

Abstract
The (p)ppGpp-mediated stringent response is important for bacterial survival in nutrient limiting conditions. For maximal effect, (p)ppGpp interacts with the cofactor DksA, which stabilizes (p)ppGpp's interaction with RNA polymerase. We previously demonstrated that (p)ppGpp was required for the virulence of Haemophilus ducreyi in humans. Here, we constructed an H. ducreyi dksA mutant and showed it was also partially attenuated for pustule formation in human volunteers. To understand the roles of (p)ppGpp and DksA in gene regulation in H. ducreyi, we defined genes potentially altered by (p)ppGpp and DksA deficiency using transcriptome sequencing (RNA-seq). In bacteria collected at stationary phase, lack of (p)ppGpp and DksA altered expression of 28% and 17% of H. ducreyi open reading frames, respectively, including genes involved in transcription, translation, and metabolism. There was significant overlap in genes differentially expressed in the (p)ppGpp mutant relative to the dksA mutant. Loss of (p)ppGpp or DksA resulted in the dysregulation of several known virulence determinants. Deletion of dksA downregulated lspB and rendered the organism less resistant to phagocytosis and increased its sensitivity to oxidative stress. Both mutants had reduced ability to attach to human foreskin fibroblasts; the defect correlated with reduced expression of the Flp adhesin proteins in the (p)ppGpp mutant but not in the dksA mutant, suggesting that DksA regulates the expression of an unknown cofactor(s) required for Flp-mediated adherence. We conclude that both (p)ppGpp and DksA serve as major regulators of H. ducreyi gene expression in stationary phase and have both overlapping and unique contributions to pathogenesis.
INTRODUCTION
Haemophilus ducreyi is the causative agent of chancroid, a sexually transmitted genital ulcer disease that facilitates both the transmission and acquisition of HIV-1 (1). Chancroid has a short duration of infectivity and is maintained only in populations with high sex partner change rates, such as commercial sex workers (2). Due to widespread implementation of syndromic management of genital ulcers, the epidemiology of chancroid is poorly defined, but its prevalence has declined in many areas where chancroid formerly was endemic (2). However, reports of chancroid persist from several countries in Africa and Asia, implying that these regions have clinical reservoirs of infected sex workers (3–8).
Although non-sexually-transmitted cutaneous ulcers in children in the South Pacific islands and equatorial Africa are usually attributed to Treponema pallidum subsp. pertenue, recent studies performed as part of a World Health Organization-directed yaws eradication campaign suggest that H. ducreyi is a major cause of this syndrome. In three large cross-sectional community surveys, the proportion of ulcers in which H. ducreyi DNA was detected greatly exceeded that of T. pallidum DNA (9–11). Considering the global prevalence of yaws, infections due to H. ducreyi may be much more common than was previously recognized. Thus, understanding the pathogenesis of H. ducreyi remains important from the point of view of public health.
In order to study H. ducreyi pathogenesis, we developed a human inoculation model in which healthy adult volunteers are infected on the skin of the upper arm with the strain 35000HP, which was originally isolated from a patient with chancroid (12, 13). Phylogenetic analysis of cutaneous ulcer strains show that they are highly related to strain 35000HP, suggesting that the human inoculation model is relevant to both chancroid and cutaneous ulcers (14). In natural chancroid and experimental infection, H. ducreyi is found within abscesses surrounded by macrophages and polymorphonuclear leukocytes and is likely subject to a variety of stresses, including antimicrobial peptides, reactive oxygen species, and nutrient limitation (15, 16).
One mechanism used by bacteria to adapt to stress is the stringent response mediated by GTP 3′-diphosphate (pppGpp) and GDP 3′-diphosphate (ppGpp), collectively referred to here as (p)ppGpp. During the stringent response, (p)ppGpp interacts with a cofactor known as the DnaK suppressor protein or DksA. DksA helps to stabilize the interaction between (p)ppGpp and the RNA polymerase (RNAP), thus enhancing (p)ppGpp-controlled transcription (17). In Escherichia coli, DksA overexpression can compensate for the loss of (p)ppGpp synthesis (18, 19). The compensated phenotypes include restoration of defects in amino acid auxotrophy and cell-cell aggregation.
In several pathogenic bacterial species, DksA regulates the transcription of genes involved in virulence. DksA regulates motility, expression of the hemagglutinin protease, and production of cholera toxin in Vibrio cholerae, all processes related to virulence (20). DksA is also involved in the posttranscriptional control of rhamnolipids and LasB elastase, which are involved in quorum sensing in Pseudomonas aeruginosa (21). In Shigella flexneri, DksA is essential for regulation of hfq, which is an important regulator of S. flexneri virulence genes (22). However, the role of DksA in bacterial pathogenesis has never been directly studied in humans.
An H. ducreyi ΔrelA ΔspoT mutant, which is unable to synthesize (p)ppGpp, is partially attenuated for pustule formation in human volunteers (23). The (p)ppGpp null [(p)ppGpp°] mutant has increased expression of dksA transcripts, which may have compensated for the loss of (p)ppGpp and contributed to its partial attenuation (23). To investigate the potential role of DksA in H. ducreyi pathogenesis, here we constructed a dksA deletion mutant and compared its virulence to that of the parent strain in human inoculation experiments. We also defined genes whose expression is altered by (p)ppGpp and DksA deficiency in H. ducreyi by using transcriptome sequencing (RNA-seq). We used the results of the transcriptome analysis to direct further characterization of the dksA and (p)ppGpp° mutants in assays relevant to H. ducreyi pathogenesis.
MATERIALS AND METHODS
Bacterial strains, plasmids, and growth conditions.
The bacterial strains and plasmids used in this study are listed in Table S1 in the supplemental material. H. ducreyi strains were grown on chocolate agar plates supplemented with 1% IsoVitaleX at 33°C with 5% CO2 or in gonococcal (GC) broth supplemented with 5% fetal bovine serum, 1% IsoVitaleX, and 50 μg/ml hemin (Aldrich Chemical Co.) at 33°C. H. ducreyi 35000HP was grown to mid-log phase (optical density at 660 nm [OD660] = 0.2), transition (OD660 = 0.35), or stationary phase (OD660 = 0.5); mutant strains were harvested when the OD660 of strain 35000HP was in the appropriate range for each phase of growth. E. coli strains were grown in Luria-Bertani medium at 37°C with the exception of strain DY380, which was maintained in low-salt broth or agar at 32°C and grown at 42°C for induction of the lambda red recombinase. When necessary, medium was supplemented with kanamycin (20 μg/ml for H. ducreyi; 50 μg/ml for E. coli) or spectinomycin (200 μg/ml for H. ducreyi; 50 μg/ml for E. coli). For H. ducreyi strains containing a pACYC177 backbone, medium was supplemented with 30 μg/ml kanamycin.
Characterization of the dksA gene.
The Basic Local Alignment Search Tool (BLAST) was used to identify putative homologues of dksA in H. ducreyi (GenBank accession no. AE017143). Reverse transcriptase PCR (RT-PCR) was conducted to determine whether dksA was in an operon with its surrounding genes using primers P1-P6 (see Table S2 in the supplemental material).
Construction and complementation of an unmarked, in-frame dksA deletion mutant.
An unmarked, in-frame dksA deletion mutant was constructed using the lambda red and FLP recombinase method described previously using primers P7-P14 (24, 25). Quantitative RT-PCR (qRT-PCR) determined that deletion of dksA did not affect transcription of the downstream gene pcnB (data not shown).
To complement the dksA deletion mutant, the gene was expressed under the control of a constitutive cat promoter from pACYC184 in the expression vector pACYC177 as described previously using primers P15-P18 (23). The final construct, designated pCH24, was confirmed by PCR and sequence analysis and electroporated into 35000HPΔdksA mutant strain to yield 35000HPΔdksA(pCH24). For controls, strains 35000HP and 35000HPΔdksA were also electroporated with pACYC177, and the resulting strains were designated 35000HP(pACYC177) and 35000HPΔdksA(pACYC177), respectively. Although all transformed strains grew well on antibiotic-supplemented plates, and strain 35000HPΔdksA(pCH24) grew normally in broth containing kanamycin, the H. ducreyi strains containing pACYC177 grew poorly in antibiotic-supplemented broth. The strains transformed with pACYC177 could be grown in broth without antibiotics for only 6 h before losing the plasmid. Thus, use of the complemented strains was limited to those assays using cells that had been grown or maintained in broth without antibiotics for 6 h or those that had been grown on plates supplemented with antibiotics.
Human inoculation experiments.
Human inoculation experiments were conducted according to the guidelines of the U.S. Department of Health and Human Services and the Institutional Review Board (IRB) of Indiana University. Seven adult volunteers (5 females and 2 males; 2 African Americans and 5 European Americans; mean age, 36 years) participated in the study. All volunteers gave written, informed consent for participation and HIV serology. The procedures for the human inoculation experiments, including calculation of the estimated delivered dose (EDD), are described in detail elsewhere (26). Papule and pustule formation rates for parent and mutant inoculation sites were compared using logistic regression with generalized estimating equations (GEE) as previously described (27). Ninety-five percent confidence intervals (95% CI) for papule and pustule formation rates were calculated using GEE-based sandwich standard errors.
