

Abstract
Background
Leucomeris decora and Nouelia insignis (Asteraceae) are narrowly and allopatrically distributed species, separated by the important biogeographic boundary Tanaka Line in Southwest China. Previous morphological, cytogenetic and molecular studies suggested that L. decora is sister to N. insignis. However, it is less clear how the two species diverged, whether in full isolation or occurring gene flow across the Tanaka Line. Here, we performed a molecular study at the population level to characterize genetic differentiation and decipher phylogeographic history in two closely related species based on variation examined in plastid and nuclear DNAs using a coalescent-based approach.
Results
These morphologically distinct species share plastid DNA (cpDNA) haplotypes. In contrast, Bayesian analysis of nuclear DNA (nDNA) uncovered two distinct clusters corresponding to L. decora and N. insignis. Based on the IMa analysis, no strong indication of migration was detected based on both cpDNA and nDNA sequences. The molecular data pointed to a major west-east split in nuclear DNA between the two species corresponding with the Tanaka Line. The coalescent time estimate for all cpDNA haplotypes dated to the Mid-Late Pleistocene. The estimated demographic parameters showed that the population size of L. decora was similar to that of N. insignis and both experienced limited demographic fluctuations recently.
Conclusions
The study revealed comprehensive species divergence and phylogeographic histories of N. insignis and L. decora divided by the Tanaka Line. The phylogeographic pattern inferred from cpDNA reflected ancestrally shared polymorphisms without post-divergence gene flow between species. The marked genealogical lineage divergence in nDNA provided some indication of Tanaka Line for its role as a barrier to plant dispersal, and lent support to its importance in promoting strong population structure and allopatric divergence.
Electronic supplementary material
The online version of this article (doi:10.1186/s12862-015-0374-5) contains supplementary material, which is available to authorized users.
Keywords: Leucomeris decora, Nouelia insignis, Plastid DNA, Nuclear DNA, Genetic divergence, Migration, Tanaka line
Background
The Quaternary climate fluctuation largely influenced the distribution ranges of plants, leading to range expansion, contraction and/or population extinction [1]. Range expansion can result in contacts between previously geographically separated species and lead to profound consequences for lineage or species evolution, as they may cause introgression between species and haplotypes sharing across species. Range contraction of widespread species may have left isolated populations in marginal areas, providing chances for peripheral speciation of regional endemics [2–5]. It is assumed that gene transfer events may have occurred between two species in previously sympatry or parapatry populations, when species-specific haplotypes are found in another with allopatric distribution [4, 6]. In addition, sharing of haplotypes across species can not only be caused by introgression but also by incomplete lineage sorting. The boundaries between species can be blurred due to the existence of introgression and/or retention of ancestral polymorphism [7].
Generally, in case of incomplete lineage sorting, we would expect the shared cytoplasmic-nuclear haplotypes to be randomly distributed in a mosaic pattern over the species geographic range [8, 9]. In contrast, if haplotypes sharing between both species arises as a function of introgression, we would expect co-occurring populations of the two species are more similar than randomly chosen population pairs [5, 10, 11]. Recent coalescent methods like IM model (Isolation with Migration) based on sequencing loci from different inherited systems can also help to address these questions by estimating the extent of genetic divergence and revealing demographic dynamics by distributional changes, which are applied to within species or between recently divergent taxa [12, 13].
Nouelia insignis Franch. and Leucomeria decora Kurz, both belonging to the Mutisieae (Asteraceae) [14], are characterized by woody growth form and rare in China. N. insignis is a monotypic genus endemic to southwest China and Leucomeris contain two extant species, one of which occurred in Yunnan China. N. insignis and L. decora are not sharply divergent morphologically but differ most prominently in flora traits. L. decora has capitula in a dense terminal cyme and tubular florets, whereas N. insignis is characterized with numerous, solitary, terminal and radiate capitula, and fertile florets with marginal uniseriate, bilabiate florets, and central tubular florets [15]. Despite the little distinctively morphological differentiation, they are closely related phylogenetically: Leucomeris and Nouelia were considered to be independent and treated as Nouelia group based on morphological characters [14, 16]; and then molecular studies based on chloroplast DNA variation in Panero and Funk showed they clustered together within a clade that also contains the South American genera Hyalis and Ianthopappus [17]; later Funk et al. considered them a ‘Leucomeris’ clade [18]. Particularly, a previous study inferred a sister relationship between these two species based on molecular phylogeny (cpDNA and nDNA) of eastern Asian Mutisieae [19]. Moreover, the two species have the same chromosome numbers (2n = 54), and this character distinguishes them from the other genera in the tribe Mutisieae [20]. Together, all these suggest that L. decora and N. insignis are tightly related, differing from other Asian or American species of Asteraceae.
L. decora and N. insignis are allopatric over their natural ranges. L. decora has a large distribution range, extending from south Asian countries like Myanmar, Thailand and Vietnam to China, whereas N. insignis has a more restricted distribution in Yunnan and Sichuan in China [21]. They both can grow in valleys with dry and hot environmental condition; however, in contrast, L. decora also prefers the edge of the forest and isolated mountaintops [22, 23]. In particular, the distribution ranges are found to coincide with known the biogeographic boundary, i.e. Tanaka Line, which separates two environmentally divergent subkingdoms, that is the ‘Sino-Himalayan Forest’ to the west and the ‘Sino-Japanese Forest’ to the east [24–26]. Although it is not an apparent physical barrier like mountain ranges, river systems or sea straits, deep divergences between the boundary have been reported (such as [27–30]). Taking the reason into account, one recent phylogeographic study supported that the dramatic climate changes during the (Late) Pleistocene, when the presently differing monsoon regimes on either side of the Tanaka Line established, may be a reasonable explanation for the divergence [31]. However, for studies with sufficient sampling from areas proximal to and/or along the Tanaka Line are still lacking, assessing the feature as a genetic boundary remains largely hypothetical.
Based on the distribution range and closely related relationship, these two species may provide a model to study the present genetic variation and assess whether there is long-term population isolation without migration across the Tanaka Line. Although, previous study using molecular sequences hypothesized the recent origins and phenotypic evolution via adaptation to dry and cool habitats during the Quaternary glaciation; however, only a small number of populations of L. decora and N. insignis and only one individual per population were sampled in the above molecular investigation. Thus, more comprehensive analyses based on a sufficient population sampling of both species are needed to address the population genetic differentiation and to investigate historical demography of the two species. In the present study, we compared sequence variation at both cpDNA fragment and the low-copy nuclear gene, across a large number of individuals and populations covering the natural ranges of L. decora and N. insignis in China. Specifically, we aim to determine the amount of sequence divergence and possible gene flow after divergence between L. decora and N. insignis, and to disentangle the extant genealogical lineage patterns possibly associated with the Tanaka Line, and attempt to explore their demographic history. Together, these analyses should add our understanding of allopatric species divergence across Tanaka Line and the possible explanation for the Tanaka Line as a biogeographic boundary in southwest China.
Methods
Population sampling
Fresh leaves of L. decora and N. insignis were collected in the field and dried directly with silica gel. We sampled 11 natural populations of L. decora and 16 of N. insignis, respectively, covering the distributional range of the two species in China. In total, 251 individuals from 27 populations were surveyed for chloroplast and nuclear DNA variations. Voucher specimens were collected and deposited in the Herbarium of Kunming Institute of Botany, Chinese Academy of Sciences (KUN). Detailed information on the geographical distribution of the two species and the localities of the populations sampled in this study is shown in Additional file 1: Table S1 and Additional file 2: Table S2.
DNA extraction, PCR amplification, DNA sequencing and cloning
Genomic DNA was extracted from silica-dried leaves using the CTAB method [32] with some modifications. One chloroplast DNA intergenic spacer rpl32-trnL(UAG) [33] was amplified and sequenced. We followed our previous study in sequencing chloroplast DNA [34]. In addition, we selected one single or low-copy nuclear locus to estimate the genetic diversity and geographic structure of the two species. The granule-bound starch synthase gene (GBSSI or WAXY) has been reported to be single–copy in diploid grasses [35, 36] and several dicots [37, 38]. Recently, it has been broadly used in molecular study [39–41].
