

Abstract
Much research has gone into the development of hybrid gene delivery systems that combine the broad tropism and efficient transduction of adenoviral vectors with the ability to achieve stable expression of cargo genes. In addition to gene therapy applications, such a system has considerable advantages for studies of gene function in vivo, permitting fine-tuned genetic manipulation with higher throughput than can be achieved using standard transgenic and DNA targeting techniques. Existing strategies are limited, however, by low integration efficiencies, small cargo capacity, and/or a dependence on target cell division. The utility of this approach could be enhanced by a system that provides all of the following: (1) efficient delivery, (2) stable expression in a high percentage of target cells (whether mitotic or not), (3) large cargo capacity, (4) flexibility to use with a wide range of additional experimental conditions, and (5) simple experimental technique. Here we report the initial characterization of a hybrid system that meets these criteria by utilizing piggyBac (PB) transposition to achieve genomic integration from adenoviral vectors. We demonstrate stable expression of an adenovirus (Ad)-PB-delivered reporter gene in ∼20–40% of hepatocytes following standard tail vein injection. Its high efficiency and flexibility relative to existing hybrid adenoviral gene delivery approaches indicate a considerable potential utility of the Ad-PB system for therapeutic gene delivery and in vivo studies of gene function.
Introduction
Viral vectors are a ubiquitous tool in molecular biology, primarily used for delivery and expression of transgenes in vivo and in vitro. Recombinant adenovirus (Ad) is a particularly common vector, as it provides broad tropism, large cargo capacity, efficient transduction of dividing or nondividing cells, and simple production of high titer preparations.1 One major limitation of Ad vectors, however, is that aside from very rare instances of genomic integration,2,3 expression of cargo genes is transient. Sustained expression depends on the maintenance of episomal viral genomes, which are nonreplicative and thus get progressively diluted with each cell division. Additionally, most recombinant Ad vectors retain some viral genes along with the transgene, the expression of which may contribute to clearance of transduced cells by the host immune system in vivo.
To overcome the limitations imposed by transient expression of cargo genes, several hybrid Ad vector systems have been developed with various integrative elements incorporated into the viral genome. This approach retains the advantages that Ad vectors provide in terms of delivery to target cells, while also allowing transgene expression to be maintained long-term. Hybrid Ad systems have been described with elements incorporated from adeno-associated virus (AAV), retrovirus, sleeping beauty (SB) transposons, L1 retrotransposons, or PhiC31 integrase.4 While each of these strategies successfully facilitates genomic integration of cargo genes, efficiencies are typically below 1% of transduced cells.5–8 A hybrid Ad-lentiviral system has achieved an in vivo stable transduction rate of 20–30% in mouse liver,9 although the efficiency was likely influenced by the immunodeficient nature of the mice that were used. This system is also limited by the cargo capacity of lentiviral vectors—packaging and integration efficiencies drop steeply with increasing transgene size above ∼4 kilobases (kb).10 Stable transduction rates up to 80% have been reported in cell culture experiments with hybrid Ad-foamy virus constructs,11,12 although the multiplicity of infection (MOI) required to attain this efficiency exceeds what can reliably be achieved in vivo. Additionally, foamy virus is only capable of integration in actively dividing cells.13 Given the limitations of existing hybrid Ad vector gene delivery approaches, a straightforward system capable of stably integrating large transgenes in a high percentage of target cells would be of significant benefit to the research community.
We have developed a novel gene delivery system that improves on existing approaches by constructing a recombinant Ad vector containing a piggyBac (PB) transposon (termed Ad-PB). This design allows transgenes to be integrated into the genome of target cells expressing PB transposase (PBase), which can be incorporated into the model system or codelivered. Based on its high excision/integration activity relative to other transposase enzymes,14 along with an ability to efficiently mobilize transposons larger than 100 kb,15 we hypothesized that PBase would be an effective tool for stable integration of Ad-delivered transgenes. We present here initial characterization of the Ad-PB system's activity, reporting stable PB-mediated transgene integration from recombinant Ad vectors both in cultured cells and in vivo with high efficiency relative to existing hybrid Ad approaches. Delivery of the Ad-PB system via standard tail vein injection yielded stable expression of a reporter gene in livers of wild-type mice over a period of 20 weeks, thus demonstrating its suitability for applications requiring stable gene delivery in vivo.
Materials and Methods
Vector construction
Lentiviral expression vectors encoding EF1α promoter-driven mPB-IRES-puroR and hyPBase-IRES-puroR were generated using the pHAGE2 self-inactivating, nonreplicative lentiviral vector previously described.16 A shuttle vector was generated containing the functional elements of Ad-T/mCherry flanked by homology arms for recombination into the pAd-nGFP adenoviral genome backbone.17 Homology arms were PCR-amplified using 10 ng pAd-nGFP as template with the following primers: 3′ homology arm (3′HArm_F: 5′–ACATGTAAGCGACGGATGTG–3′ and 3′HArm_R: 5′–GATCCAAGCTTCGACTGAC–3′), 5′ homology arm (flox5′HArm_F: 5′–GATCTCCCGATCCCCTATG–3′ and flox5′HArm_R: 5′–GCGTATATCTGGCCCGTA CA–3′). Homologous recombination was performed using a previously described recombineering system18 to insert 3′HArm-PB-EF1α-mCherry-pA-5′HArm into the pAd-nGFP vector backbone. Ad vectors were packaged by transfecting 1 μg of PacI-digested pAd-T/mCherry into 293FT cells at 50% confluency in a 10 cm dish. After 14 days of growth, cells were resuspended and subjected to three freeze–thaw cycles for lysis. Viral supernatant was submitted to the University of Iowa Gene Transfer Vector Core for purification and titration. The AAV-hyPBase was generated by cloning the EF1α-hyPBase cassette into a baculovirus-AAV packaging construct. This vector was then used to generate recombinant AAV2/9 serotype virus by the University of Iowa Gene Transfer Vector Core.
