

Summary
Non-typeable Haemophilus influenzae is an opportunistic pathogen of the human upper respiratory tract and is often found to cause inflammatory diseases that include sinusitis, otitis media and exacerbations of chronic obstructive pulmonary disease. To persist in the inflammatory milieu during infection, non-typeable H. influenzae must resist the antimicrobial activity of the human complement system. Here, we used Tn-seq to identify genes important for resistance to complement-mediated killing. This screen identified outer membrane protein P5 in evasion of the alternative pathway of complement activation. Outer membrane protein P5 was shown to bind human complement regulatory protein factor H directly, thereby, preventing complement factor C3 deposition on the surface of the bacterium. Furthermore, we show that amino acid variation within surface-exposed regions within outer membrane P5 affected the level of factor H binding between individual strains.
Introduction
Non-typeable Haemophilus influenzae (NTHi) is a Gram-negative opportunistic bacterial pathogen that usually resides asymptomatically on the mucosal surface of the human nasopharynx (Poole et al., 2013). However, NTHi is also a leading cause of otitis media (OM) (Stol et al., 2013), sinusitis (Brook, 2011) and exacerbations in patients with chronic obstructive pulmonary disease (COPD) (Sethi et al., 2007). In rare cases, NTHi can even cause severe diseases such as sepsis and meningitis (Hallstrom et al., 2010). In all these situations, NTHi encounters the antimicrobial activity of the host’s immune system and, therefore, has developed mechanisms to survive and multiply during infection.
The human complement system is part of the innate immune system and plays an important role in clearing pathogens. For NTHi to survive in areas where sufficient levels of complement are present requires mechanisms that prevent recognition by opsonizing antibodies, complement deposition or assembly of the membrane-attack complex (Hallstrom and Riesbeck, 2010). Resistance to complement-mediated killing is highly variable among NTHi isolates, with increased resistance found in strains obtained from the middle ear fluid of children with OM (Langereis et al., 2012) and sputum from patients with COPD (Nakamura et al., 2011).
Complement activation is initiated via three different pathways: (i) the classical, (ii) the lectin and (iii) the alternative pathway, of which often more than one are activated by pathogen recognition by for instance, IgG, IgM, C-reactive protein (CRP) or mannose binding lectin. All three pathways eventually result in a cleavage of complement factor C3 leading to deposition of C3b on the bacterial surface and subsequent opsonophagocytosis by phagocytes, or direct killing through recruitment of additional complement components and the formation of the membrane-attack complex (Hallstrom and Riesbeck, 2010). Additionally, complement cleavage products C3a and C5a are potent activators of the innate immune response attracting phagocytic immune cells (Klos et al., 2009).
Because complement activation has such dramatic effects on immune activation, it is highly regulated to prevent damage to host tissue. Complement activation is regulated by complement control proteins, which include, among others, C4b-binding protein (C4BP) and factor H (fH). C4b-binding protein inhibits activation of both the classical and the lectin complement pathway by acting as a cofactor for factor I, which mediates degradation of C4b and thereby accelerates the decay of the C3 convertase C4b2a (Blom et al., 2004). Factor H regulates activation of the alternative pathway by inhibiting the formation and accelerating the decay of the alternative pathway C3 convertase C3bBp. In addition, fH also acts as a cofactor for the factor I-mediated cleavage of C3b (Rodriguez de Cordoba et al., 2004).
Many pathogenic bacteria exploit complement control proteins to prevent complement deposition on their cell surface. NTHi was shown to bind C4BP, which increased its resistance to complement-mediated killing (Hallstrom et al., 2007). In addition, NTHi was also shown to bind fH, which prevents activation of the alternative complement pathway (Hallstrom et al., 2008).
We used Tn-seq (van Opijnen et al., 2009) analysis in the identification of genes that contribute to resistance to complement-mediated killing. We identified that, among others, transposon insertions in the ompP5 gene coding for outer membrane protein P5 (OmpP5) attenuated survival of serum-resistant NTHi strain R2866 in RPMI medium containing active human complement. OmpP5 is a member of the OmpA protein family that has shown to bind carcino-embryonic antigen-related cell adhesion molecule 1 (CEACAM-1) on human epithelial cells (Hill et al., 2001). Recently, Rosadini et al. showed decreased factor H binding to NTHi strain NT127 lacking OmpP5 by flow cytometry, which contributed to increased complement-mediated killing (Rosadini et al., 2014). In this study, we confirmed a direct role for fH binding to resistance to complement-mediated killing. In addition, we present evidence that the heterogeneity in OmpP5 exposed outer membrane loops determines strain-specific levels of fH binding.
Results
NTHi OmpP5 contributes to complement resistance
In an effort to identify NTHi genes important for resistance to complement-mediated killing, a library of mariner transposon mutants of a serum-resistant NTHi strain R2866 were incubated with 10% normal human serum (NHS) containing active complement or heat-inactivated (HI)-NHS to remove complement activity. Genes contributing to complement resistance were identified by transposon sequencing (Tn-seq). A total of 12 genes with at least 2 transposon insertions and 500 pseudoreads in the control condition were under-represented in the library after exposure to active complement (Table 1), indicating that these genes contribute to resistance to complement-mediated killing. R2866_0112 was found to contribute most significantly to survival of NTHi strain R2866 in the presence of active complement, which was in accordance with our previous genome-wide mariner transposon mutants screen with genomic array footprinting (GAF) as read-out (Langereis et al., 2012). Other genes (lgtF, lgtC, losB2, lic2A, neuA, siaP, siaT and mlaE) that were identified in our screen, were earlier found to be involved in resistance to complement-mediated killing (Hood et al., 1999; 2004; 2010; Ho et al., 2007; Nakamura et al., 2011; Morey et al., 2013). Furthermore, two genes were identified for which a role in complement resistance was not established previously (lapB [R2866_1281] and clpB [R2866_1533]). Interestingly, gene R2866_1237, coding for outer membrane protein P5 (OmpP5) (also known as OmpA), was shown to have a prominent role in resistance to complement-mediated killing (Table 1).
Table 1.
R2866 mutant library challenged with or without active human complement for 2 h.
| Total pseudoreads
|
Fold change | No. insertions | Adj. P-value | Gene locus | Gene name | Product | |
|---|---|---|---|---|---|---|---|
| HI-NHS | NHS | ||||||
| 19 239 | 186 | −104.2 | 21 | 7.0E-39 | R2866_0112 | – | Hypothetical protein |
| 1 813 | 25 | −72.2 | 12 | 5.0E-28 | R2866_1237 | ompP5 | Outer membrane protein P5 |
| 12 248 | 240 | −51.0 | 15 | 1.6E-13 | R2866_1822 | lgtF | UDP-glucose-lipooligosaccharide beta 1–4 glucosyltransferase |
| 50 115 | 1 099 | −45.6 | 33 | 2.3E-31 | R2866_0326 | lgtC | 1,4-Alpha-galactosyltransferase (LgtC) |
| 21 445 | 594 | −36.1 | 11 | 7.7E-22 | R2866_1281 | lapB | Hypothetical protein |
| 100 524 | 5 027 | −20.0 | 32 | 9.1E-23 | R2866_1533 | clpB | ATP-dependent Clp protease ATPase subunit |
| 4 239 | 274 | −15.5 | 9 | 2.3E-18 | R2866_0441 | siaP | Sialic acid TRAP transporter, periplasmic-binding protein SiaP |
| 62 078 | 4 213 | −14.7 | 25 | 2.2E-21 | R2866_1524 | losB2 | D-glycero-D-manno-heptosyltransferase LosB2 |
| 36 319 | 2 792 | −13.0 | 13 | 3.3E-14 | R2866_1314 | mlaE | Phospholipid ABC transporter, permease component |
| 29 391 | 2 315 | −12.7 | 7 | 1.5E-17 | R2866_0861 | neuA | N-acylneuraminate cytidylyltransferase |
| 3 355 | 304 | −11.0 | 20 | 1.6E-15 | R2866_0033 | lic2A | Lipooligosaccharide biosynthesis protein Lic2A |
| 46 566 | 4 572 | −10.2 | 28 | 6.3E-16 | R2866_0440 | siaT | Sialic acid TRAP transporter, fused permease protein SiaT |
Genes with > 10-fold decrease in NHS total pseudoreads compared to HI-NHS P < 0.05 and > 2 transposon insertions with 500 pseudoreads were selected.
In order to validate the role for OmpP5 in resistance to complement-mediated killing, we replaced the complete ompP5 gene with a spectinomycin cassette, which was confirmed by PCR (data not shown). The R2866 OmpP5 protein (358 amino acids) showed a similar migration as the highly abundant OmpP2 protein (369 amino acids), therefore, the lack of OmpP5 expression could not be determined by Coomassie stain, but Western blot analysis with specific α-OmpP5 antibody showed absence of the OmpP5 protein (Fig. 1A). No difference in growth or lipooligosaccharide (LOS) profile was observed between R2866ΔompP5 and R2866 WT (data not shown). However, deletion of the ompP5 gene rendered NTHi strain R2866 significantly more sensitive to complement-mediated killing in 10% NHS containing active complement compared to incubation in 10% HI-NHS (Fig. 1B). Taken together, these results show that OmpP5 has an important role in resistance to complement-mediated killing by NHS.
Fig. 1.

Outer membrane protein P5 contributes to complement resistance of NTHi strain R2866. The presence of OmpP5 on NTHi strains R2866 and R2866ΔompP5 mutant was detected by Coomassie stain and Western analysis with rabbit polyclonal α-OmpP5 serum (A). NTHi R2866 and R2866ΔompP5 mutant (1·105 ml−1) were incubated in 10% normal human serum (NHS) or 10% heat inactivated (HI)-NHS for 1 h. Colony-forming unit (cfu) counts were determined by plating serial dilutions on sBHI agar plates (n = 4) and % survival in NHS compared to HI-NHS was calculated (B). Statistical significance was determined with an unpaired Student’s t-test. *P < 0.05.