To ensure that there was no cross contamination of samples, colony hybridization was performed on colonies derived from the inocula, surface cultures, and biopsy specimens. Probes specific for dksA and dnaE were designed, and the DIG DNA labeling kit (Roche Applied Sciences) was used to label the probes with digoxigenin. The DIG Easy Hyb protocol was performed according to the manufacturer's instructions (Roche Applied Sciences).
RNA isolation and quality assessment.
Total RNA was extracted from strains 35000HP, 35000HPΔrelAΔspoT, and 35000HPΔdksA in the mid-log, transition, and stationary growth phases using TRIzol reagent (Invitrogen) according to the manufacturer's protocol. RNA isolation was performed on four independent bacterial cultures for each strain in each growth phase. RNA was treated twice with Turbo DNA-free DNase (Ambion). The integrity and concentration of RNA were determined using the Agilent 2100 bioanalyzer (Agilent Technologies) and the NanoDrop ND2000 spectrophotometer (Thermo Scientific), respectively. The efficacy of DNase treatment was confirmed by reverse transcriptase PCR analysis of dnaE with the primers P19-P20 (see Table S2 in the supplemental material).
RT-PCR and qRT-PCR.
cDNA from strains 35000HP and 35000HPΔdksA was synthesized from total RNA using Advantage RT-for-PCR kit (Clontech). Reverse transcriptase PCR was performed using the cDNA and FastStart PCR master mix (Roche).
Quantitative RT-PCR was performed using the QuantiTect SYBR green RT-PCR kit (Qiagen) and a Mastercycler ep realplex 4 (Eppendorf). All primer pairs (see Table S2 in the supplemental material) had greater than 95% amplification efficiency. Relative expression was calculated as EtargetΔCTtarget/EreferenceΔCTreference, where E is the amplification efficiency (10−1/slope) and the ΔCT is the change in cycle threshold. dnaE was amplified to normalize the expression levels of target genes.
mRNA enrichment.
The removal of 23S, 16S, and 5S rRNA from total RNA was performed with the Ribo-Zero Magnetic kit for Gram-negative bacteria (Epicentre Biotechnologies) by following the manufacturer's instructions. The Agilent 2100 bioanalyzer confirmed removal of rRNA from total RNA.
Preparation of RNA-seq libraries and sequencing.
The TruSeq Stranded mRNA sample preparation kit (Illumina) was used to prepare stranded RNA-seq libraries by following the manufacturer's instructions. Briefly, approximately 400 ng of the enriched mRNA was fragmented and randomly primed for first-strand cDNA synthesis. Second-strand synthesis incorporated dUTP in place of dTTP, which prevents second-strand synthesis during subsequent amplification and results in a stranded library. The cDNA was end repaired, adenylated, and ligated to adapters. The adapter-ligated cDNA library was then PCR enriched. Finally, the enriched RNA-seq library was validated with the Agilent 2100 bioanalyzer and qRT-PCR. Clusters were generated on the cBOT automated cluster-generating system with the TruSeq PE cluster kit (Illumina). Libraries were sequenced with the Illumina HiSeq 2500 sequencer with the TruSeq SBS kit (Illumina) for single-end sequencing with read lengths of 100 bp in the Biomedical Genomics Core facility at Nationwide Children's Hospital (Columbus, OH). Image analysis and base calling were performed with the HiSeq Control software and the Real Time Analysis software. Demultiplexing was performed with the Illumina CASAVA software.
Sequence mapping and quantification of transcript levels.
The sequenced reads were mapped to the H. ducreyi 35000HP genome (GenBank accession no. AE017143) with the Burrows-Wheeler Alignment tool (28) allowing up to two base mismatches. Reads that failed to map to any gene in the chromosome and reads that mapped to multiple locations in the genome were removed before quantifying the transcript levels. The total number of reads corresponding to the coding region of each gene was determined with the NGSUtils suite (29). The percentage of total reads aligned with the reference genome from all samples ranged from 82.89 to 95.87% (see Table S3 in the supplemental material).
Identification of differentially expressed genes.
Differential expression of genes across all three strains and growth phases was determined with edgeR software, a Bioconductor package, as previously described (30). To favor true identification of differentially expressed genes of biological significance, we used a prespecified falsediscovery rate of ≤0.1 and a 2-fold change as a threshold. The differentially expressed genes were functionally classified using the annotations and pathway information from the sequenced H. ducreyi genome (R. S. Munson, Jr., unpublished data), and KEGG (Kyoto Encyclopedia of Genes and Genomes) (31). We also determined whether any biological pathways were enriched among the genes differentially expressed in the (p)ppGpp° and dksA mutants compared to strain 35000HP by using pathway annotations from BioCyc (32). We used the functional annotation-clustering algorithm of the DAVID (Database for Annotation, Visualization, and Integrated Discovery) bioinformatics resources (http://david.abcc.ncifcrf.gov/) to identify the biological pathways enriched in the (p)ppGpp° and dksA mutants relative to the 35000HP strain (33). A pathway was considered enriched if the Fisher's exact P value for the cluster was less than 0.05, the enrichment score for the cluster was greater than 2, and the cluster involved greater than 5% of the genes on the submitted list (34).
Phenotypic comparisons.
Lipooligosaccharides (LOS) and outer membrane proteins (OMP) were isolated from strains 35000HP and 35000HPΔdksA and analyzed as described previously (35, 36). Serum bactericidal assays were performed on organisms grown on plates as described previously (37). To determine the expression of Flp1/2 and peptidoglycan-associated lipoprotein (PAL), Western blots were performed on whole-cell lysates and purified outer membranes and probed with antisera or monoclonal antibody specific to each protein. The antisera for Flp1/2 (kindly provided by E. Hansen, University of Texas Southwestern) and the PAL-specific monoclonal antibody 3B9 are described elsewhere (36, 50).
Phagocytosis assays.
Human peripheral blood mononuclear cells (PBMCs) were isolated from blood obtained from five healthy adult volunteers. Informed consent was obtained in accordance with an IRB-approved protocol. PBMCs were isolated by Ficoll-Paque Plus purification, and CD14+ cells were isolated (Miltenyi Biotec). The isolated CD14+ cells were differentiated into monocyte-derived macrophages (MDMs) in X-vivo 15 medium (Lonza) supplemented with 1% human AB serum (Invitrogen) for 5 days. The phagocytosis assays were performed as previously described (23). The percentage of bacterial uptake was calculated as the percentage of bacteria within the lysed MDMs after 1 h coculture compared to the initial CFU. To determine intracellular bacterial survival, the cocultures were incubated in antibiotic-free medium containing 10% fetal bovine serum for an additional 5 h and quantitatively cultured. Survival was determined by calculating the ratio of recoverable bacteria after 6 h of coculture compared to the initial uptake.
Oxidative stress assays.
As previously described, bacteria were treated with hydrogen peroxide (H2O2) to mimic extracellular oxidative stress (23). Percent survival was calculated as the ratio of recovered bacteria to the input CFU. For assays using complemented strains, bacterial cells were grown for 6 h in broth without antibiotics, collected, and exposed to H2O2 as described previously.
Adherence assays.
Adherence to human foreskin fibroblasts (HFF) was performed as described previously (40). Briefly, 24-well tissue culture plates (Costar) were seeded with 105 HFF cells and allowed to reach confluence. H. ducreyi strain 35000HP, 35000HPΔdksA (35000HP strain with the dksA gene deleted), 35000HPΔrelAΔspoT (35000HP strain with the relA and spoT genes deleted), and 35000HPΔflp1-3 (35000HP strain with the flp-1 to flp-3 genes deleted) were grown to mid-log and stationary phase, harvested, and added to confluent HFF cells at a multiplicity of infection (MOI) of 10:1 at 35°C for 2 h. After a wash, the HFF cells were lysed with 0.2% saponin (Sigma-Aldrich) and quantitatively cultured. Percent adherence of H. ducreyi to HFF cells was determined by calculating the ratio of HFF-adhered bacteria to the initial CFU.
Statistical analysis.
All data are expressed as means ± standard deviations. Oxidative stress, serum bactericidal, attachment, and phagocytosis data were analyzed using a mixed-model analysis of variance (ANOVA) followed by Tukey's honestly significant difference test. qRT-PCR and densitometry data were analyzed using Student's t test. An adjusted two-sided P value of ≤0.05 was considered statistically significant.
RNA-seq data accession number.
The data from these RNA-seq experiments were deposited at the NCBI Gene Expression Omnibus (GEO) database (http://www.ncbi.nlm.nih.gov/geo/) under accession number GSE67202.
RESULTS
Characterization of the dksA genomic locus.
