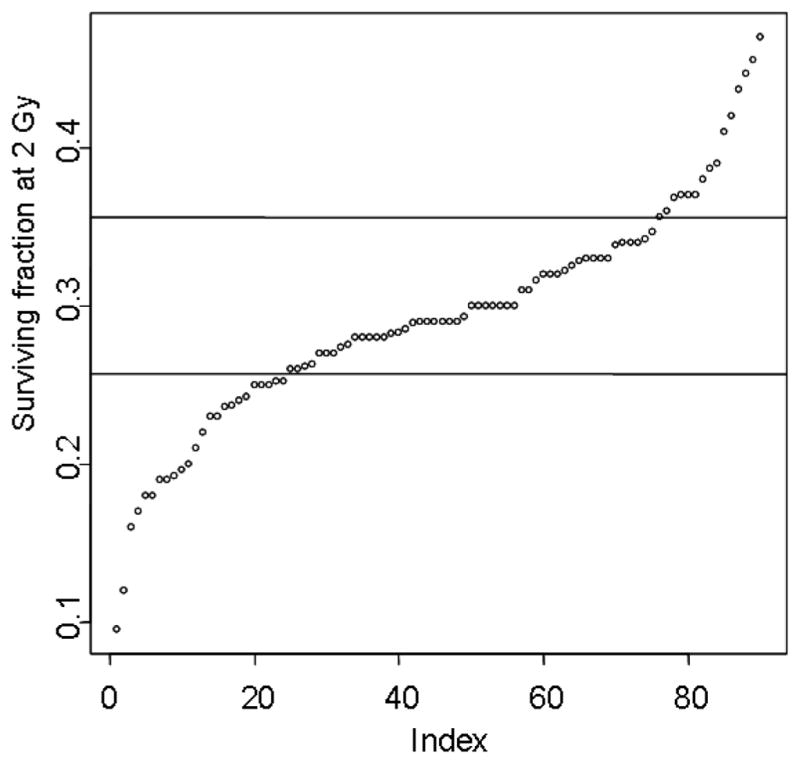
Radiosensitivity of 90 skin fibroblast cell lines as measured by SF2. Horizontal lines demarcate groups based upon the inflections in the shape of a fitted curve, not shown, and then characterized according to radiosensitivity: radiosensitive, normal, and radioresistant.
Early passage fibroblasts from skin samples were assayed for radiosensitivity by clonogenic assay according to Geara et al. (1992a, 1992b). Most importantly, for good statistical validity, coefficients of variation for independent replicate experiments were required to be <25%. K-Means cluster analysis was used to triage cell lines into normal, radiosensitive, and radioresistant cohorts based upon SF2 values. As shown in Fig. 2, the most radiosensitive cohort consists of cell lines with SF2 < 0.26, comprising 24 of the 90 cell lines in the panel. The most radioresistant cells are those with SF2 > 0.36, of which there are 15 cell lines. The remaining 51 cell lines are of average radiosensitivity and form the bulk of the population.
One hypothesis tested was the notion that DNA repair was an underlying factor in the differences in intrinsic cellular radiosensitivity between fibroblast cell lines. Based upon prior results it was determined that the assay used, pulsed field gel electrophoresis (Blocher et al. 1989; Story et al. 1994; Kurimasa et al. 1999), could discriminate cell lines based upon DSB rejoining capacity (Fig. 3). However, the differences in DSB rejoining capacity between cell lines having large differences in SF2 were small and would not adequately separate out the small but likely meaningful differences in DSB rejoining capacity that would be seen in fibroblasts with small differences in SF2. Therefore, a more sensitive alternative assay that alsodetermined whether DNA repair fidelity would be a significant factor in the radiosensitivity of fibroblasts. The technique introduced by Lobrich and colleagues (Lobrich et al. 1995, 2000; Alsbeih et al. 2003a) which measured the fidelity of radiation-induced DSB repair was adopted and applied to several fibroblast cell lines. Measurement of misrepair is achieved by quantifying the damage and restoration of a 3.2 Mbp NotI restriction fragment on pulsed-field electrophoresis gels. Because the target is small the doses required for this fidelity assay are quite large (80 Gy). However, it is clear for at least the S23 and C42 cell lines that the bulk rejoining of DSBs is intermediate between a repair competent C29 cell line and the radiosensitive AT cell line, however, for both S23 and C42 DSB repair fidelity is substantially reduced, with the C42 response being nearly equivalent to that of AT. Using another assay it was also determined that C42 did not process chromatid breaks as efficiently as normal fibroblasts (Alsbeih et al. 2003b).
Fig. 3.