For the GBSSI gene, it was amplified and sequenced using the pair of primer GBSSI2F (5’-ACA TTG CYT ACC AAG GNA GA-3’) and GBSSI2R (5’-AAC TGA ATG AGA CCA CAM GG-3’) in the preliminary phase of the study, designed from the complete coding sequence of Ipomoea batatas (Conolvulaceae, AB071976) [42] and its homologous sequences using the program Primer Premier 5.0. The obtained sequences were blasted for sequence homology to ensure that they were GBSSI genes encoding granule-bound starch synthase. Then one pair of primer GBSSIF: (5’-TTG ATT TCA TTG TTG ATG GGT-3’) and GBSSIR (5’-GGG CCA GTT GCA CAT TGA ATT TAG C-3’) was designed according to the previously obtained sequences for amplification and sequencing, which was conducted in a similar way to that described by previous study [34].
As for the nuclear gene region, some individuals with only one heterozygous site were directly split into two alleles through haplotype subtraction [43], while products from individuals with more than 2 heterozygous sites (according to the first sequencing round) were re-sequenced to eliminate sequencing errors. Products from any other individuals that could not be directly sequenced or had more than 2 heterozygous sites according to the second sequencing round were cloned.
Analyses of variation for plastid and nuclear DNAs
Multiple alignments of the sequences were obtained using Clustal X 1.81 [44] and then improved manually by BioEdit [45]. Molecular diversity indices, including the number of haplotypes, haplotype diversity (Hd), nucleotide diversity (π) [46] were estimated for the species and each population using DnaSP program version 3.95 [47]. The total gene diversity (HT) and the average gene diversity within populations (HS) were calculated for L. decora and N. insignis for cpDNA markers. Two measures of genetic differentiation within two species, GST (coefficient depending only on the frequencies of the haplotypes) and NST (coeffient of genetic variation influenced by both haplotype frequencies and genetic distances between haplotypes) were estimated following Pons and Petit [48] as implemented in the program PERMUT. Genetic differentiation between the two species was assessed in two ways. First, hierarchical partitioning of diversity among species, populations and individuals was estimated on the basis of AMOVA in Arlequin 3.1 with significance tested using 1000 permutations [49]. Secondly, a Bayesian clustering analysis using STRUCTURE ver. 2.3 was conducted to assess population structure at nuclear locus for all samples. An admixed model was used, and the correlated allele frequencies among clusters were assumed. To estimate the number of clusters (K), values of K from 1 to 10 was set using 20 independent runs per K to test the stability of the results. The optimal K was estimated according to the model value (ΔK) following the procedure described by [50].
Phylogenetic analyses and dating TMRCA of cpDNA lineages
In order to infer the interspecific relationships between the two species, cpDNA and nDNA haplotypes were reconstructed using statistical parsimony as implemented in TCS with the connection limit set to 95 % and gaps treated as fifth character [51]. For this analysis, substitution and indels were assumed to evolve with equal possibility although they may exhibit different mutation rates. And indels longer than 1 bp were coded as substitutions and mononucleotide repeats were removed due to their high degree of homoplasy [52]. The time of the most recent common ancestor was estimated using Bayesian inference on the cpDNA sequence rpl32-trnL haplotypes as implemented in BEAST v.1.5.4 [53]. We used a HKY substitution model, selected by jModelTest [54], a strict molecular clock and a Yule process as tree prior. We assumed minimum and maximum values of a range of average mutation rates reported for synonymous sites of plant chloroplast genes [i.e., 1.2 and 1.7 × 10-9 substitutions per site per year (s/s/y)] for the BEAST analysis [55]. Three separate MCMC runs were performed, each of 1 × 107 generations, with sampling every 1000th generation, following a burn-in of the initial 10 % cycles. Convergence of the parameters sampled was checked in TRACER v.1.5 and combined with Log-Combiner v.1.5.4 [56].
Neutrality tests and mismatch distribution analyses
In order to test whether historical expansion events have ever occurred in L. decora or N. insignis, two neutrality tests were performed: (1) Tajima’s D considering the frequency of mutation (segregating sites) [57], and (2) Fu’s Fs based on the haplotype distribution [58]. The demographic history of a population could be inferred by comparing such neutrality tests, given that a range expansion is suggested when Tajima’s D and Fu’s Fs are significantly negative. The statistical significance of these estimates was calculated with 1000 permutations. In addition, the molecular distances among sequences were estimated through pairwise differences. The distribution of these differences (mismatch distribution) was used to test two population expansion models, in which cases unimodal distributions are expected [59]. We tested the null hypotheses of a spatial expansion and a pure demographic expansion through the sum of squares deviation (SSD) between the observed and the expected distributions and through the Harpending’s raggedness index, which were used to test the goodness-fit of the observation mismatch distribution to the expectation of expansion models. All the above analyses were implemented in Arlequin.3.1 [49].
Demographic parameters of the IM model
Divergence time between the two species and effective population sizes as well as interspecific gene flow by calculating relative migration rates between species were estimated using an isolation-with-migration (IM) model in IMa version 12/19/2009 [60]. This model uses Markov chain Monte Carlo (MCMC) to estimate the posterior probability densities of estimated parameters. It involves several simplifying assumptions such as neutrality and no recombination within loci, lack of genetic contribution from unsampled populations, and random mating in ancestral and descendent populations. [60, 61]. It is difficult to satisfy all of these assumptions in an empirical study. We used the program IMgc [62] to analyze recombination within the nuclear locus, and the only maximally informative and recombination-free datasets identified were included in the final analysis. IMa was run for the entire dataset, including all sampled nuclear sequences without recombination and chloroplast sequences. We performed preliminary simulations to assess convergence of the MCMC chains on the data stationary distribution and only estimates whose posterior distribution dropped to zero within the prior intervals investigated were trusted. For each analysis, burn-in was set to 10 million steps, and a total of 50 million steps were conducted. To verify convergence on the same parameter values, we ran the analysis three times with different random seeds. Demographic parameters were scaled to the generation time and neutral mutation rate. We used a generation time of five years in both L. decora and N. insignis as observed in the field and under cultivation. In the absence of a well-calibrated estimate in L. decora and N. insignis, we applied a generic average mutation rate of 1.45 × 10-9 substitution per site per year (1.2 and 1.7 × 10-9) for cpDNA and 6.1 × 10-9 (5.1 and 7.1 × 10-9) for nDNA [55]; and the geometric average mutation rate of the two marker sets was used to scale the outputs to demographic units.
Results
Chloroplast haplotype lineages and dating TMRCA
One indel and three substitutions were detected from the cpDNA region rpl32- trnL(UAG) (894-900 bp excluding the mononucleotide repeats) of L. decora and N. insignis samples, and were recoved 5 chloroplast haplotypes (C1-C5). All haplotype sequences were deposited in GenBank databases under the accession numbers JF915764–JF915768. The most common haplotype C2 (frequency 62.1 %) was shared by the two species, and 37 %, 78 % of the investigated individuals carried this haplotype in L. decora and N. insignis, respectively. C3 (24.7 %) only occurred in L. decora and the other haplotypes (C1, C4, C5) were only found in N. insignis with low frequencies ranging from 1.95 % to 5.98 % (Additional file 1:Table S1 and Additional file 2: Table S2). The distribution and network of cpDNA haplotypes C1–C5 are shown in Fig. 1. The network of chloroplast haplotypes indicated that C2 was possibly ancestor to the others and away from the remaining haplotypes by a single mutation (Fig. 1b). And it was fixed in most of N. insignis populations located in the Jinsha drainage and northwestern populations of L. decora (Fig. 1). All the remaining populations of L. decora in centre/southwest Yunnan were fixed for C3 except one population SP. C1 and C4 were restricted to the Nanpan drainage (CJ, HN and ML) and C5 was only found in population DY and YM. The total cpDNA diversity was slightly higher in L. decora (HT = 0.487) than in N. insignis (HT = 0.422). Between-population differentiation levels estimated by both GST and AMOVA are relatively low within N. insignis (GST = 0.849, FST = 0.896) than in L. decora (GST = 0.862, FST = 0.907) (Tables 1, 2). The Bayesian TMRCA analysis indicated that all sampled cpDNA haplotypes of L. decora and N. insignis coalesce at about 0.54 Ma (95 % confidence interval: 0.01–1.32 Ma) or 0.38 Ma (95 % confidence interval: 0.01–0.93 Ma), assuming minimum and maximum (average) rates of synonymous substitution in cpDNA, respectively. This suggests that the divergence between L. decora and N. insignis falls into the mid-late Pleistocene.