The H. ducreyi homologue of dksA (HD0603) is in a putative operon with the gene order tbpA → dksA → pcnB → folK. RT-PCR analysis indicated that dksA is cotranscribed with tbpA, pcnB, and folK (data not shown).
dksA is required for pustule formation in human volunteers.
To determine whether DksA contributes to virulence in humans, we constructed an unmarked, in-frame dksA deletion mutant. As required by our clinical protocol, we investigated whether deletion of dksA affected the expression of outer membrane components of H. ducreyi. LOS and OMP profiles isolated from the dksA mutant showed no qualitative differences compared to the parent strain. In bactericidal assays, the dksA mutant was as resistant to killing by 50% normal human serum as the parent was (data not shown).
Mutations in dksA can profoundly impair growth (41, 42). Relative to H. ducreyi 35000HP, deletion of dksA did not result in a growth defect in broth (see Fig. S1 in the supplemental material). Moreover, similar to the (p)ppGpp° mutant, the dksA mutant had increased survival at 24 h compared to the parent strain. Thus, the virulence of the dksA mutant should not be affected by impaired growth.
We inoculated volunteers in three iterations with strain 35000HP and with strain 35000HPΔdksA. In the first iteration, three volunteers were inoculated at three sites with an EDD of 82 CFU of strain 35000HP and at one site each with an EDD of 52, 104, and 207 CFU of strain 35000HPΔdksA. In the second iteration, two volunteers were each inoculated at three sites with an EDD of 150 CFU of strain 35000HP and with the ΔdksA mutant at three sites with EDDs of 87, 174, and 348 CFU. In the third iteration, two volunteers were inoculated with an EDD of 77 CFU of strain 35000HP at three sites and an EDD of 137 CFU of the ΔdksA mutant at three sites. Overall, papules formed at 90.5% (95% CI, 79.3 to 99.9%) of the parent-inoculated sites and at 81.0% (95% CI, 63.0 to 98.9%) of the mutant-inoculated sites (P = 0.094) (Table 1). After 24 h of infection, the mean surface area of the papules was 9.1 ± 9.2 mm2 at parent sites and 4.6 ± 6.9 mm2 at mutant sites (P = 0.012). Pustules formed at 42.9% (95% CI, 14.2 to 71.5%) of parent sites and 14.3% (95% CI, 2.1 to 26.5%) of mutant sites (P = 0.043). Thus, the dksA mutant was partially attenuated for pustule formation in humans.
TABLE 1.
Response to inoculation of live H. ducreyi strains
| Volunteer (gender)a | Observation period (no. of days) | Strainb | Dose(s) (CFU)c | No. of initial papules | Final outcome of papules |
|
|---|---|---|---|---|---|---|
| No. of pustules | No. of papules that resolved | |||||
| 454 (F) | 7 | P | 82 | 2 | 0 | 2 |
| M | 52–207 | 2 | 0 | 2 | ||
| 455 (M) | 9 | P | 82 | 3 | 1 | 2 |
| M | 52–207 | 3 | 1 | 2 | ||
| 456 (F) | 5 | P | 82 | 3 | 0 | 3 |
| M | 52–207 | 2 | 0 | 2 | ||
| 457 (F) | 7 | P | 150 | 3 | 3 | 0 |
| M | 87–348 | 3 | 1 | 2 | ||
| 458 (F) | 5 | P | 150 | 2 | 0 | 2 |
| M | 87–348 | 1 | 0 | 1 | ||
| 459 (M) | 8 | P | 77 | 3 | 1 | 2 |
| M | 137 | 3 | 1 | 2 | ||
| 460 (F) | 12 | P | 77 | 3 | 3 | 0 |
| M | 137 | 3 | 0 | 3 | ||
Volunteers 454, 455, and 456 were inoculated in the first iteration. Volunteers 457 and 458 were inoculated in the second iteration. Volunteers 459 and 460 were inoculated in the third iteration. F, female; M, male.
P, parent strain 35000HP; M, mutant strain 35000HPΔdksA.
Doses (single doses) inoculated at three sites. 52–207, one dose each of 52, 104, and 207 CFU; 87–348, one dose each of 87, 174, and 348 CFU.
Three volunteers (455, 457, and 459) developed pustules at both mutant and parent sites. For each subject, one parent site and one mutant site were biopsied, and the biopsy specimen was divided in half and semiquantitatively cultured or stained with hematoxylin-eosin and anti-CD3 antibodies as previously described (43). All samples contained epidermal micropustules and dermal CD3 cells (data not shown). Histopathologically, pustules formed at mutant sites were indistinguishable from pustules formed at parent sites.
All colonies recovered from the inocula, surface cultures, and biopsy specimens were tested for the presence of dksA and dnaE sequences by colony hybridization. The dnaE probe hybridized to all the colonies tested from both parent (n = 108) and mutant (n = 107) inocula, while the dksA probe hybridized only to colonies from the parent inocula. At least one positive surface culture for H. ducreyi was obtained during follow-up visits from 19.5% of the parent inoculation sites and 4.8% of the mutant inoculation sites. The dnaE probe hybridized to colonies from both parent (n = 106)- and mutant (n = 29)-inoculated sites, while the dksA probe bound only to colonies from the parent sites. All three paired biopsy specimens of mutant and parent pustules yielded H. ducreyi. The dnaE probe hybridized to all of the colonies, both parent (n = 107) and mutant (n = 69), obtained from the biopsy specimens, while the dksA probe hybridized only to colonies obtained from the parent biopsy specimens. Thus, there was no evidence of cross contamination between the inocula and mutant and parent inoculation sites.
DksA- and (p)ppGpp-deficient transcriptomes significantly overlap at stationary phase.
Loss of dksA and (p)ppGpp can have unique, overlapping, and pleiotropic effects on transcription. To better understand their respective contributions to pathogenesis, we next determined the effects of DksA and (p)ppGpp deficiency on gene expression. We compared the transcriptomes of strains 35000HP, 35000HPΔdksA, and 35000HPΔrelAΔspoT, which is (p)ppGpp°. Four biological replicates were included for each strain harvested at the mid-log, transition, and stationary phases of growth (see Fig. S1 in the supplemental material), for a total of 36 samples. We calculated the fold change in the expression of genes in the 35000HPΔdksA mutant or 35000HPΔrelAΔspoT mutant compared to strain 35000HP. As described previously, we used a false discovery rate (FDR) of ≤0.1 and a twofold change as criteria for differential transcript expression (44). A positive fold change indicates that expression of the gene is higher in the mutant strain, while a negative fold change signifies that the expression is higher in the parent strain (see Table S4 in the supplemental material).
Comparison of the transcriptomes of the (p)ppGpp° mutant to the parent in mid-log, transition, and stationary phase yielded 149, 107, and 494 differentially expressed genes, respectively; approximately equal numbers of genes were up- and downregulated (Fig. 1). Comparison of the transcriptomes of the dksA mutant to the parent in mid-log, transition, and stationary phase yielded 58, 184, and 304 differentially expressed genes, respectively; the majority of genes were upregulated (Fig. 1).
FIG 1.
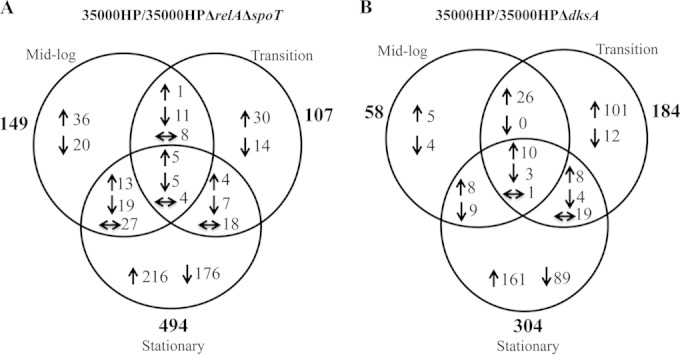
Venn diagrams showing the number of genes differentially regulated by (p)ppGpp or DksA deficiency at different phases of growth. (A) H. ducreyi 35000HPΔrelAΔspoT mutant compared to strain 35000HP and (B) 35000HPΔdksA mutant compared to strain 35000HP. The total number of genes or operons differentially regulated in different phases of growth is indicated in bold type outside the circles of the Venn diagram. Upregulated (↑) and downregulated (↓) genes and genes regulated differently in different growth phases (← and →) are indicated.
To identify the overlap of the genes that were differentially regulated by the loss of (p)ppGpp or DksA, we plotted the log10-transformed fold changes in the mutant 35000HPΔrelAΔspoT/35000HP strain against mutant 35000HPΔdksA/35000HP strain. Comparison of the genes differently regulated by loss of DksA to those differentially regulated by loss of (p)ppGpp in cells harvested from mid-log, transition, and stationary phase yielded 11, 11, and 222 overlapping genes, respectively. At mid-log and transition phase, (p)ppGpp and dksA deficiency primarily altered unique sets of genes, while at stationary phase, the differentially expressed genes significantly overlapped and were coordinately regulated (chi-square = 367.1903; P < 0.001) (Fig. 2). These data showed that in stationary phase, (p)ppGpp and DksA deficiencies primarily alter expression of similar targets. As (p)ppGpp and DksA primarily respond to nutrient stress, we focused our analysis on the stationary-phase transcriptomes.
