

Abstract
DOCK8 deficiency in humans and mice leads to multiple defects in immune cell numbers and function. Patients with this immunodeficiency have a high morbidity and mortality, and are distinguished by chronic, cutaneous viral infections, including those caused by herpes simplex virus (HSV). The underlying mechanism of the specific susceptibility to these chronic cutaneous viral infections is currently unknown, largely because the effect of DOCK8-deficiency has not been studied in suitable models. A better understanding of these mechanisms is required to underpin the development of more specific therapies. Here we show that DOCK8 deficient mice have poor control of primary cutaneous herpes simplex lesions and this is associated with increased virus loads. Furthermore, DOCK8-deficient mice showed a lack of CD4+ T cell infiltration into HSV-infected skin.
Introduction
Mutations in DOCK8 as a cause of primary human immunodeficiency was first described in 2009. Patients present with recurrent sinopulmonary bacterial infections and cutaneous viral infections; most prominently human papilloma virus (HPV), herpes simplex virus (HSV) and molluscum contagiosum 1, 2. Some of these patients were also found to have markedly elevated levels of IgE antibodies and had previously been described as having autosomal recessive hyper IgE syndrome 1, 2. In addition, these patients would usually have eczema and also food and environmental allergies 3. Due to the poor prognosis, the current recommendations are that patients with DOCK8 immunodeficiency undergo bone marrow transplantation 4. The cutaneous viral infections found in human DOCK8 immunodeficiency are severe, extensive and treatment-resistant with one survey finding HSV, varicella zoster virus (VZV), HPV and molluscum contagiosum in 95% of patients 3. The viral infections remit with bone marrow transplantation 5 but this procedure is associated with a high risk of morbidity and mortality particularly in the context of uncontrolled viral infection. More recently the unusual herpes simplex viral infections have been shown to respond to high doses of subcutaneous interferon alpha therapy 6, 7 and this treatment has also been used for papilloma virus infection 8.
Several aspects of DOCK8 deficiency have been modeled in mice. DOCK8pri/pri and other DOCK8 deficient mice produced by mutagenesis with N-Ethyl-N-Nitrosourea (ENU) have a marked decrease in naïve T cells and decreased numbers of NKT cells and MZ B cells 9, 10. B and T cells in these mice have cell-intrinsic defects in immunological synapse formation and there are failures in generation of long-lived antibodies and persistence of CD8+ T cell memory 11, 9. Finally, the migration efficiency of DOCK8-/- dendritic cells was decreased in a DOCK8 knockout model12 and a striking cell death phenotype has been noted for lymphocytes migrating in confined matrices and tissues such as epidermis 13. Despite these apparently profound defects, DOCK8-deficient mouse models had normal virus control and primary anti-viral CD8+ T cell responses to infections with influenza virus and the highly attenuated MVA strain of vaccinia virus 13. By contrast a recent report found poor control of HSV infection associated with DOCK8 deficiency and this was associated with a defect in CD8+ T cells able to migrate into the skin and become resident memory (TRM) 13. However, this paper did not examine other lymphocytes nor was a direct link made between the loss of TRM and poor control of primary HSV infection. Here we confirm the poor control of HSV disease in DOCK8-deficient mice, add biological data and find a defect in migration of CD4+ T cells to skin during infection.