Cell culture
TIB-73 cells (BNL CL.2, ATCC) were maintained in high-glucose DMEM (Life Technologies) supplemented with 10% fetal bovine serum. Lentiviral vectors were transfected into 293FT cells along with plasmids encoding VSV-G envelope and viral packaging proteins using Effectene transfection reagent (Qiagen). TIB-73 cell lines with stable expression of mPBase and hyPBase were generated by lentiviral transduction followed by antibiotic selection with 1 μg/ml puromycin. For viral gene delivery efficiency assays, 6-well plates were seeded with 100,000 cells/well and transduced with virus at the indicated MOI.
Excision PCR
Genomic DNA was extracted from transduced TIB-73 cells and liver tissue samples using the GenElute Mammalian Genomic DNA Miniprep kit (Sigma-Aldrich). PCR was performed using 50 ng DNA as template to amplify fragments from the Ad-PB vector. Primers were designed to amplify either an excision-specific product (Ad-PB_Ex_F: 5′–ACCGTTTACGTGGAGACTCG–3′ and Ad-PB_Ex_R: 5′–GCCGATATACTATGCCGATGA–3′) or a portion of the Ad genome present regardless of excision (Ad-PB_F: 5′–CACCAAAATCAACGGGACTT–3′ and Ad-PB_R: 5′–CATGGTGGAGCCTGCTTT–3′).
Flow cytometry
Cells from confluent 6-well plates were rinsed with PBS and detached with 5% trypsin-EDTA (Life Technologies). Cells were pelleted by centrifugation at 1000×g for 5 min, washed with PBS, and pelleted once more. Pellets were resuspended in 1.5 ml PBS+1% FBS on ice before mCherry expression analysis at the University of Iowa Flow Cytometry Facility using a Becton Dickinson LSR II cytometer.
In vivo transduction
A cohort of ROSA26-CreERT2 mice (The Jackson Laboratory; stock number 008463) was used for the in vivo transduction experiments. This mouse strain was chosen to provide the potential to inactivate transgene expression from intact Ad-T/mCherry virus via Cre recombination (see Fig. 1a). Six mice received 1×1012 vector genomes (VG) of AAV-hyPBase in 150 μl sterile PBS via tail vein injection. Ten days later, 21 mice (including the 6 that received AAV-hyPBase) received 1×108 PFU of Ad-T/mCherry in 200 μl sterile PBS via tail vein injection. Livers were harvested at 2, 4, 6, 8, 10, 12, and 20 weeks after Ad injection. All animal studies were approved and monitored by the Institutional Animal Care and Use Committee at the University of Iowa.
FIG. 1.

PiggyBac (PB) transposon excision from recombinant adenovirus. (a) Structure of the Ad-T/mCherry vector. A PB transposon containing an Ef1α-mCherry expression cassette was inserted into the adenoviral genome upstream and in opposite orientation to a CMV-nGFP expression cassette. Black triangles indicate loxP recognition sites. Primer binding sites for detection of excision-specific and excision-independent products are indicated with gray and black arrows, respectively. (b) PCR on DNA extracted from TIB-73 cells transduced with Ad-T/mCherry. The adenoviral genome was detected regardless of hyPBase expression, while transposon excision was detected only in cells expressing hyPBase. Depicted results are representative of three independent biological replicates. Ad, adenovirus; CMV, cytomegalovirus.
Immunohistochemistry
Liver samples collected from the left lobe were fixed in 10% neutral-buffered formalin. Samples were then paraffin perfused, embedded, sectioned to a thickness of 4 μm, and baked onto glass slides. Slides were baked at 60°C for an additional 30 min, deparaffinized in xylenes, rehydrated, and treated with citrate antigen unmasking solution (Vector Laboratories). Endogenous peroxidase activity was blocked by incubation with 3% hydrogen peroxide for 5 min. The mouse-on-mouse (M.O.M.) kit was used for immunolabeling with primary antibody anti-mCherry (Abcam 1C51). Primary antibody was diluted 1:200 and incubated with samples for 1 hr at room temperature. The ImmPACT DAB kit (Vector Laboratories) was used for detection. Sections were counterstained with hematoxylin QS (Vector Laboratories) and mounted with mount-quick (Daido Sangyo Co., Ltd.) for light microscopy.
Slides were viewed under a light microscope at 10×magnification. For each sample, four random fields were imaged. An ImageJ macro was developed to quantify the ratio of DAB-positive area to DAB-negative, hematoxylin-positive area. Two color thresholds were set to select DAB-positive pixels and DAB-negative cellular pixels, and the ratio of DAB-positive to total cellular area was calculated for each image. All images were captured and quantified with identical microscope and color threshold settings.
