

Summary
Background
In response to DNA double-strand breaks (DSBs), eukaryotic cells rapidly phosphorylate histone H2A isoform H2AX at a C-terminal serine (to form γ-H2AX) and accumulate repair proteins at or near DSBs. To date, these events have been defined primarily at the resolution of light microscopes, and the relationship between γ-H2AX formation and repair protein recruitment remains to be defined.
Results
We report here the first molecular-level characterization of regional chromatin changes that accompany a DSB formed by the HO endonuclease in Saccharomyces cerevisiae. Break induction provoked rapid γ-H2AX formation and equally rapid recruitment of the Mre11 repair protein. γ-H2AX formation was efficiently promoted by both Tel1p and Mec1p, the yeast ATM and ATR homologs; in G1-arrested cells, most γ-H2AX formation was dependent on Tel1 and Mre11. γ-H2AX formed in a large (ca. 50 kb) region surrounding the DSB. Remarkably, very little γ-H2AX could be detected in chromatin within 1–2 kb of the break. In contrast, this region contains almost all the Mre11p and other repair proteins that bind as a result of the break.
Conclusions
Both Mec1p and Tel1p can respond to a DSB, with distinct roles for these checkpoint kinases at different phases of the cell cycle. Part of this response involves histone phosphorylation over large chromosomal domains; however, the distinct distributions of γ-H2AX and repair proteins near DSBs indicate that localization of repair proteins to breaks is not likely to be the main function of this histone modification.
Introduction
DSBs threaten the replication and stable transmission of the genome, and all organisms have evolved strategies that respond to and repair them. In eucaryotes, this response is centered around phosphatidylinositol-3 kinase-like kinases (PI3KKs). These include the mammalian ATM (ataxia-telangiectasia-mutated) and ATR (ATM-and Rad3-related) proteins, the corresponding budding yeast proteins Tel1p and Mec1p, and DNA-PK (DNA-dependent protein kinase), which is found in mammals but not in yeast. Upon damage recognition, these PI3KKs initiate a kinase cascade that ultimately results in activation of DNA damage repair systems, cell cycle blockage until damage is repaired, and induction of cell death when DNA damage is not repaired [1–3].
Studies in mammals and in yeast have provided considerable information about the nature and control of the downstream response that occurs on a nucleus- or cell-wide basis. Less is known about the initial recognition of and response to DSBs. DSB recognition both by ATR/Mec1p and by ATM/Tel1p is thought to be mediated by partner proteins that recognize break-associated DNA or chromatin structures. ATRIP/Ddc2p, a tight binding partner of ATR/Mec1p, is thought to bind to damage-associated single-stranded regions [2]. DSB recognition and kinase activation of ATM/Tel1p appears to be mediated through a looser interaction with the mammalian MRE11-RAD50-NBS1 and the yeast Mre11-Rad50-Xrs2 protein complex [1, 4, 5]. This complex, hereafter referred to as the Mre11 complex, has roles in both damage recognition and DSB repair and has been shown to bind DNA ends in vitro [6]. Studies of PI3KK mutants suggest that the main responder to DSB-inducing ionizing radiation (IR) is ATM in mammals but the ATR homolog Mec1p in budding yeast [1, 2, 7]. Although the distinction between mammalian and yeast responses may reflect real differences in intrinsic PI3KK activity or the lesions recognized, differences may also exist between mammals and yeast in terms of the amplification of signals by downstream kinases and in final kinase targets.
Two general strategies have been used to examine the initial DNA damage response. The first uses cytology to determine the intra-nuclear localization of target proteins in response to DSB-generating treatments such as IR. DSB-induced chromatin modifications and repair protein movement to damage sites are detected as concentrated foci of the protein of interest ([8–12] and references within). Although this approach has been fruitful, caution must be used in distinguishing functioning repair assemblages from dead-end, irreparable lesions (for discussion, see [11]). The second strategy, used mainly in budding yeast, examines breaks formed by site-specific endonucleases and has been used to define events at the DNA level [13–17], to examine protein binding to DSBs [4, 12, 18–27], and to examine the response of the DNA damage checkpoint system to a single DSB [28–32]. This latter approach has advantages in that the site of damage is known, signals emanate from a single lesion, and complications due to differential damage sensitivity in mutants are mostly avoided.
These and other studies have identified three types of events that occur soon after DSB formation and that therefore must be considered part of the initial response. The first type, observed to date only in mammals, involves ATM, which undergoes autophosphorylation and a dimer-to-monomer transition within minutes of damage formation, but which does not appear to remain at DNA damage sites [33]. The second type, seen in both yeast and mammals, involves movement of the Mre11 complex and other checkpoint and repair proteins into foci soon after DNA damage [8, 9, 12, 19, 34–37]. The third type, seen in many organisms, involves the in situ modification of both histone and non-histone chromatin proteins [38–40]. The most dramatic of these is the phosphorylation of H2AX, a histone H2A isoform that makes up 5%–10% of the histone H2A in mammals but about 95% in yeast [41]. The yeast majority isoform will be called H2AX in this paper. H2AX phosphorylation occurs at a serine residue in a consensus PI3KK target sequence (SQE) in the C-terminal tail [38]. γ-H2AX, as the phosphorylated form will be called here, forms within minutes of DSB formation and can cover large (thousands to millions of base pairs) regions of chromatin [42, 43].
DNA repair proteins often colocalize with γ-H2AX foci [44], although such colocalization can be absent at early times after damage formation [45]. Mice lacking H2AX suffer elevated genome instability and show modest defects in repair, recombination, and DSB-induced cell cycle arrest (reviewed in [46, 47]); similar defects are seen in S. cerevisiae mutants in which alanine replaces the target serine (S129) [43, 48]. These and other findings have led to the suggestion that γ-H2AX helps recruit damage response and repair proteins to break sites. For the most part, however, the spatial and temporal relationships among DNA damage, γ-H2AX formation, and repair/response protein binding have been examined only by light microscopy. The low spatial resolution afforded by this technique precludes firm conclusions about relationships among these entities.
To examine the early response to DSBs at greater resolution, we used chromatin immunoprecipitation to probe γ-H2AX formation and Mre11 repair protein recruitment at a unique DSB in the Saccharomyces cerevisiae genome. We examined not only the timing and spatial distribution of γ-H2AX and Mre11 formation and binding but also the relative contribution of the two yeast PI3KK homologs Tel1p and Mec1p to these events. We show that H2AX phosphorylation and Mre11p binding are among the earliest events to occur at DSB sites, but their nonoverlapping distributions argue against a direct role for γ-H2AX in targeting repair proteins to break ends. We also provide evidence that Mec1p and Tel1p play different roles in the initial DSB response in different stages of the cell cycle.
