

Abstract
Several HIV envelope-targeting (Env-targeting) antibodies with broad and potent neutralizing activity have been identified and shown to have unusual features. Of these, the PG9 antibody has a long heavy chain complementarity determining region 3 (HCDR3) and possesses unique structural elements that interact with protein and glycan features of the HIV Env glycoprotein. Here, we used the Rosetta software suite to design variants of the PG9 antibody HCDR3 loop with the goal of identifying variants with increased potency and breadth of neutralization for diverse HIV strains. One variant, designated PG9_N100FY, possessed increased potency and was able to neutralize a diverse set of PG9-resistant HIV strains, including those lacking the Env N160 glycan, which is critical for PG9 binding. An atomic resolution structure of the PG9_N100FY fragment antigen binding (Fab) confirmed that the mutated residue retains the paratope surface when compared with WT PG9. Differential scanning calorimetry experiments revealed that the mutation caused a modest increase in thermodynamic stability of the Fab, a feature predicted by the computational model. Our findings suggest that thermodynamic stabilization of the long HCDR3 in its active conformation is responsible for the increased potency of PG9_N100FY, and strategies aimed at stabilizing this region in other HIV antibodies could become an important approach to in silico optimization of antibodies.
Keywords: AIDS/HIV, Autoimmunity, Immunology, Infectious disease
Introduction
Recent studies have described the isolation of a number of human mAbs directed to the HIV envelope (Env) that exhibit broad and potent neutralizing activity, many of which exhibit unusual features (1–5). One of the most interesting mAbs described to date is PG9, which possesses an antibody heavy chain complementarity determining region 3 (HCDR3) loop with a “hammerhead” structure formed by stabilizing interactions within the loop (2). PG9 and the related antibody PG16 interact with HIV Env using similar binding modes but diverge in amino acid sequences with 24% amino acid differences in their antibody heavy chain variable regions, and 33% amino acid differences in their HCDR3 sequences (6). Therefore, we hypothesized that the HCDR3 loop of PG9 could be redesigned to achieve improved affinity of binding, increased potency, and increased breadth of neutralization for diverse HIV strains. The large number of potential variants of the loop made systematic synthesis and testing of all possible variants resource intensive, if not prohibitive. Therefore, we used a hybrid method for optimization, in which we coupled structure-based computational design and experimental validation to identify candidate variant antibodies with minimal alteration that exhibited increased potency and breadth. Remarkably, we found PG9 variant antibodies with single amino acid changes in noncontact regions of the HCDR3 loop that exhibited increased affinity and neutralizing activity for HIV Env, and they achieved these enhanced functional activities without altering the paratope surface of the HCDR3. Instead, the variant amino acid caused an increase in thermodynamic stability of the Fab. The crystal structure of the variant Fab and direct measurements of antibody thermodynamic stability are in agreement with the model. The results suggest an exciting approach to improving the functional activity of HIV-neutralizing mAbs with long HCDR3s through thermodynamic stabilization of the HCDR3 in its active conformation. The work also illustrates the power of integrating Rosetta computational design with experimental verification for rapid design of antibodies with enhanced properties.
Results
Design process.
We retrieved the atomic resolution structure of the mAb PG9 in complex with the HIV CAP45.2.00.G3 variant V1/V2 scaffold from the Protein Data Bank (PDB ID: 3U4E) (2). The structure reveals interesting interaction features of the complex (Supplemental Video 1). Next, we used RosettaDesign for a complete redesign of the HCDR sequence by simultaneously optimizing predicted thermodynamic stability of the antibody/HIV Env complex as well as predicted binding affinity (7–10). RosettaDesign works by fixing the conformation of the backbone coordinates and optimizing the side-chains and amino acid identities to minimize the Rosetta energy function. The Rosetta energy function (10) approximates the free energy by computing a weighted sum of a variety of individual terms, such as van der Waals interaction, desolvation, or hydrogen bonding. Most energy terms are derived using the PDB as a knowledge base and applying Boltzmann’s relation. Using RosettaDesign for PG9, the native amino acids present in the original PG9 sequence were recovered for a majority of the amino acid sequence positions (Figure 1A). However, the design process suggested different amino acids at antibody positions 100F, 100L, and 101 (Kabat numbering) (11). These mutations were predicted to increase HCDR3 stability and/or affinity in the antibody/HIV Env interaction (Figure 1, A and B, and Supplemental Figure 1; supplemental material available online with this article; doi:10.1172/JCI80693DS1). We visually inspected the 7 best-scoring single-point mutants (Figure 1B) to rationalize the effects of the proposed changes and choose variants for further study. Specifically, we did not pursue further evaluation of designs that appeared to compromise the structural integrity of the HCDR3 loop; we rejected Y100AF and N100FF on this basis (Supplemental Figure 1). We also rejected M101E, as it was predicted to possess favorable design features only in the context of additional mutations (Supplemental Figure 1). Accordingly, we chose to further study 3 single-point mutations, N100FY, N100FL, and D100LN, which each were predicted to be beneficial on their own (Figure 1, B and C, and Supplemental Video 2). Further, we selected a variant containing more than one mutation (A96S and Y100QN; designated here PG9_2MUT), and one variant with 4 mutations (A96S, N100FL, D100LN, and Y100QN; designated here PG9_4MUT).
Figure 1. Rosetta predicts mutations that enhance PG9 fitness.