FIG 2.
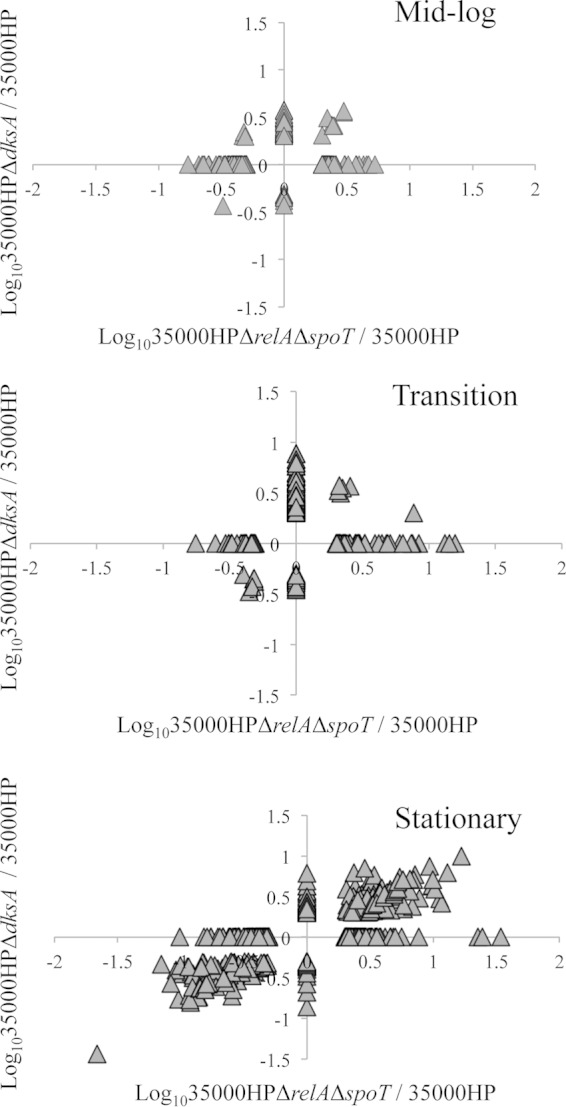
Scatter plots showing fold changes in the expression of genes differentially expressed in 35000HPΔrelAΔspoT and 35000HPΔdksA mutant strains compared to 35000HP strain. The scatter plots were generated by plotting the log10-transformed fold changes in 35000HPΔrelAΔspoT mutant versus 35000HP strain against 35000HPΔdksA mutant versus 35000HP strain at different growth phases. Each triangle in the graph indicates a single gene.
We validated selected differentially regulated genes using qRT-PCR. We focused on the (p)ppGpp° mutant transcriptome, as it had the largest number of differentially expressed genes. The genes were grouped into three categories based on their expression levels (low, medium, and high), grouped into up- and downregulated targets, and further subgrouped based on their fold change ranges (2.0-fold to 5.0-fold, 5.1-fold to 10.0-fold, and 10.1-fold to 50.0-fold). Representative genes were selected from each category; a total of 15 genes were selected for qRT-PCR validation using primers P23-P52 (see Table S2 in the supplemental material). qRT-PCR analysis confirmed the differential expression of 14/15 genes identified by RNA-seq (Fig. 3). However, hfq expression was 11.67-fold upregulated by RNA-seq but unchanged (0.97) by qRT-PCR (Fig. 3B); the reason for this discrepancy is unclear. In general, the fold changes derived from RNA-seq were in good agreement with those obtained from qRT-PCR (R2 = 0.902).
FIG 3.

qRT-PCR validation of the RNA-seq data. (A) Fold change in the expression of target genes in 35000HPΔrelAΔspoT mutant relative to 35000HP strain in stationary phase. The criteria used for selecting the targets for qRT-PCR validation are outlined in the figure. The expression levels of target genes were normalized to that of dnaE. The data represent the means plus standard deviations (SD) (error bars) from four independent experiments. (B) Correlation between the fold changes derived from qRT-PCR and RNA-seq.
Functional classification of genes altered by deficiency of (p)ppGpp° or dksA.
Using annotations and pathway information from the sequenced 35000HP genome (Munson, unpublished) and KEGG, the identified differentially expressed genes were classified into multiple functional categories, including energy metabolism, biosynthesis, transcription, translation, cell membrane, and substrate binding (Table 2). In both mutants, pathway enrichment analysis with annotations from both BioCyc and DAVID bioinformatics resources showed that genes encoding proteins involved in pilus formation, ion transport, oxidative reduction/phosphorylation, carbohydrate transport, and cytochrome complex assembly were enriched (data not shown). The (p)ppGpp° mutant also showed enrichment in genes encoding proteins involved in regulation of transcription and translation.
TABLE 2.
Functional classification of genes or operons differentially regulated by (p)ppGpp or DksA in H. ducreyi
| Functional category | No. of genes differentially expressed |
|||
|---|---|---|---|---|
| 35000HPΔrelAΔspoT/35000HPa |
35000HPΔdksA/35000HPb |
|||
| Upregulated | Downregulated | Upregulated | Downregulated | |
| Amino acid biosynthesis | 2 | 4 | 5 | 1 |
| Amino acid transport and metabolism | 6 | 6 | 5 | 2 |
| Amino sugar and nucleotide metabolism | 4 | 12 | 2 | 7 |
| Cellular carbohydrate biosynthetic process | 7 | 5 | 3 | 1 |
| Carbohydrate metabolism | 1 | 2 | 3 | 0 |
| Cell division | 2 | 5 | 1 | 2 |
| Cell membrane | 11 | 15 | 9 | 7 |
| Cellular homeostasis | 4 | 2 | 2 | 1 |
| Cellular response to stress | 0 | 2 | 3 | 0 |
| DNA binding | 3 | 1 | 4 | 2 |
| DNA metabolic process | 8 | 4 | 8 | 1 |
| Fatty acid biosynthesis and metabolism | 7 | 3 | 4 | 2 |
| Intracellular trafficking and secretion | 1 | 11 | 1 | 5 |
| Ion transport | 3 | 7 | 2 | 4 |
| Iron sulfur cluster binding | 2 | 6 | 0 | 3 |
| Lipid biosynthesis and metabolism | 3 | 4 | 5 | 2 |
| Metal ion binding | 10 | 8 | 7 | 5 |
| Nucleotide binding | 8 | 19 | 4 | 11 |
| Protein fate | 7 | 9 | 5 | 5 |
| RNA binding | 4 | 0 | 1 | 0 |
| RNA processing | 9 | 8 | 5 | 3 |
| Transcription | 10 | 9 | 2 | 3 |
| Translation | 15 | 27 | 12 | 6 |
| Transport of proteins and carbohydrates | 7 | 11 | 2 | 6 |
| Uncharacterized conserved protein | 6 | 10 | 7 | 1 |
| Hypothetical proteins | 75 | 89 | 30 | 92 |
Genes differentially expressed in 35000HPΔrelAΔspoT mutant versus 35000HP strain at stationary phase. Fifty-two percent of the genes were downregulated genes, and 48% were upregulated genes.
Genes differentially expressed in 35000HPΔdksA mutant versus 35000HP strain at stationary phase. Sixty-two percent of the genes were downregulated genes, and 38% were upregulated genes.
(p)ppGpp° and dksA deficiency leads to dysregulation of virulence determinants required for human infection.
Since both the dksA and (p)ppGpp° mutants were partially attenuated in humans, we determined the effects of their deficiencies on the expression of genes required for human infection. (p)ppGpp deficiency in strain 35000HP resulted in decreased expression of genes in the flp-tad operon, lspB-lspA2 operon, and the lspA1, hgbA, and csrA genes, which are all required for pustule formation in humans (Table 3) (26, 40, 45). Loss of (p)ppGpp increased the expression of fgbA, which is required for virulence (26). These data are consistent with the partial attenuation and some phenotypes of the (p)ppGpp° mutant reported previously (23).
TABLE 3.