Results
Rapid Mre11 Binding and γ-H2AX Formation at the MAT Double-Strand Break
We examined H2AX phosphorylation and Mre11p recruitment at an HO endonuclease-catalyzed DSB formed at a unique site in the MAT locus [49]. HO gene transcription was controlled by a galactose-inducible promoter, allowing rapid and high-level induction [13]. HO-catalyzed DSBs at MAT are normally repaired by gene conversion from the silent HML and HMR loci about 1 hr after formation [13, 14]. The strains we used contained deletions of both silent loci and thus precluded break repair by recombination. Consequently, the events examined here are expected to reflect the initial DSB response and not events occurring during subsequent break repair.
DSB ends undergo 5′-to-3′ resection, exposing single-stranded regions ending 3′ at the DSB break. We used denaturing gels [14] to monitor HO-cutting at MAT and the extent of resection (Figure 1A). γ-H2AX formation and Mre11p recruitment were measured by chromatin immunoprecipitation (ChIP, see Experimental Procedures). Relative protein occupancy in a region was measured in quantitative multiplex PCR reactions that contained a primer pair specific to regions of interest near the DSB. All reactions also contained a control primer pair specific for sequences 66 kb centromere-proximal to MAT, which allowed us to control for DSB-independent effects on protein occupancy, as well as experiment-to-experiment variation in efficiencies of crosslinking and immunoprecipitation. We monitored events by using two different culture conditions: in unsynchronized growing cells and in cells arrested in G1 by α-mating pheromone. In growing cultures, about two-thirds of cells were in G1 during the first 2 hr after HO induction (determined by microscopy and flow cytometry, not shown).
Figure 1. Single-Strand DNA Formation, γ-H2AX Formation, and Mre11p/Rad51p Binding in Response to a DSB at the MAT Locus.

HO cutting at MAT was induced either in asynchronously growing cells (“cycling,” H1069) or in cells arrested by exposure to α-mating pheromone (“G1,” H1072), and samples were collected at the appropriate time points (see Experimental Procedures).
(A) Map of the region immediately centromere-distal to the MAT DSB site. The map shows the method used to detect DSB formation and 5′-to-3′ resection. DNA was digested with SspI and separated on alkaline agarose gels, and gel blots were hybridized with a single-strand probe specific to the unresected strand (ss probe). HO-cut and uncut chromosomes produce 0.9 kb and 1.2 kb fragments, respectively; 5′-to-3′ resection past SspI sites eliminates cutting at these sites and thus produces larger SspI fragments detected by the probe.
(B) Southern blot illustrating this analysis. Bands labeled r1 through r6 are the products of resection through SspI sites 0.9, 1.6, 3.5, 4.7, 5.9, and 6.5 kb from the HO-cut site; * denotes a cross-hybridizing band that was used as a loading control.
(C) Quantitative analysis of the blot in (B) and of one replicate. (Filled squares, filled circles) Percent of chromosomes with DSBs; (open squares, open circles) percent of DSB-containing chromosomes that have had at least 0.9 kb resected. (filled squares, open squares) Data from cycling cells; (filled circles, open circles) data from G1-arrested cultures.
(D) Examples of multiplex PCR products of ChIP reactions used to determine relative amounts of γ-H2AX formed and of Mre11p bound near the MAT DSB. All reactions contained primer pairs for a sequence 66 kb from the break (“con”) and experimental sequences (“dsb”) 5.1 kb to the left for γ-H2AX and 0.02 kb to the right for Mre11p. (+) ChIP prepared with the indicated primary antibody; (−) treated identically but without primary antibody; (DNA) PCR reactions for which genomic DNA was used.
(E) Timing of γ-H2AX formation (filled squares, open squares) and Mre11p binding (filled circles, open circles) in cycling (filled symbols) and G1-arrested (open symbols) cultures. Data are from panels in (D) and 3–4 replicate experiments. For normalizing DSB-specific relative ChIP values (dsb/control band intensity ratios, see Supplemental Experimental Procedures), the maximum value obtained for each time course was set to unity. Symbols and error bars report the average and standard deviation for these normalized values.
DSB induction was rapid and efficient. In growing cultures, the MAT locus was cut in more than 90% of cells within 15 min of galactose addition. DSB induction was less rapid in G1-arrested cells but had still occurred in more than 90% of cells by 30 min after HO induction (Figures 1B and 1C). DSB end resection has been reported to be slow (about 1–2 nucleotides/sec [28]) and to initiate asynchronously after HO-catalyzed DSB formation in both cycling cells and G1-arrested cultures [15]. We observed similarly slow and asynchronous end resection. At times when both Mre11p and γ-H2AX ChIP had reached maximum levels, less than 10% of break ends had resected past an SspI site 0.9 kb from the HO-cut site, although a larger fraction is likely to have undergone less-extensive resection.
DSBs were followed closely by Mre11p binding and H2AX phosphorylation. In cycling cells, Mre11p and γ-H2AX ChIP signals were present above background by 15 min after HO induction (Figure 1D). Both achieved near-maximal levels by 30 min (Figure 1E) and did not significantly increase with further HO expression (up to 6 hr, data not shown). G1-arrested cells showed similar timing of Mre11p binding and γ-H2AX formation, with significant ChIP levels detectable by 30 min, and maximum levels were reached by 1 hr after HO induction (Figure 1E). This confirms previous findings that H2AX phosphorylation occurs shortly after DSB formation and also indicates that Mre11 complex binding is an equally rapid if not more rapid response [45]. It stands in contrast to the binding kinetics seen for the recombination protein Rad51. In cycling cells, break-associated Rad51 ChIP signal is not detected until 30 min, and it increases for several hours after HO induction [18], whereas in G1-arrested cells, Rad51p association is greatly reduced [50]. Similar findings have been made in cytological studies of DSB-induced focus formation by Mre11p, Rad51p, and other DNA repair proteins [12, 19, 51].
Contrasting Distributions of γ-H2AX and of Repair Proteins
Quantitative studies have estimated that enough γ-H2AX is formed per break to cover thousands of kb in mammalian cells and about 5 kb in S. cerevisiae [38, 41]. We determined the actual region over which break-induced histone phosphorylation occurs in budding yeast by quantitative multiplex ChIP (Figure 2). γ-H2AX was present in a 40–50 kb region around the MAT DSB site at the earliest time it could be reliably detected in both asynchronous cycling cells (15 min) and G1-arrested cells (30 min). The amount of γ-H2AX within this region increased and its distribution broadened by 60 min after HO induction (Figure 2A,B), but neither changed substantially at later times (data not shown). At all times, the greatest enrichment for γ-H2AX occurred 3–5 kb on either side of the break, but levels were elevated above background as much as 20–30 kb away. Given the estimates cited above, our yeast data are consistent with previous suggestions that one-fifth to one-tenth of the H2AX in sequences surrounding a DSB are phosphorylated [52]. However, γ-H2AX ChIP levels were conspicuously low in sequences 1–2 kb to either side of the break. In growing cells, sequences within 1 kb of the DSB displayed no enrichment over background (Figure 2A). In G1-arrested cells, the break-adjacent region of γ-H2AX reduction was somewhat narrower, but the overall picture was similar, with elevated γ-H2AX levels up to 30 kb from the DSB (Figure 2B).
