

Cao et al. solved the structure of SIRT1 in complex with resveratrol and an AMC-containing peptide. The structural information provides valuable insights into regulation of SIRT1 activity and should benefit the development of authentic SIRT1 activators.
Keywords: histone/protein deacetylase, resveratrol, sirtuins, structure
Abstract
Sirtuins with an extended N-terminal domain (NTD), represented by yeast Sir2 and human SIRT1, harbor intrinsic mechanisms for regulation of their NAD-dependent deacetylase activities. Elucidation of the regulatory mechanisms is crucial for understanding the biological functions of sirtuins and development of potential therapeutics. In particular, SIRT1 has emerged as an attractive therapeutic target, and the search for SIRT1-activating compounds (STACs) has been actively pursued. However, the effectiveness of a class of reported STACs (represented by resveratrol) as direct SIRT1 activators is under debate due to the complication involving the use of fluorogenic substrates in in vitro assays. Future efforts of SIRT1-based therapeutics necessitate the dissection of the molecular mechanism of SIRT1 stimulation. We solved the structure of SIRT1 in complex with resveratrol and a 7-amino-4-methylcoumarin (AMC)-containing peptide. The structure reveals the presence of three resveratrol molecules, two of which mediate the interaction between the AMC peptide and the NTD of SIRT1. The two NTD-bound resveratrol molecules are principally responsible for promoting tighter binding between SIRT1 and the peptide and the stimulation of SIRT1 activity. The structural information provides valuable insights into regulation of SIRT1 activity and should benefit the development of authentic SIRT1 activators.
Sirtuins are NAD-dependent protein deacetylases catalyzing the removal of the acetyl group of an acetylated lysine residue concomitant with the cleavage of the nicotinamide group from NAD, resulting in reaction products of a deacetylated protein, nicotinamide, and a novel compound (2′-O-acetyl-ADP-ribose) (Imai et al. 2000; Landry et al. 2000; Smith et al. 2000; Tanner et al. 2000; Sauve et al. 2001; Tanny and Moazed 2001). Crystal structures of several archael, yeast, and human sirtuins have been solved, and they revealed a conserved catalytic core composed of a Rossmann-fold large lobe and a zinc-containing small lobe (Finnin et al. 2001; Min et al. 2001; Avalos et al. 2002; Zhao et al. 2003, 2013; Schuetz et al. 2007; Jin et al. 2009; Pan et al. 2011; Davenport et al. 2014). NAD is bound in the cleft formed between the two lobes, with the nicotinamide group pointing inside of the cleft. The substrate acetyl-lysine approaches the nicotinamide group of NAD via a hydrophobic channel. The combined biochemical and structural analyses have yielded considerable understandings of the catalytic mechanism of sirtuins (Sauve et al. 2006; Sanders et al. 2010; Feldman et al. 2012).
The two best-characterized sirtuins are yeast Sir2 and human SIRT1. While Sir2 primarily deacetylates Lys16 of histone H4 (H4K16), a variety of SIRT1 substrates—including histones, p53, FOXOs, and PGC-1α—have been reported (Luo et al. 2001; Vaziri et al. 2001; Brunet et al. 2004; Motta et al. 2004; Vaquero et al. 2004; Rodgers et al. 2005). Accordingly, SIRT1 has been reported to function in many physiological and pathological processes, such as aging and the development of metabolic, neurodegenerative, and cardiovascular diseases (Guarente 2011). Therefore, modulating the SIRT1 activities by small molecules might serve as an effective therapeutic strategy toward the aforementioned diseases, and there has been great interest in finding SIRT1-activating compounds (STACs) (Baur 2010; Sinclair and Guarente 2014). Large-scale screenings have yielded a number of possible STACs, among which is a class of polyphenol compounds, notably represented by resveratrol, an ingredient of red wine (Howitz et al. 2003; Sauve et al. 2005; Milne et al. 2007; Bemis et al. 2009). However, several studies questioned the effectiveness of resveratrol and related compounds as direct SIRT1 activators, pointing out that the observed stimulatory effect depended on the presence of a fluorescent group in the substrates used in in vitro assays (Borra et al. 2005; Kaeberlein et al. 2005; Pacholec et al. 2010). Nevertheless, it is possible that the artificial fluorescent group in the substrate peptide may mimic an aromatic residue in certain natural substrates of SIRT1 (Dai et al. 2010; Hubbard et al. 2013; Lakshminarasimhan et al. 2013; Sinclair and Guarente 2014).
Regardless of the nature of the substrates, it was found that the stimulatory effect of resveratrol requires the presence of an N-terminal domain (NTD) in addition to the catalytic domain (CD) of SIRT1 (Milne et al. 2007; Hubbard et al. 2013). There are seven sirtuins in humans, and SIRT1 distinguishes itself from others in having sizable N-terminal and C-terminal extensions beyond the conserved catalytic core (Frye 2000). In vitro studies showed that three regions of the 747-residue human SIRT1 are required to recapitulate known biochemical properties of the full-length protein. They include the ∼260-residue CD located in the middle, an ∼25-residue C-terminal ESA sequence, and an ∼50-residue NTD (Fig. 1A; Kang et al. 2011; Pan et al. 2012; Hubbard et al. 2013). In particular, Glu230 in NTD was shown to be critical for stimulation by resveratrol (Hubbard et al. 2013).
Figure 1.
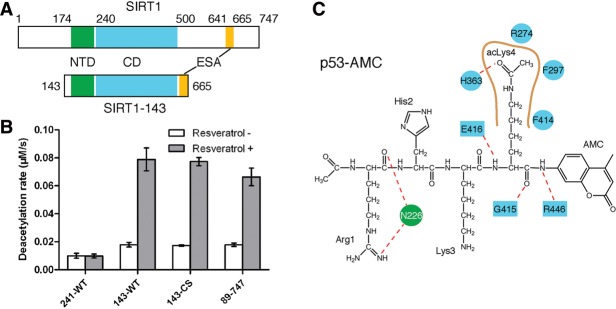
SIRT1 fragments and their resveratrol-dependent deacetylation activities. (A) A schematic diagram showing functional domains in full-length SIRT1 (top) and a truncation variant (SIRT1-143) used for crystallization (bottom). (B) A bar diagram showing deacetylase activities of the indicated SIRT1 fragments and the stimulatory effects of 0.5 mM resveratrol. (C) The p53-AMC peptide used for cocrystallization and deacetylation assays. The single-letter residue symbol together with the residue number indicates SIRT1 residues involved in protein–peptide interactions via main chain (filled circles) or side chain (filled boxes) groups. The red dashed lines denote hydrogen bonds, the brown thick curve indicates the acetyl-lysine-binding pocket in SIRT1-CD, and the nearby indicated residues represent ones contacting the acetyl-lysine via hydrophobic and/or van der Waals interactions. Cyan and green filled shapes indicate residues in CD and NTD, respectively.
