

Abstract
Certain intracellular bacteria use the host cell cytosol as the replicative niche. Although it has been hypothesized that the successful exploitation of this compartment requires a unique metabolic adaptation, supportive evidence is lacking. For Francisella tularensis, many genes of the Francisella pathogenicity island (FPI) are essential for intracellular growth, and therefore, FPI mutants are useful tools for understanding the prerequisites of intracytosolic replication. We compared the growth of bacteria taken up by phagocytic or nonphagocytic cells with that of bacteria microinjected directly into the host cytosol, using the live vaccine strain (LVS) of F. tularensis; five selected FPI mutants thereof, i.e., ΔiglA, ΔiglÇ ΔiglG, ΔiglI, and ΔpdpE strains; and Listeria monocytogenes. After uptake in bone marrow-derived macrophages (BMDM), ASC−/− BMDM, MyD88−/− BMDM, J774 cells, or HeLa cells, LVS, ΔpdpE and ΔiglG mutants, and L. monocytogenes replicated efficiently in all five cell types, whereas the ΔiglA and ΔiglC mutants showed no replication. After microinjection, all 7 strains showed effective replication in J774 macrophages, ASC−/− BMDM, and HeLa cells. In contrast to the rapid replication in other cell types, L. monocytogenes showed no replication in MyD88−/− BMDM and LVS showed no replication in either BMDM or MyD88−/− BMDM after microinjection. Our data suggest that the mechanisms of bacterial uptake as well as the permissiveness of the cytosolic compartment per se are important factors for the intracytosolic replication. Notably, none of the investigated FPI proteins was found to be essential for intracytosolic replication after microinjection.
INTRODUCTION
Bacteria and other microbes have developed an ability to invade host cells and use them as a principal habitat for replication. These so-called intracellular pathogens are able to trigger their uptake by mammalian cells, by phagocytosis when the host cells are professional phagocytes, e.g., monocytes or macrophages, or by triggered phagocytosis in the case of nonprofessional phagocytic host cells, such as epithelial or endothelial cells, hepatocytes, and fibroblasts (1, 2). After internalization, virulence factors produced by the intracellular pathogen modulate the intracellular environment to facilitate microbial survival (3, 4). For protection against intracellularly located microorganisms, the immune system is dependent on pattern recognition receptors (PRR) that identify conserved microbial components (5). The best-characterized family of PRR is the one of Toll-like receptors (TLR), a group of integral membrane proteins that recognize microbial components, such as lipopolysaccharide, bacterial lipoprotein, and CpG DNA (6, 7). Triggering of TLR leads to rapid initiation of an antimicrobial proinflammatory response (8, 9). These innate defense mechanisms are normally sufficient to mediate the eradication of phagocytosed extracellular pathogens but not to control those capable of intracellular replication.
Many intracellular bacteria, e.g., Mycobacterium, Brucella, Salmonella, Legionella, and Chlamydia spp., reside and replicate inside phagosomal compartments after subverting their composition, whereas others, such as Listeria, Francisella, Shigella, and Rickettsia spp., show further specialization and manage to escape from the confined intracellular compartments to directly use the cytoplasm as their replicative habitat (10). To combat the latter group, the macrophage utilizes cytosolic sensors belonging to the Nod-like receptor (NLR) or AIM2-like receptor families (11, 12). Engagement of these receptors leads to the formation of the inflammasome, a multiprotein complex composed of a sensor protein belonging to the NLR or AIM2-like families, an adaptor protein, ASC, and caspase-1 (13). The inflammasome activation leads to macrophage death, normally beneficial to the host since it eliminates the pathogen's normal habitat. Upon microinjection into the host cytosol, bacteria capable of phagosomal escape, unlike extracellular bacteria or normally vacuole-confined intracellular pathogens, show cytosolic replication (14). This finding implies that, despite the fact that the cytosol is a nutrient-rich compartment, access to the cytosol of mammalian cells per se is not sufficient for replication. Therefore, it was hypothesized that bacteria which successfully replicate in the cytosol harbor a metabolic machinery that is adapted to this niche in order to utilize available nutrients (14). However, there is accumulating evidence that the metabolic requirements may be similar for bacteria residing within the eukaryotic cytosol and bacteria residing extracellularly (15–19), thus indicating that modulation of the cytosolic composition, e.g., by deprivation of the availability of metabolites, may be an important factor to control replication of intracellular bacteria. To add further complexity, there is recent evidence that manipulation of autophagy is used as a means by pathogens to acquire energy and nutrients. With regard to Francisella tularensis, indirect evidence implies that it uses autophagy to increase the cytosolic nutrient pool and thereby provides energy for its rapid cytosolic replication (20, 21).
Two facultative intracellular pathogens with distinct intracellular behaviors and disease outcome are Listeria monocytogenes and F. tularensis. The former bacterium is the causative agent of the food-borne disease listeriosis, which rarely presents as a gastrointestinal disease but instead leads to meningitis with high fatality rates, particularly in immunocompromised individuals. Once taken up by macrophages, the bacterium escapes the hostile environment of the phagosomal compartment via hemolysin- and phospholipase-mediated lysis of the vacuole and reaches the cytosol, where it rapidly multiplies and from which it can move to adjacent cells by exploiting the host actin cytoskeleton (22). Also, F. tularensis replicates in the cytosol of macrophages, and it is the etiological agent of the zoonotic disease tularemia (23). The disease is relatively infrequent in humans, although there are areas of endemicity in the world with high incidence, most notably in Finland and Sweden (23). Outbreaks occur predominantly among rabbits, hares, and small rodents. The bacterium is highly infectious, and strains of the subspecies Francisella tularensis subsp. tularensis are highly virulent and cause a potentially life-threatening disease (24). An often-used surrogate F. tularensis strain is the live vaccine strain (LVS), which was derived by attenuation through plate passaging. Although not licensed for use in the general population, it has been employed as a vaccine for high-risk groups like laboratory personnel since the 1960s (23). LVS is highly virulent in mice and thus a useful strain for studies of experimental infection.
The intramacrophage survival and replication of F. tularensis are intimately dependent on the expression of most of the proteins expressed by the Francisella pathogenicity island (FPI), a locus that comprises 17 to 19 genes which are highly conserved among different subspecies (25). Exactly how Francisella executes its unique intracellular lifestyle is, however, not well understood. It has been hypothesized that the FPI proteins constitute a type VI secretion system (T6SS); there is accumulating evidence that this is indeed the case, and a number of secreted proteins have recently been identified (26–28). Bioinformatic analysis has demonstrated that the FPI gene cluster forms a group evolutionarily distinct from other described T6SSs (29). The best-characterized FPI proteins are encoded by the iglABCD operon, and it has been found that the four encoded Igl proteins are required for escape from the phagosome and for replication within the macrophage cytosol (30–33). Whereas IglC and IglD appear to be unique to F. tularensis, IglA and IglB are conserved components that constitute the sheath of the T6SS tubular structure (33–35). The IglG and IglI proteins are other examples of components that contribute to the phagosomal escape; however, they are not essential for this process, since the corresponding mutants are both capable of delayed phagosomal escape and at least the former is also capable of intact cytosolic replication (27). In contrast, the FPI component PdpE is not required for escape or intracellular growth (27, 28). Interestingly, IglC, IglI, and PdpE, but not IglA or IglG, are secreted in a T6SS-dependent manner during infection (28). Thus, the spectrum of phenotypes observed for FPI mutants renders them useful tools to understand the prerequisites of cytosolic growth since some show intact phagosomal escape and intracellular replication, whereas others are defective for both or show delayed escape but intact intracellular replication (25, 36).
The macrophage defense strategy to control Francisella and Listeria infection is unusual, since it fully or partially depends on AIM2, but not on any NLR (3, 37–40). Accordingly, AIM2-, ASC-, or caspase-1-knockout mice are highly susceptible to F. tularensis and L. monocytogenes infections (38, 39, 41). However, before reaching the cytosol, these bacteria interact during the phagosomal phase with TLR2, the principal TLR responsible for their recognition (42–47). Therefore, TLR2- and TLR-adaptor MyD88-deficient mice are highly susceptible to infections with F. tularensis and L. monocytogenes (45, 48–50), and the macrophage inflammatory responses to both pathogens are critically dependent on MyD88 (48, 49, 51). Interestingly, the MyD88- and ASC-signaling pathways appear to interact, since MyD88-dependent TLR2 activation is necessary for the rapid AIM2-inflammasome-mediated responses during infection with Francisella (44).