Fig. 1.
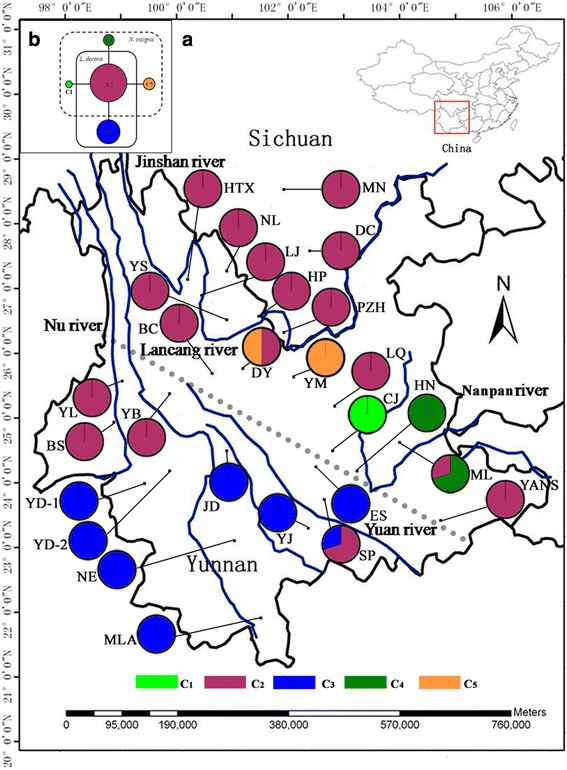
Geographic distribution and network of cpDNA haplotypes. a. Distribution of 5 chloroplast DNA (cpDNA) haplotypes detected within and among 27 populations of Leucomeris decora and Nouelia insignis. Full names of the abbreviations for the populations are shown in Additional file 1: Table S1 and Additional file 2: Table S2. b. Statistical parsimony network of genealogical relationships between five cpDNA haplotypes. Letters in/around circles represent haplotypes at each locus. The size of the circles corresponds to the frequency of each haplotype and each solid line represents one mutational step
Table 1.
Different hierachical types of analysis of molecular variance (AMOVA) of Leucomeris decora and Nouelia insignis
| Markers | Source of variation | d.f. | Sum of squares | Variance | % Total | Fixation index |
|---|---|---|---|---|---|---|
| cpDNA | Between species | 1 | 24.9 | 0.18 | 35.5 | FCT = 0.355** |
| Among populations | 25 | 70.9 | 0.30 | 58.7 | ||
| Within populations | 224 | 6.7 | 0.03 | 5.8 | FST = 0.942** | |
| L. decora | Among populations | 10 | 21.1 | 0.23 | 90.7 | FST = 0.907** |
| Within populations | 88 | 2.1 | 0.02 | 9.3 | ||
| N. insignis | Among populations | 15 | 49.9 | 0.35 | 89.6 | FST = 0.896** |
| Within populations | 136 | 5.5 | 0.04 | 10.4 | ||
| nDNA | Between species | 1 | 239.0 | 0.98 | 68.4 | FCT = 0.684** |
| Among populations | 25 | 78.3 | 0.15 | 10.6 | ||
| Within populations | 475 | 143.0 | 0.30 | 21.0 | FST = 0.79** | |
| L. decora | Among populations | 10 | 33.9 | 0.16 | 26.3 | FST = 0.263** |
| Within populations | 187 | 86.0 | 0.46 | 73.7 | ||
| N. insignis | Among populations | 15 | 44.5 | 0.15 | 42.4 | FST = 0.424** |
| Within populations | 288 | 57 | 0.20 | 57.6 |
d.f., degree of random; **P < 0.001
Nuclear DNA variation and haplotype lineages
Among the 251 individuals of L. decora and N. insignis, 214 and 37 were homozygotes and heterozygotes, respectively. A total of 17 genotypes (alleles) were recognized based on 9 substitutions in the sequence alignment (688 bp). All haplotype sequences were deposited in GenBank databases under the accession numbers JF915747–JF915763. The distribution and network of nDNA haplotypes H1-H17 are shown in Fig. 2. In the network, several closed loops, perhaps resulted from recombination, were resolved using the rules given by Templeton and Sing [63] and Posada and Crandall [64]. Seven haplotypes (H1–H7) were only found in N. insignis in which H1 was the most common haplotypes with frequency larger than 60 %. The rest of the haplotypes (H8–H17) all occurred in L. decora except for H15, which was also observed in one individual in the population ML of N. insignis. The dominant haplotype H9 had widespread distribution in 9 populations of L. decora and accounted for 44.4 % of the total haplotypes identified in the species (Fig. 2b). In addition, the network without recombination within this locus was also constructed and a similar genealogy was found, with approximately two clusters with haplotypes being species-specific except Hap5 (Additional file 3: Fig. S1).
Fig. 2.
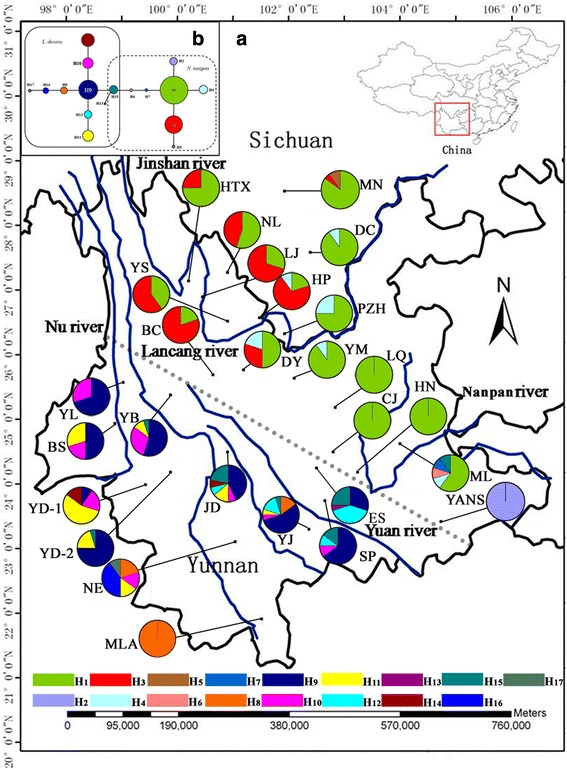
Geographic distribution and network of nDNA haplotypes. a. Distribution of 17 nuclear DNA (nDNA) haplotypes detected within and among 27 populations of Leucomeris decora and Nouelia insignis. Full names of the abbreviations for the populations are shown in Additional file 1: Table S1 and Additional file 2: Table S2. b. Statistical parsimony network of genealogical relationships between 17 nDNA haplotypes. Letters in/around circles represent haplotypes at each locus. The size of the circles corresponds to the frequency of each haplotype and each solid line represents one mutational step
Genetic differentiation and population structure
The nucleotide diversity (π) and haplotype diversity (Hd) of each population from both markers in the two species was generally low (Additional file 1: Tables S1 and Additional file 2: Table S2). At the species scale, L. decora had relatively higher nucleotide diversity and haplotype diversity than those of N. insignis (Table 2). The AMOVA analyses of cpDNA and nDNA datasets showed significant genetic differences between species as well as among populations (Table 1). For example, the difference between the two species explained 68.4 % of the total nDNA variation and 35.5 % of cpDNA. Based on the cpDNA data, in L. decora 90.7 % was attributed to differences among populations and 9.3 % was attributed to differences among individuals within the population; and in N. insignis, 92.5 % was attributed to differences among populations and 7.5 % was attributed to differences among individuals within the population. In the Bayesian clustering implemented by STRUCTURE, the most likely number of clusters was two when the ΔK statistic of Evanno et al. [50] was applied (Additional file 4: Fig. S2). When K = 2, all individuals of L. decora and N. insignis formed two separate clusters, except for admixed individuals present in several populations, especially one individual (population 26) of N. insignis (Fig. 3). These patterns of individual assignments were consistent with the result of genealogy.