(Left) Bulk rejoining of DSBs in genomic DNA in three fibroblast cell lines from the fibroblast panel and an AT cell line measured as fraction of activity released (FAR). (Right) DSB repair fidelity in a 3.2 Mbp DNA fragment in these same four cell lines. SF2 values used in figure are:
| Normal cell line | SF2 | Radiosensitive cell line | SF2 |
|---|---|---|---|
| C29 | 0.36 | S23 | 0.16 |
| C42 | 0.14 | ||
| AT | 0.14 |
With DNA repair and DNA repair fidelity identified as the explanation for the radiosensitivity of these cell lines, this analysis was extended across 14 fibroblast cell lines of varying radiosensitivity selected from the three groups in order to develop a more global picture of readioresponse. Principal component analysis was used to place these 14 cell lines into a representative three-dimensional space based upon whole genome gene expression analysis (Fig. 4). Interestingly, while the normal and radioresistant cell lines tended to cluster together, the radiosensitive cell lines can each be found in their own space and as seen in Fig. 4 the genes that segregate the radiosensitive cells lines were unique to each cell line (C42 and C34 given as representative examples). This suggests that radiosensitivity may not be a continuum of slight genomic alterations but that there are many ways for molecular signaling to deviate from normal. It also suggests that signal transduction analysis may help identify specific pathways where there is altered signaling either through chromosomal alteration, mutation, or more likely an enhanced number of SNPs in genes within specific molecular signaling pathways. This result along with the development of a 41-gene signature for radiosensitivity based upon basal gene expression patterns (Williams et al. 2011) suggests that basal gene expression measurements are sufficient.
Fig. 4.

(Top) Principal component analysis of gene expression in 14 representative fibroblast cell lines. A false discovery rate of 0.01 was used to restrict the number of genes used in the principal component analysis. (Bottom) Correlations were measured between the component for each gene from the whole genome and the projection of C34 or C42 against each principal component. The adjusted p value of less than 0.01 was considered as significantly correlated after a Bonferroni multiple testing correction. Genes correlated with the PCA patterns (138 genes in C34 and 38 genes in C42) exhibited low expression levels in the corresponding cell lines.
It is worth reiterating here that the outwardly obvious phenotype for radiosensitivity is not the primary focus. It is the identification of the subtle changes in gene expression and protein function that regulate individual differences in radiosensitivity that may have health, therapy, or regulatory consequences.
CARCINOGENESIS
While fibroblasts may provide substantial information for biomarkers of radiosensitivity, they are less likely to provide information regarding carcinogenesis. To that end, a battery of normal lung epithelial cells from 60 individuals has been developed through the University of Texas Southwestern SPORE in Lung Cancer. These lung epithelial cells are immortal via the overexpression of CDK4 and hTERT, however, they do not form tumors when implanted in immune-compromised mice (Ramirez et al. 2004; Delgado et al. 2011). Like fibroblasts, there is variability in radiosensitivity, although it is not as striking as that of the fibroblast panel given that there was no bias in sampling. Fig. 5 shows representative SF2 values for a few of the HBEC cell lines. In addition, the radiosensitivity of one HBEC cell line (HBEC3 KT) has been examined across the linear energy transfer (LET) spectrum using particles of various energies. As expected, an increased effectiveness for cell killing (Fig. 6) with increasing LET was seen. It is expected that with LETs beyond 200 keV μm−1 a reduction in particle effectiveness, first shown by Barendsen et al. (1963) will be evident.
Fig. 5.
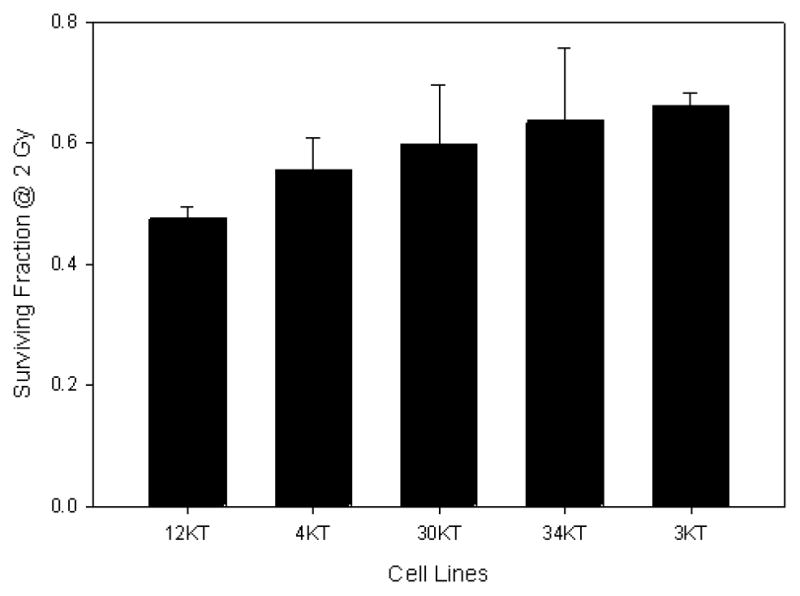
Range of relative survival of representative nononcogenically immortalized HBEC cell lines as measured by SF2.
Fig. 6.

Relative biological effectiveness (RBE) in HBEC3 KT cells as a function of increasing LET (keV μm−1) at a relative cellular survival of 0.368. Energies associated with each particle are nominal energy/atomic mass unit.