Here, we investigated the requirement for the intracellular growth of F. tularensis and the prototypic intracytosolic pathogen L. monocytogenes and specifically asked if the FPI proteins IglA, IglC, IglG, IglI, and PdpE are necessary for the intracytosolic replication of the former pathogen. This was performed by comparing the intracellular growth rates of wild-type bacteria and of bacterial mutants lacking either of the FPI proteins after normal phagocytic uptake or after microinjection into the cytosol of macrophages or epithelial cells. Our data indicate that efficient cytosolic growth appears to be intricately dependent on both host and pathogen factors and that none of the investigated FPI proteins are essential for intracytosolic replication.
MATERIALS AND METHODS
Bacterial strains and plasmids.
Bacterial strains used in this study are listed in Table S1 in the supplemental material. F. tularensis was cultivated on modified GC agar base, with 10 μg/ml of kanamycin when appropriate. L. monocytogenes was grown on brain heart infusion (BHI) agar with 10 μg/ml erythromycin at 37°C. A green fluorescent protein (GFP)-expressing plasmid, pKK289Km (32) or pNF8 (52), was introduced into F. tularensis or L. monocytogenes, respectively, by electroporation.
Cultivation and infection of macrophages and HeLa cells.
The J774 macrophage-like cell line (ATCC TIB-67), bone marrow-derived macrophages (BMDM), and the HeLa (ATCC CCL-2) human epithelial carcinoma cell line were used in cell infection assays. Cells were cultured and maintained in Dulbecco modified Eagle medium (DMEM; Gibco BRL, Grand Island, NY) with 10% heat-inactivated fetal bovine serum (FBS; Gibco) at 37°C and 5% CO2. BMDM were isolated by flushing cells from femurs and tibias of C57BL/6 or MyD88−/− or ASC−/− knockout mice, and the extracted cells were cultured for 4 days in DMEM containing 10% FBS, 2 μg/ml gentamicin, and 10% conditioned medium from L929 cells (ATCC catalog no. CCL-1). After 4 days, cells were cultivated in conditioned medium without gentamicin. One day prior to infection, cells were seeded in tissue culture plates in medium appropriate for the respective cell type. Next day, cells were washed and reconstituted with fresh culture medium and allowed to recover for at least 30 min prior to infection. Cells were infected with a multiplicity of infection (MOI) of 200 for 2 h (J774 and BMDM) or 5 h (HeLa). After infection, cells were washed and kept in DMEM with 10% FBS and 5 μg/ml gentamicin. Cells were lysed with 0.1% deoxycholate after indicated time points, and lysates were plated on modified GC base agar to determine viable counts.
All animal experiments were approved by the Ethical Committee on Animal Research, Umeå, approval no. A100-11, and the Stockholm North Region Ethical Animal Research Committee, approval no. 487/11.
Microinjection of macrophages and HeLa cells.
J774, BMDM, or HeLa cells were seeded at a density of 2 × 105 cells in glass-bottom petri dishes (MatTek Corporation, Ashland, MA) in appropriate medium. Prior to injection, medium was replaced by fresh DMEM with appropriate supplements. The cells were allowed to recover for 60 to 90 min. Plate-grown GFP-expressing bacteria were resuspended in phosphate-buffered saline (PBS) to 1 × 109 CFU/ml and mixed with the dye rhodamine dextran (RD; 25 mM in PBS, pH 7.5; Sigma-Aldrich). The dye allowed visualization of injected cells. Injections were carried out with Femtotips II (Eppendorf, Hamburg, Germany) with an injection pressure of 40 hPa supplied by an automated InjectMan NI 2 micromanipulator (Eppendorf). This pressure was carefully selected to avoid damage of cells during the injection process. According to information from the manufacturer, the maximal volume is approximately 300 fl using the standard procedure. We routinely shortened the capillary tip to enable bacteria to pass freely, and the maximal volume per injection was therefore estimated to be at most 500 fl, allowing 2 to 20 bacteria and RD to be delivered into the cell cytosol. Injected cells were washed with DMEM containing 2 μg/ml of gentamicin and 1 μg/ml of cytochalasin D (53) and incubated at 37°C for 1 h. The former was added to eradicate extracellular bacteria resulting from unsuccessful injection, and the latter was added to avoid phagocytosis of such bacteria during the incubation. The medium was then changed to DMEM with 10% FBS and with or without l-glutamine, and cultures were incubated at 37°C with 5% CO2. For each injection experiment, triplicate samples were used and 30 to 50 cells per strain and sample were injected. Out of these, typically 70 to 100% of the cells were successfully microinjected. At 2 h, colocalization of bacteria and RD was confirmed and pictures were taken using a live-cell microscope (Nikon Eclipse Ti-E equipped with an Andor iXon+ electron-multiplying charge-coupled device [EMCCD] camera). At 24 h, microscopic counting was performed on RD-positive cells only and an approximate number of bacteria per infected cell was determined, resulting in the three categories of 0 to 20, 20 to 100, and 100 to 1,000 bacteria/infected cell. The average number of bacteria of each strain per cell was calculated by multiplying the mean number of bacteria and number of cells for each category, calculating the sum of the three categories, and dividing the sum by the number of total infected cells. Typically, 100 to 150 cells were counted per bacterial strain for each cell type, with the exception of HeLa cells, for which 50 to 100 cells were counted.
Statistical analysis.
For statistical evaluation of intracellular replication after phagocytosis, a two-sided Student t test with equal variance was used. For pairwise comparisons between bacterial numbers after microinjection, the chi-square test was used in most instances since the power of the chi-square test is higher than that of Fisher's exact test. However, when the data sets were very unbalanced, i.e., the classification categories were unevenly represented, the chi-square test was not appropriate, and in these instances, Fisher's exact test was used. Bacterial replication was assessed by comparing initial values after washing of the cell cultures, denoted as the 2-h time point, with those at the 24-h time point.
RESULTS
Requirement of FPI proteins for replication after phagocytic uptake.
Previous studies have demonstrated that many of the FPI proteins are necessary for replication in J774 cells. Thus, the ΔiglA and ΔiglC mutants do not escape from the phagosome and show deficient intramacrophage replication (30, 31, 33, 54). The ΔiglG mutant replicates efficiently in the J774 macrophage cell line and in primary macrophages, whereas the ΔiglI mutant replicates only in the former cell type (27). We previously noted, however, that the latter two mutants induced much less prominent host cell cytopathogenic effects than did the parental strain, suggesting a requirement for the encoded proteins in modulating the host cell death pathway induced by F. tularensis (27, 55). In contrast, the ΔpdpE mutant is one of the few FPI mutants that exhibits wild-type phenotypes with regard to replication and cytopathogenicity in monocytic cells (27). In view of these previously published findings, the abovementioned 5 FPI mutants were included in this study together with L. monocytogenes, since the latter is a prototypic bacterium with regard to intracytosolic replication (14, 56).
To study the prerequisites of intracellular growth, we included the murine J774 macrophage-like cell line, since it has been widely used to investigate various aspects of L. monocytogenes and F. tularensis host cell infections in the past and therefore will serve as a comparison with previously published studies (14, 56–63). The available evidence indicates, however, that J774 cells do not possess as potent an antimicrobial capacity as do various forms of primary macrophages (64), and in support of this view, we have previously observed that the ΔiglI mutant replicated readily in J774 macrophages but not in BMDM (27). Therefore, we also investigated how the bacterial strains replicated in murine BMDM. In addition, there is very limited knowledge regarding the phenotypes of FPI mutants upon infection of nonphagocytic cell types, and to this end, HeLa cells were included as a model in the study.
After phagocytosis, L. monocytogenes showed rapid replication, between 2 and 3 log10 after 8 h, in all five types of cells infected, and the rapid replication led to extensive host cell death already within 24 h, resulting in a corresponding decrease of the bacterial numbers (see Fig. S1 in the supplemental material; also data not shown).
After phagocytosis, we observed that the ΔiglG, ΔiglI, and ΔpdpE mutants replicated very effectively in J774 cells and, in fact, slightly better than did LVS (Table 1; see also Fig. S2 in the supplemental material), whereas the ΔiglA and ΔiglC mutants showed no growth, which was in agreement with previously published data (27, 55). Significant growth of LVS and the ΔiglG and ΔpdpE mutants was observed also in BMDM, whereas none of the other three mutants showed any replication (see Fig. S3).
TABLE 1.