Results and Discussion
DOCK8 deficiency in mice leads to increased disease and virus load during HSV infection
Cohorts of DOCK8pri/pri and wild-type mice were inoculated by tattoo with HSV-1 strain KOS on the flank of shaved and depilated mice14. In this model of primary HSV infection, virus moves to the innervating dorsal root ganglia (DRG) concurrent with the initial skin infection. Replication in the peripheral nervous system leads to virus spreading back to the skin at sites within the inoculated dermatome that are distinct from the site of inoculation producing a characteristic ‘zosteriform’ lesion (Fig 1A) 15, 16. In immunocompetent mice, HSV is limited to the skin and innervating peripheral nervous system and acute infection is controlled by 7-8 days post infection (dpi), but loss of control can lead to central nervous system involvement or dissemination15. HSV lesions in DOCK8pri/pri mice initially formed at a similar rate to those in wild type littermates, but then continued to increase in size until 8 dpi, reaching a significantly larger size (Fig. 1A & B). By contrast wild type littermates began to control lesions by 6 dpi, such that the peak size was lower and lesions resolved more quickly. Despite the difference in lesion size there was no significant difference in weight-loss sustained by the DOCK8pri/pri mice compared with wild type controls and no mice succumbed to disease (Fig 1C). Qualitatively the lesions in DOCK8pri/pri and wild type mice looked similar with the exception of size, and the tumourous lesions found in DOCK8 deficient human patients were not seen. We speculate that this might be due to the mouse not recapitulating the atopic features of DOCK8-deficiency in humans, for example DOCK8-deficient mice do not exhibit hyper-IgE, even when aged (KLR, unpublished). While the lesions in DOCK8 deficient patients are clearly distinct from those associated with eczema herpeticum, it is possible that the superimposition of these two conditions leads to a unique lesion morphology.
Figure 1. Pathogenesis of HSV infection in DOCK8 deficient mice.
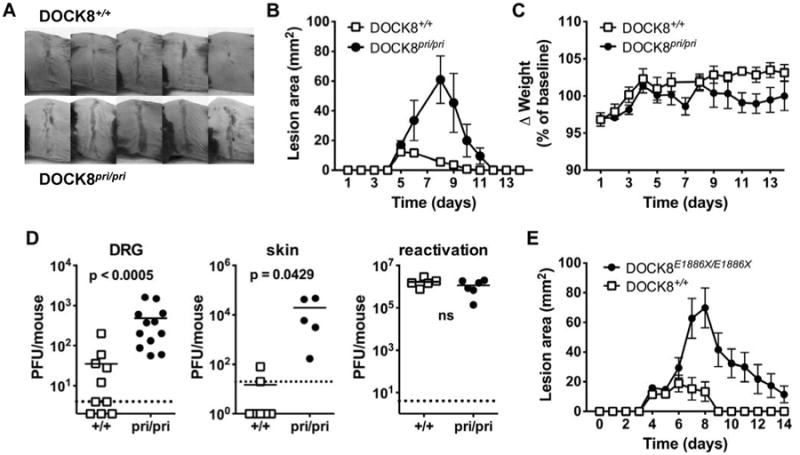
Cohorts of DOCK8 deficient mice and matched DOCK8+/+ littermates were inoculated with HSV-1 strain KOS on the flank by tattoo. (A) Lesion morphology at 7 dpi at the peak of the infection. (B) Lesion size and (C) weight change in groups of 5 DOCK8pri/ipri mice and 4 DOCK8+/+ littermates. Data have been independently repeated twice. (D) Left and middle, virus titers in DRG and skin at 7 dpi, right is the amount of virus obtained from latently-infected DRG after 5 days of explant culture to induce reactivation. In all cases, data are combined from two independent experiments, each point represents a single mouse and lines indicate means. (E) Lesion size in groups of 7 Dock8E1886X/E1886X and 4 matched wild-type mice. Statistical significance (Mann Whitney) is noted with a p value or ns for p>0.05).
The larger lesions seen in DOCK8pri/pri mice may have been due to increases in virus replication or immunopathology. To dissect these possible causes we examined the amounts of infectious virus in skin and DRG, choosing a time past the peak of infection to see if control of HSV was delayed in DOCK8pri/pri mice, as suggested by the kinetics of lesion development (Fig 1D). Significantly more infectious virus was found at both sites in DOCK8pri/pri compared with wild type mice 7 dpi. As expected by this time most wild type mice had cleared infectious virus in the skin to below the limit of detection, but by contrast all DOCK8pri/pri mice had easily detected titers of virus irrespective of site. During acute infection, HSV establishes latency in DRG and this remains for the life of the animal. This latency is exceptionally stable in mice in vivo, but removing DRG and placing them in culture causes reactivation and production of virus that can be measured in standard assays. Using these methods we found that HSV was able to establish and reactivate from latency equally well in DOCK8pri/pri and wild type mice (Fig 1D, right). We conclude that the larger lesions with delayed healing in DOCK8pri/pri mice are due to impaired clearance of HSV and this is most evident in the skin.