Figure 2. γ-H2AX Formation and Repair Protein Binding Induced by the MAT DSB.

(A) Distribution of γ-H2AX in cycling cells (H1069). DSB-specific relative ChIP values (band intensity ratios, indicated experimental locus/control locus 66 kb from the DSB; see Supplemental Experimental Procedures) are for samples taken 15 min (dotted line), 30 min (dashed line), and 60 min (solid line) after HO induction.
(B) Distribution of γ-H2AX in G1-arrested cells (H1072). DSB-specific relative ChIP values are for samples taken 30 min (dashed line) and 60 min (solid line) after HO induction, as in (A).
(C) Distribution of Mre11p (this work) and Rad51p (reproduced from Sugawara et al. [18] for comparative purposes) bound in the region around the MAT DSB. DSB-specific relative ChIP values for Mre11p (left-hand Y axis; filled circles, cycling cells; open circles, G1-arrested cells) and for Rad51p (closed triangles, right-hand Y axis) are from samples taken 60 min after HO induction.
A very different picture emerged when the same cross-linked chromatin was probed for Mre11p. Consistent with findings that the Mre11 complex binds to DNA ends in vitro, Mre11p ChIP signals were greatest adjacent to the DSB and decreased rapidly with increasing distance from the break site (Figure 2C). Similar maximum ChIP levels, but a somewhat narrower distribution, were seen in G1-arrested cells. A similar distribution of the homologous recombination protein Rad51 was also seen at this time ([18], reproduced in Figure 2C). Given the limits of resolution of ChIP (an average shear size of 0.5 kb, but many longer DNA molecules are present), our results are consistent with these repair proteins being bound at, or within a few hundred nucleotides of, the DSB. Thus, in the first 45 min to 1 hr after DSB formation, the distributions of γ-H2AX and of repair proteins are the inverse of each other.
The very limited break end resection observed makes it unlikely that this could be responsible for the low γ-H2AX levels seen in sequences within 2 kb of the DSB because the ChIP patterns observed most likely reflect a substantially greater fraction of cells than have undergone this much resection (less than 10% have resected 0.9 kb or more when γ-H2AX formation is maximal; see Figure 1). However, it remained possible that other mechanisms of nucleosome removal resulted in the absence of a substrate for γ-H2AX formation in this region. This was tested with ChIP directed against histone H2B, which tightly associates with H2A in nucleosomes and nucleosome precursors [53]. Virtually identical H2B ChIP levels were observed in chromatin from cells before or 1 hr after HO induction (Figure 3). Thus, it appears that the initial break response does not involve substantial histone depletion in sequences near a DSB, at least at the resolution and sensitivity afforded by ChIP. The efficient detection of H2B in break-associated sequences also makes it unlikely that general masking of chromatin is responsible for the reduced γ-H2AX ChIP seen in break-adjacent sequences.
Figure 3. DSB Formation is Not Accompanied by Histone Depletion.
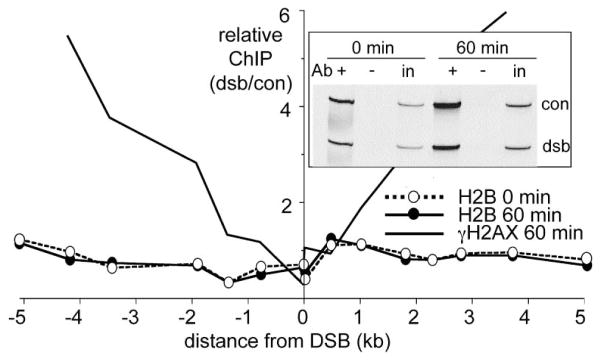
Relative histone H2B ChIP values (band intensity ratios, indicated experimental locus/ control locus 66 kb from the DSB; see Supplemental Experimental Procedures) were measured in samples taken from cycling cells (H1069) before (0 min, open circles) or 60 min after (closed circles) HO induction; a strain in which about half of all histones H2B are tagged with a single HA epitope (H1069) was used. For comparative purposes, relative γ-H2AX ChIP levels in a 60 min sample are reproduced from Figure 2A. (Inset) Representative multiplex PCR reactions with H2B ChIP samples: +, ChIP samples; −, primary antibody omitted; in, 100-fold dilution of input lysate. DSB-specific PCR primers were for sequences 0.02 kb from the DSB.
Different Checkpoint Kinase Requirements for γ-H2AX Formation in Growing and G1-Arrested Cells
Various studies have implicated each of the damage response PI3KKs (ATR, ATM, and DNA-PK in mammals; Mec1p and Telp in yeast) as being responsible for γ-H2AX formation [9, 43, 48, 54–56]. To identify the PI3KK responsible for γ-H2AX formation in our experiments, we examined strains lacking each of the two central kinases (mec1Δ and tel1Δ), as well as strains lacking genes thought to act downstream of these kinases (Figure 4B). In unsynchronized growing cultures, the DSB-specific γ-H2AX signal was eliminated only in mec1Δ tel1Δ double mutants. Mutants lacking kinases that are activated by Mec1p and Tel1p (rad53Δ, chk1Δ, or dun1Δ) displayed a robust γ-H2AX response (Figure 4B), as did mutants (rad17Δ, rad9Δ; data not shown) lacking proteins thought to recruit downstream factors to DSB sites. Consistent with the high levels of γ-H2AX seen in tel1Δ cells, substantial γ-H2AX ChIP occurred in mre11Δ mutants. These data support the view that both the Mec1 (ATR) and Tel1 (ATM) kinases can form γ-H2AX.
Figure 4. Checkpoint Proteins Required for γ-H2AX Formation.

(A) Schematic diagram of functional relationships among DNA damage response kinases. PI3KKs and their binding partners are shown in the top line; target downstream kinases are shown in the second. Arrows indicate the direction in which activating phosphorylation signals can be transmitted.
(B) Both Mec1 and Tel1 kinases can promote γ-H2AX formation in cycling cells. Panels contain products of multiplex PCR reactions on samples taken 60 min after galactose addition; primer pairs 66 (con) and 5.1 (dsb) kb from the break were used. (DNA) PCR with purified genomic DNA. Values below each panel are the dsb/control band intensity ratio. All strains are isogenic to JKM179 (wild-type) with the indicated additional mutations (details in supplementary Table S1).