Table 2.
The total rpl32-trnL and GBSSI variation, genetic diversity and differentiation parameters in all populations of Leucomeris decora and Nouelia insignis. π: nucleotide diversity and Hd: haplotype diversity
| rpl32-trnL | GBSSI | |||||||
|---|---|---|---|---|---|---|---|---|
| Species | π × 10-3 | Hd | HT | HS | GST | NST | π × 10-3 | Hd |
| L. decora | 0.53 | 0.473 | 0.487 (0.0935) | 0.042 (0.0424) | 0.913 (0.0850) | 0.913 (0.0850) | 1.67 | 0.751 |
| N. insignis* | 0.38 | 0.32 | 0.422 (0.1376) | 0.064 (0.0438) | 0.849 (0.1085) | 0.862 (0.1009) | 0.97 | 0.550 |
Standard error is shown in parentheses. *indicates the loci which exhibited significantly larger NST than GST
Fig. 3.
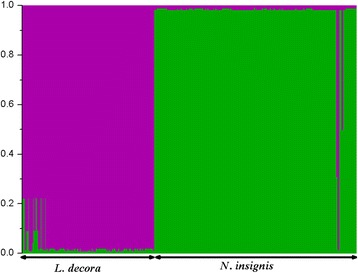
Clustering analysis of nDNA for Leucomeris decora and Nouelia insignis populations using STRUCTURE. Each bar of the plot represents one individual, with estimated likelihood assignment on the y-axis, when K = 2 cluster is assumed
Neutrality tests and mismatch distribution analyses
Neither the Tajima’s D nor Fu’s Fs neutrality tests yielded significantly negative results for all L. decora populations or N. insignis populations (Table 3). Similarly, the mismatch distribution was multimodal for L. decora populations with uniformly significant SSD and HRag values. However, the unimodal distribution pattern with non-significant SSD and HRag values was presented by all N. insignis populations under the demographic expansion model (Fig. 5). All these observations indicate the absence of expansion events.
Table 3.
Summary of mismatch distribution parameters and neutrality tests for all populations of Leucomeris decora and Nouelia insignis, respectively
| rpl32-trnL | ||||||
|---|---|---|---|---|---|---|
| Species | Tajima’s D | Fu’s Fs | SSDa | HRag a | SSDb | HRag b |
| L. decora | 1.659 | 2.187 | 0.017* | 0.227* | 0.017* | 0.227* |
| N. insignis | -0.083 | 0.793 | 0.005* | 0.194 | 0.012 | 0.194 |
SSD, the sum of square deviation; HRag, the raggedness index; *P < 0.05
aThe indices under the spatial expansion model; bThe indices under the sudden demographic expansion model
Estimates were obtained under models of spatial expansion or pure demographic expansion using Arlequin
Fig. 5.
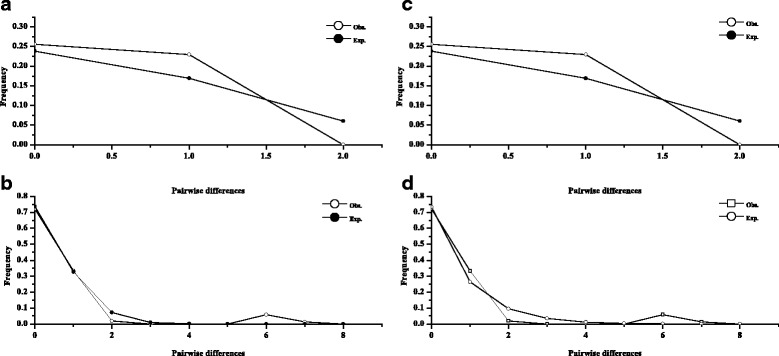
Mismatch distribution for cpDNA sequence data in Leucomeris decora (a, b) and Nouelia insignis (c, d), under the spatial expansion model (a, c) and demographic expansion mode (b, d), respectively. The line with hollow dot shows observed values, while the line with solid dot represents expected values under the model of spatial/demographic expansion
Demographic parameters of the IM model
For three independent runs of IMa, consistent unambiguous marginal posterior probability distributions of most demographic parameters were obtained for the two species (Fig. 4). However, the marginal posterior probability distributions of the parameter t did not drop to zero when sufficient high values were reached. This estimated result was likely due to small data set that may not be enough to achieve convergence [65]. The maximum-likelihood estimates (MLEs) and 90 % highest posterior densities (HPDs) were shown in Additional file 5: Table S3. Effective population sizes of L. decora and for N. insignis were not significantly different and larger than that of the ancestral species, indicating that both L. decora and N. insignis have undergone marked population expansion. The estimates of migration between L. decora and N. insignis were low and asymmetric. The gene flow from N. insignis to L. decora was larger than zero: m2 = 0.228 (90 % HPD interval: 0.008–1.563), while there was almost no evidence for gene flow in the opposite direction: m1 = 0.01 (90 % HPD interval: 0.01–1.41). The population migration rate was 2N2m2 = 0.049 from N. insignis to L. decora and 2N1m1 = 0 from L. decora to N. insignis, respectively.
Fig. 4.
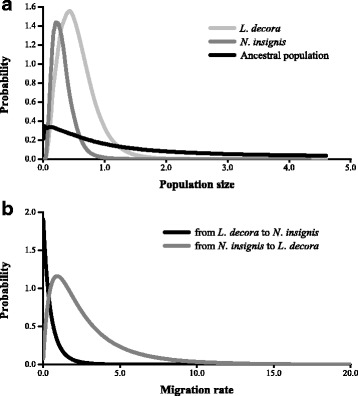
Marginal distribution of posterior probabilities for demographic parameters estimated by IM model. The IM analysis was performed for combined cpDNA and nDNA sequences. a. Population sizes of Leucomeris decora, Nouelia insignis and their ancestral population; a. migration rate between Leucomeris decora and Nouelia insignis
Discussion
Haplotype lineage and cytoplasmic-nuclear incongruence
Our results showed that the nDNA haplotypes (Fig. 2) were more species specific in comparison with the cpDNA sequences that we analysed (Fig. 1). Only H15 was shared by both species, which was frequent in L. decora, but found in only one individual of N. insignis. The network without recombination within the nuclear locus revealed a similar pattern, in which L. decora and N. insignis comprised different haplotypes and no haplotypes were shared except Hap5 (Additional file 3: Fig. S1). However, the cpDNA analyses provided an unexpected result with that one dominant haplotype (C2) detected from cpDNA and shared by L. decora and N. insignis (Fig. 1). There are three main causes of species sharing haplotypes: convergence, the persistence of ancestral polymorphism [66], or cryptic hybridization/introgression [67, 68].
Convergence appears to be the least likely explanation in this case as identical mutations are rare events, and the common haplotype with high frequency of more than 60 % of the individuals was present in the two species. Gene exchange caused by ancient and/or current hybridization between L. decora and N. insignis, resulting in the cpDNA capture, is possible. Sharing of haplotypes between two potentially hybridizing species only in areas where they are sympatric would lend support to the local hybridization hypothesis. However, in the process of thoroughly sampled populations of the two species, no overlapping distribution areas and no signs of intermediate morphology for species-specific taxonomic traits were found either at present; current hybridization between these two species seems unlikely to have played a major important role in causing the sharing of cpDNA haplotypes across them [69]. An alternative scenario is that hybridization and chloroplast capture may have occurred in the past when these two species came into contact with each other. Nevertheless, in the thoroughly sampled populations, chloroplast types were mostly distinct in the two species where they occurred allopatrically and the shared haplotype C2 was not restricted to some areas but broadly distributed in the populations of the two species. In fact, in 27 populations, we found only one haplotype was shared, and other haplotypes were in a relatively derived position in the network. Thus, as mostly interior haplotypes, if at all, are shared among species and shared chloroplast types also occur between species without overlapping distribution areas, persisting ancient polymorphisms seem to be the most likely explanation for the extant chloroplast patterns in N. insignis and L. decora. The finding of old cpDNA haplotypes shared may also indicate a recent divergence between these two closely related taxa and underwent an allopatric phase of divergence.
