

Abstract
Slowing down DNA translocation speed in a nanopore is essential to ensuring reliable resolution of individual bases. Thin membrane materials enhance spatial resolution but simultaneously reduce the temporal resolution as the molecules translocate far too quickly. In this study, the effect of exposed graphene layers on the transport dynamics of both single (ssDNA) and double-stranded DNA (dsDNA) through nanopores is examined. Nanopore devices with various combinations of graphene and Al2O3 dielectric layers in stacked membrane structures are fabricated. Slow translocations of ssDNA in nanopores drilled in membranes with layers of graphene are reported. The increased hydrophobic interactions between the ssDNA and the graphene layers could explain this phenomenon. Further confirmation of the hydrophobic origins of these interactions is obtained through reporting significantly faster translocations of dsDNA through these graphene layered membranes. Molecular dynamics simulations confirm the preferential interactions of DNA with the graphene layers as compared to the dielectric layer verifying the experimental findings. Based on our findings, we propose that the integration of multiple stacked graphene layers could slow down DNA enough to enable the identification of nucleobases.
1. Introduction
The concept of using nanopores as impedance based biosensors has emerged as an attractive and versatile tool for detection and analysis of charged biomolecules. The detection of target molecules is achieved by electrophoretically driving the molecules through nanometer-sized pores in biological or synthetic membranes and simultaneously monitoring the modulation of nanopore ionic current.[1–3] These temporary fluctuations in the ionic current can yield information on the biopolymer length, orientation, and sequence. The need for improvements in speed and cost of sequencing has prompted a great deal of interest in nanopore-based next generation DNA sequencing technology for being a single molecule, label-free, amplification-free approach that promises low cost and high-speed reading throughput.[1–3] The transport of RNA and DNA homopolymer molecules through a biological nanopore α-hemolysin was first demonstrated by Kasianowicz et al.[4] Even though biological nanopores have the advantage of remarkable reproducibility and have been demonstrated to distinguish individual nucleobases in a static strand in a nanopore,[5] sequencing has remained challenging due to the high velocity of DNA translocation. Significant progress in the engineering of biological nanopores, including mutagenesis and targeted chemical modification by incorporation of enzymes, has enabled relatively slow and controlled transport of DNA molecules in discrete steps.[6–9] Recent work combining the use of polymerase enzymes with engineered octameric protein channel MspA, which has a shorter and narrower constriction (0.6 nm long and 1.2 nm wide), has enabled a significant enhancement in nucleotide discrimination.[10,11] Despite the progress that has been made with biological nanopores, solid-state nanopores are still attractive as they promise higher stability, greater control on a range of pore dimensions, multiplexing and integration with alternative detection modalities that could improve both sensitivity and resolution of nanopore-based sequencing.[12]
Various dielectrics and metals have been used to make synthetic membranes for solid-state nanopores.[13,14] However, the finite thickness (usually above 10 nm) of the fabricated membranes presents a limit on the spatial resolution of the measurements, making single nucleotide resolution difficult to achieve. Graphene, as a single layer material with mechanical stability[15] and the same order of thickness as the nucleotide separation in a DNA strand, is an attractive solution to this problem.[1] DNA transport through nanopores in freely suspended graphene membranes has been reported by various groups.[16–18] These thin membranes demonstrate high sensitivity in conductivity modulation with change in diameter of translocating polymer.[19] The high 1/f noise in graphene nanopores reduces the signal-to-noise ratio making single nucleotide distinction using ionic current challenging.[20] However graphene as a material with interesting electronic properties[21,22] has opened avenues to combine ionic current-based measurements with transverse sensing modalities by incorporating structures such as nanoribbons,[23,24] nanogaps,[25] and tunneling electrodes.[26] But adding, transverse sensing functionalities in a graphene system is challenging due to potential leakage paths created and thus stacked systems with various dielectrics might be needed to achieve reliable and robust integration with transverse electrodes.[27] Another major roadblock presented by these thin membranes is the low temporal resolution of ionic current measurements, and thus slowing down DNA translocations remains critical to achieve sequencing using graphene nanopores. Various methods have been suggested in solid-state nanopores to slow DNA translocation speed including the use of stick-slip interactions by using dielectric materials with high surface charge density like Al2O3 and HfO2.[28,29] Other proposed techniques include the use of different ionic solutions such as LiCl,[30] increasing solution viscosity with glycerol,[31] optoelectronic control,[32] fluidic gating,[33] reducing nanopore diameter,[34] use of pressure gradients,[35] thicker membranes,[36] and temporary hydrogen bonding.[37] Recently, the potential for DNA–graphene hydrophobic interactions to induce ssDNA translocations in single-nucleotide steps was discussed.[38,39] ssDNA translocation experiments with standalone graphene membranes have been demonstrated by coating the graphene surface with a hydrophilic layer[40] or by performing the experiments at highly alkaline pH, which significantly reduce DNA–graphene interactions.[19] Our experiments are performed at a lower pH value (pH = 7.6) which enhances DNA adsorption on graphene[41] and is the first demonstration of the effect of DNA–graphene interactions on DNA translocation. We have previously demonstrated the fabrication of nanopores in stacked layers of graphene and Al2O3 and demonstrated translocation of DNA molecules.[20] The bottom graphene layer was used primarily as the supporting base. In this study, we add functionality to the structure by using the hydrophobic interactions between single or multiple graphene layers and ssDNA molecules to slow DNA transport. We compare translocation properties of the stacked graphene structures with a membrane made of Al2O3, a dielectric with a highly charged surface that binds to DNA through electrostatic interactions.[28] A significant reduction in translocation speed of ssDNA is observed in membranes with single or multiple graphene layers integrated when compared to the standalone dielectric membranes. In addition, the translocation of dsDNA through our stacked graphene–dielectric–graphene structure demonstrates significantly reduced interactions between dsDNA molecules and graphene, which is manifested in much faster dsDNA translocation speed.
2. Results and Discussion
2.1. Device Fabrication and Overview
The details of fabrication of supporting the membrane structures and subsequent formation of stacked layers of graphene and Al2O3 dielectric have been described previously.[20,27] The transport of DNA and DNA-protein complexes through these structures has been previously demonstrated.[20] In this study, we fabricate structures to study and compare the effects of exposed graphene layers on DNA transport speed. The supporting membrane structures consist of stacked layers of 50 nm Al2O3, 200 nm SiNx, and 10 nm Al2O3 fabricated using deep reactive ion etching (DRIE). A 300 nm diameter hole is drilled using focused ion beam (FIB) onto these freely suspended membranes (see Section 4). Figure 1A shows the schematic for the graphene–dielectric–graphene membrane. Graphene is grown by chemical vapor deposition (CVD) using a process described in an earlier article.[27] The first graphene layer is transferred onto the FIB drilled hole in the supporting membrane structure and covers the entire hole forming a freely suspended graphene sheet. Then we deposit 2 nm of Al which acts as the seed layer for the subsequent deposition of 24 nm of Al2O3 on graphene using atomic layer deposition (ALD).[42] The seed layer is essential for uniform and dense growth of the dielectric layer. The superiority of Al2O3 as the membrane material over SiO2 and Si3N4 in terms noise performance has been previously established and is the choice of dielectric in this study.[43,44] The thickness of dielectric is established based on our leakage studies with a similar structure and has been kept constant across all the structures described in this paper.[27] Another layer of graphene is transferred on top of the dielectric layer. The schematic of the membrane structure (Figure 1A) shows the thickness of the various layers. The motivation for this design is to study the interactions of the translocating polymer with exposed graphene, to which the ssDNA molecules are expected to adhere to, on both sides of the membrane. To compare the translocation characteristics of this membrane and understand the effect of DNA–graphene interactions on DNA translocation properties, two additional membrane structures are studied. Figure 1B shows the graphene–dielectric structure, where the same process as described above is used for sequential transfer of graphene followed by Al2O3 deposition, leaving only one graphene layer for the translocating DNA molecule to interact with. Finally, Figure 1 C (i–iv) shows our process to fabricate a purely dielectric membrane. First, a sacrificial graphene layer is transferred on the FIB-drilled hole, which acts as the support for the deposition of the Al2O3 dielectric layer on top. Subsequently, the backside trench (drain) of the device is exposed to an oxygen plasma process (see Experimental Section). The process removes all the exposed graphene on the 300 nm hole area drilled by FIB. After oxygen plasma processing, the nanopore is drilled in this membrane. As a result the translocating DNA strand can only interact with the dielectric layer as there is no exposed graphene in the pore region.
