

Abstract
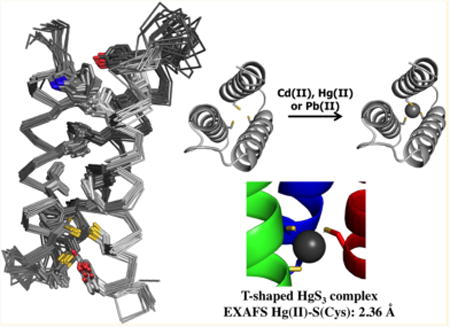
De novo protein design is a biologically relevant approach that provides a novel process in elucidating protein folding and modeling the metal centers of metalloproteins in a completely unrelated or simplified fold. An integral step in de novo protein design is the establishment of a well-folded scaffold with one conformation, which is a fundamental characteristic of many native proteins. Here, we report the NMR solution structure of apo α3DIV at pH 7.0, a de novo designed three-helix bundle peptide containing a triscysteine motif (Cys18, Cys28, and Cys67) that binds toxic heavy metals. The structure comprises 1067 NOE restraints derived from multinuclear multidimensional NOESY, as well as 138 dihedral angles (ψ, φ, and χ1). The backbone and heavy atoms of the 20 lowest energy structures have a root mean square deviation from the mean structure of 0.79 (0.16) Å and 1.31 (0.15) Å, respectively. When compared to the parent structure α3D, the substitution of Leu residues to Cys enhanced the α-helical content of α3DIV while maintaining the same overall topology and fold. In addition, solution studies on the metalated species illustrated metal-induced stability. An increase in the melting temperatures was observed for Hg(II), Pb(II), or Cd(II) bound α3DIV by 18–24 °C compared to its apo counterpart. Further, the extended X-ray absorption fine structure analysis on Hg(II)-α3DIV produced an average Hg(II)–S bond length at 2.36 Å, indicating a trigonal T-shaped coordination environment. Overall, the structure of apo α3DIV reveals an asymmetric distorted triscysteine metal binding site, which offers a model for native metalloregulatory proteins with thiol-rich ligands that function in regulating toxic heavy metals, such as ArsR, CadC, MerR, and PbrR
Metalloproteins acquire metal ions to perform the most essential reactions in nature including respiration, photosynthesis, and nitrogen fixation.1 Metalloregulatory proteins are a subset of metalloproteins used by microorganisms to control the levels of essential transition metal ions (Fe, Cu, Zn, and Mn) or decrease levels of toxic metals (Hg, As, Pb, and Cd) within their cells.1,2 Toxic heavy metals threaten biological pathways that use proteins with thiol-rich metal binding sites by displacing existing metal ions in proteins that function in catalytic pathways or in the storage or transport of essential metals.3,4 Understanding the structure–function relationship found in the overall fold and at the metal centers of these metalloproteins is critical to gaining insight into their chemical properties and roles in biological systems. This knowledge can be applied to developing ecological strategies aimed at managing the serious threat that toxic heavy metals in our environment pose to all forms of life.
One biologically relevant approach used to study the concept of the structure–function relationship in native metalloproteins is de novo protein design.5–7 Using what is known about the biophysical and biochemical properties of amino acids, this emerging approach involves designing and synthesizing a polypeptide scaffold from scratch. It allows one to tailor a sequence that is designed to form the proper hydrophobic, electrostatic, and hydrogen bonding interactions that will manifest into a unique and well-defined fold, which is a defining characteristic of native proteins.5 Therefore, de novo protein design provides a novel approach in studying the mechanisms behind protein folding and modeling the active sites of metalloproteins in a simplified or unrelated fold. Many groups have actively used this approach by incorporating the core components of the desired metal center in order to achieve a specific metal–ligand coordination or function, such as catalysis or redox activity.6,7
DeGrado and co-workers made a significant impact in the field of de novo protein design through the design, preparation, and characterization of α3D, a 73-residue peptide (Table 1) with a single conformation in solution and a unique native-like fold.8 A common de novo designed peptide uses a heptad repeat sequence that self-assembles into a parallel three-stranded coiled coil (3SCC) tertiary structure, such as the TRI9 and coil-Ser (CS)10 peptide family. In contrast to the 3SCC analogues, α3D is a single polypeptide chain that folds into a three-helix bundle, a fold that is ubiquitous in the molecular recognition domain of immunoglobulin G, DNA binding proteins, and various enzymes.11 Using the sequence of CS as a foundation, DeGrado and co-workers isolated α3D through an iterative process of modifying helix-capping interactions to dictate the topology of the bundle (clockwise vs counterclockwise), electrostatic (or salt-bridging) interactions to force the desired helix–helix pairing, and hydrophobic interactions to achieve a well-packed core.8,12 The result of this process led to a counterclockwise three-helix bundle fold that contains every natural amino acid residue except cysteine. It possesses the physical properties similar to native proteins13–16 and has a solved NMR structure (PDB 2A3D),8 offering a novel opportunity to add function to this de novo designed framework.
Table 1. Amino Acid Sequence of de novo Designed Peptidesa.
| Peptide | Sequence | Reference | |||
|---|---|---|---|---|---|
| abcdefg | abcdefg | abcdefg | abcdefg | ||
| CoilSer | AC-E WEALEKK | LAALESK | LQALEKK | LEALEHG -NH2 | 10 |
| abcdefg | abcdefg | abcdef | loop | ||
| MGSWAEFKQR | LAAIKTR | LQAL | GGS | 8 | |
| α3D | EAELAAFEKE | IAAFESE | LQAY | KGKG | |
| NPEVEALRKE | AAAIRDE | LQAYRHN | 23 | ||
| MGSWAEFKQR | LAAIKTR | CQAL | GGS | ||
| α3DIV | EAECAAFEKE | IAAFESE | LQAY | KGKG | |
| NPEVEALRKE | AAAIRDE | CQAYRHN | |||
| α3DH3 | MGSWAEFKQR | LAAIKTR | HQAL | GGS | 56 |
| α3DH3 | EAEHAAFEKE | IAAFESE | LQAY | KGKG | |
| NPEVEALRKE | AAAIRDE | HQAYRVN | GSGA |
The sequences are prepared in heptads. Residues that are underlined and bolded were changed from the previous design.
We redesigned the sequence of α3D by introducing a triscysteine motif that is found in the metal binding site of metalloregulatory proteins MerR,17–19 ArsR/SmtB,20 and CadC/CmtR.20–22 Three Leu to Cys mutations (Leu18Cys, Leu28Cys, and Leu67Cys) at the C-terminal end of α3D were performed to produce α3DIV.23 Utilizing several spectroscopic methods, we demonstrated that apo α3DIV is stable and well folded in solution at a pH range between 5 and 9. The coordination mode of Hg(II), Pb(II), and Cd(II) bound α3DIV is pH dependent. From a linear mercury complex, [Hg(II)-S2(SH)], a thiol group is deprotonated to form a trigonal [Hg(II)S3]−, and the thiol group has a pKa of 7.1 (0.1). Therefore, a pH condition below and above this pKa value leads to the linear or a trigonal complex, respectively. The formation of a trigonal pyramidal [Pb(II)S3]− and pseudotetrahedral [Cd(II)S3(N/O)]− complex was determined to require a simultaneous deprotonation of two Cys thiol groups, yielding pKa2 values of 10.6 (0.1) and 10.2 (0.1), respectively. A pH condition greater than 6 results in the formation of a [Pb(II)S3]− and [Cd(II)S3(N/O)]− complex. The [Pb(II)S3]−complex was determined to have a lower limit binding constant of 2.0 × 107 M−1, and the [Cd(II)S3(N/O)]− complex has the corresponding value of 3.1 × 107 M−1. In this work, we present the solution structure of apo α3DIV, solved at pH 7.0. This is the first reported structure that incorporates a triscysteine metal binding site in an antiparallel three-helix bundle fold of α3D through the modifications of stabilizing core hydrophobic residues to introduce a new function. The solution structure of α3DIV possesses the same overall topology and counterclockwise bundle as α3D, and the incorporation of Cys residues increased the helical content of the scaffold. Overall, the α3DIV structure provides evidence that the framework of α3D is amenable to mutations that involve removing Leu residues that were thought to be essential in inducing hydrophobic interactions. This structure offers insight into how the protein is preorganized before metal binding, which is essential for utilizing this framework in designing future functional metallopeptides.
Experimental Procedures
Protein Expression and Purification
A pET15b recombinant DNA plasmid (Celtek Genes) that contains the gene for α3DIV was transformed and expressed in Escherichia coli BL21(DE3) (Life Technologies). To overexpress 15N-labeled and 15N,13C- labeled α3DIV, E. coli cells were grown in M9 minimum media that contained 1 g/L 15NHCl4 (Cambridge Isotope Laboratories) or 1 g/L 15NHCl4 and 2 g/L [U-13C]glucose (Cambridge Isotope Laboratories), respectively. The cells were grown at 37 °C to an OD600 of 0.6, induced with 1 mM isopropyl β-d-1-thiogalactopyranoside (Life Technologies), and incubated at 18 °C overnight. The cells were lysed by sonication, and the soluble protein was obtained after heat denaturation (50 °C) and lyophilization. The protein powder was redissolved in 10% acetic acid and purified on a reversed-phase C18 HPLC, using a flow rate of 20 mL/min and a linear gradient of 0.1% TFA in water to 0.1% TFA in 9:1 CH3CN/H2O over 45 min. Using an electronspray mode on a Micromass LCT Time-of-Flight mass ionization spectrometer, the peptide mass was determined to be 7946.9 Da, which accounts for 72 of the 73 amino acids as Met1 is cleaved post-translation.
NMR Sample Preparation
Stock peptide concentrations were determined from the absorption band of the aromatic residues (Trp4, Tyr45, and Ty70) at 280 nm. Experiments that involved coherence transfer from the backbone amide protons required a 9:1 H2O-to-2H2O solution, which contained 1.0 mM 15N, 13C labeled peptide, 100 mM sodium chloride, 0.8 mM tris(2-carboxyethyl) phosphine (Fisher), 0.05 mM phenyl-methylsulfonyl fluoride (Fisher), and 0.5% sodium azide (Sigma-Aldrich). Samples for carbon-filtered aromatic to aliphatic NMR experiments were prepared in a 100% 2H2O solution and incubated overnight, containing the reagents listed above. The pH of the apoprotein samples used in the structure was adjusted at 7. The Pb(II)-15N-α3DIV sample was prepared by adding 1.0 equiv of PbCl2 to a 1 mM apo 15N-α3DIV solution, containing 20 mM BIS-TRIS buffer set at pH 7.0 in a 9:1 H2O-to-2H2O solution. The Hg(II)-15N-α3DIV sample was prepared under similar conditions but contained no buffer, and the pH was adjusted to 8.6 after the addition of 1.0 equiv of HgCl2. Control apo samples were also prepared to match the sample conditions of their metalated counterparts.
NMR Data Collection and Processing
NMR experiments for the structure determination were performed at 25 °C on a Varian (Agilent) INOVA 800 MHz NMR spectrometer, equipped with a triple resonance cold probe. The NMR acquisition parameters for the 800 MHz spectrometer are reported in Supporting Information, Table 1. 15N-HSQC spectra were collected for the metalated α3DIV species and their corresponding apo controls. These experiments were performed on a Bruker Avance 500 MHz with room temperature triple resonance probes and a Varian VNMRS 500 MHz equipped with a switchable probe. The N-HSQC experiments for the Pb(II)- N-α3DIV and its apo control were collected at 25 °C, while the Hg(II)- N-α3DIV and its apo control were obtained at 9 °C. The N-HSQC experiments were also performed at 25 °C for apo N-α3DIV with pH conditions of 5.8 and 8.6. The spectra were processed using NMRPIPE24 and then visualized and analyzed with SPARKY.25
Assignments
Backbone assignments were determined from a series of complementary three-dimensional (3D) triple resonance transverse relaxation optimized spectroscopy (TROSY)27–29 experiments: HNCO/HN(CA)CO,30 HN-(CO)CA/HNCA,31 HN(CO)CACB/HNCACB,32,33 and HN-(CA)HA34 Since the sequence of α3DIV is known, the backbone assignments were manually completed and then successively confirmed with an autoassignment program SAGA35 Ser24, Thr16, and Ser40 were used as landmarks in the manual assignment because the β-carbons of Ser and Thr residues are downfield of their α-carbons. Next, aliphatic assignments were obtained from the combined inphase/antiphase spectra of 3D HC(C)H-correlation spectroscopy (COSY)36 and (H)CCH-total correlation spectroscopy (TOCSY) 37,38 experiments. These assignments were further confirmed with 3D (1H, 15N, 1H) nuclear Overhauser effect spectroscopy (NOESY)-TROSY39 and (13C, 15N, 1H) hetero-nuclear multiple-quantum coherence (HMQC)-TROSY experiments, providing a through-space validation of the COSY and TOCSY assignments. Chemical shift assignments for residues 3–73 were then compiled and used to assign sequential-intra and inter-residue NOE upper distance limits (upl).