High levels of genetic differentiation and little gene flow between L. decora and N. insignis
Strong genetic differentiation across the Tanaka Line has been reported previously, however, it should be noted that such molecular data were derived from one species or species complexes; comparative analyses of closely related species distributed along the Tanaka Line remain still tenuous. Based on our data set, despite the sharing of one high frequent cpDNA haplotype, we detected high levels of genetic differentiation between N. insignis and L. decora populations (Table 1). Firstly, some cpDNA and GBSSI haplotypes that were frequent in N. insignis (e.g., C5, H1 and H3) were not found in L. decora and vice versa. Secondly, AMOVA produce that 35.5 % and 68.4 % of the total cpDNA and nDNA genetic variation could be attributed to differences between the two species, respectively. These observed genetic differentiation coefficients were relatively high by comparison with those of closely related species (see [70–72]). Moreover, in the STRUCTURE analyses, the absence of genetic admixture and robust population clustering in the two species also indicate high genetic differentiation (Fig. 3).
The likelihood ration test from coalescent-based analyses (IM model) indicated no gene flow from L. decora to N. insignis but very little gene flow from N. insignis to L. decora for most of the pairwise comparisons. Introgression events between closely related species are widespread in plants. Introgression within genera was far more common than that between genera [73]. Recent phylogenetic studies have also discovered intergenetic gene transfer, which played an important role in lineage evolution [74–76]. Gene flow between the two species would have seem likely because the distance between some of our samples, like HN and ES, is only about 15 km. The seeds with the light pappus may disperse by wind across the Tanaka Line. Moreover, even if the two species are currently allopatric, the possibility that the two species may have sympatric distributions and one of them went extinct in the sympatric areas is not completely excluded. Accordingly, they may not have diverged from each other in geographic isolation or subsequently always been allopatrically distributed. However, although there was unidirectional gene flow observed in the two species that are currently allopatric, the population migration rate (2N2m2) from N. insignis to L. decora was very low (0.049 < 1.0), suggesting that historical dispersal between populations seemed rare (Fig. 4, Additional file 5: Table S3). The recovered signals of close to zero migration, therefore, also indicated that the high levels of cpDNA haplotype sharing observed are most likely due to incomplete lineage sorting and widespread sharing of ancestral polymorphism, and the two species have been isolated genetically and well-differentiated since their divergence [77].
Generally, physical barriers like mountain ranges and river systems may act as physical barrier to past or contemporary dispersal. However, deep interspecific genetic divergences along the Tanaka Line without apparent physical barriers have not been reported. Most frequently, biogeographic boundaries are explained to be associated with some combination of historical and ecological processes [78], causing lineage divergence and local endemism (such as [79]). It has been proposed that monsoon characterized the two sides of the Tanaka Line (i.e., the west affected by monsoons from both the Indian and Pacific oceans and the east mainly affected by Pacific oceans) may result in the separation of species population. Presently, N. insignis and L. decora populations occupy different habitats on the two sides of the Tanaka Line. N. insignis prefers drier and hotter conditions such as valleys in the east, whereas L. decora is widely distributed in the west. It is feasible that the establishment of this biogeographic boundary corresponds with formation of the monsoon systems between the two sides, which promoted initial divergence between these two species (see [31] and discussion below).
Phylogeographic history of L. decora and N. insignis
The coalescent parameters of the divergence time with the MLEs and HPD weakly defined peaks and with undefined upper boundaries, cannot be used for scaling the precise divergence time between L. decora and N. insignis, perhaps due to the small data set that did not result in their convergence [80]. However, our crude estimated time frame (c. 0.38–0.54 Ma for cpDNA) for the TMRCA of these two species could date to Mid-Late Pleistocene, when Southwest China has experienced climatic changes such as establishment of differing monsoon regimes on either side of the Tanaka Line [27, 31, 81]. Although the main factors initiating the west-east split between the two species remain unclear, it is possible that the Pleistocene-originated ecological barriers associated with the Tanaka Line may have initiated the process of divergence between the two species without an apparent geographical barrier in this region. Under such scenario, small number of the ancestral populations of L. decora and N. insignis originally isolated of the boundary accumulated genetic differences. Following their splitting, the two species underwent past population growth as supported by joint analysis with IMa, suggesting larger effective population sizes in N. insignis and L. decora compared to the ancestor. The southwestern region of China located southeast of the Tibetan Plateau, is known to be a biodiversity hotspot due to its complex topology and unusual environmental conditions [82] and provided glacial refugia for various plant species in the Quaternary glaciations [28, 83, 84]. This region may provide sheltering for the surviving species, including the ancestor of L. decora and N. insignis [19, 23], over the periodical glaciations. Conditions during the following Quaternary glacial periods, especially the Pleistocene glacial-interglacial cycles, may have acted as stimulus to promote historically demographic shifts such as population expansions. We caution, however, that the recent spatial or demographic expansion or bottleneck for L. decora and N. insignis were not detected in the mismatch distribution estimates (Table 3, Fig. 5), which indicated their relatively stable population sizes. The different results underlie the two methods arose probably because mismatch analyses estimates the timing of sudden change in population sizes relying on parametric model of growth or decline, whereas in IMa analyses, there is longer time scale since the time of splitting and it is uncertain when the demographic changes happened in the long evolutionary history. In addition, the estimate of ancestral population sizes probably reflects post-divergence condition, resulting in biased down-estimate, for dynamics within the ancestral populations such as contractions or extinctions are not counted in IMa [85]. Further studies, particularly those employing multiple loci in coalescence times among alleles from a population to estimate population sizes through time such as extended Bayesian skyline analysis, are needed to improve examining population size change.
Conclusions
In summary, this study provides insight into the phylogeographic patterns of two closely related species distributed along two sides of the Tanaka Line in southwest China. Although our study is based on a single chloroplast locus and one nuclear locus, we have been able to gain useful insights into the clear allopatric divergence between the two species on the population level. The extant geographic structure in the chloroplast diversity patterns observed in N. insignis and L. decora is most likely interpreted as reflecting ancestrally shared polymorphism but not post-divergence gene flow between species. In addition, our present genealogy support a major phylogeographic break in nuclear DNA between N. insignis and L. decora associated with the Tanaka Line. This phylogeographic boundary appears to represent an environmental barrier to dispersal/migration for species as previously hypothesized [31]. The establishment of differing monsoon regime during the Pleistocene on either side of the Tanaka Line probably contributed to species divergences based on our crude estimates. However, the coalescent-based methods (IMa) suffered from a lack of convergence of the lineage divergence parameter (t) based on the present data, thus, efforts need to be undertaken to obtain more precise estimates with multi-locus approach and perhaps parameter-rich evolutionary model implemented with Approximate Bayesian Computation to resolve their complicated evolutionary history [86].
Availability of supporting data
The data sets of DNA sequences supporting the results of this article are available in the GenBank. GenBank accessions numbers for chloroplast DNA haplotypes are JF915764–JF915768; GenBank accessions numbers for nuclear DNA haplotypes are JF915747–JF915763.
Acknowledgements
We thank Qitai Zhang and Guoping Yang for their assistance with field sampling. We are grateful to Xueqing Yang for help with drawing the maps. This work was funded by the National Basic Research Program of China (973 Program: 2007CB411600) and the National Natural Science Foundation of China (31400324).
Additional files
Details of sample locations, sample sizes, cpDNA and GBSSI variation of Leucomeris decora. n: sample sizes, π: nucleotide diversity and Hd: haplotype diversity.
Details of sample locations, sample sizes, cpDNA and GBSSI variation of Nouelia insignis. n: sample sizes, π: nucleotide diversity and Hd: haplotype diversity.