(A) Redesigned PG9 sequences for the HCDR3 loop are shown as sequence logo. The height of the logo, in bits, indicates how many times an amino acid was preferred at that position in 100 simulations using Rosetta. The WT sequence, with 3 HCDR3 position numbering schemes (top: the sequentially numbered position as each residue appears in the PDB file; middle: IMGT, ref. 47; and bottom: Kabat, ref. 11) is shown on the x-axis. (B) Mutational energy analysis for each residue in the HCDR3, shown using Kabat numbering. Mutational fitness due to stability and binding energy ± STD (y-axis) is shown as a function of individual mutations (x-axis); a more negative fitness energy score is preferred. The mutations are grouped and colored by position. For mutations that were made experimentally, a panel shows the WT PG9 sequence in ball-and-stick representation, where the carbons match the color of the bar in the graph, nitrogens are shown in deep-blue, and oxygens are shown in red. The HCDR3 residues are shown in blue. Residues within 5 Å are shown as stick representation. The V1/V2 scaffold (PDB ID: 3U4E) (2) is shown in yellow with glycans at positions 156 and 160 (HIV strain HXBc2 numbering) shown in surface representation. Non-HCDR3 heavy chain amino acids are shown in white. Light chain amino acids are shown in pink. (C) Alignment of HCDR3 sequences of the 5 PG9 variants that were characterized experimentally. The WT PG9 sequence is shown at the top, with Kabat numbering.
Binding studies.
WT PG9 neutralizes some HIV variants for which it lacks binding to gp120 monomer, because it binds to the gp160 homo-trimer but not the gp120 monomer for those strains (12). However, it is also known that WT PG9 binds to gp120 monomers of some viral strains (2, 4, 5, 12). Therefore, we used a panel of representative gp120 monomers from HIV clades B and C to perform initial screening for binding of PG9 variants to Env (13, 14). Preliminary binding studies of the PG9_2MUT variant with computationally designed mutations produced a markedly lower signal than that of WT PG9, and this variant was not considered further (data not shown). For the remaining PG9 variants, we calculated half maximal effective concentration (EC50) values. For WT PG9 binding to gp120 monomers, the results were in agreement with previous studies (2, 15). For each gp120 monomer tested, the PG9 variants N100FL and N100FY exhibited 2.3- to 14.2-fold stronger binding than WT PG9 (Figure 2 and Supplemental Figure 2, P value 0.002), while PG9_D100LN exhibited binding affinities mostly comparable to WT PG9 (P value 0.323). PG9_4MUT exhibited 2- to 100-fold reduced binding affinities (P value 0.002). We also determined the EC50 for binding of these PG9 variants to a recombinant form of native gp140 trimer that is recognized by PG9, termed BG505-SOSIP.664 (16–18). In these assays, PG9_N100FL and PG9_N100FY exhibited 3.5- or 5.9-fold stronger binding affinity, respectively, when compared with WT PG9. The variant PG9_N100FY showed a binding curve similar to that of the glycan-specific mAb 2G12, the antibody that was used to affinity purify the trimer (Supplemental Figure 2 and ref. 18).
Figure 2. Rosetta redesigned mutants exhibit increased breadth and potency against HIV.

Binding and neutralization profiles for the characterized PG9 mutants. Antibody mutants are shown using Kabat numbering. The virus name is shown in the left column, and amino acid sequence for position 160 (HIV strain HXBc2 numbering) is shown in the second column. Strains lacking a potential N-linked glycosylation site are indicated with bold rows. A yellow-to-green color scale indicates ranges of values, from less than 0.01 μg/ml to greater than 100 μg/ml, respectively, for concentrations needed for half maximum signal in ELISA (EC50) in the left panel. A yellow-to-green scale from 0.4 ng/ml to greater than 33 μg/ml, respectively, is shown for half-maximal concentration needed to inhibit virus infection (IC50), in the right panel. Each ELISA experiment was done in triplicate, and neutralization was done in duplicate. A Wilcoxon test was used to determine if the distributions for binding and neutralization were statistically significant from WT PG9. The P value for comparison of each variant against WT PG9 (comparing the pattern of results in a single variant antibody column against the results in the left column for WT PG9) is shown at the bottom of the panels and reflects a 2-sided significance at a level of 0.05.
Neutralization assays.
We next tested the panel of redesigned PG9 variants and WT PG9 for neutralizing activity against a panel of PG9-susceptible or -resistant viruses using a TZM-bl neutralization assay (19). PG9_N100FY exhibited increased neutralization potency for all viruses tested, including viral variants for which WT PG9 did not have activity (i.e., had neutralization concentration >33 μg/ml; Figure 2). We also tested HIV strains that lacked the glycan at Env position N160, using naturally occurring or engineered knockouts for Env sequences (20). Remarkably, PG9_N100FY neutralized 7 out of the 9 viruses tested that lacked N160 glycan, with inhibitory activity at concentrations as low as 2.7 μg/ml (Figure 2). PG9_N100FL also exhibited an increase in potency against HIV strains compared with WT PG9, although not at the same level as PG9_N100FY (P value < 0.001). Although the magnitude of the improvement was modest in some cases, the improvement was consistent over a wide variety of HIV strains (P value < 0.001, geometric mean of 0.64 PG9_N100FY vs. WT PG9 2.31). We also found a decrease in potency to be statistically significant for the virus panel tested for PG9_D100LN (P value < 0.001).
Crystal structure of PG9_N100FY.