(C) Most γ-H2AX formation in G1-arrested cells is TEL1 and MRE11 dependent. Strains isogenic to yXW1 (wild-type) were arrested in G1, and HO was induced as described in the Experimental Procedures. All other details are as in panel (B).
(D) Mre11p binding is TEL1 and MEC1 independent. Mre11-ChIP reactions from G1-arrested cells were analyzed as described above with primer pairs 66 (con) and 0.02 (dsb) kb from the break.
Studies in mammalian cells suggest that ATM and ATR make different contributions to the DNA damage response during different cell cycle phases [57]. To test the possibility that the observed dual MEC1/TEL1 dependence of γ-H2AX formation in unsynchronized cells (Figure 4B) reflects differences in Mec1 and Tel1 kinase activity in different cell cycle phases, we measured DSB-induced γ-H2AX formation in G1-arrested cells (Figure 4C). In these cells, the bulk of γ-H2AX formed near the MAT DSB was TEL1 dependent and MEC1 independent, whereas a minor fraction appeared to be MEC1 dependent. DSB-specific γ-H2AX ChIP levels (Figure 4C) and distributions (data not shown) were unaffected in mec1Δ, were reduced 4-fold in tel1Δ, and were reduced to background in mec1Δ tel1Δ mutants. No DSB-specific ChIP signal could be detected in G1-arrested mre11Δ mutant cells, although the relatively high DSB-independent background in these cells (Figure 4C and data not shown) precluded determining whether less DSB-specific γ-H2AX was formed than in tel1Δ mutants. Conversely, G1-phase DSB-specific Mre11 protein binding was Tel1- and Mec1- independent and, in fact, was substantially greater in tel1Δ mutants than in wild-type (Figure 4D). These data indicate that the Tel1 kinase, like its mammalian homolog ATM, can recognize and vigorously respond to DSBs, and this response most likely requires Mre11 complex recognition of DSB ends.
Discussion
This study examined the temporal and spatial distribution of γ-H2AX formation and Mre11p binding, two events previously identified as part of the initial response to DNA double-strand breaks. Our data identify distinct differences between Mre11p recruitment and γ-H2AX formation, as well as differences in the cell cycle-dependence of kinases responsible for γ-H2AX formation. These differences are informative both about mechanisms of DSB recognition and about the initial cellular response to DSBs during different phases of cell growth.
Two aspects of the experimental approach used contributed to these findings. The first is the rapid and synchronous formation of a DSB in the vast majority of cells at a single site in the genome. The second feature, made possible by the use of a unique DSB site, is the use of ChIP to examine a large number of sites in an extended chromosomal region. Both ChIP and cytological approaches have previously been used to examine the DSB response, but the limited coverage of most previous ChIP studies and the limits of resolution of light microscopy precluded a comprehensive molecular-level picture of the events that occur soon after break formation. We anticipate that the future integration of these approaches should greatly enhance understanding of the mechanism of the recognition of and response to DNA damage.
ATM Homolog Tel1p and the Mre11 Complex Respond to DSBs in G1-Arrested Cells
Mammals have a strong G1 damage checkpoint [57], but the importance of this checkpoint in yeast remains in question. Although damage-induced G1/S arrest can occur under certain circumstances [58, 59], past findings have been interpreted as indicating that the S- and G2-phase checkpoints are more robust. Irradiated yeast cells arrest primarily in S phase and at the G2/M boundary, and this arrest is normally mediated by the ATR homolog Mec1p, with little contribution from the ATM homolog Tel1p [60–62]. Furthermore, a single unrepaired DSB in S- or G2-phase yeast cells triggers phosphorylation of the downstream Rad53 kinase but does not do so in G1-phase cells [30]. By contrast, we find that a single DSB formed in G1-phase cells provokes γ-H2AX formation at high levels in a manner that is Tel1 and Mre11 dependent. Thus, like ATM, Tel1p can efficiently recognize a single DSB and mount a robust initial response. We suggest that observed differences between the yeast and mammalian G1-phase damage response are not due to differences in the initial recognition and response to DNA damage but rather are due to differences in transduction of the signal to downstream factors and/or its subsequent amplification. It is worth noting in this regard that Tel1p promotes an efficient S-phase checkpoint in rad50S and sae2 mutants [63] and is also reported to promote an S-phase checkpoint response to phleomycin [64].
Implications of the Differing Distributions of Mre11p and γ-H2AX
At the molecular level, γ-H2AX and break repair proteins occupy distinct and, for the most part, nonoverlapping regions around a DSB. Mre11 ChIP signals were greatest in DSB-adjacent sequences, as are ChIP signals for both single-strand DNA binding protein and the Rad51 recombinase (which also binds single-strand DNA) [18, 19]. By contrast, we find that, as soon as it can be detected, γ-H2AX is present in a broad (ca. 40–50 kb total) region spanning the DSB but is reduced or absent from sequences within 1–2 kb of the break itself. Similar γ-H2AX distributions are seen at DSBs induced elsewhere in the genome, indicating that this pattern is not specific to the MAT locus (J.-A. Kim and J.E.H., unpublished observations).
These findings reinforce the need for caution in interpreting the numerous reports of colocalization of foci of γ-H2AX and DSB repair/response proteins detected by light microscopy. The nonoverlapping distributions of γ-H2AX and DNA repair proteins, the absence of γ-H2AX near break ends, and the efficient Mre11p recruitment seen in G1-arrested tel1Δ and tel1Δ mec1Δ mutants combine to support the suggestion that γ-H2AX may not play a central role in recruiting repair proteins to breaks themselves [45, 47, 64, 65]. If interactions with γ-H2AX do help concentrate repair/response proteins in sequences near DSBs, such interactions must be weak and/or transient enough to escape detection by ChIP. We note, in this regard, that yeast H2A mutants that convert the PI3KK target serine to an alanine are either insensitive to or only mildly sensitive to many forms of DNA damage [43, 48]. These mutants also switch mating type with normal kinetics (E. Unal, A.A.-E., U. Sattler, R.S., M.L., J.E.H., and D. Koshland, submitted) and undergo meiosis efficiently, with wild-type kinetics and wild-type levels of spore viability, gene conversion, and crossing over (R.S. and M.L., unpublished observations).
Instead, we think it likely that γ-H2AX promotes chromosome structural changes that occur farther away from breaks but that play a role in promoting efficient damage repair. Consistent with this are recent observations that the presence of γ-H2AX is essential for the DSB-induced recruitment of cohesin to a wide region that occurs around breaks in G2/M-phase arrested yeast cells (E. Unal, A.A.-E., U. Sattler, R.S., M.L., J.E.H., and D. Koshland, submitted). DSB-induced cohesin loading promotes sister chromatid cohesion (L. Ström, H. Betts-Lindroos, K. Shirahige, and Camilla Sjögren, submitted), which in turn could hold broken chromosome ends in register while leaving the break itself available for repair factors. It has also been suggested that γ-H2AX forms an anchor for proteins that directly hold the two DSB ends together [47]. Although our findings make it less likely that the proteins these authors suggested (the MRN/MRX complex) play this role, they do not preclude a role for other proteins in this function.