Regulation of genes required for virulence in humans by either (p)ppGpp or DksA
| Gene(s)a | Function | 35000HPΔrelAΔspoT/35000HPb,c | 35000HPΔdksA/35000HPb,d |
|---|---|---|---|
| Fully attenuated | |||
| flp-1–flp-2–flp-3 | Adherence and microcolony formation | −3.72 | −2.33e |
| tadA | Adherence and microcolony formation | −6.86 | – |
| dsrA | Major role in serum resistance | – | – |
| lspA1, lspA2 | Escape from phagocytosis | −2.75, −5.36f | −2.57f |
| ncaA | Collagen binding | – | – |
| hgbA | Heme and/or iron uptake | −2.54 | −2.37 |
| pal | Outer membrane stability | – | – |
| hfq | RNA binding chaperone | 11.67 | 2.64 |
| sapB-sapC | Resistance to antimicrobial peptides | – | – |
| cpxA | Two-component sensor kinase | – | – |
| Partially attenuated | |||
| dltA | Partial role in serum resistance | – | 4.98 |
| wecA | Initiates synthesis of putative glycoconjugate | – | – |
| luxS | Quorum sensing | – | – |
| fgbA | Fibrinogen binding | 2.04 | – |
| sapA | Resistance to antimicrobial peptides | – | – |
| csrA | Posttranscriptional regulation | −2.34 | – |
| dksA | DnaK repressor; (p)ppGpp cofactor | – | NA |
| relA, spoT | (p)ppGpp synthetase and hydrolase; stringent response | NA | 2.09 (spoT) |
Strains with mutated genes that have been tested in human volunteers and classified as fully or partially attenuated.
Fold change of the first differentially regulated gene in the operon. –, no change in expression; NA, not applicable.
Fold change in 35000HPΔrelAΔspoT mutant relative to 35000HP strain in stationary phase.
Fold change in 35000HPΔdksA mutant relative to 35000HP strain in stationary phase.
Fold change of flp-3 only.
Fold change of the lspB-lspA2 operon.
DksA deficiency resulted in decreased expression of flp-3, hgbA, and lspB, which would favor decreased virulence (26). The dksA mutant also had increased expression of hfq, dltA, and spoT, which would favor increased virulence (23, 30). These results suggest that the partial attenuation of the dksA mutant might be due to conflicting phenotypes.
The set of transcripts altered by (p)ppGpp deficiency significantly overlaps with the sets of transcripts controlled by Hfq and CpxRA.
Hfq is a major regulator of H. ducreyi stationary-phase gene expression and contributes to the positive regulation of proteins required for virulence such as LspB, DsrA, and Flp1 (30). Given that both mutants upregulated hfq (Table 3) and given the discrepancy in the hfq expression levels determined by different methods for the (p)ppGpp° mutant, we examined whether the transcriptomes of the (p)ppGpp° and DksA mutants significantly overlapped with that of the hfq mutant (30). The transcriptomes of the (p)ppGpp° and hfq mutants were negatively correlated and overlapped significantly (chi-square = 38.172; P < 0.001), while those of the dksA and hfq mutants did not. Thus, it is likely that (p)ppGpp deficiency results in upregulation of hfq transcription.
(p)ppGpp and DksA deficiencies resulted in 7.19- and 5.89-fold upregulation of cpxR, respectively (see Table S4 in the supplemental material). We therefore compared the effects of dksA and (p)ppGpp deficiencies on transcription to those produced by activation of CpxRA, defined as the transcription effects of a CpxR-activating mutant compared to a cpxR deficient mutant in stationary phase (44). The differentially expressed genes in the dksA mutant did not overlap with those of CpxRA. The effects of (p)ppGpp deficiency positively correlated and significantly overlapped with the effects of activation of CpxRA (chi-square = 17.070; P < 0.001). Since activation of the CpxRA system is associated with loss of virulence, these data are consistent with the partial attenuation of the (p)ppGpp° mutant (23).
Deletion of dksA increased uptake and reduced survival within macrophages of H. ducreyi grown to stationary phase.
The (p)ppGpp° mutant had several in vitro phenotypes that favored attenuation, such as decreased survival in macrophages and increased sensitivity to oxidative stress (23). However, the (p)ppGpp° mutant also exhibited phenotypes that favored virulence, such as increased resistance to uptake by human macrophages and prolonged survival in stationary phase (23). Since the genes affected by DksA and (p)ppGpp deficiency overlapped, we focused on characterizing virulence-associated phenotypes of the dksA mutant in similar assays.
During human infection, H. ducreyi associates with neutrophils and macrophages but remains extracellular (16, 46). The ability of H. ducreyi to evade phagocytosis is due to the secretion of the antiphagocytic proteins LspA1 and LspA2 by LspB (47, 48). Transcriptome analysis indicated that deletion of dksA downregulated expression of lspB at stationary phase, suggesting that the dksA mutant might be more readily phagocytosed than the parent strain. We compared the uptake and intracellular survival of dksA mutant cells harvested at mid-log and stationary phase by primary human macrophages. No differences were found in uptake of the dksA mutant harvested at mid-log phase compared to the parent (Fig. 4A). The dksA mutant tended toward being taken up at a higher rate than the parent when the bacteria were harvested at stationary phase (P = 0.055).
FIG 4.
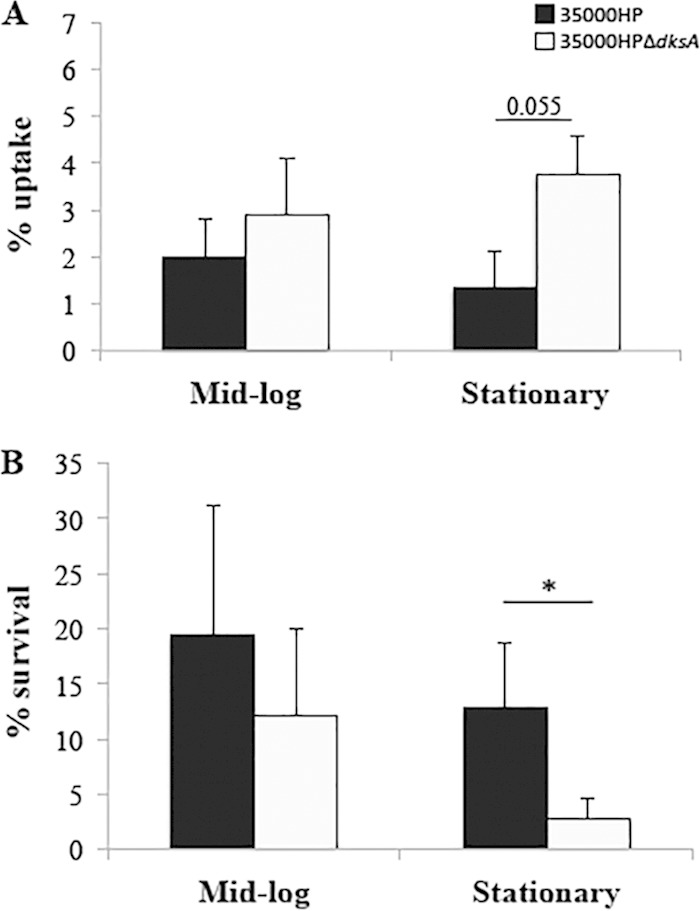
Uptake of H. ducreyi by and survival within human macrophages. (A) Percent uptake of 35000HP strain and 35000HPΔdksA mutant strain by human monocyte-derived macrophages (MDMs). MDMs were infected with opsonized H. ducreyi at an MOI of 10:1. The percent uptake was calculated as follows: (geometric mean CFU of gentamicin-protected bacteria at 1 h/geometric mean CFU of input bacteria) × 100. (B) Percent survival of 35000HP strain and 35000HPΔdksA mutant by MDMs. To determine survival, the MDMs were incubated an additional 5 h, and bacteria were collected. Survival was calculated as follows: (geometric mean CFU of bacteria collected at endpoint/geometric mean CFU of gentamicin-protected bacteria at 1 h) × 100]. Data are mean ± SD from 5 independent donors. Values that are significantly different (P ≤ 0.05) are indicated by a bar and asterisk.
We next examined whether deletion of dksA affected H. ducreyi survival within phagocytes. No differences were found for the intracellular survival of the mutant compared to the parent harvested at mid-log phase (Fig. 4B). However, a significant reduction in the intracellular survival of the dksA mutant (P = 0.023) was shown for cells harvested at stationary phase. Thus, DksA plays a crucial role in resistance to phagocytic killing in vitro.
DksA is important for resistance to oxidative stress.
The reduced survival of H. ducreyi within macrophages is linked to increased sensitivity to oxidative stress (23, 45). We compared the survival of H. ducreyi 35000HP and 35000HPΔdksA after incubation with hydrogen peroxide. For cells grown to mid-log phase, the dksA mutant survived significantly less than the parent at 2 mM H2O2 (P = 0.0005) with a trend toward significance at 0.2 mM (P = 0.052) (Fig. 5A). When stationary-phase bacteria were incubated with 0.2 mM or 2 mM H2O2, the dksA mutant survived significantly less than the parent strain (P = 0.023 and P = 0.0002, respectively) (Fig. 5A). Complementation of the dksA mutant in trans with plasmid pCH24 partially restored resistance to 2 mM H2O2 (P = <0.0001) in cells harvested at mid-log phase (Fig. 5B). Deletion of dksA did not affect expression of csrA or sodC, both of which are associated with increased sensitivity to oxidative stress (see Table S4 in the supplemental material) (45, 49). Therefore, DksA contributes to H. ducreyi survival during in vitro oxidative stress through an uncharacterized mechanism.