The structure of the CD of SIRT1 with and without the C-terminal ESA polypeptide has been determined previously (Zhao et al. 2013; Davenport et al. 2014). The structure of the catalytic core resembles that of other sirtuins, and the ESA polypeptide folds into a β hairpin, which is juxtaposed with the parallel β strands of the Rossmann-fold domain and forms an extended, mixed β sheet (Davenport et al. 2014). However, little is known about the structure of the SIRT1 NTD. This structural information is vital for understanding the stimulatory effect of resveratrol regardless of whether it works for natural substrates, as it will provide valuable insights into how SIRT1 activity is modulated, and this knowledge should be useful for the development of authentic, effective activators of SIRT1.
Results
Deacetylase activities of SIRT1 fragments
Since resveratrol was shown to stimulate the deacetylase activity of SIRT1 toward a four-residue acetylated p53 peptide, termed p53-AMC (7-amino-4-methylcoumarin), which carries a C-terminal AMC fluorophore, we intended to cocrystallize SIRT1 with resveratrol and the p53-AMC peptide. To achieve crystallization, we engineered a truncated version of SIRT1, termed SIRT1-143, spanning residues 143–665 but with the intervening sequences (amino acids 513–640) between the CD and ESA removed and the remaining segments spliced together without an exogenous linker (Fig. 1A). Judging by a continuous, enzyme-coupled microplate assay (Smith et al. 2009), SIRT1-143 exhibited an enzymatic activity level comparable with that of a longer SIRT1 fragment lacking only 88 N-terminal residues (SIRT1-89-747) using p53-AMC as the substrate (Fig. 1B,C). Furthermore, the deacetylase activity of SIRT1-143 is stimulated by resveratrol in a manner similar to that of the full-length protein, while a truncated form of SIRT1-143, SIRT1-241, lacking the noncatalytic NTD, was not stimulated by the addition of resveratrol (Fig. 1B). Therefore, a SIRT1-143 structure would reveal the salient features of SIRT1 activation by resveratrol. We succeeded in crystallizing a quadruple cysteine-to-serine mutant (C253S, C268S, C501S, and C502S) of SIRT1-143, termed SIRT1-143CS, which shares the enzymatic properties of the parental fragment (Fig. 1B), in complex with resveratrol and the p53-AMC peptide, and a 3.2-Å structure was solved by molecular replacement (Table 1; Supplemental Fig. S1).
Table 1.
Statistics of crystallographic analysis

Overall structure
The structure of SIRT1-143CS in complex with resveratrol and p53-AMC revealed a conserved catalytic core with an extended central β sheet formed by the juxtaposition of the β hairpin from the ESA sequence (Fig. 2A; Zhao et al. 2013; Davenport et al. 2014). This arrangement of the ESA β hairpin is reminiscent of that of the C-terminal β hairpin in yeast Sir2, indicating a largely structural role of the ESA sequence in the Sir2–SIRT1 clade of sirtuins (Fig. 2C; Hsu et al. 2013). The SIRT1 NTD is mainly composed of a core of three helices (amino acids 184–229)—a feature that can also be identified in Sir2 (although the Sir2 NTD has two extra helices)—and, when bound by another protein (Sir4) assumes a distinct spatial position (Fig. 2C,D; Hsu et al. 2013). Apart from covalent joining of the two domains, there are very few direct contacts between the NTD and CD, with the notable exception of an interdomain hydrogen bond between Glu230 and Arg446 (Fig. 2A,B).
Figure 2.

Structure of the SIRT1 ternary complex. (A) Overall structure of SIRT1 with the protein domains color-coded as in Figure 1A. A pair of SIRT1 residues involved in NTD–CD interdomain interactions are shown in a stick model, with the hydrogen bonds indicated by white dashed lines. The acetylated p53-AMC peptide (carbon colored wheat) and resveratrol molecules (carbon colored magenta) are shown as a stick model. (B) A detailed view of protein–resveratrol and resveratrol–peptide interactions. (C) Superposition of SIRT1 and Sir2 structures aligned via their CDs. The ribbon structure of Sir2 is shown in light pink, and the structure of the SIRT1 complex is color-coded the same as in A, with the exception that the p53-AMC peptide is shown as a light-blue stick model. The SIR4 structure was removed for viewing clarity, but the blue-shaded region indicates its general location. (D) Superposition of the N-terminal helical domains of SIRT1 and Sir2.
The p53-AMC peptide is bound in a cleft formed between the NTD and CD of SIRT1 (Fig. 2). Like in all sirtuins, a substrate-binding channel between the large and small lobes in the CD binds the target acetyl-lysine of p53-AMC. Additional direct interactions between the CD and the peptide involve three hydrogen bonds between main chain carbonyl and amino groups of three SIRT1 residues (Gly415, Glu416, and Arg446) and the peptide (Fig. 1C). These hydrogen bonds are conserved in the structures of other sirtuin–peptide complexes known to date (Avalos et al. 2002; Zhao et al. 2003). Unique to the SIRT1–p53-AMC complex, Arg1 of the peptide interacts with Asn226 of the SIRT1 NTD via two hydrogen bonds: one between the carbonyl of Arg1 and the side chain amino group of Asn226 and another between one ω-amino group of Arg1 and the side chain hydroxyl group of Asn226 (Fig. 1C). The coumarin group of p53-AMC projects away from the SIRT1 CD, with the ring plane roughly perpendicular to the side chain of acLys4 (Fig. 2B).