The distribution of γ-H2AX around the MAT DSB is even more striking when one considers that activation of Tel1p and Mec1p, the kinases responsible for γ-H2AX formation, is thought to depend on partner proteins (the Mre11 complex and Ddc2p, respectively) that themselves bind primarily to sequences close to the DSB itself (this work and [4, 66, 67]). The recruitment of Mre11p and Ddc2p to DSBs within minutes (this work and [8, 12]), combined with the failure of their corresponding PI3KKs to respond in their absence (this work and [4, 66, 67]) further reinforce the suggestion that initial DSB recognition by PI3KKs occurs through interactions with partner proteins bound to break ends. Similar conclusions have been made with regard to ATM and ATR [11, 67].
How can break-end recognition be reconciled with the γ-H2AX distribution observed, in which chromatin immediately adjacent to the break is not phosphorylated? Because histones remain present in DSB-adjacent sequences (Figure 3), the low levels of γ-H2AX in this region cannot be due to the absence of a substrate for phosphorylation. Instead, other reasons must be considered—either γ-H2AX is never formed in break-adjacent sequences, perhaps as a result of occlusion by other DSB-response proteins, or γ-H2AX forms but is rapidly removed, either by phosphatase activity [68] or by chromatin remodeling complexes that promote either general nucleosome turnover near DSBs or the specific removal of γ-H2AX-containing nucleosomes. In this regard, it is intriguing that yeast mutants lacking chromatin modification and remodeling complex components display DNA damage sensitivity [69–71].
A second issue remaining is how DSB end recognition results in the phosphorylation of chromatin that is tens of kilobase pairs (in yeast) and millions of base pairs (in mammals) from the break [41]. If PI3KKs remain associated with break ends, movement of the DSB end within a limited nuclear domain [72] could produce the broad γ-H2AX distributions we observe. However, accounting for differences between species in the amount of chromatin phosphorylated per break would require mammalian chromatin to be significantly more mobile in situ than is yeast chromatin, which seems unlikely. Alternatively, it has been suggested that PI3KKs are activated upon encountering a DSB but do not remain break associated [33]; encounters between chromatin and the activated kinase as it diffuses away would result in a broad distribution of γ-H2AX. Differences between species in the extent of chromatin phosphorylated might reflect differences in the time that kinase molecules remain active after break recognition.
In conclusion, the finding of distinct and virtually nonoverlapping distributions for γ-H2AX and repair proteins in the region surrounding a DSB reflect the partitioning, in space, of the initial DNA damage response. Processes directly involved in DNA damage repair are expected to occur in sequences immediately adjacent to the lesion, whereas processes involving higher-order chromatin or chromosome structural changes that facilitate damage repair are expected to occur in a much larger region. We expect that the application of molecular-level approaches similar to those used here should illuminate both the underlying basis of this partition, as well as the distinct events that occur in different regions subsequent to DNA damage.
Experimental Procedures
Yeast Strains
Congenic strains were constructed from JKM179 [29], JKM139 [19], and yXW1 [19] by transformation or genetic crosses. All strains were ho hml::ADE1 leu2,3-112 hmr::ADE1 ade1 lys5 trp1::hisG ura3-52 ade3::GAL10::HO. Strain names and additional genotypes are listed in Table S1, available in the Supplemental Data online.
Chromatin Immunoprecipitation and DSB Analysis
Cells were grown to mid-log phase (0.5 × 107 to 1 × −107 cells/ml) in YEPL broth [73] with vigorous aeration. G1-arrested bar1 cultures were grown to 0.5 × 107 cells/ml in YEPL broth, α-mating pheromone (Sigma) was added to reach a final concentration of 200 nM, and cells were incubated for 3 hr, at which point >95% were unbudded schmoos. DSB induction [28, 30], chromatin immunoprecipitation [74], and DSB analysis on alkaline agarose gels [14] were as described with modifications (described in detail in Supplemental Experimental Procedures). PCR primers used are listed in Supplemental Table S2. To control for experiment-to-experiment variation in crosslinking and immunoprecipitation efficiencies, we determined break-induced Mre11p and γ-H2AX occupancy and H2B occupancy by calculating the ratio of band intensities for experimental (regions near the break) and control (a region 66 kb from the break) products from multiplex PCR reactions with ChIP samples, divided by the same ratio for PCR products from genomic DNA. These are the relative ChIP ratios reported in Figures 2–4. Absolute ChIP values (ChIP/input) ranged from 0.01% to 6.2% for γ-H2AX, from <0.1% to 1% for Mre11p, and from 1.1% to 4.6% for HA-H2B.
Supplementary Material
Acknowledgments
We thank Namrita Dhillon and Rohinton Kamakaka for strains, Michael Lisby, Rodney Rothstein, and Camilla Sjögren for communicating results in advance of publication, Neal Sugawara for helpful discussion and permission to reproduce previously published data, and Carl Wu, Christophe Redon, Alastair Goldman, Dhruba Chattoraj, Cyril Buhler, and other members of our laboratories for helpful discussions. This work was supported, in part, by National Institutes of Health grants GM20056 and GM61766 to J.E.H and National Institutes of Health grants GM59413 and HFSPO to J.H.P.
Footnotes
Supplemental data include two tables and Supplemental Experimental Procedures and are available at http://www.current-biology.com/cgi/content/full/14/19/1703/DC1/.