Statistical parsimony network of genealogical relationships between 9 haplotypes derived from nDNA sequences of Leucomeris decora and Nouelia insignis without recombinations. Letters in/around circles represent haplotypes at each locus. The size of the circles corresponds to the frequency of each haplotype and each solid line represents one mutational step.
The plots of delta K value and number of clusters (K) implemented by STRUCTURE.
Maximum likelihood estimates (MLEs) of demographic parameters of the IM model between Leucomeris decora and Nouelia insignis.
Footnotes
Competing interests
The authors declare that they have no competing interests.
Author’s contributions
XG conceived of the study, and participated in experimental design. YJZ carried out the molecular genetic studies, participated in the data analyses and drafted the manuscript. All authors read and approved the final manuscript.
Contributor Information
Yu-Juan Zhao, Email: zyy36920@126.com.
Xun Gong, Email: gongxun@mail.kib.ac.cn.
References
- 1.Avise JC. Phylogeography: the history and formation of species. Cambridge, Massachusetts: Harvard University Press; 2000. [Google Scholar]
- 2.Palmé AE. Chloroplast DNA variation, postglacial recolonization and hybridization in hazel, Corylus avellana. Mol Ecol. 2002;11:1769–79. doi: 10.1046/j.1365-294X.2002.01581.x. [DOI] [PubMed] [Google Scholar]
- 3.Du FK, Petit RJ, Liu JQ. More introgression with less gene flow: chloroplast vs. mitochondrial DNA in the Picea asperata complex in China, and comparison with other Conifers. Mol Ecol. 2009;18:1396–407. doi: 10.1111/j.1365-294X.2009.04107.x. [DOI] [PubMed] [Google Scholar]
- 4.Kikuchi R, Jae-Hong P, Takahashi H, Maki M. Disjunct distribution of chloroplast DNA haplotypes in the understory perennial Veratrum album ssp. oxysepalum (Melanthiaceae) in Japan as a result of ancient introgression. New Phytol. 2010;188:879–91. doi: 10.1111/j.1469-8137.2010.03398.x. [DOI] [PubMed] [Google Scholar]
- 5.Qi XS, Chen C, Comes HP, Sakaguchi S, Liu YH, Tanaka N, et al. Molecular data and ecological niche modelling reveal a highly dynamic evolutionary history of the East Asian Tertiary relict Cercidiphyllum (Cercidiphyllaceae) New Phytol. 2012;196:617–30. doi: 10.1111/j.1469-8137.2012.04242.x. [DOI] [PubMed] [Google Scholar]
- 6.Dixon CJ, Schönswetter P, Schneeweiss GM. Traces of ancient range shifts in a mountain plant group (Androsace halleri complex, Primulaceae) Mol Ecol. 2007;16:3890–901. doi: 10.1111/j.1365-294X.2007.03342.x. [DOI] [PubMed] [Google Scholar]
- 7.Poudel RC, Möller M, Gao LM, Ahrends A, Baral SR, Liu J, et al. Using morphological, molecular and climatic data to delimitate yews along the Hindu Kush-Himalaya and adjacent regions. PLoS One. 2012;7 doi: 10.1371/journal.pone.0046873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Wang J, Abbott RJ, Peng YL, Du FK, Liu JQ. Species delimitation and biogeography of two fir species (Abies) in central China: cytoplasmic DNA variation. Heredity. 2011;107:362–70. doi: 10.1038/hdy.2011.22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Cortés-Rodríguez N, Jacobsen F, Hernandez-Baños BE, Navarro-Siguenza AG, Peters JL, Omland KE. Coalescent analyses show isolation without migration in two closely related tropical orioles: the case of Icterus graduacauda and Icterus chrysater. Ecol Evol. 2013;3:4377–87. doi: 10.1002/ece3.768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Mims MC, Darrin Hulsey C, Fitzpatrick BM, Streelman JT. Geography disentangles introgression from ancestral polymorphism in Lake Malawi cichlids. Mol Ecol. 2010;19:940–51. doi: 10.1111/j.1365-294X.2010.04529.x. [DOI] [PubMed] [Google Scholar]
- 11.Levin DA. Ecological speciation: crossing the divide. Syst Bot. 2004;29:807–16. doi: 10.1600/0363644042451134. [DOI] [Google Scholar]
- 12.Yu JJ, Kuroda C, Gong X. Natural hybridization and introgression in sympatric Ligularia species (Asteraceae, Senecioneae) J Syst Evol. 2011;49:438–48. doi: 10.1111/j.1759-6831.2011.00150.x. [DOI] [Google Scholar]
- 13.Rosenberg NA, Nordborg M. Genealogical trees, coalescent theory and the analysis of genetic polymorphisms. Nat Rev Genet. 2002;3:380–90. doi: 10.1038/nrg795. [DOI] [PubMed] [Google Scholar]
- 14.Hind DJN. Tribe Mutisieae. In: Kadereit JW, Jeffrey C, editors. Families and genera of vascular plants, vol 8 Flowing plants-eudicots-Asterales. Berlin: Springer; 2007. pp. 90–123. [Google Scholar]
- 15.Roque N, Funk VA. Morphological characters add support for some members of the basal grade of Asteraceae. Bot J Linn Soc. 2013;171:568–86. doi: 10.1111/boj.12000. [DOI] [Google Scholar]
- 16.Bremer K. Asteraceae: cladistics and classification. Portland: Timber Press; 1994. [Google Scholar]
- 17.Panero JL, Funk VA. The value of sampling anomalous taxa in phylogenetic studies: major clades of the Asteraceae revealed. Mol Phylogen Evol. 2008;47:757–82. doi: 10.1016/j.ympev.2008.02.011. [DOI] [PubMed] [Google Scholar]
- 18.Funk VA, Susanna A, Steussy TF, Bayer RJ. Systematics, evolution, and biogeography of Compositae. Vienna, Austria: International Association for Plant Taxonomy; 2009. [Google Scholar]
- 19.Lin NN. PhD Thesis. Kun Ming: Kunming Institute of Botany, Chinese Academy of Sciences; 2007. Phylogeny and biogeography of the tribe Mutisieae in Eastern Asia. [Google Scholar]
- 20.Peng YL, Sun H, Gu ZJ. Cytological study on Nouelia and Leucomeris (Compositae) Acta Bot Yunnan. 2002;24:82–6. [Google Scholar]
- 21.Wang HS. Origins of endemic genera of vascular flora of China. Acta Bot Yunnan. 1989;11:11–6. [Google Scholar]
- 22.Zhao YJ, Gong X. Genetic structure of the endangered Leucomeris decora (Asteraceae) in China inferred from chloroplast and nuclear DNA markers. Conserv Genet. 2012;13:271–81. doi: 10.1007/s10592-011-0281-2. [DOI] [Google Scholar]
- 23.Gong X, Luan SS, Hung KH, Hwang CC, Lin CJ, Chiang YC, et al. Population structure of Nouelia insignis (Asteraceae), an endangered species in southwestern China, based on chloroplast DNA sequences: recent demographic shrinking. J Plant Res. 2011;124:221–30. doi: 10.1007/s10265-010-0363-0. [DOI] [PubMed] [Google Scholar]
- 24.Li XW, Li J. On the validity of Tanaka Line & its significance viewed from the distribution of Eastern Asiatic genera in Yunnan. Acta Bot Yunnan. 1992;14:1–12. [Google Scholar]
- 25.Li XW, Li J. The Tanaka-Kaiyong Line–an important floristic line for the study of the flora of East Asia. Ann Mo Bot Gard. 1997;84:888–92. doi: 10.2307/2992033. [DOI] [Google Scholar]
- 26.Tanaka T. Species problem in Citrus. Ueno, Tokyo: Japanese Society for the Promotion of Science; 1954. [Google Scholar]
- 27.Mitsui Y, Chen ST, Zhou ZK, Peng CI, Deng YF, Setoguchi H. Phylogeny and biogeography of the Genus Ainsliaea (Asteraceae) in the Sino-Japanese region based on nuclear rDNA and plastid DNA sequence data. Ann Bot. 2008;101:111–24. [DOI] [PMC free article] [PubMed]