Ligation-mediated PCR
A previously described method for detection of integrated SB transposons was modified to facilitate detection of PB transposons.19 To remove background sequences from the Ad-PB genome, an EcoRI digest was performed after adapter ligation. Primary PCRs were performed using 3′PB_ITR (5′–CGTCACAATATGATTATCTTTCTAGGG–3′), 5′PB_ITR: (5′-TTTTACGCAGACTATCTTTCTAGGG–3′) and linker (5′-GTAATACGACTCACTATAGGGC-3′) primers. Nested secondary PCRs were performed with barcoded primers and pooled samples were submitted to the University of Iowa DNA Facility for sequencing using the Illumina HiSeq 2000 platform.
Quantitative PCR
During the flow cytometry experiment, cells were pelleted and DNA was extracted at each passage. Presence of the mCherry-containing transposon was assessed via PCR using the following primers: EF1a.qR (5′–TGGAATTTGCCCTTTTTGAG–3′) and mCherry.qF (5′–AAGCGCATGAACTCCTTGAT–3′). Presence of the adenoviral genome was assessed using the following primers: Ad-PB_F (5′–CACCAAAATCAACGGGACTT–3′) and Ad-PB_R (5′–CATGGTGGAGCCTGCTTT–3′). Presence of the AAV-hyPBase vector was assessed using the following primers internal to the hyPBase transgene: PBase-L (5′–CAGCAAGTACGGCATCAAGA–3′) and PBase-R (5′–ACGATGGTCAGCTTGTAGGG–3′). A control primer set was also included to ensure quality of each template: B2MqPCR-L (5′–AGTCGAGAGAAAATGGGCTTGG–3′) and B2MqPCR-R (5′–GACAAGAACCATCCCTTCCACA–3′). The Ad genome (Ad-PB_F+Ad-PB_R) and excision-specific (Ad-PB_Ex_F+Ad-PB_Ex_R) primer pairs were also used to perform quantitative PCR on DNA extracted from liver tissue samples. A Kruskal–Wallis ANOVA test was performed to test the compare signal intensity ranks and identify significant differences between experimental groups with the underlying assumption that the samples within each group have the same distribution.
Results
Design of a hybrid adenoviral-piggyBac vector system
We initially generated a shuttle vector containing a PB transposon transgene that expresses the mCherry fluorescent reporter from a human elongation factor-1 alpha (EF1α) promoter (Fig. 1a). LoxP sites were included flanking the transposon to allow deletion of transgenes within the Ad genome upon activation of Cre recombinase. This design provides the potential to more easily assess the extent of transgene expression specifically from transposons integrated into the host cell genome. Recombineering was used to insert the shuttle vector into a replication-defective ΔE1-E3 Ad5 genome upstream of a cytomegalovirus (CMV) promoter-driven nuclear green fluorescent protein (nGFP) transgene to generate the Ad-T/mCherry vector (Fig. 1a). The recombinant Ad-T/mCherry adenoviral genome was then linearized and packaged in 293FT cells.
Transposition efficiency in cultured cells
We next sought to determine the efficiency of transposon mobilization from the Ad-T/mCherry viral genome in cultured cells. This process is mediated by the PBase enzyme, which can be provided in trans. For the studies described here, we evaluated two versions of PBase—a mammalian codon-optimized version (mPBase)20 and a hyperactive version generated from mPBase by the introduction of seven amino acid substitutions (hyPBase).21 Cell culture experiments to assess transposition and transduction efficiencies of the Ad-T/mCherry virus under various conditions were performed in the immortalized mouse hepatocyte cell line TIB-73.
In order to qualitatively assess whether PBase is capable of excising transposons from adenoviral genomic DNA, we used lentiviral transduction to generate a pooled population of TIB-73 cells stably expressing hyPBase. These cells, as well as a population lacking PBase expression, were transduced with Ad-T/mCherry at an MOI of two. Three days later, genomic DNA was isolated from transduced cells. To assess transposition activity, excision PCR was performed with primers flanking the transposon within the Ad-T/mCherry virus (Fig. 1a). Cycling conditions were such that extension time was insufficient to amplify a product from the intact virus because of the large distance between primers (∼3 kb). Following PBase-mediated transposon excision, however, the primer binding sites are brought into close proximity (248 bp), allowing a product to be amplified. As shown in Fig. 1b, this excision-specific product was detected only in transduced cells expressing hyPBase. A separate 227 bp product amplified from a distant region of the Ad-T/mCherry virus was detected in both samples, verifying successful transduction for each cell population. The excision PCR assay confirmed that PBase is able to mobilize transposons from Ad, an encouraging result given that similar experiments using SBase failed to detect transposition from a linear Ad genome.8
We next indirectly measured transposon integration efficiency by determining the prevalence of stable mCherry expression in cells transduced with Ad-T/mCherry. Parental, mPBase-expressing, and hyPBase-expressing TIB-73 cells were transduced with Ad-T/mCherry at MOIs of 10 and 20. We also tested the efficacy of delivering hyPBase with an adeno-associated viral serotype 9 construct (AAV-hyPBase). In addition to the experiments with stable cell lines, parental TIB-73 cells were co-transduced with AAV-hyPBase (MOI 10,000 or 100,000) and Ad-T/mCherry (MOI 10 or 20). Following transduction, cells were passaged for 4 weeks (8 passages), at which point mCherry expression was analyzed using flow cytometry. No expression of mCherry was detected in parental TIB-73 cells transduced with Ad-T/mCherry after eight passages. By contrast, all conditions having PBase displayed some degree of expression (Fig. 2b). mCherry was detected in ∼8% of hyPBase-expressing cells transduced with Ad-T/mCherry at an MOI of 20, the highest efficiency observed. The degree of stable expression was lower when hyPBase was delivered by AAV; cells co-transduced with AAV-hyPBase and Ad-T/mCherry at MOIs of 100,000 and 20, respectively, displayed transposition efficiencies of approximately 1%.