References
- 1.Kurz EU, Lees-Miller SP. DNA damage-induced activation of ATM and ATM-dependent signaling pathways. DNA Repair (Amst) 2004;3:889–900. doi: 10.1016/j.dnarep.2004.03.029. [DOI] [PubMed] [Google Scholar]
- 2.Shechter D, Costanzo V, Gautier J. Regulation of DNA replication by ATR: signaling in response to DNA intermediates. DNA Repair (Amst) 2004;3:901–908. doi: 10.1016/j.dnarep.2004.03.020. [DOI] [PubMed] [Google Scholar]
- 3.Burma S, Chen DJ. Role of DNA-PK in the cellular response to DNA double-strand breaks. DNA Repair (Amst) 2004;3:909–918. doi: 10.1016/j.dnarep.2004.03.021. [DOI] [PubMed] [Google Scholar]
- 4.Nakada D, Matsumoto K, Sugimoto K. ATM-related Tel1 associates with double-strand breaks through an Xrs2-dependent mechanism. Genes Dev. 2003;17:1957–1962. doi: 10.1101/gad.1099003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Costanzo V, Paull T, Gottesman M, Gautier J. Mre11 assembles linear DNA fragments into DNA damage signaling complexes. PLoS Biology. 2004;2:e110. doi: 10.1371/journal.pbio.0020110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Stracker TH, Theunissen JWF, Morales M, Petrini JHJ. The Mre11 complex and the metabolism of chromosome breaks: the importance of communicating and holding things together. DNA Repair (Amst) 2004;3:845–854. doi: 10.1016/j.dnarep.2004.03.014. [DOI] [PubMed] [Google Scholar]
- 7.Aylon Y, Kupiec M. DSB repair: the yeast paradigm. DNA Repair (Amst) 2004;3:797–815. doi: 10.1016/j.dnarep.2004.04.013. [DOI] [PubMed] [Google Scholar]
- 8.Nelms BE, Maser RS, MacKay JF, Lagally MG, Petrini JH. In situ visualization of DNA double-strand break repair in human fibroblasts. Science. 1998;280:590–592. doi: 10.1126/science.280.5363.590. [DOI] [PubMed] [Google Scholar]
- 9.Paull TT, Rogakou EP, Yamazaki V, Kirchgessner CU, Gellert M, Bonner WM. A critical role for histone H2AX in recruitment of repair factors to nuclear foci after DNA damage. Curr Biol. 2000;10:886–895. doi: 10.1016/s0960-9822(00)00610-2. [DOI] [PubMed] [Google Scholar]
- 10.Lisby M, Antunez de Mayolo A, Mortensen UH, Rothstein R. Cell cycle-regulated centers of DNA double-strand break repair. Cell Cycle. 2003;2:479–483. [PubMed] [Google Scholar]
- 11.Petrini JHJ, Stracker TH. The cellular response to DNA double-strand breaks: defining the sensors and mediators. Trends Cell Biol. 2003;13:458–462. doi: 10.1016/s0962-8924(03)00170-3. [DOI] [PubMed] [Google Scholar]
- 12.Lisby M, Barlow JH, Burgess RC, Rothstein R. Choreography of the DNA damage response: spatiotemporal relationships among checkpoint and repair proteins. Cell. 2004;118:699–713. doi: 10.1016/j.cell.2004.08.015. [DOI] [PubMed] [Google Scholar]
- 13.Connolly B, White CI, Haber JE. Physical monitoring of mating type switching in Saccharomyces cerevisiae. Mol Cell Biol. 1988;8:2342–2349. doi: 10.1128/mcb.8.6.2342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.White CI, Haber JE. Intermediates of recombination during mating type switching in Saccharomyces cerevisiae. EMBO J. 1990;9:663–673. doi: 10.1002/j.1460-2075.1990.tb08158.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Frank-Vaillant M, Marcand S. Transient stability of DNA ends allows nonhomologous end joining to precede homologous recombination. Mol Cell. 2002;10:1189–1199. doi: 10.1016/s1097-2765(02)00705-0. [DOI] [PubMed] [Google Scholar]
- 16.Neale MJ, Ramachandran M, Trelles-Sticken E, Scherthan H, Goldman AS. Wild-type levels of Spo11-induced DSBs are required for normal single-strand resection during meiosis. Mol Cell. 2002;9:835–846. doi: 10.1016/s1097-2765(02)00498-7. [DOI] [PubMed] [Google Scholar]
- 17.Aylon Y, Liefshitz B, Bitan-Banin G, Kupiec M. Molecular dissection of mitotic recombination in the yeast Saccharomyces cerevisiae. Mol Cell Biol. 2003;23:1403–1417. doi: 10.1128/MCB.23.4.1403-1417.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Sugawara N, Wang X, Haber JE. In vivo roles of Rad52, Rad54, and Rad55 proteins in Rad51-mediated recombination. Mol Cell. 2003;12:209–219. doi: 10.1016/s1097-2765(03)00269-7. [DOI] [PubMed] [Google Scholar]
- 19.Wang X, Haber JE. Role of Saccharomyces single-stranded DNA-binding protein RPA in the strand invasion step of double-strand break repair. PLoS Biology. 2004;2:e21. doi: 10.1371/journal.pbio.0020021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Wolner B, van Komen S, Sung P, Peterson CL. Recruitment of the recombinational repair machinery to a DNA double-strand break in yeast. Mol Cell. 2003;12:221–232. doi: 10.1016/s1097-2765(03)00242-9. [DOI] [PubMed] [Google Scholar]
- 21.Melo JA, Cohen J, Toczyski DP. Two checkpoint complexes are independently recruited to sites of DNA damage in vivo. Genes Dev. 2001;15:2809–2821. doi: 10.1101/gad.903501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kondo T, Wakayama T, Naiki T, Matsumoto K, Sugimoto K. Recruitment of Mec1 and Ddc1 checkpoint proteins to double-strand breaks through distinct mechanisms. Science. 2001;294:867–870. doi: 10.1126/science.1063827. [DOI] [PubMed] [Google Scholar]
- 23.Lisby M, Mortensen UH, Rothstein R. Colocalization of multiple DNA double-strand breaks at a single Rad52 repair centre. Nat Cell Biol. 2003;5:572–577. doi: 10.1038/ncb997. [DOI] [PubMed] [Google Scholar]
- 24.Martin SG, Laroche T, Suka N, Grunstein M, Gasser SM. Relocalization of telomeric Ku and SIR proteins in response to DNA strand breaks in yeast. Cell. 1999;97:621–633. doi: 10.1016/s0092-8674(00)80773-4. [DOI] [PubMed] [Google Scholar]
- 25.Evans E, Sugawara N, Haber JE, Alani E. The Saccharomyces cerevisiae Msh2 mismatch repair protein localizes to recombination intermediates in vivo. Mol Cell. 2000;5:789–799. doi: 10.1016/s1097-2765(00)80319-6. [DOI] [PubMed] [Google Scholar]