Complement-mediated killing of R2866ΔompP5 is dependent on both classical and alternative complement pathway activation
Activation of the classical pathway by NTHi is largely dependent on recognition by IgG or IgM and binding of CRP to phosphorylcholine on the surface of the bacteria. Therefore, we compared binding of IgG, IgM and the presence of phosphorylcholine for the R2866 WT and R2866ΔompP5 mutant strains. Binding of IgG, IgM and the incorporation of phosphorylcholine, as detected by the monoclonal antibody TEPC-15 using flow cytometry, were not significantly different between the R2866 WT and R2866ΔompP5 mutant strains (Fig. 2A–C). However, in accordance with increased complement-mediated killing of the R2866ΔompP5, we observed a significant approximately threefold increase in C3 deposition on the R2866ΔompP5 mutant compared to the R2866 WT strain (Fig. 2D).
Fig. 2.

Effect of outer membrane protein P5 on IgM, IgG, phosphorycholine and C3 binding to the bacterial surface. NTHi R2866 and R2866ΔompP5 mutant were incubated for 15 min in media alone (Ab control) or in media containing 5% NHS or TEPC-15 that recognizes phosphorylcholine. Binding of IgG (n = 7) (A), IgM (n = 6) (B), TEPC-15 (n = 7) (C) or complement factor C3 (n = 6) (D) to the bacterial surface was determined by flow cytometry and compared by measurement of geometric mean fluorescence intensity (MFI) in arbitrary units (AU). NTHi R2866 and R2866ΔompP5 mutant were incubated for 15 min in 5% NHS, 5% C1q-depleted NHS or 5% factor B-depleted NHS and binding of C3 was determined by flow cytometry and compared by measurement of geometric mean fluorescence intensity (MFI) in arbitrary units (AU) (n = 4) (E). Statistical significance was determined with an unpaired Student’s t-test (A–D) or with a two-way ANOVA and the Bonferroni post hoc test (E). NS, not significant; *P < 0.05.
To further analyse the contribution of the different pathways of complement activation, we determined C3 binding with NHS, C1q-depleted NHS (to eliminate the classical complement pathway) and fB-depleted NHS (to eliminate the alternative complement pathway). The relative binding of IgG or IgM was different depending on the source of serum used, but was not significantly different between R2866 WT or R2866ΔompP5 mutant strain for each serum source tested (Supplemental Fig. S1). Depletion of C1q from NHS completely abrogated C3 deposition on the bacterial surface of strains R2866 WT and R2866ΔompP5 mutant, indicating that activation of the classical complement pathway is essential in deposition of C3 on the bacterial surface. Serum depleted of fB showed C3 binding to the bacterial surface, but no longer demonstrated increased C3 binding to the R2866ΔompP5 mutant as compared to the R2866 WT (Fig. 2E). These data show that initial C3 deposition on the bacterial surface of R2866 WT and R2866ΔompP5 mutant is equal, which corresponds with similar levels of IgG and IgM binding. However, deletion of ompP5 gene renders NTHi more sensitive to alternative complement pathway amplification, which increases C3 deposition and subsequent complement-mediated killing.
NTHi ΔompP5 strains show decreased factor H binding and complement resistance
Since increased C3 deposition on the surface of the R2866ΔompP5 mutant strain was dependent on activation of the alternative pathway of complement, we addressed whether OmpP5 might bind fH, an important regulator of alternative complement pathway activation that was reported to bind NTHi previously (Hallstrom et al., 2008). The level of fH binding as quantified by flow cytometry varied between NTHi isolates and binding reached saturation with 5% and 10% NHS (Fig. 3A), whereas OmpP5 protein expression levels as detected by Western blot analysis were not significantly different (Supplemental Fig. 2). Binding of fH to various pathogenic bacteria has been shown to be human specific (Ngampasutadol et al., 2008; Granoff et al., 2009). For mouse fH binding experiments, we used strain 11P6H because of its high binding capacity, thereby, increasing assay sensitivity. Whereas purified human fH was able to bind NTHi strain 11P6H, no binding of recombinant mouse fH was detected to this strain (Fig. 3B). Therefore, we concluded that, similar to other bacterial species, host specificity is an important determinant for fH binding to NTHi.
Fig. 3.

Human factor H binds NTHi in a strain-dependent manner and contributes to complement resistance. Surface binding of factor H to strains Rd, R2866, 3655 and 11P6H was determined by incubating bacteria in media with 1%, 2%, 5% or 10% HI-NHS for 1 h and levels of fH binding was measured by flow cytometry and compared by measurement of geometric mean fluorescence intensity (MFI) in arbitrary units (AU) (n = 5) (A). NTHi strain 11P6H was incubated 1 h with purified human fH (5 μg ml−1) or recombinant mouse fH (5 μg ml−1) and binding was measured by flow cytometry and compared by measurement of geometric mean fluorescence intensity (MFI) in arbitrary units (AU) (n = 4) (B). Outer membrane protein P5 mutants were constructed in strains Rd, R2866, 3655 and 11P6H. Surface binding of factor H was determined by incubating bacteria in medium with 10% HI-NHS for 1 h and detected by flow cytometry and compared by measurement of geometric mean fluorescence intensity (MFI) in arbitrary units (AU) (n = 4) (C). Resistance to complement-mediated killing was determined by incubating the WT and ΔompP5 mutant (1·105 ml−1) in media with 5% NHS or 5% HI-NHS for 1 h. Colony-forming unit (cfu) counts were determined by plating serial dilutions on sBHI agar plates and % survival in NHS compared to HI-NHS was calculated (n = 3) (D). Statistical significance was determined with an unpaired Student’s t-test. NS, not significant; *P < 0.05; **P < 0.01; ***P < 0.001.
In order to confirm that expression of OmpP5 is needed for fH binding and complement resistance, we measured fH binding to WT and ΔompP5 mutants (absence of OmpP5 was confirmed by Western blotting analysis, Supplemental Fig. S3) of strains Rd, R2866, 3655 and 11P6H. Deletion of the ompP5 gene from strains Rd, R2866, 3655, 11P6H decreased fH binding significantly (Fig. 3C). Interestingly, fH binding to strains 3655 and 11P6H was only decreased by ~ 50%, indicating that other strain-specific factors besides OmpP5 may contribute to fH binding.
Next, we determined whether complement resistance correlated with OmpP5 expression among strains. For these assays, we used 5% NHS because resistance to complement-mediated killing of the different strains varied extensively. Consistent with the observed decrease in fH binding, ΔompP5 mutants for NTHi strains Rd, R2866, 3655 and 11P6H showed diminished resistance to complement-mediated killing (Fig. 3D).
Binding of factor H to OmpP5 is independent of incorporation of sialic acid into the LOS structure
Phase-variable incorporation of sialic acid into the LOS of NTHi increases resistance to alternative complement pathway activation (Figueira et al., 2007) and has been implicated in affecting the binding of fH by other pathogens, including Histophilus somni (Inzana et al., 2012), Neisseria meningitidis (Lewis et al., 2012) and Neisseria gonorrhoeae (Ram et al., 1998). In order to assess the role for incorporation of sialic acid into the LOS on complement resistance and binding of fH, we grew NTHi in RPMI medium with or without N-Acetylneuraminic acid (Neu5Ac). For these experiments, we selected strain 3655 because the ΔompP5 mutant showed residual fH binding when grown in sBHI (Fig. 3C) and significant decrease in complement resistance (Fig. 3D). NTHi 3655 WT and the 3655ΔompP5 mutant strains grown in RPMI with Neu5Ac showed significantly increased resistance to complement-mediated killing in 5% NHS compared to strains grown in RPMI without Neu5Ac (Fig. 4A). This observation indicated that incorporation of sialic acid increased resistance to complement-mediated killing independently of the presence of OmpP5. However, no difference in fH binding was observed for both the 3655 WT and the 3655ΔompP5 mutant strains grown with or without Neu5Ac (Fig. 4B), indicating that sialylation of the LOS does not affect fH binding. Taken together, growth of NTHi in RPMI with Neu5Ac increased resistance to complement-mediated killing in an fH-independent manner.
Fig. 4.
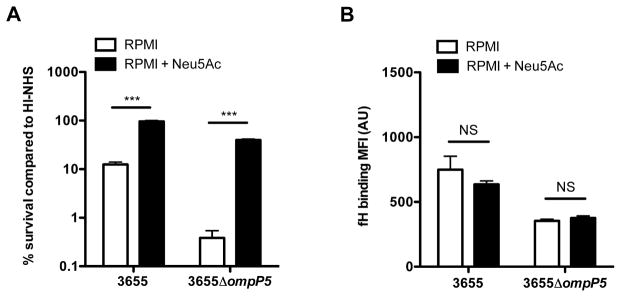
Binding of fH is independent of the incorporation of sialic acid. NTHi strains 3655 and 3655ΔompP5 mutant were grown in RPMI with or without 100 μg ml−1 N-acetylneuraminic acid (Neu5Ac). Resistance to complement-mediated killing was determined by incubating the 3655 WT and 3655 ΔompP5 mutant (1·105 ml−1) in media with 5% NHS or 5% HI-NHS for 1 h. Colony-forming unit (cfu) counts were determined by plating serial dilutions on sBHI agar plates and % survival in NHS compared to HI-NHS was calculated (n = 3) (A). Surface binding of factor H was performed by incubating bacteria in media with 10% HI-NHS for 1 h and was detected by flow cytometry and compared by measurement of geometric mean fluorescence intensity (MFI) in arbitrary units (AU) (n = 3) (B). Statistical significance was determined with a two-way ANOVA and the Bonferroni post hoc test. NS, not significant; ***P < 0.001.