To validate the Rosetta models for the structure of the PG9_N100FY antibody, we determined the crystal structure for its Fab at 2.3 Å resolution (Supplemental Table 1). All residues of the HCDR3 could be resolved in the electron density, including the N100FY mutation (Figure 3A). Overall, the HCDR3 of PG9_N100FY Fab adopts the same hammerhead conformation as in the WT PG9-gp120 V1/V2 complex structure, with a Cα root mean square deviation (RMSD) of 0.43 Å for this loop compared with the complexed structure (PDB ID: 3U4E), and a Cα RMSD of 0.68 Å against the Rosetta complexed model (Figure 3B), i.e., the Fab crystal structure and its modeled structure adopt the same conformation. We also note that the side of the HCDR3 hammerhead that harbors the N100FY mutation has increased B values but is significantly more ordered than in the previous crystal structure of WT PG9, in which the HCDR3 hammerhead could not be resolved (PDB ID: 3U36). We interpret in this observation that the N100FY mutation provides additional stability to the HCDR3 conformation for recognition of the glycoprotein by forming π-π stacking interactions with the WT residues P99 and Y100A. The PG9_N100FY Fab electron density, albeit weak, indicates that the substituted tyrosine at this position does not possess an associated sulfation. Two sulfated tyrosines (indicated in this manuscript as YS — specifically YS100G and YS100H) in WT PG9 remain sulfated in the variant Fab PG9_N100FY.
Figure 3. PG9_N100FY Fab crystal structure and Rosetta models.

(A) Unliganded PG9_N100FY HCDR3 crystal structure. 2Fo – Fc electron density is shown as a mesh grid contoured at 1σ. Electron density shows tyrosine sulfation at YS100G and YS100H but not at N100FY. N100FY stacks against P99 and Y100A, thereby providing increased stability. (B) Unliganded crystal structure aligned with Rosetta complexed model has an all-atom RMSD of 0.68 Å. Rosetta top-scoring model modeled from PDB ID: 3U4E complex is shown in orange. V1/V2 antigen from the model is shown as a gray surface. Crystal structure of the HCDR3 loop of PG9_N100FY is shown in pink. The N100FY mutation, highlighted in red, adopts the same conformation in the crystal structure as in the Rosetta model. (C) Rosetta scores for HCDR3 binding, total binding, HCDR3 stabilization, or total stabilization (scores shown as REU for 1000 simulations ± STD). Only significant differences where the means were below that of WT PG9 are shown. All other comparisons were not significant with a confidence of 0.05 or the means were above that of WT PG9 using a Student’s 2-sided t test. (D) Models for WT PG9 and the top scoring variants N100FL and N100FY are shown in a sphere and surface representation. The atom types are colored for carbon (gray), oxygen (red), or nitrogen (blue). The V1/V2 epitope is indicated in green, the antibody HCDR3 loop in dark blue, the N160 complex glycan in dark green, and non-HCDR3 portions of the antibody heavy chain in black.
Mechanism of improved binding.
We sought to understand the molecular basis for the increased potency and breadth of these PG9 variants using Rosetta analysis of variants with single mutations. N100F was mutated in WT PG9 to N100FY, and the interaction with Env was compared with that of WT PG9 using the Rosetta scoring function (Figure 3C). We calculated stabilization and binding energy for the HCDR3, stabilization of the full HCDR3 loop, and binding energy for the antibody-Env interaction (further explained in Supplemental Table 2). For each metric calculated, we observed statistically significant improvements in HCDR3 only for N100FY (P < 0.001). Consistent with the WT PG9 structure, antibody position 100F is located on an antiparallel β-sheet at the apical tip of the HCDR3, forming a hydrophobic pocket near the interface of the antigen (Figure 3D). The pocket is formed by Y100A, YS100G, and P99 of the antibody heavy chain. In addition, the asparagine at position 167 on the antigen face of gp120 contacts this pocket. The structural models reveal that a large, hydrophobic amino acid at position 100F is predicted to fill the hydrophobic pocket there, thereby likely stabilizing the HCDR3 conformation for interaction with the HIV V1/V2 epitope (Figure 3D). In addition, the polar group on the designed N100FY mutant points into accessible solvent space. Both the attractive forces of the hydrophobic bulk and the improved solvation potential were reflected in the Rosetta scoring function (Supplemental Figure 3). Further, the PG9_N100FY adds a π-π stacking interaction with YS100G that is not present with the asparagine side chain in WT PG9 (Supplemental Figure 3).
Differential scanning calorimetry.
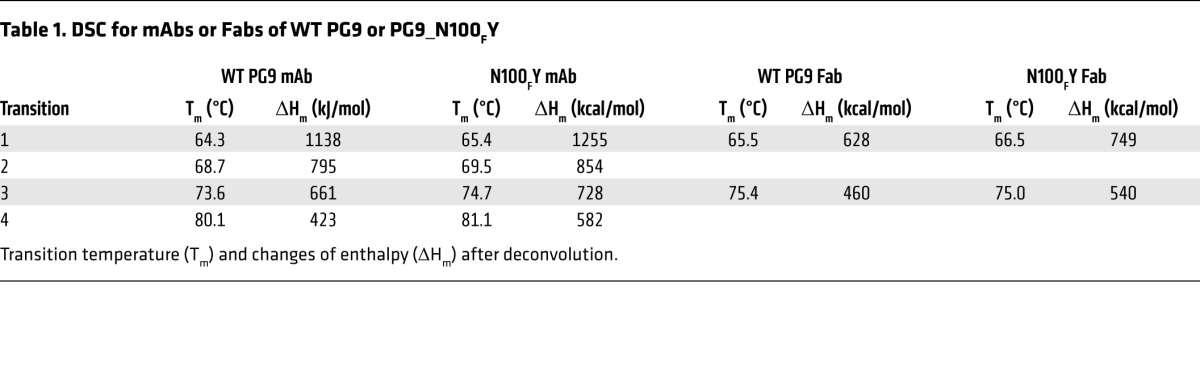
The structure of the PG9_N100FY Fab suggested that the observed increase in breadth and neutralization potency resulted not from an alteration in the contact surface of the HCDR3, but rather from a stabilization of the loop through increased thermodynamic stability. To test this parameter directly, we compared the WT PG9 and PG9_N100FY variant Fabs and full-length IgG mAbs using differential scanning calorimetry (DSC). The DSC curves of the Fabs showed a biphasic transition, while the full-length IgG mAbs showed a quadriphasic transition (Table 1 and Supplemental Figure 4). The 1st and 2nd peaks observed from Fab denaturation appeared to correspond to the 1st and 3rd peaks observed from mAb denaturation. The thermo-denaturation is irreversible under the conditions tested, which has been observed in many other antibody unfolding experiments (21–24); hence, the melting temperature and enthalpy deconvolution reported are apparent values. The apparent melting temperature (Tm) derived from the 1st peak from WT PG9 mAb/Fab was about 1°C lower than that from PG9_N100FY mAb/Fab. Deconvolution of the DSC thermogram showed higher apparent calorimetric enthalpy changes for PG9_N100FY during thermal denaturation, which indicates that more heat was absorbed during denaturation (24). The difference in apparent calorimetric enthalpy indicates that the PG9_N100FY mutation causes an altered transition state during thermo-denaturation. Taking the shift in apparent Tm by 1°C and higher denaturation heat together, the DSC results suggested that PG9_N100FY is thermodynamically more stable than WT PG9.