Resveratrol binding
Three resveratrol molecules are found in the structure, and they are denoted as Res1 to Res3. Res1 and Res2 make hydrogen bonds with both the SIRT1 NTD and p53-AMC, while Res3 contacts the SIRT1 CD and the peptide (Fig. 2B). One hydroxyl group of the dihydroxyphenyl moiety of Res1 bonds to the side chain of Glu230, and the other hydroxyl group contacts the main chain amino and carbonyl groups of Lys3 of p53-AMC. The phenyl ring of the hydroxyphenyl moiety of Res1 stacks with the coumarin ring in parallel and also enjoys hydrophobic interactions with several residues located in α2 and α3 of the SIRT1 NTD. Note that Glu230 of the SIRT1 NTD also interacts with Arg446 in the CD (Fig. 2B). Res2 is located ∼4–5 Å away from Res1, with its hydroxyl group of the hydroxyphenyl moiety making hydrogen bonds to Gln222 and Asn226 of the SIRT1 NTD and also with the carbonyl of Arg1 of the peptide. The dihydroxyphenyl ring of Res2 stacks edge to edge with the coumarin ring and also contacts both the NTD and CD via hydrophobic and van de Waals interactions. Res3 is located next to the SIRT1 CD, on the opposite side of the coumarin ring with respect to the locations of Res1 and Res2 (Fig. 2B). The hydroxyl groups of the dihydroxyphenyl moiety of Res3 interact with Asp292 and Asp298 in the CD. The distal hydroxyl group of Res3 bonds the carbonyl of Lys444, and the hydroxyphenyl ring stacks with the coumarin face to face. Res3 also interacts with Res2 via a hydrogen bond (Fig. 2B).
To date, several other sirtuin structures in complex with p53-AMC and resveratrol or its analogs, 4′-bromo-resveratrol and piceatannol, have been determined (Gertz et al. 2012; Nguyen et al. 2013). Resveratrol was reported to stimulate the activity of SIRT5 and inhibit SIRT3 using the fluorogenic “Fluor-de-Lys” (FdL; identical to p53-AMC) peptide as the substrate (Gertz et al. 2012). The structure of SIRT5 in complex with resveratrol shows that the only resveratrol molecule is bound in a location near the binding site of Res3 in SIRT1 (Fig. 3A,B), although both the resveratrol molecule and the peptidyl coumarin moiety adopt significantly different orientations in the SIRT5 complex. In the SIRT3 complexes, the bromo-resveratrol molecule is bound further away from the fluorogenic peptide (Fig. 3C), while the piceatannol molecule occupies a position similar to that taken by Res2 in the SIRT1 complex (Fig. 3D). It should be noted that neither SIRT3 nor SIRT5 have an extended NTD as in SIRT1. The contrasting binding modes of resveratrol and its analogs in different sirtuin complexes reflect distinct binding conformation of the peptides and the local environments in separate sirtuins.
Figure 3.
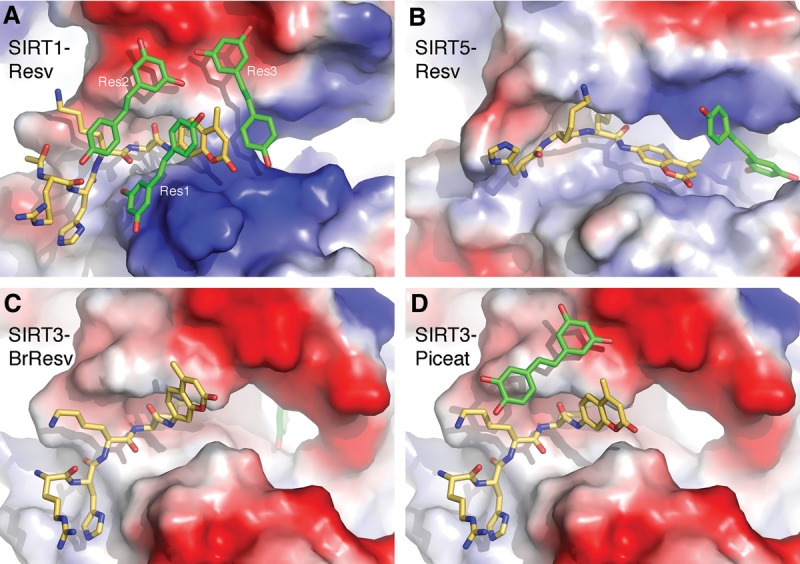
Comparison of resveratrol-binding modes. (A) A surface representation (with electrostatic potential distribution) of SIRT1 with the bound p53-AMC peptide and resveratrol. The NTD of SIRT1 was removed for viewing clarity. (B) The structure of SIRT5 with the bound peptide and resveratrol (Protein Data Bank [PDB] ID 4HDA). (C) The structure of SIRT3 with the bound peptide and bromo-resveratrol (PDB ID 4C7B). (D) The structure of SIRT3 with the bound piceatannol (PDB ID 4HD8). Both the peptides (carbon colored gold) and resveratrol or its analog (carbon colored green) are shown as a stick model.
Stimulatory effect of individual resveratrol molecules
To determine the contribution of each resveratrol molecule, we separately mutated the SIRT1 residues contacting each resveratrol and compared the deacetylase activities of the SIRT1 mutants in the presence and absence of 0.2 mM resveratrol using the continuous enzyme-coupled microplate assay. Note that the mutations were intended to disrupt the hydrogen bonds between the resveratrol and the protein, which may or may not completely abolish the binding of relevant resveratrol molecules. Hence, strictly speaking, the change in deacetylase activity is a consequence of perturbations to the observed resveratrol-binding mode. In agreement with previous observations, changing Glu230 to a lysine (E230K) or an alanine (E230A) significantly reduced the stimulatory effect of resveratrol, indicating that the binding of Res1 is crucial for the elevated SIRT1 activity toward the p53-AMC peptide (Fig. 4A). The Q222A/N226A double mutant of SIRT1 displayed a conspicuous level of reduction in response to the addition of resveratrol, confirming the role of Res2 in the stimulation of SIRT1 activity. By comparison, a D292A/D298A mutant had a mild effect on SIRT1 activity in the presence of resveratrol, suggesting an auxiliary role of Res3 in the stimulation of SIRT1 activity (Fig. 4A). Isothermal titration calorimetry (ITC) measurements revealed that resveratrol increased the binding between SIRT1 and the p53-AMC peptide, from an undetectable level without resveratrol to a KD of 7.3 μM in the presence of 0.5 mM resveratrol (Fig. 4B). Under the same binding conditions, the E230K, Q222A/N226A, and D292A/D298A mutants of SIRT1 bound the peptide with KD values of 19.5 μM, 18.2 μM, and 6.5 μM, respectively (Fig. 4C–E). The binding for the E230A mutant was too weak to be reliably measured. The observed binding strengths appear to correlate with the level of resveratrol stimulation.