Factor H binds OmpP5 directly
Although it is clear that fH binding to ΔompP5 mutants is significantly decreased, we wanted to establish whether fH binds directly to ompP5. We determined whether a highly specific polyclonal antibody (no detection of NTHi proteins for ΔompP5 mutants in Fig. 1A and Supplemental Fig. S3) recognizing OmpP5 was able to block fH binding. Incubation of NTHi strain 11P6H with increasing amounts of α-OmpP5 antibody showed increased binding of rabbit IgG antibody to the bacterial surface, as measured by flow cytometry (Fig. 5A). Increasing amounts of bound α-OmpP5 antibody decreased fH binding to 11P6H, but not for 11P6HΔompP5 (Fig. 5B), indicating that fH binding to OmpP5 is blocked specifically.
Fig. 5.

Outer membrane protein P5 binds human fH directly. NTHi strain 11P6H was incubated for 30 min with different concentrations rabbit polyclonal α-OmpP5 serum and surface binding of factor H was subsequently determined by incubating bacteria in media with 10% HI-NHS for 1 h. Binding of rabbit IgG (n = 3) (A) and human factor H (n = 3) (B) was determined by flow cytometry and compared by measurement of geometric mean fluorescence intensity (MFI) in arbitrary units (AU). Recombinant R2866 OmpP5 protein (indicated with *) or bovine serum albumin (BSA) (indicated with #) as control was separated by SDS-PAGE and fH binding was detected by far-Western analysis. For blocking studies, NHS was pre-incubated with heparin sulphate (C) or recombinant R2866 OmpP5 protein (D). Resistance to complement-mediated killing was determined by incubating the bacteria (1·105 ml−1) in media with 10% NHS, which was pre-incubated for 15 min with recombinant R2866 OmpP5 protein, for 1 h. Colony-forming unit (cfu) counts were determined by plating serial dilutions on sBHI agar plates and % survival in NHS compared to HI-NHS was calculated (n = 6) (E). Statistical significance was determined with one-way analysis of variance and the Tukey post hoc test. NS, not significant; **P < 0.01; ***P < 0.001.
In order to confirm that fH binds OmpP5 directly, we used recombinant R2866 OmpP5 protein in far-Western analysis. We observed clear binding of fH to recombinant R2866 OmpP5 protein, whereas no binding was observed to similar amounts of the bovine serum albumin control (Fig. 5C and D). Pre-incubation of NHS with heparin sulphate (Fig. 5C), previously shown to compete with fH binding (Hallstrom et al., 2008), or recombinant R2866 OmpP5 protein (Fig. 5D) blocked binding of fH to recombinant R2866 OmpP5 protein in the far-Western analysis. From these observations, we conclude that OmpP5 is sufficient to bind human fH.
Next, we wondered whether recombinant OmpP5 protein could compete with factor H in human serum by competing with factor H binding to the bacterial surface and decreasing resistance to complement-mediated killing. As shown in Fig. 5E, pre-incubation of NHS with recombinant OmpP5 protein increased complement-mediated killing of the R2866 WT, whereas no effect was seen on killing of the R2866ΔompP5 mutant strain. Therefore, we conclude that recombinant OmpP5 can compete with factor H binding to the bacterial surface and thereby blocks its effect on resistance to complement-mediated killing.
Heterogeneity in OmpP5 influences binding of fH, but not resistance to complement-mediated killing for strain Rd
Binding of fH varied among different NTHi strains, an effect that might be due to heterogeneity in amino acid sequence of the four predicted surface-exposed loops (Duim et al., 1997; Webb and Cripps, 1998). In order to determine whether differences in fH binding are dependent on differences in amino acid sequence variation of OmpP5 proteins (see Supplemental Fig. S4), we expressed OmpP5 from strains Rd, R2866 and 3655 on a low-copy plasmid in the OmpP5-deficient strain RdΔompP5. Expression levels of OmpP5 from strains Rd and complemented RdΔompP5 mutants differed less than 1.5-fold, whereas no expression was observed for RdΔompP5 (Fig. 6A). Although RdΔompP5 complemented with ompP5 gene from R2866 and 3655 showed the lowest OmpP5 protein expression, these bound significantly more fH compared to the Rd WT or RdΔompP5 complemented with OmpP5 from Rd (Fig. 6B), indicating that amino acid differences in the OmpP5 protein affect the level of fH binding.
Fig. 6.

Heterogeneity in outer membrane protein P5 contributes to differences on factor H binding among NTHi strains. The gene encoding outer membrane protein P5 from strains Rd, R2866 and 3655 were cloned into low-copy plasmid pACYC177 and expressed in RdΔompP5. The presence of OmpP5 was detected by Western analysis with rabbit polyclonal α-OmpP5 serum and Coomassie stain as loading control (A). OmpP5 spot intensity was quantified with ImageJ software (Schneider et al., 2012) and relative spot intensity was calculated by dividing the spot intensities with the spot intensity of Rd WT (n = 3). Surface binding of factor H was performed by incubating bacteria in media with 10% HI-NHS for 1 h and detected by flow cytometry and compared by measurement of geometric mean fluorescence intensity (MFI) in arbitrary units (AU) (n = 6) (B). Resistance to complement-mediated killing was determined by incubating the bacteria (1·105 ml−1) in media with 5% NHS or 5% HI-NHS for 30 min. Colony-forming unit (cfu) counts were determined by plating serial dilutions on sBHI agar plates and % survival in NHS compared to HI-NHS was calculated (n = 6) (C). Statistical significance was determined with an unpaired Student’s t-test. NS, not significant; *P < 0.05; **P < 0.01; ***P < 0.001.
Next, we determined resistance to complement-mediated killing of the Rd WT, RdΔompP5 mutant and the complemented RdΔompP5 mutant strains with 5% NHS for 30 min. As observed before, the RdΔompP5 mutant was more sensitive to complement-mediated killing (Fig. 6C). Complementation of the RdΔompP5 mutant with OmpP5 from strains Rd, R2866 and 3655 increased survival significantly. Interestingly, no statistical differences in survival was observed between the three different OmpP5 proteins, indicating that activation of the alternative complement pathway was already inhibited by the level of fH binding to OmpP5 from Rd, and increased fH binding to OmpP5 from R2866 and 3655 could not increase survival of strain Rd any further. Taken together, our data show that OmpP5 is necessary for fH binding for strain Rd. Moreover, our findings show that heterogeneity in OmpP5 protein is sufficient to affect the level of fH binding.
OmpP5 outer membrane loop 1 and 2 contribute to factor H binding to strain R2866
It is expected that fH binds to the exposed outer membrane loops of OmpP5. In order to determine binding of fH to specific outer membrane loops of OmpP5, we truncated the recombinant R2866 OmpP5 protein (Fig. 7A). Far Western analysis showed decreased fH binding to R2866 OmpP5 lacking outer membrane loop 1 (R2866_OmpP5Δ1) and R2866 OmpP5 lacking loop 2 through 4 (R2866_OmpP5Δ2–4), as compared to the R2866 full-length OmpP5, whereas binding to R2866 OmpP5 lacking loops 3 and 4 (R2866_OmpP5Δ3–4) showed similar binding compared to the full-length R2866 OmpP5 (Fig. 7B). These data indicate that outer membrane loops 1 and 2, but not loop 3 and 4, contribute to fH binding to the R2866 OmpP5 protein as detected by far Western analysis.
Fig. 7.

Outer membrane loop 1 and 2 contributes to factor H binding to R2866 OmpP5. Schematic representation of the R2866 OmpP5 proteins used in the far Western and complementation experiments (A). Recombinant R2866 OmpP5 proteins and bovine serum albumin (BSA) as control were separated by SDS-PAGE and fH binding was detected by far-Western analysis. Western blot with α-His6 antibody was used as loading control. Factor H binding to recombinant R2866 OmpP5 proteins was quantified with ImageJ software (Schneider et al., 2012) and relative spot intensity was calculated by dividing the fH spot intensity with the His6 spot intensity and normalized to R2866 OmpP5 WT protein (n = 7) (B). The presence of OmpP5 was detected by Western analysis with rabbit polyclonal α-OmpP5 serum and Coomassie stain as loading control. OmpP5 spot intensity was quantified with ImageJ software (Schneider et al., 2012) and relative spot intensity was calculated by dividing the spot intensities with the spot intensity of Rd WT (n = 3) (C). Surface binding of factor H was performed by incubating bacteria in media with 10% HI-NHS for 1 h and detected by flow cytometry and compared by measurement of geometric mean fluorescence intensity (MFI) in arbitrary units (AU) (n = 6) (D). Statistical significance was determined with an unpaired Student’s t-test. NS, not significant; *P < 0.05; **P < 0.01; ***P < 0.001.
In order to show that increased fH binding to R2866 compared to Rd was dependent on outer membrane loop 1 and 2, we complemented the RdΔompP5 with a chimeric OmpP5 consisted of R2866 loop 1–2 and Rd loop 3–4 (R2866(1–2)-Rd(3–4)) (Fig. 7A). Complementation of the RdΔompP5 showed similar OmpP5 protein expression as Rd or R2866 WT (Fig. 7C). Binding of fH to RdΔompP5 complemented with chimeric R2866(1–2)-Rd(3–4) was increased compared to the Rd WT or RdΔompP5, but similar as that of RdΔompP5 complemented with R2866 or the R2866 WT strain (Fig. 7D), indicating that loop 1 and 2 are sufficient to bind fH to a similar level as the R2866 OmpP5 protein. Altogether, we show that increased fH binding to R2866 compared to Rd OmpP5 is dependent on outer membrane loop 1 and loop 2.