Replication of F. tularensis strains and L. monocytogenes upon phagocytic uptake or microinjection into various cell types
| Bacterium | Growth in cell line afterd: |
|||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Phagocytosisa |
Microinjectionb |
|||||||||
| J774 | BMDM | ASC−/− | MyD88−/− | HeLa | J774 | BMDM | ASC−/− | MyD88−/− | HeLa | |
| Listeria monocytogenes | +++c | +++c | +++c | +++c | +++c | +++ | +++ | +++ | − | +++ |
| F. tularensis strains | ||||||||||
| LVS | ++ | ++ | ++ | +++ | ++ | ++ | ± | ++ | ± | +++ |
| ΔiglA mutant | − | − | − | − | − | ++ | ++ | +++ | +++ | ++ |
| ΔiglC mutant | − | − | − | − | − | ++ | + | +++ | +++ | +++ |
| ΔiglG mutant | +++ | ++ | ++ | + | +++ | +++ | ++ | +++ | ++ | +++ |
| ΔiglI mutant | +++ | − | + | + | ++ | ++ | ++ | +++ | +++ | +++ |
| ΔpdpE mutant | +++ | +++ | +++ | +++ | +++ | +++ | ++ | +++ | +++ | +++ |
The actual numbers of bacteria are shown in Fig. S1 to S6 in the supplemental material.
The estimated number of bacteria per cell and the differences indicated are based on statistical evaluations as summarized in Table 2.
Due to rapid growth, the number of L. monocytogenes bacteria was determined at 8 h.
Bacterial numbers were recorded after 24 h and are represented as a sliding scale ranging from very significant replication (+++) to no replication (−).
We hypothesized that the efficient control effectuated by the BMDM on bacterial growth would be dependent on MyD88 or ASC since each of them performs such essential immune functions against F. tularensis (38, 39, 41, 45, 48–50). Thus, the ability of the mutants to replicate after phagocytic uptake was investigated in ASC−/− and MyD88−/− BMDM. The ΔpdpE mutant consistently showed efficient replication, in fact, significantly better than LVS at both 24 h and 48 h. The ΔiglA, ΔiglC, ΔiglG, and ΔiglI mutants showed minimal or no growth in both of the deficient BMDM lines at 24 h (Table 1; see also Fig. S4 and S5 in the supplemental material), although the latter two replicated as well as LVS in ASC−/− BMDM at 48 h (see Fig. S4). In contrast, the ΔiglG and ΔiglI mutants showed less replication than LVS in MyD88−/− BMDM at 48 h (see Fig. S5) and the ΔiglA and ΔiglC mutants showed no replication. In fact, the numbers of the latter two decreased significantly at 48 h, suggesting that they were being killed (see Fig. S5). Interestingly, ASC appeared to be significant for the control of ΔiglI after phagocytosis, since this mutant replicated well in the ASC−/− BMDM but not in the wild-type BMDM (see Fig. S3 and S4).
We also investigated the ability of each mutant to grow within HeLa cells. The LVS strain replicated in HeLa cells, although with protracted uptake compared to macrophages (see Fig. S6 in the supplemental material). Again, the ΔiglG mutant, ΔpdpE mutant, and, to some extent, also the ΔiglI mutant all replicated effectively, while the ΔiglA and ΔiglC mutants did not replicate (see Fig. S6).
Bacterial replication within J774 macrophages after microinjection.
Several of the FPI mutants, e.g., the ΔiglA and ΔiglC mutants, have been demonstrated to not escape from the phagosome after phagocytic uptake (30–33). Since these mutants never reach the cytosolic compartment where the intracellular growth occurs, it has not been possible to determine whether the corresponding FPI proteins also play a role for the latter process. To circumvent the step of phagosomal escape, we used microinjection. Only limited information exists regarding replication of bacteria in the cytosol of phagocytic cells after microinjection (14). Thus, we developed a protocol for microinjection of GFP-labeled bacteria using automated injection equipment to compare the growth of microinjected L. monocytogenes and F. tularensis strains in various cell types. The dye RD was coinjected with bacteria to identify the injected cells. After 24 h, microscopic counting was performed on RD-positive cells and an approximate number of bacteria per infected cell was determined for each strain, resulting in the three categories of 0 to 20, 20 to 100, and 100 to 1,000 bacteria/infected cell (see Fig. S7 in the supplemental material). The mean number of the bacteria per cell was used as a measure of the propensity of intracytosolic replication.
After injection into J774 cells, L. monocytogenes replicated rapidly, and it occupied most of the cytosol within 24 h and started to spread to neighboring cells (Fig. 1), which is in agreement with previously published data (14). Of the L. monocytogenes-infected cells, the majority contained high bacterial numbers (Fig. 1; Table 2). Due to the rapid cell-to-cell spread of the bacterium, a considerable proportion of L. monocytogenes-infected cells were not stained with RD (Fig. 1). Injection of F. tularensis into J774 cells demonstrated that all of the five FPI mutants were capable of efficient intracytosolic replication during the 24-h period, equivalent to or even better than LVS (Fig. 2; Table 2).
FIG 1.
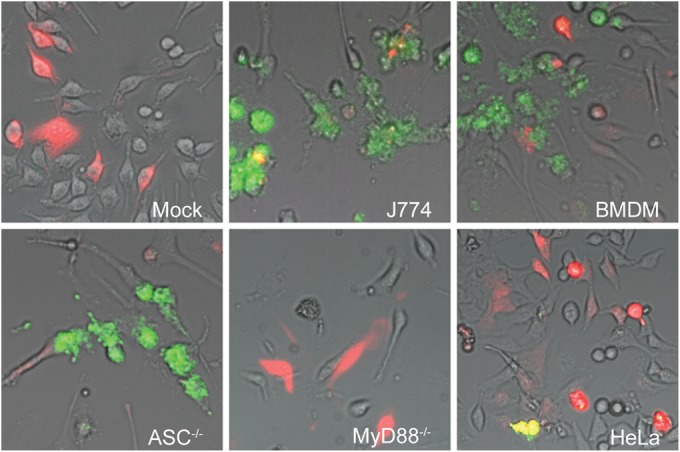
Microinjection of L. monocytogenes into indicated cell types. Pictures were taken at 24 h after injection with a live-cell imaging microscope equipped with an EMCCD camera. Colocalization of injected cells containing RD (red) and GFP-expressing bacteria (green) resulted in yellow signals, although due to the rapid cell-to-cell spread of L. monocytogenes, many of the infected cells did not contain RD. Representative pictures shown from at least three independent experiments. “Mock” indicates uninfected cells, i.e., cells injected with RD only.
TABLE 2.
Mean number of bacteria per cell in indicated cell types
| Cell type | Time (h) | Mean no. of bacteria of strain or species/cella |
||||||
|---|---|---|---|---|---|---|---|---|
|
F. tularensis |
Listeria monocytogenes | |||||||
| LVS | ΔiglA mutant | ΔiglC mutant | ΔiglG mutant | ΔiglI mutant | ΔpdpE mutant | |||
| J774 | 2 | 15 | 15.3 | 13.6 | 16.1 | 14.5 | 19.4 | 13.8 |
| 24 | 63.2* | 73.7*** | 95.8*** | 129.6*** | 97.8*** | 144.8*** | 335.8*** | |
| BMDM | 2 | 22.7 | 18.3 | 16.2 | 18.1 | 18.6 | 20.6 | 15.6 |
| 24 | 33.9 | 90.1*** | 56.0*** | 84.6*** | 99.5*** | 66.0*** | 155.5*** | |
| ASC−/− | 2 | 28.3 | 25.4 | 26.5 | 30.1 | 27.7 | 28.4 | 11.6 |
| 24 | 63.7*** | 128.7*** | 113.6*** | 119.3*** | 146.6*** | 153.8*** | 218.7*** | |
| MyD88−/− | 2 | 15.2 | 17.3 | 15.0 | 17.5 | 16.1 | 18.4 | 10.8 |
| 24 | 27.2 | 123.2*** | 93.0*** | 51.0** | 129.6*** | 78.2*** | 12.9 | |
| HeLa | 2 | 12.9 | 17.1 | 18.6 | 13.3 | 10 | 15.8 | 15.6 |
| 24 | 162.7*** | 135.5* | 302.5*** | 259.5*** | 237.7*** | 225.2*** | 199.4*** | |
Mean bacterial numbers of each strain at 2 h were compared to the mean numbers at 24 h, and the P values determined by the chi-square test or Fisher's exact test are indicated by asterisks (*, P ≤ 0.05; **, P ≤ 0.01; ***, P ≤ 0.001). Significant differences (P ≤ 0.05) between each of the F. tularensis mutants and LVS for the 24-h time point are indicated in bold.
FIG 2.