A second DOCK8-deficient mouse strain shows reduced control of HSV disease
To ensure that the response to HSV infection seen in DOCK8pri/pri mice was not unique to this particular mutation of DOCK8, we repeated the experiments with a mouse strain with a different ENU-induced DOCK8 mutation. The DOCK8E1886X/E1886X strain contains a G to T point mutation induced by ethylnitrosurea (ENU) at position 5778 in cDNA in exon 44, resulting in a premature STOP codon at position 1886 (Supplementary Figure 1A). The DOCK8E1886X/E1886X mouse strain reproduces the cellular phenotypes seen in other DOCK8 mutant mouse strains 11 (Supplementary Fig. 1B-D). Similar to the experiments above with DOCK8pri/pri, when the DOCK8E1886X/E1886X strain was infected with HSV, significantly larger lesions with delayed healing were observed when compared with wild type littermates (Figure 1E). These data indicate that loss of DOCK8 function in general leads to increased HSV pathogenesis.
HSV-infected skin in DOCK8pri/pri mice shows decreased CD4+ T cell infiltration
The timing of the difference in lesions between DOCK8pri/pri and wild-type mice suggests poor adaptive immunity to HSV. In the spleen, CD4+ and CD8+ T cells were significantly reduced in DOCK8-deficient compared with wild type mice at 7 dpi when expressed as a percent of splenocytes (Fig. 2A left) or as total number per spleen (Fig 2A right). Next, looking in the infected skin at 7 dpi, the amount of infiltration was reduced, reflected by the fraction of cells recovered bearing the pan-leukocyte marker CD45.2 (Fig. 2B left). More striking was the observation that CD4+ T cells were reduced in the skin, both as a fraction of CD45.2+ cells and in total (Fig. 2B middle and right). Surprisingly, CD8+ T cell infiltration was not significantly different between DOCK8pri/priand wild type mice either in total number of as a percent of CD45.2+ cells (Fig. 2B middle and right). Further, in spleen, peripheral blood and skin, HSV glycoprotein B (gB) dextramer+ cells were at a similar frequency in total CD8+ T cells (Fig. 2C left) irrespective of DOCK8 genotype. Only in the spleen were total numbers of gB+, CD8+ cells reduced (Fig. 2C right). We also looked at the effector differentiation of anti-viral CD8+ T cells. No significant difference was seen between DOCK8pri/pri and wild type mice in the ability of their CD8+ T cells to make IFNγ after a brief in vitro stimulation with gB peptide or the fraction of gB-dextramer+ cells making and storing granzyme B (Fig 2D left and middle). However, the total number of cells with these functions in the spleen were reduced in DOCK8pri/pri mice (Fig 2D, right), as expected due to the reduction in total CD8+ T cells noted previously. Upon resolution of infection, CD8+ and CD4+ T cell numbers and percents in spleen return to values similar to the baseline shown for uninfected DOCK8-deficient and sufficient mice (e.g. Supplementary Fig 1D and data not shown). Together these data suggest that despite the general lymphopenia in DOCK8pri/primice, CD8+ T cells were primed adequately by HSV infection and access the skin at the peak of the acute response. By contrast, CD4+ T cell recruitment to the skin was very substantially reduced. The intact primary CD8+ T cell responses echo findings for influenza virus and MVA in DOCK8 deficient mice 13. At face value our data are in contrast with other recent findings 13, but where we have looked at relatively crude preparations of cells from whole skin at the peak of infection, Zhang et al looked for CD8+ T cells in the epidermis and specifically at the formation of TRM. Thus the findings are complimentary rather than in conflict. The importance of CD4+ T cells in control of primary skin infection with HSV infection has long been known and so poor migration of these in the skin would likely contribute to loss of control of HSV in DOCK8 deficient patients 17-20, 14. The defect we find might be intrinsic to CD4+ T cells or could be related to impaired migration of an antigen presenting cell 21, 12, 22. It is also important to note that other players in anti-HSV immunity such as NK and NKT cells might be damaged by DOCK8 deficiency, but we have not examined these in this study 23, 24, 13. Finally, we have focused on primary HSV infection because this is most faithfully modeled in mice. We speculate that control of recurrence will be further compromised by DOCK8 deficiency due to problems associated with poor persistence of CD8+ T cell memory cells and in particular the profound CD8+ TRM defect 9, 25, 26, 23, 24, 27, 13, 14.