- 28.Qiu YX, Guan BC, Fu CX, Comes HP. Did glacials and/or interglacials promote allopatric incipient speciation in East Asian temperate plants? Phylogeographic and coalescent analyses on refugial isolation and divergence in Dysosma versipellis. Mol Phylogen Evol. 2009;51:281–93. doi: 10.1016/j.ympev.2009.01.016. [DOI] [PubMed] [Google Scholar]
- 29.Zhang L, Li QJ, Li HT, Chen J, Li DZ. Genetic diversity and geographic differentiation in Tacca chantrieri (Taccaceae): an autonomous selfing plant with showy floral display. Ann Bot. 2006;98:449–57. doi: 10.1093/aob/mcl123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hubisz MJ, Falush D, Stephens M, Pritchard JK. Inferring weak population structure with the assistance of sample group information. Mol Ecol Resour. 2009;9:1322–32. doi: 10.1111/j.1755-0998.2009.02591.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Fan DM, Yue JP, Nie ZL, Li ZM, Comes HP, Sun H. Phylogeography of Sophora davidii (Leguminosae) across the ‘Tanaka-Kaiyong Line’, an important phytogeographic boundary in Southwest China. Mol Ecol. 2013;22:4270–88. doi: 10.1111/mec.12388. [DOI] [PubMed] [Google Scholar]
- 32.Doyle J. DNA protocols for plants—CTAB total DNA isolation. In: Hewitt GM, Johnston A, editors. Molecular techniques in taxonomy. Berlin: Springer; 1991. [Google Scholar]
- 33.Shaw J, Lickey EB, Schilling EE, Small RL. Comparison of whole chloroplast genome sequences to choose noncoding regions for phylogenetic studies in angiosperms: the tortoise and the hare III. Am J Bot. 2007;94:275–88. doi: 10.3732/ajb.94.3.275. [DOI] [PubMed] [Google Scholar]
- 34.Wang JF, Gong X, Chiang YC, Kuroda C. Phylogenetic patterns and disjunct distribution in Ligularia hodgsonii Hook. (Asteraceae) J Biogeogr. 2013;40:1741–54. doi: 10.1111/jbi.12114. [DOI] [Google Scholar]
- 35.Clark JR, Robertson M, Ainsworth CC. Nucleotide sequence of a wheat (Triticum aestivum L.) cDNA clone encoding the waxy protein. Plant Mol Biol. 1991;16:1099–101. doi: 10.1007/BF00016086. [DOI] [PubMed] [Google Scholar]
- 36.Wang ZY, Wu ZL, Xing YY, Zheng FG, Guo XL, Zhang WG, et al. Nucleotide sequence of rice waxy gene. Nucleic Acids Res. 1990;18:5898. doi: 10.1093/nar/18.19.5898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Leij FR, Visser RGF, Ponstein AS, Jacobsen E, Feenstra WJ. Sequence of the structural gene for granule-bound starch synthase of potato (Solarium tuberosum L.) and evidence for a single point deletion in the amf allele. Mol Gen Genet. 1991;228:240–8. doi: 10.1007/BF00282472. [DOI] [PubMed] [Google Scholar]
- 38.Wang SJ, Yeh KW, Tsai CY. Molecular characterization and expression of starch granule-bound starch synthase in the sink and source tissues of sweet potato. Physiol Plant. 1999;106:253–61. doi: 10.1034/j.1399-3054.1999.106301.x. [DOI] [Google Scholar]
- 39.Levin RA, Miller JS. Relationships within tribe Lycieae (Solanaceae): paraphyly of Lycium and multiple origins of gender dimorphism. Am J Bot. 2005;92:2044–53. doi: 10.3732/ajb.92.12.2044. [DOI] [PubMed] [Google Scholar]
- 40.Levin RA, Myers NR, Bohs L. Phylogenetic relationships among the “spiny solanums” (Solanum subgenus Leptostemonum, Solanaceae) Am J Bot. 2006;93:157–69. doi: 10.3732/ajb.93.1.157. [DOI] [Google Scholar]
- 41.Yang HQ, Yang JB, Peng ZH, Gao J, Yang YM, Peng S, et al. A molecular phylogenetic and fruit evolutionary analysis of the major groups of the paleotropical woody bamboos (Gramineae: Bambusoideae) based on nuclear ITS, GBSSI gene and plastid trnL-F DNA sequences. Mol Phylogen Evol. 2008;48:809–24. doi: 10.1016/j.ympev.2008.06.001. [DOI] [PubMed] [Google Scholar]
- 42.Kimura T, Ideta O, Saito A. Identification of the gene encoding granule-bound starch synthase I in sweet potato (Ipomoea batatas (L.) Lam.) Plant Biotechnol. 2000;17:247–52. doi: 10.5511/plantbiotechnology.17.247. [DOI] [Google Scholar]
- 43.Clark AG. Inference of haplotypes from PCR-amplified samples of diploid populations. Mol Biol Evol. 1990;7:111–22. doi: 10.1093/oxfordjournals.molbev.a040591. [DOI] [PubMed] [Google Scholar]
- 44.Thompson JD, Gibson TJ, Plewniak F, Jeanmougin F, Higgins DG. The CLUSTAL_X windows interface: flexible strategies for multiple sequence alignment aided by quality analysis tools. Nucleic Acids Res. 1997;25:4876–82. doi: 10.1093/nar/25.24.4876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Hall TA. BioEdit: a user-friendly biological sequence alignment editor and analysis program for Windows 95/98/NT. Nucleic Acids Symp Ser. 1999;41:95–8. [Google Scholar]
- 46.Nei M. Molecular evolutionary genetics. New York, USA: Columbia University Press; 1987. [Google Scholar]
- 47.Rozas J, Rozas R. DnaSP version 3: an integrated program for molecular population genetics and molecular evolution analysis. Bioinformatics. 1999;15:174–5. doi: 10.1093/bioinformatics/15.2.174. [DOI] [PubMed] [Google Scholar]
- 48.Pons O, Petit RJ. Measuring and testing genetic differentiation with ordered versus unordered alleles. Genetics. 1996;144:1237–45. doi: 10.1093/genetics/144.3.1237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Excoffier L, Laval G, Schneider S. Arlequin (version 3.0): an integrated software package for population genetics data analysis. Evol Bioinforma Online. 2005;1:47–50. [PMC free article] [PubMed] [Google Scholar]
- 50.Evanno G, Regnaut S, Goudet J. Detecting the number of clusters of individuals using the software structure: a simulation study. Mol Ecol. 2005;14:2611–20. doi: 10.1111/j.1365-294X.2005.02553.x. [DOI] [PubMed] [Google Scholar]
- 51.Clement M, Posada D, Crandall KA. TCS: a computer program to estimate gene genealogies. Mol Ecol. 2000;9:1657–9. doi: 10.1046/j.1365-294x.2000.01020.x. [DOI] [PubMed] [Google Scholar]
- 52.Ingvarsson PK, Ribstein S, Taylor DR. Molecular evolution of insertions and deletion in the chloroplast genome of Silene. Mol Biol Evol. 2003;20:1737–40. doi: 10.1093/molbev/msg163. [DOI] [PubMed] [Google Scholar]
- 53.Drummond AJ, Rambaut A. BEAST: Bayesian evolutionary analysis by sampling trees. BMC Evol Biol. 2007;7:214. doi: 10.1186/1471-2148-7-214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Posada D. jModelTest: phylogenetic model averaging. Mol Biol Evol. 2008;25:1253–6. doi: 10.1093/molbev/msn083. [DOI] [PubMed] [Google Scholar]
- 55.Graur D, Li WH. Fundamentals of molecular evolution. Sunderland, Massachusetts: Sinauer Associates; 2000. [Google Scholar]
- 56.Drummond AJ, Rambaut A. Tracer-MCMC trace analysis tool. Oxford, UK: University of Oxford; 2004. [Google Scholar]