FIG. 2.

Ad-PB transposition efficiency in cultured cells. TIB-73 cells were transduced with Ad-PB-mCherry (a) at multiplicity of infections (MOIs) of 10 and 20 in triplicate. Five groups of cells were used: parental, cells stably expressing mPBase or hyPBase, and cells co-transduced with AAV-hyPBase at MOIs of 10,000 and 100,000. (b) After 4 weeks of passaging (8 separate passages), the percentage of mCherry-expressing cells was quantified by flow cytometry. Percentages are plotted for each condition, with error bars representing 95% CI. (c) The fold increase in mCherry-expressing cells relative to control parental cells is shown for each treatment group (error bars=95% CI). Quantitative PCR was performed to estimate the amount of (d) adenoviral genomes or (e) transposons remaining in each cell population after 1,2,3 or 7 passages; primer binding sites are indicated in panel (a) with black and gray arrows, respectively. In each case, the fold-enrichment was calculated relative to TIB-73 parental cells at passage 7 (error bars=SEM). AAV, adeno-associated virus.
Prior characterizations have demonstrated that the transposition efficiency of cut-and-paste transposon systems (such as PB) is significantly impacted by the expression level and relative activity of the transposase enzyme, as well as the copy number of donor transposons.14,20 Consistent with what has been reported for PB, we observed an increase in the frequency of stable transgene expression with increasing transposon copy number (i.e., Ad-T/mCherry MOI 10 vs. 20). Similarly, increasing the expression (AAV-hyPBase MOI 10,000 vs. 100,000) or activity (mPBase vs. hyPBase) of the PBase enzyme resulted in higher levels of stable mCherry expression (Fig. 2c). These results indicate that the integration activity of the Ad-PB system can be improved by increasing both the transposon and transposase components, suggesting that additional optimization may provide further increases in stable transduction efficiency.
In order to gain insight into the dynamic process of stable transduction with Ad-PB, we extracted genomic DNA from TIB-73 cells at passages one, two, three, and seven following transduction with Ad-T/mCherry. Quantitative real-time PCR was performed with primers designed to amplify products from within the transposon and from a distant region of the adenoviral genome (Fig. 2a). As expected, the PCR signal from the adenoviral genome was lost with increasing passages in parental cells, as well as in cells expressing hyPBase either stably or via AAV co-transduction (Fig. 2d). While the transposon and adenoviral genome PCR signals were lost with similar kinetics in parental TIB-73 cells, hyPBase-expressing cells retained significantly higher transposon signal after seven passages (Fig. 2e). This result is consistent with hyPBase-mediated transfer of the transposon transgene from the adenoviral genome, which is ultimately lost, to the cellular genome, where it is maintained. Together with the detection of transposon excision (Fig. 1b) and maintenance of mCherry fluorescence (Fig. 2b), these results suggest that long-term transgene expression is achieved via a PBase-mediated transposition mechanism.
Transposition efficiency in vivo
Experiments in TIB-73 cells indicated that the Ad-PB system could achieve long-term, stable transgene expression in a modest percentage of cells. We next sought to determine the efficiency of gene transfer in hepatocytes in vivo. Based on our cell culture experiments, we speculated that accumulation of hyPBase in the nucleus prior to delivery of the Ad-T/mCherry virus would facilitate the most efficient PB transposition activity (Fig. 2b and c). To achieve this in vivo, we delivered 1×1012 VG of AAV-hyPBase to six immunocompetent mice via tail vein injection. Based on prior work, we anticipated that peak expression of the hyPBase enzyme would occur roughly 10 days postinjection.22 Therefore, mice received a second tail vein injection containing 1×108 PFU of the Ad-T/mCherry virus at this time point. A control cohort lacking hyPBase was generated by injecting 15 immunocompetent mice with 1×108 PFU of the Ad-T/mCherry virus only.
In the absence of transposition, the Ad-T/mCherry virus should behave like other recombinant Ads by driving transgene expression that fades over a period of several weeks. To verify this trend, livers were collected from control mice at various time points following adenoviral injection. Following formalin fixation and paraffin embedding, liver sections were subjected to immunohistochemistry with a monoclonal antibody against mCherry. As expected, nearly all hepatocytes expressed the transgene two weeks after injection (Supplementary Fig. S1; Supplementary Data are available online at www.liebertpub.com/hum). Expression was lost at varying rates in independent animals—mCherry staining was nearly absent by 8 weeks postinjection in some mice, while others had lingering expression in a minority of cells up to 20 weeks postinjection (Supplementary Fig. S1). This variability may result from differences in the dynamic rates of immune-mediated adenoviral clearance in individual mice.