Figure 2. Immune responses during HSV infection in DOCK8 deficient mice.
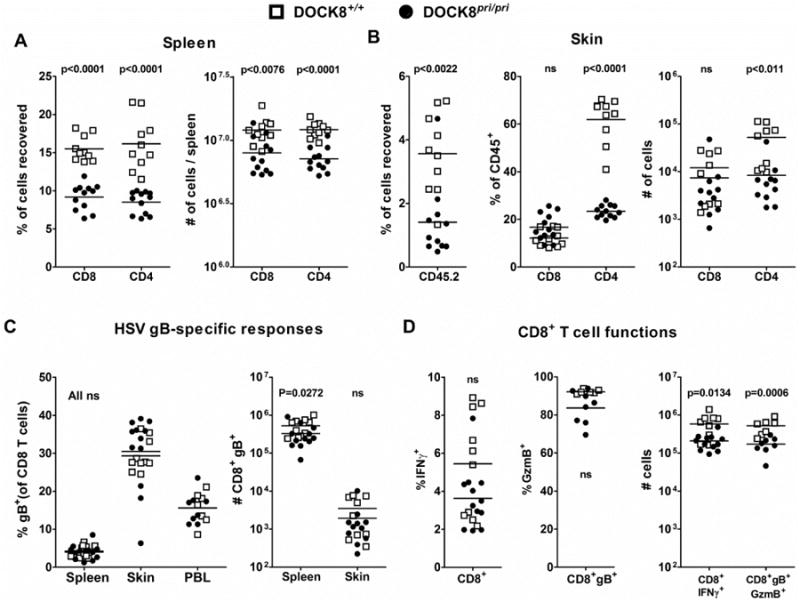
Cohorts of DOCK8pri/ipri mice and 4 DOCK8+/+ littermates were inoculated with HSV-1 strain KOS on the flank by tattoo and T cell responses were assessed at 7 dpi. (A) Percentages (left) and numbers (right) of CD8+ and CD4+ T cells in spleens. (B) Left, infiltration of leukocytes in the skin, indicated by number of CD45.2+ cells as a fraction of all cells recovered. Middle and right, CD4+ and CD8+ T cell infiltration into infected skin shown as a percent of CD45.2+ cells and as total number recovered, respectively. (C) HSV-specific CD8+ T cells, shown as the fraction of all CD8+ T cells (left) or total number (right) marked with a HSV gB-dextramer+ in spleen, skin and peripheral blood (PBL). (D) Activation of HSV-specific CD8+ T cells in the spleen (bottom) shown by the percent making IFNγ in response to stimulation with gB498 peptide (left) and the percentage of gB-dextramer+ CD8+ T cells staining for intracellular granzyme B (middle). On the right, total numbers of granzyme B+ gB-specific and IFNγ+ CD8+ T cells are shown. All graphs include data combined from two independent experiments, each point is a single animal and lines indicate the mean. Statistical significance (Mann Whitney) is noted with a p value or ns for p>0.05).
Methods
Viruses and cell lines
HSV-1 strain KOS was kindly provided by F. Carbone (The University of Melbourne, Parkville, Victoria, Australia). HSV-1 was grown and titrated by standard methods using Vero cells grown in Minimal Essential Medium supplemented with 10% FBS, 2 mM L-glutamine.
Mice
Mice were used according to ethical requirements under approval from the ANU animal ethics and experimentation committee. DOCK8pri/pri mice were generated by ENU mutagenesis as described in 11. DOCK8E1886X/E1886X mice were generated after a mutation introducing a premature stop codon in DOCK8 (19.25188409 G to T (assembly GRCM 38)) was chosen from a list of random single nucleotide variants generated by ENU mutagenesis (http://databases.apf.edu.au/mutations) and bred to homozygosity at generation 3 28.