In order to identify differences in the molecular response to heavy particle exposures, global gene expression of the HBEC3 KT cell line up to 24 h after both low- and high-LET radiations was performed. The radiation doses used were chosen based upon equal physical dose or equivalent biological endpoint (cell death): for example, 0.5 Gy of 28Si vs. 1 Gy of gamma ray; or 1 Gy of 56Fe vs. 3 Gy of gamma ray. The analysis suggests that there is a very common signaling response to either low- or high-LET radiation that is driven by radiation-induced p53 signaling. For gamma ray, silicon and iron particle (1,000 Mev μm−1) exposures, functional pathways such as ATM, AKT/PI3K, DNA replication and repair and cell-cycle checkpoint control, particularly CDKN1a signaling, are common between the radiation types. However, as seen in Fig. 7, there are also distinct patterns of gene expression that are driven by the radiation species and the time post-irradiation, but not by the dose, at least in the range used here.
Fig. 7.

Principal component analysis of gene expression using the differences in gene expression for all genes when compared to unirradiated controls, for gamma ray, silicon, or iron (1 Gev μm−1). There are three independent replicates for each data point. The ellipses were generated by measuring three standard deviations around the centroid for each group using Partek Genomics Suite.
Whether these differences in gene expression seen in HBEC3 KT cells over the 24 h period post irradiation provide clues to subsequent oncogenic events is unknown. Initiation events may be less important than subsequent events that promote transformation and ultimately oncogenesis. Equally important is whether the mutations or other genomic alterations responsible for the aberrant molecular signaling that leads to lung cancer will be unique to ionizing radiation. Furthermore, it will be important to tease out the role that smoking plays in lung carcinogenesis as the molecular characterization and clinical epidemiology of lung cancer in nonsmokers appears to be different from that of lung cancer in smokers (Sun et al. 2007; Brambilla and Gazdar 2009; Samet et al. 2009).
Irradiation of immortalized HBECs by low- or high-LET particles was first reported by Hei and colleagues (Hei et al. 1994, 1996; Franken et al. 2011; Williams et al. 2011) using the BEP2D system. However, in these early experiments the cells were immortalized using human papilloma virus (HPV) and after irradiation were highly tumorigenic (4 × 10−7 after a 30 cGy alpha-particle dose). Subsequent studies by Hei using alpha and iron particles identified the loss of Betaig-h3 as causally linked to tumorigenesis (Story et al. 1994; Kurimasa et al. 1999). In addition, Hei et al. (1996) detected the overexpression of mutated p53 as well as cyclins D1 and D2 in tumorigenic HBEC cells. These earlier studies set the stage for identifying biomarkers of radioresponse after low- and high-LET radiations. However, there is one major distinction between these early experiments and the current experiments. The rate of cellular transformation is approximately 105-fold higher in HPV immortalized HBECs (10−2 – 10−1) when compared to hTERT/p16 immortalization (10−7 – 10−6) although neither culture is oncogenic, that is, they will not generate tumors in nude mice. This suggests that HPV immortalized HBECs are more oncogenically progressed and that subsequent events such as radiation exposure may be more effective at generating oncogenic cells from HPV-immortalized cells. For example, as described above after a 30 cGy alpha-particle dose the frequency of tumorigenic BEP2D cells was estimated at 4 × 10−7. HBEC3 KT cells, on the other hand, have a spontaneous cellular transformation frequency of approximately 4 × 10−7, but not a single tumorigenic cell has been identified to date at any dose of gamma ray, silicon or iron particles.
There is a roughly fourfold increase in cellular transformation seen with low doses of 1 GeV silicon or iron and the examination of commonly mutated genes in lung cancer, p53, and KRAS in 12 transformed HBEC3 KT cells has identified one clone heterozygous for the kRAS v12 mutation and no p53 mutations in exons 5–8. Western analysis of p53, MDM2, and Betaig-h3 shows an enhanced expression of MDM2 and a reduced expression of Betaig-h3 in ~25% of the transformed HBEC3-KT clones. Given the fact that no tumors have been generated from these transformed clones it suggests that MDM2 up-regulation and Betaig-h3 down-regulation are early steps in progression to malignancy and that other alterations are needed.
Another concern for high-LET particles is low doses and rate of exposure. There exists significant evidence of enhanced rates of solid tumor carcinogenesis with low doses and low dose rates of high-LET particles (Ullrich et al. 1977, 1987; Fry et al. 1983; Lubin et al. 1995; Weil et al. 2009; Shuryak et al. 2011). The subpopulation with the greatest concern for carcinogenic risk at very low dose rates are the members of the various astronaut corps. Ongoing and future studies will explore low dose and low dose rate exposures of HBEC cells to HZE particles for transformation rate and oncogenic potential. The goal is to develop markers of progression and oncogenesis that can then be tested across a battery of normal HBECs that vary by gender, donor smoking status, and other biologic endpoints. Ultimately, the biomarkers developed will be characterized and risk coefficients developed by comparing against the biomarkers for lung tumors from both smokers and never-smokers.
SUMMARY
The data described provides cellular and molecular evidence that normal individuals may respond differently to radiation exposures. The response may be overt or subtle and being able to identify individuals at greatest risk for adverse clinical responses or at high risk for carcinogenesis has a medical and economic impact on both the individual and society at-large. Developing credible biomarkers of radiation risk or response is ongoing by a number of groups, however validation of putative biomarkers is challenging. Each assay described above has limitations and all will likely be useful in generating, validating, or defining molecular mechanism. Furthermore, this review has focused on genomic analysis, however both proteomic and epigenomic analyses are also being used to develop biomarkers.