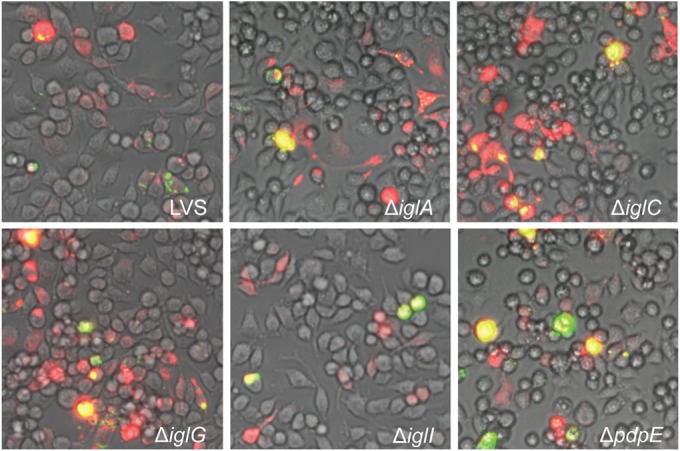
Microinjection of indicated F. tularensis strains into J774 cells. Pictures were taken at 24 h after injection with a live-cell imaging microscope equipped with an EMCCD camera. Colocalization of injected cells containing RD (red) and GFP-expressing bacteria (green) resulted in yellow signals. Representative pictures for each strain from at least three independent experiments are shown.
Collectively, our results demonstrate an ability of all tested F. tularensis strains and L. monocytogenes to replicate in the J774 cytosol upon microinjection. Notably, the absence of IglA or IglC did not have a negative impact on growth after microinjection, which is in contrast to the inability of the corresponding mutants to replicate after phagocytic uptake.
Replication within BMDM after microinjection.
The available evidence indicates that BMDM demonstrate better control of intracellular infection after phagocytosis than do J774 cells. For example, we have observed that with the exception of the ΔpdpE strain, other FPI mutants do not replicate, or show delayed replication, in BMDM, as demonstrated here and in previous publications (see Fig. S2 in the supplemental material) (27). After microinjection of bacteria, however, we observed that L. monocytogenes and all F. tularensis mutants showed efficient replication within 24 h and all five mutants showed significantly better replication than did LVS (Fig. 1 and 3), which, in fact, showed no significant increase between 2 and 24 h (Table 2). Thus, our results indicate that the key mechanisms required for cytosolic replication in BMDM as well as J774 cells are present in all mutant strains of F. tularensis as well as L. monocytogenes, whereas the LVS strain showed no significant replication in the former cell type.
FIG 3.
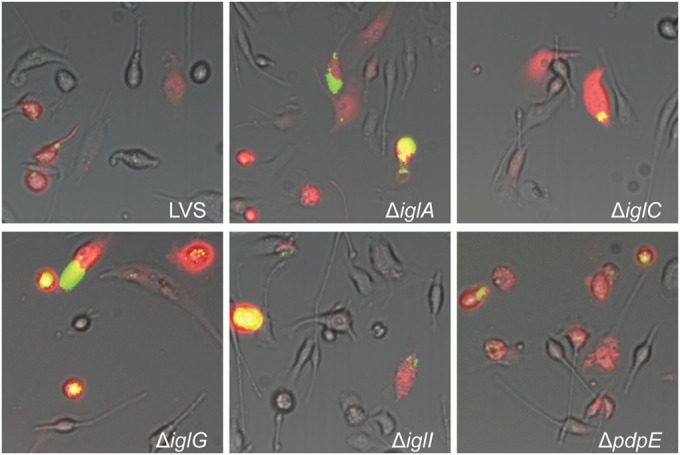
Microinjection of indicated F. tularensis strains into BMDM. Pictures were taken at 24 h after injection with a live-cell imaging microscope equipped with an EMCCD camera. Colocalization of injected cells containing RD (red) and GFP-expressing bacteria (green) resulted in yellow signals. Representative pictures for each strain from at least three independent experiments are shown.
Roles of ASC and MyD88 for control of the intracytosolic replication after microinjection.
Since ASC and MyD88 each perform such essential immune functions against F. tularensis and L. monocytogenes, their importance for intracellular growth upon microinjection was assessed. To this end, the replication of bacteria was followed after microinjection in ASC−/− or MyD88−/− BMDM. We observed that all F. tularensis strains were capable of efficient replication in ASC−/− BMDM (Fig. 4). In most instances, the mean values of the number of bacteria per cell were only marginally different between the strains, with the exception of the lower mean value for the LVS strain (Table 2). Also, in MyD88−/− BMDM, the mutants showed efficient intracellular replication, whereas LVS showed no significant replication (Fig. 5; Table 2). Thus, the cytosolic replication of the microinjected F. tularensis mutants was rapid also in the absence of MyD88 or ASC. Notably, LVS showed significant replication in ASC−/− BMDM but not in MyD88−/− BMDM (Table 2). Replication of L. monocytogenes was very rapid in ASC−/− BMDM; however, remarkably, there was essentially no replication in MyD88−/− BMDM after microinjection, even at 48 h (Fig. 1; also data not shown).
FIG 4.
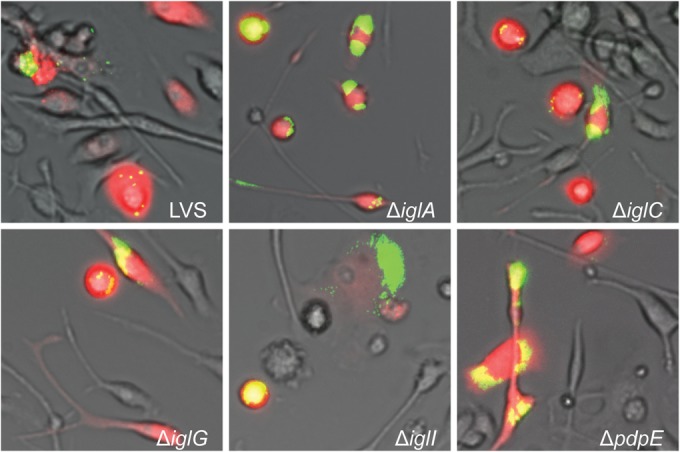
Microinjection of indicated F. tularensis strains into ASC−/− BMDM. Pictures were taken at 24 h after injection with a live-cell imaging microscope equipped with an EMCCD camera. Colocalization of injected cells containing RD (red) and GFP-expressing bacteria (green) resulted in yellow signals. Representative pictures for each strain from at least three independent experiments are shown.
FIG 5.
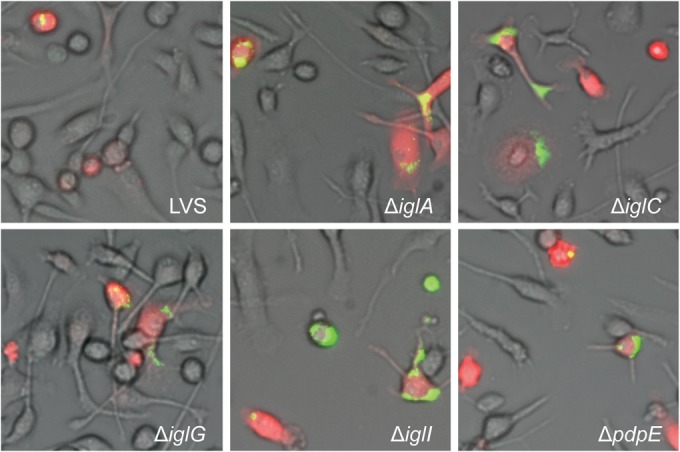
Microinjection of indicated F. tularensis strains into MyD88−/− BMDM. Pictures were taken at 24 h after injection with a live-cell imaging microscope equipped with an EMCCD camera. Colocalization of injected cells containing RD (red) and GFP-expressing bacteria (green) resulted in yellow signals. Representative pictures for each strain are shown from at least three independent experiments.
Replication within HeLa cells after microinjection.
The knowledge regarding the behavior of FPI mutants in nonphagocytic cells is very limited, and the number of published studies on the subject is low (65, 66). For this reason, HeLa cells were included in the study to investigate if differences in the cytosolic milieus of professional phagocytic cells and nonphagocytic cells would affect the fate of the bacteria after microinjection. Within 24 h, L. monocytogenes showed very rapid replication upon microinjection (Fig. 1 and Table 2) and also all the F. tularensis strains replicated efficiently (Fig. 6). The mean numbers were generally higher than those in the phagocytic cells and were for all strains between 130 and 300 bacteria per cell (Table 2). Thus, the cytosol of HeLa cells is very permissive to bacterial replication upon microinjection.
FIG 6.