Figure 1.

Schematic of the membrane structures whose DNA interaction and translocation properties are compared in this study. A) (i) Sequential transfer of graphene followed by atomic layer deposition (ALD) of 24 nm Al2O3 is followed by another graphene layer transfer. (ii) The nanopore region is magnified and dimensions of the dielectric layer are indicated. (iii) IV curve for an ≈3.5 nm pore in a graphene–dielectric–graphene membrane. (iv) TEM image of the nanopore. B) (i) Graphene transfer was followed by ALD deposition of Al2O3. (ii) The nanopore region is magnified. (iii) Open pore IV curve for an ≈4 nm pore in a graphene–dielectric membrane. (iv) TEM image of the nanopore. C) [(i)–(iv)] Graphene transfer was followed by ALD deposition of Al2O3. Reactive ion etching in an oxygen plasma environment is then used to etch the exposed graphene leaving only the oxide layer. (v) The nanopore region is magnified. (vi) Open pore IV curve for an ≈3.3 nm pore in a dielectric membrane. (vii) TEM image of nanopore. The nanopore diameters (scale bar 5 nm) are comparable and the ionic conductivity is almost similar in all three cases, essential for direct comparison of DNA translocation properties. All experiments were done in 1 M KCl, 10 × 10−3 M Tris, 1 × 10−3 M EDTA at pH 7.6. 100nt ssDNA is threaded from source to drain as indicated for all the cases studied here.
2.2. ssDNA Translocation Experiments
Nanopores are drilled using convergent beam electron diffraction (CBED) mode in a transmission electron microscope (TEM). Subsequently the chips are assembled in a custom-built fluidic setup and then buffer solution of 1 M KCl, 10 × 10−3 M Tris, 1 × 10−3 M EDTA at pH 7.6 is inserted in both the chambers. Open-pore conductance curves for all nanopores compared in this study show fairly similar conductivity (Figure 1). Since our CVD-grown graphene has been demonstrated to be mostly monolayer or bilayer thick,[27] we do not expect a significant difference in nanopore length and conductivity across all three membrane structures.[45] Upon introduction of 100nt ssDNA molecules, current blockades are observed. In all the three structures shown in Figure 1, the DNA is threaded from the topside (source) to the backside trench (drain). Translocation data at transmembrane voltage of 300 mV (drain with respect to source) for all three membrane structures are presented in Figure 2. Figure 2A–C shows sample current traces for typical events for nanopores in graphene–dielectric–graphene, grapheme–dielectric, and dielectric membrane systems, respectively. The translocation histograms for each corresponding case are presented to the right of sample traces (Figure 2 D–F). A mono-exponential decay function is fitted to the dwell time distribution and the mean values for translocation time is indicated next to the histograms. We see a slight increase in the observed translocation time when graphene layer is present on both sides of the membrane as we expect the DNA to stick to the graphene. ssDNA can stretch under the influence of the electrical field giving lengths up to 0.6 nm per base.[46] Given the thickness of the membrane, the DNA strand is expected to interact with only one graphene layer at a time during translocation for the grapheme–dielectric–graphene membranes (Supporting Information, Figure S1). This might explain the minor variation in dwell time observed while comparing translocation characteristics of nanopores in grapheme–dielectric—graphene, and grapheme–dielectric membranes.
Figure 2.
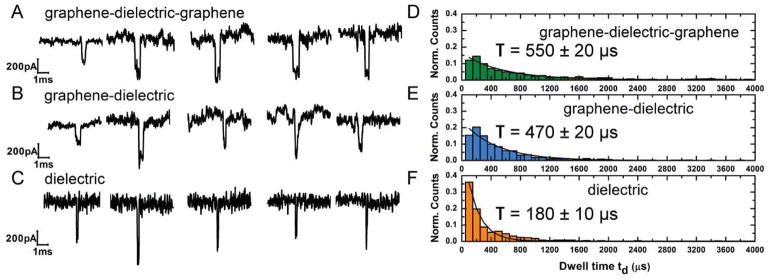
Experiments indicating the effect of graphene layers in slowing ssDNA translocation. A–C) Sample current blockades for 100 nt ssDNA for each membrane system of graphene–dielectric–graphene, graphene–dielectric, and dielectric respectively. All experiments were performed in 1 M KCl, 10 × 10−3 M Tris, 1 × 10−3 M EDTA at pH 7.6 and a transmembrane voltage of 300 mV. The sample traces show considerable slowing down with the introduction of graphene layers at the membrane area. D–F) Translocation time histograms for the cases A–C, respectively. With 100nt ssDNA we find average translocation times of T = 550 ± 20 μs, T = 470 ± 20 μs, T = 180 ± 10 μs for graphene–dielectric-graphene, graphene–dielectric, and dielectric, respectively. Graphene DNA hydrophobic interactions reduce the translocation velocity of the DNA molecule by about 3 times when compared to translocation properties of the purely dielectric membrane.