In contrast to what was observed for control animals, mice that received AAV-hyPBase prior to Ad-T/mCherry injection appeared to maintain mCherry expression in a large percentage of hepatocytes over time (Supplementary Fig. S1). The difference between cohorts was most pronounced at 20 weeks postinjection with Ad-T/mCherry, at which point mCherry expression in control mice had decreased to near-background levels (Fig. 3a–d). While occasional control livers displayed expression in up to 10% of cells (Fig. 3d), most had only rare mCherry-positive cells remaining at the 20-week time point (Fig. 3a–c). Overall, quantitative colorimetric analysis of images acquired from immunostained liver sections did not detect a significant difference between these sections and those collected from mice not injected with the Ad-T/mCherry virus (Fig. 3i). Contrastingly, this analysis revealed that mice receiving both AAV-hyPBase and Ad-T/mCherry (Fig. 3e–h) retained mCherry expression in a significantly higher percentage of cells (Fig. 3i). In fact, the proportion of positively stained hepatocytes in these mice was not significantly different from what was observed for liver sections harvested just two weeks after Ad-T/mCherry injection (Fig. 3i).
FIG. 3.

Ad-PB transposition efficiency in vivo. Immunohistochemistry detecting mCherry in mouse liver sections at 20 weeks posttransduction with Ad-T/mCherry, in the absence (a–d) or presence (e–h) of AAV-hyPBase. (i) Percentage of hepatocytes expressing mCherry at 2 or 20 weeks posttransduction. Four images were captured for each liver and quantified computationally. A standard box-and-whiskers plot depicts the 25–75% percentile values (box) along with the mean (line). The maximum and minimum values are represented by the whisker plots extending above and below each box.
The frequent, stable expression of mCherry expression in vivo was somewhat unexpected given the efficiency we observed in cell culture assays (Fig. 2). To rule out the possibility of enhanced adenoviral genome maintenance in mice pretreated with AAV-hyPBase, we performed quantitative PCR (qPCR) analysis on DNA from liver samples harvested at early (6–8 weeks) or late (20 weeks) time points following viral injection. The qPCR signal from the recombinant adenoviral genome was significantly lower after 20 weeks in mice that received both AAV-hyPBase and Ad-T/mCherry viruses when compared with those receiving only Ad-T/mCherry (Fig. 4a). This result indicates that stable transgene expression in vivo does not come from retained Ad. As expected, quantitative detection of transposon excision from the adenoviral genome shows signal only in the mice that received both transposon (Ad-T/mCherry) and transposase (AAV-hyPBase) (Fig. 4b and c).
FIG. 4.

Molecular analysis of liver samples from mice treated with AAV-hyPBase and Ad-T/mCherry. Quantitative real-time PCR analysis was performed on genomic DNA isolated from liver biopsies harvested from control mice (Ad-T/mCherry) or experimental mice (Ad-T/mCherry, AAV-hyPBase) at both early and late time points to detect total recombinant adenoviral genome (a) or adenoviral genomes that have undergone transposon excision (b). In some cases, DNA samples from nonadjacent liver sections were isolated from the same animal and treated as independent samples (indicated by brackets). A Kruskal–Wallis test showed that the adenoviral genome signal was significantly reduced in the Ad-T/mCherry, AAV-hyPBase livers after 20 weeks when compared with the Ad-T/mCherry samples at 6 weeks (a). As expected, transposon excision is specific to the Ad-T/mCherry, AAV-hyPBase livers (b). (c) A representative gel image shows the corresponding PCR products. The molecular marker indicates size in base pairs.
The immunohistochemical staining and molecular analyses suggest that transposase-mediated transfer of the mCherry transgene from the Ad to the cellular genome occurred to provide stable expression over long time periods. In this case, inserted PB transposons should be present in cellular genomic DNA from livers of mice injected with both AAV-hyPBase and Ad-T/mCherry viruses. To verify the presence of integrated transposons, we modified our recently developed ligation-mediated PCR (LM-PCR) method for detecting SB transposon insertions in normal somatic mouse tissues23 to allow detection of inserted PB transposons.
Genomic DNA was extracted from 20-week postinjection liver samples collected from three mice that received both AAV-hyPBase and Ad-T/mCherry viruses and three control mice that received only Ad-T/mCherry. One normal liver sample collected from a mouse not receiving either virus was also included as a negative control to evaluate the specificity of our method. Barcoded amplicon libraries were prepared for each of the seven samples using a previously described method.19 Briefly, genomic DNA was sheared, end-repaired, and ligated to a double-stranded adaptor. Two rounds of PCR were then performed in which the second amplification added the barcodes and adaptor sequences for direct amplicon sequencing on the Illumina platform. Following sequencing, the Integration Analysis System24 was used to parse, trim, and map transposon junctions to the mouse reference genome. The analysis pipeline reported junctions beginning with a TTAA sequence (the recognition site for PB integration) that were supported by at least 50 independent reads. We identified a total of 135 PB transposon integration sites that met these criteria in the three samples taken from mice injected with both AAV-hyPBase and Ad-T/mCherry (Fig. 5 and Supplementary Table S1).
FIG. 5.