For HZE particles the task is even more daunting. Data suggest that while there is a generic response to radiation there are also distinct molecular pathways that are over-represented based upon the particle to which HBECs are exposed. Whether this transmits from one cell type to another is unknown, whether this transmits from one individual to another is unknown. Also unknown is how particle quality, the interplay between particle charge and particle energy, impacts cellular and molecular responses. Addressing these questions is important for the proper implementation of HZE particle radiotherapy.
Acknowledgments
The research described was funded by the following grants. NIH/NCI: PO1 CA 06294, PI, Ang; and CA 70907, PI, Minna. NASA: NAG9-1522, PI, Story; NNX11AC54G, PI, Minna, and NNJ05HD36G, PI, Minna. We are indebted to the personnel at the Brookhaven National Laboratory NASA Space Radiation Laboratories for their efforts.
References
- Alizadeh AA, Eisen MB, Davis RE, Ma C, Lossos IS, Rosenwald A, Boldrick JC, Sabet H, Tran T, Yu X, Powell JI, Yang L, Marti GE, Moore T, Hudson J, Lu L, Lewis DB, Tibshirani R, Sherlock G, Chan WC, Greiner TC, Weisenburger DD, Armitage JO, Warnke R, Levy R, Wilson W, Grever MR, Byrd JC, Botstein D, Brown PO, Staudt LM. Distinct types of diffuse large B-cell lymphoma identified by gene expression profiling. Nature. 2000;403:503–511. doi: 10.1038/35000501. [DOI] [PubMed] [Google Scholar]
- Alsbeih G, Brock WA, Terry N, Story MD. Analysis of radiation-induced DNA double-strand breaks misrepair is not compromized by broken DNA in human fibroblasts. Radiat Environm Biophys. 2003a;42:107–111. doi: 10.1007/s00411-003-0197-4. [DOI] [PubMed] [Google Scholar]
- Alsbeih G, Story MD, Maor MH, Geara FB, Brock WA. Chromosomal fragility syndrome and family history of radiosensitivity as indicators for radiotherapy dose modification. Radiother Oncol. 2003b;66:341–344. doi: 10.1016/s0167-8140(02)00327-4. [DOI] [PubMed] [Google Scholar]
- Amundson SA, Do KT, Shahab S, Bittner M, Meltzer P, Trent J, Fornace AJ. Identification of potential mRNA biomarkers in peripheral blood lymphocytes for human exposure to ionizing radiation. Radiat Res. 2000;154:342–346. doi: 10.1667/0033-7587(2000)154[0342:iopmbi]2.0.co;2. [DOI] [PubMed] [Google Scholar]
- Barendsen GW, Walter HM, Fowler JF, Bewley DK. Effects of different ionizing radiations on human cells in tissue culture. III. Experiments with cyclotron-accelerated alpha-particles and deuterons. Radiat Res. 1963;18:106–119. [PubMed] [Google Scholar]
- Bartosiewicz M, Trounstine M, Barker D, Johnston R, Buckpitt A. Development of a toxicological gene array and quantitative assessment of this technology. Arch Biochem Biophys. 2000;376:66–73. doi: 10.1006/abbi.2000.1700. [DOI] [PubMed] [Google Scholar]
- Bentzen SM, Tucker SL. Individualization of radiotherapy dose prescriptions by means of an in vitro radiosensitivity assay. Radiother Oncol. 1998;46:216–218. doi: 10.1016/s0167-8140(97)00225-9. [DOI] [PubMed] [Google Scholar]
- Blocher D, Einspenner M, Zajackowski J. CHEF electrophoresis, a sensitive technique for the determination of DNA double-strand breaks. Int J Radiat Biol. 1989;56:437–448. doi: 10.1080/09553008914551591. [DOI] [PubMed] [Google Scholar]
- Bonnen PE, Story MD, Ashorn CL, Buchholz TA, Weil MM, Nelson DL. Haplotypes at ATM identify coding-sequence variation and indicate a region of extensive linkage disequilibrium. Am J Human Genet. 2000;67:1437–1451. doi: 10.1086/316908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brambilla E, Gazdar A. Pathogenesis of lung cancer signalling pathways: roadmap for therapies. Eur Respiratory J. 2009;33:1485–1497. doi: 10.1183/09031936.00014009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brock WA, Tucker SL, Geara FB, Turesson I, Wike J, Nyman J, Peters LJ. Fibroblast radiosensitivity versus acute and late normal skin responses in patients treated for breast cancer. Intl J Radiat Oncol Biol Phys. 1995;32:1371–1379. doi: 10.1016/0360-3016(95)00068-A. [DOI] [PubMed] [Google Scholar]