HSV infections
Mice were anesthetized by i.p. injection of Avertin (20 μl/g of body weight). HSV (1×108 PFU/ml) was tattooed into a 0.5×0.5 cm2 area of shaved, depilated skin on the left flank. Body weight and lesion progression were measured daily.
Measurement of infectious virus
Skin and the DRG innervating the infected dermatome were removed at day 7 dpi and collected in 1 ml of MEM supplemented with 2% FBS and 4 mM L-glutamine. Samples were homogenized, freeze-thawed three times and viral titers were determined using standard plaque assays on Vero cells 29.
Immunological analyses
Spleens were pressed through a 70 μm cell strainer using the plunger end of a syringe and red cells lysed. For analysis of IFNγ production, splenocytes were incubated with gB498 peptide (SSIEFARL) (or no peptide as a negative control) for four hours in the presence of brefeldin A 30. Skin was digested with collagenase/DNase for 30 min at 37°C and washed through a 70 μm cell strainer with cold PBS containing 1% FBS. Heparinised whole blood (20 μl) was also used. Cells from infected mice were stained with one or more of the following panels of mAbs: 1) anti-CD8-PE (clone 53-6.7; BioLegend) and anti-CD4-APC (clone GK1.5; BioLegend), anti-CD45.2-FITC (clone 104; BioLegend) was included for cells from skin; 2) H-2Kb/gB-PE dextramer, anti-CD8-FITC and in some experiments anti-GzmB-Alexa647 (clone GB11). A dextramer with an irrelevant peptide was used as a background control; 3) On peptide stimulated splenocytes, anti-CD8α-PE followed by intracellular staining with anti-IFN-γ-APC (clone XMG1.2). Cells from naïve mice were stained with (from BD Pharmingen unless otherwise specified) FITC-conjugated anti-CD21 (clone 7G6), PE-conjugated anti-CD23 (B3G4), PerCP-conjugated anti-B220 (RA36B2), APC-conjugated anti-CD44 (1M7), A700-conjugated anti-CD4 (RM4-5, Biolegend) and APC Cy7-conjugated anti-CD8 (53-6.7, Biolegend). Cells from spleen and skin were fixed with 1% paraformaldehyde before and data acquired on a LSRII flow cytometer (BD biosciences). For whole blood, 200 μl FACS lysing solution (BD Biosciences) was added per sample and incubated for 15 min at RT after mixing before acquisition on the flow cytometer within 2 hours. Analysis was done using Flowjo software (Tree Star Inc.). Events were gated for live lymphocytes on FSC × SSC and appropriate parameters examined after doublet exclusion. Data were further analysed using Prism (GraphPad).
Supplementary Material
Acknowledgments
This work was funded by NIH grant U19 AI100627, NHMRC Project grants GNT1049760 and GNT1022922 and Australia Fellowship 585490 and ARC Future Fellowship FT110100310. We thank Anselm Enders for helpful comments on the manuscript. We also thank APF and RSB animal services for mouse husbandry, the Australian Phenomics Facility for DNA preparation, genotyping and exome analysis, and the Immunogenomics Laboratory Bioinformatics Group for exome sequence analysis.