- 26.Mills KD, Sinclair DA, Guarente L. MEC1-dependent redistribution of the Sir3 silencing protein from telomeres to DNA double-strand breaks. Cell. 1999;97:609–620. doi: 10.1016/s0092-8674(00)80772-2. [DOI] [PubMed] [Google Scholar]
- 27.Naiki T, Wakayama T, Nakada D, Matsumoto K, Sugimoto K. Association of Rad9 with double-strand breaks through a MEC1-dependent mechanism. Mol Cell Biol. 2004;24:3277–3285. doi: 10.1128/MCB.24.8.3277-3285.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lee SE, Moore JK, Holmes A, Umezu K, Kolodner RD, Haber JE. Saccharomyces Ku70, Mre11/Rad50 and RPA proteins regulate adaptation to G2/M arrest after DNA damage. Cell. 1998;94:399–409. doi: 10.1016/s0092-8674(00)81482-8. [DOI] [PubMed] [Google Scholar]
- 29.Lee SE, Pellicioli A, Demeter J, Vaze MP, Gasch AP, Malkova A, Brown PO, Botstein D, Stearns T, Foiani M, et al. Arrest, adaptation, and recovery following a chromosome double-strand break in Saccharomyces cerevisiae. Cold Spring Harb Symp Quant Biol. 2000;65:303–314. doi: 10.1101/sqb.2000.65.303. [DOI] [PubMed] [Google Scholar]
- 30.Pellicioli A, Lee SE, Lucca C, Foiani M, Haber JE. Regulation of Saccharomyces Rad53 checkpoint kinase during adaptation from DNA damage-induced G2/M arrest. Mol Cell. 2001;7:293–300. doi: 10.1016/s1097-2765(01)00177-0. [DOI] [PubMed] [Google Scholar]
- 31.Lee SE, Pellicioli A, Vaze MB, Sugawara N, Malkova A, Foiani M, Haber JE. Yeast Rad52 and Rad51 recombination proteins define a seond pathway of DNA damage assessment in response to a single double-strand break. Mol Cell Biol. 2003;23:8913–8923. doi: 10.1128/MCB.23.23.8913-8923.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Toczyski DP, Galgoczy DJ, Hartwell LH. CDC5 and CKII control adaptation to the yeast DNA damage checkpoint. Cell. 1997;90:1097–1106. doi: 10.1016/s0092-8674(00)80375-x. [DOI] [PubMed] [Google Scholar]
- 33.Bakkenist CJ, Kastan MB. DNA damage activates ATM through intermolecular autophosphorylation and dimer dissociation. Nature. 2003;421:499–506. doi: 10.1038/nature01368. [DOI] [PubMed] [Google Scholar]
- 34.Mirzoeva OK, Petrini JH. DNA damage-dependent nuclear dynamics of the Mre11 complex. Mol Cell Biol. 2001;21:281–288. doi: 10.1128/MCB.21.1.281-288.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Chen HT, Bhandoola A, Difilippantonio MJ, Zhu J, Brown MJ, Tai X, Rogakou EP, Brotz TM, Bonner WM, Ried T, et al. Response to RAG-mediated V(D)J cleavage by NBS1 and gamma-H2AX. Science. 2000;290:1962–1965. doi: 10.1126/science.290.5498.1962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Stewart GS, Wang B, Bignell CR, Taylor AM, Elledge SJ. MDC1 is a mediator of the mammalian DNA damage checkpoint. Nature. 2003;421:961–966. doi: 10.1038/nature01446. [DOI] [PubMed] [Google Scholar]
- 37.Lisby M, Rothstein R, Mortensen UH. Rad52 forms DNA repair and recombination centers during S phase. Proc Natl Acad Sci USA. 2001;98:8276–8282. doi: 10.1073/pnas.121006298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Rogakou EP, Pilch DR, Orr AH, Ivanova VS, Bonner WM. DNA double-stranded breaks induce histone H2AX phosphorylation on serine 139. J Biol Chem. 1998;273:5858–5868. doi: 10.1074/jbc.273.10.5858. [DOI] [PubMed] [Google Scholar]
- 39.Yazdi PT, Wang Y, Zhao S, Patel N, Lee EY, Qin J. SMC1 is a downstream effector in the ATM/NBS1 branch of the human S-phase checkpoint. Genes Dev. 2002;16:571–582. doi: 10.1101/gad.970702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kim ST, Xu B, Kastan MB. Involvement of the cohesin protein, Smc1, in ATM-dependent and independent responses to DNA damage. Genes Dev. 2002;16:560–570. doi: 10.1101/gad.970602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Redon C, Pilch D, Rogakou E, Sedelnikova O, Newrock K, Bonner W. Histone H2A variants H2AX and H2AZ. Curr Opin Genet Dev. 2002;12:162–169. doi: 10.1016/s0959-437x(02)00282-4. [DOI] [PubMed] [Google Scholar]
- 42.Rogakou EP, Boon C, Redon C, Bonner WM. Megabase chromatin domains involved in DNA double-strand breaks in vivo. J Cell Biol. 1999;146:905–916. doi: 10.1083/jcb.146.5.905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Redon C, Pilch DR, Rogakou EP, Orr AH, Lowndes NF, Bonner WM. Yeast histone 2A serine 129 is essential for the efficient repair of checkpoint-blind DNA damage. EMBO Rep. 2003;4:1–7. doi: 10.1038/sj.embor.embor871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Fernandez-Capetillo O, Celeste A, Nussenzweig A. Focusing on foci: H2AX and the recruitment of DNA-damage response factors. Cell Cycle. 2003;2:426–427. [PubMed] [Google Scholar]
- 45.Mirzoeva OK, Petrini JHJ. DNA replication-dependent nuclear dynamics of the Mre11 complex. Mol Cancer Res. 2003;1:207–218. [PubMed] [Google Scholar]
- 46.Fernandez-Capetillo O, Lee A, Nussenzweig M, Nussenzweig A. H2AX: the histone guardian of the genome. DNA Repair (Amst) 2004;3:959–967. doi: 10.1016/j.dnarep.2004.03.024. [DOI] [PubMed] [Google Scholar]
- 47.Bassing CH, Alt FW. H2AX may function as an anchor to hold broken chromosomal DNA ends in close proximity. Cell Cycle. 2004;3:149–153. doi: 10.4161/cc.3.2.689. [DOI] [PubMed] [Google Scholar]
- 48.Downs JA, Lowndes NF, Jackson SP. A role for Saccharomyces cerevisiae histone H2A in DNA repair. Nature. 2000;408:1001–1004. doi: 10.1038/35050000. [DOI] [PubMed] [Google Scholar]
- 49.Kostriken R, Strathern JN, Klar AJ, Hicks JB, Heffron F. A site-specific endonuclease essential for mating-type switching in Saccharomyces cerevisiae. Cell. 1983;35:167–174. doi: 10.1016/0092-8674(83)90219-2. [DOI] [PubMed] [Google Scholar]