- Buchholz TA, Wu XF, Hussain A, Tucker SL, Mills GB, Haffty B, Bergh S, Story M, Geara FB, Brock WA. Evidence of haplotype insufficiency in human cells containing a germline mutation in BRCA1 or BRCA2. Intl J Cancer. 2002;97:557–561. doi: 10.1002/ijc.10109. [DOI] [PubMed] [Google Scholar]
- Buchholz TA, Weil MM, Ashorn CL, Strom EA, Sigurdson A, Bondy M, Chakraborty R, Cox JD, McNeese MD, Story MD. A Ser49Cys variant in the ataxia telangiectasia, mutated, gene that is more common in patients with breast carcinoma compared with population controls. Cancer. 2004;100:1345–1351. doi: 10.1002/cncr.20133. [DOI] [PubMed] [Google Scholar]
- Burnet NG, Nyman J, Turesson I, Wurm R, Yarnold JR, Peacock JH. Prediction of normal-tissue tolerance to radiotherapy from in-vitro cellular radiation sensitivity. Lancet. 1992;339:1570–1571. doi: 10.1016/0140-6736(92)91833-t. [DOI] [PubMed] [Google Scholar]
- Burnet NG, Johansen J, Turesson I, Nyman J, Peacock JH. Describing patients’ normal tissue reactions: concerning the possibility of individualising radiotherapy dose prescriptions based on potential predictive assays of normal tissue radiosensitivity. Steering Committee of the BioMed2 European Union Concerted Action Programme on the Development of Predictive Tests of Normal Tissue Response to Radiation Therapy. Intl J Cancer. 1998;79:606–613. doi: 10.1002/(sici)1097-0215(19981218)79:6<606::aid-ijc9>3.0.co;2-y. [DOI] [PubMed] [Google Scholar]
- Burnet NG, Nyman J, Turesson I, Wurm R, Yarnold JR, Peacock JH. The relationship between cellular radiation sensitivity and tissue response may provide the basis for individualising radiotherapy schedules. Radiother Oncol. 1994;33:228–238. doi: 10.1016/0167-8140(94)90358-1. [DOI] [PubMed] [Google Scholar]
- Chen F, Demers LM, Vallyathan V, Lu Y, Castranova V, Shi X. Impairment of NF-κB activation and modulation of gene expression by calpastatin. Am J Physiol. 2000;279:C709–C716. doi: 10.1152/ajpcell.2000.279.3.C709. [DOI] [PubMed] [Google Scholar]
- Colantuoni C, Purcell AE, Bouton CM, Pevsner J. High throughput analysis of gene expression in the human brain. J Neurosci Res. 2000;59:1–10. doi: 10.1002/(sici)1097-4547(20000101)59:1<1::aid-jnr1>3.0.co;2-2. [DOI] [PubMed] [Google Scholar]
- Cuneo A, Bigoni R, Rigolin GM, Roberti MG, Milani R, Bardi A, Minotto C, Agostini P, De Angeli C, Narducci MG, Sabbioni S, Russo G, Negrini M, Castoldi G. Acquired chromosome 11q deletion involving the ataxia teleangiectasia locus in B-cell non-Hodgkin’s lymphoma: correlation with clinicobiologic features. J Clin Oncol. 2000;18:2607–2614. doi: 10.1200/JCO.2000.18.13.2607. [DOI] [PubMed] [Google Scholar]
- de Vries CJ, van Achterberg TA, Horrevoets AJ, ten Cate JW, Pannekoek H. Differential display identification of 40 genes with altered expression in activated human smooth muscle cells. Local expression in atherosclerotic lesions of smags, smooth muscle activation-specific genes. J Biologic Chem. 2000;275:23939–23947. doi: 10.1074/jbc.M910099199. [DOI] [PubMed] [Google Scholar]
- Delgado O, Kaisani AA, Spinola M, Xie XJ, Batten KG, Minna JD, Wright WE, Shay JW. Multipotent capacity of immortalized human bronchial epithelial cells. PloS one. 2011;6:e22023. doi: 10.1371/journal.pone.0022023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dikomey E, Dahm-Daphi J, Brammer I, Martensen R, Kaina B. Correlation between cellular radiosensitivity and non-repaired double-strand breaks studied in nine mammalian cell lines. Intl J Radiat Biol Relat Stud Phys Chem Med. 1998;73:269–278. doi: 10.1080/095530098142365. [DOI] [PubMed] [Google Scholar]
- Dikomey E, Brammer I, Johansen J, Bentzen SM, Overgaard J. Relationship between DNA double-strand breaks, cell killing, and fibrosis studied in confluent skin fibroblasts derived from breast cancer patients. Intl J Radiat Oncol Biol Phys. 2000;46:481–490. doi: 10.1016/s0360-3016(99)00335-1. [DOI] [PubMed] [Google Scholar]
- Fornace AJ, Amundson SA, Bittner M, Myers TG, Meltzer P, Weinsten JN, Trent J. The complexity of radiation stress responses: analysis by informatics and functional genomics approaches. Gene Exp. 1999;7:387–400. [PMC free article] [PubMed] [Google Scholar]