Figure 4.

Deacetylase activities and peptide-binding affinities of SIRT1 variants. (A) Deacetylation rates of the indicated SIRT1 variants toward p53-AMC in the absence and presence of 0.2 mM resveratrol. (B–G) ITC measurements of binding affinities of the p53-AMC peptide to the indicated SIRT1 variants in the presence of 0.5 mM resveratrol.
The structure also shows that Glu230 makes an interdomain contact with Arg446 in the SIRT1 CD (Fig. 2) in addition to direct binding to reseveratrol. We hypothesized that stabilization of the interdomain arrangement configured by the Glu230–Arg466 interaction might play an important role for the resveratrol-dependent effect. To test this hypothesis, we made a R446A mutation, which should disrupt the Glu230–Arg446 interaction without directly affecting hydrogen bonding between Glu230 and resveratrol, and measured its binding affinity and deacetylase activity toward the p53-AMC peptide. The R446A mutant bound the peptide with a KD of 17.7 μM in the presence of 0.5 mM resveratrol and displayed a level of resveratrol stimulation similar to that of the E230K mutant (Fig. 4A,F). This result supports the notion that a proper interdomain arrangement is important for the stimulatory effect of resveratrol. To understand whether maintaining the interdomain interaction alone is sufficient for the resveratrol effect, we generated an E230R/R446E mutant swapping the two residues, with the expectation that the reciprocal change of amino acid residues will maintain a stable interdomain configuration. Evidently, the mutant had an impaired ability to stimulate SIRT1 activity and displayed a significantly weaker binding to the p53-AMC peptide (Fig. 4A,G). The result indicates that either the interdomain configuration is not restored by the amino acid swap, or a precise positioning of the two amino acids is required for the resveratrol effect.
Resveratrol effect on native peptides
Altogether, the structure shows that SIRT1 residues important for binding Res1 and Res2 are crucial for resveratrol-dependent stimulation of deacetylation of p53-AMC and are all located in the NTD. It is interesting that direct interactions between the peptide and the SIRT1 NTD are scarce, with their contacts mainly mediated by two resveratrol molecules (Figs. 1C, 2B). It was previously reported that resveratrol and other purported SIRT1 activators interact with fluorophore-attached p53 peptides weakly (Borra et al. 2005; Pacholec et al. 2010). The structure shows that resveratrol interacts with both the peptide and SIRT1, its NTD in particular. We attempted to measure the binding affinities between resveratrol and the p53-AMC peptide as well as that between resveratrol and the SIRT1 NTD (using isolated protein domain) by ITC, but they were too weak to be reliably quantitated. Nevertheless, it is clear that resveratrol and the NTD synergistically increased the binding affinities of the fluorophore-attached peptide to SIRT1. The structural, biochemical, and biophysical results are consistent with an adaptor role of resveratrol to contour the surface of the SIRT1 NTD for optimal binding of the p53-AMC peptide.
Although the resveratrol effect with the artificial p53-AMC peptide is clear, it is controversial with native peptides bearing no fluorophore. It was reported that resveratrol could stimulate SIRT1's activity toward acetylated peptides with a bulky aromatic residue at the +1 position, such as PGC-1α and FOXO3a (Hubbard et al. 2013). We also measured the effect of resveratrol on fluorophore-free PGC-1α and FOXO3a peptides using the enzyme-coupled microplate assay. We first determined the kinetic parameters of SIRT1 for these peptides: an 11-mer PGC-1α peptide that contains both the +1 tyrosine and the +6 phenylalanine suggested to have a beneficial effect on resveratrol activation, a hexameric FOXO3a peptide that has a tryptophan at the +1 position, and, for comparison, the p53-AMC peptide (Fig. 5A–E). While the addition of resveratrol significantly lowered the Km value for the p53-AMC peptide, the changes with PGC-1α and FOXO3a are less pronounced (Fig. 5E). For the two native peptides, we found no stimulation of SIRT1 activity with the addition of resveratrol under nonsaturation conditions in our assay using both the crystallized and the nearly full-length SIRT1 fragments (Fig. 5F). This result is consistent with the structural findings, as the AMC group in the p53-AMC peptide occupies the path of the main chain position of native peptides as seen in the structures of sirtuin–peptide complexes, and, in this manner, the side chain of the +1 residue in the SIRT1 complex with a native peptide cannot bind the protein in a way similar to that of the AMC group (Fig. 5G; Avalos et al. 2002).
Figure 5.

Resveratrol effect on native peptides. (A–D), Titration curves for determination of Km values of SIRT1-143CS toward the indicated peptides in the absence (black curves) and presence (red curves) of 0.2 mM resveratrol. An enzyme concentration of 200 nM in B, as opposed to 400 nM in the rest, was used to slow down the reaction for better precision measurement. (E) A summary of measured Michaelis-Menten kinetic parameters for the indicated peptides in the absence and presence of 0.2 mM resveratrol. (F) Deacetylase activities of SIRT1 fragments 143-CS and 89–747 toward the indicated peptides measured at 400 nM and 50 μM enzyme and peptide concentrations, respectively, in the absence and presence of 0.2 mM resveratrol. (G) Superposition of the fluorophoreless acetylated p53 peptide (carbon colored yellow) from an archaeal sirtuin complex (PDB ID 1MA3) with the SIRT1 ternary complex. Residue positions with respect to the acetylated lysine are indicated.
Discussion
SIRT1 differs from other mammalian sirtuins in having an extended NTD, which has been suggested to be a target for allosteric activation of SIRT1 activities. While it has been demonstrated that it indeed mediates the activation toward artificial peptides bearing a fluorophore group at the +1 position, there is considerable confusion with regard to activation of SIRT1 activity toward natural substrates. However, it is not unprecedented that an NTD of sirtuin harbors an allosteric activation mechanism toward native substrates. For example, binding of Sir4, principally through the NTD of Sir2, stimulates deacetylation of H4K16 by Sir2 (Tanny et al. 2004; Hsu et al. 2013), and binding by AROS and Lamin A, suggested via the NTD of SIRT1, potentiates the activity of SIRT1 toward p53 (Kim et al. 2007; Liu et al. 2012). Therefore, it is possible that, by modeling the protein–protein interaction effect with small molecules, allosteric activation of SIRT1 activation may be achieved. To learn about possible mechanisms, the structure of the NTD-containing SIRT1 complex with resveratrol and the fluorogenic peptide can serve as a model for mechanistic analyses.