Discussion
Resistance to complement-mediated killing varies largely between NTHi isolates. Complement resistance is determined by a variety of mechanisms that for instance result in decreased binding of IgG by incorporation of phosphorylcholine (Clark et al., 2012), decreased binding of IgM through exclusion of phospholipids from the outer leaflet of the bacterial membrane or modulation of its LOS structure (Nakamura et al., 2011; Langereis et al., 2012), or recruitment of host complement regulatory proteins such as C4BP and factor H resulting in decreased C3 deposition (Hallstrom et al., 2007; 2008). Complement resistance of NTHi is associated with the ability to survive the inflammatory response at the mucosa of the middle ear cavity and the lungs of COPD patients (Nakamura et al., 2011; Langereis et al., 2012). This is supported by in vivo experiments where complement sensitive NTHi strains showed decreased ability to cause otitis media in chinchillas (Figueira et al., 2007) as well as in mice (Langereis et al., 2012) compared to more complement resistant strains. We have previously used genomic array footprinting (GAF) to identify genes important for NTHi strain R2866 to withstand complement-mediated killing in 40% normal human serum (NHS) (Langereis et al., 2012). In this present study, we have exposed the R2866 mutant library to 10% NHS for 2 h and identified the genes contributing to withstand complement-mediated killing by Tn-seq (Table 1). Tn-seq has several advantages over the use of GAF. First, the transposon insertion site can accurately be determined as opposed to GAF in which the region of insertion (~ 200 bp) can be identified, which makes Tn-seq more precise in the identification of the transposon-disrupted genes. Also, the sequence read counts obtained with Tn-seq are a better quantitative measure compared to cDNA detection on microarrays for GAF, which makes Tn-seq more sensitive and enables a better prioritization of the identified genes important for resistance to complement-mediated killing. Twenty-four out of 57 genes identified in the previous GAF analysis were found to be attenuated at least twofold with a P < 0.01 in the Tn-seq analysis. Various genes related to LOS biosynthesis (lgtF, lgtC, losB2 and lic2A), incorporation of sialic acid in the LOS structure (neuA, siaP and siaT) and phospholipid ABC transporter permease component (mlaE) that were previously shown to be involved in resistance to complement-mediated killing (Hood et al., 1999; 2004; 2010; Ho et al., 2007; Nakamura et al., 2011; Morey et al., 2013) were identified in this screen, which shows the power of this genome-wide screening technique. The role for the genes lapB (R2866_1281) or clpB (R2866_1533) in resistance to complement-mediated killing is currently not known.
The R2866 WT strain bound substantial levels of C3 as detected by flow cytometry and C3 binding was significantly increased for the R2866ΔompP5 mutant (Fig. 2D). Therefore, it appears that C3 deposition on the bacterial surface of R2866 WT is not sufficient to initiate complement-mediated lysis of the R2866 WT strain, whereas increased C3 deposition on the R2866ΔompP5 mutant exceeded the threshold needed to initiate complement-mediated killing. Binding of C3 to the bacterial surface of the R2866ΔompP5 mutant appears to be dependent on activation of the classical complement pathway and amplification through the alternative complement pathway. This is supported by our experiments where depletion of C1q, which is an essential factor in activation of the classical complement pathway, abrogated C3 deposition to the bacterial surface of the R2866 WT and R2866ΔompP5 mutant strains completely. Inactivation of alternative complement pathway activation by fB depletion showed C3 deposition on the bacterial surface of both the R2866 WT and R2866ΔompP5 mutant through activation of the classical complement pathway, but no increase in C3 deposition was observed for the R2866ΔompP5 mutant. Therefore, we conclude that OmpP5 contributes in evasion of the alternative pathway of complement activation, which prevents amplification of C3 deposition on the bacterial surface.
Deletion of ompP5 decreased fH binding to strains Rd, R2866, 3655 and 11P6H significantly, which prompted us to determine whether OmpP5 might bind complement control protein fH. Hallstrom et al. was the first to show that binding of fH to the surface of H. influenzae decreased complement activation (Hallstrom et al., 2008). Two proteins of ~ 32 and 40 kDa were shown to bind radiolabelled fH in a far Western analysis, of which one is likely OmpP5. Since we detected significant residual binding of fH to strains 3655 and 11P6H, it is possible that another unknown factor contributes to fH binding in these strains.
We questioned whether sialic acid incorporation might be contributing because it has been implicated in the binding of fH by other pathogens (Ram et al., 1998; Inzana et al., 2012; Lewis et al., 2012) and phase-variable incorporation of sialic acid into the LOS of NTHi increases resistance to activation of the alternative complement pathway (Figueira et al., 2007). However, fH binding to strain 3655 was independent of incorporation of sialic acids into the LOS structure, whereas increased complement resistance was increased. Therefore, at least for NTHi strain 3655, sialylation of the LOS has no effect on fH binding.
Recently, Rosadini et al. presented evidence that OmpP5 is required for resistance to complement-mediated killing initiated via the classical and alternative complement pathway (Rosadini et al., 2014). In this study, we present evidence that (i) OmpP5 contributes in evasion of the alternative pathway of complement activation, (ii) OmpP5 is sufficient for fH binding, and (iii) heterogeneity in OmpP5 exposed outer membrane loops 1 and 2 determines the level of fH binding. In accordance with the study by Rosa-dini et al., we observed decreased complement resistance for the ompP5 gene deletion in strain Rd. However, in contrast to the study by Rosadini et al., we observed a small decrease in fH binding to the OmpP5 mutant in Rd (Fig. 4A). This might be explained by longer incubation time (60 min versus 30 min) for the bacteria with a higher proportion of NHS (10% versus 5%) and the use of different antibodies for flow cytometry. In contrast to the observations described by Rosadini and co-workers, we have not observed differences in IgM binding between the R2866 WT and ompP5 gene mutant strain, which might be due to the use of a different NTHi strain in our study.
Although we provide evidence that outer membrane loop 1 and loop 2 of OmpP5 contribute to fH binding to strain R2866, the exact amino acids responsible for this interaction remains unknown. As reported before (Webb and Cripps, 1998), there is substantial heterogeneity in amino acid sequence for the different predicted outer membrane loops of strains Rd, R2866, 3655 and 11P6H (Supplemental Fig. S4). The first 37 amino acids among the four strains used in this study were highly conserved (Supplemental Fig. S4). However, part of the first extracellular loop of strain Rd (LRALAREYKV) was substantially different and smaller compared to those from strain 3655 (INNNGAIKEALTSASY), strain R2866 (INNNGAI-KEDLSLGY) and strain 11P6H (INNNGAIKNDLLELTSY). In addition, 4 of the 19 amino acids of the predicted Rd loop 2 are non-conserved compared to R2866, 3655 or 11P6H. Heterogeneity in OmpP5 among NTHi strains has previously shown to determine CEACAM1 binding (Hill et al., 2001). Binding of CEACAM1 to strain NT2 was absent, but introduction of OmpP5 gene from strain Rd in the OmpP5 deletion mutant of NT2 increased binding of soluble CEACAM1, as well as adhesion to CHO cells expressing CEACAM1 (Hill et al., 2001). Therefore, it is tempting to speculate that the amino acid sequence or size of loop 1 and loop 2 determines the level of fH binding. This is supported by our far Western analysis and complementation experiments where we used three wild-type or R2866(1–2)-Rd(3–4) chimeric OmpP5 proteins expressed in RdΔompP5 as genetic background. We observed significantly increased fH binding to OmpP5 from strains R2866, 3655 and the R2866(1–2)-Rd(3–4) chimeric protein compared to the Rd wild-type OmpP5 protein, despite the similar level of OmpP5 protein expression.
Other respiratory pathogens such as Streptococcus pneumoniae and Moraxella catarrhalis also bind fH from human serum (Neeleman et al., 1999; Bernhard et al., 2014). Recently, M. catarrhalis was shown to bind fH to Opa-like protein A (OlpA) (Bernhard et al., 2014), whereas fH binding to S. pneumoniae is mediated through pneumococcal surface protein C (PspC) (Dave et al., 2001). Although the binding site of fH for all these pathogens appears to be short consensus repeat (SCR)20 (Meri et al., 2013), no common similarities on the bacterial side are found, pointing to distinct mechanisms for immune evasion evolved for the different bacterial species.
Although NTHi is rarely found in the blood causing invasive disease, it has developed strategies to utilize fH in preventing complement activation, indicating that these mechanisms might also contribute during non-invasive disease or colonization. Factor H is found to be present in the middle ear fluid of patients with OM at levels similar or higher than found in NHS (Narkio-Makela et al., 2001). Therefore, binding of fH might not only prevent complement-mediated killing in NHS, but also when present at inflammatory sites.
Since resistance to complement-mediated killing seems to be an important virulence mechanism for NTHi, it is considered an interesting target for antimicrobial compounds and vaccines. For instance, in the case for N. meningitidis type b, the fH-binding protein (fHbp) is one of the four protein components in the 4CMenB (Bexsero) vaccine (Serruto et al., 2012). The antibody response induced by the 4CMenB vaccine appears to have two modes of action: (i) direct activation of the classical complement pathway and (ii) preventing binding of fH on the bacterial surface (Serruto et al., 2012). Interestingly, for NTHi, OmpP5 is considered a vaccine target (Novotny et al., 2008), which is increasingly attractive with the understanding that OmpP5 contributes to complement resistance through binding of human fH. Our identification of factors responsible for complement resistance, including OmpP5, might identify other novel target for antimicrobial therapeutics.