NOESY Experiments
In order to attain structural distance restraints, several 3D- 13C-edited and -resolved NOESY experiments were completed. Aliphatic–aliphatic NOEs were acquired from (13C-edited) HC(C)H-NOESY (inphase/anti-phase), which were collected at 60 and 200 ms mixing times. Aromatic to aliphatic NOEs were obtained from a 3D NOESY 13CHSQC experiment, while aromatic to amide NOEs were collected from 3D NOESY- 13C(resolved)-TROSY and the 3D NOESY (13C resolved)-TROSY. Further, non-intraresidue amide proton NOEs were acquired from 3D (1H, 15N, 1H) NOESY-TROSY and (13C, N, H) HMQC-NOESY-TROSY experiments. The upper-limit NOE restraints, which contain intraresidue, intrahelical, and interhelical NOEs, were then determined from the manual chemical shift assignments made on the COSY and TOCSY spectra.
Dihedral Angles and Hydrogen Bonds Restraints
TALOS-N41 was used to generate φ, ψ, and χ1 dihedral angle restraints from 1H, 1Hα, 14N, 13C, 13Cα, and 13Cβ chemical shift assignments. Dihedral φ and ψ angles that were classified as “strong” (residues 2–20, 25–45, and 45, 48, 51–70) were included in all the structure calculations, and χ1 angles for selected aliphatic residues were then included in the final rounds of calculations. TALOS-N also predicted the secondary structure and order parameters for each amino acid residue, which provided the structured regions: residues 5–20, 25–44, and 51–70. Furthermore, backbone hydrogen bonds in most of the structured regions were later incorporated in the upl restraints, excluding residues in the loop-turn regions. Upper limit distance restraints of 2.0 and 3.0 Å were given to Oi to Hi+4 and Oi to Ni+4, respectively.
Structure Calculation
CYANA 2.142 was used in the structure calculations and the input files comprised of the upper limit restraints obtained from 3D 15N- and 13C-edited/resolved-NOESY spectra, dihedral angle restraints (φ, ψ, and χ1), and backbone hydrogen bond restraints in the structured regions of the sequence. In the initial rounds of structure calculations, φ and ψ dihedral angle restraints were used along with the NOE upper limit distances, which were set at 3 or 5 Å. These values were then adjusted in the later rounds of calculations. That is, intraresidue upper limit distances were modified to have a 3–7 Å range, increasing the restraints between pseudoatom–pseudoatom contacts. For sequential–intrahelical distances, a 3–4.5 Å range was applied following Wuethrich's 1H-1H short-to-medium-range distances for an α-helical secondary structure. Long sequential–intrahelical distances, 1Hi to 1Hi+4, were given a 5–7 Å range. After, interhelical upl distances were adjusted to a 4–7 Å range, where the lower ends distances were based off the intensity in the NOESY spectra, and higher ends were set to again compensate for pseudoatom–pseudoatom correlations. In the final rounds of calculations, χ1 dihedral angle restraints were added and were followed by the incorporation of backbone hydrogen bonds. No lower limit distances were used. Of the 100 calculated structures, only the 20 lowest structures are reported here.
Circular Dichroism Experiments
Circular dichorism (CD) spectra were collected on an AVIV CD spectrometer at 25 °C, and each sample was scanned between 260 and 195 nm. CD samples contained 10 μM peptide and 10 mM potassium phosphate buffer. The metalated samples were prepared by adding 1.0 equiv of HgCl2, CdCl2, or PbCl2 to an apo solution. The pH was adjusted at 8.2 for the apo, Cd(II)-, Pb(II)-, and Hg(II)-α3DIV. The pH conditions were chosen so that the metalated samples formed the appropriate complex: [Hg(II)S3]−, [Pb(II)S3]−, and [Cd(II)S3(N/O)]−. All the samples were purged with Ar(g) and prepared in triplicates. The mean residue ellipticities (MRE) were determined using equation:
where θobs is the measured ellipticity in millidegrees, l is the cell path length in centimeters, c is the concentration in M, and n is the number of residues in the structured regions. Fifty-five and 59 residues were used in the MRE calculation for α3DIV (apo and metalated species) and α3D, respectively.
Thermal Denaturation Experiments
Thermal denaturation studies were performed on a nano differential scanning calorimetry (N-DSC TA Instruments model 602001). The samples contained 0.13 mM peptide (1.0 mg/mL), 30 mM MOPS or HEPES buffer solution, and 100 mM NaCl. The pH for the samples containing MOPS was adjusted at 7.0, while the HEPES solution had a pH of 8.2. The metalated samples were prepared by adding 1.0 equiv of HgCl2, CdCl2, or PbCl2 to an apoprotein solution. All the samples were prepared in an anaerobic environment and were degassed prior to injection. In each experiment, 300 μL of a peptide-buffer solution and its corresponding buffer solution were injected in the sample cell and reference cell, accordingly. Experiments were performed in duplicates or triplicates. The experimental methods involved a heating cycle that originated at 25 °C and ended at 110 °C, using a 1 or 2 °C/min scan rate, and spectra were collected in an anaerobic Coy Box environment. Thermograms were blanked with the appropriate control (buffer or metal buffer) scans, baseline corrected, and normalized with protein concentration. NanoAnalyze Data Analysis (version 2.4.1 by TA Instruments) was used to fit the thermograms to determine the melting temperatures and thermodynamic parameters (Supporting Information, Figure 1).
Figure 1.
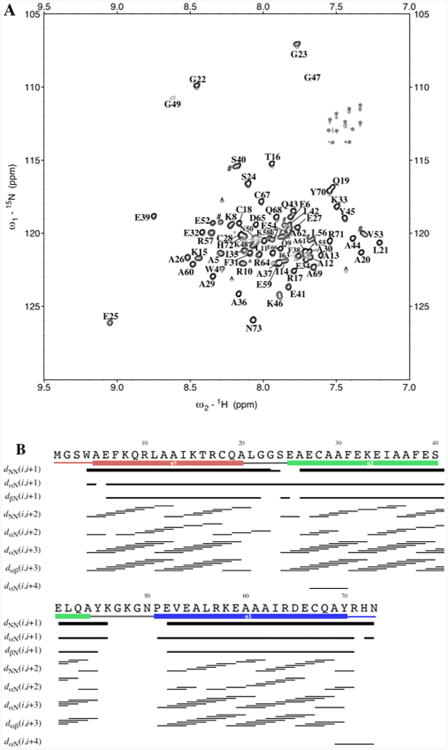
(A) 15N-TROSY spectrum of 15N-labeled α3DIV. The assignments are adjacent to their corresponding peaks. Of the 73 residues, 69 were assigned, with residues 1–3 and Pro51 not observed in the spectrum. The residual Gln and Asn side-chain peaks were not assigned and are marked with an asterisk. Further, aliased and noise peaks are respectively given a pound and circumflex symbol. (B) Summary of sequential NOEs for apo α3DIV, which were determined from 3D 15N- NOESY-TROSY and 13C-NOESY-HSQC spectra.
X-ray Absorption Spectroscopy (XAS)
Samples for XAS were prepared with final concentrations of 2.5 mM HgCl2, 2.7 mM α3DIV, 30 mM TRIS buffer, and 40% glycerol at pH 8.7. Samples were then loaded into lucite XAS sample cells wrapped in Kapton tape, flash frozen in liquid N2, and stored at 77 K until data collection. XAS data were collected at the National Synchrotron Light Source (NSLS) on beamline X3A. This beamline utilized a Si[220] single crystal monochromator equipped with a harmonic rejection mirror. During data collection, samples were maintained at 24° K using a He Displex Cryostat. Protein fluorescence excitation spectra were collected using a 13-element solid-state Ge detector with a Ga fluorescence filter (0.3 μM in width) placed between the cryostat and detector to remove lower energy photons. XAS spectra were measured in 5 eV increments in the pre-edge regions (12200–12270 eV), 0.25 eV increments in the edge regions (12270–12350 eV), and 0.05 Å−1 increments in the extended X-ray absorption fine structure (EXAFS) region (to k = 14 Å−1), integrating from 1s to 25 s in a k3 weighted manner for a total scan length of approximately 50 min. X-ray energies were individually calibrated by collecting Au foil absorption spectra simultaneously with protein data. The first inflection point of the Au foil spectrum was assigned to 11 919 eV. Each fluorescence channel of each scan was examined for spectral anomalies, and data represent the average of 20 scans.
XAS data were processed using the Macintosh OS X version of the EXAFSPAK program suite44 integrated with the Feff v8 software45 for theoretical model generation. Data reduction utilized a Gaussian function in the pre-edge region and a three-region cubic spline throughout the EXAFS region. Data were converted to k-space using a mercury E0 value of 12 284 eV. The k cubed weighted EXAFS was truncated at 1.0 and 12 Å−1 for filtering purposes. This k range corresponds to a spectral resolution of ca. 0.14 Å for all mercury–ligand interactions; therefore, only independent scattering environments >0.14 Å were considered resolvable in the EXAFS fitting analysis. EXAFS simulation analysis was performed on filtered and then on raw/unfiltered data; results listed in Supporting Information, Table 2 were from simulating unfiltered data. EXAFS data were fit using single scattering amplitude and phase functions calculated with the program Feff v8. Single scattering theoretical models were calculated for Hg-oxygen and Hg-sulfur coordination to simulate mercury nearest-neighbor ligand environments. Scale factors (Sc) and E0 values used during the simulations were calibrated by fitting crystallographically characterized Hg models; specific values include a scale factor of 0.95, and E0 values for O and S of 0 and 1.5 eV, respectively. During the simulation, only the bond lengths and Debye–Waller factors were allowed to freely vary, adjusting coordination number values during the simulations in a nonvaried incremental fashion. Criteria for judging the best-fit simulation utilized both the lowest mean square deviation between data and fit (F′), corrected for the number of degrees of freedom and a reasonable Debye–Waller factor. 46, 47
Table 2. Structural Statistics for apo α3DIVa.
| Restraints | ||
|---|---|---|
| Total NOE | 1067 | |
| intraresidue | 395 | |
| short-to-medium range (1 < |i − j| < 5) | 367 | |
| long range (|i − j| ≥ 5) | 305 | |
| Total dihedral anglesb | 138 | |
| Φ | 60 | |
| Ψ | 61 | |
| χ1 | 17 | |
| hydrogen bondsc | 39 × 2 | |
| Average CYANA target function (kcal mol−1) Residual distance restraint violationsd | 1.9 (0.4) | |
| average no. of violations >0.35 Å | 0.00 (0.00) | |
| average of maximal violations (Å) Residual dihedral angle restraint violationsd | 0.24 (0.12) | |
| average no. of violations >5.0° | 0.00 (0.00) | |
| average of maximal violations (deg) van der Waals violationsd | 1.94 (0.67) | |
| average no. of violations >0.35 Å | 0.00 (0.00) | |
| average of sum violations (Å) | 0.28 (0.07) | |
| Ramachandran statistics | CYANAe | PSVSf |
| favored (%) | 90.1 | 90.1 |
| allowed (%) | 7.8 | 7.8 |
| generously allowed (%) | 2.0 | 2.0 |
| disallowed (%) | 0.1 | 0.1 |
| RMSD from the mean structure (Å) | CYANA | PSVS |
| backbone (N, Cα, C) | 0.79 (0.16)g | 0.8h |
| heavy atoms (all non-H atoms) | 1.31 (0.15)g | 1.3h |
| ordered backbone | 0.49 (0.12)i | 0.6j |
| ordered heavy atoms | 0.97 (0.11)i | 1.0j |
| Close Contacts and Deviations from Ideal Geometry (PDB validation software) | PSVS | |
| number of close contacts within 1.6 Å for H atoms and 2.2 Å for heavy atoms | 0 | |
| RMS deviation for bond angles (deg) | 0.2 | |
| RMS deviation for bond lengths (Å) | 0.001 | |
Summary from CYANA42 structure calculation. The ensemble of structures did not exhibit distance violations of >0.60 Å or dihedral angle violations >5°.
Dihedral angle restraints were obtained from a TALOS-N41 analysis.
Upper-limit hydrogen bond distance restraints were used in the ordered regions of the sequence.
Violations were obtained from CYANA.
Ramachandran plot summary of Procheck-style analysis on CYANA, 1–73 residues.
Ramachandran plot summary for residues 1–73 from PSVS51 analysis.
Residues 3–73.
Residues 1–73.
Structured regions: residues 5–20, 25–44, and 51–70.
Structured regions from PSVS analysis: residues 4–20, 25–42, and 51–70.
Result
Structure of Apo α3DIV
Overall Structure
Apo α3DIV has an α-helical fold as indicated by the pattern in the chemical shift dispersion in the 15N-TROSY spectrum (Figure 1A) and sequential NOE correlations (Figure 1B). The 15N-TROSY spectrum of apo α3DIV at pH 7.0 exhibit core residues with well-dispersed chemical shifts and a pattern that is typical for a well-folded α-helical protein. 48,49 Chemicals shift assignments for 69 of the 72 residues, determined from triple resonance NMR experiments, are indicated in Figure 1A, and the total number of peaks validate the amino acid sequence of α3DIV. Resonances for residues Met1, Gly2, and Ser3 are not observed in the spectrum. Met1 is cleaved post-translation, Gly2 and Ser3 are in the dynamic region of the structure. Even after three Leu residues in α3D were substituted for Cys, the 15N-TROSY spectrum contains single peaks for every residue, indicating that α3DIV exhibits a single conformation in solution or an ensemble of conformations interconverting at a sub-microsecond time scale.