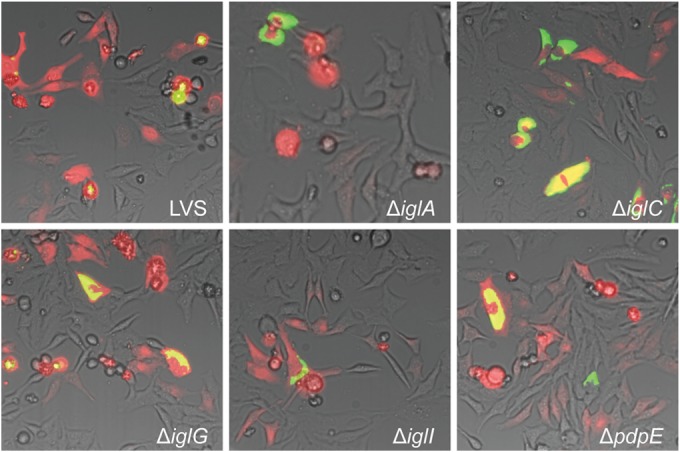
Microinjection of HeLa cells with LVS or isogenic FPI mutants thereof. Pictures were taken at 24 h after injection with a live-cell imaging microscope equipped with an EMCCD camera. Colocalization of injected cells containing RD (red) and GFP-expressing bacteria (green) resulted in yellow signals. Representative pictures for each strain from at least three independent experiments are shown.
Effect of coinjection of bacteria on intracytosolic replication.
Since the replication of LVS upon microinjection was unexpectedly low in comparison to the FPI mutants, we asked whether coinfection would affect the cytosol and make it more permissive for intracytosolic growth. Previously, the ΔiglG mutant has been shown to display a distinct phenotype with intact intracellular replication in J774 cells and BMDM but impaired modulation of the inflammatory response (27). Therefore, we performed coinjection with non-GFP-labeled ΔiglG bacteria together with GFP-labeled LVS. The experiments were performed as described previously, and injected J774 and BMDM cells were followed over 24 h. As determined by microscopic counting, again the LVS strain showed only limited replication (see Fig. S8 in the supplemental material; also data not shown). Thus, we conclude that potential changes in the cytosolic environment due to the replication of the ΔiglG mutant did not have an impact on the replication of the LVS strain in the tested cell types.
Relative permissiveness of the investigated cell types.
The overall numbers of the intracellular bacteria were categorized for each cell type and bacterial strain (Table 1; also see Fig. S9 in the supplemental material). The mean numbers of the F. tularensis mutants in phagocytic cells after 24 h were in all cases very significantly increased compared to the numbers after 2 h and ranged between 51 and 154 bacteria/cell, whereas the number of LVS bacteria varied from 27 to 64 and the latter showed no significant replication in BMDM and MyD88−/− BMDM (Table 2). There were high numbers of F. tularensis bacteria in HeLa cells; means ranged from 136 to 303, and these numbers were as high as, or higher than, those in any of the phagocytic cell types (Table 2). The mean numbers of L. monocytogenes bacteria were high in J774 cells, BMDM, ASC−/− BMDM, and HeLa cells, between 156 and 336, whereas they were only 13 in MyD88−/− BMDM, suggesting that Listeria is unable to replicate within this cell type (Table 2).
DISCUSSION
Much is unknown regarding the prerequisites for cytosolic replication of intracellular bacteria. Accumulating evidence indicates that the nutrient composition of the cytosol is a critical denominator, since deficiencies in metabolic pathways render intracytosolic bacteria incapable of intracellular replication (15–18, 67–69). In addition, evidence indicates that primary phagocytic cells, such as BMDM, generally control infection caused even by pathogens that replicate in the cytosol better than most macrophage-derived cell lines. Although this indirectly implies that the cytosol of certain phagocytic cells would inherently be less permissive for replication, the direct evidence for this hypothesis is essentially lacking. Our present findings demonstrate that the cytosolic compartments of different cell types exhibited discrete abilities to control growth of microinjected bacteria, most distinctly that the cytosol of HeLa cells was more permissive than those of phagocytic cells. Even in the latter cell types, there was, however, significant replication, and notably, even the lack of fundamental innate immune factors such as ASC or MyD88 did not markedly affect the cytosolic permissiveness for the F. tularensis mutants. This was somewhat unexpected but indicates that the successful adaptation to the cytosolic compartment is dependent on both host factors and bacterial factors and that the lack of expression of the investigated FPI proteins does not affect intracytosolic replication.
There were several notable findings regarding the phenotypes of the investigated FPI mutants, one of which was the lack of replication of the ΔiglA and ΔiglC mutants after uptake in the phagocytic cells as well as in HeLa cells, a cell type that was otherwise highly permissive for intracellular replication, whereas both mutants showed efficient replication after microinjection. IglC has also been shown to be translocated into the cytosol of J774 macrophages, which indicates the importance of that protein for phagosomal escape (27, 55). In addition, we observed that the ΔiglG mutant replicated efficiently after phagocytic uptake in all tested cell types and also after microinjection. Despite this, we have previously observed that it induces much less pronounced cytopathogenic effects and specifically modulates a host cell death pathway (27, 55). Thus, IglG seems to play a critical role in the interaction with the intracellular environment, although it is not essential for growth after phagocytosis or microinjection. After phagocytosis, the ΔiglI mutant was found to lack replication in BMDM, in agreement with previous studies using BMDM and peritoneal cells, but demonstrated intact replication in J774 cells (27). Here, we found that ΔiglI effectively replicated after microinjection. Thus, IglI appears to perform important roles for the intracellular survival of F. tularensis after phagocytosis (27), but, like IglA and IglC, it is dispensable for cytosolic replication. Based on our results, the mutants fall into three categories: (i) those that replicated efficiently upon normal infection as well as after microinjection, i.e., the ΔpdpE mutant; (ii) those that replicated to a variable degree after phagocytosis and consistently after microinjection, i.e., the ΔiglG and ΔiglI mutants; and (iii) those that did not replicate upon phagocytosis but did after injection into the cell cytosol, i.e., the ΔiglA and ΔiglC mutants. Notably, the LVS strain showed somewhat impaired growth compared to most of the mutants after microinjection, which may emphasize the importance of the phagosomal escape step for intracellular bacteria to efficiently adapt to the host cell environment. Wehrly et al. identified both early and late induction events of FPI genes, a finding consistent with a need for FPI proteins during the early phagosomal stage and at the end of the cytosolic replication stage (70). This could indicate that it may not be beneficial for the bacterium to have an active T6SS during the intermediate stage; perhaps, this increases the risk for host-mediated clearance. Based on this reasoning, LVS may be more easily recognized by the host than any FPI mutant that carries an inactive or defective T6SS, which then will result in a growth disadvantage for LVS upon microinjection; this was most notable in BMDM and MyD88-deficient macrophages.
The essential role of ASC as the adaptor protein for the inflammasome-mediated recognition of F. tularensis and L. monocytogenes has been thoroughly documented, and the critical role of MyD88 for the early inflammatory response to F. tularensis and Listeria is well established, although bacterial replication is not increased in MyD88-deficient macrophages (38, 41, 48, 50, 51). Therefore, we analyzed whether these innate immune pathways contributed to the bacterial control in the cytosol; however, after microinjection, the pattern of replication for the F. tularensis mutants was essentially indistinguishable between the wild-type macrophages and ASC−/− BMDM, whereas the MyD88−/− BMDM generally were slightly less permissive, resulting in lower bacterial numbers, and in fact, LVS showed no significant growth. Neither did LVS show any significant replication in BMDM. The most notable finding regarding the role of MyD88 was related to Listeria. In contrast to its very rapid replication in all other cell types after phagocytosis or microinjection, the bacterium exhibited essentially no replication in MyD88-deficient macrophages. Although the finding appears paradoxical in view of the important role of MyD88 for innate immune functions, it is possible that its absence affects the cytosolic response such that the cytosol will not be permissive for LVS and Listeria, e.g., by the lack of essential metabolites. Recent studies have identified that the release of bacterial components, e.g., DNA, from the phagosome induces a specific cytosolic response (11, 12, 51, 71, 72). Although it is often assumed that the sensing of bacterial products is a prerequisite for the control of intracellularly located bacteria, it is possible that successful intracellular pathogens have developed means to exploit this host response to their advantage and that, in fact, it is a necessity for the intracytosolic bacterial replication in certain cell types. Moreover, this implies that signaling after the microinjection will not trigger the same type of cytosolic responses and thereby the cytosolic environment may be distinct after phagocytic uptake versus microinjection. In addition, it was obvious that the average increase of bacterial numbers was generally lower after microinjection than after phagocytosis, and we cannot exclude the possibility that the microinjection technique per se may physically affect the host cell in such a way that the bacterial intracellular replication becomes adversely affected.
The present findings demonstrate the utility of the microinjection technique for delineating the prerequisites of intracytosolic growth. Although it provides direct evidence for the ability of bacteria to replicate in the cytosol, the technique has rarely been used (14). The previous study concluded that replication in the cytosolic compartment occurred only for bacteria that normally inhabit this niche, and it was hypothesized that successful cytosolic replication requires a metabolic adaptation that is not present in extracellular bacteria or intracellular vacuole-enclosed bacteria (14). Our findings imply, in addition, that the metabolic adaptation of intracytosolic microbes is a necessary, but alone not sufficient, factor for their replication and that an intricate host-bacterium interaction is required for the intracytosolic replication. In fact, using the microinjection technique, we previously demonstrated its utility to directly elucidate metabolic requirements for cytosolic replication, since it was demonstrated that a biotin biosynthesis mutant of Francisella novicida was incapable of intracytosolic growth unless biotin was added to the culture medium (19).