FIG 5.

H. ducreyi survival after oxidative stress. (A) Percent survival of strains 35000HP and 35000HPΔdksA following treatment with either 0.2 or 2 mM H2O2 at mid-log or stationary phase. (B) Percent survival of mid-log-phase strain 35000HP(pACYC177), strain 35000HPΔdksA(pACYC177), and the complemented strain 35000HPΔdksA(pCH24) following incubation with 0.2 mM or 2 mM H2O2 for 1 h. All values for percent survival were calculated as follows: (geometric mean CFU after treatment/geometric mean before treatment) × 100]. The data are means ± SD from five independent experiments. *, P ≤ 0.05.
DksA compensates for loss of (p)ppGpp in resistance to oxidative stress.
In H. ducreyi, loss of (p)ppGpp coincided with an increase in transcript levels of dksA (23). The (p)ppGpp° mutant also exhibited increased sensitivity to oxidative stress. In E. coli, dksA overexpression can compensate for loss of (p)ppGpp (18, 19). We therefore determined whether overexpression of dksA could compensate for loss of (p)ppGpp. To this end, we generated 35000HPΔrelAΔspoT(pCH24), which overexpresses dksA but does not synthesize (p)ppGpp. We used strain 35000HPΔrelAΔspoT(pCH30), the ΔrelA ΔspoT complemented strain as a control for the assay (23). The sensitivity of strain 35000HPΔrelAΔspoT(pACYC177) to 2 mM hydrogen peroxide was significantly restored in both 35000HPΔrelAΔspoT(pCH30) (P = 0.0002) and 35000HPΔrelAΔspoT(pCH24) (P = 0.0029) strains (Fig. 6). Therefore, DksA partially compensates for the loss of (p)ppGpp in resistance to oxidative stress in vitro.
FIG 6.

Complementation of the 35000HPΔrelAΔspoT mutant strain by dksA. Percent survival of mid-log-phase strain 35000HP(pACYC177), strain 35000HPΔrelAΔspoT(pACYC177), and strains 35000HPΔrelAΔspoT(pCH30) (pCH30 is the relA spoT double complementation plasmid) and 35000HPΔrelAΔspoT(pCH24) (pCH24 is the dksA complementation plasmid), following incubation with 0.2 mM or 2 mM H2O2 for 1 h. All values for percent survival were calculated as follows: (geometric mean CFU after treatment/geometric mean before treatment) × 100. The data are means plus SD from five independent experiments. *, P ≤ 0.05.
DksA is important for adherence of H. ducreyi to HFF cells by a Flp-Tad-independent mechanism.
The flp-tad operon is composed of a Tad secretion system that secretes three fimbria-like proteins, Flp1 to Flp3, which mediate adherence of the bacterium to HFF cells and are associated with virulence (40). The flp-tad operon was downregulated approximately 12-fold in the (p)ppGpp° mutant; only flp-3 was downregulated in the dksA mutant (Table 3). We compared the adherence of strains 35000HP, 35000HPΔdksA, 35000HPΔrelAΔspoT, and 35000HPΔflp1-3 (strains harvested in stationary phase) to HFF cells. The 35000HPΔflp1-3 mutant served as a negative control for the assay. As expected, both the (p)ppGpp° (66.7% ± 9.5%) and 35000HPΔflp1-3 (4.6% ± 1.6%) mutants exhibited reduced attachment compared to the parent (115.8% ± 21.5%) (P = 0.0002 and P = <0.0001, respectively) (Fig. 7A). Surprisingly, the dksA mutant also attached to HFF cells at significantly lower levels than the parent (15.9% ± 5.2%; P = <0.0001) (Fig. 7A).
FIG 7.

H. ducreyi adherence to HFF cells and Flp protein expression. (A) Percent adherence of H. ducreyi 35000HP, 35000HPΔdksA, 35000HPΔrelAΔspoT, and 35000HPΔflp1-3 to HFF cells calculated as follows: (geometric mean CFU of HFF-adherent bacteria/geometric mean CFU of initial bacteria added per well) × 100. The data are means plus SD from 5 independent experiments. *, P ≤ 0.05. (B and C) Western blot analysis of whole-cell lysates (B) and purified outer membranes (C) analyzed by SDS-PAGE. Samples were probed with Flp1/2 antibody. MAb 3B9 was used to verify equivalent loading. Data are representative of four independent experiments. Lanes 1, 35000HP; lanes 2, 35000HPΔdksA; lane 3, 35000HPΔrelAΔspoT; lanes 4, 35000HP.400, the tadA mutant; lanes 5, 35000HPΔflp1-3, the flp mutant.
The decreased attachment of the dksA mutant could have resulted from decreased secretion of the Flp proteins (Flps) by the Tad secretion system, decreased expression of the Flp proteins, or decreased expression of a cofactor(s) required for Flp-dependent adherence (50). To determine whether the decreased adherence of the dksA mutant was a result of decreased expression or altered localization of the Flp proteins, we probed whole-cell lysates and outer membranes with Flp1/2 antiserum by Western blotting. The tadA mutant and the 35000HPΔflp1-3 mutant were used as controls for the assay. As expected, the tadA mutant expressed the Flp proteins as seen in whole-cell lysates, but it did not secrete the proteins to the outer membrane (Fig. 7B and C). The (p)ppGpp° mutant expresses reduced levels of the Flp proteins (Fig. 7B). In contrast, both the wild type and the dksA mutant expressed the Flp proteins in whole cells and outer membranes (Fig. 7B and C). Thus, the adherence defect in the dksA mutant cannot be attributed to decreased expression or lack of localization of the Flp proteins to the outer membrane; these data suggest that DksA regulates the expression of unknown cofactor(s) that is required for Flp-mediated adherence in vitro.
DISCUSSION
The ability to respond to stress is a critical survival mechanism for bacterial pathogens. During human infection, H. ducreyi is found in the hostile environment of an abscess and utilizes several mechanisms to resist stress. We previously showed that (p)ppGpp, a molecule involved in the stress response to nutrient deprivation, was necessary for virulence of H. ducreyi in humans (23). In E. coli, DksA is a transcription factor that acts by binding directly to RNAP and amplifies the effects of (p)ppGpp to enhance the stringent response. In addition to its critical role in the stringent response, DksA can interact with RNAP and regulate gene transcription independently of (p)ppGpp. Here we showed that an H. ducreyi dksA mutant is also partially attenuated for virulence in human volunteers, indicating that DksA also plays an important role in H. ducreyi pathogenesis. We provided evidence that (p)ppGpp and DksA primarily contribute to the coordinated regulation of gene expression in stationary phase but also control some unique targets. Taken together, these data suggest an important role for (p)ppGpp and DksA in controlling H. ducreyi stationary phase and virulence gene expression.
In cells harvested from stationary phase, loss of (p)ppGpp and DksA led to differential expression of 28% and 17% of the H. ducreyi open reading frames, respectively, and significantly overlapped. We found that many of the differentially regulated genes identified in our study have been previously associated with the stringent response (19, 51). (p)ppGpp and DksA deficiency in H. ducreyi resulted in altered expression of genes involved in transcription, translation, biosynthesis of macromolecules, and energy metabolism. This is consistent with findings in E. coli that the (p)ppGpp/DksA system regulates genes necessary for adaptation to nutritional stress (18, 19, 51). However, 27% of the genes regulated by DksA deficiency were independent of those regulated by (p)ppGpp deficiency. This suggests that H. ducreyi is similar to other organisms in that DksA functions both in concert with and independently of (p)ppGpp (18, 19).
Although the loss of DksA upregulated the expression of hfq transcripts, there was no significant overlap in the genes controlled by these regulators. In contrast, the loss of (p)ppGpp appeared to upregulate the expression of hfq, and the genes controlled by these factors significantly overlapped. (p)ppGpp deficiency also upregulated expression of cpxR transcripts and genes associated with CpxR activation. In H. ducreyi, Hfq positively regulates the expression of several virulence determinants, including lspB-lspA2, flp1-3 (flp-1 to flp-3), and dsrA (30). In contrast, activation of the CpxRA system downregulates the expression of lspB, flp1-3, and dsrA (44). (p)ppGpp deficiency resulted in decreased expression of lspB-lspA2 and flp1-3, while dsrA expression was unchanged. These data suggest that upregulation of cpxR or another (p)ppGpp-dependent target overcomes the potential effects of overexpression of hfq on these virulence determinants in the (p)ppGpp-deficient mutant.