An ensemble of the 20 lowest energy structures is visualized in Figure 2A using PYMOL50. The structure of α3DIV encompasses 1067 NOE restraints, which include 395 intraresidue, 367 short-to-medium, and 305 long-range NOEs (Table 2). There are ∼15 NOE restraints per residue for 70 residues. In addition, 138 dihedral angles restraints were utilized in the structure, which were derived from the chemical shifts of 1H, 1Hα, 14N, 13Cα, and 13Cβ atoms for residues 4–73 using TALOS-N. These restraints included 60 φ and 61 ψ angle restraints, as well as 17 χ1 angles. The majority of the χ1 restraints (10 restraints) were designated to core aliphatic and aromatic residues. The rotamers largely had a trans or gauche(−) conformation, characteristic rotamers of nonpolar-aliphatic residues. Finally, 39 hydrogen bond restraints (total of 78) were added in the structured regions. The ensemble has an averaged CYANA energy function of 1.9 (0.4) kcal mol−1. Two distance restraints were greater than the 0.35 Å cut off; however, no dihedral angles >5° or van der Waals contacts >0.35 Å were violated. The Ramachandran statistics for the 20 structures show that 90.1% of the backbone stereochemistry is in the favored regions (Supporting Information, Figure 2) and agrees well with the Ramachandran analysis by Protein Structure Validation Suite51 (PSVS) (Supporting Information, Table 3). There are 55 residues in the structured regions (residues 5–20, 25–44, and 51–70) of α3DIV. The backbone (N, Cα, C) root mean square deviation (RMSD) of the 20 structures for residues 3–73 and the structured regions is 0.79 (0.16) and 0.49 (0.12) Å, respectively. Likewise, these RMSD value corroborates with the structure quality analysis by PSVS (see Supporting Information, Table 4 for the global quality scores).
Figure 2.
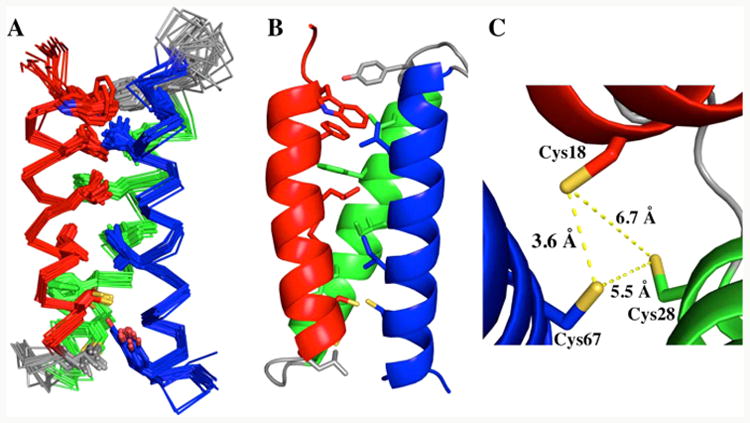
(A) An overlay of the 20 lowest-energy structures of apo α3DIV, calculated with CYANA 2.1 and visualized with PYMOL.50 Showing residues 1–73 (red: helix1, green: helix 2, blue: helix 3 and gray: loops 1 and 2). The structures were calculated from 1067 experimental NOE experimental restraints, 138 dihedral angles generated from the chemical shift index, and 78 backbone hydrogen bonds, added after the initial structure was obtained. The backbone and heavy atom RMSD values for residues 3–73 are 0.79 (0.16) Å and 1.31 (0.15) Å, respectively. For the structured regions residues 5–20, 25–44, and 51–70, the backbone and heavy atom RMSD values are 0.49 (0.12) Å and 0.97 (0.11) Å, respectively. (B) Lowest energy structure, where the side chains of core residues are visualized. (C) Top down view of the metal binding site: Cys18, Cys28, and Cys67.
Table 3. Circular Dichroism and Thermal Denaturation Parametersa.
| sample | −[θ]222 nm (deg cm2 dmol−1 res−1) | [θ]222 nm/[θ]208 nm | Tm (°C) | ΔHcalb (kcal mol−1) | ΔHcal/ΔHvan't Hoff |
|---|---|---|---|---|---|
| α3Dc | 25213 (306) | 1.01 | 89.6 (0.3) | 49.9 (4.6) | 1.2 (0.3) |
| α3DIV | 29231 (672)c | 1.03 | 64.4 (0.6)d | 50.0 (0.2) | 2.4 (0.4) |
| 60.2 (0.1)c | 44.9 (1.2) | 1.9 (0.2) | |||
| Hg-α3DIVc | 27589 (421) | 1.51 | 84.0 (1.7) | 65.1 (3.5) | 6.7 (2.4) |
| Pb-α3DIVc | 26940 (2420) | 1.01 | 83.4 (3.3) | 59.2 (6.9) | 4.5 (2.3) |
| Cd-α3DIVc | 27600 (487) | 1.07 | 78.1 (0.7) | 52.7 (4.3) | 4.2 (0.5) |
All the averaged values were determined from triplicate or duplicate experiments.
Calculated excess heat capacity.
pH 8.2.
pH 7.0.
Table 4. Hg EXAFS Fitting Analysis of α3DIV Compared to Hg–S Bonds of Relevant Model Compounds and Proteins.
| complex | CN | geometry | Hg–S R (Å) | σ × 103 (Å2) | F | pH | ref |
|---|---|---|---|---|---|---|---|
| Hg-α3DIVa | 2 | 2.36 | 3.54 | 2.24 | 8.7 | this work | |
| 2.5 | 2.36 | 4.97 | 2.21 | ||||
| 3 | 2.36 | 6.20 | 2.21 | ||||
| Hg-TRIL16Cb | 2 3 | 2.324 2.443 | 5.5 9.5 | 71 | |||
| Hg-MerRb | 3 | 2.43 | 7.5 | 19 | |||
| average of five modelsc | 2 | linear | 2.348 | 72 | |||
| average of 11 modelsc | 3 | trigonal | 2.462 | 72 | |||
| average of three modelsc | 3 | trigonal T-shaped | 2.497 avg two shortest bonds: 2.372 | 72 |
Data fit over a k range of 1 to 12 Å−1.
Independent metal–ligand scattering environment at R < 3.0 Å. Scattering atoms: S (sulfur). F = number of degrees of freedom weighted mean square deviation between data and fit.
Hg(II)–S bond lengths determined from EXAFS.
Hg(II)–S bond lengths determined from an X-ray crystal structure (see Supporting Information).
The lowest energy structure of α3DIV is illustrated in Figure 2B and has a CYANA energy function of 1.2 kcal mol−1. The three-helix bundle adopts a counterclockwise orientation, which is in complete agreement with α3D. The interhelical-tilt angles, which were calculated using QHELIX,52 further confirmed this counterclockwise topology: Ω1,2 = −149°, Ω1,3 = 21°, and Ω2,3 = −156. These angles decreased by 16°, 7°, and 15° from the Ω1,2, Ω1,3, and Ω2,3 values reported for α3D, respectively. However, when the same QHELIX analysis was performed on α3D and compared again to α3DIV, a much more modest change was observed. The tilt angles of α3D reduced by 11° (Ω1,2), 8° (Ω1,3), and 8° (Ω2,3).
The triscysteine metal binding site has Sγ–Sγ distances of 6.7, 5.5, and 3.6 Å between Cys18–Cys28, Cys28–Cys68, and Cys18–Cys67, respectively (Figure 2C). It should be noted that these distances were not restrained in the calculations. This hydrophobic plane has an area of 9.82 Å2 and can accommodate large heavy metals like Cd(II), Hg(II), and Pb(II). In addition, the χ1 dihedral angles (N–Cα–Cβ–Sγ) for the Cys residues are 168°, −52.7°, and −68.1° for Cys18, Cys28, and Cys67, respectively. Both Cys28 and Cys67 have χ1 angles that are close to the most common rotamer for Cys (−65°).53
Analysis of Metalated α3DIV
15N-HSQC Spectra of Metalated α3DIV
Our subsequent objective was to determine the chemical shift perturbation in the 15N-HSQC spectrum of α3DIV after the addition of Hg(II) and Pb(II). These results provide a qualitative assessment of the change in the peptide fold in the presence of a metal–ligand complex, as well as the opportunity of collecting further 3D NMR experiments in order to solve a metalated structure of α3DIV. However, the chemical shift peak dispersions in the 15N-HSQC spectrum of Pb(II)- and Hg(II)-α3DIV were observed to be significantly perturbed.
In Figure 3, a superimposition of the 15N-HSQC spectrum of an apo control and Pb(II)-α3DIV shows a significant decrease in number of cross peaks for the Pb(II) spectrum. The pH of the apo and metalated species was set at 7.0 in order to achieve a trigonal pyramidal lead complex23 The apo spectrum displays 68 of the 69 expected peaks (Trp4 not observed), again demonstrating a well-defined conformation of the apo-peptide. Upon the addition of 1.0 equiv of Pb(II), only about 57 peaks could be observed in the Pb(II)-α3DIV spectrum, and they were compared to corresponding peaks in the assigned apo spectrum. Of these identified peaks, several core residues, including Cys18, Phe38, Val53, and Ile63 overlay well with or slightly deviate by about ±0.2 ppm in 1H from their corresponding apo peaks. The peaks for other residues, such as Leu21, Phe31, Leu56, and Cys67, are either broadened beyond detection or have significantly shifted, resulting to an incomplete assignment of the chemical shifts for Pb(II)-α3DIV. Dramatic perturbation was also observed for the 15N-HSQC spectrum of Hg-α3DIV at pH 8.6 (Supporting Information, Figure 3), which forms a trigonal complex at this condition. The 15N-HSQC spectrum of Hg-α3DIV contained only 47 observable peaks, while the spectrum of its apo counterpart only has 55 of the 68 expected resonance peaks at the same pH. The resonances in the Hg-α3DIV spectrum were not assigned.
Figure 3.
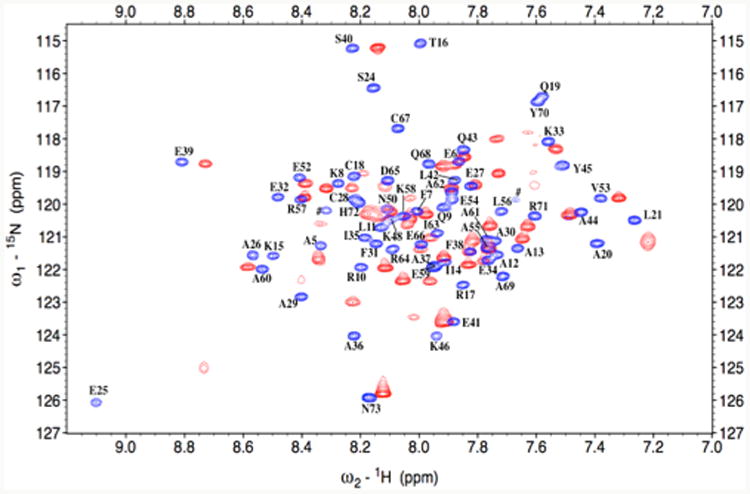
Enlargement of the N-HSQC spectra of apo α3DIV (blue) and Pb(II)-α3DIV (red), both at pH 7.0. Spectra were collected on a 500 MHz Bruker Avance NMR spectrometer at 25 °C. The assignments in the apo spectrum are adjacent to their corresponding peak. In this view, the apo spectrum displays 63 of 68 assigned peaks, while the Pb spectrum α3DIV shows 54 of its 68 expected peaks.
CD Analysis of Metalated α3DIV
The CD spectra of Hg(II)-, Pb(II)-, and Cd(II)-α3DIV at pH 8.2 are compared to apo α3DIV, as well as to α3D in Supporting Information, Figure 4A. Like its apo counterpart and α3D (both at pH 8.2), the metalated spectra display double minima at 208 and 222 nm, displaying a CD profile of a well-folded α-helical system. This is further supported by the large negative molar ellipticity values (Table 3). Apo α3DIV has the largest −[θ]222 nm value of 29 231 (672) deg cm2 dmol−1 res−1, while Hg(II)-α3DIV, Pb(II)-α3DIV, and Cd(II)-α3DIV had values of 27 589 (421), 26 940 (2420) and 27 600 (487) deg cm2 dmol−1 res−1, respectively. α3D had the lowest −[θ]222 nm value of 25 213 (306) deg cm2 dmol−1 res−1, indicating that α3DIV has a higher helical content than its parent structure. Furthermore, the [θ222]/[θ208] ratio for all the samples were ∼1.0, which is representative of a bundled or coiled-coil tertiary structure. These results show that binding heavy metal ions to α3DIV does not lead to the unfolding or destabilization of its overall structure; instead, α3DIV still retains an α-helical fold.
Figure 4.