Experimental procedures
Bacterial strains and growth conditions
NTHi strains used in this study are listed in Table 2. NTHi was routinely grown shaking at 37°C in brain heart infusion (BHI) broth (Becton Dickinson) supplemented with 10 μg ml−1 haemin (Sigma-Aldrich) or 2% Fildes enrichment (Thermo Scientific) and 2 μg ml−1 β-nicotinamide adenine dinucleotide (Merck) (sBHI). Growth on plates was performed on sBHI agar at 37°C + 5% CO2. For experiments with N-Acetylneuraminic acid (Neu5Ac) (Sigma-Aldrich) supplementation, bacteria were grown in supplemented RPMI medium as described previously (Coleman et al., 2003) for ~ 8 generations before use in experiments. Viable bacterial counts were determined by plating serial dilutions in phosphate-buffered saline (PBS) on sBHI agar plates. Optical density was measured at 620 nm. Escherichia coli was grown in Luria–Bertani (LB) medium (Becton Dickinson). For NTHi mutant libraries, gene deletion mutants, and complemented gene deletion mutants were grown in sBHI containing 100 μg ml−1 spectinomycin (Calbiochem) and/or 7 μg ml−1 kanamycin (Fisher Bioreagents). E. coli containing plasmids were grown in LB containing 50μg ml−1 kanamycin.
Table 2.
List of strains, primers and plasmids used in this study.
| Strains/primers/plasmids | Description | Reference |
|---|---|---|
| Strains | ||
| R2866 | NTHi clinical isolate from septic patient | Williams et al. (2001) |
| R2866ΔompP5 | R2866 with ompP5 gene replaced by Specr cassette | This study |
| 3655 | NTHi clinical isolate from patient with acute otitis media | Melhus et al. (1998) |
| 3655ΔompP5 | 3655 with ompP5 gene replaced by Specr cassette | This study |
| 11P6H | NTHi clinical isolate from sputum of an adult COPD patient during an acute exacerbation | Yi et al. (1997) |
| 11P6HΔompP5 | 11P6H with ompP5 gene replaced by Specr cassette | This study |
| Rd | Non-encapsulated type D strain | Fleischmann et al. (1995) |
| RdΔompP5 | Rd with ompP5 gene replaced by Specr cassette | This study |
| RdΔompP5::pACYC177 | Rd with ompP5gene replaced by Specr cassette with empty pACYC177 plasmid | This study |
| RdΔompP5::pACYC177-ompP5_Rd | Rd with ompP5 gene replaced by Specr cassette with pACYC177-ompP5_Rd plasmid | This study |
| RdΔompP5::pACYC177-ompP5_R2866 | Rd with ompP5 gene replaced by Specr cassette with pACYC177-ompP5_R2866 plasmid | This study |
| RdΔompP5::pACYC177-ompP5_3655 | Rd with ompP5 gene replaced by Specr cassette with pACYC177-ompP5_3655 plasmid | This study |
| RdΔompP5::pACYC177-ompP5_ R2866(1–2)-Rd(3–4) | Rd with ompP5 gene replaced by Specr cassette with pACYC177-ompP5_ R2866(1–2)-Rd(3–4) plasmid | This study |
| E. coli JM109 | recA, supE, hsR, Δ(lac-pro) | Yanisch-Perron et al. (1985) |
| E. coli JM109 pACYC177 | E. coli JM109 with empty pACYC177 plasmid | This study |
| E. coli JM109 pACYC177-ompP5_Rd | E. coli JM109 with pACYC177-ompP5_Rd plasmid | This study |
| E. coli JM109 pACYC177-ompP5_R2866 | E. coli JM109 with pACYC177-ompP5_R2866 plasmid | This study |
| E. coli JM109 pACYC177-ompP5_3655 | E. coli JM109 with pACYC177-ompP5_3655 plasmid | This study |
| E. coli JM109 pACYC177-ompP5_R2866(1–2)-Rd(3–4) | E. coli JM109 with pACYC177-ompP5_R2866(1–2)-Rd(3–4) plasmid | This study |
| Primers | ||
| R2866_1237_L1 | TCAACAGAATTGACCGCACT | This study |
| R2866_1237_L2 | CCACTAGTTCTAGAGCGGCCGCTGCTAAACCAGCAACTA | This study |
| R2866_1237_R1 | CTATGGCTTGGCAGGCTTAC | This study |
| R2866_1237_R2 | GCGTCAATTCGAGGGGTATCTTATCGCTTGTCTTGCTCCA | This study |
| R2866_1237_C | GGAAAGATCCTTGACCAGCTT | This study |
| PBpR412_L | GCCGCTCTAGAACTAGTGG | Burghout et al. (2007) |
| PBpR412_R | GATACCCCTCGAATTGACGC | Burghout et al. (2007) |
| PBMrTn9 | CAATGGTTCAGATACGACGAC | de Vries et al. (2013) |
| PBGSF23 | CAAGCAGAAGACGGCATACGAAGACCGGGGACTTATCATCCAACCTGT | de Vries et al. (2013) |
| PBGSF31 | AATGATACGGCGACCACCGAGATCTACACTCTTTCCCTACACGACGCTCTTCCGATCT | de Vries et al. (2013) |
| R2866_1237_F_NheI | GCTAGCATGAAAAAAACTGCAATCGCATT | This study |
| R2866_1237_R_SalI | GTCGACTTATTTAGTACCGTTTACCGCG | This study |
| R2866_1237_F_HincII | CAACGTTGACCTCAAAAGAATTTGATCTTTTCAAT | This study |
| R2866_1237_R_PstI | AAAAGCTGCAGAAACTTGATTAAAATGCTTGGTTTT | This study |
| Plasmids | ||
| pGSF8 | Donor for marinerT7-MmeI, Specr | de Vries et al. (2013) |
| pET28a | Kanr vector with N-terminal His-tag | Novagen |
| pET28a-R2866-ompP5 | pET28a with R2866 ompP5 gene cloned into NheI and SalI sites | This study |
| pET28a-R2866-ompP5Δ1 | pET28a with R2866 ompP5 gene lacking outer membrane loop 1 | This study |
| pET28a-R2866-ompP5Δ3–4 | pET28a with R2866 ompP5 gene lacking outer membrane loops 3 and 4 | This study |
| pET28a-R2866-ompP5Δ2–4 | pET28a with R2866 ompP5 gene lacking outer membrane loops 2 through 4 | This study |
| pACYC177 | Kanr, ampr low-copy cloning vector | Chang and Cohen (1978) |
| pACYC177-ompP5_Rd | pACYC177 with Rd ompP5 gene cloned into HincII and PstI sites | This study |
| pACYC177-ompP5_R2866 | pACYC177 with R2866 ompP5 gene cloned into HincII and PstI sites | This study |
| pACYC177-ompP5_3655 | pACYC177 with 3655 ompP5 gene cloned into HincII and PstI sites | This study |
| pACYC177-ompP5_R2866(1–2)-Rd(3–4) | pACYC177 with ompP5 loops 1 and 2 from R2866 and loops 3 and 4 from Rd | This study |
Generation of the NTHi R2866 transposon mutant library
Genomic DNA was isolated from mid-log-phase cultures with the Qiagen Genomic-tip 20/G (Qiagen). The NTHi R2866 marinerT7-MmeI transposon mutant library was generated as described previously for NTHi 86-028NP (Langereis et al., 2013). Briefly, 1 μg of NTHi R2866 genomic DNA was incubated in the presence of purified HimarC9 transposase and 0.5 μg of plasmid pGSF8 as a donor of the marinerT7-MmeI transposon conferring spectinomycin resistance. After repair of the resulting transposition products with E. coli DNA ligase and T4 DNA polymerase, 500 ng mutagenized DNA was used for NTHi transformation by the method of Herriott et al. (1970). For mutant libraries, the required number of colonies was scraped from the plates, pooled, grown to mid-log phase in sBHI medium supplemented with spectinomycin, and stored in 15% glycerol at −80°C.
Identifying NTHi genes essential for survival in 10% normal human serum
All experiments were conducted with the same batch of pooled normal human serum (NHS) containing active complement obtained from GTI Diagnostics (catalogue No. PHS-N100). Normal human serum was heat-inactivated by incubating 20 min at 56°C.
The NTHI R2866 marinerT7-MmeI transposon mutant library was diluted to 2 × 106 cfu ml−1 in HEPES-buffered RPMI medium containing 1 μg ml−1 haemin and 2 μg ml−1 β-nicotinamide adenine dinucleotide and incubated for 2 h in the presence of 10% heat-inactivated normal human serum (control) or 10% normal human serum in quadruplicate at 37°C. After challenge, the mutant library was grown in sBHI to an OD620 ~ 0.2 (~ 4 h) and chromosomal DNA was isolated for Tn-seq analysis. Generation times were confirmed by viable bacterial counts.
Readout of the mutant library was performed with Tn-seq as described previously (van Opijnen et al., 2009) with minor modifications. Briefly, a 200 μl solution with 2 μg mutant library genomic DNA in NEBuffer 4 (New England Biolabs) with 50 μM S-adenosylmethionine was digested with 10 U MmeI (New England Biolabs) for 4 h at 37°C and dephosphorylated with 1 U calf intestine alkaline phosphatase (Invitrogen) for 30 min at 50°C. Next, the reaction was successively extracted with 200 μl phenol : chloroform : isoamyl alcohol (25:24:1), extracted with 200 μl chloroform : isoamyl alcohol (24:1), ethanol-precipitated, and the dried DNA pellet was dissolved in 20 μl H2O. Tn-seq adapters with a 6 bp barcode were prepared by combining 5 nmol of two matching oligonucleotides in 1× TE and 50 mM NaCl in a total volume of 50 μl, a 10 min denaturation step at 95°C, and an annealing step in which the reaction was slowly cooled to room temperature. A 20 μl solution with 200 pmol adapter was phosphorylated with T4 polynucleotide kinase (3′ phosphatase minus) (New England Biolabs) in T4 DNA ligase buffer (New England Biolabs) for 5 min at 37°C, and heat-inactivated for 10 min at 70°C. Ligation of 100 ng dephosphorylated MmeI restriction fragments with 2 pmol phosphorylated adapter was performed in the presence of T4 DNAligase buffer with 2 U T4 DNAligase (New England Biolabs) in a total volume of 20 μl for 1 h at 16°C. Immediately after the ligation, Tn-seq DNA probes were generated by PCR with 2.5 μl ligation reaction as template, 20 pmol PBGSF23 and PBGSF31 primers (Table 2), HF buffer, 0.2 mM dNTP mix, and 1 U Phusion DNApolymerase in a total volume of 50 μl. PCR cycling conditions were as follows: 72°C for 1 min, 98°C for 30 s; 25 cycles of 98°C for 30 s, 57°C for 30 s, and 72°C for 10 s; and 72°C for 5 min. The resulting PCR product of ~ 130 bp were purified from the PCR reaction with the Minelute Reaction Cleanup Kit (Qiagen). After pooling of samples with up to 12 different 6 bp barcodes, typically 9 fmol Tn-seq DNA probes was loaded on a Genome Analyser II (Illumina) for sequence analysis with the manufacturer’s protocols, using the Genomic DNA Sequencing Primer (Illumina), and 36 sequencing cycles.