Experimentally, the live vaccine strain (LVS) of F. tularensis is widely used as a model of virulent strains of the species, although it is very significantly attenuated in the mouse model. Dysregulation of the iron uptake is the major mechanism behind its attenuation (73, 74). Despite this dysregulation, LVS proliferates as effectively as virulent strains within resting macrophages, presumably because there is an ample supply of iron under in vitro conditions. However, upon activation, the intracellular replication of LVS is constrained, and it is obvious that LVS is much more susceptible to certain intracellular killing mechanisms than the highly virulent strain SCHU S4 (75, 76). In addition, there are multiple immunomodulatory mechanisms executed by SCHU S4 that appear to be missing in LVS (77, 78). Collectively, these distinctions between virulent strains and LVS imply that the biological relevance of our current findings for F. tularensis strains in general will ultimately require that the work be validated using virulent strains. Further, it was recently shown that the ΔiglC mutant of F. novicida U112 was unable to replicate upon delivery into the cytosol of mammalian cells (79), in contrast to our findings after microinjection. Thus, phenotypic differences appear to exist between the different subspecies of F. tularensis that affect their intracellular survival.
Based on our findings, we conclude that the mode of uptake, the location in and escape from the phagosomal compartment, and the intracellular milieu of the host cell cytoplasm control the fate of intracellular F. tularensis and Listeria. Moreover, our study shows that several FPI proteins are important for successful replication after phagocytosis but not to facilitate growth in the cytosol. The findings illustrate some previously unanticipated requirements for intracytosolic replication in phagocytic cells and provide a basis for the future exploration of how the intracytosolic microbes successfully exploit the intracellular environment as their replicative niche.
Supplementary Material
ACKNOWLEDGMENTS
This work was supported by grants 2006-3426 (to J.E.B.), 2013-4581, and 2013-8621 (to A.S.) from the Swedish Research Council and a grant from the Medical Faculty, Umeå University, Umeå, Sweden, and the JC Kempe Memorial Foundation (to L.M.). The work was performed in part at the Umeå Centre for Microbial Research (UCMR).
We are grateful to Patrik Rydén for support with the statistical analysis. Also, we thank Jörgen Johansson for the L. monocytogenes EGD-GFP strain and Igor Golovliov and Marios Stylianou for assistance with some of the Listeria experiments.
Footnotes
Supplemental material for this article may be found at http://dx.doi.org/10.1128/IAI.00416-15.
REFERENCES
- 1.Underhill DM, Ozinsky A. 2002. Phagocytosis of microbes: complexity in action. Annu Rev Immunol 20:825–852. doi: 10.1146/annurev.immunol.20.103001.114744. [DOI] [PubMed] [Google Scholar]
- 2.Pluddemann A, Mukhopadhyay S, Gordon S. 2011. Innate immunity to intracellular pathogens: macrophage receptors and responses to microbial entry. Immunol Rev 240:11–24. doi: 10.1111/j.1600-065X.2010.00989.x. [DOI] [PubMed] [Google Scholar]
- 3.Broz P, Monack DM. 2011. Molecular mechanisms of inflammasome activation during microbial infections. Immunol Rev 243:174–190. doi: 10.1111/j.1600-065X.2011.01041.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Mostowy S, Cossart P. 2012. Virulence factors that modulate the cell biology of Listeria infection and the host response. Adv Immunol 113:19–32. doi: 10.1016/B978-0-12-394590-7.00007-5. [DOI] [PubMed] [Google Scholar]
- 5.Underhill DM. 2007. Collaboration between the innate immune receptors dectin-1, TLRs, and Nods. Immunol Rev 219:75–87. doi: 10.1111/j.1600-065X.2007.00548.x. [DOI] [PubMed] [Google Scholar]
- 6.Kawai T, Akira S. 2010. The role of pattern-recognition receptors in innate immunity: update on Toll-like receptors. Nat Immunol 11:373–384. doi: 10.1038/ni.1863. [DOI] [PubMed] [Google Scholar]
- 7.Palm NW, Medzhitov R. 2009. Pattern recognition receptors and control of adaptive immunity. Immunol Rev 227:221–233. doi: 10.1111/j.1600-065X.2008.00731.x. [DOI] [PubMed] [Google Scholar]
- 8.Takeuchi O, Akira S. 2010. Pattern recognition receptors and inflammation. Cell 140:805–820. doi: 10.1016/j.cell.2010.01.022. [DOI] [PubMed] [Google Scholar]
- 9.Martinez FO, Helming L, Gordon S. 2009. Alternative activation of macrophages: an immunologic functional perspective. Annu Rev Immunol 27:451–483. doi: 10.1146/annurev.immunol.021908.132532. [DOI] [PubMed] [Google Scholar]
- 10.Ray K, Marteyn B, Sansonetti PJ, Tang CM. 2009. Life on the inside: the intracellular lifestyle of cytosolic bacteria. Nat Rev Microbiol 7:333–340. doi: 10.1038/nrmicro2112. [DOI] [PubMed] [Google Scholar]
- 11.Kofoed EM, Vance RE. 2012. NAIPs: building an innate immune barrier against bacterial pathogens: NAIPs function as sensors that initiate innate immunity by detection of bacterial proteins in the host cell cytosol. Bioessays 34:589–598. doi: 10.1002/bies.201200013. [DOI] [PubMed] [Google Scholar]
- 12.Franchi L, Munoz-Planillo R, Nunez G. 2012. Sensing and reacting to microbes through the inflammasomes. Nat Immunol 13:325–332. doi: 10.1038/ni.2231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Gross O, Thomas CJ, Guarda G, Tschopp J. 2011. The inflammasome: an integrated view. Immunol Rev 243:136–151. doi: 10.1111/j.1600-065X.2011.01046.x. [DOI] [PubMed] [Google Scholar]
- 14.Goetz M, Bubert A, Wang G, Chico-Calero I, Vazquez-Boland JA, Beck M, Slaghuis J, Szalay AA, Goebel W. 2001. Microinjection and growth of bacteria in the cytosol of mammalian host cells. Proc Natl Acad Sci U S A 98:12221–12226. doi: 10.1073/pnas.211106398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Alkhuder K, Meibom KL, Dubail I, Dupuis M, Charbit A. 2009. Glutathione provides a source of cysteine essential for intracellular multiplication of Francisella tularensis. PLoS Pathog 5:e1000284. doi: 10.1371/journal.ppat.1000284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ramond E, Gesbert G, Rigard M, Dairou J, Dupuis M, Dubail I, Meibom K, Henry T, Barel M, Charbit A. 2014. Glutamate utilization couples oxidative stress defense and the tricarboxylic acid cycle in Francisella phagosomal escape. PLoS Pathog 10:e1003893. doi: 10.1371/journal.ppat.1003893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Gesbert G, Ramond E, Tros F, Dairou J, Frapy E, Barel M, Charbit A. 2015. Importance of branched-chain amino acid utilization in Francisella intracellular adaptation. Infect Immun 83:173–183. doi: 10.1128/IAI.02579-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Gesbert G, Ramond E, Rigard M, Frapy E, Dupuis M, Dubail I, Barel M, Henry T, Meibom K, Charbit A. 2014. Asparagine assimilation is critical for intracellular replication and dissemination of Francisella. Cell Microbiol 16:434–449. doi: 10.1111/cmi.12227. [DOI] [PubMed] [Google Scholar]
- 19.Napier BA, Meyer L, Bina JE, Miller MA, Sjöstedt A, Weiss DS. 2012. Link between intraphagosomal biotin and rapid phagosomal escape in Francisella. Proc Natl Acad Sci U S A 109:18084–18089. doi: 10.1073/pnas.1206411109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Chong A, Wehrly TD, Child R, Hansen B, Hwang S, Virgin HW, Celli J. 2012. Cytosolic clearance of replication-deficient mutants reveals Francisella tularensis interactions with the autophagic pathway. Autophagy 8:1342–1356. doi: 10.4161/auto.20808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Steele S, Brunton J, Ziehr B, Taft-Benz S, Moorman N, Kawula T. 2013. Francisella tularensis harvests nutrients derived via ATG5-independent autophagy to support intracellular growth. PLoS Pathog 9:e1003562. doi: 10.1371/journal.ppat.1003562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Schnupf P, Portnoy DA. 2007. Listeriolysin O: a phagosome-specific lysin. Microbes Infect 9:1176–1187. doi: 10.1016/j.micinf.2007.05.005. [DOI] [PubMed] [Google Scholar]