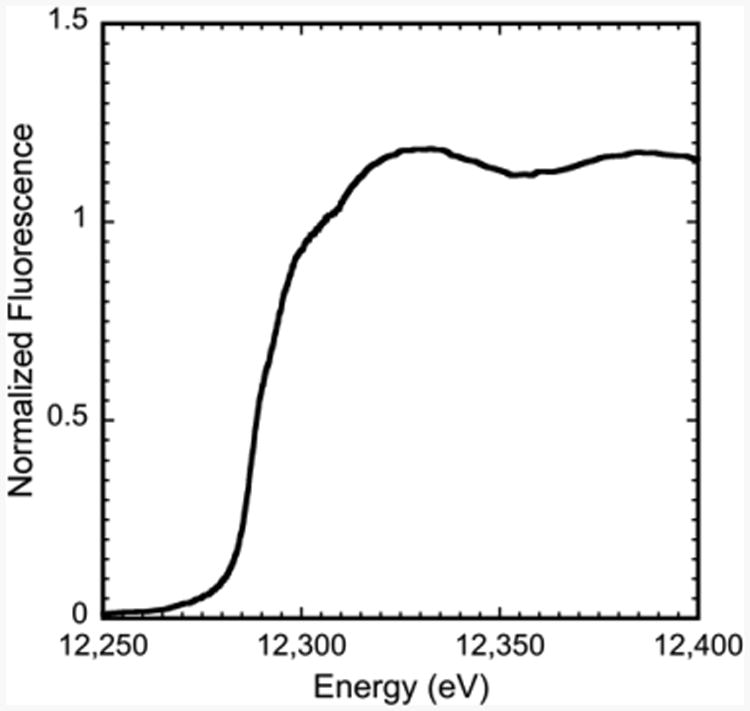
Normalized XANES spectrum for average of all Hg-α3DIV spectra.
DSC Studies of Metalated α3DIV
The thermograms were collected on apo, Hg(II)-α3DIV, Pb(II)-α3DIV, Cd(II)-α3DIV, and α3D with a differential scanning calorimetry (DSC) apparatus. The thermal denaturation profile of α3D, apo samples (at pH 7.0 and 8.2), and metalated α3DIV are overlaid in Supporting Information, Figure 4B and their respective thermodynamic parameters are listed in Table 3. Apo α3DIV at pH 7.0 displays a broad melting curve, which is resolved in the thermogram of apo α3DIV at pH 8.2. The pH 7.0 thermograms fitted well to a two-peak model and the higher pH to a three-peak model. The first peak was identified to be the primary melting temperature of α3DIV. The primary peak at pH 7.0 has a melting temperature of 64.4 (0.6) °C, while the melting maxima at pH 8.2 decreased to 60.2 (0.1) °C, exhibiting a pH-dependence on the thermal-induced denaturation. Both species yielded similar ΔHcal (excess heat capacity) values of 50.0 (0.2) and 49.9 (4.6) kcal mol−1 for pH 7.0 and 8.2, respectively. The ΔHvan't Hoff was determined (Supporting Information Table 4), and the ΔHcal/ΔHvan't Hoff ratio is listed in Table 3. A value of 1 is indicative of a two-state unfolding model, while a value above or below 1 signifies self-association (e.g., as dimer, trimer, etc.) or unfolding through one or more intermediate states, respectively. This ratio for the parent structure, α3D, is 1.2. Apo α3DIV has value of 2.4 and 1.9 at pH 7.0 and 8.2, respectively. The metalated structures have ΔHcal/ΔHvan't Hoff ratio values >2.0.
A significant shift in the melting temperatures was observed for the metalated species of α3DIV, demonstrating that the metal-ligand complex provides extra stability against thermal denaturation. The melting curve of Hg(II)-α3DIV and Cd(II)-α3DIV fitted well to a one-peak model, whereas Pb(II)-α3DIV was fitted with two peaks. The Tm values for the metalated species increased by a range of 18–24 °C, with Hg(II)-α3DIV exhibiting the largest Tm. This trend was also present in the enthalpy values, where an 8–20 kcal mol−1 growth was observed. In addition, the thermogram of α3D at pH 8.2 was collected and compared with α3DIV. The Tm value for α3D was determined to be 89.6 (0.3), which is consistent with previously reported values. This Tm is ∼30 °C higher than apo α3DIV but only about 6–12 °C greater than the metalated species. Nevertheless, the ΔHcal values for apo α3DIV and α3D are within a similar range and, most importantly, the metalated α3DIV generated values that were 3–15 kcal mol−1 greater than α3D. Overall, these stability studies using CD and DSC signify that binding a metal ion in the triscysteine binding site of α3DIV does not destabilize or unfold its structure. In fact, from the DSC analysis, we illustrate that metal binding further stabilizes the structure of α3DIV.
XAS Analysis of Hg-α3DIV
XAS was utilized to determine the metrical parameters for mercury bound to α3DIV. Several spectra were collected on independent reproducible samples and spectra were averaged independently to verify reproducibility. The spectrum presented in Figure 4 represents an average of the data for optimal signal/noise ratio. The X-ray absorption near edge structure (XANES) spectrum in Figure 4 is consistent with those typically observed for Hg(II)-S complexes.55 The Hg EXAFS could only be fit with a single Hg–S scattering environment constructed by a coordination number between 2.0 and 3.0 (±0.5) and centered at a bond length of 2.36 Å (Supporting Information, Figure 5). There is no evidence for Hg–O/N scattering in this area. Long-range scattering could not be deconvoluted from noise signals in that region of the data. The averaged mercury–sulfur bond length in Hg-α3DIV is compared to relevant Hg(II)-S complexes, including protein systems and model compounds in Table 4.
Figure 5.
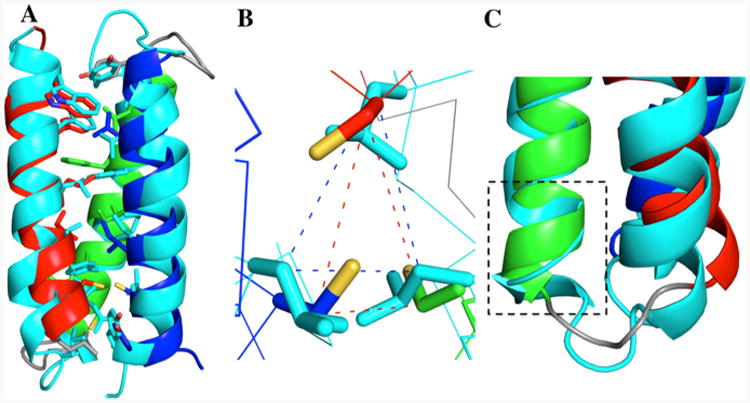
(A) An overlay of α3DIV (helix 1: red, helix 2: green, and helix 3: blue) and α3D (cyan). The backbone (N, Cα, C,O) rms was determined on PYMOL to be 1.75 Å. (B). Top-down view of the mutation site (18, 28, and 67), displaying superimposed Cys/Leu residues. (C) Gain of helical content in helix 2 for residues 26–28.
Discussion
Peptide α3DIV was prepared in our effort to functionalize the framework of α3D by incorporating a metal binding site.23 NMR and stability studies that examined the dynamic behavior and malleability of α3D revealed that the C-terminal end was the most amenable to mutations. In addition, this region of the bundle contained apolar residues that formed a hydrophobic “box” region, which offered a suitable location for a metal binding site. For these reasons, we converted Leu to Cys residues to produce a triscysteine motif and demonstrated its binding properties to the heavy metal ions Hg(II), Pb(II), and Cd(II). Subsequently, we prepared an α3DIV related construct that substituted the same leucine residues with histidine in order to form a ZnN3(H2O) that served as an excellent analogue of carbonic anhydrase (Table 1).56 Despite these successes, we felt it was critical to assess how amino acid substitution at this important position impacted the structure of the apo protein (e.g., was a stable, preformed metal binding site realized by these changes).
Comparison to the Structural Statistics of α3D
The structural statistics comparison of α3DIV and its parent structure, α3D, are found in Table 5. There are notable differences in the NOE and dihedral angle restraints used in the structure calculation of both structures and the empirical origins of these restraints. The solution structure of α3D was based on 1143 NOE restraints,8 incorporating 70 more NOE restraints than the structure of α3DIV. The majority of its NOEs for α3D were classified in the short-to-medium range (45%), whereas both structures have roughly the same percentage (∼30%) of short-to-medium and long-range restraints. Unlike the structure of α3D, α3DIV included φ, ψ, and χ1 dihedral angle restraints that were generated from a TALOS-N analysis, which employed the chemical shift assignments of 1H, 1Hα, 14N, 13C, 13Cα, and 13Cβ atoms to predict dihedral angle restraints. α3D only contained φ angle restraints that were derived from a triple resonance HNHA experiment, as well as χ1 angles, which were determined from NOE patterns. Moreover, the comparison of the backbone and heavy atoms RMSD from the mean structure, for the same residue range and number of calculated structures, illustrated that the ensemble of the 13 lowest energy structures for α3DIV is lower than α3D. The RMSD for α3DIV is ∼0.3 Å lower than the 13 structures reported for α3D, for both the backbone and heavy atoms in the structured regions and the backbone atoms for residues 1–73.
Table 5. Structural Statistics of α3DIV Compared to α3D.
| RMSD from the mean structure (Å) | α3D | α3DIV | |
|---|---|---|---|
| backbone (residues 1–73, N, Cα, C) | 1.06a | 0.78 (0.15)a | 1.08 (0.31)b |
| backbone (residues 4–21, 24–45, and 51–70, N, Cα, C) | 0.75a | 0.48 (0.11)a | 0.53 (0.15)b |
| heavy atoms (residues 1–73) | 1.61a | 1.25 (0.13)a | 1.58 (0.32)b |
| heavy atoms (residues 4–21, 24–45, and 51–70) | 0.92 (0.10)a | 0.99 (0.10)b | |
Thirteen structures.8
ψ angles excluded from the structure calculation.
The α-helical regions for the two structures slightly differ, with α3D (residues 4–21, 24–45, and 51–70) covering two more residues than α3DIV (5–20, 25–44, and 51–70) in helix 1 and 2. The structure of α3D incorporated Trp4, Leu21, Ser24, and Tyr45 in its structured regions, but these residues, from a TALOS-N analysis, were designated to have a coiled (Trp4) or loop (Leu21, Ser24, and Tyr45) secondary structure.
Comparison of the 13 Lowest Energy Structures
The 13 lowest energy structures of α3DIV were calculated in order to properly compare the minimized model to the reported structural statistics of α3D,8 and we found that our overall ensemble is better ordered. The backbone (N, Cα, CO) and heavy atom RMSD values for the 13 structures of α3DIV were observed to be lower by ∼0.3 Å, even though the α3D contained more intraresidue and short-to-medium range NOE restraints (Table 5). We attributed this difference to the dihedral angle and NOE sequential restraints used in the structure calculations. Specifically, the α3DIV structure contains φ and ψ backbone dihedral angles, while α3D only incorporated φ angle restraints. The presence of both dihedral angles limits the range of interaction between Ni-Ni+1 and Ci-Ci+1 atoms in the structured regions, thereby eliminating unfavorable sterics and could result in better ordered α-helical chains. This was tested in an analysis that involved removing ψ angle restraints in the structure calculation of α3DIV. It was determined that the RMSD from the mean structure values of the backbone and heavy atoms for residues 1–73 in α3DIV increased to 1.08 (0.31) from 0.78 (0.15) Å and to 1.58 (0.32) from 1.25 (0.13), respectively, which are almost equal to the same parameters determined for α3D. However, for the structured regions, a very modest increase of 0.02 Å was observed, which is well within the statistical error (∼ ± 0.10 Å).
In addition, the increased unity in the 13 structures of α3DIV can be further justified by the amount of sequential NOE restraints between 1H backbone atoms used in the calculations. The α3D structure has a proper extent of HNi-HNi+1 and Hαi- Hiβi+3 sequential NOE correlations in all three helices and strong Hiβi-H i+1 NOE correlations in helix 3 for residues 50–70. Nevertheless, it significantly lacked Hα-HNi+1, Hα-HNi+3, and Hβi-HNi+1 in helix 1 and 2 correlations, NOEs that are typically observed in α-helical structures since the distances between these 1H atoms are within the experimental limit of 5 Å. On the other hand, in the structured regions, α3DIV contained most of the NOE correlations mentioned above in a sequential manner. Additionally, the sequential NOE pattern in the structure of α3DIV can be viewed as compensating restraints since only a modest increase in the RMSD values were observed in the structured regions after ψ dihedral angle restraints were removed.
Comparison to the Lowest Energy Structure of α3D
The lowest energy structure of α3DIV and α3D are superimposed in Figure 5A. The two structures were aligned via backbone (N, Cα, C, O) atoms of residues 1–73, and this alignment yielded an overall heavy atom RMS value of 1.75 Å. From the overlay, α3DIV possesses the same overall topology as α3D, with a counterclockwise bundle that was expected in the design process of α3D. Figure 5B is an overlay of the Leu-to-Cys mutation site, positions 18, 28 and 67, of both structures. The Cβ distances between the Leu–Leu and Cys–Cys were measured, and the triangular plane that forms between these residues has an area of 15.5 Å2 in α3D and 14.1 Å2 in α3DIV. This slight deviation demonstrates that the fold at the C-terminal end was not significantly affected even after removing Leu residues that provide stabilizing and packing interactions in α3D. Regardless, incorporating Cys residues with polar, uncharged thiol groups to form a metal-binding site in α3DIV led to two significant structural differences as a result of a more packed core.
First, the α3DIV structure has lower Ω1,2, Ω2,3, and Ω1,3 angles by 16°, 15°, and 7°, respectively. This deviation from α3D could stem from better packing of the apolar layers above the triscysteine site (Supporting Information, Figure 6). The first layer, which is composed of a plane between the Cβ atoms of Ile14, Phe31, and Ile63, has an area of 12.9 Å2 in α3DIV, whereas in α3D this plane is 9.0 Å2 greater. This trend is also observed in the two subsequent layers 2 (Leu11, Ile35, and Ala60) and 3 (Phe7, Phe38, and Leu56), which has an area of 7.8 Å2 (16.1 Å2) and 7.3 Å2 (17.7 Å2) lower than α3D.