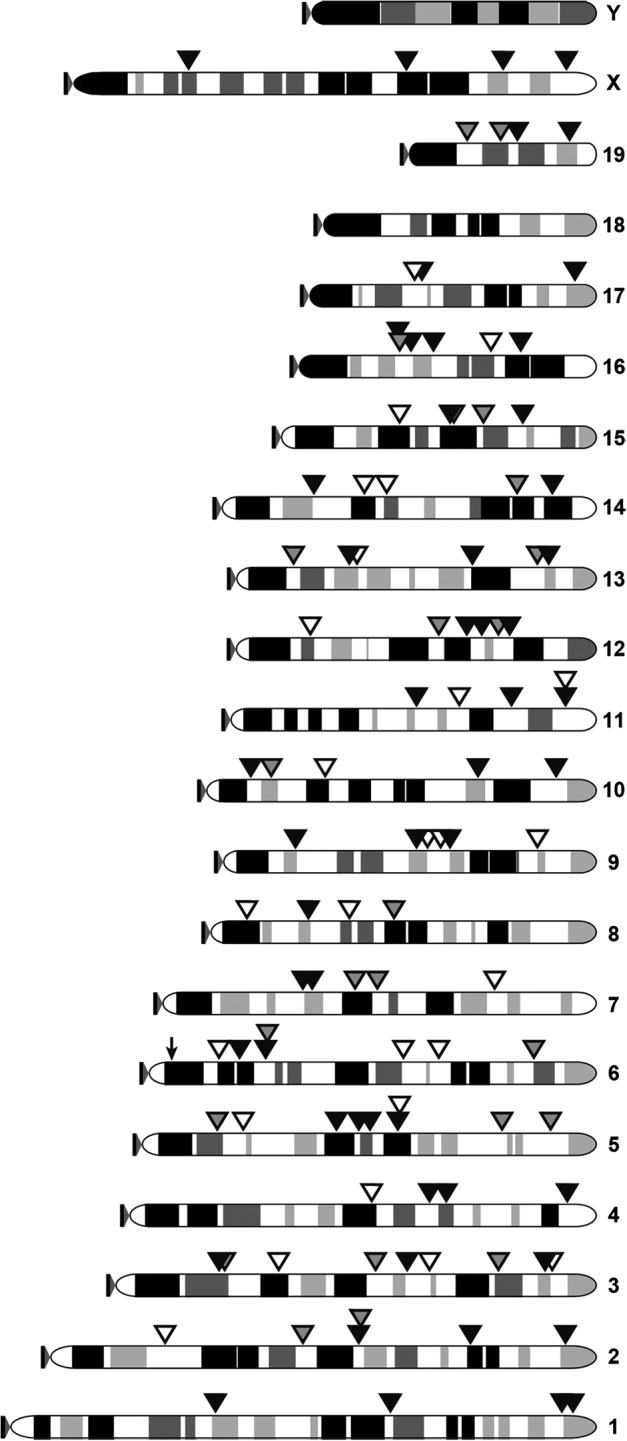
PB transposon insertion sites identified in mice receiving AAV-hyPBase and Ad-T/mCherry. A total of 135 integration sites were identified in three liver samples taken from mice injected with both AAV-hyPBase and Ad-T/mCherry (filled triangles). A single background event was detected in a control sample taken from a mouse that received only the Ad-T/mCherry virus (arrow).
A single transposition event was detected in one control sample that received only the Ad-T/mCherry virus, indicating the presence of a low but detectable background signal for the analytical methods that were utilized. Based on a random integration profile, ∼37.54% of insertions would be expected within RefSeq genes, with 2.46% and 35.08% in exons and introns, respectively. In contrast, 59.26% (80/135) of the insertions that we detected were within genes, with 6.67% (9/135) located in exons. This observed enrichment for genic insertions is consistent with the known integration site selection bias of the PB system.25–27 Also consistent is our observation that another 8.15% (11/135) of insertions occurred within 5 kb upstream of a transcription start site, which is higher than the 4.59% expected for random distribution. These results confirm that PBase-mediated transposition from adenoviral DNA to cellular genomic DNA occurs in vivo.
Discussion
Here we have described the development and characterization of a hybrid adenoviral-PB gene delivery system (Ad-PB) capable of stable transgene integration in cultured cells and in vivo. When administered to wild-type mice via standard tail vein injection, this system achieved stable expression of a reporter transgene in ∼20–40% of liver cells that was maintained for at least 5 months. This in vivo efficiency is among the highest described to date for a hybrid Ad gene delivery system. A hybrid Ad-lentiviral vector was reported to achieve comparable efficiency in vivo,9 although that study's use of immunocompromised mice and an experimental endpoint of four weeks postinfection make a direct comparison to our results difficult. While other strategies based on genomic integration of Ad-encoded transgenes for stable expression typically provide less than 1% efficiency,5–8 two independent approaches utilizing nonintegrative methods for stable maintenance of Ad-delivered transgenes have shown recent success in vivo,28,29 though efficiencies were not directly calculated.
We noted a substantial difference in the rate of stable transgene expression between cell culture and in vivo experiments. While additional characterization is required to better understand the behavior of the Ad-PB system, our cell culture experiments provide some insight into a possible explanation for this outcome. The TIB-73 hepatocytes that we used proliferate at a high rate and thus will begin to rapidly dilute the intracellular recombinant adenoviral genomes after infection because of distribution among daughter cells during mitosis. Consequently, the transposon copy number delivered by the Ad is diminished rapidly, limiting the number of substrates available for transposition. Similarly, cell division dilutes the AAV-hyPBase genomes present within each cell (Supplementary Fig. S2), leading to a rapid decline in hyPBase protein levels with cell passaging. Our experiments show that suboptimal levels of PBase or transposon substrate can cause a substantial reduction in the efficiency of long-term transgene expression. Because of the relatively low rate of hepatocyte proliferation in vivo, as compared with that of cultured cells, viral DNA does not get diluted as quickly (Supplementary Fig. S2), and therefore delivery of a similar viral titer in vivo is likely to result in a more prolonged incubation period in which both the PBase enzyme and transposon substrates are present at sufficient levels for efficient transposition to occur.