Tn-seq data analysis
For Tn-seq data analysis FASTQ files with 35 bp sequences were imported in the web-based interface ESSENTIALS (Zomer et al., 2012). Of note, the first nucleotide of the Genome Analyser II (Illumina) 36 bp sequence reads often has a poor quality and was omitted. As a result, only the last 5 bp of the 6 bp barcode sequence were available for data analysis. To identify these barcodes, a mismatch of 2 bp was allowed. After removal of the barcode and transposon sequences, the length of the remaining transposon-flanking genomic sequence was set at ≤ 17 bp, and alignment of this sequence with the forward strand of the NTHi R2866 reference genome should give a match of at least 16 bp. Count data (i.e. pseudo-reads) were generated per unique sequence read and per gene, and locally weighted scatterplot smoothing corrected for the bias in Tn-seq data caused by the increase in available DNA close to the origin of replication. Normalization factors were calculated using trimmed mean of M-values. Pseudoreads in the control and target samples were tested for significant differences (P ≤ 0.001) by quantile-adjusted conditional maximum likelihood method assuming moderated tagwise dispersion of replicates. The prior.n value to determine the amount of smoothing of tagwise dispersions was set at 10. P-value adjustment (adj. P ≤ 0.05) was based on Benjamini and Hochberg. Gene essentiality was determined by comparing the expected number of reads per gene (based on the number of insertion sites per gene, the mutant library size and the sequencing depth) and the measured number of reads per gene. Significantly under-represented genes were considered essential and omitted from the data analysis. Analysis data can be found at http://bamics2.cmbi.ru.nl/websoftware/essentials/essentials_run.php?session=essentials%2Ffactorh.
Generation of NTHi-directed gene deletion mutants
Bacterial genomic DNA was isolated by cetyltrimethylammonium bromide (CTAB) extraction method (van Soolingen et al., 1994). Directed NTHi gene deletion mutants were generated by allelic exchange of the target gene with a spectinomycin resistance cassette, as described previously (Langereis et al., 2013). Briefly, overlap extension PCR was performed, which inserted the spectinomycin resistance cassette amplified from the pGSF8 plasmid with primers PBpR412_L and PBpR412_R (Table 2) between the two ~1000 bp flanking sequences surrounding the target gene. NTHi mutants were obtained by transformation of the final PCR product by the method of Herriott et al. (1970), selected by plating on sBHI agar plates containing 150 μg ml−1 of spectinomycin, and validated by PCR with primer sets R2866_1237_L1 + R2866_1237_C (Table 2) and R2866_1237_L1 + PBMrTn9 (Table 2) that control for the presence of the gene or spectinomycin cassette respectively. Gene deletions were crossed back to the wild-type strain by using chromosomal DNA of the mutant strains as donor during transformation. All primers (Biolegio, Nijmegen, the Netherlands or Integrated DNA technologies Coralville, IA, USA) used in this study are listed in Table 2.
Flow cytometric analysis
Surface binding of factor H (fH) was performed by incubating bacteria with heat-inactivated normal human serum (NHS) (GTI Diagnostics), 5 μg ml−1 purified human fH (CompTech) or 5 μg ml−1 recombinant mouse fH (produced by HEK293 cells) in Hank’s buffered salt solution (HBSS) without phenol red containing Ca2+/Mg2+ for 1 h at 37°C. Bacteria were fixed in 2% paraformaldehyde, and incubated with 1:100 diluted sheep anti-human fH (Abcam) or 1:100 diluted mouse anti-mouse fH clone 1A2 mAb (Hycult) in HBSS without phenol red containing Ca2+/Mg2+ + 5% FCS for 30 min at 4°C, followed by an incubation with 1:200 diluted donkey anti-sheep-FITC-conjugated antibodies (KPL) or 1:500 diluted goat anti-mouse-Alexa488-conjugated antibodies (Invitrogen) in HBSS without phenol red containing Ca2+/Mg2+ + 5% FCS for 30 min at 4°C. Bacteria were washed and taken up in PBS for flow cytometry.
For human IgG, human IgM, human C3 or TEPC-15 binding, bacteria were incubated with 5% NHS (GTI Diagnostics), 5% C1q-depleted NHS (Quidel), 5% fB-depleted NHS (Quidel) or 1:500-diluted TEPC-15 (Sigma-Aldrich) in HBSS without phenol red containing Ca2+/Mg2+ for 15 min at 37°C. Bacteria were fixed by 20 min incubation with 2% paraformaldehyde. Subsequently, bacteria were washed and incubated with 1:500 diluted FITC-labelled polyclonal goat anti-human C3 (MP biomedicals), 1:100 diluted FITC-labelled Fc-specific goat anti-human IgG (Sigma-Aldrich), 1:50 diluted FITC-labelled μ-chain-specific goat anti-human IgM (Sigma-Aldrich) or 1:100 diluted FITC-labelled α-chain-specific goat anti-mouse IgA (Sigma-Aldrich) for TEPC-15 in HBSS without phenol red containing Ca2+/Mg2+ + 5% FCS for 30 min at 4°C. Bacteria were washed and taken up in PBS for flow cytometry.
For blocking experiments, bacteria were pre-incubated with α-OmpP5 rabbit polyclonal antibody for 15 min at 37°C. Flow cytometry was performed using a FACS LSR II (BD Biosciences). Data were analysed using FlowJo version 7.6.3. Differences in mean fluorescence intensity between experiments were caused by the use of two different FACS LSR II machines and different secondary antibodies.
Western blot analysis
One millilitre of culture of OD620 = 0.6 was washed once with PBS and boiled 5 min in samples buffer (60 mM Tris-HCl pH 6.8, 2% SDS, 2% β-mercaptoethanol, trace bromophenol blue). Whole cell lysates were analysed on Tris-glycine SDS-PAGE gel in a Protean II xi cell electrophoresis system (Bio-Rad) and visualized by Coomassie staining or transferred to nitrocellulose for Western blotting. Membranes were blocked with 5% BSA in PBS for 1 h, incubated 2 h with 1:2000 diluted α-OmpP5 rabbit polyclonal antibody with 0.5% BSA in PBS, washed five times for 5 min with Tris buffered saline + 0.05% Tween-20, incubated with 1:5000 diluted HRP-labelled donkey anti-rabbit Ig (GE Healthcare) and washed five times for 5 min with Tris buffered saline + 0.05% Tween-20. Binding was detected with ECL Western blotting substrate (Pierce) with a Fujifilm LAS-1000 scanner in an intelligent dark box II (Fuji Film). Densitometry analysis was performed with ImageJ (Schneider et al., 2012). Relative spot intensity was calculated by dividing the intensity of the OmpP5 protein as determined by Western blot by the intensity of all proteins in the Coomassie stain as loading control.
Serum survival assays
NTHi was grown in supplemented BHI medium to an OD620 ~0.6, washed once with PBS and diluted to an OD620 of 0.1 in HBSS without phenol red containing Ca2+/Mg2+ + 0.1% gelatin. Samples were finally diluted 10 000-fold in HBSS without phenol red containing Ca2+/Mg2+ + 0.1% gelatin to obtain a concentration of ~ 20 000 cfu ml−1. Fifty microlitres of the bacterial culture were mixed with 50 μl NHS or heat-inactivated (HI)-NHS, diluted in HBSS without phenol red containing Ca2+/Mg2+ + 0.1% gelatin and incubated 30 min or 1 h at 37°C. Serial dilutions were plated on sBHI plates and incubated overnight at 37°C in 5% CO2. Survival was determined by dividing the cfu counts in 5% NHS with the cfu count in HI-NHS after incubation.
ompP5-His6 synthesis and purification
The ompP5 open reading frame was amplified from R2866 genomic DNA with primers containing NheI and SalI restriction enzyme sites (Table 2). The PCR product and pET28a expression vector (Novagen) were digested with NheI and SalI, gel purified and ligated overnight with T4 DNA ligase. The ligation mix was transformed into E. coli BL21 and transformants selected with 50 μg ml−1 kanamycin and checked by sequencing. Expression plasmids with truncated ompP5 were constructed by digestion of pET28a-R2866_ompP5 with NheI + DraI (pET28a-ompP5_R2866Δ1), AflII + NotI (pET28a-ompP5_R2866Δ3–4) or DraI + NotI (pET28a-ompP5_R2866Δ2–4), treated with T4 DNA polymerase (New England Biolabs), gel purified and ligated overnight with T4 DNA ligase (New England Biolabs). The ligation mix was transformed into E. coli BL21 and transformants selected with 50 μg ml−1 kanamycin and confirmed by sequencing. His-tagged NTHi OmpP5 was expressed and purified as described previously (Webb and Cripps, 1999).