Figure 6.
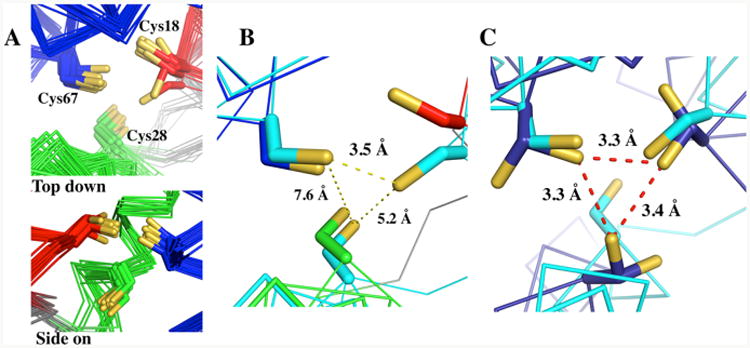
(A) Preorganization of the triscysteine metal binding site of the 20 lowest energy structures (Cys18: red, Cys28: green and Cys67: blue). (B) A partially preformed trigonal site for metal binding in structure 16 (cyan) of 20 aligned with the lowest energy structure, structure 1. The Sγ–Sγ distances are measured for structure 16 of α3DIV. (C) Structure 16 (cyan) aligned with the Cys “a” sites of CSL9C (PDB: 3LJM) (blue), which contain two conformations.61 The Sγ–Sγ distances are measured for CSL9C.
Next, we found that the incorporation of Cys residues did not disrupt the overall α-helical framework of α3D, as demonstrated in the 15N-TROSY and CD studies. Unexpectedly, the alignment of α3D and α3DIV revealed that the addition of a Cys residue in the 28th position in helix 2 improved its helical content (Figure 5C). In α3D, the helicity in the second strand breaks after Ala26 and becomes continuous again at Ala29. Residues Glu27 and Leu28 appear to have a more coil-like secondary structure. In contrast, this disruption in the helicity between residues 26–28 is not observed in the structure of α3DIV, which demonstrates an increase in the α-helical content. Additionally, the analysis from two web-based structural determination programs, TALOS-N41 and volume area dihedral angle reporter (VADAR),57 as well as the CD comparison of apo α3DIV with α3D supports this observation. TALOS-N uses a database of 580 proteins with almost complete backbone NMR chemical shifts (1H, 1Hα, 14N, 13C, 13Cα, and 13Cβ) and an additional database of 9523 high-resolution X-ray crystal structures (without experimental chemical shifts) to predict dihedral angles φ, ψ, and χ1, as well as the secondary structures from experimental NMR chemical shifts. Similar to the dihedral angle prediction, the TALOS-N secondary structure classification system involves a two-system artificial neural network (ANN) and categorized residues 26–28 into an α-helical secondary structure, with a probability score of ∼0.96. VADAR analysis on the PDB coordinates of the lowest energy structure of α3DIV structure also characterized residues 26–28 to be in an α-helical structure, while the same analysis for α3D resulted in a coiled secondary structure. Furthermore, the molar ellipticity value for apo α3DIV is about −4200 deg cm2 dmol−1 res−1 greater than α3D. This again indicates that the replacement of Leu residues with a Cys at the 28th position may have increased the overall helical content of the α3D framework.
Preorganization of the Triscysteine Site
Native metalloproteins have evolved to possess metal coordination environments that are unique to their function, and two limiting extremes of metal binding sites have been described. The first group is typified by zinc finger proteins, which adopt a stable fold only after a metal has bound to the endogenous ligands of the sequence.58 In this case, metal binding defines protein structure. Less extreme, but still within this category, are cases where metal binding leads to important protein conformational changes that define function. Examples of such behavior can be seen with metalloregulatory proteins such as MerR or ArsR.1,2 The second group is illustrated by the cupredoxins that have a well-defined, preorganized binding site. The concept of a racked-induced bonding or entatic state model has been proposed for such systems to elucidate the unique properties of these electron transfer (ET) proteins.59 The rack-induced bonding model explicates that the ligand environment (CysHis2 site and one or two axial weakly bound axial ligands) in a cupredoxin fold forces the copper ion, regardless of the oxidation state, into a strict distorted-tetrahedral geometry. This feature in cupredoxins and redox-active metalloproteins was discovered to be one of the fundamental driving forces in attaining efficient electron transfer activity. Ultimately, it is of great importance in the de novo design of metallopeptides to be able to predict accurately the extent of metal binding site preorganization prior to the introduction of a metal ion in order to achieve the desired affinity, selectivity and function.
The sequence of α3D was designed to contain diverse sets of heptad repeats (defined as “abcdefg”), where nonpolar residues are incorporated at the “a” and “d” positions to provide core stabilizing interactions and charged residues at the “e” and “g” to form salt-bridges between helices.9 Our previous work on parallel three-stranded coiled-coil (TRI and coil-Ser) systems showed that the subtle difference between the “a” and “d” positions can produce distinctive outcomes in heavy metal binding affinity and geometry, which can be attributed to the preorganization of the sulfur ligands prior to metal binding.9,60 We found that the substitution of Leu to Cys residues in “a” sites tend to favor the formation of a trigonal metal binding site, where three Sγ atoms point into the core (Supporting Information, Figure 7A). For instance, apo CSL9C (PDB 3LJM)61 comprises “a” site Sγ atoms with distances of 3.3 and 3.4 Å. Conversely, “d” site substitutions orient Sγ atoms away from the core and toward the helical interface forming a much larger cavity (Supporting Information, Figure 7B). Apo CSL19C (PDB 2X6P)61 contains “d” site Cys residues with Sγ–Sγ distances of 3.4 and 4.6 Å. Initial examination of the α3D NMR structure indicated that a preorganized metal binding site could be carved into a hydrophobic box region of the protein. Therefore, it was for these structural reasons that leucines in the “a” positions of α3D were targeted for modification; however, one should recognize that the side chain orientations are expected to differ in this antiparallel α3DIV framework.
Figure 7.
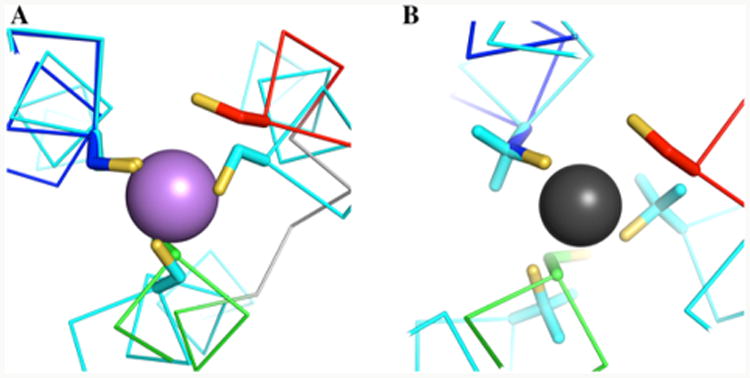
(A) Structure 1 of α3DIV superimposed over the As-Cys3 site of As(CSL9C)3 (PDB 2JGO at 1.81 Å resolution).76 The Cα and Cβ of Cys67 was visually aligned with a Cys residue (B) Structure 1 of α3DIV superimposed over the Hg-Penicillamine3 site of [Hg(II)]S-[Zn(II)(H2O/OH−)]N(CSL9PenL23H)3n+ (PDB 3PBJ at 2.20 Å resolution).77 The Cα and Cβ of Cys67 were also visually aligned with a penicillamine residue.
In α3DIV, we incorporated “a” site Cys residues and expected the Sγ atom of Cys18 and Cys67 to point into the core to initiate the generation of a preformed trigonal plane of sulfurs starting with these two ligands. Cys28 was intended to provide a more adaptable ligand, which was predicted to orient toward the C-terminal end since this residue is on the antiparallel strand. Our objective with the design of α3DIV was to create a single-polypeptide scaffold that produces a more asymmetric or a distorted triscysteine metal binding site than the symmetric Cys sites found in TRI or coil-Ser analogues. As a result, the more native-like properties of α3DIV could provide a better model for native metalloregulatory proteins or metallochaperones that sequester toxic heavy metals in nature.
The triscysteine metal binding site of the 20 lowest energy structures of apo α3DIV is superimposed in Figure 6A. Overall, the 20 conformations of the three Cys residues illustrate a well-defined metal binding site. For instance, the Cβ and Sγ atoms of Cys28 are uniformly oriented toward the C-terminal end of the bundle (Figure 6A, side-on view), while the Sγ atoms of Cys18 are pointing to the interhelical interface between helix 1 and 3 in 18 of the 20 structures. It should be pointed out that the Cys18 χ1 dihedral angle restraint was not constrained, but this corresponding angle was restrained in Cys28 and Cys67. Despite this definition of the average structure, we believe the site is not rigid, because the amide proton N-TROSY signals for the three cysteine residues and their sequential neighbors are ∼30% more intense than the average, indicating motion on the nanosecond time scale for these residues. With this caveat, we further describe the (time) averaged structure of the metal binding site in the following.
The sulfur atoms of Cys28 and Cys67 are observed to have set orientations, while Cys18 contains two conformations that diverge from its ensemble. The Cβ and Sγ atoms of Cys28 on the antiparallel strand are directed toward the C-terminal end of the bundle (Figure 6A, side-on view). This orientation was also predicted to occur for the Cβ atoms of noncoded D-amino acids, such as D-Pen or D-Leu, in 3SCC analogues.62, 63 The change in chirality in these two D derivatives were expected to direct their respective thiol and isopropyl moeity toward the C-terminal end. Consequently, we show here in our α3DIV structure that an l-Cys in an antiparrallel strand provides the same effect as a D-amino acid. For Cys67, its Sγ atoms are oriented inside the core of the bundle, which was predicted for this “a” site substitution. Lastly, the collective Sγ atoms of Cys18 result in a more “d” site-like orientation, where the Sγ atoms are directed at the interhelical interface between helix 1 and 3. Cys18 possesses two structures (16 and 18) that deviate from the ensemble. In structure 14, the “d” site conformation of the Sγ atom of Cys18 is oriented toward the opposite direction, at helix 1 and 2. Structure 16 contains a Cys18 Sγ atom that points inside the core, providing a structure that presents a metal binding site with two “a” site ligands at Cys18 and Cys67 (Figure 6B). When compared to apo CSL9C (Figure 6C), which contain “a” site residues (with two conformations), the Cβ and Sγ atoms of Cys18 and Cys67 in structure 16 overlay well with two Cys9 residues. As a result, structure 16 demonstrates a partially preformed trigonal cavity for metal ions that coordinate in a 3 or 4 coordination environment, such as Hg(II), Pb(II), and Cd(II).
Implications on Metal Binding
With a solution structure in hand of apo α3DIV, one may next consider the consequence of metal binding to the system. The apo structure allowed us to investigate the global, as well as local structural changes that occur when a protein scaffold is functionalized with Cys residues to form a metal binding site. A metalated structure would afford a concise comparison with the apo structure to assess the extent of preorganization of the site, which could provide a mechanistic model for metal ion acquisition in native systems. In terms of de novo protein design, both structures offer a base for future design of a metal binding site with mixed residues/ligands or with identical residues that require secondary interactions (such as hydrogen bonding) in able to achieve redox or catalytic activity with transition metals Fe(II/III), Cu(I/II), or Zn(II).
The spectroscopic properties of Hg(II), Pb(II), and Cd(II) bound α3DIV were previously reported23 to form the predicted complexes but a much closer inspection of the solution stability of metalated α3DIV was necessary to illustrate that the addition of a metal ion does not have a unfavorable effect on its fold. We collected 15N-HSQC spectra of metalated α3DIV and performed both circular dichroism and thermal denaturation studies to illustrate that heavy metal binding reinforces and improves the solution stability of α3DIV. In addition, the Hg-α3DIV species was analyzed by XAS to obtain structural metrics on a metalated species.
Circular dichroism studies have been a central tool in investigating and confirming metal-induced (transition and/or heavy metal ions) folding in native proteins, such as zinc finger proteins64 and calmodulin65, as well as in de novo designed peptides including isoleucine zipper66 and BABY67 3SCC peptides. The CD fingerprint of α-helical proteins and peptides exhibit two minima at 208 and 222 nm, which arise from π−π* and n−π* transitions in the amide atoms.68 When compared to apo α3DIV, the CD spectra of the metalated species also contain a double band profile at 208 and 222 nm (Supporting Information, Figure 4A). These two absorption bands are a characteristic feature in α-helical folds. The calculated molar ellipticity values ([θ222]) for the metalated species fall within the range of the apo, and these large negative [θ] values are typical for a well-folded α-helical fold. Furthermore, the CD results show that the apo form has a high α-helical content, which was the intended target in the original design of α3D. In this peptide system, metal binding to the C-terminal end of the bundle was not necessarily predicted to increase the helicity of the peptide because the overall fold is highly α-helical. Metal binding was expected to perturb that region of the peptide (positions 18, 28, and 67 at the C-terminal end), resulting in a slight decrease in the helicity of the metalated species. This corroborates well with DSC results (which is discussed subsequently) because, although a loss in helicity was observed, metal binding provides M-thiolate bonds that enhance the tertiary stability of the structure, thus much higher melting temperatures when compared to the apo. In addition, the replacement of Leu to Cys residues, to build a metal binding site, removes the stabilizing hydrophobic interaction within the α3D fold, and binding a metal ion into this site counters this loss of stability.