References
- 1.Engelhardt KR, McGhee S, Winkler S, Sassi A, Woellner C, Lopez-Herrera G, et al. Large deletions and point mutations involving the dedicator of cytokinesis 8 (DOCK8) in the autosomal-recessive form of hyper-IgE syndrome. J Allergy Clin Immunol. 2009;124:1289–302 e4. doi: 10.1016/j.jaci.2009.10.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Zhang Q, Davis JC, Lamborn IT, Freeman AF, Jing H, Favreau AJ, et al. Combined immunodeficiency associated with DOCK8 mutations. N Engl J Med. 2009;361:2046–55. doi: 10.1056/NEJMoa0905506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Chu EY, Freeman AF, Jing H, Cowen EW, Davis J, Su HC, et al. Cutaneous manifestations of DOCK8 deficiency syndrome. Arch Dermatol. 2012;148:79–84. doi: 10.1001/archdermatol.2011.262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.McDonald DR, Massaad MJ, Johnston A, Keles S, Chatila T, Geha RS, et al. Successful engraftment of donor marrow after allogeneic hematopoietic cell transplantation in autosomal-recessive hyper-IgE syndrome caused by dedicator of cytokinesis 8 deficiency. J Allergy Clin Immunol. 2010;126:1304–5 e3. doi: 10.1016/j.jaci.2010.07.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Gatz SA, Benninghoff U, Schutz C, Schulz A, Honig M, Pannicke U, et al. Curative treatment of autosomal-recessive hyper-IgE syndrome by hematopoietic cell transplantation. Bone Marrow Transplant. 2011;46:552–6. doi: 10.1038/bmt.2010.169. [DOI] [PubMed] [Google Scholar]
- 6.Keles S, Jabara HH, Reisli I, McDonald DR, Barlan I, Hanna-Wakim R, et al. Plasmacytoid dendritic cell depletion in DOCK8 deficiency: rescue of severe herpetic infections with IFN-alpha 2b therapy. J Allergy Clin Immunol. 2014;133:1753–5 e3. doi: 10.1016/j.jaci.2014.03.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Papan C, Hagl B, Heinz V, Albert MH, Ehrt O, Sawalle-Belohradsky J, et al. Beneficial IFN-alpha treatment of tumorous herpes simplex blepharoconjunctivitis in dedicator of cytokinesis 8 deficiency. J Allergy Clin Immunol. 2014;133:1456–8. doi: 10.1016/j.jaci.2014.02.008. [DOI] [PubMed] [Google Scholar]
- 8.Al-Zahrani D, Raddadi A, Massaad M, Keles S, Jabara HH, Chatila TA, et al. Successful interferon-alpha 2b therapy for unremitting warts in a patient with DOCK8 deficiency. Clin Immunol. 2014;153:104–8. doi: 10.1016/j.clim.2014.04.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Randall KL, Chan SS, Ma CS, Fung I, Mei Y, Yabas M, et al. DOCK8 deficiency impairs CD8 T cell survival and function in humans and mice. J Exp Med. 2011;208:2305–20. doi: 10.1084/jem.20110345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Crawford G, Enders A, Gileadi U, Stankovic S, Zhang Q, Lambe T, et al. DOCK8 is critical for the survival and function of NKT cells. Blood. 2013;122:2052–61. doi: 10.1182/blood-2013-02-482331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Randall KL, Lambe T, Johnson AL, Treanor B, Kucharska E, Domaschenz H, et al. Dock8 mutations cripple B cell immunological synapses, germinal centers and long-lived antibody production. Nature Immunol. 2009;10:1283–91. doi: 10.1038/ni.1820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Harada Y, Tanaka Y, Terasawa M, Pieczyk M, Habiro K, Katakai T, et al. DOCK8 is a Cdc42 activator critical for interstitial dendritic cell migration during immune responses. Blood. 2012;119:4451–61. doi: 10.1182/blood-2012-01-407098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Zhang Q, Dove CG, Hor JL, Murdock HM, Strauss-Albee DM, Garcia JA, et al. DOCK8 regulates lymphocyte shape integrity for skin antiviral immunity. J Exp Med. 2014;211:2549–66. doi: 10.1084/jem.20141307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Russell TA, Stefanovic T, Tscharke DC. Engineering herpes simplex viruses by infection–transfection methods including recombination site targeting by CRISPR/Cas9 nucleases. J Virol Met. 2015;213:18–25. doi: 10.1016/j.jviromet.2014.11.009. [DOI] [PubMed] [Google Scholar]