Table 1. DSC for mAbs or Fabs of WT PG9 or PG9_N100FY.
Absence of autoreactivity for PG9_N100FY.
Antibodies with long HCDR3s are thought to exhibit a high frequency of autoreactivity (25). Indeed, we have shown previously that the length of antibody genes in B cells encoding long HCDR3s usually are generated at the time of original recombination, not by insertional mutagenesis during somatic hypermutation; however, their frequency is reduced as B cells move from the naive B cell compartment to memory B cell compartments (26). Additionally, some broadly neutralizing antibodies against HIV have been shown to be autoreactive (27). Thus, we considered whether the mutation that we made synthetically in PG9_N100FY might inadvertently increase autoreactivity, and therefore the increase in breadth of binding could be attributed to nonspecific binding. We compared the binding to human HEp-2 cell antigens of PG9wt, PG9_N100FY, 4E10 (an HIV mAb with known autoreactive properties), or a heterologous control antibody, motavizumab, directed to the respiratory syncytial virus fusion protein (Supplemental Figure 5 and ref. 28). PG9_N100FY exhibited a comparable level of reactivity as PG9 and a lower level of reactivity than the other control antibodies, suggesting the increased thermodynamic stability of the HCDR3 did not cause undesired acquisition of autoreactive or nonspecific binding.
Discussion
These results have important implications for antibody design because they reveal the potential of computational modeling to design antibodies with improved function. The crystal structure shows that the mutations recovered from RosettaDesign are pointed away from the antigen V1/V2 strand-C face. Based on the DSC results, we conclude that the increased potency and breadth of the PG9 variant might result from a thermal stabilization of the HCDR3, which allows the loop to adopt a more lock-and-key–like binding mode, limiting the entropic cost when engaging the antigen. Our ability to resolve the HCDR3 of an unliganded PG9 variant — in contrast to WT PG9 — also is consistent with increased thermal stabilization of the HCDR3, although we cannot rule out contributions from a different crystal packing in the PG9 variant. This finding of increased loop thermostability was obtained in the context of a holistic model and facilitated identification of a variant antibody that bound in a glycan-independent manner and exhibited increased potency. This finding is consistent with recent mutagenesis experiments showing that noncontact residues are essential for antigen recognition by many broadly neutralizing antibodies to HIV (29). Noncontact residues in antibody frameworks contribute to high-affinity binding by facilitating formation and stability of a preconfigured, low-energy binding site (30–33). Optimally configured binding sites form ordered paratopes that pay a smaller entropic penalty upon forming antibody-antigen complexes (34). We cannot rule out the possibility that, in addition to increasing thermostability of the CDRH3, the N100FY mutation in PG9 also contributes additional interactions with the antigen at the trimer apex; these interactions are yet to be understood in the absence of a high-resolution PG9-Env structure.
This work also demonstrates the potential of combining computational design with expert knowledge and validation experiments. For a HCDR3 loop of 28–amino acids, 532 single-point mutants are possible, and the theoretical number of variants with more than one mutation (2028) is enormous. From this large potential set of mutated antibodies, Rosetta identified a focused panel of candidate PG9 variants, from which a small subset was considered favorable, and 2 of 5 experimentally tested variants exhibited enhanced potency and breadth of neutralization. The computational experiments provided enrichment for variants with improved binding, leading to an efficient laboratory process. As expected, the predictions were not completely accurate. For example, although the predictive model suggested that D100LN would have the greatest increase in fitness (indicated by a more negative score; Figure 1 and Supplemental Figure 1), this variant was not improved in activity. We hypothesize that this discrepancy is due to approximations made in the Rosetta energy function, incompleteness of the conformational sampling, or the absence of a starting trimeric crystal structure complexed with PG9. In summary, 2 of 5 designed variants that were tested showed improved binding, one showed comparable binding, and 2 of 5 exhibited reduced binding.
The design of more potent PG9 and PG16 antibodies have been previously reported (5). For example, 3 PG16-specific residues (arginine, serine, and histidine designated RSH) were shown to be critical for binding sialic acid on complex-type glycans, and introduction of these residues into a PG9 backbone produced a chimeric antibody with enhanced HIV-1 neutralization. That work involved previously reported crystal structures to inform chimerization experiments. In contrast, in the work presented here, computational design yielded a more potent and broadly neutralizing PG9 variant that bound and neutralized in a glycan-independent fashion.