We compare the translocation properties observed in our stacked layers with a positively charged (at experimental pH) dielectric layer of Al2O3 which has been reported to slow DNA translocations through electrostatic interactions.[28] We report a ssDNA translocation speed of 180 ± 10 μs which is in the same range as expected from literature for such oxide membranes.[29] The alumina membrane nanopore shows a three times faster translocation speed than the graphene–dielectric–graphene case. We model the pore as having a truncated double conical structure (Figure S1, Supporting Information) similar to that reported for Alumina nanopores.[43] The pore conductance is given by Equation (1)
| (1) |
where Gpore is the pore conductance calculated by adding the pore resistance derived through geometric arguments.[47] In this equation σ is the ionic conductivity of 1 M KCl buffer solution (measured to be 112.8 mS cm−1). d is the diameter of the pore and h is the height of the membrane (≈24 nm) and δ = (h – heff)/d. Assuming effective channel length heff= h/3 and a cone angle of α = 30° we calculate expected nanopore diameters based on observed conductance values (Figure S1, Supporting Information). This is consistent with previous work on Al2O3 nanopores from our lab using aluminum oxide nanopores and the same electron microscope instrument.[43] The calculated pore diameters are reported in Figure 1 (insets). The expected and observed pore diameter values fit well to the conductance model for the graphene and graphene–dielectric membranes. The geometric model however does not take into account the presence of different materials in the three membrane structures which could have a surface charge-based contribution to the ionic flow and slightly different geometric shapes based on different sputtering rate of stacked materials.[48] Translocation statistics are reported to be sensitive to variations in pore diameter when translocating polymer and nanopore have comparable diameters.[34] For ssDNA translocations as pore diameter increases above 3 nm, translocation velocity is expected to saturate as a result of decreased van der Waals interactions with the pore walls.[49,50] All our nanopores are approximately 3 times the diameter of the ssDNA molecule and significant variation in pore-DNA interactions with minor changes (≈0.4 nm) in pore diameter is not expected. Additionally, the graphene–dielectric membrane pore shows a larger current than the dielectric pore but still shows significantly longer translocation times indicating that the observations are not due to variations in the pore diameter. We attribute the observed changes to specific interactions between the DNA molecule and the membrane materials. We hypothesize the possible cause for slow ssDNA translocations in the graphene embedded membranes to be hydrophobic interactions between ssDNA and the graphene layers. Nanopore experiments in Al2O3 membranes[28,43,44] indicate an order of magnitude reduction in translocation speeds as compared to Si3N4 or SiO2 based nanopores. The addition of graphene layers makes the pore hydrophobic (Supporting Information, Figure S5). ssDNA–graphene interactions due to hydrophobic attraction is well known. The aromatic purines and pyrimidine bases of ssDNA have been observed to freely adsorb on graphene surfaces.[51]
We observe material inhomogeneity in the vicinity of the nanopore as seen in the contrast around the nanopore in the TEM images (Figure 1). Changes in local stoichiometry and crystallization of material have been reported for Alumina membranes due to preferential sputtering of Oxygen atoms.[28] We have observed such material inhomogeneities in our previous study with similar stacked structures.[20] The possibility of graphene damage due to TEM convergent beam has also been addressed to confirm the presence of graphene in the pore vicinity for these structures (Supporting Information, Figure S1). We simulated the same beam conditions on a pure graphene membrane. While the pore nucleates very quickly, the pore expansion saturates if the beam is not moved to the edges of the pore. Intensity profiles for the electron beam probes used in these studies have been reported to have a tail approximating a radius of 5 nm.[28] Consistent with those results, even long exposures (well beyond those used for the pores in the fluidic experiments) do not expand the pore to beyond 7 nm of radius. The protocol to minimize graphene damage has been described in the Experimental Section. Also graphene layers are expected to adhere well to oxides due to van der Waals interactions and we expect the graphene damage reported here to be an upper bound for the possible graphene damage in the stacked grapheme–dielectric structures.[52] While local graphene crystal structure changes or amorphization under the influence of electron irradiation cannot be ruled out,[53] hydrophobic interactions of DNA with different forms of carbon have been shown.[54] Given the length of the DNA strand used in these experiments, we expect graphene to have a significant influence on translocation characteristics (Supporting Information, Figure S1).
The enhanced pore/membrane and DNA interactions produce translocation rates of ≈5.5 μs bp−1 at 300 mV and ≈10.1 μs bp−1 at 200 mV. This compares favorably to studies on ssDNA translocation through other materials such as HfO2,[29] SiNx,[55] and bare graphene membranes, when operated in experimental conditions where the DNA–graphene interactions are minimized.[19,40] In addition, graphene–DNA interactions are known to be nucleobase specific. The specificity of these interactions have been applied to a variety of optical sensors and for separation of single and double-stranded DNA.[41,56,57] The binding energy of the four nitrogenous nucleobases with graphene is known to vary[58,59] with guanine known to have the strongest binding affinity. The difference in interaction could result in different translocation speeds and provide a basis of nucleotide separation essential for sequencing applications. The addition of dielectric layers increases the thickness of the membrane and thus can reduce the spatial resolution of the measurement if ionic currents through the pore are expected to resolve the DNA sequence. We envision the eventual integration of these dielectric–graphene-based membrane systems with an embedded graphene ribbon to produce a stack of graphene–dielectric–graphene–dielectric–graphene. In such a device, the top and bottom graphene layers can interact and slow the DNA molecule, while the middle graphene layer can be patterned into a ribbon to sense DNA.[23] Our previous studies show that the electrical integrity of embedded graphene layer is not damaged by the TEM beam used to drill the nanopore.[27] While measurement of DNA charge-based current fluctuations in graphene nanoribbon current is expected to have very high intrinsic bandwidth, the fabrication of these structures can be highly challenging as they need ribbons with nanoscale widths and constrictions of less than 5 nm.[24] Other mechanisms of sensing have been reported with larger graphene ribbons up to 100 nm in width.[23] In this study, the graphene current was measured using a 400 kHz current amplifier, whose temporal resolution would be in the same range as demonstrated by our graphene–dielectric–based membrane systems shown here. In such devices, further slowing DNA transport would allow the use of filters with lower bandwidth, thus improving signal-to-noise ratio and enhance the reliability of nucleotide separation.
The necessary controls and detailed analysis for both graphene–dielectric–graphene and dielectric nanopore experiments are presented in Figure 3. The scatter plots (Figure 3A,B) display similar blockade levels in both cases. The slightly higher blockade levels for the dielectric membrane are indicative of a slightly smaller nanopore. Additionally, we speculate that the higher degree of freedom for the ssDNA molecules in the absence of graphene layers and specific interactions can lead to formation of secondary structures and higher current blockades. The significant shift in the event duration densities of the blockade events (as borne out by the translocation time histograms) for both membrane systems seems to indicate significant differences in the DNA–membrane material interactions. In addition, translocation histograms at two different transmembrane voltages have been presented to indicate the expected increase in average dwell time with applied voltage, confirming the observed blockades to be due to translocations instead of random collisions. The additional scatter plots and histograms (Supporting Information, Figure S3) also provide similar conclusions. The broad distribution of translocation times in all cases indicate significant interaction with the pore surface.[28,34] Expected blockade ratio for ssDNA (dDNA = 1.2 nm) blocking a 3.5 nm pore can roughly be estimated by Equation (2)
Figure 3.

Control experiments for grapheme–dielectric–graphene, and dielectric nanopores for translocation experiments with 100nt ssDNA. For these experiments, a 3.5 nm pore was drilled in the graphene–dielectric–graphene membrane and a 3.3 nm pore was drilled in a dielectric membrane. A,B) Scatter diagram for Blockade ratio versus Dwell time for both the membrane systems. C,D) Translocation time histograms for the graphene—dielectric—graphene, and dielectric nanopores showing an increase in Dwell time with increased transmembrane voltage indicating the events observed to be due to DNA translocation. All experiments were performed in 1 M KCl, 10 × 10−3 M Tris, 1 × 10−3 M EDTA at pH 7.6. The blockade ratios are in the same range and a significant difference is observed in the translocation time.
| (2) |
The expected blockade ratio for unfolded ssDNA translocation is 0.12. The blockade histograms (Supporting Information, Figure S2A) for a 3.5 nm pore in graphene–dielectric–graphene membrane displays a mean blockade level of ΔI ≈ 326.4 ± 7.3 pA at 300 mV. This indicates a mean blockade level of Br = ΔI/I = 0.14, where I is the open pore current, in good agreement with expected blockade for unfolded DNA translocation. Also the average blockade level reduces as expected with decreasing voltage (Supporting Information, Figure S2B); however, the blockade ratio (Br) is similar.