Both the dksA mutant and (p)ppGpp° mutant were partially attenuated for pustule formation in human volunteers. While the (p)ppGpp° mutant formed papules whose sizes were not significantly different than those of the parent, the dksA mutant caused significantly smaller papules than the parent did. Although we have not compared the two mutants directly in human volunteers, the data suggest that DksA and (p)ppGpp might have different effects on virulence. The dksA mutant was taken up more readily than the parent by macrophages, while the (p)ppGpp° mutant was phagocytized less readily than the parent was. Both the dksA mutant and (p)ppGpp° mutant exhibited increased sensitivity to oxidative stress. This increased sensitivity could be linked to the decreased survival within macrophages seen in both the dksA and (p)ppGpp° mutants (Fig. 4B and unpublished data). Both the dksA mutant and (p)ppGpp° mutant exhibited decreased attachment to HFF cells; however, the decreased attachment likely resulted from different mechanisms. Taken together, the data suggest that DksA and (p)ppGpp have both distinct and overlapping functions.
The ability of H. ducreyi to attach to human foreskin fibroblasts has also been correlated with virulence, and the dksA mutant showed decreased attachment to HFF cells compared to the parent. Similarly, studies of pathogenic and nonpathogenic E. coli show that dksA mutants tend to have decreased attachment to eukaryotic cells as well as decreased expression of genes involved in the regulation of attachment (18, 19, 52). We found no evidence for decreased expression or altered localization of the Flp proteins in the dksA mutant. Taken together, these data suggest that DksA affects adherence to HFF cells through another mechanism, such as regulating one or more yet to be defined cofactors that are required for Flp-mediated adherence.
In summary, we show that DksA and (p)ppGpp likely serve as major contributors of virulence and stationary-phase gene regulation in H. ducreyi. We show that despite similar gene expression patterns, the dksA and (p)ppGpp° mutants are phenotypically distinct. Future studies will focus on identifying (p)ppGpp and/or DksA-dependent proteins to better understand the unique intersecting and diverging roles the two regulators play in H. ducreyi virulence.
Supplementary Material
ACKNOWLEDGMENTS
We thank Julia van Rensburg, Dharanesh Gangaiah, and Margaret Bauer for their thoughtful criticism of the manuscript and the volunteers who participated in the trial.
This work was supported by the National Institutes of Allergy and Infectious Diseases (AI27863 and AI27863S1 to S.M.S.) and a Diversity Supplement (AI27863S2 to C.L.H.). The human challenge trials were also supported by the Indiana Clinical and Translational Sciences Institute and the Indiana Clinical Research Center (UL RR052761).
We declare that we have no conflicts of interest.
Footnotes
Supplemental material for this article may be found at http://dx.doi.org/10.1128/IAI.00692-15.
REFERENCES
- 1.Spinola SM. 2008. Chancroid and Haemophilus ducreyi, p 689–699. In Holmes KK, Sparling PF, Stamm WE, Piot P, Wasserheit JN, Corey L, Cohen MS, Watts DH (ed), Sexually transmitted diseases, 4th ed McGraw-Hill, New York, NY. [Google Scholar]
- 2.Steen R, Wi TE, Kamali A, Ndowa F. 2009. Control of sexually transmitted infections and prevention of HIV transmission: mending a fractured paradigm. Bull World Health Organ 87:858–865. doi: 10.2471/BLT.08.059212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Phiri S, Zadrozny S, Weiss HA, Martinson F, Nyirenda N, Chen CY, Miller WC, Cohen MS, Mayaud P, Hoffman IF. 2013. Etiology of genital ulcer disease and association with HIV infection in Malawi. Sex Transm Dis 40:923–928. doi: 10.1097/OLQ.0000000000000051. [DOI] [PubMed] [Google Scholar]
- 4.Fayemiwo SA, Odaibo GN, Oni AA, Ajayi AA, Bakare RA, Olaleye DO. 2011. Genital ulcer diseases among HIV-infected female commercial sex workers in Ibadan, Nigeria. Afr J Med Med Sci 40:39–46. [PubMed] [Google Scholar]
- 5.Maan MA, Hussain F, Iqbal J, Akhtar SJ. 2011. Sexually transmitted infections in Pakistan. Ann Saudi Med 31:263–269. doi: 10.4103/0256-4947.81541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bhutto AM, Shah AH, Ahuja DK, Solangi AH, Shah SA. 2011. Pattern of sexually transmitted infections in males in interior Sindh: a 10-year-study. J Ayub Med Coll Abbottabad 23(3):110–114. [PubMed] [Google Scholar]
- 7.Weiss HA, Paz Bailey G, Phiri S, Gresenguet G, LeGoff J, Pepin J, Lewis DA, Belec L, Hoffman IF, Miller WC, Mayaud P. 2011. Episodic therapy for genital herpes in sub-Saharan Africa: a pooled analysis from three randomized controlled trials. PLoS One 6:e22601. doi: 10.1371/journal.pone.0022601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ray K, Bala M, Gupta SM, Khunger N, Puri P, Muralidhar S, Kumar J. 2006. Changing trends in sexually transmitted infections at a regional STD centre in north India. Indian J Med Res 124:559–568. [PubMed] [Google Scholar]
- 9.Mitjà O, Lukehart SA, Pokowas G, Moses P, Kapa A, Godornes C, Robson J, Cherian S, Houinei W, Kazadi W, Siba P, de Lazzari E, Bassat Q. 2014. Haemophilus ducreyi as a cause of skin ulcers in children from a yaws-endemic area of Papua New Guinea: a prospective cohort study. Lancet Global Health 2:e235–e241. doi: 10.1016/S2214-109X(14)70019-1. [DOI] [PubMed] [Google Scholar]
- 10.Marks M, Chi KH, Vahi V, Pillay A, Sokana O, Pavluck A, Mabey DC, Chen CY, Solomon AW. 2014. Haemophilus ducreyi associated with skin ulcers among children, Solomon Islands. Emerg Infect Dis 20:1705–1707. doi: 10.3201/eid2010.140573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ghinai R, El-Duah P, Chi KH, Pillay A, Solomon AW, Bailey RL, Agana N, Mabey DC, Chen CY, Adu-Sarkodie Y, Marks M. 2015. A cross-sectional study of ‘yaws’ in districts of Ghana which have previously undertaken azithromycin mass drug administration for trachoma control. PLoS Negl Trop Dis 9:e0003496. doi: 10.1371/journal.pntd.0003496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Spinola SM, Wild LM, Apicella MA, Gaspari AA, Campagnari AA. 1994. Experimental human infection with Haemophilus ducreyi. J Infect Dis 169:1146–1150. doi: 10.1093/infdis/169.5.1146. [DOI] [PubMed] [Google Scholar]
- 13.Al-Tawfiq JA, Thornton AC, Katz BP, Fortney KR, Todd KD, Hood AF, Spinola SM. 1998. Standardization of the experimental model of Haemophilus ducreyi infection in human subjects. J Infect Dis 178:1684–1687. doi: 10.1086/314483. [DOI] [PubMed] [Google Scholar]
- 14.Gaston JR, Roberts SA, Humphreys TL. 2015. Molecular phylogenetic analysis of non-sexually transmitted strains of Haemophilus ducreyi. PLoS One 10:e0118613. doi: 10.1371/journal.pone.0118613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hays RC, Mandell GL. 1974. pO2, pH, and redox potential of experimental abscesses. Proc Soc Exp Biol Med 147:29–30. doi: 10.3181/00379727-147-38275. [DOI] [PubMed] [Google Scholar]
- 16.Bauer ME, Goheen MP, Townsend CA, Spinola SM. 2001. Haemophilus ducreyi associates with phagocytes, collagen, and fibrin and remains extracellular throughout infection of human volunteers. Infect Immun 69:2549–2557. doi: 10.1128/IAI.69.4.2549-2557.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Perederina A, Svetlov V, Vassylyeva MN, Tahirov TH, Yokoyama S, Artsimovitch I, Vassylyev DG. 2004. Regulation through the secondary channel–structural framework for ppGpp-DksA synergism during transcription. Cell 118:297–309. doi: 10.1016/j.cell.2004.06.030. [DOI] [PubMed] [Google Scholar]
- 18.Magnusson LU, Gummesson B, Joksimovic P, Farewell A, Nystrom T. 2007. Identical, independent, and opposing roles of ppGpp and DksA in Escherichia coli. J Bacteriol 189:5193–5202. doi: 10.1128/JB.00330-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Aberg A, Fernandez-Vazquez J, Cabrer-Panes JD, Sanchez A, Balsalobre C. 2009. Similar and divergent effects of ppGpp and DksA deficiencies on transcription in Escherichia coli. J Bacteriol 191:3226–3236. doi: 10.1128/JB.01410-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Pal RR, Bag S, Dasgupta S, Das B, Bhadra RK. 2012. Functional characterization of the stringent response regulatory gene dksA of Vibrio cholerae and its role in modulation of virulence phenotypes. J Bacteriol 194:5638–5648. doi: 10.1128/JB.00518-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Jude F, Kohler T, Branny P, Perron K, Mayer MP, Comte R, van Delden C. 2003. Posttranscriptional control of quorum-sensing-dependent virulence genes by DksA in Pseudomonas aeruginosa. J Bacteriol 185:3558–3566. doi: 10.1128/JB.185.12.3558-3566.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Sharma AK, Payne SM. 2006. Induction of expression of hfq by DksA is essential for Shigella flexneri virulence. Mol Microbiol 62:469–479. doi: 10.1111/j.1365-2958.2006.05376.x. [DOI] [PubMed] [Google Scholar]