- 57.Tajima F. Statistical method for testing the neutral mutation hypothesis by DNA polymorphism. Genetics. 1989;123:585–95. doi: 10.1093/genetics/123.3.585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Fu YX. Statistical tests of neutrality of mutations against population growth, hitchhiking and background selection. Genetics. 1997;147:915–25. doi: 10.1093/genetics/147.2.915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Rogers AR, Harpending H. Population growth makes waves in the distribution of pairwise genetic differences. Mol Biol Evol. 1992;9:552–69. doi: 10.1093/oxfordjournals.molbev.a040727. [DOI] [PubMed] [Google Scholar]
- 60.Hey J, Nielsen R. Integration within the Felsenstein equation for improved Markov chain Monte Carlo methods in population genetics. Proc Natl Acad Sci USA. 2007;104:2785–90. doi: 10.1073/pnas.0611164104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Hey J, Nielsen R. Multilocus methods for estimating population sizes, migration rates and divergence Time, With applications to the divergence of Drosophila pseudoobscura and D. persimilis. Genetics. 2004;167:747–60. doi: 10.1534/genetics.103.024182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Woerner AE, Cox MP, Hammer MF. Recombination-filtered genomic datasets by information maximization. Bioinformatics. 2007;23:1851–3. doi: 10.1093/bioinformatics/btm253. [DOI] [PubMed] [Google Scholar]
- 63.Templeton AR, Sing CF. A cladistic analysis of phenotypic associations with haplotypes inferred from restriction endonuclease mapping. IV. nested analyses with cladogram uncertainty and recombination. Genetics. 1993;134:659–69. doi: 10.1093/genetics/134.2.659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Posada D, Crandall KA. Intraspecific gene genealogies: trees grafting into networks. Trends Ecol Evol. 2001;16:37–45. doi: 10.1016/S0169-5347(00)02026-7. [DOI] [PubMed] [Google Scholar]
- 65.Hey J, Won YJ, Sivasundar A, Nielsen R, Markert JA. Using nuclear haplotypes with microsatellites to study gene flow between recently separated Cichlid species. Mol Ecol. 2004;13:909–19. doi: 10.1046/j.1365-294X.2003.02031.x. [DOI] [PubMed] [Google Scholar]
- 66.Jakob SS, Blattner FR. A chloroplast genealogy of Hordeum (Poaceae): long-term persisting haplotypes, incomplete lineage sorting, regional extinction, and the consequences for phylogenetic inference. Mol Biol Evol. 2006;23:1602–12. doi: 10.1093/molbev/msl018. [DOI] [PubMed] [Google Scholar]
- 67.Palme AE, Su Q, Palsson S, Lascoux M. Extensive sharing of chloroplast haplotypes among European birches indicates hybridization among Betula pendula, B. pubescens and B. nana. Mol Ecol. 2004;13:167–78. doi: 10.1046/j.1365-294X.2003.02034.x. [DOI] [PubMed] [Google Scholar]
- 68.Abbott R, Albach D, Ansell S, Arntzen JW, Baird SJE, Bierne N, et al. Hybridization and speciation. J Evol Biol. 2013;26:229–46. doi: 10.1111/j.1420-9101.2012.02599.x. [DOI] [PubMed] [Google Scholar]
- 69.Yih D. Land bridge travelers of the Tertiary: the Eastern Asian-Eastern North American floristic disjunction. Arnoldia. 2012;69:14–23. [Google Scholar]
- 70.Zheng XM, Ge S. Ecological divergence in the presence of gene flow in two closely related Oryza species (Oryza rufipogon and O. nivara) Mol Ecol. 2010;19:2439–54. doi: 10.1111/j.1365-294X.2010.04674.x. [DOI] [PubMed] [Google Scholar]
- 71.Remington DL, Robichaux RH. Influences of gene flow on adaptive speciation in the Dubautia arborea–D. ciliolata complex. Mol Ecol. 2007;16:4014–27. doi: 10.1111/j.1365-294X.2007.03447.x. [DOI] [PubMed] [Google Scholar]
- 72.Caroline SS, Dick CW, Caron H, Vendramin GG, Guichoux E, Buonamici A, et al. Phylogeography of a species complex of lowland Neotropical rain forest trees (Carapa, Meliaceae) J Biogeogr. 2013;40:676–92. doi: 10.1111/j.1365-2699.2011.02678.x. [DOI] [Google Scholar]
- 73.Whitney KD, Ahern JR, Campbell LG, Albert LP, King MS. Patterns of hybridization in plants. Perspect Plant Ecol Evol Syst. 2010;12:175–82. doi: 10.1016/j.ppees.2010.02.002. [DOI] [Google Scholar]
- 74.Fehrer J, Gemeinholzer B, Chrtek J, Jr, Brätigam S. Incongruent plastid and nuclear DNA phylogenies reveal ancient intergeneric hybridization in Pilosella hawkweeds (Hieracium, Cichorieae, Asteraceae) Mol Phylogen Evol. 2007;42:347–61. doi: 10.1016/j.ympev.2006.07.004. [DOI] [PubMed] [Google Scholar]
- 75.Yuan YW, Olmstead RG. A species-level phylogenetic study of the Verbena complex (Verbenaceae) indicates two independent intergeneric chloroplast transfers. Mol Phylogen Evol. 2008;48:23–33. doi: 10.1016/j.ympev.2008.04.004. [DOI] [PubMed] [Google Scholar]
- 76.Triplett JK, Clark LG, Fisher AE, Wen J. Independent allopolyploidization events preceded speciation in the temperate and tropical woody bamboos. New Phytol. 2014;204:66–73. doi: 10.1111/nph.12988. [DOI] [PubMed] [Google Scholar]
- 77.Wright S. Evolution in mendelian populations. Genetics. 1931;16:97–159. doi: 10.1093/genetics/16.2.97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Glor RE, Warren D. Testing ecological explanations for biogeographic boundaries. Evolution. 2011;65:673–83. doi: 10.1111/j.1558-5646.2010.01177.x. [DOI] [PubMed] [Google Scholar]
- 79.Pearson RG, Raxworthy CJ. The evolution of local endemism in madagascar: watershed versus climatic gradient hypotheses evaluated by null biogeographic models. Evolution. 2009;63:959–67. doi: 10.1111/j.1558-5646.2008.00596.x. [DOI] [PubMed] [Google Scholar]
- 80.McIntire EJB, Fajardo A. Facilitation as a ubiquitous driver of biodiversity. New Phytol. 2014;201:403–16. doi: 10.1111/nph.12478. [DOI] [PubMed] [Google Scholar]
- 81.Wang WM. Paleofloristic and paleoclimatic implications of Neogene palynofloras in China. Rev Palaeobot Palynol. 1994;82:239–50. doi: 10.1016/0034-6667(94)90078-7. [DOI] [Google Scholar]
- 82.Myers N, Mittermeier RA, Mittermeier CG, Fonseca G, Kent J. Biodiversity hotspots for conservation priorities. Nature. 2000;403:854–8. doi: 10.1038/35002501. [DOI] [PubMed] [Google Scholar]
- 83.Wang HW, Ge S. Phylogeography of the endangered Cathaya argyrophylla (Pinaceae) inferred from sequence variation of mitochondrial and nuclear DNA. Mol Ecol. 2006;15:4109–22. doi: 10.1111/j.1365-294X.2006.03086.x. [DOI] [PubMed] [Google Scholar]
- 84.Gong W, Chen C, Dobes C, Fu CX, Koch MA. Phylogeography of a living fossil: Pleistocene glaciations forced Ginkgo biloba L. (Ginkgoaceae) into two refuge areas in China with limited subsequent postglacial expansion. Mol Phylogen Evol. 2008;48:1094–105. doi: 10.1016/j.ympev.2008.05.003. [DOI] [PubMed] [Google Scholar]
- 85.Won YJ, Sivasundar A, Wang Y, Hey J. On the origin of Lake Malawi cichlid species: A population genetic analysis of divergence. Proc Natl Acad Sci USA. 2005;102:6581–6. doi: 10.1073/pnas.0502127102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Gugger PF, Cavender-Bares J. Molecular and morphological support for a Florida origin of the Cuban oak. J Biogeogr. 2013;40:632–45. doi: 10.1111/j.1365-2699.2011.02610.x. [DOI] [Google Scholar]