Our structure shows that two of the three bound resveratrol molecules mediate the interaction between the p53-AMC peptide and the NTD of SIRT1. The NTD contains a core of three helices, which are arranged in a manner similar to that of three of the five helices in the NTD of yeast Sir2, although the NTDs are positioned quite differently with respect to their respective CDs in ligand-bound structures. A common feature of the two structures is that there are rarely any direct contacts between the NTD and CD in both proteins. It has been previously suggested and confirmed by molecular dynamics simulation that the two domains in Sir2 are not stably held together in the absence of Sir4 (Hsu et al. 2013). It is expected that the same is true for SIRT1 when the peptide and resveratrol are not bound. The structure shows that resveratrol serves as an adaptor to bridge the NTD and the peptide together by principally interacting with the peptidyl AMC moiety on one side and via specific NTD residues on the other side. The structural findings are consistent with previous as well as our enzyme kinetics and binding results that resveratrol lowered the Km value of the fluorogenic peptide and increased the binding affinity between the peptide and the enzyme (Howitz et al. 2003; Borra et al. 2005; Kaeberlein et al. 2005).
As with native peptides carrying no fluorophore, we did not detect significant changes of Km values with and without resveratrol and observed no stimulation of deacetylase activity of SIRT1 upon addition of resveratrol in our experiments. This is in agreement with several previous reports (Borra et al. 2005; Kaeberlein et al. 2005; Beher et al. 2009; Pacholec et al. 2010; Liu et al. 2012) and contrasts with some others (Dai et al. 2010; Hubbard et al. 2013). Significantly, the two native peptides used in our experiments all have an aromatic residue at the +1 position, and the PGC-1α peptide also contains an additional aromatic residue at the +6 position, which has been suggested to further stimulation of SIRT1 activity by STACs (Hubbard et al. 2013). From a structural point of view, the inability of resveratrol to stimulate SIRT1 activity toward native peptides may stem from the difference in the positioning of the +1 aromatic residue in native peptides and the coumarin group in p53-AMC. However, the mechanism of SIRT1 activation learned here—namely, resveratrol serves as an adaptor to strengthen the binding between the fluorogenic peptide and the NTD of SIRT1—should work for native peptides in principle if suitable compounds can be identified to mediate proper interactions between the substrate and SIRT1. The structural information obtained here may facilitate a computational search of such compounds. The structural property of SIRT1, together with further information on protein–protein interaction, should also benefit the understanding of new activation mechanisms of SIRT1 involving additional binding factors, such as Lamin A, which is reported to synergize with resveratrol to stimulate SIRT1 activity (Liu et al. 2012).
Materials and methods
Protein expression and purification
Human SIRT1 fragments were all expressed as his-sumo-tagged proteins in the BL21(DE3) strain of Escherichia coli using a pET28a-smt3 vector. The fragments used for crystallization studies—SIRT1-143 and SIRT1-143CS—encompass the NTD, the CD, and the C-terminal ESA region (amino acids 143–512 and 641–665). SIRT1-143CS differs from the wild-type SIRT1-143 by substitution of four cysteine residues to serine (C253S, C268S, C501S, and C502S) to improve the protein property for crystallization. SIRT1-241 and SIRT1-241CS are corresponding truncation variants lacking 240 N-terminal residues (amino acids 241–512 and 641–665). The longest SIRT1 fragments used in this study lack 88 N-terminal residues (amino acids 89–747). All SIRT1 fragments were expressed and purified following a protocol similar to that described below. Cells reaching a density of OD600 ∼0.6–0.8 were induced with 0.3 mM IPTG for ∼18 h at 16°C. Harvested cells were resuspended in a buffer containing 20 mM Tris (pH 8.0), 500 mM NaCl, 5% glycerol, and 1 mM PMSF and were lysed by sonication. The cell lysate was clarified by centrifugation, and the supernatant was applied to a Ni-IDA column and eluted via a 0∼100 mM imidazole gradient in 20 mM Tris (pH 8.0), 200 mM NaCl, and 5% glycerol. Eluted fractions enriched with the recombinant protein were pooled and treated with sumo protease for 2 h, and then the sample was diluted and loaded onto the Ni-IDA column again to remove the his-sumo tag. Afterward, the tag-free protein was concentrated and loaded onto a Superdex HiLoad 75 16/60 size exclusion column (GE Healthcare) in a buffer containing 20 mM HEPES (pH 7.5), 150 mM NaCl, 10% glycerol, and 0.1% β-mercaptoethanol. High-purity fractions were pooled and concentrated to ∼36 mg/mL, and the freshly purified proteins were used for crystallization.
Peptides
The p53-AMC peptide carrying the sequence of [acR]HK[Kac]-AMC, where [acR] stands for Nα-acetylated arginine, and [Kac] stands for Nε-acetylated lysine, was purchased from Sangong Biotech (Shanghai). Remaining peptides—PGC-1α (TKS[Kac]YDSLDFD) and FOXO3a (QLS[Kac]WP)—were purchased from Scilight Biotechnology.
Crystallization, data collection, structure determination, and refinement
The p53-AMC peptide was dissolved in water at 100 mg/mL, and a stock solution of 50 mM resveratrol (Sigma) in 50% DMSO was prepared. The peptide was added at 2:1 and resveratrol was added at 5:1 molar ratios with respect to SIRT1, and the mixture was incubated overnight at 4°C and centrifuged before setting up for crystallization. Crystals of the SIRT1-143CS, p53-AMC, and resveratrol complex were grown at 16°C by hanging-drop vapor diffusion using 1.0 μL of the protein–peptide–resveratrol mixture (∼32–36 mg/mL) and 1.0 μL of reservoir solution (0.9 M sodium malonate, 0.5% Jeffamin M-600, 0.1 M HEPES at pH 7.8).