Despite the large-scale efforts in recent years to isolate HIV antibodies, it is apparent that further antibody optimization is still possible. For example, using the BG505 SOSIP Env, novel antibodies to the V1/V2 loop region, including the potent PGDM1400 antibody, were isolated (35). Additional antibody exploration efforts may be worthwhile to identify antibodies of interest with which to design epitope mimetic vaccines, as has been successfully implemented recently in other systems (36–38). Our findings suggest that in silico thermodynamic stabilization of long HCDR3s in their active conformation could become an important approach to optimization of antibodies for enhanced potency and breadth.
Methods
Redesign of PG9 HCDR3.
Using the RosettaDesign algorithm (9), iterative rounds of design, docking, and minimization were applied to each position in the HCDR3, with a small energetic bonus applied to recovery of the native sequence. The small energetic bonus is commonly applied to redesigned models to reduce complexity of sequence space for large designs (9). 100 models were generated using this protocol (http://www.crowelab.com/protocol-captures). For each mutated position, mutations seen in greater that 10% of the models and conveying an energetic bonus of greater than 1.0 Rosetta energy units (REU) were inspected manually using PyMOL (Version 1.5.0.4; Schrödinger LLC) and compared with the native sequence along with the native PG9 structure (PDB ID: 3U4E) (2). The change in fitness for any mutation was determined to be the sum of the contribution to binding energy, as compared with WT for each mutation (ΔΔΔG) (39), and increased thermodynamic stability energy compared with WT PG9 (ΔΔG). Both energies were approximated by the Rosetta scoring function.
Antibody and gp120 expression.
We used recombinant expression in mammalian cells to recover natural posttranslational modifications, as previously described (40). Briefly, the mAb PG9 heavy- and light-chain genes were cloned into the pEE6.4 and pEE12.4 vectors, respectively (Lonza). BsiWI and XhoI cloning sites were generated at amino acid position 93 and 103, respectively. Variant HCDR3 sequences were synthesized and cloned into the PG9 backbone (GeneArt) using the unique cloning sites. The DNA was cotransfected at a 1:1 heavy-light ratio into FreeStyle 293-F cells (Human Embryonic Kidney; Invitrogen) using 25 kDa linear polyethylenimine (PEI; Polysciences Inc.) transfection reagent at a ratio 2:1 of PEI to DNA. 1L of culture was used for each variant, and the supernatant was collected on day 5. Antibody was purified and concentrated from supernatant on a protein G column (GE Healthcare). The expression levels for all PG9 variants were comparable to that of WT PG9.
A cDNA encoding each gp120 was cloned into pCDNA3.4 (Invitrogen) using HindIII and EcoRI restriction sites. A CD5 signal peptide or 8X His tag was cloned onto the 5′ or 3′ end, respectively. The DNA was transfected into FreeStyle 293-F cells (Human Embryonic Kidney; Invitrogen) using PEI at a ratio of 2:1. On day 5, the supernatant was clarified and the protein was purified on a 5 ml HisTALON cobalt column (Clontech), according to the manufacturer’s specifications. The protein was concentrated using Amicon Ultra centrifugal filters with a 100 kD cutoff (Millipore) and further purified on a Superdex column (GE Healthcare) using size exclusion.
BG505 SOSIP.664 trimer protein was received as a gift from John P. Moore, the Weill Medical College of Cornell University, New York, New York, USA.
PG9 variant characterization.
ELISA plates were coated with 3 μg/ml of gp120 and incubated overnight at 4°C. The wells were washed with phosphate-buffered saline with 0.05% Tween (PBS-T) in the following steps. The uncoated sites on the wells were blocked with 2% milk and 1% goat serum in PBS-T for 2 hours at room temperature. All antibodies were diluted serially in 2-fold steps starting from 25 μg/ml, for 24 dilutions. Horseradish peroxidase–conjugated goat anti‑human IgG (catalog 2040-05; Southern Biotechnology Associates) was added to each well, allowed to incubate for 1 hour at 37°C, and color developed with 3,3′,5,5′-tetramethylbenzidine (Thermo). The reaction was stopped with 1N HCl and read at 450 nM. The concentration and absorbance were fit with a nonlinear robust fit using Prism default parameters to generate an EC50. The EC50 of each PG9 variant was compared with that of the WT PG9 positive control.
For BG505 SOSIP.664 trimer, ELISAs were performed according the protocol as previously described (18). Maxisorp 96-well plates (Nunc) were coated overnight with Ab D7324 (product code D7324; Aalto Bio Reagents) at 5 μg/ml in 0.1 M NaHCO3, pH 8.6 (100 μl/well). After the washing and blocking steps, purified D7324-tagged BG505 Env proteins were added at 800 ng/ml in PBS and 2% milk for 2 hours at ambient temperature, and the unbound Env proteins were washed away. WT PG9 and PG9 variants were diluted to 25 μg/ml in PBS with 10% sheep serum/2% milk, diluted serially 2-fold, and then allowed to incubate for 2 hours at room temperature followed by 3 washes with PBS-T. Horseradish peroxidase–conjugated goat anti-human IgG was added for 1 hour at a 1:3,000 dilution (final concentration 0.33 μg/ml) in 10% sheep serum/2% milk, followed by 5 washes with PBS-T. Color development, optical density measurement, and EC50 calculations were done as above.
Neutralization assays.