- 50.Ira G, Pellicioli A, Balijja A, Wang X, Fiorani S, Carotenuto W, Liberi G, Bressan D, Wan L, Hollingsworth NM, Haber JE, Foiani M. CDK1 requirement in DNA end resection, homologous recombination and DNA damage checkpoint activation. Nature. 2004 doi: 10.1038/nature02964. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Miyazaki T, Bressan DA, Shinohara M, Haber JE, Shinohara A. In vivo assembly and disassembly of Rad51 and Rad52 complexes during double-strand break repair. EMBO J. 2004;23:939–949. doi: 10.1038/sj.emboj.7600091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Pilch DR, Sedelnikova OA, Redon C, Celeste A, Nussenzweig A, Bonner WM. Characteristics of γ-H2AX foci at DNA double-strand breaks. Biochem Cell Biol. 2003;81:123–129. doi: 10.1139/o03-042. [DOI] [PubMed] [Google Scholar]
- 53.Jackson V. Deposition of newly synthesized histones: new histones H2A and H2B do not deposit in the same nucleosome with new histones H3 and H4. Biochemistry. 1987;26:2315–2325. doi: 10.1021/bi00382a037. [DOI] [PubMed] [Google Scholar]
- 54.Burma S, Chen BP, Murphy M, Kurimasa A, Chen DJ. ATM phosphorylates histone H2AX in response to DNA double-strand breaks. J Biol Chem. 2001;276:42462–42467. doi: 10.1074/jbc.C100466200. [DOI] [PubMed] [Google Scholar]
- 55.Ward IM, Chen J. Histone H2AX is phosphorylated in an ATR-dependent manner in response to replicational stress. J Biol Chem. 2001;276:47759–47762. doi: 10.1074/jbc.C100569200. [DOI] [PubMed] [Google Scholar]
- 56.Stiff T, O’Driscoll M, Rief N, Iwabuchi K, Lobrich M, Jeggo PA. ATM and DNA-PK function redundantly to phosphorylate H2AX after exposure to ionizing radiation. Cancer Res. 2004;64:2390–2396. doi: 10.1158/0008-5472.can-03-3207. [DOI] [PubMed] [Google Scholar]
- 57.Melo J, Toczyski D. A unified view of the DNA-damage checkpoint. Curr Opin Cell Biol. 2002;14:237–245. doi: 10.1016/s0955-0674(02)00312-5. [DOI] [PubMed] [Google Scholar]
- 58.Siede W, Friedberg A, Friedberg E. RAD9-dependent G1 arrest defines a second checkpoint for damaged DNA in the cell cycle of Saccharomyces cerevisiae. Proc Natl Acad Sci USA. 1993;90:7985–7989. doi: 10.1073/pnas.90.17.7985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Gerald JNF, Benjamin JM, Kron SJ. Robust G1 checkpoint arrest in budding yeast: dependence on DNA damage signaling and repair. J Cell Sci. 2002;115:1749–1757. doi: 10.1242/jcs.115.8.1749. [DOI] [PubMed] [Google Scholar]
- 60.Weinert TA, Kiser GL, Hartwell LH. Mitotic checkpoint genes in budding yeast and the dependence of mitosis on DNA replication and repair. Genes Dev. 1994;8:652–665. doi: 10.1101/gad.8.6.652. [DOI] [PubMed] [Google Scholar]
- 61.Greenwell PW, Kronmal SL, Porter SE, Gassenhuber J, Obermaier B, Petes TD. TEL1, a gene involved in controlling telomere length in S. cerevisiae, is homologous to the human ataxia telangiectasia gene. Cell. 1995;82:823–829. doi: 10.1016/0092-8674(95)90479-4. [DOI] [PubMed] [Google Scholar]
- 62.Morrow DM, Tagle DA, Shiloh Y, Collins FS, Hieter P. TEL1, an S. cerevisiae homolog of the human gene mutated in ataxia telangiectasia, is functionally related to the yeast checkpoint gene MEC1. Cell. 1995;82:831–840. doi: 10.1016/0092-8674(95)90480-8. [DOI] [PubMed] [Google Scholar]
- 63.Usui T, Ogawa H, Petrini JH. A DNA damage response pathway controlled by Tel1 and the Mre11 complex. Mol Cell. 2001;7:1255–1266. doi: 10.1016/s1097-2765(01)00270-2. [DOI] [PubMed] [Google Scholar]
- 64.Nakada D, Shimomura T, Matsumoto K, Sugimoto K. The ATM-related Tel1 protein of Saccharomyces cerevisiae controls a checkpoint response following phleomycin treatment. Nucleic Acids Res. 2003;31:1715–1724. doi: 10.1093/nar/gkg252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Celeste A, Fernandez-Capetillo O, Kruhlak MJ, Pilch DR, Staudt DW, Lee A, Bonner RF, Bonner WM, Nussenzweig A. Histone H2AX phosphorylation is dispensable for the initial recognition of DNA breaks. Nat Cell Biol. 2003;5:675–679. doi: 10.1038/ncb1004. [DOI] [PubMed] [Google Scholar]
- 66.Rouse J, Jackson SP. Lcd1p recruits Mec1p to DNA lesions in vitro and in vivo. Mol Cell. 2002;9:857–869. doi: 10.1016/s1097-2765(02)00507-5. [DOI] [PubMed] [Google Scholar]
- 67.Zou L, Elledge SJ. Sensing DNA damage through ATRIP recognition of RPA-ssDNA complexes. Science. 2003;300:1542–1548. doi: 10.1126/science.1083430. [DOI] [PubMed] [Google Scholar]
- 68.Nazarov IB, Smirnova AN, Krutilina RI, Svetlova MP, Solovjeva LV, Nikiforov AA, Oei SL, Zalenskaya IA, Yau PM, Bradbury EM, et al. Dephosphorylation of histone gamma-H2AX during repair of DNA double-strand breaks in mammalian cells and its inhibition by Calyculin A. Radiat Res. 2003;160:309–317. doi: 10.1667/rr3043. [DOI] [PubMed] [Google Scholar]
- 69.Shen X, Mizuguchi G, Hamiche A, Wu C. A chromatin remodelling complex involved in transcription and DNA processing. Nature. 2000;406:541–544. doi: 10.1038/35020123. [DOI] [PubMed] [Google Scholar]
- 70.Mizuguchi G, Shen X, Landry J, Wu WH, Sen S, Wu C. ATP-driven exchange of histone H2AZ variant catalyzed by SWR1 chromatin remodeling complex. Science. 2004;303:343–348. doi: 10.1126/science.1090701. [DOI] [PubMed] [Google Scholar]
- 71.Bird AW, Yu DY, Pray-Grant MG, Qiu Q, Harmon KE, Megee PC, Grant PA, Smith MM, Christman MF. Acetylation of histone H4 by Esa1 is required for DNA double-strand break repair. Nature. 2002;419:411–415. doi: 10.1038/nature01035. [DOI] [PubMed] [Google Scholar]
- 72.Gasser SM. Visualizing chromatin dynamics in interphase nuclei. Science. 2002;296:1412–1416. doi: 10.1126/science.1067703. [DOI] [PubMed] [Google Scholar]
- 73.Rudin N, Haber JE. Efficient repair of HO-induced chromosomal breaks in Saccharomyces cerevisiae by recombination between flanking homologous sequences. Mol Cell Biol. 1988;8:3918–3928. doi: 10.1128/mcb.8.9.3918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Strahl-Bolsinger S, Hecht A, Luo K, Grunstein M. SIR2 and SIR4 interactions differ in core and extended telomeric heterochromatin in yeast. Genes Dev. 1997;11:83–93. doi: 10.1101/gad.11.1.83. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.