For X-ray data collection, the crystals were flash-frozen in liquid nitrogen using a cryoprotectant prepared from the reservoir solution supplemented with 15% ethylene glycol. Diffraction data were collected at Beamline BL17U of the Shanghai Synchrotron Radiation Facility (SSRF) using a Quantum 315r CCD detector (ADSC) at a wavelength of 0.9792 Å, and the data were processed using HKL2000 (Otwinowski and Minor 1997). The crystal belonged to the P41 space group, and there were three complexes per asymmetric unit. The structure was solved by molecular replacement with the Phaser software (McCoy et al. 2007) using the structure of the CD of human SIRT1 (PDB ID 4I5I) as the search model. The remaining structure was manually built using Coot (Emsley and Cowtan 2004), and refinement was carried out using Refmac (Murshudov et al. 1997) and Phenix (Adams et al. 2010). The refined structure has a R-work and R-free of 20.7% and 25.2%, respectively. The density for one of the three SIRT1 molecules (chain C) in the asymmetric unit was poor, as also reflected in its higher average B factor (156.7 Å2 compared with 74.9 Å2 and 78.1 Å2 for chain A and chain B, respectively). The region spanning residues 157–173 of SIRT1 was disordered in the model, preventing us from reliably connecting an N-terminal fragment (142–156) to the major body of SIRT1. Detailed statistics for data collection and refinement are in Supplemental Table S1.
Atomic coordinates and diffraction data have been deposited with PDB under accession code 5BTR.
Enzyme-coupled deacetylation assay
The deacetylase activity of SIRT1 was measured using a continuous, enzyme-coupled microplate assay (Smith et al. 2009). In brief, the reaction product nicotinamide was converted to nicotinic acid and ammonia by nicotinamidase PncA. Next, glutamate dehydrogenase converted ammonia, α-ketoglutarate, and NADH to glutamate and NAD+. The oxidation of NADH was measured spectrophotometrically at 340 nm using a Multilabel reader (Enspire, PerkinElmer), with the extinction coefficient for NADH of 6.22 mM−1 cm−1.
Determination of Km values and measurements of SIRT1 deacetylase activities were performed with individual peptides at various concentrations but with the rest of the conditions fixed unless explicitly stated otherwise; i.e., 2 mM NAD, 400 nM SIRT1-143CS, 0.2 mM NADH, 3.3 mM α-ketoglutarate, 1 μM PncA, 3 U of bovine liver glutamate dehydrogenase (Sigma), 1 mM DTT, and 2% DMSO in PBS buffer (pH 7.4). Recombinant PncA was prepared according to a published protocol (Garrity et al. 2007). The reactions were carried out in 100 μL with a flat-bottom 96-well plate. The assay mixtures without NAD were first incubated for 5–10 min at 37°C until the absorbance at 340 nm reached equilibrium. Next, the reactions were initiated by the addition of NAD (2 μL of stock solution at 100 mM), and changes of absorbance at 340 nm were recorded and analyzed continuously for 15 min. Readings from control reactions lacking NAD were taken as the background and subtracted from that of reactions containing NAD.
For determination of Km values, individual peptides with concentrations ranging from 10 to 800 μM were titrated in the absence or presence of 0.2 mM resveratrol. A SIRT1 concentration of 200 nM, instead of 400 nM as in other measurements, was used in the measurement of Km value of the p53-AMC peptide with resveratrol.
The data were first analyzed using Excel and then fitted to the Michaelis-Menten equation using GraphPad Prism (GraphPad Software, Inc.). The kinetic parameters were calculated according to the Lineweaver-Burk plot. Deacetylation activities were measured in the absence and presence of 0.2 mM reseveratrol and peptide concentrations at 100 μM for p53-AMC and 50 μM for PGC-1α and FOXO 3a. The data were also analyzed using Excel and GraphPad Prism, and the deacetylation rates were obtained from the velocity calculated from the linear portion of the curves. All of the measurements above were repeated three times.
ITC measurements
All ITC measurements were carried out at 25°C using an ITC200 calorimeter (MicroCal LLC). Except for the experiment with the E230R/R446E sample, a total of 20 injections of 1.5 mM peptide, with a 0.5-μL first drop (data not used) and 2-μL subsequent drops, was titrated into a 200-μL well of 0.1 mM protein solution. For the E230R/R446E mutant, a peptide concentration of 4 mM was used. The reference power was set at 5 μCal/sec, initial delay was set at 60 sec, and the sample cell was stirred at 600 rpm. Injections were carried out at 0.5 μL/sec with an interval of 2 min between injections. The titrant and the protein well solution were in the same buffer (20 mM HEPES at pH 7.5, 150 mM NaCl, 10% glycerol), all with or without 0.5 mM resveratrol. Buffer control for each experiment was performed under the same conditions without peptide in the titrant, and the measured background heat was subtracted from the integrated data. All of the data were integrated and analyzed using a one-site model with the Origin software (Origin Laboratory).
Supplementary Material
Acknowledgments
We thank SSRF beamline scientists for help with data collection, Dr. Jorge Escalante-Semerena at the University of Wisconsin-Madison for the generous gift of the E. coli expression plasmid of PncA, Litao Sun for participation at an early stage of the work, Yuanyuan Chen for help with ITC experiments, and Zhensheng Xie and Lili Niu for help with MS analyses. We also thank grant support from the Chinese Ministry of Science and Technology (2012CB910702), the Natural Science Foundation of China (31430018 and 31210103914), the Strategic Priority Research Program (XDB08010100), and the Key Research Program (KJZD-EW-L05) of the Chinese Academy of Sciences, the National Key New Drug Creation and Manufacturing Program of China (2014ZX09507002), and Pfizer. N.Y. is also supported by the Youth Innovation Promotion Association of the Chinese Academy of Sciences.
Footnotes
Supplemental material is available for this article.
Article is online at http://www.genesdev.org/cgi/doi/10.1101/gad.265462.115.