2.3. MD Simulations
To elucidate the microscopic interactions that give rise to the experimentally observed variation of the DNA translocation rate, we construct two atomic-scale models of the experimental systems. Each model, shown in Figure 4A,B, contains a 50-nucleotide fragment of a poly(dT) strand threaded through a double-cone nanopore of 1.7 nm minimum diameter cut in a 5.5 nm-thick amorphous silica membrane. One of the systems has a graphene sheet added on each side of the membrane; the pore was cut in graphene to match the size and shape of the nanopore openings. Following our earlier work,[60] we use slightly positively charged amorphous silica as a model of alumina. The systems are solvated and populated with ions to produce a 1 M KCl solution. The systems equilibrate under constant pressure for over 80 ns. Following that, a 500 mV bias is applied across each system forcing ssDNA to move from one side of the membrane to the other through the nanopore (see Animations M1 and M2, Supporting Information). Further details on the construction and equilibration procedures are given in the Experimental section.
Figure 4.
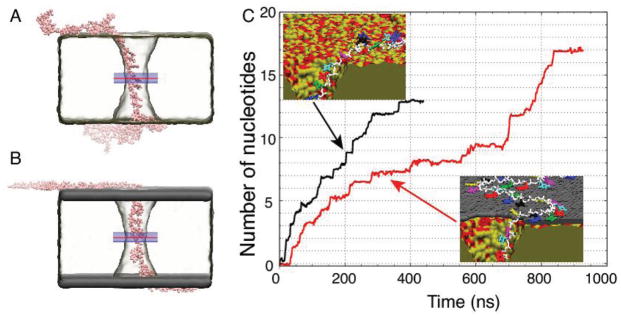
All-atom MD simulations of ssDNA translocation through nanopores. A,B) Representative conformations of a poly(dT)50 strand in MD simulations of nanopore transport through dielectric. A) and grapheme—dielectric–graphene. B) membranes. The minimum diameter of the nanopore in each membrane was 1.7 nm. C) The number of nucleotides transported through the midplane of the membrane by a 500 mV bias versus simulation time. The midplane’s location is indicated by a red line in panels A and B. The average permeation rates are 0.0324 and 0.0195 nucleotides/ns for the dielectric (black trace) and graphene–dielectric–graphene (red trace) membranes, respectively. The insets depict interactions of ssDNA with the surface of the dielectric (top) and graphene–dielectric–graphene (bottom) membranes. The bases do not strongly interact with the surface of silica, but stack strongly to graphene.
During equilibration, ssDNA is observed to adhere to the surface of the membrane in both systems. However, the microscopic conformations of ssDNA in the two systems are qualitatively different: hydrophobic adhesion of DNA bases is observed only in the case of the graphene–dielectric–graphene membrane, Figure 4B. Such differences are seen to affect the rate of ssDNA transport through the nanopore. In Figure 4C, we plot the number of nucleotides permeated through the membrane’s midplane under a 500 mV transmembrane bias. The transport of ssDNA through the nanopore in the graphene covered membrane is considerably slower than that in the bare dielectric membrane. To calculate the average permeation rate, we divide the total number of permeated nucleotides by the duration of the simulation. The graphene–silica–graphene pore is simulated for 951 ns, and the permeation rate is measured to be 0.0195 nt ns−1, considerably slower than in the case of a free-standing graphene membrane.[38] The silica-only pore is simulated for 464 ns, and the observed permeation rate is 0.0324 nt ns−1. Thus, the presence of the graphene sheets contributes to approximately a 40% reduction of the DNA permeation rate, which is in excellent agreement with the experimental measurements reported above. For comparison, the translocation rate of dsDNA in a bare dielectric membrane under identical conditions (but using a larger 3.8 nm diameter pore) is 4.9 bp ns−1, two orders of magnitude faster than the ssDNA permeation rate.
Two types of interactions determine the rate of ssDNA permeation through the nanopores. The first one is stacking of the DNA bases to the graphene sheets. The abundance of such hydrophobic contacts can be appreciated from the close-up views of the nanopore edge shown in the inset to Figure 4C. All DNA bases outside the nanopore adsorb to graphene but only few are adsorbed to the surface of the bare dielectric membrane. The second type is an attraction of the DNA backbone to silica. Examples of such interactions can be seen in the snapshots as Movies M1 and M2, Supporting Information: the base of a nucleotide points toward the center of the pore as the backbone attaches to the wall.
Figure 5 provides further details on the process of ssDNA translocation. Snapshots in Figure 5A illustrate the conformation of ssDNA at the beginning (left) and the end (right) of the 951 ns MD trajectory. The DNA strand appears to be under tension. The average distance between the nucleotides along the pore axis in the top (inlet) and bottom (outlet) halves of the pore is about 0.59 and 0.54 nm, respectively, which corresponds to a stretching force of approximately 80 and 40 pN.[61,62] Lower tension in the bottom part of the DNA strand permits for intermittent accumulation of DNA nucleotides at the exit opening of the nanopore.
Figure 5.
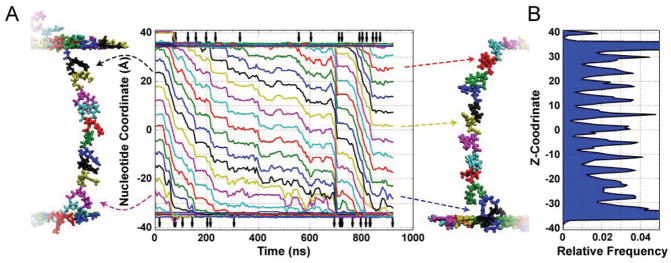
Stepwise transport of ssDNA through graphene–dielectric–graphene membrane. A) Z-coordinates of DNA nucleotides (measured as the center of mass of each nucleotide’s backbone) versus simulation time. The snapshots illustrate the conformation of ssDNA at the beginning (left) and the end (right) of the MD simulation. The color of the nucleotides corresponds to the color of the Z-coordinate traces; dashed arrows relate select traces to the nucleotides they represent. Vertical arrows at the top and bottom of the graph indicate the moments of DNA base unbinding from the top layer of graphene (top) or binding of a DNA base to the bottom layer of graphene (bottom). The traces demonstrate long periods of very little motion punctuated by short quick movements of approximately the length of one nucleotide. B) The distribution of the DNA nucleotide along the length of the nanopore averaged over the course of the ≈950 ns MD simulation. Repetitive placement of DNA nucleotides within the same region of the pore may be advantageous for DNA sequencing applications.