A thermal denaturation analysis was performed to further assess the stability of metalated α3DIV and to explore the concept that metal binding to proteins could lead to greater stability. This rise in thermal stability has been previously observed in ligand-binding studies on native proteins. For example, the melting temperature (Tm) of a copper metallochaperone BsSco was determined to increase by 23 °C when bound to Cu(II).69 Ca(II) and Zn(II) ion binding to coagulation factors IX binding protein increased its melting temperature by 4–5 °C and calculated enthalpic parameter (ΔHcal) by more than 100 kcal mol−1.70 The DSC results illustrate that metal bound α3DIV is more stable than its apo counterpart in thermally induced denaturations. The metalated species have much higher melting temperatures, by a range of 18–24 °C, and the calculated enthalpic parameters (ΔHcal) are greater by 8–20 kcal mol−1 (Table 3). The enhancement of both thermodynamic values shows that the metal–thiolate bonds (Cd–S, Hg–S, or Pb–S) supplements the weak-nonbonding forces (hydrophobic interactions, hydrogen bonds, and salt-bridges) that are essential in protein folding and disruptive during the denaturation process. In addition, the Tm value of parent structure α3D is still greater than apo but slightly higher than the metalated α3DIV species, which indicates that the incorporation of a triscysteine site in its apo or metal-bound form cannot entirely reproduce the hydrophobic interactions that are provided by Leu residues in a protein fold. This finding is further supported by a chemical denaturation performed on apo α3DIV, where we observed a 2.5 kcal mol−1 decrease in the ΔGUF from the reported value of α3D (5.0 kcal mol−1).23 Moreover, the ΔHcal/ΔHvan't Hoff ratios (Table 3) reveal the thermal-induced denaturation process and a value of 1 indicates a two-state model. α3D has a ratio closest to this model of 1.2, which matches well with the reported value.8 Apo and metalated α3DIV have values >2.0, and the denaturation curve of these species are much broader than the corresponding curve of α3D, demonstrating a self-associating unfolding process. This result was not unexpected as the metal binding site contains three thiolate ligands that could induce the formation of external disulfide bonds at higher temperatures.
The transition from a two-coordinate (2 C. N.) [Hg(II)-S2(SH)] to a three-coordinate (3 C. N.) [Hg(II)S3]− complex in α3DIV has an apparent pKa of 7.1,23 and this transition was examined by 199Hg-NMR and UV–vis. We found that α3DIV forms a linear [Hg(II)S2(SH)] 2 C.N. below pH 6.0 and a three-coordinate trigonal [Hg(II)S3]− complex above pH 8.5 with a single chemical shift at −938 and −244 ppm, respectively. Further, under intermediate pH conditions (pH ≈ 7.5), two chemical shifts were observed at −240 and −926 ppm, which demonstrate a mixture of both species. The three-coordinate Hg-α3DIV species (at pH 8.7) was further analyzed with XAS to obtain structural parameters on metalated α3DIV, as well as to confirm previous findings. The EXAFS analysis on the Hg(II)-α3DIV is dominated by nearest-neighbor scattering typical of sulfur ligation (Supporting Information, Figure 5) and yielded an average Hg(II)–S bond length at 2.36 Å and a 2.5 C. N. This bond length falls in between reported values for 2 C. N. and 3 C. N. mercury–sulfur complexes. Therefore, in order to correlate this EXAFS analysis to the coordination environment of the [Hg(II)S3]− complex of α3DIV, we compared our result to the Hg(II)–S distances of protein and de novo designed peptide systems derived from EXAFS analysis and model compounds measured from X-ray crystal structures in Table 4 and Supporting Information, Table 5. The native protein, MerR, which was determined to form a trigonal mercury complex, has an EXAFS derived Hg(II)–S bond length at 2.43 Å.19 Similarly, the EXAFS analysis on Hg(II)-TRIL16C provides Hg(II)–S distances of 2.32 and 2.44 Å for the corresponding 2 and 3 coordinate species.71 Table 4 also compares our EXAFS result to 2 C. N. and 3 C. N. compounds from the Cambridge Structural Database.72 The five model compounds with a 2 C. N. have an average Hg(II)–S bond length of 2.348 (0.023) Å. The three-coordinate compounds (11 total) produced an average Hg(II)–-S distance of 2.462 (0.044) Å. Three of these compounds exhibit a T-shaped coordination environment, which consists of two short bonds with an average bond length of 2.372 (0.01) Å and a single longer bond at 2.497 (0.071) Å. These structure based Hg(II)–S bond lengths corroborate well with the EXAFS analysis of Hg(II)–TRIL16C and Hg(II)–MerR.
The two most straightforward interpretations of the observed 2.36 Å value for Hg-α3DIV is either an approximately equimolar mixture of two- and three-coordinate mercury sites or a single, distorted T-shaped structure similar to those of small molecule models. The mixed speciation behavior was previously observed in our EXAFS analysis of Hg(II)–TRIL16C. A mixture of 2 and 3 C. N. species has a 2.37 Å Hg(II)–S bond length. However, this mixture of two- and three-coordinate Hg(II)-TRIL16C simultaneously show 199Hg-NMR resonance peaks at ∼−180 ppm and ∼−830 ppm73 indicative of both species, whereas Hg-α3DIV has a single resonance at −244 ppm. Furthermore, the apparent pKa for the formation of Hg(II)(TRIL16C)3 was calculated to be 7.8, and a 9.5 pH condition was used to obtain a pure 3 C. N species in the EXAFS analysis. This pKa value is nearly 1 log unit higher than what was determined for the [Hg(II)S3]− complex of Hg(II)-α3DIV. Thereby, our pH condition of 8.7, which is 1.6 log unit higher than its apparent pKa, favors a 3 C. N. species instead of a mixture with a 2 C. N. complex as observed in Hg(II)-TRIL16C. Finally, the absorbance values in the ultraviolet between 2–4 coordinate mercury–sulfur chromophores are distinctive, where Hg(SR)2 complexes have a λmax band <220 nm and Hg(SR)3 and Hg(SR)4 compounds exhibit absorption λmax bands between 230–340 nm.74 The absorption characteristics of Hg(II)-α3DIV at pH 8.6 is more consistent with a 3 C. N. complex. For these reasons, we believe that the best interpretation of the XAS data collectively with the 199Hg NMR and UV–vis is to assign a T-shaped geometry with two short Hg(II)-S bonds and one longer (∼2.8–3.0 Å) bond.
Together, the results from the CD and DSC studies demonstrate that metal binding did not disrupt the overall secondary structure of α3DIV; instead it provides further stability to the framework. In addition, the EXAFS analysis validates Hg(II) binding to the triscysteine site and provides structural metrics on the [HgS3]− complex. Therefore, the loss of cross peaks in the 15N-HSQC spectrum of Pb(II)- and Hg(II)-α3DIV is not due to an unfolding of α3DIV. Instead, it indicates that metal binding quenches the more rapid dynamics of the metal-site (see above) and selects conformations that interconvert on a milli-microsecond time scale, leading to line-broadening and loss of the corresponding 15N-HSQC NMR cross peaks. Nonetheless, in the case of Pb(II)-α3DIV, we were still able to identify 57 of the 68 resonance peaks and the perturbation of many residues, such as K8, Cys18, Phe38, Ala44, and Val53, can be identified. The missing peaks are due to residues localized near the metal binding site or at the C-terminal end, which was already determined to be more dynamic than the rest of the fold in α3D. For example, the chemical shift change for residues that are located at this end of the bundle, involving Gln19, Ser24, Ala29, Gln68, and Tyr70, cannot be easily assigned suggesting severe line broadening or overlap with a neighboring signal. For Hg(II)-α3DIV, the 15N-HSQC spectrum has seven fewer peaks than Pb(II)-α3DIV and has eight less than its apo counterpart. In contrast to the Pb spectrum, the loss in peaks seems to be global and cannot be assigned to any specific region of the sequence. Like the Pb spectrum, we can attribute the some of peak loss to Hg(II) binding, but the more dramatic effect could be largely associated with the pH environment. As pH increases from 6 to 9, the average lifetime of the exchangeable hydrogen groups, including backbone amide protons, increases from ms to μs range, thereby decreasing the observable resonance peaks in a 15N-HSQC spectrum. This pH effect is exhibited in the comparison of the 15N-HSQC spectra of apo α3DIV at pH 5.8 and 8.6 (Supporting Information, Figure 8). The higher pH spectrum contains 13 less peaks than the lower pH, demonstrating that the significant loss in cross peaks in the Hg(II)-α3DIV spectrum is mainly due to the pH condition.
Figure 8.
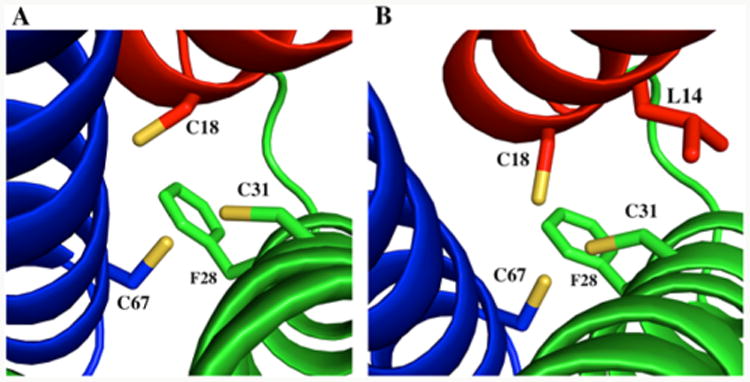
(A) Model of α3DIV C28F, F31C, where the Cys residue on helix 2 is moved one layer above its original position in order to form a triscysteine plane that is more perpendicular with the bundle. (B) Model of α3DIV I14L, C28F, F31C, where a bulky Ile is replaced with a Leu residue in able generating space for the Cys residues to ultimately form a preformed metal binding site.
An alternative explanation for the significant change in 15N-HSQC results for the metalated species could stem from the asymmetric nature of the triscysteine site and leading to structural dynamics on micro-milli second time scale resulting in line broadening and peak loss. The side-on view of the triscysteine site of structure 1 in Figure 6A shows that the Cβ and Sγ atoms of Cys28 points toward the C-terminal end of the bundle. Upon metal binding, Cys28 requires a rotation at the N–Cα–Cβ bonds of about 56° toward the N-terminal end in order to achieve the proper trigonal plane for Hg(II) or Pb(II). In the same manner, the “d” site-like conformation of Cys18 will have to adopt an “a” site conformation as seen in structure 16 of α3DIV or translate toward the core (Figure 6B). To further illustrate this process, apo α3DIV was overlaid to the X-ray crystal structures of metalated de novo designed peptides, As(III)(CSL9C)3 (at 1.81 Å resolution)76 and [Hg(II)]S[Zn-(II)(H2O/OH−)]N(CSL9PenL23H)3n+ (at 2.20 Å resolution) in Figure 7.
Similar to Pb(II), As(III) has a sterochemically active lone pair of electrons and binds to a triscysteine environment in a trigonal pyramidal geometry. There are several possible orientations for the As(III) ion, with the most symmetric being endo and exo.78 In the energetically preferred endo conformation, the As(III) ion and all three Cβ atoms are on the same side of the plane of S atoms. In the exo conformation, As(III) and the Cβ atoms are on opposite sides of this plane. It is also possible to have a mixed conformation, where one or two Cβ atoms are on the same side and one or two are on the opposite side. The crystal structure of As(III)(CSL9C)3 reveals an endo conformation (Figure 7A). For Pb(II)-α3DIV in solution, it is possible that the Pb atom forms a distorted, asymmetric triscysteine site that undergoes a exchange between these conformations; thereby, influencing the chemical shift dispersion in the N-HSQC spectrum. In addition, the 207Pb-NMR spectrum of Pb(II)-α3DIV contained no resonances in the range observed for the more symmetric Pb(II)-S3 complexes in coil-Ser, BABY, and GRAND parallel three-stranded coiled coils. UV–vis studies confirm that Pb(II) does bind in a trigonal pyramidal geometry, so the absence of the 207Pb NMR signal supports the idea that Pb(II) is in a very dynamic environment. 207Pb-NMR is much more sensitive to the motion in the coordination environment than UV–vis spectroscopy and can provide insight into the dynamic behavior of a metal–ligand complex
Hg(II) can form two- and three-coordinate sulfur complexes depending on the pH. In the 3 C. N. complex, two geometries are possible: a trigonal planar complex with equal Hg–Sγ bond lengths (∼2 Å), and a T-shaped complex with two short Hg–-Sγ bonds (2.1–2.3 Å) and one long (∼3 Å) bond. Our EXAFS analysis on the 3 C. N. complex of Hg(II)-α3DIV illustrates a T-shaped coordination environment; therefore, the triscysteine site of α3DIV was aligned with the X-ray crystal structure of the T-shaped Hg(II)-trispenicillamine of [Hg(II)]S[Zn(II)(H2O/OH-)]N(CSL9PenL23H)3n+ (Figure 7B). This alignment shows that the Sγ atom of Cys67 has the proper orientation to form a short Hg–Sγ bond; however Cys18 and Cys28 would require ample conformational changes to align with this T-shaped mercury-complex in [Hg(II)]S[Zn(II)(H2O/OH-)]N-(CSL9PenL23H)3n+. Thus, like the Pb(II) complex, the distorted T-shaped geometry of Hg(II)-α3DIV could form different conformations, where the one long bond can alternate between Cys18 and Cys28. The resonance peak in the 199Hg-NMR spectrum of 199Hg-α3DIV can elucidate this dynamic process. This nucleus is exceptionally sensitive to a slight perturbation in the Hg-ligand bond length and angle. A 0.01 Å deviation in a Hg–Cl bond length and 10° change in Cl–Hg–Cl bond angle was calculated to change their corresponding chemical shift values by ∼50 and 100 ppm.80 For Hg(II)-α3DIV in a 3 C. N. complex, its 199Hg-NMR chemical shift value was experimentally determined to be −240 ppm. This value falls in between the 199Hg-NMR signals reported for symmetric Cys “a” and “d” sites in 3SCC peptides with chemical shifts of ∼−185 and ∼−316 ppm, respectively. This deviation by +55 ppm from an “a” and −76 ppm from a “d” site could be representative of a distorted trigonal thiolato [HgS3]− T-shaped complex of Hg(II)-α3DIV with metal–ligand bond lengths and angles that deviate from its 3SCC counterparts.