References
- Adams PD, Afonine PV, Bunkoczi G, Chen VB, Davis IW, Echols N, Headd JJ, Hung LW, Kapral GJ, Grosse-Kunstleve RW, et al. 2010. Phenix: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr D Biol Crystallogr 66: 213–221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Avalos JL, Celic I, Muhammad S, Cosgrove MS, Boeke JD, Wolberger C. 2002. Structure of a Sir2 enzyme bound to an acetylated p53 peptide. Mol Cell 10: 523–535. [DOI] [PubMed] [Google Scholar]
- Baur JA. 2010. Biochemical effects of SIRT1 activators. Biochim Biophys Acta 1804: 1626–1634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beher D, Wu J, Cumine S, Kim KW, Lu SC, Atangan L, Wang M. 2009. Resveratrol is not a direct activator of SIRT1 enzyme activity. Chem Biol Drug Des 74: 619–624. [DOI] [PubMed] [Google Scholar]
- Bemis JE, Vu CB, Xie R, Nunes JJ, Ng PY, Disch JS, Milne JC, Carney DP, Lynch AV, Jin L, et al. 2009. Discovery of oxazolo[4,5-b]pyridines and related heterocyclic analogs as novel SIRT1 activators. Bioorg Med Chem Lett 19: 2350–2353. [DOI] [PubMed] [Google Scholar]
- Borra MT, Smith BC, Denu JM. 2005. Mechanism of human SIRT1 activation by resveratrol. J Biol Chem 280: 17187–17195. [DOI] [PubMed] [Google Scholar]
- Brunet A, Sweeney LB, Sturgill JF, Chua KF, Greer PL, Lin Y, Tran H, Ross SE, Mostoslavsky R, Cohen HY, et al. 2004. Stress-dependent regulation of FOXO transcription factors by the SIRT1 deacetylase. Science 303: 2011–2015. [DOI] [PubMed] [Google Scholar]
- Dai H, Kustigian L, Carney D, Case A, Considine T, Hubbard BP, Perni RB, Riera TV, Szczepankiewicz B, Vlasuk GP, et al. 2010. SIRT1 activation by small molecules: kinetic and biophysical evidence for direct interaction of enzyme and activator. J Biol Chem 285: 32695–32703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davenport AM, Huber FM, Hoelz A. 2014. Structural and functional analysis of human SIRT1. J Mol Biol 426: 526–541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Emsley P, Cowtan K. 2004. Coot: model-building tools for molecular graphics. Acta Crystallogr D Biol Crystallogr 60: 2126–2132. [DOI] [PubMed] [Google Scholar]
- Feldman JL, Dittenhafer-Reed KE, Denu JM. 2012. Sirtuin catalysis and regulation. J Biol Chem 287: 42419–42427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Finnin MS, Donigian JR, Pavletich NP. 2001. Structure of the histone deacetylase SIRT2. Nat Struct Biol 8: 621–625. [DOI] [PubMed] [Google Scholar]
- Frye RA. 2000. Phylogenetic classification of prokaryotic and eukaryotic Sir2-like proteins. Biochem Biophys Res Commun 273: 793–798. [DOI] [PubMed] [Google Scholar]
- Garrity J, Gardner JG, Hawse W, Wolberger C, Escalante-Semerena JC. 2007. N-lysine propionylation controls the activity of propionyl-CoA synthetase. J Biol Chem 282: 30239–30245. [DOI] [PubMed] [Google Scholar]
- Gertz M, Nguyen GT, Fischer F, Suenkel B, Schlicker C, Franzel B, Tomaschewski J, Aladini F, Becker C, Wolters D, et al. 2012. A molecular mechanism for direct sirtuin activation by resveratrol. PLoS One 7: e49761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guarente L. 2011. Franklin H. Epstein lecture: sirtuins, aging, and medicine. N Engl J Med 364: 2235–2244. [DOI] [PubMed] [Google Scholar]
- Howitz KT, Bitterman KJ, Cohen HY, Lamming DW, Lavu S, Wood JG, Zipkin RE, Chung P, Kisielewski A, Zhang LL, et al. 2003. Small molecule activators of sirtuins extend Saccharomyces cerevisiae lifespan. Nature 425: 191–196. [DOI] [PubMed] [Google Scholar]
- Hsu HC, Wang CL, Wang M, Yang N, Chen Z, Sternglanz R, Xu RM. 2013. Structural basis for allosteric stimulation of Sir2 activity by Sir4 binding. Genes Dev 27: 64–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hubbard BP, Gomes AP, Dai H, Li J, Case AW, Considine T, Riera TV, Lee JE, E SY, Lamming DW, et al. 2013. Evidence for a common mechanism of SIRT1 regulation by allosteric activators. Science 339: 1216–1219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Imai S, Armstrong CM, Kaeberlein M, Guarente L. 2000. Transcriptional silencing and longevity protein Sir2 is an NAD-dependent histone deacetylase. Nature 403: 795–800. [DOI] [PubMed] [Google Scholar]
- Jin L, Wei W, Jiang Y, Peng H, Cai J, Mao C, Dai H, Choy W, Bemis JE, Jirousek MR, et al. 2009. Crystal structures of human SIRT3 displaying substrate-induced conformational changes. J Biol Chem 284: 24394–24405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaeberlein M, McDonagh T, Heltweg B, Hixon J, Westman EA, Caldwell SD, Napper A, Curtis R, DiStefano PS, Fields S, et al. 2005. Substrate-specific activation of sirtuins by resveratrol. J Biol Chem 280: 17038–17045. [DOI] [PubMed] [Google Scholar]
- Kang H, Suh JY, Jung YS, Jung JW, Kim MK, Chung JH. 2011. Peptide switch is essential for Sirt1 deacetylase activity. Mol Cell 44: 203–213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim EJ, Kho JH, Kang MR, Um SJ. 2007. Active regulator of SIRT1 cooperates with SIRT1 and facilitates suppression of p53 activity. Mol Cell 28: 277–290. [DOI] [PubMed] [Google Scholar]
- Lakshminarasimhan M, Rauh D, Schutkowski M, Steegborn C. 2013. Sirt1 activation by resveratrol is substrate sequence-selective. Aging (Albany NY) 5: 151–154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Landry J, Sutton A, Tafrov ST, Heller RC, Stebbins J, Pillus L, Sternglanz R. 2000. The silencing protein SIR2 and its homologs are NAD-dependent protein deacetylases. Proc Natl Acad Sci 97: 5807–5811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu B, Ghosh S, Yang X, Zheng H, Liu X, Wang Z, Jin G, Zheng B, Kennedy BK, Suh Y, et al. 2012. Resveratrol rescues SIRT1-dependent adult stem cell decline and alleviates progeroid features in laminopathy-based progeria. Cell Metab 16: 738–750. [DOI] [PubMed] [Google Scholar]