Because the strand is under tension, displacements of the nucleotides in the nanopore are correlated, which can be clearly seen from the main panel of Figure 5A that plots the coordinates of individual nucleotides in the nanopore. For translocation to occur, the bases must desorb from graphene. Vertical arrows at the top of Figure 5 indicate the times at which a base desorbs from the graphene and enters the nanopore. This unbinding process is likely the reason for slower translocation kinetics of ssDNA in graphene–dielectric–graphene membrane in comparison to otherwise identical bare dielectric system. The higher extent of DNA adsorption at low pH has been reported for graphene oxide.[41] The single nucleotide traces in Figure 5A also clearly show that the translocation process is not only stepwise, but also that the DNA nucleotides visit the same locations during the translocation, Figure 5B. Repetitive placement of DNA nucleotides within the same region of the pore may be advantageous for DNA sequencing applications.
2.4. dsDNA Translocation Experiments
The degree and effect of these hydrophobic interactions can be further understood by comparing ssDNA and dsDNA translocation properties. The dsDNA translocation experiments are conducted with long dsDNA molecules (850 bp) in an approximately 6 nm pore through a graphene–dielectric–graphene membrane. Even though the nanopore size is larger than those used for ssDNA translocation experiments, we believe the direct comparison is still valid, as translocation velocity and DNA–pore wall interactions have been calculated to be fairly constant for large nanopores.[49] The diameter of the pore is more than twice the diameter of dsDNA. In all our studies, the nanopore diameter is much larger than the molecule size thus the major contributing factor to interactions slowing down DNA molecules are expected to be interactions between the membrane surface and DNA strand. The results of dsDNA experiments and translocation histograms at transmembrane voltages of 300 mV and 500 mV have been presented in Figure 6B,C. The scatter diagrams indicating the expected trend of increasing translocation speed with transmembrane voltage and current blockade histograms indicating expected blockade levels (Supporting Information, Figure S4A–D) validate the observed blockades to be true translocations rather than random collisions.
Figure 6.
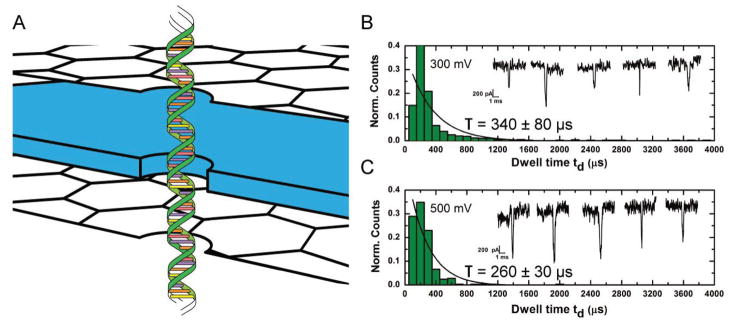
Nanopore experiments indicating the effect of graphene layers on double-stranded DNA translocation. A) Schematic of graphene–dielectric–graphene membrane used in studying translocation of 850bp dsDNA. A 6 nm pore is used in this experiment. B,C) Translocation histograms for dsDNA translocations at transmembrane voltages of 300 mV and 500 mV, respectively. All experiments were performed in 1 M KCl, 10 × 10−3 M Tris, 1 × 10−3 M EDTA at pH 7. Insets: Sample current traces of dsDNA blockade events for both values of transmembrane voltages. The smaller timescale of translocation of a much longer dsDNA (compared to ssDNA used in this study) indicates significantly reduced hydrophobic interactions in between graphene and dsDNA as compared to graphene and ssDNA.
The translocation speed for the dsDNA strand is observed to be an order of magnitude faster than that for the ssDNA experiments. Table 1 shows a summary of translocation time and speeds for ssDNA and dsDNA molecules. While this may appear surprising at first glance, as the dsDNA molecules used here are much longer than the ssDNA molecules, the dsDNA molecule is expected to have significantly less interaction with graphene.[51] For dsDNA, the nucleobases are effectively shielded within the helical structure protected by a charged phosphate backbone. In addition, the persistence length for ssDNA molecules is around 2–4 nm which, owing to a larger degree of freedom, is significantly smaller than that for dsDNA (50 nm) molecules.[63,64] The stiffer dsDNA molecule has weak interactions with the membrane surface and could be expected to be linearly oriented (Figure 6A) under the influence of the strong local electric field and translocate much faster than the ssDNA molecules. On the other hand, a ssDNA molecule can easily orient along the graphene surface and enter the nanopore through a 2D diffusion process which would significantly slow down DNA translocations as confirmed by our results.[38,39]
Table 1.
Summary of all nanopore measurements conducted at transmembrane voltage of 300 mV. The translocation rate is calculated from the Dwell time measured for different molecules listed here.
| Membrane material [thickness, t ≈24 nm] | Pore diameter [d] [nm] | Dwell time, τ [transmembrane voltage 300 mV] [μs] | DNA length | Translocation rate [@300 mV] |
|---|---|---|---|---|
| Graphene–Al2O3–graphene | 3.5 | 550 | 100nt – ssDNA | 5.5 μs nt −1 |
| Graphene-Al2O3 | 4.0 | 470 | 100nt – ssDNA | 4.7 μs nt −1 |
| Al2O3 | 3.3 | 180 | 100nt – ssDNA | 1.8 μs nt −1 |
| Graphene–Al2O3– graphene | 6 | 340 | 850bp – dsDNA | 0.4 μs bp −1 |
3. Conclusions
In summary, we investigated the interaction effects of DNA and graphene as a means of slowing DNA transport through a nanopore. We find that a reduced rate of ssDNA translocation can be induced by the presence of exposed graphene layers on dielectric membranes. The results can be explained by the possibility of hydrophobic interactions between the nucleobases and graphene layers. The adsorption of nucleobases to DNA and subsequent desorption necessary for translocation provides the impeding force for DNA translocations. This is evidenced through direct comparison with dielectric membranes consisting of materials with highly charged surface at experimental pH conditions. Also the significant difference in translocation characteristics of ssDNA and dsDNA observed in these experiments validates our conclusions regarding the hydrophobic origins of these interactions.
4. Experimental Section
Nanopore Fabrication and Fluidic Measurements
The fabrication of the supporting membrane structure stack of Al2O3, SiNx, Al2O3, and chemical vapor deposition of graphene has been explained in detail previously.[20,27] The graphene–dielectric–graphene, grapheme–dielectric membranes are fabricated sequentially as described earlier. The dielectric (Al2O3) thickness of 24 nm is deposited using atomic layer deposition (ALD). The deposition is done at a platen temperature of 250 °C using tetramethylaluminum (TMA) as the metal precursor and water vapor as the oxygen precursor. A seed layer Al (2 nm thick) is deposited on graphene using a CHA SEC-600 electron-beam evaporator prior to deposition of both dielectric layers. For the pure dielectric membrane, a graphene–dielectric membrane stack is fabricated using the same process as above. Subsequently the graphene is etched away in Plasmalab Reactive Ion Etcher using an O2 plasma at 90 W for 30 s at a flow rate of 20 sccm. All nanopores are drilled using a JOEL 2010F field-emission gun TEM operated at 200 kV in CBED mode with focused electron probe of diameter = 1.6 nm. The nanopores take ≈30–40 s to form consistent with our previous studies on stacked structures.[27] To minimize damage to the graphene layers, beam alignment is performed on the supporting membrane area, which is much thicker and we wait about 5 min to minimize beam drift. The beam is then quickly moved to a clean fresh area on the FIB hole area to drill the pore. For the experiments involving the purely dielectric membrane, a pretreatment with an O2 plasma treatment is done for 1 min at 50 W to facilitate wetting. In all other experiments the chip is assembled as is. The chip is assembled in a custom-built chamber. Ethanol is filled in both reservoirs initially to help clean the devices and promote wetting. Subsequently, the ethanol is flushed out and the reservoirs are filled with a solution of 1 M KCl, 10 × 10−3 M Tris, 1 × 10−3 M EDTA at pH 7.6. All nanopore experiments are performed with Axopatch 200B at room temperature (22 ± 2 °C). Data are low-pass-filtered at 10 kHz using the built-in 8-pole Bessel filter. The output signal is sent to a Digidata 1440A data-acquisition module (Axon Instruments, USA) and is digitized at 100 kHz and recorded using pClamp 10.2 software. DNA translocation studies involve the use of 100 nt ssDNA (Integrated DNA Technologies) and 850bp dsDNA (Thermo Scientific).