Implications on Future Designs
De novo protein design allows for an iterative approach in modifying a stable peptide framework The structure of apo α3DIV demonstrates that by mutating packing Leu to Cys residues to achieve a thiol-rich metal binding site we are still able to retain a well-folded peptide construct. With this at hand, we can now prepare new constructs that will result in a more preformed, more symmetric triscysteine metal binding site in an antiparallel three-helix bundle. In the ensemble of structures, the Sγ of Cys28 consistently orients toward the C-terminal end forming a skewed S3 plane. The first iteration involves moving the Cys28 one layer toward the N-terminal end by performing Cys28Phe and Phe31Cys mutations (Figure 8A). Placing a Cys in the 31st position produces a S3 plane that is perpendicular with the helical bundle and a Phe in the 28th provides a capping and directing force for Cys31, which yields a preformed site. The Cys28Phe/Phe31Cys α3DIV construct can then be further modified with an iteration that directs the Sγ of Cys18 to form more of an “a” orientation (Figure 8B). This can be achieved with an Ile14Leu mutation, where a smaller Leu residue affords space to form for the thiol group of Cys18 to orient toward the core.
Conclusions
Toxic heavy metals, such as Hg, Pb, As, and Cd, threaten all forms of life. By understanding the mechanism by which bacteria detoxify heavy metal through metalloregulatory proteins, one can develop strategies to combat this environmental crisis. De novo protein design is an emerging and biologically relevant approach in studying the metal binding sites of metalloregulatory proteins, as well as metalloproteins. It offers a novel approach of modeling metal centers using simple peptide scaffolds. Here, we report the solution structure of apo α3DIV, a de novo designed three-helix bundle peptide with a triscysteine metal binding that sequesters heavy metals Hg(II), Pb(II), and Cd(II). This thiol-rich environment is a ubiquitous motif in the metal binding site of metalloregulatory proteins, including ArsR, MerR, and CadC. We show that α3DIV has the same overall topology as and aligns well with its parent structure α3D. We found that the incorporating Cys in place of Leu residues, which provide stabilizing packing interactions in the core, increased the helical content of the α3D framework and resulted in a more packed core. In addition, heavy metal binding to α3DIV induces further stability. Ultimately, this structure provides a stable framework for designing future metallopeptides that could perform specific catalytic or redox reactions.
Supplementary Material
Acknowledgments
Most NMR experiments were carried out in the Program for Biophysics at the University of Michigan. DSC experiments were collected in the Department of Pharmaceutical Sciences at Wayne State University in the group of T.L.S.
Funding: V.L.P. would like to thank the National Institutes of Health (NIH) for financial support for this research (ES012236). J.S.P. thanks the Rackham Graduate School at the University of Michigan for a research fellowship. S.P.D and T.L.S. are supported by the NIH (DK101230 and DK068139, respectively). E.R.P.Z. acknowledges support of the Department of Biological Chemistry.
Footnotes
Associated Content: Supporting Information: This section contains figures of thermal denaturation curves, 15N-TROSY spectrum of Hg(II)-α3DIV overlaid with an apo control, Ramachandran plot of the 20 lowest structures, CD plots, EXAFS fit, 15N-TROSY spectra of α3DIV at pH 5.8 and 8.6 and so on. In addition, tables that contain the NMR acquisition parameters for the structural determination of apo α3DIV, EXAFS metrical parameters, Ramachandran plot summary, PSVS global quality scores for α3DIV and α3D, thermal denaturation parameters and metrical parameters for mercury-sulfur model compounds are located in this section. Atomic coordinates, structural restraints (distance and dihedral angle restraints), and NMR chemical shift assignments for α3DIV have been deposited via ADIT-NMR system (http://bmrb.wisc.edu). The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acs.biochem.5b00064.
Accession Codes: Apo α3DIV has PDB code 2MTQ, and BMRB ID is 25177 and both depositions will be released upon publication.
Notes: The authors declare no competing financial interest.
References
- 1.Waldron KJ, Rutherford JC, Ford D, Robinson NJ. Metalloproteins and Metal Sensing. Nature. 2009;460:823–830. doi: 10.1038/nature08300. [DOI] [PubMed] [Google Scholar]
- 2.Giedroc DP, Arunkumar AI. Metal sensor proteins: nature's metalloregulated allosteric switches. Dalton Trans. 2007:3107–3120. doi: 10.1039/b706769k. [DOI] [PubMed] [Google Scholar]
- 3.Ibrahim D, Froberg B, Wolf A, Rusyniak DE. Heavy Metal Poisoning: Clinical Presentations and Pathophysiology. Clin Lab Med. 2006;26:67–97. doi: 10.1016/j.cll.2006.02.003. [DOI] [PubMed] [Google Scholar]
- 4.Rooney JPK. The role of thiols, dithiols, nutritional factors and interacting ligands in the toxicology of mercury. Toxicology. 2007;234:145156. doi: 10.1016/j.tox.2007.02.016. [DOI] [PubMed] [Google Scholar]
- 5.DeGrado WF, Summa CM, Pavone V, Nastri F, Lombardi A. De novo design and structural characterization of proteins and metalloproteins. Annu Rev Biochem. 1999;68:779–819. doi: 10.1146/annurev.biochem.68.1.779. [DOI] [PubMed] [Google Scholar]
- 6.Lu Y, Yeung N, Sieracki N, Marshall NM. Design of functional metalloproteins. Nature. 2009;460:855–862. doi: 10.1038/nature08304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Yu F, Cangelosi VM, Zastrow ML, Tegoni M, Plegaria JS, Tebo AG, Mocny CS, Ruckthong L, Qayyum H, Pecoraro VL. Protein Design: Toward Functional Metalloenzymes. Chem Rev. 2014;114:3495–3578. doi: 10.1021/cr400458x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Walsh SR, Cheng H, Bryson JW, Roder H, DeGrado WF. Solution structure and dynamics of a de novo designed three-helix bundle protein. Proc Natl Acad Sci U S A. 1999;96:5486–5491. doi: 10.1073/pnas.96.10.5486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Peacock AFA, Iranzo O, Pecoraro VL. Harnessing natures ability to control metal ion coordination geometry using de novo designed peptides. Dalton Trans. 2009;9226:2271–2280. doi: 10.1039/b818306f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lovejoy B, Choe S, Cascio D, McRorie DK, DeGrado WF, Eisenberg D. Crystal structure of a synthetic triple-stranded alpha-helical bundle. Science. 1993;259:1288–1293. doi: 10.1126/science.8446897. [DOI] [PubMed] [Google Scholar]
- 11.Schneider JP, Lombardi A, DeGrado WF. Analysis and design of three-stranded coiled coils and three-helix bundles. Folding Des. 1998;3:29–40. doi: 10.1016/S1359-0278(98)00011-X. [DOI] [PubMed] [Google Scholar]
- 12.Bryson JW, Desjarlais JR, Handel TM, Degrado WF. From coiled coils to small globular proteins: Design of a native-like three-helix bundle. Protein Sci. 1998;7:1404–1414. doi: 10.1002/pro.5560070617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Walsh STR, Sukharev VI, Betz SF, Vekshin NL, DeGrado WF. Hydrophobic Core Malleability of a De Novo Designed Three-helix Bundle Protein. J Mol Biol. 2001;305:361–373. doi: 10.1006/jmbi.2000.4184. [DOI] [PubMed] [Google Scholar]
- 14.Walsh STR, Lee AL, DeGrado WF, Wand AJ. Dynamics of a De Novo Designed Three-Helix Bundle Protein Studied by 15N, 13C, and 2H NMR Relaxation Methods. Biochemistry. 2001;40:9560–9569. doi: 10.1021/bi0105274. [DOI] [PubMed] [Google Scholar]
- 15.Walsh STR, Cheng RP, Wright WW, Alonso DOV, Daggett V, Vanderkooi JM, DeGrado WF. The hydration of amides in helices; a comprehensive picture from molecular dynamics, IR and NMR. Protein Sci. 2003;12:520–531. doi: 10.1110/ps.0223003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Zhu Y, Alonso DOV, Maki K, Huang CY, Lahr SJ, Daggett V, Roder H, DeGrado WF, Gai F. Ultrafast folding of α3D: A de novo designed three-helix bundle protein. Proc Natl Acad Sci U S A. 2003;100:15486–15491. doi: 10.1073/pnas.2136623100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.O'Halloran T, Walsh C. Metalloregulatory DNA-binding protein encoded by the merR gene: isolation and characterization. Science. 1987;235:211–214. doi: 10.1126/science.3798107. [DOI] [PubMed] [Google Scholar]
- 18.Wright JG, Natan MJ, MacDonnel FM, Ralston DM, O'Halloran TV. Mercury (II)—Thiolate Chemistry and the Mechanism of the Heavy Metal Biosensor MerR. Prog Inorg Chem. 1990;38:323–412. [Google Scholar]
- 19.Wright JG, Tsang HT, Penner-Hahn JE, O'Halloran TV. Coordination chemistry of the Hg-MerR metalloregulatory protein: evidence for a novel tridentate mercury-cysteine receptor site. J Am Chem Soc. 1990;112:2434–2435. [Google Scholar]
- 20.Busenlehner LS, Weng TC, Penner-Hahn JE, Giedroc DP. Elucidation of Primary (α3N) and Vestigial (α5) Heavy Metal-binding Sites in Staphylococcus aureus pI258 CadC: Evolutionary Implications for Metal Ion Selectivity of ArsR/SmtB Metal Sensor Proteins. J Mol Biol. 2002;319:685–701. doi: 10.1016/S0022-2836(02)00299-1. [DOI] [PubMed] [Google Scholar]
- 21.Ye J, Kandegedara A, Martin P, Rosen BP. Crystal Structure of the Staphylococcus aureus pI258 CadC Cd(II)/Pb(II)/Zn(II)-Responsive Repressor. J Bacteriol. 2005;187:4214–4221. doi: 10.1128/JB.187.12.4214-4221.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Banci L, Bertini I, Cantini F, Ciofi-Baffoni S, Cavet JS, Dennison C, Graham AI, Harvie DR, Robinson NJ. NMR Structural Analysis of Cadmium Sensing by Winged Helix Repressor CmtR. J Biol Chem. 2007;282:30181–30188. doi: 10.1074/jbc.M701119200. [DOI] [PubMed] [Google Scholar]
- 23.Chakraborty S, Kravitz JY, Thulstrup PW, Hemmingsen L, DeGrado WF, Pecoraro VL. Design of a three-helix bundle capable of binding heavy metals in a triscysteine environment. Angew Chem, Int Ed Engl. 2011;50:2049–2053. doi: 10.1002/anie.201006413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Delaglio F, Grzesiek S, Vuister GW, Zhu G, Pfeifer J, Bax A. NMRPipe: a multidimensional spectral processing system based on UNIX pipes. J Biomol NMR. 1995;6:277–293. doi: 10.1007/BF00197809. [DOI] [PubMed] [Google Scholar]
- 25.Goddard, T. D. and Kneller, D. G. University of California, San Francisco.