- 23.Holley C, Gangaiah D, Li W, Fortney KR, Janowicz DM, Ellinger S, Zwickl B, Katz BP, Spinola SM. 2014. A (p)ppGpp-null mutant of Haemophilus ducreyi is partially attenuated in humans due to multiple conflicting phenotypes. Infect Immun 82:3492–3502. doi: 10.1128/IAI.01994-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Bozue JA, Tarantino L, Munson RS Jr. 1998. Facile construction of mutations in Haemophilus ducreyi using lacZ as a counter-selectable marker. FEMS Microbiol Lett 164:269–273. doi: 10.1111/j.1574-6968.1998.tb13097.x. [DOI] [PubMed] [Google Scholar]
- 25.Spinola SM, Fortney KR, Baker B, Janowicz DM, Zwickl B, Katz BP, Blick RJ, Munson RS Jr. 2010. Activation of the CpxRA system by deletion of cpxA impairs the ability of Haemophilus ducreyi to infect humans. Infect Immun 78:3898–3904. doi: 10.1128/IAI.00432-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Janowicz DM, Ofner S, Katz BP, Spinola SM. 2009. Experimental infection of human volunteers with Haemophilus ducreyi: fifteen years of clinical data and experience. J Infect Dis 199:1671–1679. doi: 10.1086/598966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Spinola SM, Fortney KR, Katz BP, Latimer JL, Mock JR, Vakevainen M, Hansen EJ. 2003. Haemophilus ducreyi requires an intact flp gene cluster for virulence in humans. Infect Immun 71:7178–7182. doi: 10.1128/IAI.71.12.7178-7182.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Li H, Durbin R. 2010. Fast and accurate long-read alignment with Burrows-Wheeler transform. Bioinformatics 26:589–595. doi: 10.1093/bioinformatics/btp698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Breese MR, Liu Y. 2013. NGSUtils: a software suite for analyzing and manipulating next-generation sequencing datasets. Bioinformatics 29:494–496. doi: 10.1093/bioinformatics/bts731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Gangaiah D, Labandeira-Rey M, Zhang X, Fortney KR, Ellinger S, Zwickl B, Baker B, Liu Y, Janowicz DM, Katz BP, Brautigam CA, Munson RS Jr, Hansen EJ, Spinola SM. 2014. Haemophilus ducreyi Hfq contributes to virulence gene regulation as cells enter stationary phase. mBio 5(1):e01081-13. doi: 10.1128/mBio.01081-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kanehisa M, Goto S. 2000. KEGG: Kyoto encyclopedia of genes and genomes. Nucleic Acids Res 28:27–30. doi: 10.1093/nar/28.1.27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Caspi R, Altman T, Dreher K, Fulcher CA, Subhraveti P, Keseler IM, Kothari A, Krummenacker M, Latendresse M, Mueller LA, Ong Q, Paley S, Pujar A, Shearer AG, Travers M, Weerasinghe D, Zhang P, Karp PD. 2012. The MetaCyc database of metabolic pathways and enzymes and the BioCyc collection of pathway/genome databases. Nucleic Acids Res 40:D742–D753. doi: 10.1093/nar/gkr1014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Huang DW, Sherman BT, Lempicki RA. 2009. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat Protoc 4:44–57. doi: 10.1038/nprot.2008.211. [DOI] [PubMed] [Google Scholar]
- 34.Raivio TL, Leblanc SK, Price NL. 2013. The Escherichia coli Cpx envelope stress response regulates genes of diverse function that impact antibiotic resistance and membrane integrity. J Bacteriol 195:2755–2767. doi: 10.1128/JB.00105-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Apicella MA, Griffiss JM, Schneider H. 1994. Isolation and characterization of lipopolysaccharides, lipooligosaccharides, and lipid A. Methods Enzymol 235:242–252. [DOI] [PubMed] [Google Scholar]
- 36.Spinola SM, Griffiths GE, Bogdan JA, Menegus MA. 1992. Characterization of an 18,000-molecular-weight outer membrane protein of Haemophilus ducreyi that contains a conserved surface-exposed epitope. Infect Immun 60:385–391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Banks KE, Fortney KR, Baker B, Billings SD, Katz BP, Munson RS Jr, Spinola SM. 2008. The enterobacterial common antigen-like gene cluster of Haemophilus ducreyi contributes to virulence in humans. J Infect Dis 197:1531–1536. doi: 10.1086/588001. [DOI] [PubMed] [Google Scholar]
- 38.Reference deleted. [Google Scholar]
- 39.Reference deleted. [Google Scholar]
- 40.Janowicz DM, Cooney SA, Walsh J, Baker B, Katz BP, Fortney KR, Zwickl B, Ellinger S, Munson RS Jr. 2011. Expression of the Flp proteins by Haemophilus ducreyi is necessary for virulence in human volunteers. BMC Microbiol 11:208. doi: 10.1186/1471-2180-11-208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Ancona V, Lee JH, Chatnaparat T, Oh J, Hong JI, Zhao Y. 2015. The bacterial alarmone (p)ppGpp activates the type III secretion system in Erwinia amylovora. J Bacteriol 197:1433–1443. doi: 10.1128/JB.02551-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Vinella D, Potrykus K, Murphy H, Cashel M. 2012. Effects on growth by changes of the balance between GreA, GreB, and DksA suggest mutual competition and functional redundancy in Escherichia coli. J Bacteriol 194:261–273. doi: 10.1128/JB.06238-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Young RS, Fortney K, Haley JC, Hood AF, Campagnari AA, Wang J, Bozue JA, Munson RS Jr, Spinola SM. 1999. Expression of sialylated or paragloboside-like lipooligosaccharides are not required for pustule formation by Haemophilus ducreyi in human volunteers. Infect Immun 67:6335–6340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Gangaiah D, Zhang X, Fortney KR, Baker B, Liu Y, Munson RS Jr, Spinola SM. 2013. Activation of CpxRA in Haemophilus ducreyi primarily inhibits the expression of its targets, including major virulence determinants. J Bacteriol 195:3486–3502. doi: 10.1128/JB.00372-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Gangaiah D, Li W, Fortney KR, Janowicz DM, Ellinger S, Zwickl B, Katz BP, Spinola SM. 2013. Carbon storage regulator A contributes to the virulence of Haemophilus ducreyi in humans by multiple mechanisms. Infect Immun 81:608–617. doi: 10.1128/IAI.01239-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Bauer ME, Spinola SM. 2000. Localization of Haemophilus ducreyi at the pustular stage of disease in the human model of infection. Infect Immun 68:2309–2314. doi: 10.1128/IAI.68.4.2309-2314.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Vakevainen M, Greenberg S, Hansen EJ. 2003. Inhibition of phagocytosis by Haemophilus ducreyi requires expression of the LspA1 and LspA2 proteins. Infect Immun 71:5994–6003. doi: 10.1128/IAI.71.10.5994-6003.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Ward CK, Mock JR, Hansen EJ. 2004. The LspB protein is involved in the secretion of the LspA1 and LspA2 proteins by Haemophilus ducreyi. Infect Immun 72:1874–1884. doi: 10.1128/IAI.72.4.1874-1884.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.San Mateo LR, Hobbs MM, Kawula TH. 1998. Periplasmic copper-zinc superoxide dismutase protects Haemophilus ducreyi exogenous superoxide. Mol Microbiol 27:391–404. doi: 10.1046/j.1365-2958.1998.00687.x. [DOI] [PubMed] [Google Scholar]
- 50.Nika JR, Latimer JL, Ward CK, Blick RJ, Wagner NJ, Cope LD, Mahairas GG, Munson RS Jr, Hansen EJ. 2002. Haemophilus ducreyi requires the flp gene cluster for microcolony formation in vitro. Infect Immun 70:2965–2975. doi: 10.1128/IAI.70.6.2965-2975.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Traxler MF, Summers SM, Nguyen HT, Zacharia VM, Hightower GA, Smith JT, Conway T. 2008. The global, ppGpp-mediated stringent response to amino acid starvation in Escherichia coli. Mol Microbiol 68:1128–1148. doi: 10.1111/j.1365-2958.2008.06229.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Nakanishi N, Abe H, Ogura Y, Hayashi T, Tashiro K, Kuhara S, Sugimoto N, Tobe T. 2006. ppGpp with DksA controls gene expression in the locus of enterocyte effacement (LEE) pathogenicity island of enterohaemorrhagic Escherichia coli through activation of two virulence regulatory genes. Mol Microbiol 61:194–205. doi: 10.1111/j.1365-2958.2006.05217.x. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.