MD Simulations
The software package NAMD[65] is used to perform all MD simulations. Periodic boundary conditions are applied to the simulation box. The simulations use particle mesh Ewald electrostatics[66] computed over a 0.11 nm grid to regulate long-range interactions. We employ multiple time-stepping[67] to calculate local interactions every time step and the full electrostatics every three time steps. SETTLE[68] and RATTLE[69] algorithms with a time step of 2 fs is used on the covalent bonds involving hydrogen atoms in water and DNA, respectively. The van der Waals forces are cut offsmoothly starting at 0.7 nm and completely cut offby 0.8 nm. The CHARMM27[70] force field is used for nucleic acids, graphene, water, and ions. Type CA atoms from CHARMM27 are used for graphene,[38] a custom force field is used for SiO2,[71] and NBFIX corrections are included for ions.[72] A langevin thermostat controls the temperature acting on the membrane atoms with a 1.0 ps−1 damping constant. In all simulations, the graphene and silica are harmonically restrained with a spring constant of 20 kcal mol−1 Å2. Atomic-scale models of annealed silica nanopores are built by adding silicon and oxygen atoms into a membrane volume, with a G-SMD force applied to expel the atoms from the pore and confine the atoms to the membrane volume. The membrane size is 10 nm × 10 nm in cross-section and 5.5 nm thick with an hourglass-shaped nanopore of minimum diameter 1.7 nm passing through the center. Because Al2O3 has a slight positive surface charge under a pH of 7.6, 39 oxygen atoms are removed from the membrane system to simulate the experimental conditions. The BKS[73] force field anneals the systems at 7000 K, 5000 K, 2000 K, and finally 300 K for 20 ps, 20 ps, 50 ps, and 50 ps respectively. For the stacked system, the inorganic builder plugin[74] of VMD (Visual Molecular Dynamics) produces a single-layer graphene sheet.[75] A pore is added to the sheet by removing atoms that satisfy the condition x2 + y2 < r2, where r = 1.75 nm is the radius of the graphene pore and is chosen to be the same as the silica pore radius at the mouth of the silica pore and custom cut to fit the rectangular simulation box. One copy of the graphene sheet is placed on top of the membrane, and another placed on the bottom. These graphene sheets are harmonically restrained in all simulations, and are not bonded to the silica. A 50 nt strand of poly(dT) ssDNA is inserted into the pore, and the system is solvated and ionized to produce an electrically neutral solution at 1 M KCl. The final dimensions of the system are 10 × 10 nm2 in cross-section, 16 nm in the z direction, and contained approximately 160 000 atoms. The systems equilibrate in the NPT ensemble for over 80 ns using a Nosé-Hoover Langevin piston pressure control[76] using a 1 atm pressure target and a 295 K temperature target. Furthermore, a small G-SMD force applied on the DNA atoms whenever they came near to the silica encourages unbinding of the DNA from the silica.[77] During the equilibration of over 80 ns, the DNA is allowed to move, and by the end of the equilibration, the DNA adheres to the graphene surface (graphene–silica–graphene) or clusters near the silica membrane (silica only). Following the equilibration, an electric field is added to the simulation in the z-direction, so that the electric potential over the entire simulation box dropped by 500 mV going from bottom to top. The stacked graphene system is simulated in the NVT ensemble with the electric field for 951 ns, and the silica only system is simulated in those same conditions for 464 ns. We calculate the center of mass of the backbone atoms for each nucleotide for each frame (approximately every 10 ps) by measuring the backbone positions over time. This position data are block averaged with a block length of 1 ns. Measurement of permeation is calculated by counting the number of nucleotides that were below a plane passing through the middle of the membrane. For nucleotides that were partly above the plane and partly below the plane, the fraction of atoms in the backbone below the plane is added to the count, so that the number permeated is not necessarily an integer.
Supplementary Material
Acknowledgments
The authors would like to acknowledge support from the National Institutes of Health (R21 CA155863), Oxford Nanopore Technologies U.K., National Science Foundation (DMR-0955959), and the National Institutes of Health (R01-HG007406) for supporting the effort. The authors also gladly acknowledge supercomputer time provided through XSEDE Allocation Grant No. MCA05S028 and the BlueWaters Sustained Petascale Computer System (UIUC).
Footnotes
Supporting Information is available from the Wiley Online Library or from the author.
Contributor Information
Shouvik Banerjee, Micro and Nanotechnology Laboratory, 208 North Wright Street Urbana, IL 61801, USA. Department of Materials Science and Engineering, University of Illinois Urbana-Champaign, Urbana, IL 61801, USA.
Dr. James Wilson, Department of Physics, University of Illinois Urbana-Champaign, Urbana, IL 61801, USA
Dr. Jiwook Shim, Micro and Nanotechnology Laboratory, 208 North Wright Street Urbana, IL 61801, USA. Department of Electrical and Computer Engineering, University of Illinois Urbana-Champaign Urbana, IL 61801, USA. Department of Bioengineering, University of Illinois Urbana-Champaign, Urbana, IL 61801, USA
Manish Shankla, Center for Biophysics and Computational Biology, University of Illinois Urbana-Champaign, Urbana, IL 61801, USA.