Neutralization was measured as a function of reduction in luciferase (Luc) reporter gene expression after a single round of infection in TZM-bl cells, as described (19, 41). This assay has been optimized and validated (42) and was performed in compliance with Good Clinical Laboratory Practices. TZM-bl cells were obtained from the NIH AIDS Research and Reference Reagent Program, as contributed by John Kappes and Xiaoyun Wu. Briefly, virus at a dose of 50,000–150,000 relative luminescence unit (RLU) equivalents was incubated with serial 3-fold dilutions of test sample in duplicate in a total volume of 150 μl for 1 hour at 37°C in 96-well flat-bottom culture plates. Freshly trypsinized cells (10,000 cells in 100 μl of growth medium containing 75 μg/ml DEAE dextran) were added to each well. One set of control wells received a combination of the cells and the virus (virus control) and another set received cells only (background control). After a 48-hour incubation, 100 μl of cells was transferred to a 96-well black solid plate (Corning Inc.) for measurements of luminescence using the Britelite Luminescence Reporter Gene Assay System (PerkinElmer). Neutralization titers are the dilution at which RLUs were reduced by 50%, compared with virus control wells, after subtraction of background RLUs. Assay stocks of molecularly cloned Env-pseudotyped viruses were prepared by transfection in 293T cells and were titrated in TZM-bl cells as described (19). For all neutralization assays, the starting concentration for PG9 and PG9 mutants was 33 μg/ml. However, for viruses BG505ΔCT/T322N, YU2, HxBC2P3.2, CAP45.2.00.G33, and BaL.26, the starting concentration for WT PG9 was 3.3 μg/ml. For RHPA.LucR.T2A.ecto, the PG9 starting concentration was 10 μg/ml. Additional details of the assay and all supporting protocols may be found at (http://www.hiv.lanl.gov/content/nab-reference-strains/html/home.htm).
Crystallization, data collection, structure determination, and refinement.
Crystallization trials with the PG9_N100FY Fab fragment were set up with the IAVI/Joint Center for Structural Genomics (JCSG)/TSRI CrystalMation robotic system (Rigaku), at a concentration of 10 mg/ml. A crystal was obtained in a condition containing 5% PEG 3000, 40% PEG 400, and 0.1M MES. Data were collected at SSRL beamline 11-1. Data were processed using XDS (43). The structure was solved using PHASER (44) with WT PG9 (PDB ID: 3U4E; ref. 2) as the initial search model. COOT (15) and PHENIX (45) were used for refinement, and the statistics are reported in Supplemental Table 1. Pymol (Version 1.5.0.4; Schrödinger LLC) and UCSF Chimera (46) were used for the rendering.
DSC.
The samples were loaded in a MicroCal VP-DSC (GE Healthcare) sample cell against the reference cell containing PBS. The samples were heated at a constant heating rate of 1°C/min. Experiments using mAbs were performed with protein concentration at 0.5 μM, while experiments using Fabs were performed with protein concentration at 1 μM. Data analysis was done using Origin for DSC package. Deconvolution of the excess heat capacity function was done using 4 “two-state” transitions for mAbs, and 2 “two-state” transitions for Fabs.
Cell staining for autoreactivity.
HEp-2 cells were plated on glass coverslips within 48 well plates 24 hours prior to staining. Cells stained for intracellular autoreactivity were fixed with freshly prepared 4% paraformaldehyde and permeablized with 3% BSA/0.3% Triton-X-100, while cells stained for cell-surface autoreactivity were fixed with 4% paraformaldehyde only. Fixed cell monolayers were incubated with 25 μg/ml of HIV mAb 4E10, which is known to exhibit autoreactive properties; WT PG9; the PG9_N100FY variant; or motavizumab, a control mAb directed to the respiratory syncytial virus fusion protein. Secondary goat antihuman IgG antibodies containing Alexa Fluor 488 (Invitrogen) were used to detect mAb staining of human antigens. TO-PRO3-iodine dye were used for detection of DNA in the cell nuclei, to identify cells. Images were captured at ×40 magnification on a Zeiss LSM 710 META Inverted laser scanning confocal microscope with a ×40/1.10 LD C-APOCHROMAT water objective and analyzed using ZEN Blue software platform (Zeiss).
Statistics and graph generation.
All statistics were calculated in the R-programming language (http://www.r-project.org) or Prism package (GraphPad Software) through the IPython interface (www.ipython.org). All graphs were generated in Prism package or the ggplot2 library (http://ggplot2.org) in the R-programming language. To facilitate comparison of both WT PG9 and variants for all viruses tested, separate Wilcoxon signed-rank tests for paired data were used to test for differences in IC50 or EC50 between WT PG9 and each of the 4 antibody variants. We compared the EC50 or IC50 of all viruses using a 2-sided significance level of 0.05. A P value of less than 0.05 was considered significant.
Supplementary Material
Acknowledgments
The authors thank Albert Cupo and John Moore from the Weill College of Medicine of Cornell for their contribution of gp140 trimer and helping us to establish a trimer-binding protocol. These studies were supported by grants from NIAID/NIH (U01 AI78407; P01 AI082362; UM1 AI100663; and R01 AI084817) and the Bill and Melinda Gates Foundation.
Footnotes
Note regarding evaluation of this manuscript: Manuscripts authored by scientists associated with Duke University, The University of North Carolina at Chapel Hill, Duke-NUS, and the Sanford-Burnham Medical Research Institute are handled not by members of the editorial board but rather by the science editors, who consult with selected external editors and reviewers.
Conflict of interest: Ian A. Wilson has an ongoing collaboration, though not in the HIV field, with the Crucell Vaccine Institute, Leiden, Netherlands.
Reference information:J Clin Invest. 2015;125(6):2523–2531. doi:10.1172/JCI80693.
See the related Commentary beginning on page 2271.