Host immune response is another complicating factor in comparing our cell culture and in vivo experiments. Transduction of mouse hepatocytes in vivo with recombinant E1-deleted Ad has been shown to elicit a strong immune response that leads to loss of transgene expression over the course of several weeks.30 We observed a similar trend in control mice injected with Ad-T/mCherry only (Fig. 3a–d and Supplementary Fig. S1). In contrast, mice that received both AAV-hyPBase and Ad-T/mCherry maintained stable transgene expression in a large percentage of hepatocytes (Fig. 3e–h). Interestingly, the abundance of recombinant adenoviral genome appears to be reduced over time in the presence of hyPBase (Fig. 4a).
We also demonstrated that PB-mediated integration of the transgene is occurring in the livers of these mice (Fig. 5); however, the extent to which the immune system influences the rate of long-term expression in our experiment is unclear at present. One possibility is that transposon excision from the adenoviral genome by PBase leads to enhanced degradation of the remaining Ad DNA, in which case a blunted immune response would be expected because of a decreased abundance of viral proteins and/or transcripts encoded by the recombinant Ad vector. The resultant prolonged presence of Ad-T/mCherry could promote increased transgene expression both directly from the adenoviral genome and by increasing the duration of the PBase enzyme's exposure to transposon substrates for integration into the cellular genome. This mechanism could also potentially be capitalized on by adapting the system to use high-capacity, helper-dependent adenoviral vectors. Because all adenoviral genes have been deleted from these vectors, they induce significantly attenuated immune responses in vivo.31 The influence of the AAV vector itself is also unclear, given that control mice received only the Ad-T/mCherry construct. Although its presence is not expected to contribute to the observed enhancement of long-term transgene expression in vivo, further characterization will be required to make a definitive conclusion regarding any mechanistic role for this factor. More work is also required to determine the impact of host immune response on the Ad-PB system's performance in vivo.
Two significant advantages of Ad-PB are its simplicity and flexibility. Ad provides an easily produced vector with broad tropism, and PB facilitates stable integration. All that is required for integration is expression of PBase, which can be incorporated into an animal system (e.g., transgenic, knock-in) or transiently coexpressed (e.g., using the AAV-hyPBase virus). Because of its simplicity, the Ad-PB system could easily be modified to accommodate a variety of transgene designs. Unlike the analogous hybrid Ad-SB system, which depends on recombinase-mediated circularization of transposon substrates for transposition to occur,8 Ad-PB transposition occurs directly from the adenoviral genome. This feature simplifies the process of stable transduction and provides an opportunity to design more complex transgenes, for instance, those that can be induced or inactivated upon expression of a recombinase enzyme. In addition to their relatively low immunogenicity, the use of high-capacity, helper-dependent adenoviral vectors would allow the design of transgenes up to ∼25 kb in length.
PBase is able to efficiently mobilize transposons of this size,15 and so the Ad-PB system has the potential to provide stable expression of much larger transgenes than can be integrated with systems using hybrid lentiviral, AAV, or SB vectors. Alternatively, multiple small transposons could be placed within a high-capacity Ad. This approach could improve the efficiency of stable transgene integration by increasing the number of transposon substrates delivered to each cell without increasing viral titer. Doubling the Ad-T/mCherry viral titer (and therefore the number of transposon transgenes) in our cell culture experiments increased the rate of stable transgene expression 2–3-fold in cells with hyPBase (Fig. 2b). The achievable MOI can be a limiting factor for in vivo adenoviral transductions. Increasing the number of delivered transposon substrates without changing MOI may therefore be a particularly effective way to improve the efficiency of stable gene transfer in vivo.
Supplementary Material
Acknowledgments
We would like to thank Yulong Zhang for his assistance with performing tail vein injections of recombinant viruses.
Author Disclosure Statement
The authors have no conflicts of interest related to this work to disclose.