- 15.van Lint A, Ayers M, Brooks AG, Coles RM, Heath WR, Carbone FR. Herpes simplex virus-specific CD8+ T cells can clear established lytic infections from skin and nerves and can partially limit the early spread of virus after cutaneous inoculation. J Immunol. 2004;172:392–7. doi: 10.4049/jimmunol.172.1.392. [DOI] [PubMed] [Google Scholar]
- 16.Kastrukoff LF, Lau AS, Takei F, Smyth MJ, Jones CM, Clarke SRM, et al. Redundancy in the immune system restricts the spread of HSV-1 in the central nervous system (CNS) of C57BL/6 mice. Virol. 2010;400:248–58. doi: 10.1016/j.virol.2010.02.013. [DOI] [PubMed] [Google Scholar]
- 17.Cunningham AL, Turner RR, Miller AC, Para MF, Merigan TC. Evolution of recurrent herpes simplex lesions. An immunohistologic study. J Clin Invest. 1985;75:226–33. doi: 10.1172/JCI111678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Manickan E, Rouse BT. Roles of different T-cell subsets in control of herpes simplex virus infection determined by using T-cell-deficient mouse-models. J Virol. 1995;69:8178–9. doi: 10.1128/jvi.69.12.8178-8179.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Cornish AL, Keating R, Kyparissoudis K, Smyth MJ, Carbone FR, Godfrey DI. NKT cells are not critical for HSV-1 disease resolution. Immunol Cell Biol. 2005;84:13–9. doi: 10.1111/j.1440-1711.2005.01396.x. [DOI] [PubMed] [Google Scholar]
- 20.Iijima N, Iwasaki A. A local macrophage chemokine network sustains protective tissue-resident memory CD4 T cells. Science. 2014;346:93–8. doi: 10.1126/science.1257530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lee HK, Zamora M, Linehan MM, Iijima N, Gonzalez D, Haberman A, et al. Differential roles of migratory and resident DCs in T cell priming after mucosal or skin HSV-1 infection. J Exp Med. 2009;206:359–70. doi: 10.1084/jem.20080601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Macleod BL, Bedoui S, Hor JL, Mueller SN, Russell TA, Hollett NA, et al. Distinct APC subtypes drive spatially segregated CD4+ and CD8+ T-cell effector activity during skin infection with HSV-1. PLoS Pathog. 2014;10:e1004303. doi: 10.1371/journal.ppat.1004303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Crawford G, Enders A, Gileadi U, Stankovic S, Zhang Q, Lambe T, et al. DOCK8 is critical for the survival and function of NKT cells. Blood. 2013;122:2052–61. doi: 10.1182/blood-2013-02-482331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Mizesko MC, Banerjee PP, Monaco-Shawver L, Mace EM, Bernal WE, Sawalle-Belohradsky J, et al. Defective actin accumulation impairs human natural killer cell function in patients with dedicator of cytokinesis 8 deficiency. J Allergy Clin Immunol. 2013;131:840–8. doi: 10.1016/j.jaci.2012.12.1568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ariotti S, Beltman JB, Chodaczek G, Hoekstra ME, van Beek AE, Gomez-Eerland R, et al. Tissue-resident memory CD8+ T cells continuously patrol skin epithelia to quickly recognize local antigen. PNAS. 2012;109:19739–44. doi: 10.1073/pnas.1208927109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Mackay LK, Stock AT, Ma JZ, Jones CM, Kent SJ, Mueller SN, et al. Long-lived epithelial immunity by tissue-resident memory T (TRM) cells in the absence of persisting local antigen presentation. PNAS. 2012;109:7037–42. doi: 10.1073/pnas.1202288109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Zhu J, Peng T, Johnston C, Phasouk K, Kask AS, Klock A, et al. Immune surveillance by CD8αα+ skin-resident T cells in human herpes virus infection. Nature. 2013;497:494–7. doi: 10.1038/nature12110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Andrews TD, Whittle B, Field MA, Balakishnan B, Zhang Y, Shao Y, et al. Massively parallel sequencing of the mouse exome to accurately identify rare, induced mutations: an immediate source for thousands of new mouse models. Open Biol. 2012;2:120061. doi: 10.1098/rsob.120061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Russell W. A sensitive and precise plaque assay for herpes virus. Nature. 1962;195:1028–9. doi: 10.1038/1951028a0. [DOI] [PubMed] [Google Scholar]
- 30.Flesch IEA, Hollett NA, Wong YC, Tscharke DC. Linear fidelity in quantification of antiviral CD8+ T cells. PLoS ONE. 2012;7:e39533. doi: 10.1371/journal.pone.0039533. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.