Dr. Elise A. Corbin, Micro and Nanotechnology Laboratory, 208 North Wright Street Urbana, IL 61801, USA. Department of Bioengineering, University of Illinois Urbana-Champaign, Urbana, IL 61801, USA
Prof. Aleksei Aksimentiev, Email: aksiment@illinois.edu, Department of Physics, University of Illinois Urbana-Champaign, Urbana, IL 61801, USA
Prof. Rashid Bashir, Email: rbashir@illinois.edu, Micro and Nanotechnology Laboratory, 208 North Wright Street Urbana, IL 61801, USA. Department of Electrical and Computer Engineering, University of Illinois Urbana-Champaign Urbana, IL 61801, USA. Department of Bioengineering, University of Illinois Urbana-Champaign, Urbana, IL 61801, USA
References
- 1.Venkatesan BM, Bashir R. Nat Nanotechnol. 2011;6:615. doi: 10.1038/nnano.2011.129. [DOI] [PubMed] [Google Scholar]
- 2.Deamer DW, Akeson M. Trends Biotechnol. 2000;18:147. doi: 10.1016/s0167-7799(00)01426-8. [DOI] [PubMed] [Google Scholar]
- 3.Clarke J, Wu HC, Jayasinghe L, Patel A, Reid S, Bayley H. Nat Nanotechnol. 2009;4:265. doi: 10.1038/nnano.2009.12. [DOI] [PubMed] [Google Scholar]
- 4.Kasianowicz JJ, Brandin E, Branton D, Deamer DW. Proc Natl Acad Sci USA. 1996;93:13770. doi: 10.1073/pnas.93.24.13770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ashkenasy N, Sánchez-Quesada J, Bayley H, Ghadiri MR. Angew Chem, Int Ed Engl. 2005;117:1425. doi: 10.1002/anie.200462114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Benner S, Chen RJ, Wilson NA, Abu-Shumays R, Hurt N, Lieberman KR, Deamer DW, Dunbar WB, Akeson M. Nat Nanotechnol. 2007;2:718. doi: 10.1038/nnano.2007.344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bayley H, Braha O, Cheley S, Gu L-Q. Nanobiotechnology. Wiley-VCH Verlag GmbH & Co. KGaA; Weinheim: 2005. p. 93. [Google Scholar]
- 8.Stoddart D, Maglia G, Mikhailova E, Heron AJ, Bayley H. Angew Chem, Int Ed Engl. 2010;49:556. doi: 10.1002/anie.200905483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Stoddart D, Heron AJ, Mikhailova E, Maglia G, Bayley H. Proc Natl Acad Sci USA. 2009;106:7702. doi: 10.1073/pnas.0901054106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Manrao EA, Derrington IM, Laszlo AH, Langford KW, Hopper MK, Gillgren N, Pavlenok M, Niederweis M, Gundlach JH. Nat Biotechnol. 2012;30:349. doi: 10.1038/nbt.2171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Derrington IM, Butler TZ, Collins MD, Manrao E, Pavlenok M, Niederweis M, Gundlach JH. Proc Natl Acad Sci USA. 2010;107:16060. doi: 10.1073/pnas.1001831107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Dekker C. Nat Nanotechnol. 2007;2:209. doi: 10.1038/nnano.2007.27. [DOI] [PubMed] [Google Scholar]
- 13.Li J, Stein D, McMullan C, Branton D, Aziz MJ, Golovchenko JA. Nature. 2001;412:166. doi: 10.1038/35084037. [DOI] [PubMed] [Google Scholar]
- 14.Healy K, Schiedt B, Morrison AP. Nanomedicine. 2007;2:875. doi: 10.2217/17435889.2.6.875. [DOI] [PubMed] [Google Scholar]
- 15.Geim AK. Science. 2009;324:1530. doi: 10.1126/science.1158877. [DOI] [PubMed] [Google Scholar]
- 16.Garaj S, Hubbard W, Reina A, Kong J, Branton D, Golovchenko JA. Nature. 2010;467:190. doi: 10.1038/nature09379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Schneider GF, Kowalczyk SW, Calado VE, Pandraud G, Zandbergen HW, Vandersypen LM, Dekker C. Nano Lett. 2010;10:3163. doi: 10.1021/nl102069z. [DOI] [PubMed] [Google Scholar]
- 18.Merchant CA, Healy K, Wanunu M, Ray V, Peterman N, Bartel J, Fischbein MD, Venta K, Luo Z, Johnson AT, Drndic M. Nano Lett. 2010;10:2915. doi: 10.1021/nl101046t. [DOI] [PubMed] [Google Scholar]
- 19.Garaj S, Liu S, Golovchenko JA, Branton D. Proc Natl Acad Sci USA. 2013;110:12192. doi: 10.1073/pnas.1220012110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Venkatesan BM, Estrada D, Banerjee S, Jin X, Dorgan VE, Bae MH, Aluru NR, Pop E, Bashir R. ACS Nano. 2012;6:441. doi: 10.1021/nn203769e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Novoselov KS, Geim AK, Morozov SV, Jiang D, Zhang Y, Dubonos SV, Grigorieva IV, Firsov AA. Science. 2004;306:666. doi: 10.1126/science.1102896. [DOI] [PubMed] [Google Scholar]
- 22.Bolotin KI, Sikes KJ, Jiang Z, Klima M, Fudenberg G, Hone J, Kim P, Stormer HL. Solid State Commun. 2008;146:351. [Google Scholar]
- 23.Traversi F, Raillon C, Benameur SM, Liu K, Khlybov S, Tosun M, Krasnozhon D, Kis A, Radenovic A. Nat Nanotechnol. 2013;8:939. doi: 10.1038/nnano.2013.240. [DOI] [PubMed] [Google Scholar]
- 24.Saha KK, Drndic M, Nikolic BK. Nano Lett. 2012;12:50. doi: 10.1021/nl202870y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Postma HW. Nano Lett. 2010;10:420. doi: 10.1021/nl9029237. [DOI] [PubMed] [Google Scholar]
- 26.Tsutsui M, Taniguchi M, Yokota K, Kawai T. Nat Nanotechnol. 2010;5:286. doi: 10.1038/nnano.2010.42. [DOI] [PubMed] [Google Scholar]
- 27.Banerjee S, Shim J, Rivera J, Jin X, Estrada D, Solovyeva V, You X, Pak J, Pop E, Aluru N, Bashir R. ACS nano. 2012;7:834. doi: 10.1021/nn305400n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Venkatesan BM, Shah AB, Zuo JM, Bashir R. Adv Funct Mater. 2010;20:1266. doi: 10.1002/adfm.200902128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Larkin J, Henley R, Bell DC, Cohen-Karni T, Rosenstein JK, Wanunu M. ACS Nano. 2013;7:10121. doi: 10.1021/nn404326f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kowalczyk SW, Wells DB, Aksimentiev A, Dekker C. Nano Lett. 2012;12:1038. doi: 10.1021/nl204273h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Fologea D, Uplinger J, Thomas B, McNabb DS, Li J. Nano Lett. 2005;5:1734. doi: 10.1021/nl051063o. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Di Fiori N, Squires A, Bar D, Gilboa T, Moustakas TD, Meller A. Nat Nanotechnol. 2013;8:946. doi: 10.1038/nnano.2013.221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Paik KH, Liu Y, Tabard-Cossa V, Waugh MJ, Huber DE, Provine J, Howe RT, Dutton RW, Davis RW. ACS Nano. 2012;6:6767. doi: 10.1021/nn3014917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Wanunu M, Sutin J, McNally B, Chow A, Meller A. Biophys J. 2008;95:4716. doi: 10.1529/biophysj.108.140475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Zhang H, Zhao Q, Tang Z, Liu S, Li Q, Fan Z, Yang F, You L, Li X, Zhang J, Yu D. Small. 2013;9:4112. doi: 10.1002/smll.201301263. [DOI] [PubMed] [Google Scholar]