References
- 1.Corti D, Lanzavecchia A. Broadly neutralizing antiviral antibodies. Annu Rev Immunol. 2013;31:705–742. doi: 10.1146/annurev-immunol-032712-095916. [DOI] [PubMed] [Google Scholar]
- 2.McLellan JS, et al. Structure of HIV-1 gp120 V1/V2 domain with broadly neutralizing antibody PG9. Nature. 2011;480(7377):336–343. doi: 10.1038/nature10696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Walker LM, et al. Broad neutralization coverage of HIV by multiple highly potent antibodies. Nature. 2011;477(7365):466–470. doi: 10.1038/nature10373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Pejchal R, et al. Structure and function of broadly reactive antibody PG16 reveal an H3 subdomain that mediates potent neutralization of HIV-1. Proc Natl Acad Sci USA. 2010;107(25):11483–11488. doi: 10.1073/pnas.1004600107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Pancera M, et al. Crystal structure of PG16 and chimeric dissection with somatically related PG9: structure-function analysis of two quaternary-specific antibodies that effectively neutralize HIV-1. J Virol. 2010;84(16):8098–8110. doi: 10.1128/JVI.00966-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Pancera M, et al. Structural basis for diverse N-glycan recognition by HIV-1-neutralizing V1-V2-directed antibody PG16. Nat Struct Mol Biol. 2013;20(7):804–813. doi: 10.1038/nsmb.2600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Fleishman SJ, et al. Computational design of proteins targeting the conserved stem region of influenza hemagglutinin. Science. 2011;332(6031):816–821. doi: 10.1126/science.1202617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Fleishman SJ, et al. RosettaScripts: a scripting language interface to the Rosetta macromolecular modeling suite. PLoS One. 2011;6(6):e20161. doi: 10.1371/journal.pone.0020161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kuhlman B, Baker D. Native protein sequences are close to optimal for their structures. Proc Natl Acad Sci USA. 2000;97(19):10383–10388. doi: 10.1073/pnas.97.19.10383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kaufmann KW, Lemmon GH, Deluca SL, Sheehan JH, Meiler J. Practically useful: what the Rosetta protein modeling suite can do for you. Biochemistry. 2010;49(14):2987–2998. doi: 10.1021/bi902153g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Kabat EA. Sequences of Proteins of Immunological Interest. Bethesda, Maryland, USA: US Department of Health and Human Services, Public Health Service, National Institutes of Health; 1991. [Google Scholar]
- 12.Walker LM, et al. Broad and potent neutralizing antibodies from an African donor reveal a new HIV-1 vaccine target. Science. 2009;326(5950):285–289. doi: 10.1126/science.1178746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Li M, et al. Human immunodeficiency virus type 1 env clones from acute and early subtype B infections for standardized assessments of vaccine-elicited neutralizing antibodies. J Virol. 2005;79(16):10108–10125. doi: 10.1128/JVI.79.16.10108-10125.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Li M, et al. Genetic and neutralization properties of subtype C human immunodeficiency virus type 1 molecular env clones from acute and early heterosexually acquired infections in Southern Africa. J Virol. 2006;80(23):11776–11790. doi: 10.1128/JVI.01730-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Emsley P, Cowtan K. Coot: model-building tools for molecular graphics. Acta Crystallogr D Biol Crystallogr. 2004;60(pt 12 pt 1):2126–2132. doi: 10.1107/S0907444904019158. [DOI] [PubMed] [Google Scholar]
- 16.Julien JP, et al. Asymmetric recognition of the HIV-1 trimer by broadly neutralizing antibody PG9. Proc Natl Acad Sci USA. 2013;110(11):4351–4356. doi: 10.1073/pnas.1217537110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Sanders RW, et al. Stabilization of the soluble, cleaved, trimeric form of the envelope glycoprotein complex of human immunodeficiency virus type 1. J Virol. 2002;76(17):8875–8889. doi: 10.1128/JVI.76.17.8875-8889.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Sanders RW, et al. A next-generation cleaved, soluble HIV-1 Env Trimer, BG505 SOSIP.664 gp140, expresses multiple epitopes for broadly neutralizing but not non-neutralizing antibodies. PLoS Pathog. 2013;9(9):e1003618. doi: 10.1371/journal.ppat.1003618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Montefiori DC. Measuring HIV neutralization in a luciferase reporter gene assay. Methods Mol Biol. 2009;485:395–405. doi: 10.1007/978-1-59745-170-3_26. [DOI] [PubMed] [Google Scholar]
- 20.Doores KJ, Doores KJ, Burton DR, Burton DR. Variable loop glycan dependency of the broad and potent HIV-1-neutralizing antibodies PG9 and PG16. J Virol. 2010;84(20):10510–10521. doi: 10.1128/JVI.00552-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Garber E, Demarest SJ. A broad range of Fab stabilities within a host of therapeutic IgGs. Biochem Biophys Res Commun. 2007;355(3):751–757. doi: 10.1016/j.bbrc.2007.02.042. [DOI] [PubMed] [Google Scholar]
- 22.Ionescu RM, Vlasak J, Price C, Kirchmeier M. Contribution of variable domains to the stability of humanized IgG1 monoclonal antibodies. J Pharm Sci. 2008;97(4):1414–1426. doi: 10.1002/jps.21104. [DOI] [PubMed] [Google Scholar]
- 23.Makhatadze GI, Privalov PL. Energetics of protein structure. Adv Protein Chem. 1995;47:307–425. doi: 10.1016/S0065-3233(08)60548-3. [DOI] [PubMed] [Google Scholar]