References
- 1.Volpers C, Kochanek S. Adenoviral vectors for gene transfer and therapy. J Gene Med 2004;6 Suppl 1:S164–S171 [DOI] [PubMed] [Google Scholar]
- 2.Harui A, Suzuki S, Kochanek S, et al. . Frequency and stability of chromosomal integration of adenovirus vectors. J Virol 1999;73:6141–6146 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Stephen SL, Montini E, Sivanandam VG, et al. . Chromosomal integration of adenoviral vector DNA in vivo. J Virol 2010;84:9987–9994 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Muther N, Noske N, Ehrhardt A. Viral hybrid vectors for somatic integration—are they the better solution? Viruses 2009;1:1295–1324 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ehrhardt A, Yant SR, Giering JC, et al. . Somatic integration from an adenoviral hybrid vector into a hot spot in mouse liver results in persistent transgene expression levels in vivo. Mol Ther 2007;15:146–156 [DOI] [PubMed] [Google Scholar]
- 6.Goncalves MA, Holkers M, Van Nierop GP, et al. . Targeted chromosomal insertion of large DNA into the human genome by a fiber-modified high-capacity adenovirus-based vector system. PLoS One 2008;3:e3084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Soifer H, Higo C, Kazazian HH Jr., et al. . Stable integration of transgenes delivered by a retrotransposon-adenovirus hybrid vector. Hum Gene Ther 2001;12:1417–1428 [DOI] [PubMed] [Google Scholar]
- 8.Yant SR, Ehrhardt A, Mikkelsen JG, et al. . Transposition from a gutless adeno-transposon vector stabilizes transgene expression in vivo. Nat Biotechnol 2002;20:999–1005 [DOI] [PubMed] [Google Scholar]
- 9.Kubo S, Kataoka M, Tateno C, et al. . In vivo stable transduction of humanized liver tissue in chimeric mice via high-capacity adenovirus-lentivirus hybrid vector. Hum Gene Ther 2010;21:40–50 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Park F. Lentiviral vectors: are they the future of animal transgenesis? Physiol Genomics 2007;31:159–173 [DOI] [PubMed] [Google Scholar]
- 11.Picard-Maureau M, Kreppel F, Lindemann D, et al. . Foamy virus—adenovirus hybrid vectors. Gene Ther 2004;11:722–728 [DOI] [PubMed] [Google Scholar]
- 12.Russell RA, Vassaux G, Martin-Duque P, et al. . Transient foamy virus vector production by adenovirus vectors. Gene Ther 2004;11:310–316 [DOI] [PubMed] [Google Scholar]
- 13.Trobridge G, Russell DW. Cell cycle requirements for transduction by foamy virus vectors compared to those of oncovirus and lentivirus vectors. J Virol 2004;78:2327–2335 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Wu SC, Meir YJ, Coates CJ, et al. . piggyBac is a flexible and highly active transposon as compared to sleeping beauty, Tol2, and Mos1 in mammalian cells. Proc Natl Acad Sci U S A 2006;103:15008–15013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Li MA, Turner DJ, Ning Z, et al. . Mobilization of giant piggyBac transposons in the mouse genome. Nucleic Acids Res 2011;39:e148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Pan H, Mostoslavsky G, Eruslanov E, et al. . Dual-promoter lentiviral system allows inducible expression of noxious proteins in macrophages. J Immunol Methods 2008;329:31–44 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Zhou Q, Brown J, Kanarek A, et al. . In vivo reprogramming of adult pancreatic exocrine cells to beta-cells. Nature 2008;455:627–632 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Liu P, Jenkins NA, Copeland NG. A highly efficient recombineering-based method for generating conditional knockout mutations. Genome Res 2003;13:476–484 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Rogers LM, Olivier AK, Meyerholz DK, et al. . Adaptive immunity does not strongly suppress spontaneous tumors in a sleeping beauty model of cancer. J Immunol 2013;190:4393–4399 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Cadinanos J, Bradley A. Generation of an inducible and optimized piggyBac transposon system. Nucleic Acids Res 2007;35:e87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Yusa K, Zhou L, Li MA, et al. . A hyperactive piggyBac transposase for mammalian applications. Proc Natl Acad Sci USA 2011;108:1531–1536 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Inagaki K, Fuess S, Storm TA, et al. . Robust systemic transduction with AAV9 vectors in mice: efficient global cardiac gene transfer superior to that of AAV8. Mol Ther 2006;14:45–53 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Riordan JD, Drury LJ, Smith RP, et al. . Sequencing methods and datasets to improve functional interpretation of sleeping beauty mutagenesis screens. BMC Genomics 2014;15:1150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Brett BT, Berquam-Vrieze Ke, Nannapaneni K, et al. . Novel molecular and computational methods improve the accuracy of insertion site analysis in sleeping beauty-induced tumors. PLoS One 2011;6:e24668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ammar I, Gogol-Doring A, Miskey C, et al. . Retargeting transposon insertions by the adeno-associated virus Rep protein. Nucleic Acids Res 2012;40:6693–6712 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.De Jong J, Akhtar W, Badhai J, et al. . Chromatin landscapes of retroviral and transposon integration profiles. PLoS Genet 2014;10:e1004250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Liang Q, Kong J, Stalker J, et al. . Chromosomal mobilization and reintegration of sleeping beauty and PiggyBac transposons. Genesis 2009;47:404–408 [DOI] [PubMed] [Google Scholar]
- 28.Gil JS, Gallaher SD, Berk AJ. Delivery of an EBV episome by a self-circularizing helper-dependent adenovirus: long-term transgene expression in immunocompetent mice. Gene Ther 2010;17:1288–1293 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Voigtlander R, Haase R, Muck-Hausl M, et al. . A novel adenoviral hybrid-vector system carrying a plasmid replicon for safe and efficient cell and gene therapeutic applications. Mol Ther Nucleic Acids 2013;2:e83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Yang Y, Nunes FA, Berencsi K, et al. . Cellular immunity to viral antigens limits E1-deleted adenoviruses for gene therapy. Proc Natl Acad Sci USA 1994;91:4407–4411 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Muruve DA, Cotter MJ, Zaiss AK, et al. . Helper-dependent adenovirus vectors elicit intact innate but attenuated adaptive host immune responses in vivo. J Virol 2004;78:5966–5972 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.