- Franken NA, ten Cate R, Krawczyk PM, Stap J, Haveman J, Aten J, Barendsen GW. Comparison of RBE values of high-LET alpha-particles for the induction of DNA-DSBs, chromosome aberrations and cell reproductive death. Radiat Oncol. 2011;6:64. doi: 10.1186/1748-717X-6-64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Freeman JL, Perry GH, Feuk L, Redon R, McCarroll SA, Altshuler DM, Aburatani H, Jones KW, Tyler-Smith C, Hurles ME, Carter NP, Scherer SW, Lee C. Copy number variation: new insights in genome diversity. Genome Res. 2006;16:949–961. doi: 10.1101/gr.3677206. [DOI] [PubMed] [Google Scholar]
- Fry RJ, Powers-Risius P, Alpen EL, Ainsworth EJ, Ullrich RL. High-LET radiation carcinogenesis. Adv Space Res. 1983;3:241–248. doi: 10.1016/0273-1177(83)90194-1. [DOI] [PubMed] [Google Scholar]
- Geara FB, Peters LJ, Ang KK, Wike JL, Brock WA. Radiosensitivity measurement of keratinocytes and fibroblasts from radiotherapy patients. Intl J Radiat Oncol Biol Phys. 1992a;24:287–293. doi: 10.1016/0360-3016(92)90683-9. [DOI] [PubMed] [Google Scholar]
- Geara FB, Peters LJ, Ang KK, Wike JL, Sivon SS, Guttenberger R, Callender DL, Malaise EP, Brock WA. Intrinsic radiosensitivity of normal human fibroblasts and lymphocytes after high- and low-dose-rate irradiation. Cancer Res. 1992b;52:6348–6352. [PubMed] [Google Scholar]
- Geara FB, Peters LJ, Ang KK, Wike JL, Brock WA. Prospective comparison of in vitro normal cell radiosensitivity and normal tissue reactions in radiotherapy patients. Intl J Radiat Oncol Biol Phys. 1993;27:1173–1179. doi: 10.1016/0360-3016(93)90540-c. [DOI] [PubMed] [Google Scholar]
- Golub TR, Slonim DK, Tamayo P, Huard C, Gaasenbeek M, Mesirov JP, Coller H, Loh ML, Downing JR, Caligiuri MA, Bloomfield CD, Lander ES. Molecular classification of cancer: class discovery and class prediction by gene expression monitoring. Science. 1999;286:531–537. doi: 10.1126/science.286.5439.531. [DOI] [PubMed] [Google Scholar]
- Gowen LC, Avrutskaya AV, Latour AM, Koller BH, Leadon SA. BRCA1 required for transcription-coupled repair of oxidative DNA damage. Science. 1998;281:1009–1012. doi: 10.1126/science.281.5379.1009. [DOI] [PubMed] [Google Scholar]
- Hei TK, Piao CQ, Willey JC, Thomas S, Hall EJ. Malignant transformation of human bronchial epithelial cells by radon-simulated alpha-particles. Carcinogenesis. 1994;15:431–437. doi: 10.1093/carcin/15.3.431. [DOI] [PubMed] [Google Scholar]
- Hei TK, Piao CQ, Sutter T, Willey JC, Suzuki K. Cellular and molecular alterations in human epithelial cells transformed by high LET radiation. Adv Space Res. 1996;18:137–148. doi: 10.1016/0273-1177(95)00800-t. [DOI] [PubMed] [Google Scholar]
- Hwang DM, Dempsey AA, Lee CY, Liew CC. Identification of differentially expressed genes in cardiac hypertrophy by analysis of expressed sequence tags. Genomics. 2000;66:1–14. doi: 10.1006/geno.2000.6171. [DOI] [PubMed] [Google Scholar]
- Iafrate AJ, Feuk L, Rivera MN, Listewnik ML, Donahoe PK, Qi Y, Scherer SW, Lee C. Detection of large-scale variation in the human genome. Nature Genet. 2004;36:949–951. doi: 10.1038/ng1416. [DOI] [PubMed] [Google Scholar]
- Johansen J, Bentzen SM, Overgaard J, Overgaard M. Evidence for a positive correlation between in vitro radiosensitivity of normal human skin fibroblasts and the occurrence of subcutaneous fibrosis after radiotherapy. Intl J Radiat Biol. 1994;66:407–412. doi: 10.1080/09553009414551361. [DOI] [PubMed] [Google Scholar]
- Johansen J, Bentzen SM, Overgaard J, Overgaard M. Relationship between the in vitro radiosensitivity of skin fibroblasts and the expression of subcutaneous fibrosis, telangiectasia, and skin erythema after radiotherapy. Radiother Oncol. 1996;40:101–109. doi: 10.1016/0167-8140(96)01777-x. [DOI] [PubMed] [Google Scholar]
- Jones LA, Scott D, Cowan R, Roberts SA. Abnormal radiosensitivity of lymphocytes from breast cancer patients with excessive normal tissue damage after radiotherapy: chromosome aberrations after low dose-rate irradiation. Intl J Radiat Biol. 1995;67:519–528. doi: 10.1080/09553009514550631. [DOI] [PubMed] [Google Scholar]
- Jones IM, Thomas CB, Xi T, Mohrenweiser HW, Nelson DO. Exploration of methods to identify polymorphisms associated with variation in DNA repair capacity phenotypes. Mutat Res. 2006 doi: 10.1016/j.mrfmmm.2006.11.005. [DOI] [PubMed] [Google Scholar]