- 36.Liu Q, Wu H, Wu L, Xie X, Kong J, Ye X, Liu L. PLoS One. 2012;7:e46014. doi: 10.1371/journal.pone.0046014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Krishnakumar P, Gyarfas B, Song W, Sen S, Zhang P, Krstic P, Lindsay S. ACS Nano. 2013;7:10319. doi: 10.1021/nn404743f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Wells DB, Belkin M, Comer J, Aksimentiev A. Nano Lett. 2012;12:4117. doi: 10.1021/nl301655d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Shankla M, Aksimentiev A. Nat Commun. 2014;5:5171. doi: 10.1038/ncomms6171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Schneider GF, Xu Q, Hage S, Luik S, Spoor JNH, Malladi S, Zandbergen H, Dekker C. Nat Commun. 2013;4:2619. doi: 10.1038/ncomms3619. [DOI] [PubMed] [Google Scholar]
- 41.Wu M, Kempaiah R, Huang PJJ, Maheshwari V, Liu J. Langmuir. 2011;27:2731. doi: 10.1021/la1037926. [DOI] [PubMed] [Google Scholar]
- 42.Kim S, Nah J, Jo I, Shahrjerdi D, Colombo L, Yao Z, Tutuc E, Banerjee SK. Appl Phys Lett. 2009;94:062107. [Google Scholar]
- 43.Venkatesan BM, Dorvel B, Yemenicioglu S, Watkins N, Petrov I, Bashir R. Adv Mater. 2009;21:2771. doi: 10.1002/adma.200803786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Chen P, Mitsui T, Farmer DB, Golovchenko J, Gordon RG, Branton D. Nano Lett. 2004;4:1333. doi: 10.1021/nl0494001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Smeets RMM, Keyser UF, Krapf D, Wu MY, Dekker NH, Dekker C. Nano Lett. 2005;6:89. doi: 10.1021/nl052107w. [DOI] [PubMed] [Google Scholar]
- 46.Dessinges MN, Maier B, Zhang Y, Peliti M, Bensimon D, Croquette V. Phys Rev Lett. 2002;89:248102. doi: 10.1103/PhysRevLett.89.248102. [DOI] [PubMed] [Google Scholar]
- 47.Kim MJ, Wanunu M, Bell DC, Meller A. Adv Mater. 2006;18:3149. [Google Scholar]
- 48.Wu MY, Smeets RMM, Zandbergen M, Ziese U, Krapf D, Batson PE, Dekker NH, Dekker C, Zandbergen HW. Nano Lett. 2008;9:479. doi: 10.1021/nl803613s. [DOI] [PubMed] [Google Scholar]
- 49.Ramachandran A, Liu Y, Asghar W, Iqbal S. Am J Biomed Sci. 2009;1:344. [Google Scholar]
- 50.Tang WY, Hu GH. RSC Adv. 2013;3:19861. [Google Scholar]
- 51.Akca S, Foroughi A, Frochtzwajg D, Postma HWC. PLoS One. 2011;6:e18442. doi: 10.1371/journal.pone.0018442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Koenig SP, Boddeti NG, Dunn ML, Bunch JS. Nat Nanotechnol. 2011;6:543. doi: 10.1038/nnano.2011.123. [DOI] [PubMed] [Google Scholar]
- 53.Teweldebrhan D, Balandin AA. Appl Phys Lett. 2009;94:013101. [Google Scholar]
- 54.Alloyeau D, Ding B, Ramasse Q, Kisielowski C, Lee Z, Jeon KJ. Chem Commun. 2011;47:9375. doi: 10.1039/c1cc13654b. [DOI] [PubMed] [Google Scholar]
- 55.Venta K, Shemer G, Puster M, RodrÍguez-Manzo JA, Balan A, Rosenstein JK, Shepard K, Drndić M. ACS Nano. 2013;7:4629. doi: 10.1021/nn4014388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Liu J. Phys Chem Chem Phys. 2012;14:10485. doi: 10.1039/c2cp41186e. [DOI] [PubMed] [Google Scholar]
- 57.Huang PJ, Liu J. Nanomaterials. 2013;3:221. doi: 10.3390/nano3020221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Varghese N, Mogera U, Govindaraj A, Das A, Maiti PK, Sood AK, Rao CNR. ChemPhysChem. 2009;10:206. doi: 10.1002/cphc.200800459. [DOI] [PubMed] [Google Scholar]
- 59.Gowtham S, Scheicher RH, Ahuja R, Pandey R, Karna SP. Phys Rev B. 2007;76:033401. [Google Scholar]
- 60.Venkatesan B, Polans J, Comer J, Sridhar S, Wendell D, Aksimentiev A, Bashir R. Biomed Microdevices. 2011;13:671. doi: 10.1007/s10544-011-9537-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Smith SB, Cui Y, Bustamante C. Science. 1996;271:795. doi: 10.1126/science.271.5250.795. [DOI] [PubMed] [Google Scholar]
- 62.Maffeo C, Ngo TTM, Ha T, Aksimentiev A. J Chem Theory Comput. 2014;10:2891. doi: 10.1021/ct500193u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Tinland B, Pluen A, Sturm J, Weill G. Macromolecules. 1997;30:5763. [Google Scholar]
- 64.Chi Q, Wang G, Jiang J. Phys A. 2013;392:1072. [Google Scholar]
- 65.Phillips JC, Braun R, Wang W, Gumbart J, Tajkhorshid E, Villa E, Chipot C, Skeel RD, Kalé L, Schulten K. J Comput Chem. 2005;26:1781. doi: 10.1002/jcc.20289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Darden T, York D, Pedersen L. J Chem Phys. 1993;98:10089. [Google Scholar]
- 67.Batcho PF, Case DA, Schlick T. J Chem Phys. 2001;115:4003. [Google Scholar]
- 68.Miyamoto S, Kollman PA. J Comput Chem. 1992;13:952. [Google Scholar]
- 69.Andersen HC. J Comput Phys. 1983;52:24. [Google Scholar]
- 70.MacKerell AD, Bashford D, Bellott M, Dunbrack RL, Evanseck JD, Field MJ, Fischer S, Gao J, Guo H, Ha S, Joseph-McCarthy D, Kuchnir L, Kuczera K, Lau FTK, Mattos C, Michnick S, Ngo T, Nguyen DT, Prodhom B, Reiher WE, Roux B, Schlenkrich M, Smith JC, Stote R, Straub J, Watanabe M, Wiórkiewicz-Kuczera J, Yin D, Karplus M. J Phys Chem B. 1998;102:3586. doi: 10.1021/jp973084f. [DOI] [PubMed] [Google Scholar]
- 71.Cruz-Chu ER, Aksimentiev A, Schulten K. J Phys Chem B. 2006;110:21497. doi: 10.1021/jp063896o. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Yoo J, Aksimentiev A. J Phys Chem Lett. 2011;3:45. [Google Scholar]
- 73.van Beest BWH, Kramer GJ, van Santen RA. Phys Rev Lett. 1990;64:1955. doi: 10.1103/PhysRevLett.64.1955. [DOI] [PubMed] [Google Scholar]
- 74.Aksimentiev A, Brunner R, Cruz-Chu E, Comer J, Schulten K. IEEE Nanotech Mag. 2009;3:20. doi: 10.1109/MNANO.2008.931112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Humphrey W, Dalke A, Schulten K. J Mol Graph. 1996;14:33. doi: 10.1016/0263-7855(96)00018-5. [DOI] [PubMed] [Google Scholar]
- 76.Martyna GJ, Tobias DJ, Klein ML. J Chem Phys. 1994;101:4177. [Google Scholar]
- 77.Comer J, Dimitrov V, Zhao Q, Timp G, Aksimentiev A. Biophys J. 2009;96:593. doi: 10.1016/j.bpj.2008.09.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.