- 23.Sjöstedt A. 2007. Tularemia: history, epidemiology, pathogen physiology, and clinical manifestations. Ann N Y Acad Sci 1105:1–29. doi: 10.1196/annals.1409.009. [DOI] [PubMed] [Google Scholar]
- 24.Oyston PC, Sjöstedt A, Titball RW. 2004. Tularaemia: bioterrorism defence renews interest in Francisella tularensis. Nat Rev Microbiol 2:967–978. doi: 10.1038/nrmicro1045. [DOI] [PubMed] [Google Scholar]
- 25.Bröms JE, Sjöstedt A, Lavander M. 2010. The role of the Francisella tularensis pathogenicity island in type VI secretion, intracellular survival, and modulation of host cell signaling. Front Microbiol 1:136. doi: 10.3389/fmicb.2010.00136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Barker JR, Chong A, Wehrly TD, Yu JJ, Rodriguez SA, Liu J, Celli J, Arulanandam BP, Klose KE. 2009. The Francisella tularensis pathogenicity island encodes a secretion system that is required for phagosome escape and virulence. Mol Microbiol 74:1459–1470. doi: 10.1111/j.1365-2958.2009.06947.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Bröms JE, Lavander M, Meyer L, Sjöstedt A. 2011. IglG and IglI of the Francisella pathogenicity island are important virulence determinants of Francisella tularensis LVS. Infect Immun 79:3683–3696. doi: 10.1128/IAI.01344-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Bröms JE, Meyer L, Sun K, Lavander M, Sjöstedt A. 2012. Unique substrates secreted by the type VI secretion system of Francisella tularensis during intramacrophage infection. PLoS One 7:e50473. doi: 10.1371/journal.pone.0050473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Bingle LE, Bailey CM, Pallen MJ. 2008. Type VI secretion: a beginner's guide. Curr Opin Microbiol 11:3–8. doi: 10.1016/j.mib.2008.01.006. [DOI] [PubMed] [Google Scholar]
- 30.Lindgren H, Golovliov I, Baranov V, Ernst RK, Telepnev M, Sjöstedt A. 2004. Factors affecting the escape of Francisella tularensis from the phagolysosome. J Med Microbiol 53:953–958. doi: 10.1099/jmm.0.45685-0. [DOI] [PubMed] [Google Scholar]
- 31.Santic M, Molmeret M, Klose KE, Jones S, Kwaik YA. 2005. The Francisella tularensis pathogenicity island protein IglC and its regulator MglA are essential for modulating phagosome biogenesis and subsequent bacterial escape into the cytoplasm. Cell Microbiol 7:969–979. doi: 10.1111/j.1462-5822.2005.00526.x. [DOI] [PubMed] [Google Scholar]
- 32.Bönquist L, Lindgren H, Golovliov I, Guina T, Sjöstedt A. 2008. MglA and Igl proteins contribute to the modulation of Francisella tularensis live vaccine strain-containing phagosomes in murine macrophages. Infect Immun 76:3502–3510. doi: 10.1128/IAI.00226-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Bröms JE, Lavander M, Sjöstedt A. 2009. A conserved alpha-helix essential for a type VI secretion-like system of Francisella tularensis. J Bacteriol 191:2431–2446. doi: 10.1128/JB.01759-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Das S, Chaudhuri K. 2003. Identification of a unique IAHP (IcmF associated homologous proteins) cluster in Vibrio cholerae and other proteobacteria through in silico analysis. In Silico Biol 3:287–300. [PubMed] [Google Scholar]
- 35.Nano FE, Schmerk C. 2007. The Francisella pathogenicity island. Ann N Y Acad Sci 1105:122–137. doi: 10.1196/annals.1409.000. [DOI] [PubMed] [Google Scholar]
- 36.Ludu JS, de Bruin OM, Duplantis BN, Schmerk CL, Chou AY, Elkins KL, Nano FE. 2008. The Francisella pathogenicity island protein PdpD is required for full virulence and associates with homologues of the type VI secretion system. J Bacteriol 190:4584–4595. doi: 10.1128/JB.00198-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Fernandes-Alnemri T, Yu JW, Juliana C, Solorzano L, Kang S, Wu J, Datta P, McCormick M, Huang L, McDermott E, Eisenlohr L, Landel CP, Alnemri ES. 2010. The AIM2 inflammasome is critical for innate immunity to Francisella tularensis. Nat Immunol 11:385–393. doi: 10.1038/ni.1859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Rathinam VA, Jiang Z, Waggoner SN, Sharma S, Cole LE, Waggoner L, Vanaja SK, Monks BG, Ganesan S, Latz E, Hornung V, Vogel SN, Szomolanyi-Tsuda E, Fitzgerald KA. 2010. The AIM2 inflammasome is essential for host defense against cytosolic bacteria and DNA viruses. Nat Immunol 11:395–402. doi: 10.1038/ni.1864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Jones JW, Kayagaki N, Broz P, Henry T, Newton K, O'Rourke K, Chan S, Dong J, Qu Y, Roose-Girma M, Dixit VM, Monack DM. 2010. Absent in melanoma 2 is required for innate immune recognition of Francisella tularensis. Proc Natl Acad Sci U S A 107:9771–9776. doi: 10.1073/pnas.1003738107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Jones JW, Broz P, Monack DM. 2011. Innate immune recognition of Francisella tularensis: activation of type-I interferons and the inflammasome. Front Microbiol 2:16. doi: 10.3389/fmicb.2011.00016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Mariathasan S, Weiss DS, Dixit VM, Monack DM. 2005. Innate immunity against Francisella tularensis is dependent on the ASC/caspase-1 axis. J Exp Med 202:1043–1049. doi: 10.1084/jem.20050977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Cole LE, Mann BJ, Shirey KA, Richard K, Yang Y, Gearhart PJ, Chesko KL, Viscardi RM, Vogel SN. 2011. Role of TLR signaling in Francisella tularensis-LPS-induced, antibody-mediated protection against Francisella tularensis challenge. J Leukoc Biol 90:787–797. doi: 10.1189/jlb.0111014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Cole LE, Shirey KA, Barry E, Santiago A, Rallabhandi P, Elkins KL, Puche AC, Michalek SM, Vogel SN. 2007. Toll-like receptor 2-mediated signaling requirements for Francisella tularensis live vaccine strain infection of murine macrophages. Infect Immun 75:4127–4137. doi: 10.1128/IAI.01868-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Jones CL, Weiss DS. 2011. TLR2 signaling contributes to rapid inflammasome activation during Francisella novicida infection. PLoS One 6:e20609. doi: 10.1371/journal.pone.0020609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Abplanalp AL, Morris IR, Parida BK, Teale JM, Berton MT. 2009. TLR-dependent control of Francisella tularensis infection and host inflammatory responses. PLoS One 4:e7920. doi: 10.1371/journal.pone.0007920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Katz J, Zhang P, Martin M, Vogel SN, Michalek SM. 2006. Toll-like receptor 2 is required for inflammatory responses to Francisella tularensis LVS. Infect Immun 74:2809–2816. doi: 10.1128/IAI.74.5.2809-2816.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Torres D, Barrier M, Bihl F, Quesniaux VJ, Maillet I, Akira S, Ryffel B, Erard F. 2004. Toll-like receptor 2 is required for optimal control of Listeria monocytogenes infection. Infect Immun 72:2131–2139. doi: 10.1128/IAI.72.4.2131-2139.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Collazo CM, Sher A, Meierovics AI, Elkins KL. 2006. Myeloid differentiation factor-88 (MyD88) is essential for control of primary in vivo Francisella tularensis LVS infection, but not for control of intra-macrophage bacterial replication. Microbes Infect 8:779–790. doi: 10.1016/j.micinf.2005.09.014. [DOI] [PubMed] [Google Scholar]
- 49.Cole LE, Laird MH, Seekatz A, Santiago A, Jiang Z, Barry E, Shirey KA, Fitzgerald KA, Vogel SN. 2010. Phagosomal retention of Francisella tularensis results in TIRAP/Mal-independent TLR2 signaling. J Leukoc Biol 87:275–281. doi: 10.1189/jlb.0909619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Edelson BT, Unanue ER. 2002. MyD88-dependent but Toll-like receptor 2-independent innate immunity to Listeria: no role for either in macrophage listericidal activity. J Immunol 169:3869–3875. doi: 10.4049/jimmunol.169.7.3869. [DOI] [PubMed] [Google Scholar]