- Kinsella TJ, Mitchell JB, McPherson S, Russo A, Tietze F. In vitro x-ray sensitivity in ataxia telangiectasis homozygote and heterozygote skin fibroblasts under oxic and hypoxic conditions. Cancer Res. 1982;42:3950–3956. [PubMed] [Google Scholar]
- Kurimasa A, Kumano S, Boubnov NV, Story MD, Tung CS, Peterson SR, Chen DJ. Requirement for the kinase activity of human DNA-dependent protein kinase catalytic subunit in DNA strand break rejoining. Molecul Cellul Biol. 1999;19:3877–3884. doi: 10.1128/mcb.19.5.3877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lobrich M, Rydberg B, Cooper PK. Repair of x-ray-induced DNA double-strand breaks in specific Not I restriction fragments in human fibroblasts: joining of correct and incorrect ends. Proc Natl Acad Sci USA. 1995;92:12050–12054. doi: 10.1073/pnas.92.26.12050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lobrich M, Kuhne M, Wetzel J, Rothkamm K. Joining of correct and incorrect DNA double-strand break ends in normal human and ataxia telangiectasia fibroblasts. Genes Chromos Cancer. 2000;27:59–68. [PubMed] [Google Scholar]
- Lubin JH, Boice JD, Jr, Edling C, Hornung RW, Howe G, Kunz E, Kusiak RA, Morrison HI, Radford EP, Samet JM, Tirmarche M, Woodward A, Yao SX. Radon-exposed underground miners and inverse dose-rate (protraction enhancement) effects. Health Phys. 1995;69:494–500. doi: 10.1097/00004032-199510000-00007. [DOI] [PubMed] [Google Scholar]
- Mohrenweiser HW, Wilson DM, 3rd, Jones IM. Challenges and complexities in estimating both the functional impact and the disease risk associated with the extensive genetic variation in human DNA repair genes. Mutat Res. 2003;526:93–125. doi: 10.1016/s0027-5107(03)00049-6. [DOI] [PubMed] [Google Scholar]
- Morgan JL, Holcomb TM, Morrissey RW. Radiation reaction in ataxia telangiectasia. Am J Diseases Children. 1968;116:557–558. doi: 10.1001/archpedi.1968.02100020561022. [DOI] [PubMed] [Google Scholar]
- Paterson MC, Anderson AK, Smith BP, Smith PJ. Enhanced radiosensitivity of cultured fibroblasts from ataxia telangiectasia heterozygotes manifested by defective colony-forming ability and reduced DNA repair replication after hypoxic gamma-irradiation. Cancer Res. 1979;39:3725–3734. [PubMed] [Google Scholar]
- Pippard EC, Hall AJ, Barker DJ, Bridges BA. Cancer in homozygotes and heterozygotes of ataxia-telangiectasia and xeroderma pigmentosum in Britain. Cancer Res. 1988;48:2929–2932. [PubMed] [Google Scholar]
- Ramirez RD, Sheridan S, Girard L, Sato M, Kim Y, Pollack J, Peyton M, Zou Y, Kurie JM, Dimaio JM, Milchgrub S, Smith AL, Souza RF, Gilbey L, Zhang X, Gandia K, Vaughan MB, Wright WE, Gazdar AF, Shay JW, Minna JD. Immortalization of human bronchial epithelial cells in the absence of viral oncoproteins. Cancer Res. 2004;64:9027–9034. doi: 10.1158/0008-5472.CAN-04-3703. [DOI] [PubMed] [Google Scholar]
- Risch N, Merikangas K. The future of genetic studies of complex human diseases. Science. 1996;273:1516–1517. doi: 10.1126/science.273.5281.1516. [DOI] [PubMed] [Google Scholar]
- Samet JM, Avila-Tang E, Boffetta P, Hannan LM, Olivo-Marston S, Thun MJ, Rudin CM. Lung cancer in never smokers: clinical epidemiology and environmental risk factors. Clinic Cancer Res. 2009;15:5626–5645. doi: 10.1158/1078-0432.CCR-09-0376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sebat J, Lakshmi B, Troge J, Alexander J, Young J, Lundin P, Maner S, Massa H, Walker M, Chi M, Navin N, Lucito R, Healy J, Hicks J, Ye K, Reiner A, Gilliam TC, Trask B, Patterson N, Zetterberg A, Wigler M. Large-scale copy number polymorphism in the human genome. Science. 2004;305:525–528. doi: 10.1126/science.1098918. [DOI] [PubMed] [Google Scholar]
- Sgroi DC, Teng S, Robinson G, LeVangie R, Hudson JR, Elkahloun AG. In vivo gene expression profile analysis of human breast cancer progression. Cancer Res. 1999;59:5656–5661. [PubMed] [Google Scholar]
- Shuryak I, Brenner DJ, Ullrich RL. Radiation-induced carcinogenesis: mechanistically based differences between gamma-rays and neutrons, and interactions with DMBA. PloS one. 2011;6:e28559. doi: 10.1371/journal.pone.0028559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Story MD, Mendoza EA, Meyn RE, Tofilon PJ. Pulsed-field gel electrophoretic analysis of DNA double-strand breaks in mammalian cells using photostimulable storage phosphor imaging. Int J Radiat Biol. 1994;65:523–528. doi: 10.1080/09553009414550611. [DOI] [PubMed] [Google Scholar]