- Luo J, Nikolaev AY, Imai S, Chen D, Su F, Shiloh A, Guarente L, Gu W. 2001. Negative control of p53 by Sir2α promotes cell survival under stress. Cell 107: 137–148. [DOI] [PubMed] [Google Scholar]
- McCoy AJ, Grosse-Kunstleve RW, Adams PD, Winn MD, Storoni LC, Read RJ. 2007. Phaser crystallographic software. J Appl Crystallogr 40: 658–674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Milne JC, Lambert PD, Schenk S, Carney DP, Smith JJ, Gagne DJ, Jin L, Boss O, Perni RB, Vu CB, et al. 2007. Small molecule activators of SIRT1 as therapeutics for the treatment of type 2 diabetes. Nature 450: 712–716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Min J, Landry J, Sternglanz R, Xu RM. 2001. Crystal structure of a SIR2 homolog–NAD complex. Cell 105: 269–279. [DOI] [PubMed] [Google Scholar]
- Motta MC, Divecha N, Lemieux M, Kamel C, Chen D, Gu W, Bultsma Y, McBurney M, Guarente L. 2004. Mammalian SIRT1 represses forkhead transcription factors. Cell 116: 551–563. [DOI] [PubMed] [Google Scholar]
- Murshudov GN, Vagin AA, Dodson EJ. 1997. Refinement of macromolecular structures by the maximum-likelihood method. Acta Crystallogr D Biol Crystallogr 53: 240–255. [DOI] [PubMed] [Google Scholar]
- Nguyen GT, Gertz M, Steegborn C. 2013. Crystal structures of Sirt3 complexes with 4′-bromo-resveratrol reveal binding sites and inhibition mechanism. Chem Biol 20: 1375–1385. [DOI] [PubMed] [Google Scholar]
- Otwinowski Z, Minor W. 1997. Processing of X-ray diffraction data collected in oscillation mode. Methods Enzymol 276: 307–326. [DOI] [PubMed] [Google Scholar]
- Pacholec M, Bleasdale JE, Chrunyk B, Cunningham D, Flynn D, Garofalo RS, Griffith D, Griffor M, Loulakis P, Pabst B, et al. 2010. SRT1720, SRT2183, SRT1460, and resveratrol are not direct activators of SIRT1. J Biol Chem 285: 8340–8351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pan PW, Feldman JL, Devries MK, Dong A, Edwards AM, Denu JM. 2011. Structure and biochemical functions of SIRT6. J Biol Chem 286: 14575–14587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pan M, Yuan H, Brent M, Ding EC, Marmorstein R. 2012. SIRT1 contains N- and C-terminal regions that potentiate deacetylase activity. J Biol Chem 287: 2468–2476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodgers JT, Lerin C, Haas W, Gygi SP, Spiegelman BM, Puigserver P. 2005. Nutrient control of glucose homeostasis through a complex of PGC-1α and SIRT1. Nature 434: 113–118. [DOI] [PubMed] [Google Scholar]
- Sanders BD, Jackson B, Marmorstein R. 2010. Structural basis for sirtuin function: what we know and what we don't. Biochim Biophys Acta 1804: 1604–1616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sauve AA, Celic I, Avalos J, Deng H, Boeke JD, Schramm VL. 2001. Chemistry of gene silencing: the mechanism of NAD+-dependent deacetylation reactions. Biochemistry 40: 15456–15463. [DOI] [PubMed] [Google Scholar]
- Sauve AA, Moir RD, Schramm VL, Willis IM. 2005. Chemical activation of Sir2-dependent silencing by relief of nicotinamide inhibition. Mol Cell 17: 595–601. [DOI] [PubMed] [Google Scholar]
- Sauve AA, Wolberger C, Schramm VL, Boeke JD. 2006. The biochemistry of sirtuins. Annu Rev Biochem 75: 435–465. [DOI] [PubMed] [Google Scholar]
- Schuetz A, Min J, Antoshenko T, Wang CL, Allali-Hassani A, Dong A, Loppnau P, Vedadi M, Bochkarev A, Sternglanz R, et al. 2007. Structural basis of inhibition of the human NAD+-dependent deacetylase SIRT5 by suramin. Structure 15: 377–389. [DOI] [PubMed] [Google Scholar]
- Sinclair DA, Guarente L. 2014. Small-molecule allosteric activators of sirtuins. Annu Rev Pharmacol Toxicol 54: 363–380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith JS, Brachmann CB, Celic I, Kenna MA, Muhammad S, Starai VJ, Avalos JL, Escalante-Semerena JC, Grubmeyer C, Wolberger C, et al. 2000. A phylogenetically conserved NAD+-dependent protein deacetylase activity in the Sir2 protein family. Proc Natl Acad Sci 97: 6658–6663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith BC, Hallows WC, Denu JM. 2009. A continuous microplate assay for sirtuins and nicotinamide-producing enzymes. Anal Biochem 394: 101–109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanner KG, Landry J, Sternglanz R, Denu JM. 2000. Silent information regulator 2 family of NAD-dependent histone/protein deacetylases generates a unique product, 1-O-acetyl-ADP-ribose. Proc Natl Acad Sci 97: 14178–14182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanny JC, Moazed D. 2001. Coupling of histone deacetylation to NAD breakdown by the yeast silencing protein Sir2: evidence for acetyl transfer from substrate to an NAD breakdown product. Proc Natl Acad Sci 98: 415–420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanny JC, Kirkpatrick DS, Gerber SA, Gygi SP, Moazed D. 2004. Budding yeast silencing complexes and regulation of Sir2 activity by protein–protein interactions. Mol Cell Biol 24: 6931–6946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vaquero A, Scher M, Lee D, Erdjument-Bromage H, Tempst P, Reinberg D. 2004. Human SirT1 interacts with histone H1 and promotes formation of facultative heterochromatin. Mol Cell 16: 93–105. [DOI] [PubMed] [Google Scholar]
- Vaziri H, Dessain SK, Ng Eaton E, Imai SI, Frye RA, Pandita TK, Guarente L, Weinberg RA. 2001. hSIR2(SIRT1) functions as an NAD-dependent p53 deacetylase. Cell 107: 149–159. [DOI] [PubMed] [Google Scholar]
- Zhao K, Chai X, Marmorstein R. 2003. Structure of the yeast Hst2 protein deacetylase in ternary complex with 2′-O-acetyl ADP ribose and histone peptide. Structure 11: 1403–1411. [DOI] [PubMed] [Google Scholar]
- Zhao X, Allison D, Condon B, Zhang F, Gheyi T, Zhang A, Ashok S, Russell M, MacEwan I, Qian Y, et al. 2013. The 2.5 A crystal structure of the SIRT1 catalytic domain bound to nicotinamide adenine dinucleotide (NAD+) and an indole (EX527 analogue) reveals a novel mechanism of histone deacetylase inhibition. J Med Chem 56: 963–969. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.