R2866-ompP5 far-Western analysis
R2866-ompP5-His6 protein or BSA were boiled 5 min in sample buffer (60 mM Tris-HCl pH 6.8, 2% SDS, 2% β-mercaptoethanol, trace bromophenol blue) and separated on a Tris-glycine SDS-PAGE gel using a Protean II xi cell electrophoresis system (Bio-Rad, Hercules, CA, USA) and visualized by Coomassie staining or transferred to nitrocellulose. Membranes were blocked with 5% BSA in PBS for 1 h, incubated 2 h with 5% NHS, or with 5% NHS pre-incubated 30 min with 100 nM heparin sulphate or recombinant R2866 OmpP5 protein, washed five times for 5 min with Tris buffered saline + 0.05% Tween-20, incubated 1 h with 1:1000 diluted α-fH sheep polyclonal antibody (Abcam) with 0.5% BSA in PBS, washed five times for 5 min with Tris buffered saline + 0.05% Tween-20, incubated with 1:5000 diluted HRP-labelled donkey anti-sheep Ig (Sigma-Aldrich) and washed five times for 5 min with Tris buffered saline + 0.05% Tween-20. Binding was detected with ECL Western blotting substrate (Pierce) with a Fujifilm LAS-1000 scanner in an intelligent dark box II (Fuji Film).
Construction ompP5 complementation plasmid
The ompP5 gene with its endogenous promotor was amplified from genomic DNAof strains Rd, R2866 and 3655 with primers containing HincII and PstI restriction enzyme sites (Table 2). The PCR product and pACYC177 plasmid were digested with HincII and PstI, gel purified and ligated overnight with T4 DNA ligase. The ligation mix was transformed into E. coli JM109 and transformants were selected with 50 μg ml−1 kanamycin. For pACYC177-ompP5_R2866(1–2)-Rd(3–4), pACYC177-ompP5_ R2866 and pACYC177-ompP5_Rd were digested with HindIII, fragments were purified by gel extraction (Qiagen), ligated with T4 ligase (New England Biolabs), transformed into E. coli JM109 and transformants were selected with 50 μg ml−1 kanamycin and confirmed by sequencing. The plasmid was isolated with the Purelink HiPure Plasmid Filter Midiprep Kit (Invitrogen) and 5 μg plasmid was used for NTHi transformation by the method of Herriott et al. (1970) or by electroporation as described previously (Mason et al., 2003). NTHi containing the plasmid was selected with 7 μg ml−1 kanamycin. All primers (Biolegio, Nijmegen, Netherlands or Integrated DNA technologies Coralville, IA, USA) and plasmids used in this study are listed in Table 2.
Sequencing and ClustalW2 alignment
The ompP5 gene was amplified from genomic DNA using primers R2866_1237_F_HincII and R2866_1237_R_PstI (Table 2) and PCR products were purified with QIAquick PCR cleanup kit (Qiagen). Two hundred nanograms of purified PCR product was mixed with 2 pM R2866_1237_F_HincII primer in 8 μl, mixed with 2 μl BigDye Terminator v3.1 Cycle Sequencing Kit (Applied Biosystems) and amplified 25 rounds 10 s at 96°C, 55 s at 55°C and 4 min at 60°C. Sequencing products were ethanol precipitated, dried briefly in a speed-vac, resuspended in Hi-Di formamide and loaded on the 3730 DNA analyser (Applied Biosystem) for capillary electrophoresis and analysis. ClustalW2 alignment was performed with online multiple sequence alignment tool (http://www.ebi.ac.uk/Tools/msa/clustalw2/).
Statistical analysis
Statistical analyses were performed with Graphpad Prism version 5.03 for Windows (GraphPad Software, San Diego, CA) where P < 0.05 was considered significant. The specific statistical tests that were used for the various experiments are specified in the figure legends.
Supplementary Material
Acknowledgments
We thank Robert Munson for the α-OmpP5 rabbit polyclonal antibody, Matthew Pickering for the mouse fH expression construct and Erika van der Maten for the recombinant mouse fH protein. This work is supported by a Dutch Lung Foundation long-term fellowship grant number 3.2.12.126FE (JDL) and grants from the US Public Health Service AI078538 and AI044231 (JNW). The authors have no conflict of interest to declare.
Footnotes
Additional supporting information may be found in the online version of this article at the publisher’s web-site.
References
- Bernhard S, Fleury C, Su YC, Zipfel P, Koske I, Nordstrom T, Riesbeck K. The outer membrane protein OlpA contributes to Moraxella catarrhalis serum resistance via interaction with factor H and the alternative pathway. J Infect Dis. 2014 doi: 10.1093/infdis/jiu241. pii: jiu241 [Epub ahead of print] [DOI] [PubMed] [Google Scholar]
- Blom AM, Villoutreix BO, Dahlback B. Functions of human complement inhibitor C4b-binding protein in relation to its structure. Arch Immunol Ther Exp (Warsz) 2004;52:83–95. [PubMed] [Google Scholar]
- Brook I. Microbiology of sinusitis. Proc Am Thorac Soc. 2011;8:90–100. doi: 10.1513/pats.201006-038RN. [DOI] [PubMed] [Google Scholar]
- Burghout P, Bootsma HJ, Kloosterman TG, Bijlsma JJ, de Jongh CE, Kuipers OP, Hermans PW. Search for genes essential for pneumococcal transformation: the RADA DNA repair protein plays a role in genomic recombination of donor DNA. J Bacteriol. 2007;189:6540–6550. doi: 10.1128/JB.00573-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang AC, Cohen SN. Construction and characterization of amplifiable multicopy DNA cloning vehicles derived from the P15A cryptic miniplasmid. J Bacteriol. 1978;134:1141–1156. doi: 10.1128/jb.134.3.1141-1156.1978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clark SE, Snow J, Li J, Zola TA, Weiser JN. Phosphorylcholine allows for evasion of bactericidal antibody by Haemophilus influenzae. PLoS Pathog. 2012;8:e1002521. doi: 10.1371/journal.ppat.1002521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coleman HN, Daines DA, Jarisch J, Smith AL. Chemically defined media for growth of Haemophilus influenzae strains. J Clin Microbiol. 2003;41:4408–4410. doi: 10.1128/JCM.41.9.4408-4410.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dave S, Brooks-Walter A, Pangburn MK, McDaniel LS. PspC, a pneumococcal surface protein, binds human factor H. Infect Immun. 2001;69:3435–3437. doi: 10.1128/IAI.69.5.3435-3437.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duim B, Bowler LD, Eijk PP, Jansen HM, Dankert J, van Alphen L. Molecular variation in the major outer membrane protein P5 gene of nonencapsulated Haemophilus influenzae during chronic infections. Infect Immun. 1997;65:1351–1356. doi: 10.1128/iai.65.4.1351-1356.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Figueira MA, Ram S, Goldstein R, Hood DW, Moxon ER, Pelton SI. Role of complement in defense of the middle ear revealed by restoring the virulence of nontypeable Haemophilus influenzae siaB mutants. Infect Immun. 2007;75:325–333. doi: 10.1128/IAI.01054-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fleischmann RD, Adams MD, White O, Clayton RA, Kirkness EF, Kerlavage AR, et al. Whole-genome random sequencing and assembly of Haemophilus influenzae Rd. Science. 1995;269:496–512. doi: 10.1126/science.7542800. [DOI] [PubMed] [Google Scholar]
- Granoff DM, Welsch JA, Ram S. Binding of complement factor H (fH) to Neisseria meningitidis is specific for human fH and inhibits complement activation by rat and rabbit sera. Infect Immun. 2009;77:764–769. doi: 10.1128/IAI.01191-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hallstrom T, Riesbeck K. Haemophilus influenzae and the complement system. Trends Microbiol. 2010;18:258–265. doi: 10.1016/j.tim.2010.03.007. [DOI] [PubMed] [Google Scholar]
- Hallstrom T, Jarva H, Riesbeck K, Blom AM. Interaction with C4b-binding protein contributes to non-typeable Haemophilus influenzae serum resistance. J Immunol. 2007;178:6359–6366. doi: 10.4049/jimmunol.178.10.6359. [DOI] [PubMed] [Google Scholar]
- Hallstrom T, Zipfel PF, Blom AM, Lauer N, Forsgren A, Riesbeck K. Haemophilus influenzae interacts with the human complement inhibitor factor H. J Immunol. 2008;181:537–545. doi: 10.4049/jimmunol.181.1.537. [DOI] [PubMed] [Google Scholar]
- Hallstrom T, Resman F, Ristovski M, Riesbeck K. Binding of complement regulators to invasive non-typeable Haemophilus influenzae isolates is not increased compared to nasopharyngeal isolates, but serum resistance is linked to disease severity. J Clin Microbiol. 2010;48:921–927. doi: 10.1128/JCM.01654-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herriott RM, Meyer EM, Vogt M. Defined nongrowth media for stage II development of competence in Haemophilus influenzae. J Bacteriol. 1970;101:517–524. doi: 10.1128/jb.101.2.517-524.1970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hill DJ, Toleman MA, Evans DJ, Villullas S, Van Alphen L, Virji M. The variable P5 proteins of typeable and non-typeable Haemophilus influenzae target human CEACAM1. Mol Microbiol. 2001;39:850–862. doi: 10.1046/j.1365-2958.2001.02233.x. [DOI] [PubMed] [Google Scholar]
- Ho DK, Ram S, Nelson KL, Bonthuis PJ, Smith AL. lgtC expression modulates resistance to C4b deposition on an invasive nontypeable Haemophilus influenzae. J Immunol. 2007;178:1002–1012. doi: 10.4049/jimmunol.178.2.1002. [DOI] [PubMed] [Google Scholar]
- Hood DW, Makepeace K, Deadman ME, Rest RF, Thibault P, Martin A, et al. Sialic acid in the lipopolysaccharide of Haemophilus influenzae: strain distribution, influence on serum resistance and structural characterization. Mol Microbiol. 1999;33:679–692. doi: 10.1046/j.1365-2958.1999.01509.x. [DOI] [PubMed] [Google Scholar]