- 51.McCaffrey RL, Fawcett P, O'Riordan M, Lee KD, Havell EA, Brown PO, Portnoy DA. 2004. A specific gene expression program triggered by Gram-positive bacteria in the cytosol. Proc Natl Acad Sci U S A 101:11386–11391. doi: 10.1073/pnas.0403215101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Mandin P, Fsihi H, Dussurget O, Vergassola M, Milohanic E, Toledo-Arana A, Lasa I, Johansson J, Cossart P. 2005. VirR, a response regulator critical for Listeria monocytogenes virulence. Mol Microbiol 57:1367–1380. doi: 10.1111/j.1365-2958.2005.04776.x. [DOI] [PubMed] [Google Scholar]
- 53.Perez B, Paquette N, Paidassi H, Zhai B, White K, Skvirsky R, Lacy-Hulbert A, Stuart LM. 2012. Apoptotic cells can deliver chemotherapeutics to engulfing macrophages and suppress inflammatory cytokine production. J Biol Chem 287:16029–16036. doi: 10.1074/jbc.M112.340489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.de Bruin OM, Ludu JS, Nano FE. 2007. The Francisella pathogenicity island protein IglA localizes to the bacterial cytoplasm and is needed for intracellular growth. BMC Microbiol 7:1. doi: 10.1186/1471-2180-7-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Lindgren M, Eneslätt K, Bröms JE, Sjöstedt A. 2013. Importance of PdpC, IglC, IglI, and IglG for modulation of a host cell death pathway induced by Francisella tularensis. Infect Immun 81:2076–2084. doi: 10.1128/IAI.00275-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Joseph B, Goebel W. 2007. Life of Listeria monocytogenes in the host cells' cytosol. Microbes Infect 9:1188–1195. doi: 10.1016/j.micinf.2007.05.006. [DOI] [PubMed] [Google Scholar]
- 57.Slaghuis J, Goetz M, Engelbrecht F, Goebel W. 2004. Inefficient replication of Listeria innocua in the cytosol of mammalian cells. J Infect Dis 189:393–401. doi: 10.1086/381206. [DOI] [PubMed] [Google Scholar]
- 58.Golovliov I, Ericsson M, Sandström G, Tärnvik A, Sjöstedt A. 1997. Identification of proteins of Francisella tularensis induced during growth in macrophages and cloning of the gene encoding a prominently induced 23-kilodalton protein. Infect Immun 65:2183–2189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Lai XH, Shirley RL, Crosa L, Kanistanon D, Tempel R, Ernst RK, Gallagher LA, Manoil C, Heffron F. 2010. Mutations of Francisella novicida that alter the mechanism of its phagocytosis by murine macrophages. PLoS One 5:e11857. doi: 10.1371/journal.pone.0011857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Holicka M, Novosad J, Kudlova M, Loudova M, Andrys C, Krejsek J. 2010. J774 macrophage-like cell line cytokine and chemokine patterns are modulated by Francisella tularensis LVS strain infection. Folia Microbiol (Praha) 55:191–200. doi: 10.1007/s12223-010-0028-3. [DOI] [PubMed] [Google Scholar]
- 61.Fuller JR, Craven RR, Hall JD, Kijek TM, Taft-Benz S, Kawula TH. 2008. RipA, a cytoplasmic membrane protein conserved among Francisella species, is required for intracellular survival. Infect Immun 76:4934–4943. doi: 10.1128/IAI.00475-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Schulert GS, Allen LA. 2006. Differential infection of mononuclear phagocytes by Francisella tularensis: role of the macrophage mannose receptor. J Leukoc Biol 80:563–571. doi: 10.1189/jlb.0306219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Lauriano CM, Barker JR, Yoon SS, Nano FE, Arulanandam BP, Hassett DJ, Klose KE. 2004. MglA regulates transcription of virulence factors necessary for Francisella tularensis intraamoebae and intramacrophage survival. Proc Natl Acad Sci U S A 101:4246–4249. doi: 10.1073/pnas.0307690101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Buchmeier NA, Heffron F. 1989. Intracellular survival of wild-type Salmonella typhimurium and macrophage-sensitive mutants in diverse populations of macrophages. Infect Immun 57:1–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Qin A, Mann BJ. 2006. Identification of transposon insertion mutants of Francisella tularensis tularensis strain Schu S4 deficient in intracellular replication in the hepatic cell line HepG2. BMC Microbiol 6:69. doi: 10.1186/1471-2180-6-69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Law HT, Sriram A, Fevang C, Nix EB, Nano FE, Guttman JA. 2014. IglC and PdpA are important for promoting Francisella invasion and intracellular growth in epithelial cells. PLoS One 9:e104881. doi: 10.1371/journal.pone.0104881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Meibom KL, Charbit A. 2010. Francisella tularensis metabolism and its relation to virulence. Front Microbiol 1:140. doi: 10.3389/fmicb.2010.00140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Brown MJ, Russo BC, O'Dee DM, Schmitt DM, Nau GJ. 2014. The contribution of the glycine cleavage system to the pathogenesis of Francisella tularensis. Microbes Infect 16:300–309. doi: 10.1016/j.micinf.2013.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Dieppedale J, Gesbert G, Ramond E, Chhuon C, Dubail I, Dupuis M, Guerrera IC, Charbit A. 2013. Possible links between stress defense and the tricarboxylic acid (TCA) cycle in Francisella pathogenesis. Mol Cell Proteomics 12:2278–2292. doi: 10.1074/mcp.M112.024794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Wehrly TD, Chong A, Virtaneva K, Sturdevant DE, Child R, Edwards JA, Brouwer D, Nair V, Fischer ER, Wicke L, Curda AJ, Kupko JJ III, Martens C, Crane DD, Bosio CM, Porcella SF, Celli J. 2009. Intracellular biology and virulence determinants of Francisella tularensis revealed by transcriptional profiling inside macrophages. Cell Microbiol 11:1128–1150. doi: 10.1111/j.1462-5822.2009.01316.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Manzanillo PS, Shiloh MU, Portnoy DA, Cox JS. 2012. Mycobacterium tuberculosis activates the DNA-dependent cytosolic surveillance pathway within macrophages. Cell Host Microbe 11:469–480. doi: 10.1016/j.chom.2012.03.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Houben D, Demangel C, van Ingen J, Perez J, Baldeon L, Abdallah AM, Caleechurn L, Bottai D, van Zon M, de Punder K, van der Laan T, Kant A, Bossers-de Vries R, Willemsen P, Bitter W, van Soolingen D, Brosch R, van der Wel N, Peters PJ. 2012. ESX-1-mediated translocation to the cytosol controls virulence of mycobacteria. Cell Microbiol 14:1287–1298. doi: 10.1111/j.1462-5822.2012.01799.x. [DOI] [PubMed] [Google Scholar]
- 73.Salomonsson E, Kuoppa K, Forslund AL, Zingmark C, Golovliov I, Sjöstedt A, Noppa L, Forsberg A. 2009. Reintroduction of two deleted virulence loci restores full virulence to the live vaccine strain of Francisella tularensis. Infect Immun 77:3424–3431. doi: 10.1128/IAI.00196-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Lindgren H, Honn M, Golovlev I, Kadzhaev K, Conlan W, Sjöstedt A. 2009. The 58-kilodalton major virulence factor of Francisella tularensis is required for efficient utilization of iron. Infect Immun 77:4429–4436. doi: 10.1128/IAI.00702-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Lindgren H, Shen H, Zingmark C, Golovliov I, Conlan W, Sjöstedt A. 2007. Resistance of Francisella tularensis strains against reactive nitrogen and oxygen species with special reference to the role of KatG. Infect Immun 75:1303–1309. doi: 10.1128/IAI.01717-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Binesse J, Lindgren H, Lindgren L, Conlan W, Sjöstedt A. 2015. Roles of reactive oxygen species-degrading enzymes of Francisella tularensis SCHU S4. Infect Immun 83:2255–2263. doi: 10.1128/IAI.02488-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Bosio CM, Bielefeldt-Ohmann H, Belisle JT. 2007. Active suppression of the pulmonary immune response by Francisella tularensis Schu S4. J Immunol 178:4538–4547. doi: 10.4049/jimmunol.178.7.4538. [DOI] [PubMed] [Google Scholar]
- 78.Ireland R, Wang R, Alinger JB, Small P, Bosio CM. 2013. Francisella tularensis Schu S4 and Schu S4 lipids inhibit IL-12p40 in primary human dendritic cells by inhibition of IRF1 and IRF8. J Immunol 191:1276–1286. doi: 10.4049/jimmunol.1300867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Wu YC, Wu TH, Clemens DL, Lee BY, Wen X, Horwitz MA, Teitell MA, Chiou PY. 2015. Massively parallel delivery of large cargo into mammalian cells with light pulses. Nat Methods 12:439–444. doi: 10.1038/nmeth.3357. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.