- Sun S, Schiller JH, Gazdar AF. Lung cancer in never smokers–a different disease. Nat Rev Cancer. 2007;7:778–790. doi: 10.1038/nrc2190. [DOI] [PubMed] [Google Scholar]
- Swift M, Sholman L, Perry M, Chase C. Malignant neoplasms in the families of patients with ataxia-telangiectasia. Cancer Res. 1976;36:209–215. [PubMed] [Google Scholar]
- Taylor AM, Harnden DG, Arlett CF, Harcourt SA, Lehmann AR, Stevens S, Bridges BA. Ataxia telangiectasia: a human mutation with abnormal radiation sensitivity. Nature. 1975;258:427–429. doi: 10.1038/258427a0. [DOI] [PubMed] [Google Scholar]
- Tucker SL, Turesson I, Thames HD. Evidence for individual differences in the radiosensitivity of human skin. Eur J Cancer. 1992;11:1783–1791. doi: 10.1016/0959-8049(92)90004-l. [DOI] [PubMed] [Google Scholar]
- Tucker SL, Geara FB, Peters LJ, Brock WA. How much could the radiotherapy dose be altered for individual patients based on a predictive assay of normal-tissue radiosensitivity? Radiother Oncol. 1996;38:103–113. doi: 10.1016/0167-8140(95)01669-4. [DOI] [PubMed] [Google Scholar]
- Tuzun E, Sharp AJ, Bailey JA, Kaul R, Morrison VA, Pertz LM, Haugen E, Hayden H, Albertson D, Pinkel D, Olson MV, Eichler EE. Fine-scale structural variation of the human genome. Nature Genet. 2005;37:727–732. doi: 10.1038/ng1562. [DOI] [PubMed] [Google Scholar]
- Ullrich RL, Jernigan MC, Storer JB. Neutron carcinogenesis. Dose and dose-rate effects in BALB/c mice. Radiat Res. 1977;72:487–498. [PubMed] [Google Scholar]
- Ullrich RL, Jernigan MC, Satterfield LC, Bowles ND. Radiation carcinogenesis: time-dose relationships. Radiat Res. 1987;111:179–184. [PubMed] [Google Scholar]
- Walker MG, Volkmuth W, Sprinzak E, Hodgson D, Klingler T. Prediction of gene function by genome-scale expression analysis: prostate cancer-associated genes. Genome Res. 1999;9:1198–1203. doi: 10.1101/gr.9.12.1198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang A, Pierce A, Judson-Kremer K, Gaddis S, Aldaz CM, Johnson DG, MacLeod MC. Rapid analysis of gene expression (RAGE) facilitates universal expression profiling. Nucleic Acids Res. 1999;27:4609–4618. doi: 10.1093/nar/27.23.4609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weil MM, Bedford JS, Bielefeldt-Ohmann H, Ray FA, Genik PC, Ehrhart EJ, Fallgren CM, Hailu F, Battaglia CL, Charles B, Callan MA, Ullrich RL. Incidence of acute myeloid leukemia and hepatocellular carcinoma in mice irradiated with 1 GeV/nucleon 56Fe ions. Radiat Res. 2009;172:213–219. doi: 10.1667/RR1648.1. [DOI] [PubMed] [Google Scholar]
- Whitney LW, Becker KG, Tresser NJ, Caballero-Ramos CI, Munson PJ, Prabhu VV, Trent JM, McFarland HF, Biddison WE. Analysis of gene expression in mutiple sclerosis lesions using cDNA microarrays. Ann Neurol. 1999;46:425–428. doi: 10.1002/1531-8249(199909)46:3<425::aid-ana22>3.0.co;2-o. [DOI] [PubMed] [Google Scholar]
- Williams PD, Owens CR, Dziegielewski J, Moskaluk CA, Read PW, Larner JM, Story MD, Brock WA, Amundson SA, Lee JK, Theodorescu D. Cyclophilin B expression is associated with in vitro radioresistance and clinical outcome after radiotherapy. Neoplasia. 2011;13:1122–1131. doi: 10.1593/neo.111398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woudstra EC, Driessen C, Konings AW, Kampinga HH. DNA damage induction and tumour cell radiosensitivity: PFGE and halo measurements. Intl J Radiat Biol. 1998;73:495–502. doi: 10.1080/095530098142031. [DOI] [PubMed] [Google Scholar]
- Wurm R, Burnet NG, Duggal N, Yarnold JR, Peacock JH. Cellular radiosensitivity and DNA damage in primary human fibroblasts. Intl J Radiat Oncol Biol Phys. 1994;30:625–633. doi: 10.1016/0360-3016(92)90949-i. [DOI] [PubMed] [Google Scholar]
- Xi T, Jones IM, Mohrenweiser HW. Many amino acid substitution variants identified in DNA repair genes during human population screenings are predicted to impact protein function. Genomics. 2004;83:970–979. doi: 10.1016/j.ygeno.2003.12.016. [DOI] [PubMed] [Google Scholar]