- Hood DW, Deadman ME, Cox AD, Makepeace K, Martin A, Richards JC, Moxon ER. Three genes, lgtF, lic2C and lpsA, have a primary role in determining the pattern of oligosaccharide extension from the inner core of Haemophilus influenzae LPS. Microbiology. 2004;150:2089–2097. doi: 10.1099/mic.0.26912-0. [DOI] [PubMed] [Google Scholar]
- Hood DW, Deadman ME, Engskog MK, Vitiazeva V, Makepeace K, Schweda EK, Moxon R. Genes required for the synthesis of heptose-containing oligosaccharide outer core extensions in Haemophilus influenzae lipopolysaccharide. Microbiology. 2010;156:3421–3431. doi: 10.1099/mic.0.041780-0. [DOI] [PubMed] [Google Scholar]
- Inzana TJ, Balyan R, Howard MD. Decoration of Histophilus somni lipooligosaccharide with N-acetyl-5-neuraminic acid enhances bacterial binding of complement factor H and resistance to killing by serum and polymor-phonuclear leukocytes. Vet Microbiol. 2012;161:113–121. doi: 10.1016/j.vetmic.2012.07.008. [DOI] [PubMed] [Google Scholar]
- Klos A, Tenner AJ, Johswich KO, Ager RR, Reis ES, Kohl J. The role of the anaphylatoxins in health and disease. Mol Immunol. 2009;46:2753–2766. doi: 10.1016/j.molimm.2009.04.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langereis JD, Stol K, Schweda EK, Twelkmeyer B, Bootsma HJ, de Vries SP, et al. Modified lipooligosaccharide structure protects nontypeable Haemophilus influenzae from IgM-mediated complement killing in experimental otitis media. mBio. 2012;3:e00079–00012. doi: 10.1128/mBio.00079-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langereis JD, Zomer A, Stunnenberg HG, Burghout P, Hermans PW. Nontypeable Haemophilus influenzae carbonic anhydrase is important for environmental and intracellular survival. J Bacteriol. 2013;195:2737–2746. doi: 10.1128/JB.01870-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lewis LA, Carter M, Ram S. The relative roles of factor H binding protein, neisserial surface protein A, and lipooligosaccharide sialylation in regulation of the alternative pathway of complement on meningococci. J Immunol. 2012;188:5063–5072. doi: 10.4049/jimmunol.1103748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mason KM, Munson RS, Jr, Bakaletz LO. Nontypeable Haemophilus influenzae gene expression induced in vivo in a chinchilla model of otitis media. Infect Immun. 2003;71:3454–3462. doi: 10.1128/IAI.71.6.3454-3462.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Melhus A, Hermansson A, Forsgren A, Prellner K. Intra- and interstrain differences of virulence among nontypeable Haemophilus influenzae strains. APMIS. 1998;106:858–868. [PubMed] [Google Scholar]
- Meri T, Amdahl H, Lehtinen MJ, Hyvarinen S, McDowell JV, Bhattacharjee A, et al. Microbes bind complement inhibitor factor H via a common site. PLoS Pathog. 2013;9:e1003308. doi: 10.1371/journal.ppat.1003308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morey P, Viadas C, Euba B, Hood DW, Barberan M, Gil C, et al. Relative contributions of lipooligosaccharide inner and outer core modifications to nontypeable Haemophilus influenzae pathogenesis. Infect Immun. 2013;81:4100–4111. doi: 10.1128/IAI.00492-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakamura S, Shchepetov M, Dalia AB, Clark SE, Murphy TF, Sethi S, et al. Molecular basis of increased serum resistance among pulmonary isolates of non-typeable Haemophilus influenzae. PLoS Pathog. 2011;7:e1001247. doi: 10.1371/journal.ppat.1001247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Narkio-Makela M, Hellwage J, Tahkokallio O, Meri S. Complement-regulator factor H and related proteins in otitis media with effusion. Clin Immunol. 2001;100:118–126. doi: 10.1006/clim.2001.5043. [DOI] [PubMed] [Google Scholar]
- Neeleman C, Geelen SP, Aerts PC, Daha MR, Mollnes TE, Roord JJ, et al. Resistance to both complement activation and phagocytosis in type 3 pneumococci is mediated by the binding of complement regulatory protein factor H. Infect Immun. 1999;67:4517–4524. doi: 10.1128/iai.67.9.4517-4524.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ngampasutadol J, Ram S, Gulati S, Agarwal S, Li C, Visintin A, et al. Human factor H interacts selectively with Neisseria gonorrhoeae and results in species-specific complement evasion. J Immunol. 2008;180:3426–3435. doi: 10.4049/jimmunol.180.5.3426. [DOI] [PubMed] [Google Scholar]
- Novotny LA, Partida-Sanchez S, Munson RS, Jr, Bakaletz LO. Differential uptake and processing of a Haemophilus influenzae P5-derived immunogen by chinchilla dendritic cells. Infect Immun. 2008;76:967–977. doi: 10.1128/IAI.01395-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Opijnen T, Bodi KL, Camilli A. Tn-seq: high-throughput parallel sequencing for fitness and genetic interaction studies in microorganisms. Nat Methods. 2009;6:767–772. doi: 10.1038/nmeth.1377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poole J, Foster E, Chaloner K, Hunt J, Jennings MP, Bair T, et al. Analysis of nontypeable haemophilus influenzae phase-variable genes during experimental human nasopharyngeal colonization. J Infect Dis. 2013;208:720–727. doi: 10.1093/infdis/jit240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ram S, Sharma AK, Simpson SD, Gulati S, McQuillen DP, Pangburn MK, Rice PA. A novel sialic acid binding site on factor H mediates serum resistance of sialylated Neisseria gonorrhoeae. J Exp Med. 1998;187:743–752. doi: 10.1084/jem.187.5.743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodriguez de Cordoba S, Esparza-Gordillo J, Goicoechea de Jorge E, Lopez-Trascasa M, Sanchez-Corral P. The human complement factor H: functional roles, genetic variations and disease associations. Mol Immunol. 2004;41:355–367. doi: 10.1016/j.molimm.2004.02.005. [DOI] [PubMed] [Google Scholar]
- Rosadini CV, Ram S, Akerley BJ. Outer membrane protein P5 is required for resistance of nontypeable Haemophilus influenzae to both the classical and alternative complement pathways. Infect Immun. 2014;82:640–649. doi: 10.1128/IAI.01224-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schneider CA, Rasband WS, Eliceiri KW. NIH Image to ImageJ: 25 years of image analysis. Nat Methods. 2012;9:671–675. doi: 10.1038/nmeth.2089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Serruto D, Bottomley MJ, Ram S, Giuliani MM, Rappuoli R. The new multicomponent vaccine against meningococcal serogroup B, 4CMenB: immunological, functional and structural characterization of the antigens. Vaccine. 2012;30(Suppl 2):B87–B97. doi: 10.1016/j.vaccine.2012.01.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sethi S, Sethi R, Eschberger K, Lobbins P, Cai X, Grant BJ, Murphy TF. Airway bacterial concentrations and exacerbations of chronic obstructive pulmonary disease. Am J Respir Crit Care Med. 2007;176:356–361. doi: 10.1164/rccm.200703-417OC. [DOI] [PubMed] [Google Scholar]
- van Soolingen D, de Haas PE, Hermans PW, van Embden JD. DNA fingerprinting of Mycobacterium tuberculosis. Methods Enzymol. 1994;235:196–205. doi: 10.1016/0076-6879(94)35141-4. [DOI] [PubMed] [Google Scholar]
- Stol K, Verhaegh SJ, Graamans K, Engel JA, Sturm PD, Melchers WJ, et al. Microbial profiling does not differentiate between childhood recurrent acute otitis media and chronic otitis media with effusion. Int J Pediatr Otorhinolaryngol. 2013;77:488–493. doi: 10.1016/j.ijporl.2012.12.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Vries SP, Burghout P, Langereis JD, Zomer A, Hermans PW, Bootsma HJ. Genetic requirements for Moraxella catarrhalis growth under iron-limiting conditions. Mol Microbiol. 2013;87:14–29. doi: 10.1111/mmi.12081. [DOI] [PubMed] [Google Scholar]
- Webb DC, Cripps AW. Secondary structure and molecular analysis of interstrain variability in the P5 outer-membrane protein of non-typable Haemophilus influenzae isolated from diverse anatomical sites. J Med Microbiol. 1998;47:1059–1067. doi: 10.1099/00222615-47-12-1059. [DOI] [PubMed] [Google Scholar]
- Webb DC, Cripps AW. A method for the purification and refolding of a recombinant form of the nontypeable Haemophilus influenzae P5 outer membrane protein fused to polyhistidine. Protein Expr Purif. 1999;15:1–7. doi: 10.1006/prep.1998.0990. [DOI] [PubMed] [Google Scholar]
- Williams BJ, Morlin G, Valentine N, Smith AL. Serum resistance in an invasive, nontypeable Haemophilus influenzae strain. Infect Immun. 2001;69:695–705. doi: 10.1128/IAI.69.2.695-705.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yanisch-Perron C, Vieira J, Messing J. Improved M13 phage cloning vectors and host strains: nucleotide sequences of the M13mp18 and pUC19 vectors. Gene. 1985;33:103–119. doi: 10.1016/0378-1119(85)90120-9. [DOI] [PubMed] [Google Scholar]
- Yi K, Sethi S, Murphy TF. Human immune response to nontypeable Haemophilus influenzae in chronic bronchitis. J Infect Dis. 1997;176:1247–1252. doi: 10.1086/514119. [DOI] [PubMed] [Google Scholar]
- Zomer A, Burghout P, Bootsma HJ, Hermans PW, van Hijum SA. ESSENTIALS: software for rapid analysis of high throughput transposon insertion sequencing data. PLoS ONE. 2012;7:e43012. doi: 10.1371/journal.pone.0043012. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.