- 26.Bax A, Grzesiek S. Methodological advances in protein NMR. Acc Chem Res. 1993;26:131–138. [Google Scholar]
- 27.Pervushin K, Rek R, Wider G, Wüthrich K. Attenuated T2 relaxation by mutual cancellation of dipole–dipole coupling and chemical shift anisotropy indicates an avenue to NMR structures of very large biological macromolecules in solution. Proc Natl Acad Sci U S A. 1997;94:12366–12371. doi: 10.1073/pnas.94.23.12366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Salzmann M, Pervushin K, Wider G, Senn H, Wüthrich K. TROSY in triple-resonance experiments: New perspectives for sequential NMR assignment of large proteins. Proc Natl Acad Sci U S A. 1998;95:13585–13590. doi: 10.1073/pnas.95.23.13585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Loria JP, Rance M, Palmer AG. Transverse-Relaxation-Optimized (TROSY) Gradient-Enhanced Triple-Resonance NMR Spectroscopy. J Magn Reson. 1999;141:180–184. doi: 10.1006/jmre.1999.1891. [DOI] [PubMed] [Google Scholar]
- 30.Muhandiram DR, Kay LE. Gradient-enhanced triple-resonance three-dimensional NMR experiments with improved sensitivity. J Mag Reson, Ser B. 1994;103:203–216. [Google Scholar]
- 31.Grzesiek S, Bax A. Improved 3D triple-resonance NMR techniques applied to a 31 kDa protein. J Magn Reson. 1992;96:432–440. [Google Scholar]
- 32.Grzesiek S, Bax A. Correlating backbone amide and side chain resonances in larger proteins by multiple relayed triple resonance NMR. J Am Chem Soc. 1992;114:6291–6293. [Google Scholar]
- 33.Grzesiek S, Bax A. An efficient experiment for sequential backbone assignment of medium-sized isotopically enriched proteins. J Magn Reson. 1992;99:201–207. [Google Scholar]
- 34.Kuboniwa H, Grzesiek S, Delaglio F, Bax A. Measurement of HN-Hα J couplings in calcium-free calmodulin using new 2D and 3D water-flip-back methods. J Biomol NMR. 1994;4:871–878. doi: 10.1007/BF00398416. [DOI] [PubMed] [Google Scholar]
- 35.Crippen G, Rousaki A, Revington M, Zhang Y, Zuiderweg ERP. SAGA: rapid automatic mainchain NMR assignment for large proteins. J Biomol NMR. 2010;46:281–298. doi: 10.1007/s10858-010-9403-2. [DOI] [PubMed] [Google Scholar]
- 36.Bax A, Clore GM, Gronenborn AM. 1H-1H correlation via isotropic mixing of 13C magnetization, a new three-dimensional approach for assigning 1H and 13C spectra of 13C-enriched proteins. J Magn Reson. 1990;88:425–431. [Google Scholar]
- 37.Fesik SW, Eaton HL, Olejniczak ET, Zuiderweg ERP, McIntosh LP, Dahlquist FW. 2D and 3D NMR spectroscopy employing 13C-13C magnetization transfer by isotropic mixing. Spin system identification in large proteins. J Am Chem Soc. 1990;112:886–888. [Google Scholar]
- 38.Wang H, Zuiderweg ERP. HCCH-TOCSY spectroscopy of 13C-labeled proteins in H2O using heteronuclear cross-polarization and pulsed-field gradients. J Biomol NMR. 1995;5:207–211. doi: 10.1007/BF00208812. [DOI] [PubMed] [Google Scholar]
- 39.Fesik SW, Zuiderweg ERP. Heteronuclear three-dimensional NMR spectroscopy. A strategy for the simplification of homonuclear two-dimensional NMR spectra. J Magn Reson. 1988;78:588–593. [Google Scholar]
- 40.Fischer MWF, Zeng L, Zuiderweg ERP. Use of 13C-13C NOE for the Assignment of NMR Lines of Larger Labeled Proteins at Larger Magnetic Fields. J Am Chem Soc. 1996;118:12457–12458. [Google Scholar]
- 41.Shen Y, Bax A. Protein backbone and sidechain torsion angles predicted from NMR chemical shifts using artificial neural networks. J Biomol NMR. 2013;56:227–241. doi: 10.1007/s10858-013-9741-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Guntert P. Automated NMR structure calculation with CYANA. In: Downing AK, editor. Methods in Molecular Biology: Protein NMR Techniques. Vol. 278. Humana Press Inc.; Clifton, NJ: 2004. pp. 353–378. [DOI] [PubMed] [Google Scholar]
- 43.Wüthrich K. NMR of Proteins and Nucleic Acids. Wiley-Interscience; New York: 1986. [Google Scholar]
- 44.George GN, George SJ, Pickering IJ. EXAFSPAK 2001 [Google Scholar]
- 45.Ankudinov AL, Rehr JJ. Relativistic calculations of spin-dependent X-ray absorption spectra. Phys Rev B. 1997;56:1712–1715. [Google Scholar]
- 46.Bencze KZ, Kondapalli KC, Stemmler TL. X-ray absorption spectroscopy. In: Scott RA, Lukehart CM, editors. Applications of Physical Methods to Inorganic and Bioinorganic Chemistry: Handbook, Encyclopedia of Inorganic Chemistry. 2nd. John Wiley & Sons, Ltd; Chichester, UK: 2007. pp. 513–528. [Google Scholar]
- 47.Cotelesage JJH, Pushie MJ, Grochulski P, Pickering IJ, George NG. Metalloprotein active site structure determination: Synergy between X-ray absorption spectroscopy and X-ray crystallography. J Inorg Biochem. 2012;115:127–137. doi: 10.1016/j.jinorgbio.2012.06.019. [DOI] [PubMed] [Google Scholar]
- 48.Wishart DS, Sykes BD, Richards FM. Relationship between nuclear magnetic resonance chemical shift and protein secondary structure. J Mol Biol. 1991;222:311–333. doi: 10.1016/0022-2836(91)90214-q. [DOI] [PubMed] [Google Scholar]
- 49.Yao J, Dyson HJ, Wright PE. Chemical shift dispersion and secondary structure prediction in unfolded and partly folded proteins. FEBS Lett. 1997;419:285–289. doi: 10.1016/s0014-5793(97)01474-9. [DOI] [PubMed] [Google Scholar]
- 50.The PyMOL Molecular Graphics System, Version 1.5.0.4 Schrodinger, LLC.
- 51.Bhattacharya A, Tejero R, Montelione GT. Evaluating protein structures determined by structural genomics consortia. Proteins. 2007;66:778–795. doi: 10.1002/prot.21165. [DOI] [PubMed] [Google Scholar]
- 52.Lee HS, Choi J, Yoon S. QHELIX: A Computational Tool for the Improved Measurement of Inter-Helical Angles in Proteins. Protein J. 2007;26:556–561. doi: 10.1007/s10930-007-9097-9. [DOI] [PubMed] [Google Scholar]
- 53.Ponder JW, Richards FM. Tertiary templates for proteins: use of packing criteria in the enumeration of allowed sequences for different structural classes. J Mol Biol. 1987;193:775–791. doi: 10.1016/0022-2836(87)90358-5. [DOI] [PubMed] [Google Scholar]
- 54.Hodges RS. De novo design of α-helical proteins: basic research to medical applications. Biochem Cell Biol. 1996;74:133–154. doi: 10.1139/o96-015. [DOI] [PubMed] [Google Scholar]
- 55.Tesmer JJG, Stemmler TL, Penner-Hahn JE, Davisson VJ, Smith JL. Preliminary X-ray analysis of E. coli GMP Synthetase: Determination of anomalous scatterring factors for a cysteinyl mercury derivative. Proteins: Struct, Funct, Genet. 1994;18:394–403. doi: 10.1002/prot.340180410. [DOI] [PubMed] [Google Scholar]
- 56.Cangelosi VM, Deb A, Penner-Hahn JE, Pecora VL. A De Novo Designed Metalloenzyme for the Hydration of CO2. Angew Chem, Int Ed Engl. 2014;53:7900–7903. doi: 10.1002/anie.201404925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Willard L, Ranjan A, Zhang H, Monzavi H, Boyko RF, Sykes BD, Wishart D. VADAR: a web server for quantitative evaluation of protein structure quality. Nucleic Acids Res. 2003;31:3316–3319. doi: 10.1093/nar/gkg565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Lee MS, Gippert GP, Soman KV, Case DA, Wright PE. Three-Dimensional Solution Structure of a Single Zinc Finger DNA-Binding Domain. Science. 1989;245:635–637. doi: 10.1126/science.2503871. [DOI] [PubMed] [Google Scholar]
- 59.Malmstrom BG. Rack-induced bonding in blue-copper proteins. Eur J Biochem. 1994;223:711–718. doi: 10.1111/j.1432-1033.1994.tb19044.x. [DOI] [PubMed] [Google Scholar]
- 60.Dieckmann GR, McRorie DK, Lear JD, Sharp KA, DeGrado WF, Pecoraro VL. The role of protonation and metal chelation preferences in defining the properties of mercury-binding coiled coils. J Mol Biol. 1998;280:897–912. doi: 10.1006/jmbi.1998.1891. [DOI] [PubMed] [Google Scholar]
- 61.Chakraborty S, Touw DS, Peacock AFA, Stuckey JA, Pecoraro VL. Structural comparisons of apo- and metalated three-stranded coiled coils clarify metal binding determinants in thiolate containing designed peptides. J Am Chem Soc. 2010;132:13240–13250. doi: 10.1021/ja101812c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Peacock AFA, Hemmingsen L, Pecoraro VL. Using diastereopeptides to control metal ion coordination in proteins. Proc Natl Acad Sci U S A. 2008;105:16566–16571. doi: 10.1073/pnas.0806792105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Peacock AFA, Stuckey JA, Pecoraro VL. Switching the chirality of the metal environment alters the coordination mode in designed peptides. Angew Chem, Int Ed in Engl. 2009;48:7371–7374. doi: 10.1002/anie.200902166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Frankel AD, Berg JM, Pabo CO. Metal-dependent folding of a single zinc finger from transcription factor IIIA. Proc Natl Acad Sci U S A. 1987;84:4841–4845. doi: 10.1073/pnas.84.14.4841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Siedlecka M, Goch G, Ejchart A, Sticht H, Bierzyǹski A. α-Helix nucleation by a calcium-binding peptide loop. Proc Natl Acad Sci U S A. 1999;96:903–908. doi: 10.1073/pnas.96.3.903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Suzuki K, Hiroaki H, Kohda D, Nakamura H, Tanaka T. Metal Ion Induced Self-Assembly of a Designed Peptide into a Triple-Stranded α-Helical Bundle: A Novel Metal Binding Site in the Hydrophobic Core. J Am Chem Soc. 1998;120:13008–13015. [Google Scholar]
- 67.Farrer BT, Harris NP, Balchus KE, Pecoraro VL. Thermodynamic Model for the Stabilization of Trigonal Thiolato Mercury(II) in Designed Three-Stranded Coiled Coils. Biochemistry. 2001;40:14696–14705. doi: 10.1021/bi015649a. [DOI] [PubMed] [Google Scholar]
- 68.Chen YH, Yang JT, Chau KH. Determination of the helix and β form of proteins in aqueous solution by circular dichroism. Biochemistry. 1974;13:3350–3359. doi: 10.1021/bi00713a027. [DOI] [PubMed] [Google Scholar]
- 69.Davidson DE, Hill BC. Stability of oxidized, reduced and copper bound forms of Bacillus subtilis Sco. Biochim Biophys Acta. 2009;1794:275–281. doi: 10.1016/j.bbapap.2008.10.005. [DOI] [PubMed] [Google Scholar]
- 70.Wu H, Xu X, Shen D, Peng L, Song J, Zhang Y. Binding of Ca + and Zn + to factor IX/X-binding protein from venom of Agkistrodon halys Pallas: stabilization of the structure during GdnHCl-induced and thermally induced denaturation. J Biol Inorg Chem. 2011;16:69–79. doi: 10.1007/s00775-010-0703-5. [DOI] [PubMed] [Google Scholar]
- 71.Matzapetakis M, Farrer BT, Weng TS, Hemmingsen L, Penner-Hahn JE, Pecoraro VL. Comparison of the Binding of Cadmium(II), Mercury(II), and Arsenic(III) to the de Novo Designed Peptides TRI L12C and TRI L16C. J Am Chem Soc. 2002;124:8042–8054. doi: 10.1021/ja017520u. [DOI] [PubMed] [Google Scholar]
- 72.See Supporting Information, Table 5.
- 73.Dieckmann GR, McRorie DK, Tierney DL, Utschig LM, Singer CP, O'Halloran TV, Penner-Hahn JE, DeGrado WF, Pecoraro VL. De novo design of mercury-binding two-and three-helical bundles. J Am Chem Soc. 1997;119:6195–6196. [Google Scholar]
- 74.Pecoraro VL, Peacock FA, Iranzo O, Luczkowshi M. Understanding the biological chemistry of mercury using a de novo protein design strategy. In: Long E, Baldwin MJ, editors. Bioinorganic Chemistry ACS Symposium Series. American Chemical Society; Washington DC: 2009. pp. 183–197. [Google Scholar]
- 75.Rule GS, Hitchens KT. Fundamentals of Protein NMR spectroscopy. Springer, Netherlands; Netherlands: 2006. pp. 313–351. [Google Scholar]
- 76.Touw DS, Nordman CE, Stuckey JA, Pecoraro VL. Identifying important structural characteristics of arsenic resistance proteins by using designed three-stranded coiled coils. Proc Natl Acad Sci U S A. 2007;104:11969–11974. doi: 10.1073/pnas.0701979104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Zastrow ML, Peacock AFA, Stuckey JA, Pecoraro VL. Hydrolytic catalysis and structural stabilization in a designed metalloprotein. Nat Chem. 2012;4:118–123. doi: 10.1038/nchem.1201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Zampella G, Neupane KP, Gioia L, Pecoraro VL. The importance of stereochemically active lone pairs for influencing Pb(II) and As(III) protein binding. Chem —Eur J. 2012;18:2040–2050. doi: 10.1002/chem.201102786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Neupane KP, Pecoraro VL. Probing a homoleptic PbS3 coordination environment in a designed peptide using 207Pb NMR spectroscopy: implications for understanding the molecular basis of lead toxicity. Angew Chem, Int Ed in Engl. 2010;49:8177–8180. doi: 10.1002/anie.201004429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Wolff SK, Ziegler T, van Lenthe E, Baerends EJ. Density functional calculations of nuclear magnetic shieldings using the zeroth-order regular approximation (ZORA) for relativistic effects: ZORA nuclear magnetic resonance. J Chem Phys. 1999;110:7689–7698. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.