- 24.Privalov PL, Khechinashvili NN. A thermodynamic approach to the problem of stabilization of globular protein structure: a calorimetric study. J Mol Biol. 1974;86(3):665–684. doi: 10.1016/0022-2836(74)90188-0. [DOI] [PubMed] [Google Scholar]
- 25.Yurasov S. Defective B cell tolerance checkpoints in systemic lupus erythematosus. J Exp Med. 2005;201(5):703–711. doi: 10.1084/jem.20042251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Briney BS, Willis JR, Crowe JE. Human peripheral blood antibodies with long HCDR3s are established primarily at original recombination using a limited subset of germline genes. PLoS One. 2012;7(5):e36750. doi: 10.1371/journal.pone.0036750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Haynes BF, et al. Cardiolipin polyspecific autoreactivity in two broadly neutralizing HIV-1 antibodies. Science. 2005;308(5730):1906–1908. doi: 10.1126/science.1111781. [DOI] [PubMed] [Google Scholar]
- 28.Ramilo O, et al. Motavizumab treatment of infants hospitalized with respiratory syncytial virus infection does not decrease viral load or severity of illness. Pediatr Infect Dis J. 2014;33(7):703–709. doi: 10.1097/INF.0000000000000240. [DOI] [PubMed] [Google Scholar]
- 29.Klein F, et al. Somatic mutations of the immunoglobulin framework are generally required for broad and potent HIV-1 neutralization. Cell. 2013;153(1):126–138. doi: 10.1016/j.cell.2013.03.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Willis JR, Briney BS, Deluca SL, Crowe JE, Meiler J. Human germline antibody gene segments encode polyspecific antibodies. PLoS Comput Biol. 2013;9(4):e1003045. doi: 10.1371/journal.pcbi.1003045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Schmidt AG, et al. Preconfiguration of the antigen-binding site during affinity maturation of a broadly neutralizing influenza virus antibody. Proc Natl Acad Sci USA. 2013;110(1):264–269. doi: 10.1073/pnas.1218256109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Wong SE, Sellers BD, Jacobson MP. Effects of somatic mutations on CDR loop flexibility during affinity maturation. Proteins. 2011;79(3):821–829. doi: 10.1002/prot.22920. [DOI] [PubMed] [Google Scholar]
- 33.Schultz PG, Yin J, Lerner RA. The chemistry of the antibody molecule. Angew Chem Int Ed Engl. 2002;41(23):4427–4437. doi: 10.1002/1521-3773(20021202)41:23<4427::AID-ANIE4427>3.0.CO;2-K. [DOI] [PubMed] [Google Scholar]
- 34.Wedemayer GJ, Patten PA, Wang LH, Schultz PG, Stevens RC. Structural insights into the evolution of an antibody combining site. Science. 1997;276(5319):1665–1669. doi: 10.1126/science.276.5319.1665. [DOI] [PubMed] [Google Scholar]
- 35.Sok D, et al. Recombinant HIV envelope trimer selects for quaternary-dependent antibodies targeting the trimer apex. Proc Natl Acad Sci USA. 2014;111(49):17624–17629. doi: 10.1073/pnas.1415789111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Correia BE, et al. Proof of principle for epitope-focused vaccine design. Nature. 2014;507(7491):201–206. doi: 10.1038/nature12966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Jardine J, et al. Rational HIV immunogen design to target specific germline B cell receptors. Science. 2013;340(6133):711–716. doi: 10.1126/science.1234150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.McLellan JS, et al. Structure-based design of a fusion glycoprotein vaccine for respiratory syncytial virus. Science. 2013;342(6158):592–598. doi: 10.1126/science.1243283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Lemmon G, Kaufmann K, Meiler J. Prediction of HIV-1 protease/inhibitor affinity using RosettaLigand. Chem Biol Drug Des. 2012;79(6):888–896. doi: 10.1111/j.1747-0285.2012.01356.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Xu R, Ekiert DC, Krause JC, Hai R, Crowe JE, Jr, Wilson IA. Structural basis of preexisting immunity to the 2009 H1N1 pandemic influenza virus. Science. 2010;328(5976):357–360. doi: 10.1126/science.1186430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Montefiori DC. Evaluating neutralizing antibodies against HIV, SIV, and SHIV in luciferase reporter gene assays. Curr Protoc Immunol. 2005;Chapter 12:Unit 12.11. doi: 10.1002/0471142735.im1211s64. [DOI] [PubMed] [Google Scholar]
- 42.Sarzotti-Kelsoe M, et al. Optimization and validation of the TZM-bl assay for standardized assessments of neutralizing antibodies against HIV-1. J Immunol Methods. 2014;409:131–146. doi: 10.1016/j.jim.2013.11.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kabsch W. Integration, scaling, space-group assignment and post-refinement. Acta Crystallogr D Biol Crystallogr. 2010;66(pt 2):125–132. doi: 10.1107/S0907444909047374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.McCoy AJ, Grosse-Kunstleve RW, Adams PD, Winn MD, Storoni LC, Read RJ. Phaser crystallographic software. J Appl Crystallogr. 2007;40(pt 4):658–674. doi: 10.1107/S0021889807021206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Adams PD, et al. PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr D Biol Crystallogr. 2010;66(pt 2):213–221. doi: 10.1107/S0907444909052925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Pettersen EF, et al. UCSF Chimera — a visualization system for exploratory research and analysis. J Comput Chem. 2004;25(13):1605–1612. doi: 10.1002/jcc.20084. [DOI] [PubMed] [Google Scholar]
- 47.Lefranc MP. IMGT-ONTOLOGY and IMGT databases, tools and Web resources for immunogenetics and immunoinformatics. Mol Immunol. 2004;40(10):647–660. doi: 10.1016/j.molimm.2003.09.006. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.